
Submitted:
26 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Recent studies revealing varied responses of infected cells to LRAs underscore the limited effectiveness of these agents and emphasize the wide array of determinants contributing to the heterogeneity of reservoirs, including virus genetic background, cell model, cell type, silencing mechanisms, tissue reservoirs, integration sites, patient, and gender specific factors. The enhancer region of the HIV-1 LTR contains two adjacent NF-κB binding sites that play a central role in mediating inducible HIV-1 gene expression. Beyond the involvement of various transcription factors, such as NF-κB, epigenetic constraints also play a pivotal role in suppressing the initiation of latent HIV transcription. Consequently, even latent viruses containing functional NF-κB sites remain unresponsive to drugs that activate NF-κB. Thus, it is evident that the activation of NF-κB alone does not suffice to trigger latent HIV, contradicting the central hypothesis of this study. The author used bioinformatics methods to analyze the viral proteins and their primer binding sites. The results show that the amino acid sequence ARG of Gag proteins of HTLV-1, HTLV-2, STLV-1 and STLV-2 match their primer binding site GGGGGCTCG in the 3'-to-5' direction and that the amino acid sequence SPR of Gag proteins of HIV-1, HIV-2, SIV and FIV match their primer binding site GGCGCCCGA in the 3'-to-5' direction. Related studies have shown that the genomic Gag/Gag-Pol complex recruits the LysRS/tRNA complex. The selective packaging of the tRNA primer requires HIV-1 Gag and Gag-Pol, and an interaction between LysRS and Gag is observed in vitro. In HIV-1, Gag/LysRS interaction depends on Gag sequences within the CTD of CA around amino acids 283-363 and motif 1 of LysRS around amino acids 208-259. It should be noted that the amino acid sequence SPR of the Gag protein is located at amino acids 148-150 within the NTD of CA, specifically at the NTD-NTD interface 1. Although this research is purely bioinformatics analysis, the relevant studies have demonstrated that Gag proteins match the HIV-1 primer binding site and possess the potential to directly activate dormant retroviruses.
Keywords:
HIV
; HTLV
; NF-κB
; LysRS
; Gag
Introduction
Acquired immunodeficiency syndrome (AIDS) is a disease caused by human immunodeficiency virus (HIV). The virus attacks immune system cells in the body and then uses their machinery to make copies of itself. However, some HIV-infected immune cells enter a state in which they do not produce new virus, which is called the resting or latent state. These cells form a latent HIV reservoir in which HIV can hide for years, resulting in the avoidance of HIV therapy. At any time, these cells can become active again and start to make more copies of the virus. [1]
A significant challenge in curing HIV infection is the virus's ability to stay dormant within specific immune cells like CD4 cells, sometimes for years. During latency, the immune system remains unaware of the virus, rendering antiretroviral therapy (ART) ineffective. Latency-reversing agents (LRAs) are being explored to reactivate latent HIV in cluster of differentiation 4 (CD4) cells, enabling ART and the immune system to combat the virus. Currently, LRAs still have not been approved by the FDA. [2]
Scientists have used this opportunity to develop methods to target these latent reservoirs and make them active such that they can be identified and targeted by HIV therapy. However, scientists at Johns Hopkins reported compounds they hoped would 'wake up' dormant reservoirs of HIV inside the immune system, but T cells have failed to achieve this effect in laboratory tests using white blood cells collected directly from patients infected with HIV. [3]
Hence, further investigation is needed to determine the applicability of this method. In this study, the author analyzed whether LRAs can be used to activate dormant virus reservoirs and proposed the use of Gag proteins to reawaken dormant HIV infection.
Methods
HIV latency is a complex phenomenon where the virus remains dormant within cells, evading the immune system and antiretroviral drugs. The promoter-proximal (enhancer) region of the HIV-1 long terminal repeat (LTR) contains two adjacent NF-κB binding sites that play a central role in mediating inducible HIV-1 gene expression. [4,5,6] This region is pivotal in mediating the inducible expression of the HIV-1 gene when the virus is activated.
Nuclear factor kappa light chain enhancer of activated B cells (NF-κB) is a transcription factor that plays a significant role in regulating immune responses and inflammation, it controls the activation of various genes involved in these processes. [7] In the context of HIV-1, the presence of NF-κB binding sites in the promoter-proximal region of the LTR is significant for several reasons.
The adjacent NF-κB binding sites allow NF-κB transcription factors to bind to the HIV-1 LTR. [8] When immune responses or other stimuli activate NF-κB, it can bind to these sites and initiate the transcription of the HIV-1 gene. This activation leads to the production of viral RNA and eventually new virus particles.
By having NF-κB binding sites in the promoter-proximal region, the virus gains a mechanism to control its replication in response to changes in the host environment. [4] This allows the virus to remain dormant when conditions are not favorable for replication, and to become active when immune responses or other factors signal an appropriate environment for replication.
The presence of NF-κB binding sites in the promoter-proximal region presents a potential target for therapeutic interventions. If researchers can manipulate NF-κB activation, they might be able to control the expression of the HIV-1 gene and potentially develop strategies to suppress viral replication or reactivate latent virus for elimination.
However, clinical trials of LRAs within the "shock and kill" strategy have so far produced unconvincing results. [9] Recent studies revealing varied responses of infected cells to LRAs underscore the limited effectiveness of these agents [10,11,12] and emphasize the wide array of determinants contributing to the heterogeneity of reservoirs, including virus genetic background [13], cell model [14], cell type [15,16,17], silencing mechanisms [18,19], tissue reservoirs [18,19,20], integration sites [21,22,23], patient [19,24], and gender [25] specific factors. Furthermore, some studies show conflicting observations on the impact of LRAs on natural killer cell [26] and cytotoxic T-cell lymphocyte [27] activity, indicating either an immunosuppressive effect or a reduced influence of LRA activity on cells sensing HIV-1 reactivation [10,27,28,29,30].
Beyond the involvement of various transcription factors, such as NF-κB, epigenetic constraints also play a pivotal role in suppressing the initiation of latent HIV transcription. Epigenetic modifications involve changes to the structure of DNA and its associated proteins, affecting gene expression without altering the DNA sequence itself. In the context of latent HIV, these modifications create a repressive environment that prevents the virus from becoming active. This repression is maintained through various mechanisms, including DNA methylation and histone modifications.
Even if the NF-κB transcription factor becomes activated, as it normally triggers immune responses and gene expression, it doesn't necessarily overcome the epigenetic barriers present in latent HIV. This means that even if the latent HIV viruses have functional NF-κB binding sites, they may remain unresponsive to drugs that specifically activate NF-κB. Essentially, the epigenetic modifications create a "brake" on the transcriptional machinery that even NF-κB activation might not be able to fully release.
Consequently, even latent viruses containing functional NF-κB sites remain unresponsive to drugs that activate NF-κB. Thus, it is evident that the activation of NF-κB alone does not suffice to trigger latent HIV, contradicting the central hypothesis of this study.
Several studies claim that AZD5582 can reawaken sleeping HIV and SIV, but the effectiveness rate was found to be only 42%. [31,32,33] Most importantly, the novel small-molecule IAP inhibitor AZD5582 has been used for the treatment of cancer and reportedly causes cIAP1 degradation and thus induces apoptosis in the MDA-MB-231 breast cancer cell line at subnanomolar concentrations in vitro. [34]
In addition to AZD5582, many studies claim that LRAs, including ciapavir [35], bryostatin-1 [36], disulfiram [37], ingenol-B [38], and prostratin [39], can be used to reawaken sleeping HIV. These LRAs have also been used for the treatment of cancer: disulfiram inhibits prostate cancer cell growth [40], bryostatin-1 exhibits potent antitumor activity in vitro and in vivo in human tumor xenografts [41], semisynthetic ingenol compounds show potent antitumor activity on all cancer cell lines evaluated [42], and prostratin exerts a potential anticancer effect through SIK3 inhibition [43].
The LRAs associated with cancer are directed towards the NF-κB binding sites. This indicates that these LRAs are specifically designed to affect or interact with the NF-κB signaling pathway, which is relevant to cancer. The LRAs target the NF-κB binding sites instead of the HIV-1 primer binding site. As a result, LRAs do not directly reawaken dormant retrovirus infections. Therefore, the author suggests that rather than using LRAs to indirectly activate NF-κB binding sites, it might be more effective to employ other agents, such as viral proteins, to directly activate the HIV-1 primer binding site.
Viral RNA is specifically packaged into virions, not NF-κB or cancer RNA. This specificity allows the virus to accurately identify its own genetic material. Consequently, viral proteins carry information that enables the recognition of viral RNA, much like how LRAs activate NF-κB binding sites. It is conceivable that a specific viral protein may possess a function analogous to NF-κB, which could be harnessed to directly identify the HIV-1 primer binding site.
It is well known that HIV-1 recruits human uncharged tRNA (Lys) to serve as the reverse transcription primer [44], and tRNA also functions as the physical link between the mRNA and the amino acid sequences of proteins [45]. Therefore, the author posits that uncharged tRNA serves as the physical link between the promoter and the protein receptors, which are recruited by RNA polymerase II. To identify the specific viral protein that matches the primer binding site, a Python program was developed to match all proteins with their respective gene sequences and visualize them graphically.
Results
Latent HIV can synthesize a 5'-3' RNA chain by transcribing the existing 3'-5' complementary DNA strand after cellular infection [46,47]. The author uses the x-axis to represent the protein and the y-axis to represent the primer binding site. Negative numbers indicate that the protein or tRNA may have rotated 180 degrees (which did not happen) or been bound in the 3'-to-5' direction (if both values are negative).
The author also expanded the analysis to include other retroviruses, including Deltaretroviruses (HTLV and STLV) and Lentiviruses (HIV, SIV, and FIV). The gene data was sourced from the GenBank database at the NCBI. The author used the following sequences for the analysis: NC_001436 (HTLV-1) [48], NC_001488 (HTLV-2) [49], NC_000858 (STLV-1) [50], NC_001815 (STLV-2) [51], NC_001802 (HIV-1) [52], NC_001722 (HIV-2) [53], NC_001549 (SIV) [54], and NC_001482 (FIV) [55].
Having 2 amino acid sequences of the matching points leads to many possibilities, and it is thus impossible to confirm which protein matches the primer binding site. When there are 4 amino acid sequences, no matching target can be found. However, when there are 3 amino acid sequences, there is exactly one perfect matching region. Different types of retroviruses are represented by different patterns and colors, and their sequences around the primer binding site are matched with their own proteins, as shown in Figure 1.
As shown in Figure 1, inside the red box, the coordinates of 8 different viruses appear at the same time and are extremely close, which means that they represent the same protein. Other locations contained either only Deltaretroviruses or only Lentiviruses, and the spacing between the different color coordinates was too large, indicating that they were not even the same protein and were therefore excluded. If the virus amino acid sequences of the protein mutated, its primer binding site remained the same, which means that it was not the matching target.
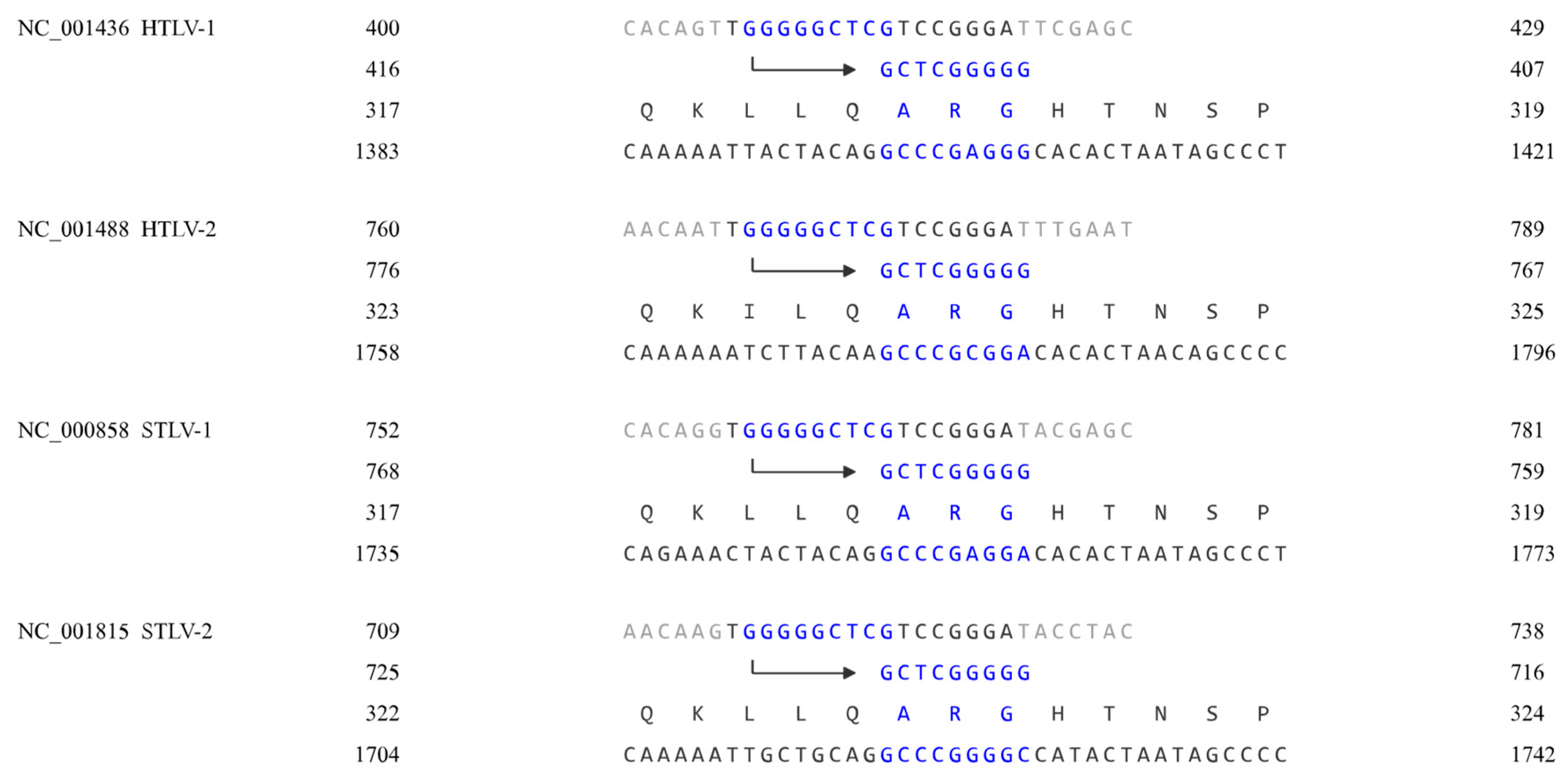
In the GenBank database, the primer binding site of the HTLV-2 (NC_001488) genome is approximately nt 766 to 783, and that of the HIV-1 (NC_001802) genome is approximately nt 182 to 199. Their primer binding sites start with TGG and end with GGGA, and after aligning the sequences, their matching points can be found in the same position, as shown in Figure 2.
As shown in Figure 2, the sequences on the second line represent the 3'-5' complementary DNA strand that gag proteins match with, and the arrow indicates the 3'-5' direction. The Gag proteins of the viruses match the same primer binding site, even though the viruses are highly different.
To determine whether this finding is a coincidence, the author analyzed the probability. Because viruses of the same type, Deltaretrovirus or Lentivirus, have the same primer binding site, one virus can be considered a mutation from another. The author used the HTLV-1 and HIV-1 genomes as templates and used Pairwise Sequence Alignment (EMBOSS Needle) to compare the genetic similarity of different viruses. The similarity of the NC_001488 (HTLV-2), NC_000858 (STLV-1) and NC_001815 (STLV-2) genomes to the NC_001436 (HTLV-1) genome was 59.2%, 89.3% and 61.0%, respectively. The similarity of the NC_001722 (HIV-2), NC_001549 (SIV) and NC_001482 (FIV) genomes to the NC_001802 (HIV-1) genome was 51.1%, 54.1% and 49.1%, respectively. The similarity of six viruses can be written as
The average probabilities of the amino acid sequences A (GCT, GCC, GCA, GCG), R (CGT, CGC, CGA, CGG, AGA, AGG), G (GGT, GGC, GGA, GGG), S (TCT, TCC, TCA, TCG, AGT, AGC) and P (CCT, CCC, CCA, CCG) remaining unchanged after a mutation are 3/63, 5/63, 3/63, 5/63, and 3/63, respectively. Thus, the average probabilities of the amino acid sequences ARG and SPR remaining unchanged after a mutation are 11/189 and 13/189, respectively. Therefore, the average probability that 3 amino acid sequences of six viruses remain unchanged after a mutation can be represented by as follows:
Assuming that each gene sequence has the same probability of mutation, the number of amino acid sequence mutations increases with increases in the diversity of the viruses. The probability that 3 amino acid sequences of different viruses match the same primer binding site is
The result shows that the probability is approximately 3.67636×10-9, which is extremely small; thus, it can be determined that it is not a coincidence.
Related studies have shown that the genomic Gag/Gag-Pol complex recruits the LysRS/tRNA complex [56]. The selective packaging of the tRNA primer requires HIV-1 Gag and Gag-Pol [57], and an interaction between LysRS and Gag is observed in vitro [58]. Relevant studies have demonstrated that Gag proteins match the HIV primer binding site and possess the potential to directly activate dormant retroviruses.
In HIV-1, Gag/LysRS interaction depends on Gag sequences within the C-terminal domain (CTD) of CA around amino acids 283-363 [59] and motif 1 of LysRS around amino acids 208-259 [58]. It should be noted that the amino acid sequence SPR of the Gag protein is located at amino acids 148-150 within the N-terminal domain (NTD) of CA, specifically at the NTD-NTD interface 1.
Discussion
Because the primer binding sites of different viruses are extremely stable, other amino acid sequences of viral proteins may also match these sites, but there is no sufficient information to confirm this hypothesis at present. In addition, the mutation rate of HIV-1 is extremely high. While analyzing those sequences around the HIV-1 long terminal repeat, the author didn't find any similarity in the adjacent NF-κB binding sites of different viruses. This suggests that the NF-κB binding sites will mutate as the virus mutates. Consequently, scientists must constantly develop new LRAs for new viruses. More importantly, the virus utilizes the NF-κB pathway to enhance its expression, but this doesn't necessarily imply that the virus must always possess an NF-κB binding site. If the NF-κB binding site mutates to become an enhancer of other genes unrelated to NF-κB, LRAs will have no effect on patients.
When comparing the use of LRAs to activate NF-κB binding sites, the utilization of Gag proteins has the advantage of directly reawakening the retrovirus, irrespective of the mutation of the NF-κB binding site at adjacent locations. This pattern is consistent in both Deltaretrovirus and Lentivirus, implying that this treatment could potentially be applied to most patients without requiring the simultaneous administration of multiple LRAs. Moreover, a noteworthy aspect is that the CRISPR-Cas9 enzyme can be employed to modify the amino acid sequences of viral proteins. This modification can prevent the recruitment of uncharged tRNAs to match the NF-κB binding site, thus avoiding the activation of cancer-related genes and mitigating unforeseen risks to patients.
Conclusion
The enhancer region of the HIV-1 LTR contains two adjacent NF-κB binding sites that play a central role in mediating inducible HIV-1 gene expression. The LRAs target the NF-κB binding sites instead of the HIV-1 primer binding site. As a result, LRAs do not directly reawaken dormant retrovirus infections. The results show that the amino acid sequence ARG of Gag proteins of HTLV-1, HTLV-2, STLV-1 and STLV-2 match their primer binding site GGGGGCTCG in the 3'-to-5' direction and that the amino acid sequence SPR of Gag proteins of HIV-1, HIV-2, SIV and FIV match their primer binding site GGCGCCCGA in the 3'-to-5' direction. Related studies have shown that the genomic Gag/Gag-Pol complex recruits the LysRS/tRNA complex. The selective packaging of the tRNA primer requires HIV-1 Gag and Gag-Pol, and an interaction between LysRS and Gag is observed in vitro. In HIV-1, Gag/LysRS interaction depends on Gag sequences within the CTD of CA around amino acids 283-363 and motif 1 of LysRS around amino acids 208-259. It should be noted that the amino acid sequence SPR of the Gag protein is located at amino acids 148-150 within the NTD of CA, specifically at the NTD-NTD interface 1. Although this research is purely bioinformatics analysis, the relevant studies have demonstrated that Gag proteins match the HIV-1 primer binding site and possess the potential to directly activate dormant retroviruses.
List of abbreviations
| LRAs | Latency-reversing agents |
| CD4 | Cluster of differentiation 4 |
| NF-κB | Nuclear factor kappa light chain enhancer of activated B cells |
| CRISPR | Clustered regularly interspaced short palindromic repeat |
| IAP | Inhibitor of apoptosis protein |
| cIAP1 | Cellular inhibitor of apoptosis protein 1 |
| SIK3 | Salt-inducible kinase 3 |
| HTLV | Human T-lymphotropic virus |
| STLV | Simian T-lymphotropic virus |
| HIV | Human immunodeficiency virus |
| SIV | Simian immunodeficiency virus |
| FIV | Feline immunodeficiency virus |
| LysRS | Lysyl-tRNA synthetase |
| PBS | Primer binding site |
| LTR | Long terminal repeat |
| CTD | C-terminal domain |
| NTD | N-terminal domain |
| Gag | Group-specific antigen |
| CA | Capsid |
Funding
None.
Ethics approval and consent to participate
Not applicable.
Consent to publish
The author gives consent for the publication of identifiable details, which can include photographs and details within the text to be published in the above Journal and Article.
Availability of data and materials
The datasets were produced by Python3, and the tool is available at https://github.com/rheast/genome. Pairwise Sequence Alignment (EMBOSS Needle) was used to identify regions of similarity between two biological sequences with the tool available at https://www.ebi.ac.uk/Tools/psa/. Nucleotides were downloaded from the NCBI database at https://www.ncbi.nlm.nih.gov/nuccore/. The sample nucleotides correspond to the accession numbers NC_001436, NC_001488, NC_000858, NC_001815, NC_001802, NC_001722, NC_001549 and NC_001482.
Contributions
S.C. wrote the manuscript.
Acknowledgments
None.
Competing interests
None.
References
- National Institute of Health. What is a Latent HIV Reservoir? NIH. 2021. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/what-latent-hiv-reservoir.
- National Institute of Health. Latency-Reversing Agents. NIH. 2021. Available online: https://clinicalinfo.hiv.gov/en/glossary/latency-reversing-agents.
- Siliciano, R.F. Drugs fail to reawaken dormant HIV infection. Johns Hopkins Medicine. 2014. Available online: https://hopkinsmedicine.org/news/media/releases/drugs_fail_to_reawaken_dormant_hiv_infection.
- Hiscott, J.; Kwon, H.; Génin, P. Hostile takeovers: viral appropriation of the NF-κB pathway. The Journal of clinical investigation 2001, 107, 143–151. [Google Scholar] [CrossRef]
- Kwon, H.; Pelletier, N.; DeLuca, C.; et al. Inducible expression of IκBα repressor mutants interferes with NF-κB activity and HIV-1 replication in Jurkat T cells. Journal of biological chemistry 1998, 273, 7431–7440. [Google Scholar] [CrossRef] [PubMed]
- Quinto, I.; Mallardo, M.; Baldassarre, F.; et al. Potent and stable attenuation of live-HIV-1 by gain of a proteolysis-resistant inhibitor of NF-κB (IκB-αS32/36A) and the implications for vaccine development. Journal of biological chemistry 1999, 274, 17567–17572. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I. NF-κB regulation in the immune system. Nature reviews immunology 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Makropoulos, V.; Brüning, T.; Schulze-Osthoff, K. Selenium-mediated inhibition of transcription factor NF-κ B and HIV-1 LTR promoter activity. Archives of toxicology 1996, 70, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; et al. Current status of latency reversing agents facing the heterogeneity of HIV-1 cellular and tissue reservoirs. Frontiers in microbiology 2020, 10, 3060. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.;Choudhary; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Spivak, A.M.; Andrade, A.; Eisele, E.; et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1–infected adults on antiretroviral therapy. Clinical infectious diseases 2014, 58, 883–890. [Google Scholar] [CrossRef]
- Elliott, J.H.; McMahon, J.H.; Chang, C.C.; et al. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet HIV 2015, 2, e520–e529. [Google Scholar] [CrossRef]
- Norton, N.J.; Mok, H.P.; Sharif, F.; et al. HIV silencing and inducibility are heterogeneous and are affected by factors intrinsic to the virus. Mbio 2019, 10, 10–1128. [Google Scholar] [CrossRef]
- Spina, C.A.; Anderson, J.; Archin, N.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS pathogens 2013, 9, e1003834. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.E.; Niessl, J.; Fromentin, R.; et al. Single-cell characterization of viral translation-competent reservoirs in HIV-infected individuals. Cell host & microbe 2016, 20, 368–380. [Google Scholar] [CrossRef]
- Grau-Expósito, J.; Luque-Ballesteros, L.; Navarro, J.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS pathogens 2019, 15, e1007991. [Google Scholar] [CrossRef]
- Kula, A.; Delacourt, N.; Bouchat, S.; et al. Heterogeneous HIV-1 reactivation patterns of disulfiram and combined disulfiram+ romidepsin treatments. JAIDS Journal of Acquired Immune Deficiency Syndromes 2019, 80, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS pathogens 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Kaiser, P.; Kim, P.; et al. HIV latency in isolated patient CD4+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Science translational medicine 2018, 10, eaap9927. [Google Scholar] [CrossRef]
- Telwatte, S.; Lee, S.; Somsouk, M.A.; et al. Gut and blood differ in constitutive blocks to HIV transcription, suggesting tissue-specific differences in the mechanisms that govern HIV latency. PLoS pathogens 2018, 14, e1007357. [Google Scholar] [CrossRef]
- Chen, H.C.; Martinez, J.P.; Zorita, E.; et al. Position effects influence HIV latency reversal. Nature structural & molecular biology 2017, 24, 47–54. [Google Scholar] [CrossRef]
- Abner, E.; Stoszko, M.; Zeng, L.; et al. A new quinoline BRD4 inhibitor targets a distinct latent HIV-1 reservoir for reactivation from other “shock” drugs. Journal of Virology 2018, 92, 10–1128. [Google Scholar] [CrossRef]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; et al. Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4+ T cells. Elife 2018, 7, e34655. [Google Scholar] [CrossRef]
- Darcis, G.; Bouchat, S.; Kula, A.; et al. Reactivation capacity by latency-reversing agents ex vivo correlates with the size of the HIV-1 reservoir. Aids 2017, 31, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Dobrowolski, C.; Luttge, B.; et al. Estrogen receptor-1 is a key regulator of HIV-1 latency that imparts gender-specific restrictions on the latent reservoir. Proceedings of the National Academy of Sciences 2018, 115, E7795–E7804. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Spivak, A.M.; Soriano-Sarabia, N.; et al. HIV latency-reversing agents have diverse effects on natural killer cell function. Frontiers in immunology 2016, 7, 356. [Google Scholar] [CrossRef] [PubMed]
- Walker-Sperling, V.E.; Pohlmeyer, C.W.; Tarwater, P.M.; et al. The effect of latency reversal agents on primary CD8+ T cells: implications for shock and kill strategies for human immunodeficiency virus eradication. EBioMedicine 2016, 8, 217–229. [Google Scholar] [CrossRef]
- Jones, R.B.; O'Connor, R.; Mueller, S.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS pathogens 2014, 10, e1004287. [Google Scholar] [CrossRef]
- Clutton, G.; Xu, Y.; Baldoni, P.L.; et al. The differential short-and long-term effects of HIV-1 latency-reversing agents on T cell function. Scientific reports 2016, 6, 30749. [Google Scholar] [CrossRef]
- Desimio, M.G.; Giuliani, E.; Ferraro, A.S.; et al. In vitro exposure to prostratin but not bryostatin-1 improves natural killer cell functions including killing of CD4+ T cells harboring reactivated human immunodeficiency virus. Frontiers in immunology 2018, 9, 1514. [Google Scholar] [CrossRef]
- National Institute of Health. NIH-supported scientists reverse HIV and SIV latency in two animal models. NIH. 2020. Available online: https://www.nih.gov/news-events/news-releases/nih-supported-scientists-reverse-hiv-siv-latency-two-animal-models.
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef]
- McBrien, J.B.; Mavigner, M.; Franchitti, L.; et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 2020, 578, 154–159. [Google Scholar] [CrossRef]
- Hennessy, E.J.; Adam, A.; Aquila, B.M.; et al. Discovery of a novel class of dimeric Smac mimetics as potent IAP antagonists resulting in a clinical candidate for the treatment of cancer (AZD5582). Journal of medicinal chemistry 2013, 56, 9897–9919. [Google Scholar] [CrossRef]
- Pache, L.; Marsden, M.D.; Teriete, P.; et al. Pharmacological activation of non-canonical NF-κB signaling activates latent HIV-1 reservoirs in vivo. Cell Reports Medicine 2020, 1. [Google Scholar] [CrossRef] [PubMed]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; et al. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nature medicine 2014, 20, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Andrade, A.; Eisele, E.; et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1–infected adults on antiretroviral therapy. Clinical infectious diseases 2014, 58, 883–890. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; et al. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+ JQ1 and ingenol-B+ JQ1 to potently reactivate viral gene expression. PLoS pathogens 2015, 11, e1005063. [Google Scholar] [CrossRef]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.S.; et al. Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. The Journal of clinical investigation 2015, 125, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Haffner, M.C.; Zhang, Y.; et al. Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. The Prostate 2011, 71, 333–343. [Google Scholar] [CrossRef]
- Philip, P.A.; Rea, D.; Thavasu, P.; et al. Phase I study of bryostatin 1: assessment of interleukin 6 and tumor necrosis factor α induction in vivo. JNCI: Journal of the National Cancer Institute 1993, 85, 1812–1818. [Google Scholar] [CrossRef]
- Silva, V.A.O.; Rosa, M.N.; Tansini, A.; et al. Cytotoxic activity of semi-synthetic ingenol derived from Euphorbia tirucalli on a large panel of human cancer cell lines. 2013. [Google Scholar] [CrossRef]
- Alotaibi, D.; Amara, S.; Johnson, T.L.; et al. Potential anticancer effect of prostratin through SIK3 inhibition. Oncology letters 2018, 15, 3252–3258. [Google Scholar] [CrossRef]
- Duchon, A.A.; St. Gelais, C.; Titkemeier, N.; et al. HIV-1 exploits a dynamic multi-aminoacyl-tRNA synthetase complex to enhance viral replication. Journal of virology 2017, 91, 10–1128. [Google Scholar] [CrossRef]
- Weiner, A.M.; Maizels, N. tRNA-like structures tag the 3'ends of genomic RNA molecules for replication: implications for the origin of protein synthesis. Proceedings of the National Academy of Sciences 1987, 84, 7383–7387. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.S.; Temin, H.M. Retroviral recombination and reverse transcription. Science 1990, 250, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Negroni, M.; Buc, H. Copy-choice recombination by reverse transcriptases: reshuffling of genetic markers mediated by RNA chaperones. Proceedings of the National Academy of Sciences 2000, 97, 6385–6390. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, C. Retroviral taxonomy, protein structures, sequences, and genetic maps. In Retroviruses; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 1997; pp. 757–805. [Google Scholar]
- Shimotohno, K.; Takahashi, Y.; Shimizu, N.; et al. Complete nucleotide sequence of an infectious clone of human T-cell leukemia virus type II: an open reading frame for the protease gene. Proceedings of the National Academy of Sciences 1985, 82, 3101–3105. [Google Scholar] [CrossRef]
- Saksena, N.K.; Hervé, V.; Sherman, M.P.; et al. Sequence and phylogenetic analyses of a new STLV-I from a naturally infected tantalus monkey from Central Africa. Virology 1993, 192, 312–320. [Google Scholar] [CrossRef]
- Van Brussel, M.; Salemi, M.; Liu, H.F.; et al. The Simian T-Lymphotropic Virus STLV-PP1664 fromPan paniscusIs Distinctly Related to HTLV-2 but Differs in Genomic Organization. Virology 1998, 243, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Martoglio, B.; Graf, R.; Dobberstein, B. Signal peptide fragments of preprolactin and HIV-1 p-gp160 interact with calmodulin. The EMBO journal 1997, 16, 6636–6645. [Google Scholar] [CrossRef]
- Kirchhoff, F.; Jentsch, K.D.; Bachmann, B.; et al. A novel proviral clone of HIV-2: biological and phylogenetic relationship to other primate immunodeficiency viruses. Virology 1990, 177, 305–311. [Google Scholar] [CrossRef]
- Fomsgaard, A.; Hirsch, V.M.; Allan, J.S.; et al. A highly divergent proviral DNA clone of SIV from a distinct species of African green monkey. Virology 1991, 182, 397–402. [Google Scholar] [CrossRef]
- Olmsted, R.A.; Hirsch, V.M.; Purcell, R.H.; et al. Nucleotide sequence analysis of feline immunodeficiency virus: genome organization and relationship to other lentiviruses. Proceedings of the National Academy of Sciences 1989, 86, 8088–8092. [Google Scholar] [CrossRef]
- Jin, D.; Musier-Forsyth, K. Role of host tRNAs and aminoacyl-tRNA synthetases in retroviral replication. Journal of Biological Chemistry 2019, 294, 5352–5364. [Google Scholar] [CrossRef]
- Mak, J.; Jiang, M.; Wainberg, M.A.; et al. Role of Pr160gag-pol in mediating the selective incorporation of tRNA (Lys) into human immunodeficiency virus type 1 particles. Journal of virology 1994, 68, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Kovaleski, B.J.; Kennedy, R.; Hong, M.K.; et al. In vitro characterization of the interaction between HIV-1 Gag and human lysyl-tRNA synthetase. Journal of Biological Chemistry 2006, 281, 19449–19456. [Google Scholar] [CrossRef] [PubMed]
- Bell, N.M.; Lever, A.M.L. HIV Gag polyprotein: processing and early viral particle assembly. Trends in microbiology 2013, 21, 136–144. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Coordinates of matched points.
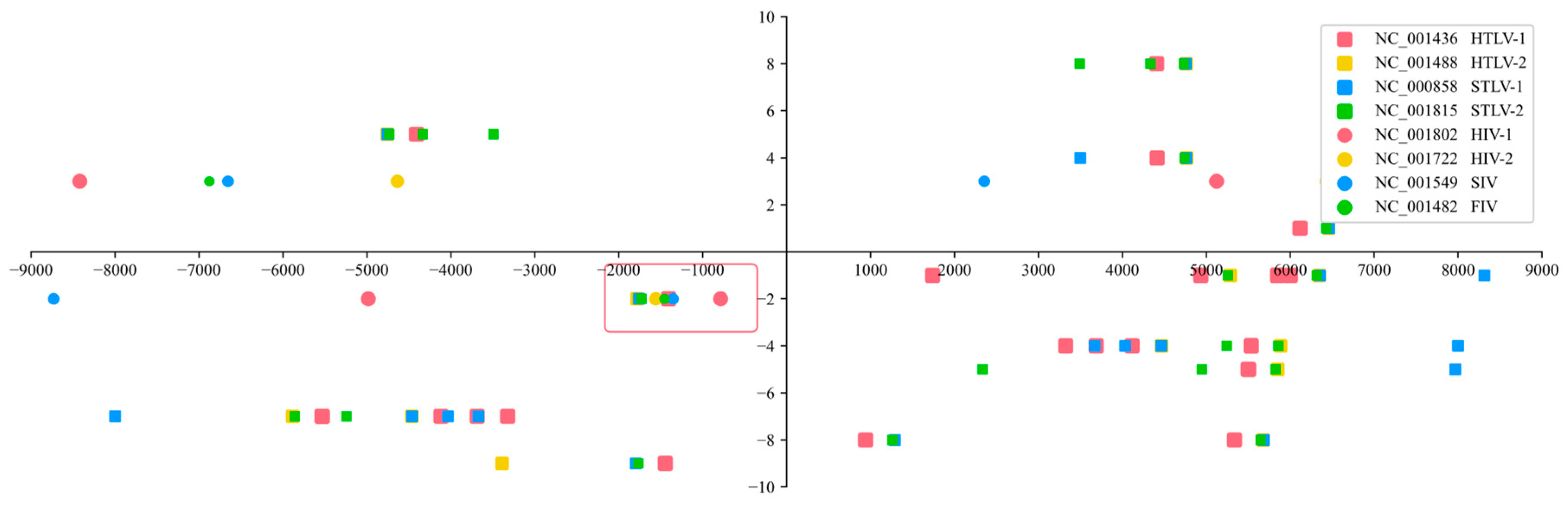
Figure 2.
Deltaretrovirus and Lentivirus.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



