
Submitted:
12 June 2023
Posted:
12 June 2023
You are already at the latest version
Preprints.org 2023 Most Popular Preprints Award Winner Collection
Abstract
Skin melanoma presents increasing prevalence and poor outcomes. Progression to aggressive stages is characterized by overexpression of the transcription factor E2F1 and activation of downstream prometastatic gene regulatory networks (GRNs). Appropriate therapeutic manipulation of the E2F1-governed GRNs holds the potential to prevent metastasis however, these networks entail complex feedback and feedforward regulatory motifs among various regulatory layers, which make it difficult to identify druggable components. To this end, computational approaches such as mathematical modeling and virtual screening are important tools to unveil the dynamics of these signaling networks and identify critical components that could be further explored as therapeutic targets. Herein, we integrated a well-established E2F1-mediated epithelial-mesenchymal transition (EMT) map with transcriptomics data from E2F1-expressing melanoma cells to reconstruct a core regulatory network underlying aggressive melanoma. Using logic-based in silico perturbation experiments of a core regulatory network, we identified that simultaneous perturbation of Protein kinase B (AKT1) and oncoprotein murine double minute 2 (MDM2) drastically reduces EMT in melanoma. Using the structures of the two protein signatures, virtual screening strategies were performed with the FDA-approved drug library. Thus, by combining drug repurposing and computer-aided drug design techniques, followed by molecular dynamics simulation analysis identified two potent drugs (Cialis and Finasteride) that can efficiently inhibit AKT1 and MDM2 protein signatures respectively, and with better therapeutic properties. We proposed that these two drugs could be considered for the development of therapeutic strategies for the management of aggressive melanoma.
Keywords:
Melanoma
; Core regulatory network
; in silico perturbationSystems pharmacology
; Boolean model
; Small molecule inhibitors
; Virtual screening
; ADME
; E2F1
1. Introduction
Cutaneous melanoma arises from melanocytes and represents the deadliest form of skin cancer, with increasing prevalence. Once it becomes metastatic, the prognosis is very unfavourable. Melanoma formation is driven by mutations in the BRAF and NRAS oncogenes [1]. However, these oncogenic aberrations are early events in melanoma genesis that alone do not seem to be sufficient to drive metastasis[2]. Over the past years, we [3,4,5] and others [6,7] have demonstrated that in addition to these driver events, melanoma progression is catalyzed by the abundant expression of E2F1, a member of the E2F transcription factor family. Although this transcription factor activates tumor-suppressive pathways at early oncogenesis, upon disease progression unbalanced E2F1 activity is rewired to deregulated cancer networks that underlie hallmarks of metastatic progression such as resistance to apoptosis, chemoresistance [3,8], neoangiogenesis [9], extravasation [7], EMT [10,11], metabolic reprogramming [12], and genomic instability [13]. By integrating logic-based network modeling and gene expression profiles of cancer cell lines from E2F1-driven tumors and patient cohorts displaying cancer aggressiveness, we identified tumor-type specific receptor signatures associated with EMT, where the combined action of highly expressed E2F1, TGFBR1, and FGFR1 triggers the most invasive phenotype [11,14]. Several other protein-coding genes, miRNA genes, and lncRNA genes have been identified as constituents of E2F1-activated prometastatic GRNs [11,12,13,15,16]. Within the E2F1-governed GRNs, non-linear feedback and feedforward regulatory motifs are formed among various regulatory network layers, entailing protein-coding and non-coding RNA genes [10,15,16]. Such regulatory motifs, which are commonly encountered in cancer networks [17,18], induce a whole range of dynamic behaviors, thereby evading the use of conventional data analysis methods [19]. As a result, the prediction of potential therapeutic targets within this network requires the aid of systems biology-based and computational methods.
Uncovering major epigenetic features and the immune contexture of melanoma has catalyzed the development of anti-melanoma therapies within less than two decades. In 2004, no systemic therapies for melanoma had been shown to provide a survival benefit. Now, at least four regimens of targeted therapy and three for immunotherapy improve overall survival and disease-free survival, with each modality presenting distinct benefits and limitations. Particularly in 2011, vemurafenib became the first BRAF-targeted therapy approved by the Food and Drug Administration (FDA) for the treatment of melanoma [20]. Unfortunately, responses to BRAF inhibitor monotherapy, although impressive and rapid, were usually transient. In most cases, this was due to the development of resistance via reactivation of the mitogen-activated protein kinase (MAPK) pathway. Combined BRAF and MEK inhibition addresses this MAPK-mediated mechanism of resistance and constitutes the current standard-of-care for targeted melanoma therapy. Regimens of BRAF plus MEK inhibitors achieve long-lasting disease control and are better tolerated than BRAF inhibitor monotherapy, but a major concern is that resistance, although delayed, is eventually developed [21,22]. Likewise, the treatment response of patients with mutant NRAS-positive metastatic melanoma to MEK inhibitors is transient and short-lived [23]. In the context of the paradigm-changing advances in cancer immunotherapy, several next-generation immune-based formulations, such as the checkpoint inhibitors ipilimumab, pembrolizumab, and nivolumab, have received FDA approval for the indication of metastatic melanoma and ensure durable responses. However, they are linked with immune-related toxicities and pose limitations for use in patients with either an overactive (autoimmune disease patients) or a suppressed (organ transplant recipients) immune system [24]. In summary, targeted therapy shows a rapid response time and generally less toxic off-target effects however, resistance can be developed. While immunotherapy offers increased durability of benefit in all patients irrespective of tumor genotype, this is also frequently associated with immune-related side-effects, especially when combinations of immunotherapeutic drugs are used [21,22]. It is therefore essential to develop both, effective and safe strategies, that specifically interfere with the complex melanoma networks. This should enrich the anti-melanoma drug arsenal with more personalized therapeutic options. The past decade’s clinical experience taught us that, in general, combination therapies may be superior in terms of efficacy and/or safety than monotherapies. Combining anticancer drugs is currently seen as the approach most likely to overcome single-agent resistance, to produce sustained clinical remissions via multi-targeting effects on distinct mechanisms of action, and to reduce unwanted side-effects by usage of lower drug doses [25,26]. In fact, the need for combinatory therapies is an inevitable consequence of the evolving nature of tumors. Clonal evolution is particularly active when tumors are under selective pressures due to medical treatments, thereby promoting resistance to therapy. Resistant cell clones often pre-exist (although undetectable) at the start of treatment, supporting the idea that early administration of combinatorial treatments stands a higher chance of eradicating such clones when their number is very low before acquired resistance is overtly diagnosed. Simultaneous targeting of the driver oncogenic mutations along with the expected secondary resistance may provide a significant advantage in survival compared with administration at relapse. However, ab initio combination therapies are challenging in the clinical oncology setting because of the narrow therapeutic window between tumor cells and host, which overall limits the number of agents that can be simultaneously tested [26,27]. With recent advances in high-throughput screening methods, a systematic evaluation of combinations among large collections of chemical compounds in vitro has become feasible. This typically requires large-scale experiments, in which the combinatorial responses are tested in various doses on cancer cell lines or patient-derived cells, resulting in dose-response matrices that capture the measured combination effects for every concentration pair in a particular sample [25,26]. For example, systematic screening of pairwise combinations of 104 FDA-approved oncology drugs has been performed in the NCI-60 panel of human tumor cell lines to produce a comprehensive platform for defining the pairs with enhanced therapeutic efficacy [25]. However, even with modern high throughput instruments, experimental screening of drug combinations can become a herculean task, as the number of conceivable drug combinations increases rapidly with the number of drugs under consideration. In addition, the inherent heterogeneity of cancer cells further challenges the experimental efforts, as the combinations need to be tested in various cell contexts and genomic backgrounds. Hence, computational methods are often recruited to guide the discovery of effective combinations that can be prioritized for further pre-clinical and clinical validations [26,28].
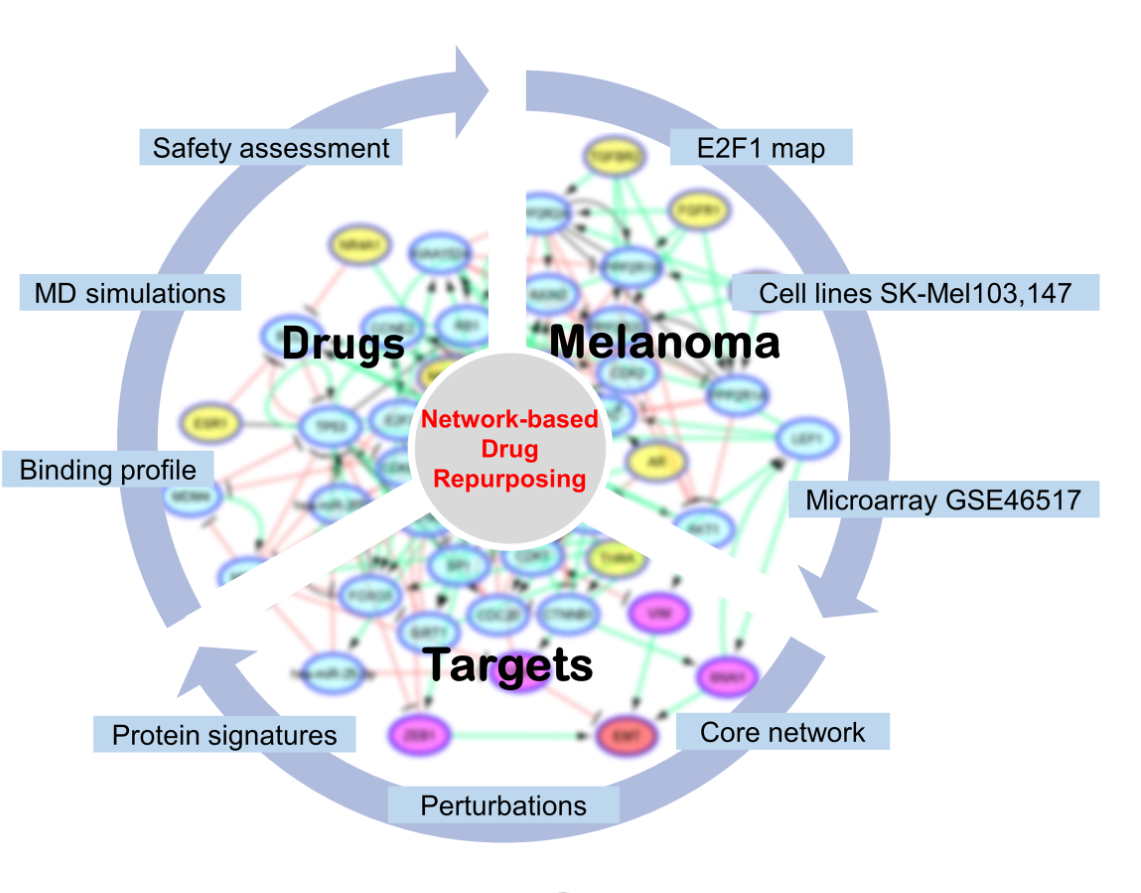
Herein, aided by in silico workflows, we sought to predict efficient and safe compounds that either alone or in combination prevent melanoma progression by specifically targeting components of the prometastatic E2F1-governed GRNs in melanoma. Using a comprehensive regulatory and functional map of E2F1 in tumor progression and metastasis [11] which contains different types of regulatory factors, including genes, proteins, microRNAs, or complexes, we identified a core regulatory network in melanoma [28,29]. The core regulatory network was subjected to logic-based modeling for detecting protein signatures which play an important role in interconnecting many of the responsive genes that are typically not identified through gene-based differential expression analysis. The subsequent virtual screening which is a major contributor to computer-aided drug design (CADD) and drug repurposing concept, are increasingly popular techniques that improve the speed and efficiency of the drug discovery process [30,31], was applied to find FDA-approved drugs against protein signatures. This combined approach allows us to take advantage of existing safety profiles and established pharmacokinetic properties of approved drugs [32]. The selected protein-drug complexes were subjected to molecular dynamics simulation studies for investigating the stability of the complex formed. Our approach supports the role of AKT1 and MDM2 protein signatures as drivers of EMT in melanoma cancer and suggests that MDM2 plus AKT1 inhibitors could be a promising combination that is worthwhile to be further investigated as a novel anti-metastatic regimen in high E2F1-expressing melanoma patients.
2. Results
2.1. Establishment of a computational pipeline for the prediction of drug-targetable components of the E2F1-governed prometastatic GRN in melanoma and in silico screening of different inhibitors, alone or in combination
Previously, we have designed a comprehensive regulatory and functional map of E2F1 in tumor progression and metastasis [11] which contains different types of regulatory factors (n= 879) including genes, proteins, microRNAs, or complexes; and interactions (m=2278) based on information retrieved from published literature and databases. The map was modularized into three E2F1 regulatory compartments such as extra-/intracellular receptor signaling, post-translational modifications, regulators of E2F1 activity; and seven functional compartments including cell cycle, quiescence, DNA repair, metabolism, apoptosis, survival, and angiogenesis/invasion. Using a computational pipeline, we used the map to unravel a tumor type-specific regulatory core and to predict receptor protein signatures in bladder and breast cancer underlying E2F1-mediated EMT transition. The E2F1 map and the previously used workflow were applied to identify a key functional module (core regulatory network) in melanoma. This core regulatory network is composed of regulatory motifs and critical molecular interactions that drive phenotype switching in melanoma. The core regulatory network for melanoma was subjected to our computational pipeline to detect protein signatures that play an important role in interconnecting many of the responsive genes that are typically not identified through gene-based differential expression analysis.
Our workflow includes (i) network-based analysis of topological parameters to characterize the pattern of factors in a networked system, (ii) mapping of the gene expression profiles from melanoma cell lines onto the E2F1 map, (iii) network reduction via a multi-objective function [34] to provide motif ranking by user-defined weights in an iterative manner, (iv) boolean modeling to analyze and predict the protein signatures linked to aggressiveness in melanoma, (v) structure-based virtual screening and molecular dynamics simulation (MDS) studies to find repurposed drugs against protein signatures that elicit measurable biological responses, and (vi) to predict ADMET behaviors and pharmacokinetic parameters of candidate drugs. The workflow in Figure 1 was used for prioritizing therapeutic targets and also to screen potential candidate drugs that may be validated for advanced melanoma tumors.
2.2. Identification of the metastatic melanoma-specific core regulatory network
We used our previously published network-based approach to construct a melanoma-specific regulatory core from the comprehensive map of E2F1 [11]. Here, we utilized the workflow and the E2F1 map to identify key network motifs and critical molecular interactions that drive a highly invasive melanoma cell phenotype. To do this, we have used the data extracted from the E2F1 map and identified important network motifs by calculating of topological and non-topological properties of each node (Figure 1A) (additional file 1a). The motifs were prioritized based on a multi-objective optimization function where the function contains parameters accounting for each property. Weights are assigned to each property in terms of their importance in a user-defined manner and then, the motifs are ranked according to the value of the objective function. The top ten high-scored motifs were selected from each weighting scenario (additional file 1b-c). Finally, we merged all the top-ranked motifs to obtain a melanoma-specific regulatory core. We expanded the regulatory core by adding receptor proteins which are the first neighbors of ranked motif nodes in the E2F1 map. We also added four well-known markers CDH1, VIM, ZEB1, and SNAI1 [10] in the core network (additional file 1d) to measure the EMT response (Figure 1B).
2.3. Boolean modeling of the melanoma-specific core regulatory network
We encoded the core-regulatory network into a boolean model for stimulus-response and perturbation analyses. Stimulus-response analysis was used to identify the effect of up/down expressed receptors on the EMT phenotype, and perturbation analysis predicted a potential drug target that can bring the phenotype to the lower possible level. In the boolean model, the state of a node is represented with two possible conditions i.e., 0 (OFF, inactive) or 1 (ON, active) [33]. The regulatory relationships between upstream nodes (i.e., sources) to downstream nodes (i.e., targets) are encoded into boolean functions using logical gates ‘NOT’, ‘OR’, and ‘AND’. Further, we calibrated the boolean functions with fold-change (FC) expression data [33] of the publicly available dataset GSE46517 [34] from Gene Expression Omnibus (GEO).
To evaluate the input-output behavior, we divided the model into three layers: 1) the input layer, containing receptor molecules, 2) the regulatory layer, comprising nodes constituting a core-regulatory network, and 3) the output layer, including EMT phenotype (Figure 1B). The input layer was initialized with an FC expression profile i.e., a node with negative FC was represented by a state 0, and a node with positive FC was represented by a state 1.
2.4. In silico perturbation simulations using boolean modeling
The model was simulated with initial values derived from the expression profile of input nodes and confirmed that the logical state of nodes in a regulatory layer represents the data (for details see additional file 2a-e). The EMT phenotype was regulated by nodes ZEB1, CDH1, VIM, and SNAI1 [10], and represented by 5 ordinal levels ranging from 0 (minimum) to 4 (maximum):
EMT = ZEB1 + NOT (CDH1) + VIM + SNAI1
For the initial condition, model simulations result in EMT of level 3, where ZEB1 and SNAI1 are active and CDH1 is inactive (see Table 1a). Further, we performed perturbation analysis of all nodes (except ZEB1, CDH1, VIM, and SNAI1) in the regulatory layer of the model to bring EMT from level 3 to a minimum level. We identified that for a single perturbation (in this case inhibition) of MDM2 or MIR25, EMT can be reduced to level 1 (Table 1). CDH1 is activated upon inhibition of MDM2 which inhibits EMT as well as inhibits CTNNB1 which subsequently inhibits SNAI1 [35,36] to further reduce EMT. A similar effect was observed upon inhibition of MIR25 [37,38]. On the other hand, a single perturbation (in this case activation) of AKT1 can increase the EMT to the highest level 4.
2.5. Assessment of protein signatures identified through boolean modeling
Our boolean model simulations suggested two key proteins AKT1 and MDM2 that upon inhibition can bring the EMT from level 3 to 1. Interestingly, AKT1 directly activates the VIM, a key marker for EMT. AKT1 also activates MDM2 which interacts with p53 to regulate the immune axis in metastatic melanoma. MDM2 also indirectly activates the EMT by downregulating another hallmark protein CDH1. We investigated the expression profiles of AKT1 and MDM2 and their impact on melanoma patient survival using the Kaplan-Meier curve (Figure 2) using the TCGA melanoma SKCM dataset. We found that higher expression of both AKT1 and MDM2 resulted in poor patient survival. These observations also confirm that the boolean model simulation was successful in predicting potential proteins that may be targeted for the treatment of metastatic melanoma.
2.6. Screening of FDA-approved drugs to block protein signatures
To identify drugs that are most likely to bind to AKT1 and MDM2 protein signatures, molecular docking was performed with the FDA-approved drug library from the ZINC database (additional file 3a-c). The information about the active sites of proteins is retrieved from the literature and PDB database. More specifically, for AKT1 we performed screening against the kinase domain (150-408) which is previously selected to identify ATP-competitive inhibitors [39,40] (Figure 1C). For MDM2, many recent studies indicate that its overexpression and subsequent deactivation of p53 result in failure of apoptosis and cancer cell survival [41,42,43]. We investigated the p53–Mdm2 interaction surface which is ~700 Å2. This druggable pocket of MDM2 where p53 binds provides a great opportunity for compound inhibitors to disrupt p53–MDM2 interaction [44] (Figure 1C).
From the achieved docked poses for each ligand-protein complex, we selected the pose with the lowest energy value (out of 9 conformations) and compared it with the crystallographic pose, RMSD ≤ 2.0 Å. Further, the best predicted binding mode and the corresponding binding affinity (in kcal/mol) are selected for each complex. In case of AKT1 Figure 3A (i-v), docking analyses revealed that the candidate drugs are packed against the residues LEU156, GLY157, PHE161, VAL164, ALA177, LYS179, GLU191, HIS194, GLU198, TYR229, ALA230, GLU234, ASP274, ASN279, MET281, ASP292, GLY294, LEU295, TYR437, PHE438, ASP439, and PHE442 and was stabilized by the hydrogen bonds, electrostatic, hydrophobic, and van der walls interactions. The best binding affinity of AKT1 is obtained with Cialis (-11.1 kcal/mol), followed by Paliperidone (-10.8 kcal/mol), Cobimetinib (-10.6 kcal/mol), Troglitazone (-10.5 kcal/mol), and Sertindole (-10.3 kcal/mol). The binding affinities and the number of interactions of these candidate drugs towards the ATP binding pocket of AKT1 are comparable to ATP competitive inhibitors [40]. Particularly, GLU234 of the protein backbone is necessary for the AKT1 biological activity. Secondly, the electrostatic interactions and hydrogen bonds to ASP292 in AKT1 are critical because this position is typically occupied by a divalent cation (Mg2+) bound to ATP [45]. Both of the residues (GLU234 and ASP292) of AKT1 were found to bind with previously known ATP-competitive kinase inhibitors [45,46] and also found with our candidate drugs. Other active site residues could be seen in binding were ALA230, GLU228, GLU278, ASN279, PHE161, and LYS179. Cialis/Tadalafil is a US FDA-approved drug for the treatment of pulmonary arterial hypertension [47]. Also, the drug shows biologic activity in human melanoma and pilot trial studies reported improved clinical outcomes in patients with head and neck cancer (HNSCC) and metastatic melanoma [48,49]. The next drug Paliperidone is an antipsychotic drug that reportedly inhibits glioblastoma growth in mice [50]. However, existing evidence is conflicting with its role as the drug increases prolactin levels, which might increase the risk of breast cancer [51]. Further in the list, Cobimetinib/GDC-0973 is FDA-approved for the treatment of patients with metastatic melanoma with a BRAF V600E or V600K mutation, in combination with vemurafenib [52]. Also, in combination with (i) chemotherapy and (ii) Niraparib, with or without atezolizumab are used as a treatment for patients with breast cancer and with advanced platinum-sensitive ovarian cancer (red) respectively [53]. The next drug hit is Troglitazone, which is a type II diabetic drug and, in clinical trials used in combination with lovastatin and their cotreatment was found to induce cell cycle arrest at the G0/G1 phase in Anaplastic thyroid cancer [54]. On the contrary, there have been concerns as it has detrimental side effects such as causing liver toxicity [55]. The last drug hit was Sertindole which exhibits antiproliferative activities in breast cancer with a potential application for the treatment of breast-to-brain metastases [56] and by inhibiting the STAT3 signaling pathway in human gastric cancer cells [57].
In the case of MDM2 Figure 3B (i-v), the molecular docking was performed into lining residues of this pocket containing amino acids (LEU54, LEU57, ILE61, MET62, TYR67, GLN72, VAL75, PHE86, PHE91, VAL93, HIS96, ILE99, TYE100, and ILE101). These residues form a hydrophobic cavity on the MDM2 protein structure and are potentially occupied by known inhibitors [58,59]. The best binding affinity of MDM2 is obtained with Finasteride (-10.7 kcal/mol), followed by Cobimetinib (-10.6 kcal/mol), Troglitazone (-10.4 kcal/mol), Loratadine (-9.8 kcal/mol), and Drospirenone (-9.7 kcal/mol). All top five candidates exhibit convincing binding mode into the druggable pocket of MDM2, and specifically hydrophobic interactions with key residues (VAL93 and LEU54) of MDM2 are obtained in all drug hits. Other important interface residues could be seen in binding were LEU57, ILE61, MET62, TYR67, GLN72, VAL75, PHE86, PHE91, HIS96, ILE99, TYE100, and ILE101. The binding affinity of Finasteride with MDM2 is in a similar range with Cobimetinib and Troglitazone; however, binding affinities of Loratadine and Drospirenone are comparatively lower. Finasteride/ Proscar is used for the treatment of alopecia and prostate cancer. In melanoma, the protective effect of finasteride on melanogenesis via downregulation of tyrosinase, TRP-1, MITF, and ACs expression has been demonstrated [60], further in vivo animal experiments are required for confirmation. Another study similar to this reported that Finasteride declines the risk of melanoma following prostate cancer [61]. The effects of Cobimetinib and Troglitazone were described above. The next drug hit is Loratadine which is routinely given to cancer patients in combination with other drugs and substantially improved survival in both breast cancer and cutaneous malignant melanoma [62]. In some instances, Loratadine has been linked to clinically apparent acute liver injury [63]. The use of Drospirenone in birth control pills and causing severe blood clots (thromboembolism) in women is conflicting [64]. Additionally, long-term use may increase the risk of breast cancer [65].
2.7. ADMET profile of top candidate repurposable drugs
The bioavailability radar plots in Figure 4(A) show that the hits are falling entirely within the physicochemical range on each axis and hence seem to fit into the bioavailability criteria. The pharmacokinetic profile in Figure 4(B) shows that all the top candidate drugs have high gastrointestinal (GI) adsorption. Except for Paliperidone and Troglitazone, all the top-hit drugs cross the blood-brain barrier (BBB) and can impair tumor development in brain metastasis. Three drug hits Cialis, Paliperidone, and Finasteride showed no violations for Lipinski, Ghose, Veber, Egan, and Muegge rules which suggests that these drugs are likely orally active drugs. Also, no alerts for PAINS and Brenk filters provide information that Cialis, Paliperidone, and Finasteride drugs didn’t contain potentially problematic fragments which are toxic or metabolically unstable. It is also essential to predict the interaction of drug hits with cytochromes P450 (CYP) and P-glycoprotein (P-gp) as these are key players in drug elimination through metabolic biotransformation [66]. Cialis, Paliperidone, and Finasteride all three are substrates of P-gp; and particularly, Cialis and Finasteride are not expected to cause unwanted adverse effects due to the lower clearance or accumulation of the drug metabolized by CYP450 isoforms (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4).
Overall, the results of molecular docking and ADMET profile suggested two potential strong-binding drug candidates (Cialis and Finasteride) of protein signatures (AKT1 and MDM2, respectively), shown to be orally bioavailable, non-toxic, and have good absorption and medicinal properties.
2.8. Molecular dynamics simulation (MDS) and docking validation
Since docking gives a static view of the binding interaction of compound hits into the active site of protein signatures, MDS gives a more clear idea about the physical movements of atoms and molecules with time by integration of Newton’s equation of motion [67]. Therefore, the two docked complexes (AKT1_ Cialis and MDM2_Finasteride), with the highest predictive binding affinity of -11.1 and -10.7 kcal/mol, respectively, along with safe therapeutic properties, were used for molecular dynamics study.
A 100 ns molecular dynamic simulation was performed to understand the molecular insights involved in the binding of Cialis in the active pocket of AKT1. The trajectory analysis (Figure 5A, (i) PL-RMSD) graph shows that the Ca atoms fluctuated in the range of 2.00–3.50 Å and stabilized after 80 ns of simulation with an RMSD value of 3.0 Å. Higher fluctuation (up to 3.50 Å) in RMSD was observed at 88 ns for the protein. The AKT1 backbone Cα experiences a gradual increase in deviation for the entirety of the simulation period. Also, Cialis in complex with AKT1 undergoes a gradual increase in deviation up to 40 ns and then decreases with minor deviations for a period of 40- 65 ns; and further maintained a deviation of ~2.20 Å for the remainder of the simulation period. The histogram (Figure 5A, (ii) P-%SSE) shows the secondary structure element (SSE) distribution for each trajectory frame over the course of the simulation. The percentages of the helix, strand, and overall secondary structure elements were found to be 25.06%, 14.63%, and 39.68% respectively. A total of 22 ligand contacts were formed with amino acids of protein (Figure 5A, (iii) PL-Contacts), from Leu156, Lys158, Gly159, Phe161 to Val164, Ala177, Lys179, Leu181, Met227, Tyr229, Ala230, Glu234, Glu278, Met281, Thr291, Asp292, Phe438, Asp439, Glu441, and Phe442. Figure 5A, (iv) Timeline graph represents that the ligand is stabilized by forming a majority of hydrophobic interactions with residues Leu156, Phe161, Val164, Ala177, Lys179, Leu181, Met227, met281, Phe438, and Phe442 with 1%–68% of simulation time. Hydrogen bonds are formed with residues Gly159, Lys179, Tyr229, and Asp292 throughout 1%–60% of the simulation trajectory. The RMSF value of the protein is coupled to the ligand in Figure 5A, (v) P-RMSF graph of simulated protein AKT1 for 100 ns, with X-axis measuring the average deviation of all protein residues (from 144-478) over time and Y-axis indicates RMSF (Å) values. The residues with higher peaks fluctuate the most during the simulation as determined by MD trajectories. Green-colored vertical bars indicate AKT1 residues that interact with Cialis and correspondingly, low RMSF values indicate the stability of the binding. Figure 5A, (vi) 2D-trajectory interaction diagram depicts that the hydrogen bond formed by the docking pose with Tyr229 is preserved in the MD trajectory pose with 59% of the total simulation time.
Further, again a 100 ns molecular dynamic simulation was performed to understand the molecular insights involved in the binding of Finasteride in the p53-MDM2 interaction surface. Figure 5B, (i) PL-RMSD graph shows that the Ca atoms fluctuated in the range of 1.00–2.25 Å and finally stabilized after 65 ns of simulation with an RMSD value of 1.5 Å. Higher fluctuation (up to 2.25 Å) in RMSD was observed at 15 ns. A slight divergence can be seen towards the end of the simulation around 92 ns. Since, the fluctuation lies under the permissible range of 1–3 Å, hence, can be considered as non-significant. The RMSD plot of Finasteride and MDM2 backbone were lying over each other. Hence, the formation of a stable complex can be inferred. Figure 5B, (ii) P-%SSE histogram shows the percentages of helix, strand, and the overall secondary structure elements to be 36.11%, 10.12%, and 46.24% respectively. Figure 5B, (iii) PL-Contacts histogram shows that a total of 20 ligand contacts were formed with amino acids of protein, from Gln18 to Ala21, Gln24, Leu54, Phe55, Ile61, Met62, Arg65, Tyr67, Asp68, Gln72 to Val75, Val93, His96, Ile99, and Tyr100. Figure 5B, (iv) Timeline representation suggested that the ligand is stabilized by forming Hydrogen bonds with Leu54, Gln72, His96, and Tyr100 with 1%-95% of simulation time. Hydrophobic interactions were formed with residues Ile19, Pro20, Leu54, Phe55, Ile61, Met62, Tyr67, Val75, Val93, and Ile99 over the course of 1%-36% simulation trajectory. Figure 5B, (v) P-RMSF graph of simulated protein MDM2 for 100 ns, with the X-axis measuring the average deviation of all protein residues (from 18-110) over time and Y-axis indicating fluctuation RMSF (Å) values. More fluctuations were seen at the C-terminal of the protein during simulation which indicates greater flexibility of the residues while other parts of the protein are more rigid and fluctuate less. Protein residues that interact with the Finasteride are marked with green colored vertical bars. Figure 5B, (vi) 2D-trajectory interaction diagram depicts that the hydrogen bonds formed by the docking pose with Tyr100 and Gln72 were preserved in the MD trajectory pose with 47% and 66% of total simulation time respectively. The RMSF value of the protein-coupled to the ligand.
3. Discussion
A common approach to anticancer drug development has been based on a workflow, whereby molecules that are designed from scratch, to specifically interfere with a certain pathway, are anticipated to target and eradicate tumors in a highly selective manner, analogous to the “lock-and-key” specificity, hence maximizing efficacy and minimizing side effects [68]. Despite their promising results in the preclinical setting, the majority of innovative drugs are proven insufficient or suboptimal when administered in clinical patients, thereby leading to unacceptably low success rates in clinical trials [69,70]. The high failure rate of this approach is the consequence of several unpredictable parameters, mainly: (a) the individual genetic background of cancer patients, which limits the therapeutic benefits only to specific patient subpopulations and necessitates treatment personalization [22]; (b) the fact that cancer-related genes are highly interconnected and regulate each other through complex loops from different pathways [71]; (c) the inherent ability of tumors to adapt and evolve, which catalyzes acquisition of resistance to therapies, especially monotherapies [27]. To address these challenges, computational methodologies including, but not limited to, algorithms and machine learning tools, are now being increasingly integrated in drug discovery programs. For example, computational approaches that 'dock' small molecules into the structures of macromolecular targets and 'score' their potential complementarity to binding sites are widely used in hit identification and lead optimization and are currently reforming the pharmacopeia landscape [72]. This approach allows for fast and comprehensive screening of the efficacy and safety profiles of a high number of leads, in the context of a particular cancer type. Prioritization of the top-resulting leads or combinations thereof could subsequently facilitate a faster introduction to clinical trials and significantly reduce the costs for drug development.
Having in mind that metastasis is linked with the activation of E2F1-governed GRNs, we applied a transcriptomics-aided bioinformatics workflow, followed by virtual drug screening to comprehensively characterize novel therapeutic targets in melanoma and predict their corresponding drug inhibitors. Due to the documented ability of targeted drugs to show superior safety and efficacy in combination schemes [22], we were particularly interested in drugs that can perturb these prometastatic GRNs when used simultaneously. Using a well-established E2F1 map [11], we derived a set of three-node FBLs (n = 44) and used a ranking scheme that applies a weighted multi-objective function integrating topological and non-topological properties of each node. Topological properties such as node degree (number of edges connected to the node) are important for network organization and molecular connections [73]. It is reported that cancer-associated proteins have large betweenness centrality as they control the communication between different components of a network [74].
Among non-topological properties, we have calculated the involvement of the motif constituents in the disease pathway, the gene prioritization score, and the average Log2 fold change for each motif based on the change in expression values of each node from non-invasive to invasive phenotypes derived from in vitro experiments. Since the network was originally constructed around E2F1, the topological properties for some nodes are expected to be higher than other nodes. Therefore, to give equal importance to all nodes, we used different weighting scenarios in the multi-objective optimization function to avoid biases and ranked motifs accordingly. The top-ranked motifs are merged to understand their combined effect on the regulation of EMT in melanoma. We further expanded the regulatory core network by adding receptor proteins the first neighbors of the ranked motif nodes and four marker proteins and their direct connections from the E2F1 map. Receptor proteins work as determinative factors and markers proteins are required to measure the EMT response. We developed a three-layered logic-based model of the regulatory core consisting of an input layer, a regulatory layer, and an output layer. We analyzed the regulatory core by using boolean logic for the input and regulatory layers, and multi-valued logic for the output layer which allows us to assess the combined effect of various network components on the EMT phenotype. Our model simulations identified two protein signatures AKT1 and MDM2 as potential drivers of EMT in melanoma. Further virtual screening of FDA-approved drugs was employed and after binding affinity analysis top five candidate drugs selected for both the proteins AKT1 and MDM2, have evaluated for their safety profile. Following the candidate drugs with safe therapeutic properties and the least binding affinity with the signatures were subjected to MD simulations to check the conformational stability of the complex and dynamics/flexibility of the protein at 100 ns. The trajectory analysis confirms that the candidate drugs (Cialis and Finasteride) stabilized in the active pocket of protein signatures (AKT1 and MDM2) over the course of the simulation (at 80 ns and 65 ns respectively). Analysis of PL-contacts histogram and 2D-trajectory diagrams indicate that the hydrogen bonds formed by the residues TYR229 (in AKT1) and, TYR100 & GLN272 (in MDM2) are critically important residues and observed to play a predominant role in drug binding, thus contributing to the high stability of the complexes and could be further explored for in-vitro or in-vivo studies.
The PI3K-AKT signaling pathway is predominantly activated in melanomas, upon mutations of crucial melanoma drivers, in parallel with the MAPK (Ras-Raf-MEK-ERK) pathway. The BRAF/MEK inhibitors, which constitute the standard of care in the targeted therapeutics of melanoma, suppress the MAPK pathway. However, an active PI3K-AKT pathway offers alternative avenues for the evasion of MAPK-targeting regimens and disease progression. Indeed, members of the PI3K/AKT pathway are implicated in melanoma progression, metastasis, and acquired resistance to MAPK-targeting therapies. AKT1 activation frequently occurs through the silencing of PTEN, a tumor-suppressor gene encoding a phosphatase that acts on lipid and protein substrates. The major PTEN substrate is PIP3, which recruits AKT to the membrane and activates it by phosphorylation. By specifically dephosphorylating PIP3, PTEN suppresses the downstream signaling of AKT and, vice-versa its loss results in increased PIP3 levels and subsequent AKT1 activation. The combination of PTEN silencing with BRAFV600E expression in vivo leads to melanoma formation, while further ectopic expression of activated AKT1 in this genetic context co-operates with PTEN silencing to accelerate metastasis to the lungs and brain [75]. Progressing melanomas, in their vast majority, develop resistance to therapy due to the reactivation of MAPK signaling, commonly via alterations in BRAF, NRAS, and MEK1/2. However, a small proportion of resistant melanomas rely on the activation of the compensatory PI3K/AKT signaling cascade. PI3K/AKT-activating mutations in melanoma do not override proliferative arrest induced by BRAF/MEK inhibition but rather enable the survival of a small dormant subpopulation of MAPK-inhibited melanoma cells. The persistent exposure of this cell subpopulation to the constant selective pressure of BRAF/MEK inhibition eventually promotes the reorganization of signaling circuits and accelerates the evolution of tumor subclones that are highly resistant to targeted therapies [76]. Upregulation of the PI3K-AKT pathway is a critical event during the early and late evolution of resistance to MAPK pathway inhibition [77]. It has been proposed that AKTi combined with BRAFi-based therapy may benefit patients with tumors harbouring BRAF mutations along with PTEN deletions or AKT mutations [78]. In agreement with these studies, our analysis highlighted inhibition of AKT1 as an attractive strategy for preventing EMT-driven metastatic progression of melanomas with high-E2F1 content.
In addition to the PI3K/AKT pathway, the manipulation of the p53-controlled pathways is emerging as an alternative therapeutic option with the potential to overcome the suboptimal response rates for MAPK-targeting therapies [79]. The p53 gene represents a well-established player in carcinogenesis. Over 50% of all tumors carry p53 loss-of-function mutations, leading to the synthesis of functionally impaired protein products that are unable to transactivate genes that induce cell cycle arrest and apoptosis in response to oncogenic stress [79]. Tumors maintaining wild-type p53 exhibit other types of downstream p53 inactivation, such as hyperactivation of its endogenous post-repressor MDM2, a ubiquitin ligase that catalyzes p53 degradation and inactivation. Most melanomas retain a wild-type p53 and exhibit low p53 mutation rates, but instead show frequent inactivation of the CDKN2A, which eventually lead to MDM2 upregulation and subsequent p53 inhibition [80]. As a result, small-molecule inhibitors that block the p53-MDM2 interaction have been pursued as a new cancer therapeutic strategy for restoring the tumor-suppressive function of p53 in wild-type tumors [81,82]. Shattuck-Brandt et al. [83] recently highlighted the therapeutic efficacy of MDM2 inhibition against p53WT melanomas with either a wild-type or a mutant BRAF background. They showed that an MDM2 antagonist (namely KRT-232) alone or in combination with BRAF and/or MEK inhibitors inhibits tumor growth in patient-derived xenografts (PDX) from 15 melanoma patients by suppressing p53 degradation. MDM2 inhibitor monotherapy was effective against BRAFV600WT tumors, while a combination of KRT-232 plus BRAF/MEK inhibitors exhibited a synergistic effect on BRAFV600E mutant PDXs [83]. On a similar note, our study revealed that a number of small-molecule MDM2 inhibitors show potential for preventing E2F1-driven metastatic progression.
Overall, our computational approach predicted AKT1 and MDM2 inhibitors as promising anti-melanoma drugs. If combined with the standard-of-care regimens for melanoma, such as BRAF and MEK inhibitors, these substances could offer appealing alternative therapeutic strategies, potentially overcoming therapeutic resistance and improving the disease-free survival of melanoma patients. Future experiments are essential to confirm the metastasis-preventing potential of such combinations in melanoma animal models, in combination with standard-of-care melanoma regimens. Given that combinations of targeted therapeutics with immunotherapeutics could produce durable responses to clinical patients [84], another significant question that is worth further investigation is whether AKT1 and/or MDM2 inhibitors effectively synergize with checkpoint inhibitors to improve patient survival.
Melanoma is a highly heterogeneous and dynamically evolving cancer type. The increased intratumoral cell diversity can accelerate somatic evolution, because a tumor consisting of a genetically heterogeneous cell population has more possibilities to respond to microenvironmental changes, to evolve, and to spread. It is noteworthy that melanomas can, in several cases, evolve through unorthodox pathways, via the same genes that can successfully inhibit melanoma proliferation. For example, while tyrosinase inhibition is seen as an approach to successfully target proliferative melanoma cells, its loss can trigger EMT-mediated melanoma progression [85]. Similarly, stabilization of the tumor-suppressor p53 can drive therapeutic resistance and it was recently suggested that inhibiting rather than activating wild-type p53 may sensitize previously resistant metastatic melanoma cells to therapy [86]. Advanced computational methods, such as artificial intelligence and machine learning are anticipated to shed more light on the roles of target genes and unveil the spatiotemporal complexity of networks underlying metastasis [87] towards developing personalized therapies.
4. Conclusions
Cancer is a disease where multiple pathways are dysregulated, and its development and progression involve both independent and overlapping molecular targets. Advanced computational methods can unravel the properties of cancer-related proteins and their interactions in the molecular networks and enable the designing of next-generation targeted therapeutics. With the computational pipeline used in this study, we were successful in the identification of key protein signatures (AKT1 and MDM2) in melanoma from a core regulatory network that is based on a published E2F1 interaction map. In this work, a hybrid approach of logic-based modeling coupled with computer-aided drug design techniques was applied for the identification of drug candidates that can modulate the protein’s activity and could be possibly used for melanoma research. From virtual screening, the top candidate drugs based on the lowest binding affinity values against protein signatures were reported and evaluated for their safety profiles. MD simulations confirmed the stability of the two candidate drugs (Cialis and Finasteride) in complex with protein signatures over the course of 100 ns trajectory analysis. In conclusion, Cialis and Finasteride were predicted to be potent drugs to target AKT1 and MDM2 respectively; and may increase cell death in melanoma cancer cells, and this effect is mediated in the presence of E2F1. These findings would facilitate the development of effective inhibitors for clinical use in melanoma metastasis.
5. Methods
5.1. Network analysis and motif identification
The Cytoscape version of the E2F1 map was downloaded from https://sourceforge.net/projects/e2f1map/files and converted into a format suitable for the Cytoscape plugin NetDS v3.0 [17]. The purpose of this was to identify important nodes and network motifs in the network. The loop length was set to three nodes and feedback motifs (n = 444) were retrieved. We then used the Cytoscape plugin NetworkAnalyzer to evaluate the topological properties of nodes [88]. More specifically, we calculated the average number of neighbors for each node in the network (degree) [89] and the density of connections among the neighbors of a node (betweenness centrality) [90] to understand the overall organization of the network. Among non-topological properties, we calculated the number of nodes in a motif involved in the KEGG melanoma pathway (KEGG: 05218), and a prioritization score for each gene from the web resource DISEASES [91].
5.2. Array data from aggressive melanoma cell lines
5.3. Motif prioritization
The regulatory motifs were prioritized using a ranking score for each motif considering key topological and non-topological properties with respect to the relevance of the melanoma phenotype. The motif ranking score is calculated using Eq. (1).
The equation uses a multi-objective function which is normalized to the maximum property value under consideration. We used a ranking scheme that is previously developed [11] by assigning different weights to various topological and non-topological parameters. In particular, the weights to two topological parameters (node degree ⟨ND⟩ and betweenness centrality ⟨BC⟩) were divided into half for avoiding overemphasis on the topological properties and assigning equal weighting factors W2j-W4j to give equal importance to other properties (disease pathway association ⟨DP⟩, gene prioritization score ⟨GP⟩, Log2 fold change ⟨|FC|⟩) in motif prioritization. The equation generates a ranking score for each motif i (1…n) depending on the sets of values chosen for the weighting scenarios j (1 to13) shown in additional file 1a-c. Later, the top 10 motifs were selected from each of the weighting scenarios (13*10=130 motifs). Furthermore, a unique set of motifs were identified and processed for the construction of a melanoma-specific core regulatory network. The optimization of multi-objective function is discussed in detail [11].
5.4. Derivation of a core regulatory network
All the top-ranked motifs identified in the previous steps were merged to create a regulatory core. Additionally, we also considered receptor proteins as critical factors determining the EMT phenotype and directly interacting/ regulating nodes present in the top-ranked motifs. In total, we found and included ten receptor proteins (AR, ESR1, FGFR1, FLT4, NR2F2, NR4A1, TGFBR1, TGFBR2, THRA, and THRB) into the regulatory core. These receptor proteins are the first neighbors of ranked motif nodes and are present in the E2F1 map. In addition, we added four EMT marker proteins (CDH1, VIM, ZEB1, and SNAI1) and direct connections with motif nodes (additional file 1d) in our regulatory core.
5.5. Logic-based modeling to derive protein signatures
To identify protein signatures in the regulatory core, the network is translated into a logic-based model, and in silico perturbation experiments were performed in the software tool CellNetAnalyzer [92]. For this, we derived Boolean rules for the input (receptor proteins) layer and propagation of signals from the input layer to the output layer through the nodes present in the regulatory layer. The network is simulated to determine the impact of the input layer vectors on the EMT phenotype (output layer). We performed single and double perturbation experiments iteratively for the initial conditions that are determined through the additional publicly available gene expression dataset (GSE46517) from Gene Expression Omnibus (GEO). The perturbation experiments were performed by changing the boolean state of each node alone and in combination with other nodes in the regulatory layer to see the impact on the invasiveness (results in additional file 2a-e). Those node(s) which upon inhibition change the EMT to minimum level or upon activation to maximum level are further evaluated as effective protein signatures associated with EMT transition in melanoma.
5.6. Virtual screening of repurposable drugs
Virtual screening was performed as follows:
5.6.1. FDA-approved drug library preparation
The FDA-approved drug library was downloaded from the ZINC12 (http://zinc.dock-ing.org/zinc/) database. Since the library contains 2D structures, Open Babel 3.1.1 (https://pypi.org/project/openbabel/3.1.1/) was used to generate 3D energy-minimized structures to be utilized for docking studies.
5.6.2. Protein structure preparation
The crystal structure of protein signatures AKT1 (PDB: 3OCB) and MDM2 (PDB: 3JZK) were downloaded from the RCSB Protein Data Bank (https://www.rcsb.org/). Proteins were pre-processed by removal of heteroatoms, adding polar hydrogens, and gasteiger charges using the AutoDock Vina [93]. Further, the coordinates of the active site residues were determined.
5.6.3. Binding affinity prediction using molecular docking
Virtual screening was carried out in PyRx v0.8 (AutoDock Vina-based) software [94]. The library compounds were first imported as SDF files in the open babel of PyRx and further energy minimization (using Universal Force Filed) of all the library compounds was performed followed by conversion into PDBQT format files. Later, a gird box was designed to cover the binding site residues within the protein signatures and then the prepared FDA-approved drug library was subjected to docking against AKT1 and MDM2. To efficiently explore the docking conformational space, the search efficiency was set at 100%. For docking calculations, 9 con-formers were generated for each ligand-protein complex. The resulting ligand-docked poses were compared with the crystallographic poses based on ≤ 2.0 Å RMSD tolerance on the heavy atoms. The best predicted binding mode and the corresponding binding affinity (or binding free energy) in kcal/mol were selected. The more negative numerical values of binding free energy represent the better binding between a ligand and a protein signature. The docked complexes and graphical visualization were done in DS Visualizer [95].
5.6.4. Safety profile assessment of candidate drugs
The candidate drugs that bind to the protein signatures (in total, 1254 with AKT1 and 1257 with MDM2 respectively) were subjected to safety profile assessment based on ADMET risk, pharmacokinetics, drug-likeness, and medicinal chemistry friendliness prediction using the SwissADME tool (http://www.swissadme.ch/) [96]. The detailed properties/score values of the candidate drugs are provided (in the additional file 3a) along with the binding affinities (in kcal/mol) for both protein signatures separately (additional file 3b-c).
5.6.5. Molecular dynamics simulation (MDS)
Schrödinger LLC Desmond software was used to simulate the docked complexes for 100 nanoseconds [97]. For this, the complexes were pre-processed in Maestro Wizard for preparation and refinement steps. Missing side chains and loops were added using Prime, optimized with ProtAssign, and minimized using the OPLS_2005 force field. Further, the System Builder tool was implemented to build a system for simulation. The system was created in an orthorhombic box and the TIP3P water solvation model was used with default boundary conditions. The system was neutralized by adding counter ions and salt concentration set to 0.15M NaCl to mimic physiological conditions. The simulation parameters were defined as follows: simulation time 100 ns, recording interval trajectory 100 ps, ensemble class NPT, pressure bar 1.01325, and temperature 300 K. The stability of the simulation was verified by comparing the protein and ligand RMSD over time. The interactions between the protein and ligand were analyzed in the Desmond simulation interaction diagram tool.
Supplementary Materials
Additional file 1: 1a: FBLs in E2F1 map and ranking score; 1b: Top-ranked FBLs in melanoma; 1c: Weighting scenarios for motif prioritization; 1d: Regulatory core interactions. Additional file 2: 2a: Single_perturbation_in data; 2b: Single_perturbation_out data; 2c: Double_perturbation_in data; 2d: Double_perturbation_out data; 2e: Selected double perturbation experiments which bring EMT to 1. Additional file 3: 3a: Prediction of ADMET risk, pharmacokinetics, drug-likeness, and medicinal friendliness score of FDA-approved candidate drugs that were docked to protein signatures AKT1 and MDM2. 3b: Binding affinity of AKT1 protein to candidate drugs. 3c: Binding affinity of MDM2 protein to candidate drugs.
Author Contributions
Conceptualization, SG, SL, LB, BP, JV; methodology, SG, NS, JV; software, SG, NS, JV; experiments, SL, NS, FK; writing—original draft preparation, NS, SL, SG, BP; writing—review and editing, OW, SG, JV, BP; visualization, NS, SG, OW, SL, BP; supervision, SG, LB, OW, BP; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the German Federal Ministry of Education and Research (BMBF) projects e:Med-MelAutim, grant numbers 01ZX1905A, 01ZX1905B, 01ZX1905D, e:Med-SASKit, grant number 01ZX1903B, Deutsche Krebshilfe, grant number 70112353, and the Deutsche Forschungsgemeinschaft (DFG), grant number PU 188/17-1.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
All datasets generated for this study are included in the article/Supporting information.
Acknowledgments
NS and SG are thankful to the CSIR–Indian Institute of Toxicology Research (IITR), Lucknow 226001, India for the facilities provided to conduct this research work. NS acknowledges Muhammad Nasir Iqbal from Dept. of Bioinformatics, The Islamia University of Bahawalpur for technical support in MDS studies.
Competing interests
The authors declare no conflict of interest.
Abbreviations
GRNs: gene regulatory networks, EMT: epithelial-mesenchymal transition, FDA: Food and Drug Administration, CADD: computer-aided drug design, ADMET: absorption, distribution, metabolism, excretion, and toxicity, PL RMSD: Protein-Ligand root mean square deviation, P RMSF: Protein root mean square fluctuation, SSE: secondary structure elements, TPSA: topological polar surface area, MW: molecular weight.
Description of gene symbols appearing in the network:
AKT1: AKT serine/threonine kinase 1, AR: androgen receptor, AXIN2: axin 2, BCL2: BCL2 apoptosis regulator, CCNA2: cyclin A2, CCNB1: cyclin B1, CCNE2: cyclin E2, CDC20: cell division cycle 20, CDH1: cadherin 1, CDK1: cyclin dependent kinase 1, CDK2: cyclin dependent kinase 2, CDKN2A: cyclin dependent kinase inhibitor 2A, CTNNB1: catenin beta 1, E2F1: E2F transcription factor 1, E2F2: E2F transcription factor 2, E2F3: E2F transcription factor 3, ESR1: estrogen receptor 1, FGFR1: fibroblast growth factor receptor 1, FLT4: fms related receptor tyrosine kinase 4, FOXO3: forkhead box O3, KIAA1524: cancerous Inhibitor Of Protein Phosphatase 2A, LEF1: lymphoid enhancer binding factor 1, MDM2: MDM2 proto-oncogene, MDM4: MDM4 regulator of p53, MYC: MYC proto-oncogene, bHLH transcription factor, NR2F2: nuclear receptor subfamily 2 group F member 2, NR4A1: nuclear receptor subfamily 4 group A member 1, PPP2R1A: protein phosphatase 2 scaffold subunit A alpha, PPP2R1B: protein phosphatase 2 scaffold subunit A beta, PPP2R2A: protein phosphatase 2 regulatory subunit B alpha, PPP2R2D: protein phosphatase 2 regulatory subunit B delta, PI3K: phosphatidylinositol 3-kinase, PIP3: phosphatidylinositol-3,4,5-triphosphate, RB1: RB transcriptional corepressor 1, SIRT1: sirtuin 1, SNAI1: snail family transcriptional repressor 1, SP1: Sp1 transcription factor, TGFBR1: transforming growth factor beta receptor 1, TGFBR2: transforming growth factor beta receptor 2, THRA: thyroid hormone receptor alpha, THRB: thyroid hormone receptor beta, TP53: tumor protein p53, VIM: vimentin, ZEB1: zinc finger E-box binding homeobox 1.
References
- Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nature Reviews Cancer. 2014, 14, 455–467. [Google Scholar] [CrossRef]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky Jr WE, et al. Braf V600E cooperates with Pten loss to induce metastatic melanoma. Nature genetics. 2009, 41, 544–552. [Google Scholar] [CrossRef]
- Alla V, Engelmann D, Niemetz A, Pahnke J, Schmidt A, Kunz M, et al. E2F1 in melanoma progression and metastasis. Journal of the National Cancer Institute. 2010, 102, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Pützer BM, Steder M, Alla V. Predicting and preventing melanoma invasiveness: advances in clarifying E2F1 function. Expert review of anticancer therapy. 2010, 10, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Wang Y, Alla V, Goody D, Gupta SK, Spitschak A, Wolkenhauer O, et al. Epigenetic factor EPC1 is a master regulator of DNA damage response by interacting with E2F1 to silence death and activate metastasis-related gene signatures. Nucleic acids research. 2016, 44, 117–133. [Google Scholar] [CrossRef]
- Rouaud F, Hamouda-Tekaya N, Cerezo M, Abbe P, Zangari J, Hofman V, et al. E2F1 inhibition mediates cell death of metastatic melanoma. Cell death & disease. 2018, 9, 1–12. [Google Scholar]
- Meier C, Spitschak A, Abshagen K, Gupta S, Mor JM, Wolkenhauer O, et al. Association of RHAMM with E2F1 promotes tumour cell extravasation by transcriptional up-regulation of fibronectin. The Journal of pathology. 2014, 234, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Vera J, Schmitz U, Lai X, Engelmann D, Khan FM, Wolkenhauer O, et al. Kinetic Modeling–Based Detection of Genetic Signatures That Provide Chemoresistance via the E2F1-p73/DNp73-miR-205 Network. Cancer research. 2013, 73, 3511–3524. [Google Scholar] [CrossRef] [PubMed]
- Engelmann D, Pützer BM. The dark side of E2F1: in transit beyond apoptosis. Cancer research. 2012, 72, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Knoll S, Fürst K, Kowtharapu B, Schmitz U, Marquardt S, Wolkenhauer O, et al. E2F1 induces miR-224/452 expression to drive EMT through TXNIP downregulation. EMBO reports. 2014, 15, 1315–1329. [Google Scholar] [CrossRef]
- Khan FM, Marquardt S, Gupta SK, Knoll S, Schmitz U, Spitschak A, et al. Unraveling a tumor type-specific regulatory core underlying E2F1-mediated epithelial-mesenchymal transition to predict receptor protein signatures. Nature communications. 2017, 8, 1–15. [Google Scholar]
- Logotheti S, Marquardt S, Gupta SK, Richter C, Edelhäuser BA, Engelmann D, et al. LncRNA-SLC16A1-AS1 induces metabolic reprogramming during Bladder Cancer progression as target and co-activator of E2F1. Theranostics. 2020, 10, 9620. [Google Scholar] [CrossRef]
- Richter C, Marquardt S, Li F, Spitschak A, Murr N, Edelhäuser BA, et al. Rewiring E2F1 with classical NHEJ via APLF suppression promotes bladder cancer invasiveness. Journal of Experimental & Clinical Cancer Research. 2019, 38, 1–16. [Google Scholar]
- Marquardt S, Solanki M, Spitschak A, Vera J, Pützer BM, editors. Emerging functional markers for cancer stem cell-based therapies: Understanding signaling networks for targeting metastasis. Seminars in cancer biology; 2018: Elsevier.
- Singh N, Eberhardt M, Wolkenhauer O, Vera J, Gupta SK. An integrative network-driven pipeline for systematic identification of lncRNA-associated regulatory network motifs in metastatic melanoma. BMC bioinformatics. 2020, 21, 1–17. [Google Scholar]
- Goody D, Gupta SK, Engelmann D, Spitschak A, Marquardt S, Mikkat S, et al. Drug repositioning inferred from E2F1-Coregulator interactions studies for the prevention and treatment of metastatic cancers. Theranostics. 2019, 9, 1490. [Google Scholar] [CrossRef] [PubMed]
- Le D-H, Kwon Y-K. NetDS: a Cytoscape plugin to analyze the robustness of dynamics and feedforward/feedback loop structures of biological networks. Bioinformatics. 2011, 27, 2767–2768. [Google Scholar] [CrossRef] [PubMed]
- Khan FM, Sadeghi M, Gupta SK, Wolkenhauer O. A network-based integrative workflow to unravel mechanisms underlying disease progression. Systems Biology: Springer; 2018. p. 247-76.
- Glass DS, Jin X, Riedel-Kruse IH. Nonlinear delay differential equations and their application to modeling biological network motifs. Nature communications. 2021, 12, 1–19. [Google Scholar]
- Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nature reviews Clinical oncology. 2011, 8, 426–433. [Google Scholar] [CrossRef]
- Smalley KS, Eroglu Z, Sondak VK. Combination therapies for melanoma: a new standard of care? American journal of clinical dermatology. 2016, 17, 99–105.
- Curti BD, Faries MB. Recent Advances in the Treatment of Melanoma. New England Journal of Medicine. 2021, 384, 2229–2240. [Google Scholar] [CrossRef]
- Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. The lancet oncology. 2013, 14, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Logotheti S, Pützer BM. STAT3 and STAT5 targeting for simultaneous management of melanoma and autoimmune diseases. Cancers. 2019, 11, 1448. [Google Scholar] [CrossRef]
- Holbeck SL, Camalier R, Crowell JA, Govindharajulu JP, Hollingshead M, Anderson LW, et al. The National Cancer Institute ALMANAC: a comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity. Cancer research. 2017, 77, 3564–3576. [Google Scholar] [CrossRef]
- Julkunen H, Cichonska A, Gautam P, Szedmak S, Douat J, Pahikkala T, et al. Leveraging multi-way interactions for systematic prediction of pre-clinical drug combination effects. Nature communications. 2020, 11, 1–11. [Google Scholar]
- Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer discovery. 2017, 7, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Singh N, Gupta SK. Recent advancement in the early detection of melanoma using computerized tools: An image analysis perspective. Skin Research and Technology. 2019, 25, 129–141. [Google Scholar] [CrossRef]
- Ribeiro DM, Zanzoni A, Cipriano A, Delli Ponti R, Spinelli L, Ballarino M, et al. Protein complex scaffolding predicted as a prevalent function of long non-coding RNAs. Nucleic acids research. 2018, 46, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Singh N, Freiesleben S, Wolkenhauer O, Shukla Y, Gupta SK. Identification of antineoplastic targets with systems approaches, using resveratrol as an in-depth case study. Current pharmaceutical design. 2017, 23, 4773–4793. [Google Scholar] [CrossRef]
- da Silva Rocha SF, Olanda CG, Fokoue HH, Sant'Anna CM. Virtual screening techniques in drug discovery: review and recent applications. Current topics in medicinal chemistry. 2019, 19, 1751–1767. [Google Scholar] [CrossRef]
- Spitschak A, Gupta S, Singh KP, Logotheti S, Pützer BM. Drug Repurposing at the Interface of Melanoma Immunotherapy and Autoimmune Disease. Pharmaceutics. 2023, 15, 83. [Google Scholar]
- Abou-Jaoudé W, Traynard P, Monteiro PT, Saez-Rodriguez J, Helikar T, Thieffry D, et al. Logical modeling and dynamical analysis of cellular networks. Frontiers in genetics. 2016, 7, 94. [Google Scholar]
- Kabbarah O, Nogueira C, Feng B, Nazarian RM, Bosenberg M, Wu M, et al. Integrative genome comparison of primary and metastatic melanomas. PloS one. 2010, 5, e10770. [Google Scholar]
- Lu X, Yan C, Huang Y, Shi D, Fu Z, Qiu J, et al. Mouse double minute 2 (MDM2) upregulates Snail expression and induces epithelial-to-mesenchymal transition in breast cancer cells in vitro and in vivo. Oncotarget. 2016, 7, 37177. [Google Scholar] [CrossRef]
- Chen Y, Wang D-D, Wu Y-P, Su D, Zhou T-Y, Gai R-H, et al. MDM2 promotes epithelial–mesenchymal transition and metastasis of ovarian cancer SKOV3 cells. British journal of cancer. 2017, 117, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Zhou Y, Hu Y, Yang M, Jat P, Li K, Lombardo Y, et al. The miR-106b∼ 25 cluster promotes bypass of doxorubicin-induced senescence and increase in motility and invasion by targeting the E-cadherin transcriptional activator EP300. Cell Death & Differentiation. 2014, 21, 462–474. [Google Scholar]
- Liu B, Li X, Li C, Xu R, Sun X. miR-25 mediates metastasis and epithelial–mesenchymal-transition in human esophageal squamous cell carcinoma via regulation of E-cadherin signaling. Bioengineered. 2019, 10, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Wu J, Limmer A, Narayanan D, Doan H, Simonette R, Rady P, et al. The novel AKT inhibitor Afuresertib suppresses human Merkel cell carcinoma MKL-1 cell growth. Clinical and experimental dermatology. 2021.
- Kallan NC, Spencer KL, Blake JF, Xu R, Heizer J, Bencsik JR, et al. Discovery and SAR of spirochromane Akt inhibitors. Bioorganic & medicinal chemistry letters. 2011, 21, 2410–2414. [Google Scholar]
- Nguyen TTT, Shingyoji M, Hanazono M, Zhong B, Morinaga T, Tada Y, et al. An MDM2 inhibitor achieves synergistic cytotoxic effects with adenoviruses lacking E1B55kDa gene on mesothelioma with the wild-type p53 through augmenting NFI expression. Cell Death & Disease. 2021, 12, 1–10. [Google Scholar]
- Fang DD, Tang Q, Kong Y, Rong T, Wang Q, Li N, et al. MDM2 inhibitor APG-115 exerts potent antitumor activity and synergizes with standard-of-care agents in preclinical acute myeloid leukemia models. Cell death discovery. 2021, 7, 1–15. [Google Scholar]
- Wang HQ, Mulford IJ, Sharp F, Liang J, Kurtulus S, Trabucco G, et al. Inhibition of MDM2 Promotes Antitumor Responses in p53 Wild-Type Cancer Cells through Their Interaction with the Immune and Stromal Microenvironment. Cancer Research. 2021, 81, 3079–3091. [Google Scholar] [CrossRef]
- Gonzalez-Lopez de Turiso F, Sun D, Rew Y, Bartberger MD, Beck HP, Canon J, et al. Rational design and binding mode duality of MDM2–p53 inhibitors. Journal of medicinal chemistry. 2013, 56, 4053–4070. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy S, Henry K, Pei H, Clayton J, Rempala M, Johns D, et al. Discovery of chiral dihydropyridopyrimidinones as potent, selective and orally bioavailable inhibitors of AKT. Bioorganic & medicinal chemistry letters. 2018, 28, 1887–1891. [Google Scholar]
- Wiechmann S, Ruprecht B, Siekmann T, Zheng R, Frejno M, Kunold E, et al. Chemical phosphoproteomics sheds new light on the targets and modes of action of AKT inhibitors. ACS Chemical Biology. 2021, 16, 631–641. [Google Scholar] [CrossRef]
- Arif SA, Poon H. Tadalafil: a long-acting phosphodiesterase-5 inhibitor for the treatment of pulmonary arterial hypertension. Clinical therapeutics. 2011, 33, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Hassel JC, Jiang H, Bender C, Winkler J, Sevko A, Shevchenko I, et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Ta dalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology. 2017, 6, e1326440. [Google Scholar] [CrossRef]
- Luginbuhl AJ, Johnson JM, Harshyne LA, Linnenbach AJ, Shukla SK, Alnemri A, et al. Tadalafil enhances immune signatures in response to neoadjuvant nivolumab in resectable head and neck squamous cell carcinoma. Clinical Cancer Research. 2022, 28, 915–927. [Google Scholar] [CrossRef]
- Liu Y-S, Huang B-R, Lin C-J, Shen C-K, Lai S-W, Chen C-W, et al. Paliperidone inhibits glioblastoma growth in mouse brain tumor model and reduces PD-L1 expression. Cancers. 2021, 13, 4357. [Google Scholar] [CrossRef]
- Pottegård A, Lash TL, Cronin-Fenton D, Ahern TP, Damkier P. Use of antipsychotics and risk of breast cancer: a Danish nationwide case–control study. British Journal of Clinical Pharmacology. 2018, 84, 2152–2161. [Google Scholar] [CrossRef]
- Grimaldi AM, Simeone E, Ascierto PA. Vemurafenib plus cobimetinib in the treatment of mutated metastatic melanoma: the CoBRIM trial. Melanoma Management. 2015, 2, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Brufsky A, Kim S, Zvirbule Ž, Eniu A, Mebis J, Sohn J, et al. A phase II randomized trial of cobimetinib plus chemotherapy, with or without atezolizumab, as first-line treatment for patients with locally advanced or metastatic triple-negative breast cancer (COLET): primary analysis. Annals of Oncology. 2021, 32, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Zhong W-B, Tsai Y-C, Chin L-H, Tseng J-H, Tang L-W, Horng S, et al. A synergistic anti-cancer effect of troglitazone and lovastatin in a human anaplastic thyroid cancer cell line and in a mouse xenograft model. International journal of molecular sciences. 2018, 19, 1834. [Google Scholar] [CrossRef]
- Watkins PB. Insight into hepatotoxicity: the troglitazone experience. LWW; 2005. p. 229-30.
- Zhang W, Zhang C, Liu F, Mao Y, Xu W, Fan T, et al. Antiproliferative activities of the second-generation antipsychotic drug sertindole against breast cancers with a potential application for treatment of breast-to-brain metastases. Scientific reports. 2018, 8, 15753. [Google Scholar] [CrossRef] [PubMed]
- Dai C, Liu P, Wang X, Yin Y, Jin W, Shen L, et al. The antipsychotic agent sertindole exhibited antiproliferative activities by inhibiting the STAT3 signaling pathway in human gastric cancer cells. Journal of Cancer. 2020, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Chukwuemeka PO, Umar HI, Iwaloye O, Oretade OM, Olowosoke CB, Elabiyi MO, et al. Targeting p53-MDM2 interactions to identify small molecule inhibitors for cancer therapy: beyond “Failure to rescue”. Journal of Biomolecular Structure and Dynamics. 2021:1-19.
- Lang L, Perez A. Binding Ensembles of p53-MDM2 Peptide Inhibitors by Combining Bayesian Inference and Atomistic Simulations. Molecules. 2021, 26, 198. [Google Scholar] [CrossRef] [PubMed]
- Seo JO, Yumnam S, Jeong KW, Kim SY. Finasteride inhibits melanogenesis through regulation of the adenylate cyclase in melanocytes and melanoma cells. Archives of pharmacal research. 2018, 41, 324–332. [Google Scholar] [CrossRef]
- Li W-Q, Han J. Reply to finasteride and dutasteride may reduce melanoma risk. Cancer. 2015, 121, 3558–3559. [Google Scholar]
- Fritz I, Wagner P, Olsson H. Improved survival in several cancers with use of H1-antihistamines desloratadine and loratadine. Translational Oncology. 2021, 14, 101029. [Google Scholar] [CrossRef]
- Bethesda M. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet].): National Institute of Diabetes and Digestive and Kidney Diseases. US National Library of Medicine. National Center for Biotechnology Information https://pubmed ncbi nlm nih gov/31643176.2012.
- Geampana, A. Pregnancy is more dangerous than the pill: A critical analysis of professional responses to the Yaz/Yasmin controversy. Social Science & Medicine. 2016, 166, 9–16. [Google Scholar]
- Lee A, Syed YY. Estetrol/drospirenone: a review in oral contraception. Drugs. 2022, 82, 1117–1125. [Google Scholar] [CrossRef]
- Testa B, Kraemer SD. The biochemistry of drug metabolism–an introduction: part 4. reactions of conjugation and their enzymes. Chemistry & biodiversity. 2008, 5, 2171–2336. [Google Scholar]
- Wu X, Xu LY, Li EM, Dong G. Application of molecular dynamics simulation in biomedicine. Chemical biology & drug design. 2022, 99, 789–800. [Google Scholar]
- Gibbs, JB. Mechanism-based target identification and drug discovery in cancer research. Science. 2000, 287, 1969–1973. [Google Scholar] [CrossRef]
- Gashaw I, Ellinghaus P, Sommer A, Asadullah K. What makes a good drug target? Drug discovery today. 2011;16(23-24):1037-43.
- Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nature biotechnology. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Wang L, Zhao H, Li J, Xu Y, Lan Y, Yin W, et al. Identifying functions and prognostic biomarkers of network motifs marked by diverse chromatin states in human cell lines. Oncogene. 2020, 39, 677–689. [Google Scholar] [CrossRef]
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature reviews Drug discovery. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Yu DH, Ruan X-L, Huang J-Y, Liu X-P, Ma H-L, Chen C, et al. Analysis of the interaction network of hub miRNAs-hub genes, being involved in idiopathic pulmonary fibers and its emerging role in non-small cell lung cancer. Frontiers in genetics. 2020, 11, 302. [Google Scholar] [CrossRef] [PubMed]
- Wei P-J, Wu F-X, Xia J, Su Y, Wang J, Zheng C-H. Prioritizing cancer genes based on an improved random walk method. Frontiers in genetics. 2020, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Cho JH, Robinson JP, Arave RA, Burnett WJ, Kircher DA, Chen G, et al. AKT1 activation promotes development of melanoma metastases. Cell reports. 2015, 13, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Irvine M, Stewart A, Pedersen B, Boyd S, Kefford R, Rizos H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis. 2018, 7, 1–11. [Google Scholar]
- Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer discovery. 2014, 4, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Lassen A, Atefi M, Robert L, Wong DJ, Cerniglia M, Comin-Anduix B, et al. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Molecular cancer. 2014, 13, 1–14. [Google Scholar]
- Lee JT, Herlyn M. MEK'ing the most of p53 reactivation therapy in melanoma. Journal of Investigative Dermatology. 2012, 132, 263–265. [Google Scholar] [CrossRef]
- Logotheti S, Marquardt S, Pützer BM. p73-governed miRNA networks: translating bioinformatics approaches to therapeutic solutions for cancer metastasis. Computational Biology of Non-Coding RNA. 2019:33-52.
- Loureiro JB, Raimundo L, Calheiros J, Carvalho C, Barcherini V, Lima NR, et al. Targeting p53 for melanoma treatment: counteracting tumour proliferation, dissemination and therapeutic resistance. Cancers. 2021, 13, 1648. [Google Scholar] [CrossRef]
- Beloglazkina A, Zyk N, Majouga A, Beloglazkina E. Recent small-molecule inhibitors of the p53–MDM2 protein–protein interaction. Molecules. 2020, 25, 1211. [Google Scholar] [CrossRef]
- Shattuck-Brandt RL, Chen S-C, Murray E, Johnson CA, Crandall H, O'Neal JF, et al. Metastatic Melanoma Patient–Derived Xenografts Respond to MDM2 Inhibition as a Single Agent or in Combination with BRAF/MEK Inhibition. Clinical Cancer Research. 2020, 26, 3803–3818. [Google Scholar] [CrossRef] [PubMed]
- Ribas A, Algazi A, Ascierto PA, Butler MO, Chandra S, Gordon M, et al. PD-L1 blockade in combination with inhibition of MAPK oncogenic signaling in patients with advanced melanoma. Nature communications. 2020, 11, 6262. [Google Scholar] [CrossRef]
- Fuerst K, Steder M, Logotheti S, Angerilli A, Spitschak A, Marquardt S, et al. DNp73-induced degradation of tyrosinase links depigmentation with EMT-driven melanoma progression. Cancer letters. 2019, 442, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Webster MR, Fane ME, Alicea GM, Basu S, Kossenkov AV, Marino GE, et al. Paradoxical role for wild-type p53 in driving therapy resistance in melanoma. Molecular cell. 2020, 77, 633–644. [Google Scholar] [CrossRef]
- Fleming, N. Computer-calculated compounds. Nature. 2018, 557, S55–S57. [Google Scholar] [CrossRef] [PubMed]
- Assenov Y, Ramírez F, Schelhorn S-E, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Wang X, Yang Q, Liu M, Ma X. Comprehensive influence of topological location and neighbor information on identifying influential nodes in complex networks. Plos one. 2021, 16, e0251208. [Google Scholar]
- Abbasi A, Hossain L, Leydesdorff L. Betweenness centrality as a driver of preferential attachment in the evolution of research collaboration networks. Journal of Informetrics. 2012, 6, 403–412. [Google Scholar] [CrossRef]
- Pletscher-Frankild S, Pallejà A, Tsafou K, Binder JX, Jensen LJ. DISEASES: Text mining and data integration of disease–gene associations. Methods. 2015, 74, 83–89. [Google Scholar] [CrossRef]
- Klamt S, Saez-Rodriguez J, Lindquist JA, Simeoni L, Gilles ED. A methodology for the structural and functional analysis of signaling and regulatory networks. BMC bioinformatics. 2006, 7, 1–26. [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Chemical biology: Springer; 2015. p. 243-50.
- Biovia DS. Discovery studio modeling environment. Release; 2017.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific reports. 2017, 7, 1–13. [Google Scholar]
- Release S. 3: Desmond molecular dynamics system, DE Shaw research, New York, NY, 2017. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY. 2017.
Figure 1.
Workflow for the identification and prioritization of therapeutic targets regulating metastatic melanoma phenotypes and virtual screening of repurposed drugs. The overall workflow is divided into four parts. (A) The E2F1 interaction map was used to derive positive/negative feedback loops followed by calculation of node properties, network reduction via a multi-objective function, and subsequently merging the top-ranked motifs to generate a network core. (B) Melanoma-specific regulatory core: The constructed core consists of 183 direct interactions (edges) involving 34 core proteins and miRNAs, 10 receptor proteins, and 4 EMT marker proteins. Regulatory directions were retrieved from the E2F1 map as activation(+1), inhibition(-1), and unidentified(0). (B) Logic-based model: The model is divided into three layers: the input layer containing receptor molecules (yellow color nodes), the middle layer comprising the regulatory network molecules (cyan color nodes) with known-marker proteins (pink color nodes), and the output layer containing the EMT phenotype (red color node). Green color edges represent activation, red color edges represent inhibition and gray edges represent neutral regulatory relationships among the nodes. (C) Two protein signatures (AKT1 and MDM2) were identified through the in silico perturbation experiments on the logic-based model. The functional binding sites of AKT1 and MDM2 are shown in the ribbon model with key amino acid residues participating in the binding pocket formation. At the bottom, the surface model of AKT1 and MDM2 are exposed. In the case of AKT1 (PDB: 3OCB_chainA) the kinase domain showing the ATP binding pocket is identified as the main binding pocket however, in the case of MDM2 (PDB: 3JZK_chain A) the binding site is identified as the main hydrophobic cavity that interacts with p53, displayed in yellow spheres respectively. (D) Virtual screening highlights various filtering steps for the identification of potential drug inhibitors from FDA-approved drug library.
Figure 1.
Workflow for the identification and prioritization of therapeutic targets regulating metastatic melanoma phenotypes and virtual screening of repurposed drugs. The overall workflow is divided into four parts. (A) The E2F1 interaction map was used to derive positive/negative feedback loops followed by calculation of node properties, network reduction via a multi-objective function, and subsequently merging the top-ranked motifs to generate a network core. (B) Melanoma-specific regulatory core: The constructed core consists of 183 direct interactions (edges) involving 34 core proteins and miRNAs, 10 receptor proteins, and 4 EMT marker proteins. Regulatory directions were retrieved from the E2F1 map as activation(+1), inhibition(-1), and unidentified(0). (B) Logic-based model: The model is divided into three layers: the input layer containing receptor molecules (yellow color nodes), the middle layer comprising the regulatory network molecules (cyan color nodes) with known-marker proteins (pink color nodes), and the output layer containing the EMT phenotype (red color node). Green color edges represent activation, red color edges represent inhibition and gray edges represent neutral regulatory relationships among the nodes. (C) Two protein signatures (AKT1 and MDM2) were identified through the in silico perturbation experiments on the logic-based model. The functional binding sites of AKT1 and MDM2 are shown in the ribbon model with key amino acid residues participating in the binding pocket formation. At the bottom, the surface model of AKT1 and MDM2 are exposed. In the case of AKT1 (PDB: 3OCB_chainA) the kinase domain showing the ATP binding pocket is identified as the main binding pocket however, in the case of MDM2 (PDB: 3JZK_chain A) the binding site is identified as the main hydrophobic cavity that interacts with p53, displayed in yellow spheres respectively. (D) Virtual screening highlights various filtering steps for the identification of potential drug inhibitors from FDA-approved drug library.
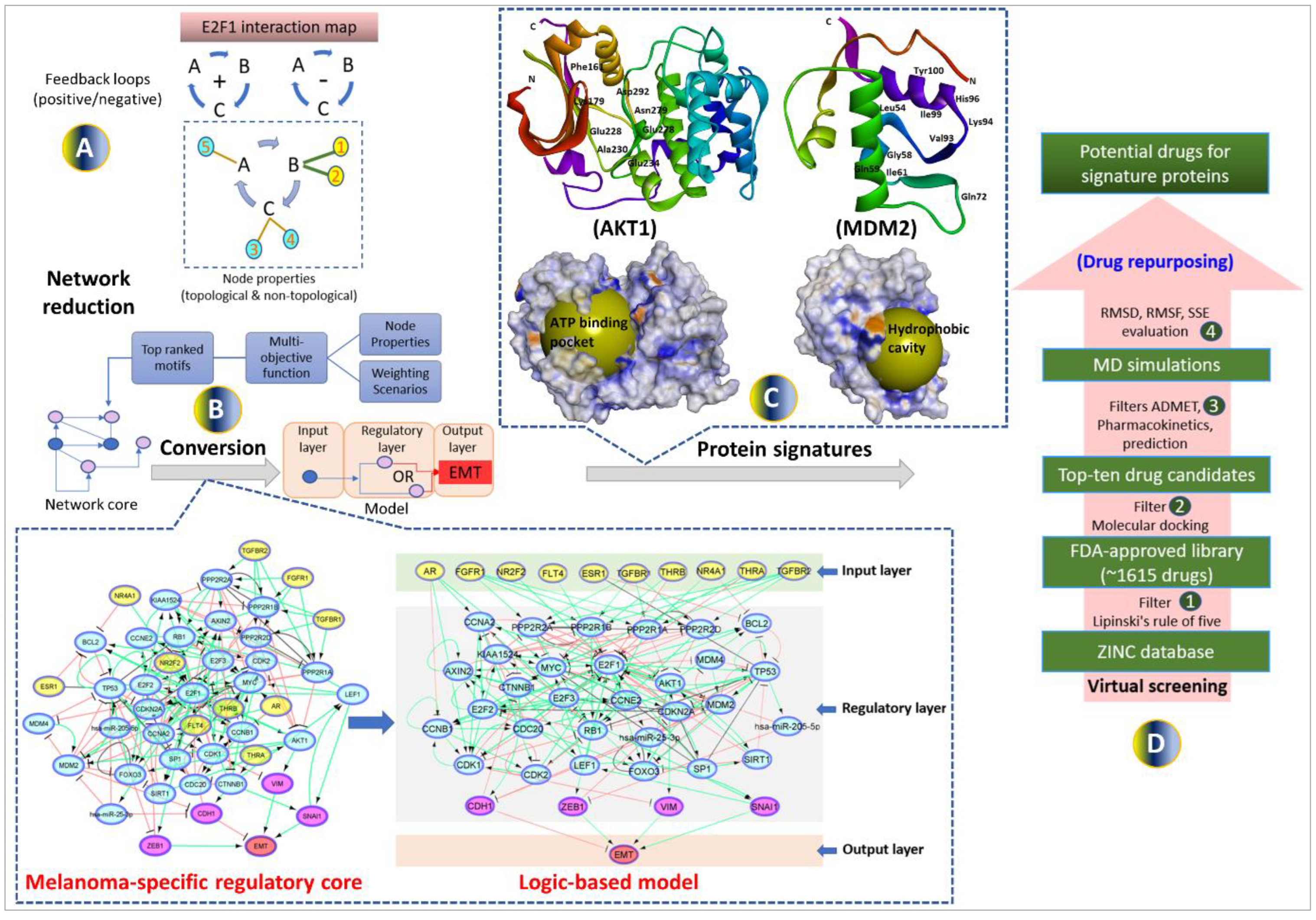
Figure 2.
Kaplan-Meier plots suggest that patients with high expression of (a) AKT1; (b) MDM2; and (c) high expression of AKT1 and MDM2 together have the worst clinical outcomes.
Figure 2.
Kaplan-Meier plots suggest that patients with high expression of (a) AKT1; (b) MDM2; and (c) high expression of AKT1 and MDM2 together have the worst clinical outcomes.
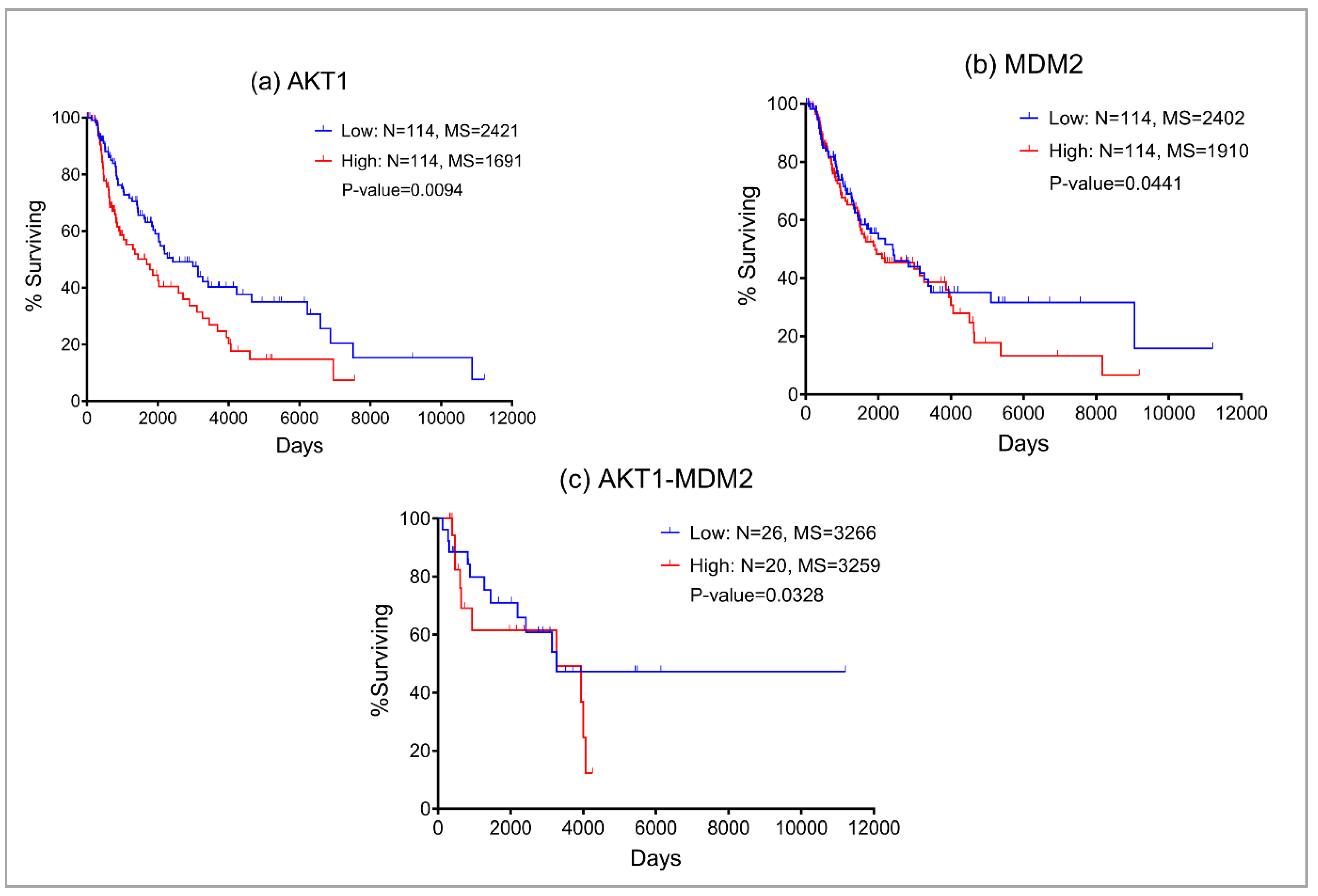
Figure 3.
2D interaction diagrams showing binding profile of both the protein signatures A(i-v) AKT1 and B(i-v) MDM2 with their top five screened candidate drugs: Cialis, Paliperidone, Sertindole, Troglitazone, Cobimetinib, Drospirenone, Finasteride, and Loratadine. The binding affinities are measured in kcal/mol (given in brackets). Intramolecular interactions are depicted as colored dashed lines between protein residues and drug atoms. The solvent-accessible surface of an interacting residue is represented by a blue halo around the residue.
Figure 3.
2D interaction diagrams showing binding profile of both the protein signatures A(i-v) AKT1 and B(i-v) MDM2 with their top five screened candidate drugs: Cialis, Paliperidone, Sertindole, Troglitazone, Cobimetinib, Drospirenone, Finasteride, and Loratadine. The binding affinities are measured in kcal/mol (given in brackets). Intramolecular interactions are depicted as colored dashed lines between protein residues and drug atoms. The solvent-accessible surface of an interacting residue is represented by a blue halo around the residue.
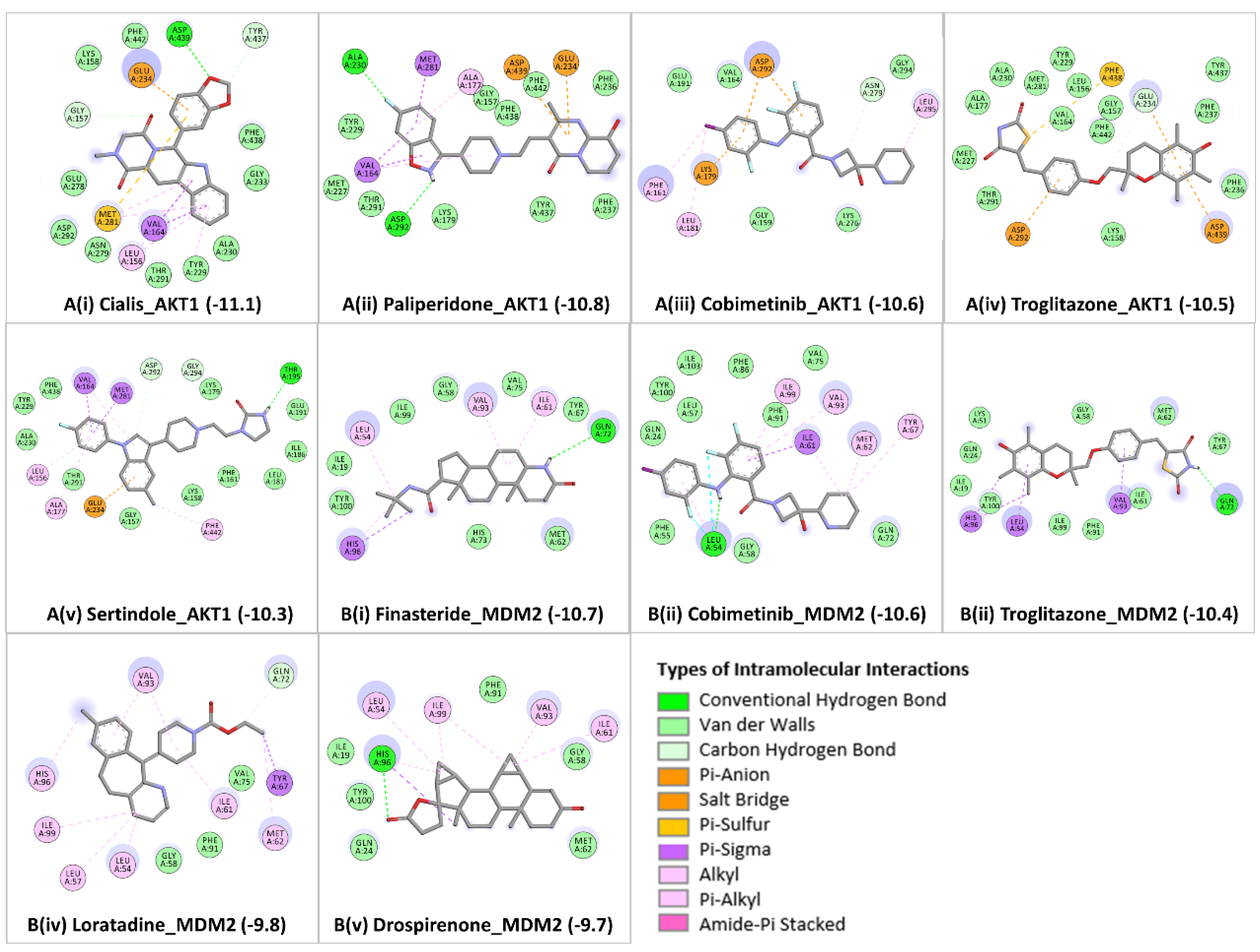
Figure 4.
Radar plots 4(a) of top screened candidate drugs for oral bioavailability based on six physicochemical properties LIPO (lipophilicity), SIZE (molecular weight), POLAR (topological polar surface area), INSOLU (insolubility), INSATU (in-saturation), and FLEX (flexibility). The pink-colored area represents the ideal range for each property i.e. XLOGP3 (− 0.7 and + 5.0), MW (150 and 500 g/mol), TPSA (20 and 130 Å2), Log S (<6), Fraction Csp3 (<1), and Rotatable bonds (<9), respectively. 4(b) Prediction of water solubility, medicinal chemistry, drug-likeness, pharmacokinetic, and toxicity profile of top screened candidate drugs.
Figure 4.
Radar plots 4(a) of top screened candidate drugs for oral bioavailability based on six physicochemical properties LIPO (lipophilicity), SIZE (molecular weight), POLAR (topological polar surface area), INSOLU (insolubility), INSATU (in-saturation), and FLEX (flexibility). The pink-colored area represents the ideal range for each property i.e. XLOGP3 (− 0.7 and + 5.0), MW (150 and 500 g/mol), TPSA (20 and 130 Å2), Log S (<6), Fraction Csp3 (<1), and Rotatable bonds (<9), respectively. 4(b) Prediction of water solubility, medicinal chemistry, drug-likeness, pharmacokinetic, and toxicity profile of top screened candidate drugs.
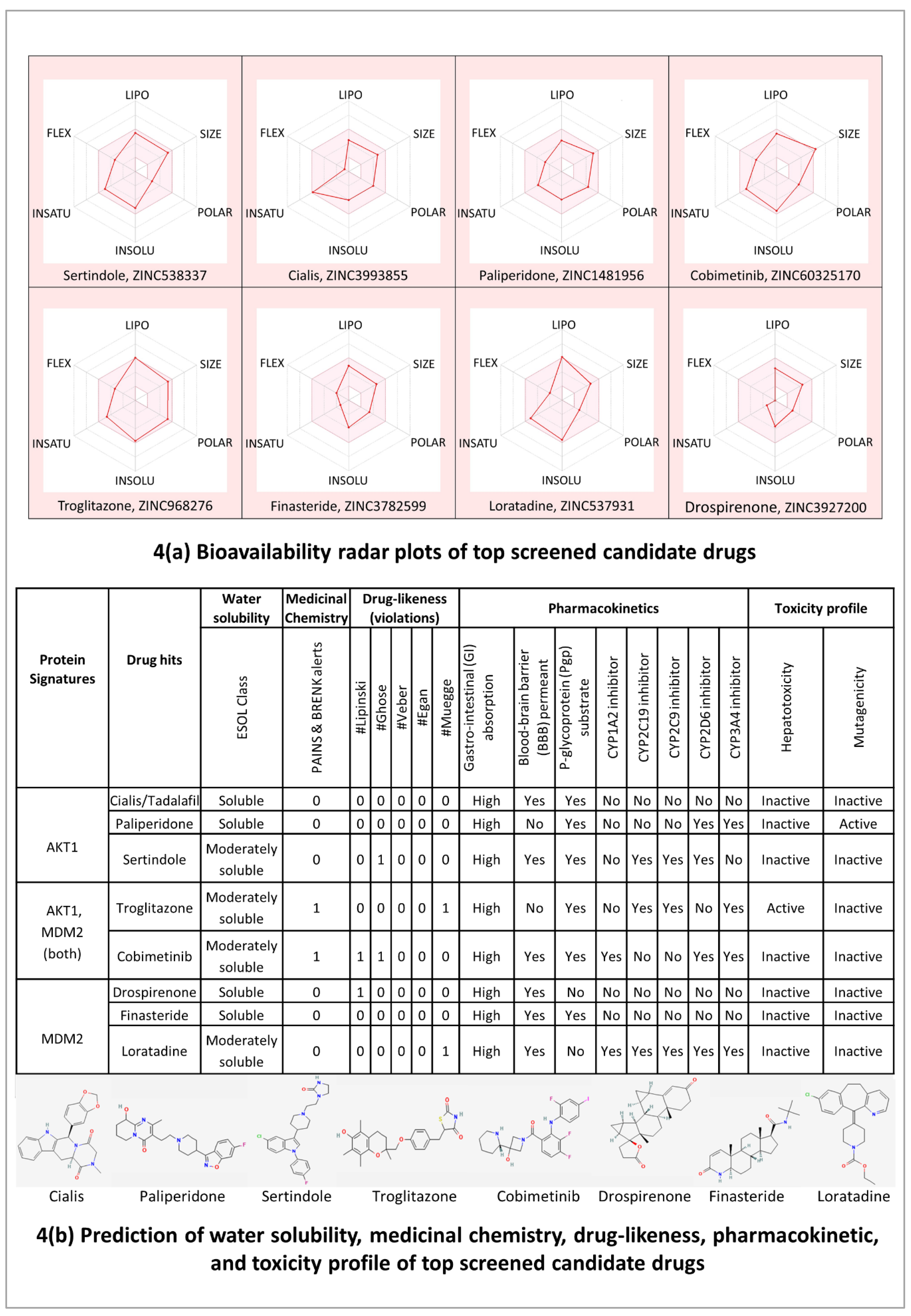
Figure 5.
MDS analysis of protein-ligand complex. 5(a) Simulation results for AKT1-Cialis complex and 5(b) Simulation results for MDM2_Finasteride complex. The graphs showing (i) PL-RMSD of simulated C-alpha atoms of protein in complex with inhibitor during 100 ns MD simulation. The X-axis shows the variation of protein RMSD through time and the Y-axis shows the variation of protein RMSD through time. (ii) P-%SSE histogram showing protein secondary structure element distribution by residue index throughout the protein structure complexed with the ligand. Red columns indicate the alpha helices and blue columns indicate the beta-strands. (iii) PL-Contacts histogram showing four types of protein interactions (H-bonds, hydrophobic, ionic, and water bridges) with the ligand throughout the simulation. The stacked bar is normalized over the course of the trajectory. (iv) Timeline representation of the interactions and contacts. The top panel shows the total number of specific protein contacts with the ligand and the below panel shows the specific residues which interact with the ligand in each trajectory frame (dense areas represent more than one contact with the ligand). (v) P-RMSF of simulated protein in complex with inhibitor during 100 ns MDS. (vi) Schematic 2D diagram of ligand atom interactions with the protein residues (interactions that occur more than 30% of the simulation time in the selected trajectory from 0 to 100 ns are only shown).
Figure 5.
MDS analysis of protein-ligand complex. 5(a) Simulation results for AKT1-Cialis complex and 5(b) Simulation results for MDM2_Finasteride complex. The graphs showing (i) PL-RMSD of simulated C-alpha atoms of protein in complex with inhibitor during 100 ns MD simulation. The X-axis shows the variation of protein RMSD through time and the Y-axis shows the variation of protein RMSD through time. (ii) P-%SSE histogram showing protein secondary structure element distribution by residue index throughout the protein structure complexed with the ligand. Red columns indicate the alpha helices and blue columns indicate the beta-strands. (iii) PL-Contacts histogram showing four types of protein interactions (H-bonds, hydrophobic, ionic, and water bridges) with the ligand throughout the simulation. The stacked bar is normalized over the course of the trajectory. (iv) Timeline representation of the interactions and contacts. The top panel shows the total number of specific protein contacts with the ligand and the below panel shows the specific residues which interact with the ligand in each trajectory frame (dense areas represent more than one contact with the ligand). (v) P-RMSF of simulated protein in complex with inhibitor during 100 ns MDS. (vi) Schematic 2D diagram of ligand atom interactions with the protein residues (interactions that occur more than 30% of the simulation time in the selected trajectory from 0 to 100 ns are only shown).
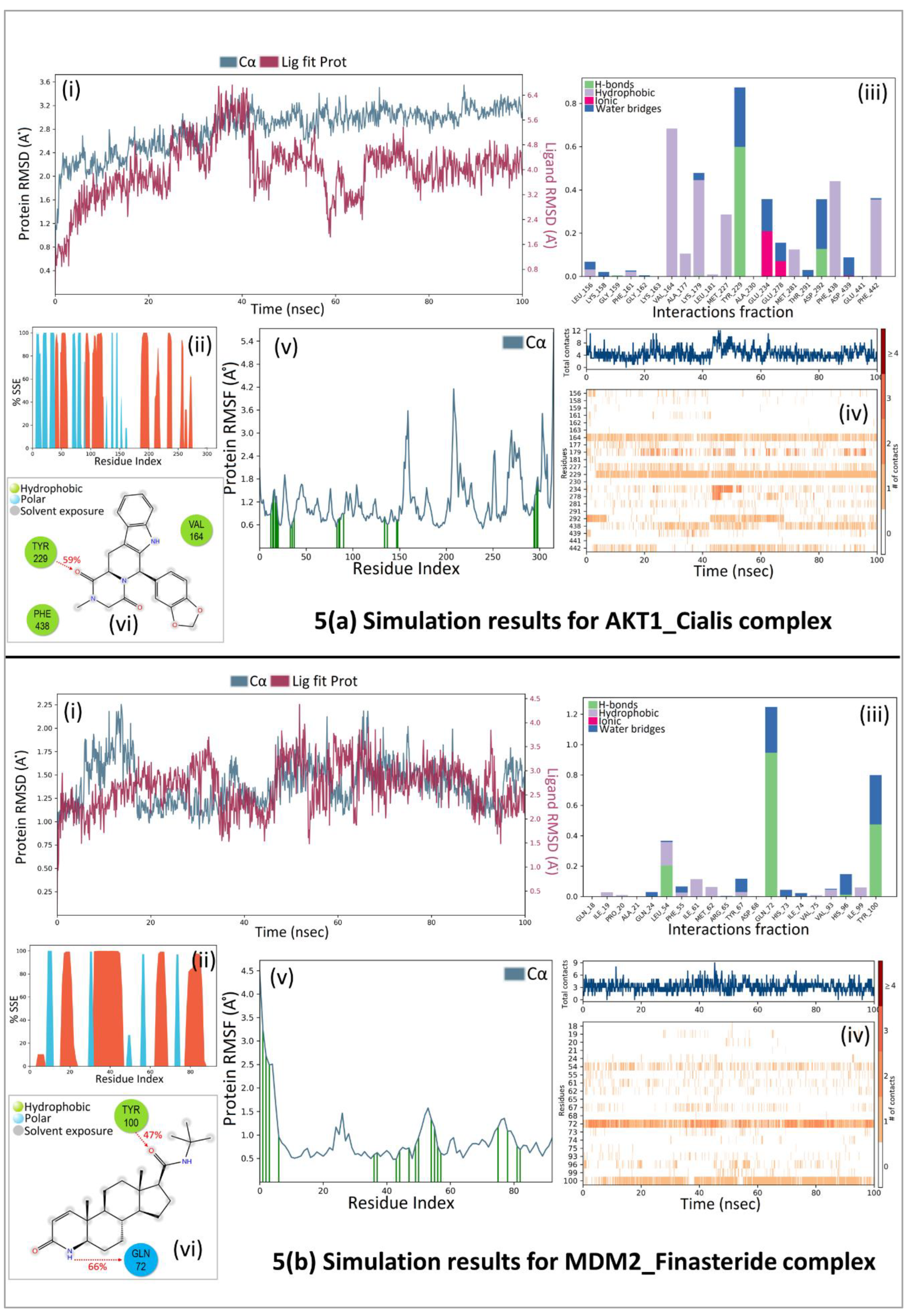

Table 1.
Stimulus-response and perturbation simulations. The underlined perturbed nodes have the most effect on the EMT phenotype.
Table 1.
Stimulus-response and perturbation simulations. The underlined perturbed nodes have the most effect on the EMT phenotype.
|
(a) Stimulus-response analysis for the initial condition. (Model simulation results of initial condition which results in higher EMT level.) | |||||||||||||||||
| AR | ESR1 | FGFR1 | FLT4 | NR2F2 | NR4A1 | TGFBR1 | TGFBR2 | THRA | THRB | MDM2 | MIR25 | AKT1 | ZEB1 | CDH1 | VIM | SNAI1 | EMT |
| NaN | 1 | 1 | NaN | 1 | 1 | 1 | NaN | 0 | NaN | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 3 |
|
(b) Single perturbations analysis (inhibition of MDM2, MIR25 and activation of AKT1) for EMT level of 3. (Single perturbation by inhibiting MDM2 or MIR25 can bring EMT from level 3 to 1, while upregulating AKT1 resulted in EMT level to 4.) | |||||||||||||||||
| AR | ESR1 | FGFR1 | FLT4 | NR2F2 | NR4A1 | TGFBR1 | TGFBR2 | THRA | THRB | MDM2 | MIR25 | AKT1 | ZEB1 | CDH1 | VIM | SNAI1 | EMT |
| NaN | 1 | 1 | NaN | 1 | 1 | 1 | NaN | 0 | NaN | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 |
| NaN | 1 | 1 | NaN | 1 | 1 | 1 | NaN | 0 | NaN | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 |
| NaN | 1 | 1 | NaN | 1 | 1 | 1 | NaN | 0 | NaN | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



