
Submitted:
19 December 2020
Posted:
21 December 2020
You are already at the latest version
Abstract
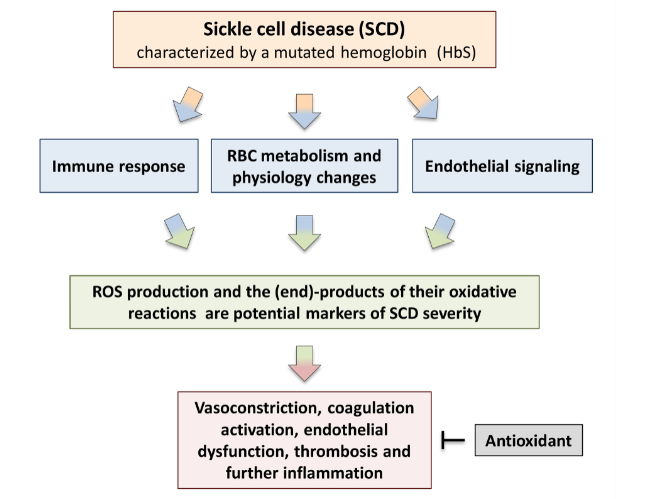
Sickle cell disease (SCD) is the most common hereditary disorder of hemoglobin (Hb) that affects approximately a millions people worldwide. It is characterized by a single nucleotide substitution on the β-globin gene, leading to the production of abnormal sickle hemoglobin with multi-system consequences. Mutated Hb leads to profound changes in: i) red blood cell metabolism and physiology; ii) endothelial signaling; and iii) immune response. Oxidative stress is an important hallmark of SCD. It plays a key role in the pathophysiology of hemolysis, vessel occlusion and the following organ damage in sickle cell patients. For this reason, reactive oxidizing species and the (end)-products of their oxidative reactions have been proposed as markers of both tissue pro-oxidant status and disease severity. Although more studies are needed to clarify their role, antioxidant agents have been shown to be effective in reducing pathological consequences of the disease by preventing oxidative damage in SCD, i.e. by decreasing the oxidant formation or repairing the induced damage. An improved understanding of oxidative stress will lead to targeted antioxidant therapies that should prevent or delay the development of organ complications in this patient population.
Keywords:
sickle cell disease
; hemoglobin
; oxidative stress
; antioxidants
; red blood cells
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



