Submitted:
22 February 2023
Posted:
22 February 2023
Read the latest preprint version here
Abstract
Aging kills 100,000 people a day - more than any other cause of death combined. The exact causes of aging have been much discussed, but the most pressing issue with regard to aging appears to me to be lipofuscin accumulation. That is, the accumulation of indigestible cellular garbage that needs to be removed from our cells, then the body. In this piece, I will explain why I think “getting rid of the garbage” should be at least one of our main goals with regard to longevity research for now.
Keywords:
Anti-aging therapy
; lipofuscin
; SENS
; oncolytic vector
; telomerase
; TFEB
; intracellular microbe
Introduction
There are many theories about why we age, but one stands out as being the most plausible based on the evolutionary and mechanistic evidence. That is the “garbage catastrophe theory of aging.” Drs. Brunk and Terman postulated years ago that the problem of aging can essentially be summed up as a “garbage disposal issue[1].” The main idea is that basically old molecules are sometimes damaged in ways that prevent the lysosomes from breaking them down properly, and over time these damaged, old molecules accumulate inside the lysosomes. Eventually, the lysosomes become full of this indigestible garbage, i.e., “lipofuscin”, and cannot perform their normal function - then there is a garbage back-up and the cell starts to decline health-wise.
Lipofuscin can eventually occupy a large portion of the cytoplasm in certain cell types. Whether or not lipofuscin itself can exert negative effects on the cell that contains it, simply accumulating to a critical level would logically cause a garbage backup. The cells may try to produce more lysosomes - but will eventually reach capacity. It also makes sense that reaching a critical threshold of lipofuscin in non-dividing cells throughout the body could accelerate the accumulation of the other six categories of age-related damage as defined by Dr. Aubrey de Grey[2]. (Intracellular aggregates, i.e., lipofuscin, is his fourth category of age-related damage.)
Dysfunctional mitochondria are a hallmark of aging[3]. With lipofuscin accumulation, damaged mitochondria - sometimes with mtDNA mutations, sometimes just with damage to their membranes, proteins, lipids, and DNA - will not be recycled as rapidly and may then start to accumulate. Mitochondrial DNA mutation accumulation is Dr. de Grey’s fifth category of age-related damage - and they can in fact accrue over time. However, I will address this on a fundamental level later.
Tau will begin to build up inside neurons due to the garbage back-up, eventually becoming hyper-phosphorylated and forming neurofibrillary tangles. Similarly, β-amyloid that is normally degraded may continue to persist, building to levels that lead to plaque formation. Hopefully, if lipofuscin is removed, plaques that have already formed could regress - in line with Le Chatelier’s Principle. In other words, if lipofuscin is cleared and β-amyloid in solution is taken up and autophagocytosed, insoluble plaques may start to dissipate back into solution as well - at which point the β-amyloid in solution would again be taken up and degraded. Transthyretin amyloid could also be another amyloid that forms in the extracellular spaces of our bodies as a result of lipofuscin accumulation in various tissues.
Furthermore, it is clear that with the extracellular matrix (ECM) not being properly cared for by its resident cells, other damage such as fragmentation, glycation, elastocalcinosis, and cross-linking could rapidly reach pathological levels. After extensive lipofuscin removal, hopefully much of this will damage prove to be reversible. If healthy proteostasis could be restored within our parenchymal cells, ECM turnover efficiency should be restored to youthful levels - although it may not be able to reverse all the damage that has accumulated in elderly individuals. Certain cross-links may be cleavable by endogenous, secreted enzymes; after treatment, said cross-link–degrading enzymes could be secreted at the appropriate levels.
Glucosepane is a cross-link that becomes prevalent with age in human tissues that is uncleavable - or at least not cleaved very efficiently/often. Exogenously administered cross-link–breaking enzymes could be helpful. However, there are almost certainly a multitude of indigestible cross-links that accumulate with age. Just like with lipofuscin, an unfeasible number of enzymes might be required to address the problem in this way. Fortunately, even if there are some cross-links that can’t be degraded by endogenous enzymes - if the ECM turnover efficiency is restored through extensive lipofuscin removal - molecules bound together by uncleavable cross-links could be excised and endocytosed by tissue-resident cells or phagocytosed by (bioengineered or regular) tissue-resident macrophages.
Importantly, ECM damage doesn’t seem to be an issue with the “immortal” Hydra vulgaris[4]. H. vulgaris has a body column with cells that continuously divide - driving cells out towards its extremities. The cells at its extremities slough off. Thus, it has a built-in mechanism for ridding itself of lipofuscin. Its layer of ECM, the mesoglea, is not shed along with the cells at its extremities; it is turned over. Without lipofuscin accumulation in its cells, and with constant ECM turnover, this organism appears not to age - or at least age very slowly. Of course, the human ECM is much more complex than that of H. vulgaris. However, the longevity of its mesoglea may indicate that if lipofuscin removal is done periodically (e.g., every decade) in humans starting at a young age, ECM damage may not really be a problem for a long time at least. If started at an older age, a large portion of the damage may be reversed, and then subsequent ECM damage accumulation would hopefully be very slow.
Additionally, cancer is more likely to initiate or progress if many microenvironments throughout the body are corrupted by lipofuscin accumulation. Along those lines, stem cell niche corruption may prevent them from replicating efficiently to replenish tissues.
Senescent cells may start to accumulate if tissue-resident immune cells are rendered inert by lipofuscin accumulation and the non-functionality of the parenchymal cells around them also caused by lipofuscin accumulation, leading to dilapidation of the ECM. Here I refer to irreversibly senescent cells, which have suffered DNA damage and can no longer function properly. However, many cells that show signs of senescence like the senescence-associated secretory phenotype[5] may be reversibly senescent. They may have entered into that state due to epigenetic damage brought on by lipofuscin accumulation. I have much hope that this epigenetic state can revert back to a youthful state once lipofuscin is removed, however.
There was a paper published recently in Nature Aging that suggests that epigenetic damage is mostly reversible at least[6]. The hallmarks of aging that it related to were mitochondrial dysfunction, nutrient sensing, and stem cell composition. If lipofuscin is removed, damaged mitochondria will be recycled and nutrient sensing processes should go back to normal. Stem cells in this study likely show a younger epigenetic age because they divide frequently and thus dilute out their lipofuscin. That relates to my hypothesis about why Yamanaka factors rejuvenate tissues[7]; I believe it may be because they transiently induce a pluripotent stem cell state. Thus, cells that normally wouldn’t divide start to divide transiently, and thereby dilute out their lipofuscin. Sox2, one of the four Yamanaka factors, also initially stimulates autophagy[8] - which decreases “false” lipofuscin. I’m defining false lipofuscin as intracellular garbage that a cell could potentially recycle if it were encouraged in some way. “Real” lipofuscin is intracellular garbage that cannot be digested no matter how we manipulate a cell’s metabolism - namely the junk that must be removed. Perhaps, however, some age-related epigenetic changes will not revert after lipofuscin removal. If so, the aforementioned rejuvenation technique, known as partial reprogramming, could help to reverse them. However, partial reprogramming may be dangerous; it poses a serious risk of causing cancer through teratoma formation.
Of course, the other types of age-related damage aside from lipofuscin can occur simply through random metabolic errors, but perhaps not very often. And damage of those sorts can often be dealt with by endogenous processes. It seems likely that eventually, the steady accumulation of lipofuscin throughout life reaches a point where it increases the rate of accumulation of other age-related damage due to decreasing autophagy/proteasomal efficiency. That may in turn increase the rate of lipofuscin accumulation as well by elevating cellular stress. Eventually, lipofuscin and other age-related damage accumulation must reach a critical threshold level in or around cells throughout our body, leading to a downward spiral, pathologically-speaking[9]. However, even then, removing the lipofuscin may halt/at least somewhat reverse this downward spiral.
I have written an article about strategies for removing lipofuscin from cells in culture to test this theory, and how we might go about removing lipofuscin in whole organisms if cell culture work indicates that it leads to substantial rejuvenation[10]. It is also important to remember that cells in culture don’t always die from causes that are reflective of in vivo aging.
To treat whole organisms in a high-throughput manner, however, we will need to have a way of delivering trash-collecting (i.e., phagocytic) intracellular microbes or proteins that induce the export of lipofuscin to all the target cells throughout the body. As this probably includes most cells in the body, that really necessitates that we use the bloodstream as a systemic delivery system. If we can get a relatively even distribution of bioengineered leukocytes into tissues throughout the body - but only at low levels - we can still simply promote their replication via small molecule while visualizing the process through HSV1-TK[11].
Systemic extravasation of bioengineered carrier macrophages:
The trouble with trying to intravenously administer large liposomes or viral vectors, which can contain much more complex payloads, is that they cannot reach many intraparenchymal target cells throughout the body because the vascular endothelium serves as a stringent barrier. Some intracellular microbes can transcytose across the intestinal epithelium - but this is not necessarily helpful for the vascular endothelium[12]. There are some pathogens that can cross the blood-brain barrier, but this is typically under conditions of systemic inflammation or bacterial-mediated cytolysis of vascular endothelial cells to cause gaps in the endothelium[13]. Basically, we do not know nearly as much about effecting safe microbial transcytosis/paracellular passage across the vascular endothelium as we do about white blood cell extravasation[14] - although we do not know all the mechanisms of leukocyte diapedesis either. Thus, in the more immediate future, to get the trash-collecting microbes across the vascular endothelium, we may have to employ a somewhat complex, two-step delivery system that involves carrier white blood cells.
The two-step system could involve first delivering a gene vector to vascular endothelial cells - this would then inducibly allow for the transmigration of bioengineered white blood cells containing the trash-collecting microbes. Basically, clinicians would seek to infect all or the majority of a patient’s vascular endothelial cells with a herpes simplex virus type 1 (HSV-1) vector. (Different pseudotypes of HSV-1 or bi-specific antibodies may be necessary to ensure that all vascular endothelial cells throughout the body are transduced; one marker may not be enough - as vascular endothelial cells in different anatomical locales do not have the exact same gene expression patterns[15].) They would then inducibly express from the viral genomes (upon small molecule administration) synthetic, luminal adherence proteins and chemokines[16]. These would allow for the attachment and transmigration of bioengineered macrophages. After transmigration, perhaps chemorepellents could be secreted abluminally to direct them away from the vascular endothelial cells, across the rest of the vascular wall, and toward target tissue. The macrophages could then randomly migrate around until binding to a cell type of interest, at which point they could (directionally) donate trash-collecting microbes to the target cell.
In fact, the trash-collecting microbes could replicate within the carrier white blood cells up to a moderate copy number[17] - restrained by AI-2-based quorum sensing, perhaps[18]. The carrier white blood cells could then continuously donate the trash-collecting microbes to target cells via microvesicular secretion, secretory autophagy, transient TNTs, or even partial cell-cell fusion; they could enter target cells, engulf their lysosomes, and escape.
Figure 1.
A) Gene vectors are administrated intravenously; they bind to vascular endothelial cells, are internalized, and transduce them to allow control over synthetic adherence proteins and chemokines that facilitate the extravasation of bioengineered white blood carrier cells. B) The bioengineered white blood cells carrying intracellular microbes are “smuggled” across the vascular endothelium in many regions throughout the body.
Figure 1.
A) Gene vectors are administrated intravenously; they bind to vascular endothelial cells, are internalized, and transduce them to allow control over synthetic adherence proteins and chemokines that facilitate the extravasation of bioengineered white blood carrier cells. B) The bioengineered white blood cells carrying intracellular microbes are “smuggled” across the vascular endothelium in many regions throughout the body.
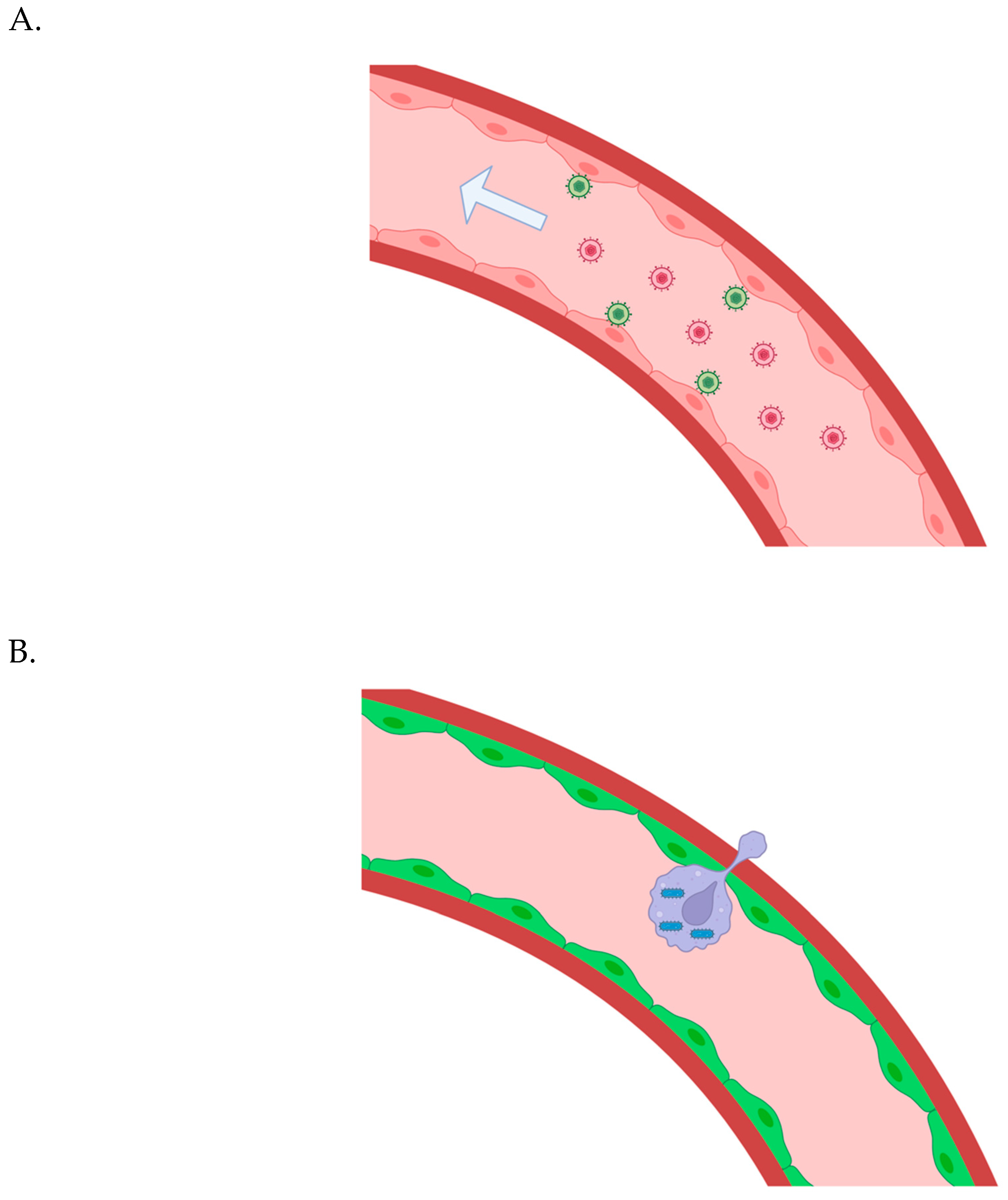
The vascular endothelial glycocalyx may make the delivery of large gene vectors difficult. AAV vectors are able to reach the vascular endothelial cells beneath the glycocalyx, at least in many regions (in mice)[19,20]. I suggested HSV, however, because I thought multiple proteins may be required to ensure extravasation of the bioengineered white blood cells carrying intracellular, trash-collecting microbes - perhaps multiple proteins that vary based on anatomical locale. But perhaps a single, synthetic chemokine will be enough. Multiple AAVs can be employed if necessary. (An adenoviral vector is also a possibility, perhaps[21].) If a larger gene vector is required and can only transduce a small number of vascular endothelial cells throughout the body - that could still be enough, or at least very helpful. Imaging via HSV1-TK could let us know when the bioengineered white blood cells have reached the tissue parenchyma in various regions - and a small molecule could be intravenously administered to promote the replication of the bioengineered white blood cells in those regions. The intracellular microbes in the bioengineered white blood cells would keep pace with that division - restrained by quorum sensing. The two-step delivery process could, in this sense, still potentially save a given patient a lot of intraparenchymal infusions in different anatomical locales.
It may not require a gene vector, however, to see the effects we want. If the patient first undergoes leukapheresis, we could then deliver liposomally-encapsulated siRNA to endothelial cells that knocks down endomucin[22], an inhibitor of endothelial cell-leukocyte adhesion and an important protein in enhancing the glycocalyx, and finally administer the bioengineered macrophages that will cross the vascular wall. Magnetic nanoparticle (MNP)-based permeabilization could also be applied for various areas that have a thicker glycocalyx, for example[23]. Endothelial cells in those areas could be bound by peptides that recognize that vascular “zip code” - conjugated with MNPs. Importantly, though, endomucin is specifically expressed by venous and capillary endothelial cells[24]. We may also need to remove lipofuscin from various cell types like smooth muscle cells in the arterial walls.
The only other option that I think makes any sense on a large scale is to manually inject parenchymal regions throughout the body with bioengineered white blood cells carrying the trash-collecting microbes. The bioengineered white blood cells could be induced to replicate up to a sufficient number once there via small molecule. That solution isn’t pretty, but if lipofuscin is truly the main culprit in age-related disease, we simply have to do whatever is required. More likely, we could combine the aforementioned two-step delivery system and intraparenchymal injections for full coverage. Please keep in mind that the small molecule-induced replication option works for the two-step system as well of course, and that in both scenarios there would be continuous donation of trash-collecting microbes from each carrier white blood cell while they randomly migrate around a given tissue.
There are two important considerations for the two-step delivery system. One is that clinicians should perhaps slowly infuse the patient with bioengineered macrophages after inducing the synthetic luminal adherence proteins and chemokines, so as not to create too much stress on the vasculature. The other is that one may need to employ superinfection exclusion (SIE)[25] in terms of the HSV-1 vector to make sure that all or the majority of vascular endothelial cells are transduced - but without ending up with too high of a vector copy number per given cell. SIE would ideally be on the level of intracellular capsid trafficking. For example, the degradation system cited here[26] could ensure the degradation of incoming capsids before they transfer their DNA to the nucleus[27]. A synthetic gene circuit imbuing network-dosage compensation could also be of use in this situation potentially[28]. Given that the average EC lifespan is >1 year[29], there is a clear therapeutic window here if the vasculature can be thoroughly transduced first. The extent of vascular endothelial transduction could be visualized via the HSV-1 thymidine kinase. And a kill switch could be included to destroy the vector genomes (based on CRISPR, for example) after lipofuscin removal treatment.
Testing this type of delivery system in mice may be more facile than actually implementing the real formulation because new mice strains can be engineered that express various foreign genes in their vascular endothelial cells. They could have a synthetic chemokine that brings across bioengineered macrophages[30], endomucin siRNA, a chemorepellent from the abluminal side of the vascular endothelium, etc. Ferritin could even potentially be overexpressed in the mouse vascular endothelial cells[31] to enable magnetic-based disruption of endothelial junctions in large regions of the body - as in the previously-cited article23.
Two remaining issues in the near future:
There are two remaining age-related issues I can think of that we should consider in the immediate future. One is telomere shortening. Telomeres are simply non-sense, repetitive sequences of DNA on both ends of every one of our chromosomes. Telomeres exist because otherwise, important chromosomal DNA would be lost every time the cell divides. To replenish our telomere lengths, stem cells - the cells that divide - use an enzyme called “telomerase”. If our telomeres shorten too much through not expressing enough telomerase, too much division, damage, or some combination of these factors, it triggers a program where the cell either undergoes apoptosis or enters a senescent state. It has been proposed that not enough telomerase is made in our stem cells, and that over time their telomeres shorten so much they become senescent and we age. (Shorter telomeres are also associated with an increased risk of cancer.) However, even though stem cell telomeres do shorten with age in some individuals, for some individuals, their telomeres stay the same length or even lengthen with age.
Blood stem cells from aged individuals can still function normally if they are “rejuvenated” ex vivo - and then transplanted into a young niche[32]. It appears as though every compound that I’ve seen used to “rejuvenate” aged stem cells (e.g., CASIN and rapamycin) actually decreases false lipofuscin in the cells through the stimulation of autophagy[33,34]. From the theoretical perspective, one could imagine that if a stem cell is slowly-dividing, it could build up lipofuscin over time even with some replication. Alternatively, a rapidly-dividing stem cell could be restrained by a niche full of lipofuscin, and then start to accumulate lipofuscin itself. Lipofuscin removal from the stem cells/their niches may make them better able to degrade damaged telomerase components and generate new ones, but it is still possible that a gene vector encoding telomerase could be needed to lengthen the telomeres of our stem cells - preventing them from becoming cancerous or senescent. To negate this potential issue, we could deliver a gene vector encoding inducible telomerase to our stem cells using essentially the same delivery system I am proposing for lipofuscin clearance - and overexpress it periodically.
Importantly, non-dividing cell telomeres shorten to some extent with age as well due to stress/damage[35]. This may not be something we have to worry about for quite some time, but eventually, telomere shortening in long-lived post-mitotic cells could become problematic. Perhaps if the telomeres of long-lived non-dividing cells like neurons reach a short enough level, they would be able to detect that and endogenously express a sufficient amount of telomerase (given periodic lipofuscin removal). If not, we could also periodically induce telomerase expression in long-lived non-dividing cell types - although much less frequently than in aged stem cells. In other words, we want to ensure that none of our cells have excessively long telomeres, as that could contribute to cancer.
Constitutive telomerase expression in many tissues from birth leads to oncogenic effects. However, periodic overexpression of telomerase in all our adult stem cells could be safe for a long period of time if (DNA repair proteins and) tumor suppressor proteins are also overexpressed. Dr. Blasco et al. demonstrated that in mice overexpressing tumor suppressor proteins, telomerase upregulation increases longevity[36]. However, it should be noted that mice have much longer telomeres than humans; their telomere biology is also different in other ways. The only other option would be to periodically reseed all of our stem cells. For some stem cell types at least, this could be rather difficult. Another protective mechanism that should probably be developed would be to assign microbes to dispense the telomerase in a manner that is carefully guarded - as I will discuss later on.
An adeno-associated virus serotype 9 (AAV9) encoding telomerase has been shown to extend the lifespan adult/old mice when injected intravenously. Additionally, cytomegalovirus (CMV) encoding telomerase had even more impressive results when administered intranasally and intraperitoneally old mice[37,38]. AAVs are non-replicating vectors, so even slowly-dividing stem cells will eventually lose the vector unless there is some sort of asymmetric inheritance process for the vector genome (even with otherwise symmetrical stem cell division). The CMV vector was a replicating vector, but the infection is cleared eventually. Some latent genome copies may remain in non-dividing cells, however. It is possible that remaining viral genomes replicate in dividing cells to a small extent, but will likely eventually be lost - perhaps in all but the slowest-dividing cells. The infection potentially uses leukocytes as trojan horses to gain access to various tissues.
Again, mouse telomere biology is different than human telomere biology, so it is still unclear how safe it is to use either approach in humans. However, there is a human case study where a 44-year-old female underwent two series of AAV9-hTERT injections - one in 2015 and one in 2020[39]. It was shown that her leukocyte telomere lengths greatly increased over time. In this approach, the idea is that a non-replicating vector is used, so it is not permanently kept in stem cells - they divide and lose the vector, and more rapidly-dividing stem cells lose the vector more quickly.
Given the approach, it would at first glance seem a little strange that her leukocyte telomeres continued to increase in length over time, as hematopoietic stem cells are rapidly-dividing, and thus would dilute out the AAV vector quickly. However, it appears as though hTERT also induces autophagy[40] and stimulates replication, at least in (embryonic) stem cells[41]; if hTERT were overexpressed for a while in her aged niche-restrained hematopoietic stem cells (HSCs) and their niches - division could then ensue or speed up once much false lipofuscin had been degraded/had been programmatically forced to be diluted out from the HSCs. If this occurred for a sufficient period of time, her HSCs would theoretically be able to divide normally again, dilute out the remainder of their lipofuscin, and maintain their own telomere lengths.
It is worth noting that AAV-mediated delivery appears to be much more effective in mice[42] than larger animals like non-human primates[43,44,45] - perhaps due to differences in extravasation. Thus, we may need to use my aforementioned delivery system to effectively deliver a gene vector encoding hTERT in humans in a manner I described in my anti-aging article. In terms of cancer, even though cell division rates and DNA mutation rates are not correlated[46], perhaps more rapidly-dividing stem cells are more prone to become malignant or at least aggressively malignant[47] - and so losing the vector in those cells prevents the increase in cancer we see when telomerase is constitutively overexpressed throughout the body[48]. But if aged stem cells can degrade damaged telomerase enzymes and create more, new ones after shrinking their lipofuscin deposits - then why is there no increase in oncogenesis? It appears as though hTERT expression can actually promote proliferation, not simply enable it to occur indefinitely - so its overexpression in already rapidly-dividing stem cells could be problematic.
The second issue is whether we should worry about memory B and T cells. Specifically, should we worry that they will eventually build up in our bodies over time through exposure to different pathogens to the point where there is no more space for naïve B and T cells? Dr. de Grey mentioned this as being an issue, especially for CMV. If the thymus and other lymphoid tissues are free of lipofuscin and still functioning properly, perhaps this will not be a problem - as the clonal expansion of memory B and T cells may be a compensatory action to counter the marked drop in the output of naïve B and T cells due to bone marrow/spleen aging and age-related regression of the thymus, respectively[49].
However, perhaps for the elderly who already suffer from a clonal expansion of memory B and T cells, removing lipofuscin from lymphoid tissues may not be enough. We may also need to eliminate the memory B and T cells that have clonally expanded and massively skewed their pathogen resistance profiles. It would be relatively simple to do that with immunotoxins against B and T cells in general or CAR T-cells against them. Perhaps we could specifically target clones that are most prevalent using a gene vector-type approach wherein sensors look for particular DNA or RNA sequences and kill the cell if they are detected. We could pre-emptively lengthen the telomeres of our hematopoietic stem cells to ready them for massive expansion after that. Notably, if memory B and T cells sit around for long periods of time without replicating - i.e., when the body is not exposed to their corresponding pathogen, they could fill with lipofuscin over time and be unable to function properly (e.g., re-enter a replicative state when necessary) after that.
Anti-aging gene therapy:
Altering endogenous gene expression or adding foreign genes to our cells - when it is not necessary - may not be great idea. Introducing DNA in general into our nuclei through gene therapy could be problematic; it could integrate at sites of double-strand breaks, for example. Also, if you mess with finely-tuned genetic circuits, it may result in cancer. If you mess with said circuits in many of our cells at once, it also may have other very harmful effects potentially. Dr. Aubrey de Grey has said as much. However, perhaps there are some good, safe targets other than telomerase that can be underexpressed or overexpressed to increase longevity.
TFEB, a protein that is considered the “master regulator of autophagy”, might also be a target that would be safe to periodically overexpress throughout the body. Notably, gene vectors may not be necessary for altering TFEB gene dosage. A company called Generian is developing a small molecule inhibitor of the E3 ligase that degrades TFEB, which would thereby boost its levels in our tissues. Perhaps overexpression of TFEB from a gene vector would be more potent, however. In mice, TFEB can safely be overexpressed in the ventral midbrain at least[50].
FOXN1 may also be a good target. FOXN1 overexpression has been shown to counter-act thymic involution[51]. I argue that this is because it increases cell division[52] as well as proteasome activity[53], which can partially compensate for declining autophagy efficiency due to lipofuscin accumulation. Maybe TFEB overexpression in the thymus would yield similar results. Perhaps the thymus accumulates lipofuscin much more rapidly than other organs because it is so metabolically active and thus its constituent cells need to divide much more frequently to dilute out the lipofuscin.
Senescent cells:
Senescent cells could possibly accumulate even if lipofuscin removal is done periodically starting from a young age. However, the aforementioned delivery system could allow for a gene vector to be delivered to cells throughout the body10. This gene vector would encode the same genetic trick that has been applied in mice - where a dimerizable caspase (Casp8) is under the control of a minimal p16Ink4a promoter[54]. It could be permanently installed into cells by DNA insertion or just be episomal. If permanent installation is desired, SIE may not be desirable, but network-dosage compensation would be important. SIE could still be employed if the template DNA replicates up to a certain copy number, restrained by a plasmid copy number control mechanism. That would provide enough template for efficient insertion. If it is episomal, it could replicate with the plasmid copy number control mechanism or an S/MAR to maintain itself in dividing cells. If permanent installation is desired, it would be important to install the relevant genes in the thymus or bone marrow first - with initial immunosuppression - to enable tolerance.
Cancer:
If lipofuscin removal is indeed such a potent rejuvenation therapy, our risk of getting cancer would dramatically decrease. If you first receive (an extensive) lipofuscin removal treatment at a very advanced age - your risk of getting cancer might be slightly higher, but I think the biological age range that we can all achieve is 25-40, which is very low risk. Cancer in that age group is possible, however, although it’s much more rare than in the elderly (e.g., through essentially random metabolic errors or excessive sunlight exposure). Notably, cancer is especially unlikely in young people who are unstressed, eat healthy, exercise regularly, and do not get excessive sun exposure. Early detection of cancer is also extremely beneficial; we can all have an ensemble of blood tests, as well as full body scans, frequently. Full body scans may not be too helpful for very early screening, but larger growths can be identified.
I have written two articles on the subject of cancer therapy about a novel approach, which I call Oncolytic Vector Efficient Replication Contingent on Omnipresent Mutation Engagement (OVERCOME)[55,56]. OVERCOME, or a variation of it that I mention, may be enough to cure cancer or at least effectively treat it without side effects in a way that can be repeated indefinitely. It would be difficult to explain how it works exactly in this piece. But you can read about the details here55,56. Basically OVERCOME involves using an intracellular microbe to detect mutations in a patient’s cancer that are ubiquitous throughout their tumors - and respond by replicating within/destroying any cancer cell containing the targeted mutations.
As opposed to invasive biopsies - we could potentially intravenously inject MNP-loaded macrophages, direct them to visible growths[57], induce them via small molecule to phagocytose some of the cells (maintaining the engulfed cell in an intact state), and then extract them to analyze the DNA of their cargo. Alternatively, the macrophages could deliver magnetotactic intracellular bacteria to the growth, where they could be induced to escape. They would then enter target cells, phagocytose nuclei from cells in the growth (maintaining their cargo in an intact state), and finally be directed to an extraction point. This type of system could also be used for delivering oncolytic vectors to the tumors.
It is perhaps possible that solid tumors will be overcome via other types of treatments like immunotherapy or targeted therapies. In terms of immunotherapy, the problem I see is that many mutations affect proteins that are internal to the cell, so they cannot be targeted externally. It is true that cells “present” a sample of the peptides inside of them to immune cells for review, to ensure that they are destroyed if they are infected or cancerous. However, cancer cells often downregulate the complex that they use to present their internal peptides - in order to escape recognition by the immune system[58]. Moreover, some cancer cells may also downregulate production of the targeted external or internal mutant protein to escape treatment - which can’t really be addressed from outside said cancer cells. OVERCOME, on the other hand, involves using an intracellular microbe to detect mutations - and mutant genes can be forced to produce their corresponding protein.
The success we’ve seen with CAR T-cells is because they can be used to eliminate entire populations of white blood cells to destroy certain blood cancers, as these white blood cells can be wiped out without killing the patient, but we clearly can’t do that with the cells in solid tissues. Unfortunately, even if a protein is targeted by CAR T-cells that is normally present on all B cells, for example, blood cancers can evolve to downregulate its expression. That’s why sometimes a patient’s blood cancer recurs after treatment with CAR T-cells[59]. The situation is even worse for solid tumors, as the microenvironment of solid tumors is often quite complex immunologically and can shut down our white blood cells - although researchers are working very hard to figure out how to bioengineer CAR T-cells to be resistant to immunosuppressive effects. Of note, there was recently a very interesting clinical trial where all 18 rectal cancer patients went into remission after treatment with an antibody against PD-1[60].
While targeted therapies can involve small molecules capable of crossing plasma membranes and binding to intracellular proteins, it is still the case that cancer cell escape variants will likely emerge - even when combinations of small molecules are used - as many genetic pathways have a lot of redundancy. I am hopeful that new treatments for cancers with various genetic/epigenetic signatures will be discovered - but it may be a slow process, taking decades before many cancers can be treated. OVERCOME, on the other hand, may be able to treat a huge number of cancers with just a bit of preliminary bioengineering work.
Maybe in many cases eliminating the majority (i.e., not even the vast majority) of a patient’s tumor cells is enough for your natural immune system to step in and finish the job and/or devastating enough so that the remaining tumor cells are simply unable to continue to proliferate after so much of the tumor or tumors have been decimated. But it is obviously better to kill as many of them as possible. Without computerized microrobots and/or nanorobots, one probably is never going to be able to eliminate every single one of a patient’s cancer cells, even if multiple ubiquitous mutations are targeted and a potent bystander effect is employed.
However, it also true that with immunotherapies like anti-PD-1 antibodies and CAR T-cells you run the risk of the patient developing cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome; with targeted small molecules there can also be serious side effects. While OVERCOME may initially require immunosuppression, which has many negative side effects as well, eventually stealth microbes can be utilized in such a way that there are essentially no side effects[61,62].
Overexpressing DNA repair and tumor suppressor proteins[63,64,65] in our cells could help stave off cancer until we develop computerized microrobots and/or nanorobots that can eliminate/prevent cancer without fail. As I mentioned earlier in terms of overexpressing longevity proteins or underexpressing proteins that accelerate aging, it is not really advisable to alter gene dosage or introduce new genes into our cells, but it may be a necessary risk here if OVERCOME is not always curative or cannot even stave off death from cancer in all cases. As an important sidenote, lipofuscin removal could potentially help treat or even cure cancer in the elderly in some cases - at least in part by re-establishing a healthy immune system.
In reality, the best type of anti-cancer strategy aside from computerized microrobots and/or nanorobots would be to pre-empt it entirely. Such an approach was conceived of by Dr. de Grey. He calls it “Whole-body Interdiction of Lengthening of Telomeres” (WILT)[66]. It involves deleting the machinery required to lengthen telomeres in all cells in our body and periodically reseeding our stem cells with similarly bioengineered cells. This would essentially completely eliminate the problem of cancer. Unfortunately, aside from telomerase there is another mechanism cancer cells can use to extend their telomeres, called “alternative lengthening of telomeres” (ALT). It has been estimated that ALT is active in 10-20% of all cancers[67]. One or more of the proteins involved in ALT may be required for other critical cellular processes - so knocking them out of our cells may not be possible. Another crucial issue is that reseeding stem cells in certain tissues at least may not be particularly facile. Dr. de Grey, however, has pointed out that the more rapidly-dividing stem cells like those of the skin, gut, and immune system seem easier to reseed than stem cells that divide more slowly, like those in the brain and heart. (My delivery system may help with reseeding - if bioengineered macrophages could carry a stem cell inside them, detect niches, and deposit their cargo there, but I’m not sure.)
Perhaps we could knock out hTERT in all of our cells and install an intracellular bacterium that replicates up to a small number inside our stem cells; they could dispense telomerase periodically (when induced via exogenous small molecule). They could have many mechanisms in place to ensure that their telomerase cannot be hijacked by the host cell. Maybe just many redundant, inducible kill switches. But then we would have to re-“infect” all of a patient’s stem cells again. The intracellular bacteria could use potentially use molecular switches to detect mRNA or protein that indicate that certain ALT proteins are required at that moment. That seems a bit difficult, but perhaps less so than reseeding stem cells in areas like the brain.
Conclusion:
Finally, four other issues must still be addressed with regard to age-related disease. However, in the next few hundred years we grant ourselves, they can be dealt with. They are mitochondrial DNA mutations (as Dr. de Grey pointed out), nuclear DNA mutations, nuclear DNA damage (as Ben Best, director of the Cryonics Society of Canada, has written about[77]), and dilapidation of our body structures (e.g., foam cell accumulation in the artery wall and aneurysm formation). Periodic lipofuscin removal should help to reverse/stave off all of these issues. Additionally, there are various stop-gap solutions and fundamental solutions to these issues. The first three issues could be stalled by DNA repair protein overexpression, and there is perhaps a way of stalling the fourth category as well - namely, by promoting extensive ECM turnover more frequently than it occurs endogenously, in combination with more even more frequent lipofuscin removal treatments than I originally envisioned (i.e., once every decade). Structural dilapidation may also be mitigated if we bioengineer tissue-resident or autologous, exogenous macrophages to patrol tissues and more actively assist with maintenance of our bodies.
Mitochondrial DNA mutations should be the easiest to fundamentally address. Dr. de Grey has suggested allotopic expression as a solution2. My delivery system could also be used to deliver microbes that engulf old mitochondria and secrete new ones or at least new mtDNA - if it can be successfully delivered to pre-existing mitochondria in the target cell. While the latter three issues might need computerized microrobots and/or nanorobots to fix fundamentally, in the hundreds or even thousands of years of healthy life we can hopefully grant ourselves with lipofuscin removal and stop-gap measures, I believe we can develop such technology[78].
We should sequence our nuclear genomes with the best sequencing technology available (100x) to have pristine reference sequences for the future[79]. We should do the same for our mitochondrial genomes. It was determined that high-level mitochondrial DNA heteroplasmy is rare in humans[80]; low-level heteroplasmy is rather prevalent, however. Hopefully, low-level heteroplasmy or situations where an inherited variant becomes dominant through expansion is not required in any of our tissues for healthy functionality - and also is not important with regard to conservation of our identities in terms of brain cell (e.g., neuronal, astrocytic, and microglial) mitochondria. In that case, we would only need to worry about sequencing our dominant, inherited mitochondrial genome. (Replacing all of our mitochondrial genomes with our dominant variants could also potentially induce an inflammatory immune response, though.)
Heteroplasmy levels can vary in cells throughout an individual’s body. To determine the dominant, inherited mitochondrial genome, we may have to take biopsy samples from many different tissue types, e.g., blood and skeletal muscle - to see which variant is the most prevalent. (For tissues like skeletal muscle, we should take samples from multiple regions of the body, as well.) Similar to the tumor nuclear DNA sampling approach I mentioned earlier, if the delivery system I proposed is developed - we can sample mitochondrial DNA from many different cell types with intracellular microbes. They can even detect miRNA with protein switches to mark themselves as having entered a particular cell type - helping us gather data on the mitochondrial genomes inside various cell types. Depending on various factors like cell type, fission may have to be promoted first via proteins like Fis1 and Drp1. Perhaps the intracellular microbes could replicate up to a small copy number, restrained by quorum sensing, in order to sample many mitochondria inside various cells. That strategy is also applicable to more rapid lipofuscin removal, although it may not be very safe unless much work is done to perfect the system.
We should replace the mitochondrial DNA in our bodies gradually - to allow the nuclear genome time to epigenetically adapt to the loss of mitochondrial DNA variants. Microbes have been shown to be able to conjugate with mitochondria[81,82]. This could be the best way to introduce the dominant mitochondrial DNA variant. Mitochondrial turnover should also be effected while the dominant variant is being introduced. The mitochondrial genome being introduced could also lack a cleavage site, and endogenous mitochondrial genomes could be cleaved - but inherited heteroplasmic differences or somatic mutations may prevent this approach from being completely effective. The fact that conplastic mice with two different, wild-type mitochondrial genomes appear to be equally healthy is good news. Thus, it might not be too important if we accidentally pick a variant that was not the originally-dominant variant in the zygote - as long as it is “healthy”. However, another study indicated that conplastic mice with different wild-type mitochondrial genomes showed differences with aging[83]. With periodic lipofuscin removal - hopefully age-related differences will not manifest.
Notably, high degrees of mitochondrial heteroplasmy (>10%) appears to cause pathology in mice[84,85]. Homoplasmic mice appear to be healthy. We can also test if typically heteroplasmic mice (i.e., around 10% heteroplasmy) can have their entire mitochondrial genome pools replaced by their dominant inherited mitochondrial genome; i.e., whether it maintains its health and if its consciousness seems to be altered due to swapping out all the neuronal mitochondria with the dominant inherited variant. More gradual replacement of neuronal mitochondria may be important to maintain one’s perceived identity. (My proposed delivery system could be used to replace mitochondria or mitochondrial DNA, as mentioned earlier.) Of course, mouse studies may not translate directly to humans, though.
Fortunately, although somatic mutations do build up in the mitochondrial genomes of humans with age, relatively few cells in a given tissue are heavily burdened[86]. Also, mutations are estimated to affect only ~32% of mtDNA molecules in 77-99-year-old humans[87]. Thus, as long as we sequence the mitochondrial genomes in a large number of cells of multiple tissue type samples, we should be able identify original variants that lack somatic mutations. If one has data from any maternal relatives, this could also help to pinpoint inherited vs. somatic mutations.
Mitochondrial DNA damage and age-related epigenetic changes may constitute two more issues to consider, but as mitochondrial DNA replacement may be harmless and facile eventually, they may not warrant any concern.
Ideally, we would sequence one’s mitochondrial and nuclear genomes at as young an age as possible, to make sure the reference sequence is as close to the original as possible. If an individual is elderly and has acquired somatic mutations in their nuclear and mitochondrial DNA, perhaps harvesting germline stem cells (GSCs) and sequencing the DNA from them would be most appropriate. They appear to have the best nuclear DNA repair mechanisms of all male cells[88] - and possibly the best mitochondrial DNA repair mechanisms. However, it is unclear whether female GSCs exist[89]. (Oocytes can be harvested up until menopause - they have very efficient DNA repair mechanisms as well[90]; there are strict selection processes for oocyte mitochondria[91].) The quality of male GSCs and female oocytes declines with age, however. Post-mitotic cells like skeletal muscle cells may also be good sources of relatively pristine nuclear DNA in the elderly[92,93]. The skeletal muscle cells can be reprogrammed back to pluripotency, and then expanded to obtain enough cells for 100x whole genome sequencing.
I am a bit concerned that the nuclear mutations in whatever cell type/anatomical region we sequence may be problematic if that genome is used as the reference for (all) other cell types/regions of body - in terms of both functionality and an inflammatory immune response. (Of course, in addition to age-related mutations, developmental somatic mosaicism can also occur.) For elderly patients at least, we may need to use the mitochondrial sampling approach I mentioned for the nuclear genome as well. The most simple way to install the new genomes would be to use intracellular microbes that can engulf old nuclei and deliver new ones. Epigenetically-speaking, this may not be feasible. Another possibility is to use reference DNA to create stem cells of the appropriate types and reseed the patient’s stem cells, then increase cellular turnover. (Epigenetic alterations due to somatic mutations or age-related changes could also possibly cause immunogenicity.) But the best approach would require medical, computerized microrobots and/or nanorobots to correct mutations and fix damage while leaving the epigenetic state largely the same. (Epigenetic age-related changes may accrue to pathological levels eventually even with lipofuscin removal treatments if a plethora of DNA mutation/damage and structural damage to our bodies accumulates before those things can be repaired. In that case, transient Yamanaka factor expression may be helpful. In fact, eventually, pathological levels of epigenetic drift may occur even with repair of all the other issues. If either of these scenarios are true, epigenetic, age-related changes would constitute another issue.)
We may also want to freeze stem cells of every type we can in case we need to expand them later and reseed them into our bodies because our old stem cells have acquired too many mutations[94] (before medical, computerized microrobots and/or nanorobots can be developed), but we also could potentially just freeze any cells in our bodies with a low mutational burden. They could be eventually reprogrammed into iPSCs and then we could differentiate them into the proper stem cell types. (Certain stem cells types at least may not accumulate mutations faster than neurons, for example[95,96],94,93, but the mutations could possibly affect them more.)
If we still need more time to fix nuclear DNA mutations and nuclear DNA damage in long-lived post-mitotic cells, we may have to increase cell turnover - maybe at least in part by setting a lower threshold of DNA damage that stimulates apoptosis. Overexpressing tumor suppressor proteins may be one way to increase the turnover of generally-mutated cells.
Proto-oncogene activation through mutagenesis can be recognized by tumor suppressor proteins. They can also recognize more general conditions of cellular stress, potentially including being riddled with mutations in non-proto-oncogenes. Thus, more heavily mutated cells overexpressing tumor suppressor proteins may undergo apoptosis or become senescent, at which point the senescent cells could be eliminated by the youthful immune system or exogenous means. However, increasing cellular turnover in general as well would probably be ideal. Increasing cellular turnover at least dramatically would be feasible for cardiac tissue and skeletal muscle perhaps - but maybe not the brain. Of course, with dramatically increased cellular turnover, we would have to induce telomerase more frequently in our stem cells.
Will we will be able to fix these issues in the brain without potentially compromising an individual’s identity until we have a much more complex understanding of the brain? Even what may be considered “damage” to neuronal nuclear DNA, for example, may be part of an individual’s identity. However, we may just have to “bite the bullet” - i.e., replace or fix these things gradually and hope we’re alright - if there isn’t enough time to fully understand all these complex philosophical, psychological, and neuronal issues with regard to identity. Notably, mitochondrial epigenetics in neurons may be important as well - with regard to mitochondrial DNA methylation, etc. Hopefully, the epigenetic state of newly introduced mitochondrial genomes will be restored through endogenous gene expression of the neuronal nuclear genomes. Again, gradual replacement may be important. (Ostensibly, mitochondrial epigenetics does not require any consideration for non-brain cells, at least.) I have mentioned neurons as being important to our identities, but other brain cells like glia could also be critical in this regard[97]. I think we will probably only have to worry about neurons, however, in terms of subtle changes to the nuclear/mitochondrial DNA. (And even with neurons, small alterations may not make a big difference.)
I believe, though, that we will have the time to understand neurobiology enough to ensure that we are essentially the same individuals. If you think about it, effective cognitive function and our memories are all that really matter. Fixing age-related issues will probably increase our cognitive functions, and we can be careful to retain synaptic connections involved in maintaining our memories.
There is one final caveat that is not related to aging per se, i.e., a steady accumulation of biomolecular damage, but is related more to random chance. Vascular issues may occur even if periodic lipofuscin clearance is started at an early age.
Clots and bleeds are unlikely in young, healthy individuals, but can still occur. It would be ideal if we could limit the damage that clots and bleeds cause before they can be fixed surgically. For clots, perhaps my proposed delivery system could deliver genes to all of our cells that ensure they do not undergo apoptosis when starved of nutrients and oxygen, but rather enter into a more dormant state. When the clot is finally broken up, eliminated, or removed, they could function normally again. For bleeds - we could bioengineer circulating white blood cells to quickly patch up the leak, as well as bioengineer tissue-resident macrophages to phagocytose red blood cells and general bloodstream biomolecules much more efficiently. Any damage that is caused regardless of these interventions can eventually be fixed through stem cell therapy (to some extent at least), tissue engineering, or possibly xenotransplantation before tissue engineering biotechnology has advanced far enough. Xenotransplantation may be more feasible earlier. (For neurons, obviously large scale tissue replacement is not really even feasible without computerized microrobots and/or nanorobots to reinstall synaptic connections, etc. Gradual neuronal tissue replacement - i.e., in multiple sessions spaced out over time - may help address the identity issue a bit at least.) Inducing faster endogenous stem cell-mediated repair could also be a viable option.
Additionally, frequent full body scans should be performed on everyone to look for vascular issues like aortic dissections, aneurysms, and arteriovenous fistulae. They might occur due to an essentially random, severe insult or, perhaps more likely, cluster of insults in a given vascular locale. Of course, that concept is applicable to any tissue region, but if a relatively small area of a major blood vessel is in jeopardy, it could cause a lot of damage.
Funding
Funding not received for the study.
Authors' contributions
M.R. wrote the paper.
Ethics approval and consent to participate
N/A
Consent for publication
N/A
Availability of data and material
N/A
Acknowledgments
Thank you very much to my family - and also my friends, especially Chris Welch and Ray Lomeli. Also, thank you to Dr. Aubrey de Grey and Michael Rae for their work in the field of anti-aging research. The figure in this piece was created with BioRender.com.
Competing interests
The author declares that he has no competing interests.
References
- Terman A and Brunk UT. Lipofuscin. The International Journal of Biochemistry & Cell Biology 2004;36(8):1400–1404. [CrossRef]
- Zealley B and de Grey ADNJ. Strategies for Engineered Negligible Senescence. Gerontology 2013;59(2):183–189. [CrossRef]
- López-Otín C, Blasco MA, Partridge L, et al. The Hallmarks of Aging. Cell 2013;153(6):1194–1217. [CrossRef]
- Aufschnaiter R, Zamir EA, Little CD, et al. In vivo imaging of basement membrane movement: ECM patterning shapes Hydra polyps. Journal of Cell Science 2011;124(23):4027–4038. [CrossRef]
- Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev 2020;34(23–24):1565–1576. [CrossRef]
- Kabacik S, Lowe D, Fransen L, et al. The Relationship between Epigenetic Age and the Hallmarks of Aging in Human Cells. Nat Aging 2022;2(6):484–493. [CrossRef]
- Lu Y, Brommer B, Tian X, et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020;588(7836):124–129. [CrossRef]
- Wang S, Xia P, Ye B, et al. Transient activation of autophagy via Sox2-mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell 2013;13(5):617–625. [CrossRef]
- Kang Y-K, Min B, Eom J, et al. Different Phases of Aging in Mouse Old Skeletal Muscle. Aging 2022;14(1):143–160. [CrossRef]
- Renteln M. Lipofuscin as the main driving force of current age-related disease: justification and strategies for removal. Molecular Biology Reports (Under Review) Preprint →. [CrossRef]
- Johnson M, Karanikolas BDW, Priceman SJ, et al. Titration of Variant HSV1-tk Gene Expression to Determine the Sensitivity of 18F-FHBG PET Imaging in a Prostate Tumor. J Nucl Med 2009;50(5):757–764. [CrossRef]
- Nikitas G, Deschamps C, Disson O, et al. Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of goblet cell accessible E-cadherin. J Exp Med 2011;208(11):2263–2277. [CrossRef]
- Le Guennec L, Coureuil M, Nassif X, et al. Strategies used by bacterial pathogens to cross the blood–brain barrier. Cellular Microbiology 2020;22(1):e13132. [CrossRef]
- Ley K, Laudanna C, Cybulsky MI, et al. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007;7(9):678–689. [CrossRef]
- Chi J-T, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, et al. Endothelial cell diversity revealed by global expression profiling. Proceedings of the National Academy of Sciences (2003) 100:10623–10628. [CrossRef]
- van Buul JD, Hordijk PL. Signaling in Leukocyte Transendothelial Migration. Arteriosclerosis, Thrombosis, and Vascular Biology (2004) 24:824–833. [CrossRef]
- Gupta M, Shin D-M, Ramakrishna L, et al. IRF8 directs stress-induced autophagy in macrophages and promotes clearance of Listeria monocytogenes. Nature Communications 2015;6:6379. [CrossRef]
- Miller ST, Xavier KB, Campagna SR, Taga ME, Semmelhack MF, Bassler BL, Hughson FM. Salmonella typhimurium recognizes a chemically distinct form of the bacterial quorum-sensing signal AI-2. Mol Cell (2004) 15:677–687. [CrossRef]
- Lipinski DM, Reid CA, Boye SL, et al. Systemic Vascular Transduction by Capsid Mutant Adeno-Associated Virus After Intravenous Injection. Hum Gene Ther 2015;26(11):767–776. [CrossRef]
- Krolak T, Chan KY, Kaplan L, et al. A High-Efficiency AAV for Endothelial Cell Transduction throughout the Central Nervous System. Nat Cardiovasc Res 2022;1(4):389–400. [CrossRef]
- Lu ZH, Kaliberov S, Zhang J, et al. The Myeloid-Binding Peptide Adenoviral Vector Enables Multi-Organ Vascular Endothelial Gene Targeting. Lab Invest 2014;94(8):881–892. [CrossRef]
- Zahr A, Alcaide P, Yang J, et al. Endomucin prevents leukocyte–endothelial cell adhesion and has a critical role under resting and inflammatory conditions. Nat Commun 2016;7(1):10363. [CrossRef]
- Qiu Y, Tong S, Zhang L, et al. Magnetic forces enable controlled drug delivery by disrupting endothelial cell-cell junctions. Nat Commun 2017;8(1):15594. [CrossRef]
- Zhang G, Yang X, Gao R. Research progress on the structure and function of endomucin. Animal Model Exp Med 2021;3(4):325–329. [CrossRef]
- Hunter M, Fusco D. Superinfection exclusion: A viral strategy with short-term benefits and long-term drawbacks. PLoS Comput Biol 2022;18(5):e1010125. [CrossRef]
- Caussinus E, Kanca O, Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nature Structural & Molecular Biology (2012) 19:117–121. [CrossRef]
- Horan KA, Hansen K, Jakobsen MR, Holm CK, Søby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol (2013) 190:2311–2319. [CrossRef]
- Acar M, Pando BF, Arnold FH, Elowitz MB, van Oudenaarden A. A general mechanism for network-dosage compensation in gene circuits. Science (2010) 329:1656–1660. [CrossRef]
- Montezano, Augusto C., Karla B. Neves, Rheure A. M. Lopes, and Francisco Rios. “Isolation and Culture of Endothelial Cells from Large Vessels.” In Hypertension: Methods and Protocols, edited by Rhian M. Touyz and Ernesto L. Schiffrin, 345–48. Methods in Molecular Biology. New York, NY: Springer, 2017. [CrossRef]
- Graham GJ, Handel TM, Proudfoot AEI. Leukocyte Adhesion: Reconceptualizing Chemokine Presentation by Glycosaminoglycans. Trends Immunol 2019;40(6):472–481. [CrossRef]
- Stanley SA, Gagner JE, Damanpour S, et al. Radio-Wave Heating of Iron Oxide Nanoparticles Can Regulate Plasma Glucose in Mice. Science 2012;336(6081):604–608. [CrossRef]
- Guidi N, Marka G, Sakk V, et al. An Aged Bone Marrow Niche Restrains Rejuvenated Hematopoietic Stem Cells. STEM CELLS 2021;39(8):1101–1106. [CrossRef]
- Landspersky T, Saçma M, Rivière J, et al. Autophagy in Mesenchymal Progenitors Protects Mice against Bone Marrow Failure after Severe Intermittent Stress. Blood 2022;139(5):690–703. [CrossRef]
- Zhang X, Chen W, Gao Q, et al. Rapamycin Directly Activates Lysosomal Mucolipin TRP Channels Independent of MTOR. PLOS Biology 2019;17(5):e3000252. [CrossRef]
- Ain Q, Schmeer C, Penndorf D, et al. Cell cycle-dependent and -independent telomere shortening accompanies murine brain aging. Aging (Albany NY) 2018;10(11):3397–3420. [CrossRef]
- Tomás-Loba A, Flores I, Fernández-Marcos PJ, et al. Telomerase Reverse Transcriptase Delays Aging in Cancer-Resistant Mice. Cell 2008;135(4):609–622. [CrossRef]
- Bernardes de Jesus B, Vera E, Schneeberger K, et al. Telomerase Gene Therapy in Adult and Old Mice Delays Aging and Increases Longevity without Increasing Cancer. EMBO Mol Med 2012;4(8):691–704. [CrossRef]
- Jaijyan DK, Selariu A, Cruz-Cosme R, et al. New Intranasal and Injectable Gene Therapy for Healthy Life Extension. Proc Natl Acad Sci U S A 2022;119(20):e2121499119. [CrossRef]
- Sewell PE. Systemic Human Htert Aav Gene Transfer Therapy And The Effect On Telomere Length And Biological Age, A Case Report. J Regen Biol Med 2022. [CrossRef]
- Ali M, Devkota S, Roh J-I, et al. Telomerase Reverse Transcriptase Induces Basal and Amino Acid Starvation-Induced Autophagy through MTORC1. Biochemical and Biophysical Research Communications 2016;478(3):1198–1204. [CrossRef]
- Yang C, Przyborski S, Cooke MJ, et al. A Key Role for Telomerase Reverse Transcriptase Unit in Modulating Human Embryonic Stem Cell Proliferation, Cell Cycle Dynamics, and In Vitro Differentiation. Stem Cells 2008;26(4):850–863. [CrossRef]
- Zincarelli C, Soltys S, Rengo G, et al. Analysis of AAV Serotypes 1-9 Mediated Gene Expression and Tropism in Mice after Systemic Injection. Mol Ther 2008;16(6):1073–1080. [CrossRef]
- Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther (2011) 19:1058–1069. [CrossRef]
- Bevan AK, Duque S, Foust KD, Morales PR, Braun L, Schmelzer L, Chan CM, McCrate M, Chicoine LG, Coley BD, et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther (2011) 19:1971–1980. [CrossRef]
- Hordeaux J, Wang Q, Katz N, Buza EL, Bell P, Wilson JM. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol Ther (2018) 26:664–668. [CrossRef]
- Cagan A, Baez-Ortega A, Brzozowska N, et al. Somatic Mutation Rates Scale with Lifespan across Mammals. Nature 2022;604(7906):517–524. [CrossRef]
- Chen X, Burkhardt DB, Hartman AA, et al. MLL-AF9 Initiates Transformation from Fast-Proliferating Myeloid Progenitors. Nat Commun 2019;10(1):5767. [CrossRef]
- Se A, S A, Mk T, et al. Constitutive Telomerase Expression Promotes Mammary Carcinomas in Aging Mice. Proceedings of the National Academy of Sciences of the United States of America 2002;99(12). [CrossRef]
- Weng N. Aging of the Immune System: How Much Can the Adaptive Immune System Adapt? Immunity 2006;24(5):495–499. [CrossRef]
- Torra A, Parent A, Cuadros T, et al. Overexpression of TFEB Drives a Pleiotropic Neurotrophic Effect and Prevents Parkinson’s Disease-Related Neurodegeneration. Mol Ther 2018;26(6):1552–1567. [CrossRef]
- Bredenkamp N, Nowell CS, Blackburn CC. Regeneration of the aged thymus by a single transcription factor. Development 2014;141(8):1627–1637. [CrossRef]
- Zook EC, Krishack PA, Zhang S, et al. Overexpression of Foxn1 attenuates age-associated thymic involution and prevents the expansion of peripheral CD4 memory T cells. Blood 2011;118(22):5723–5731. [CrossRef]
- Uddin MM, Ohigashi I, Motosugi R, et al. Foxn1-β5t transcriptional axis controls CD8+ T-cell production in the thymus. Nat Commun 2017;8(1):14419. [CrossRef]
- Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016;530(7589):184–189. [CrossRef]
- Renteln M. Conditional Replication of Oncolytic Viruses Based on Detection of Oncogenic MRNA. Gene Therapy 2018;25(1):1–3. [CrossRef]
- Renteln M. Promoting oncolytic vector replication with switches that detect ubiquitous mutations. Current Cancer Therapy Reviews (Under Review) Preprint. [CrossRef]
- Muthana M, Kennerley AJ, Hughes R, et al. Directing cell therapy to anatomic target sites in vivo with magnetic resonance targeting. Nature Communications 2015;6(1):8009. [CrossRef]
- Cornel AM, Mimpen IL, Nierkens S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers (Basel) 2020;12(7):E1760. [CrossRef]
- Park JH, Rivière I, Gonen M, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 2018;378(5):449–459. [CrossRef]
- Cercek A, Lumish M, Sinopoli J, et al. PD-1 Blockade in Mismatch Repair–Deficient, Locally Advanced Rectal Cancer. New England Journal of Medicine 2022;0(0):null. [CrossRef]
- Stritzker J, Hill PJ, Gentschev I, et al. Myristoylation negative msbB-mutants of probiotic E. coli Nissle 1917 retain tumor specific colonization properties but show less side effects in immunocompetent mice. Bioeng Bugs 2010;1(2):139–145. [CrossRef]
- Harimoto T, Hahn J, Chen Y-Y, et al. A Programmable Encapsulation System Improves Delivery of Therapeutic Bacteria in Mice. Nat Biotechnol 2022;40(8):1259–1269. [CrossRef]
- García-Cao I, García-Cao M, Martín-Caballero J, et al. ‘Super P53’ Mice Exhibit Enhanced DNA Damage Response, Are Tumor Resistant and Age Normally. EMBO J 2002;21(22):6225–6235. [CrossRef]
- Zhao Y, Burikhanov R, Qiu S, et al. Cancer Resistance in Transgenic Mice Expressing the SAC Module of Par-4. Cancer Res 2007;67(19):9276–9285. [CrossRef]
- Garcia-Cao I, Song MS, Hobbs RM, et al. Systemic Elevation of PTEN Induces a Tumor-Suppressive Metabolic State. Cell 2012;149(1):49–62. [CrossRef]
- Grey ADNJ de. Whole-Body Interdiction of Lengthening of Telomeres: A Proposal for Cancer Prevention. Front Biosci 2005;10(1–3):2420. [CrossRef]
- MacKenzie D, Watters AK, To JT, et al. ALT Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers (Basel) 2021;13(10):2384. [CrossRef]
- Streeter MD, Rowan S, Ray J, et al. Generation and Characterization of Anti-Glucosepane Antibodies Enabling Direct Detection of Glucosepane in Retinal Tissue. ACS Chem Biol 2020;15(10):2655–2661. [CrossRef]
- Furber JD. Extracellular glycation crosslinks: prospects for removal. Rejuvenation Res 2006;9(2):274–278. [CrossRef]
- Planque SA, Nishiyama Y, Hara M, et al. Physiological IgM class catalytic antibodies selective for transthyretin amyloid. J Biol Chem 2014;289(19):13243–13258. [CrossRef]
- Bruxel MA, Tavares AMV, Zavarize Neto LD, et al. Chronic whole-body heat treatment relieves atherosclerotic lesions, cardiovascular and metabolic abnormalities, and enhances survival time restoring the anti-inflammatory and anti-senescent heat shock response in mice. Biochimie 2019;156:33–46. [CrossRef]
- Hansen T, Ahlström H, Johansson L. Whole-body screening of atherosclerosis with magnetic resonance angiography. Top Magn Reson Imaging 2007;18(5):329–337. [CrossRef]
- Uebe R, Schüler D. Magnetosome biogenesis in magnetotactic bacteria. Nat Rev Microbiol 2016;14(10):621–637. [CrossRef]
- Shiratori T, Suzuki S, Kakizawa Y, Ishida K. Phagocytosis-like cell engulfment by a planctomycete bacterium. Nat Commun (2019) 10:5529. [CrossRef]
- Marques ARA, Ramos C, Machado-Oliveira G, et al. Lysosome (Dys)Function in Atherosclerosis—A Big Weight on the Shoulders of a Small Organelle. Frontiers in Cell and Developmental Biology 2021;9:658995. [CrossRef]
- Cheng C-W, Adams GB, Perin L, et al. Prolonged Fasting reduces IGF-1/PKA to promote hematopoietic stem cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014;14(6):810–823. [CrossRef]
- Best BP. Nuclear DNA Damage as a Direct Cause of Aging. Rejuvenation Research 2009;12(3):199–208. [CrossRef]
- Freitas Jr. RA. Progress in Nanomedicine and Medical Nanorobotics. In: Handbook of Theoretical and Computational Nanotechnology. (Rieth M, Schommers W. eds.) American Scientific Publishers: Stevenson Ranch, Calif; 2005.
- Anonymous. Nebula Genomics - DNA Testing. n.d. Available from: https://nebula.org/whole-genome-sequencing-dna-test/ [Last accessed: 11/16/2022].
- Stewart JB, Chinnery PF. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat Rev Genet 2021;22(2):106–118. [CrossRef]
- Yoon YG, Koob MD. Transformation of isolated mammalian mitochondria by bacterial conjugation. Nucleic Acids Res 2005;33(16):e139. [CrossRef]
- Yoon YG, Koob MD. Nonreplicating intracellular bacterial vector for conjugative DNA transfer into mitochondria. Pharm Res 2012;29(4):1040–1045. [CrossRef]
- Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016;535(7613):561–565. [CrossRef]
- Hirose M, Schilf P, Gupta Y, et al. Low-level mitochondrial heteroplasmy modulates DNA replication, glucose metabolism and lifespan in mice. Sci Rep 2018;8(1):5872. [CrossRef]
- Lechuga-Vieco AV, Latorre-Pellicer A, Calvo E, et al. Heteroplasmy of Wild-Type Mitochondrial DNA Variants in Mice Causes Metabolic Heart Disease With Pulmonary Hypertension and Frailty. Circulation 2022;145(14):1084–1101. [CrossRef]
- Keogh M, Chinnery PF. Hereditary mtDNA Heteroplasmy: A Baseline for Aging? Cell Metabolism 2013;18(4):463–464. [CrossRef]
- Shokolenko IN, Wilson GL, Alexeyev MF. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J Exp Med 2014;4(4):46–57.
- Barroca V, Fouchet P. Germline Stem Cells: The First Guards of Heredity. Cell Stem Cell 2008;2(2):108–110. [CrossRef]
- Silvestris E, Cafforio P, Felici C, et al. Ddx4+ Oogonial Stem Cells in Postmenopausal Women’s Ovaries: A Controversial, Undefined Role. Cells 2019;8(7):650. [CrossRef]
- Stringer JM, Winship A, Zerafa N, et al. Oocytes can efficiently repair DNA double-strand breaks to restore genetic integrity and protect offspring health. Proceedings of the National Academy of Sciences 2020;117(21):11513–11522. [CrossRef]
- St. John JC. Mitochondria and Female Germline Stem Cells—A Mitochondrial DNA Perspective. Cells 2019;8(8):852. [CrossRef]
- Franco I, Johansson A, Olsson K, et al. Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nat Commun 2018;9(1):800. [CrossRef]
- Abascal F, Harvey LMR, Mitchell E, et al. Somatic mutation landscapes at single-molecule resolution. Nature 2021;593(7859):405–410. [CrossRef]
- Blokzijl F, Ligt J de, Jager M, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016;538(7624):260. [CrossRef]
- Manders F, van Boxtel R, Middelkamp S. The Dynamics of Somatic Mutagenesis During Life in Humans. Frontiers in Aging 2021;2.
- Cagan A, Baez-Ortega A, Brzozowska N, et al. Somatic mutation rates scale with lifespan across mammals. Nature 2022;604(7906):517–524. [CrossRef]
- Han X, Chen M, Wang F, et al. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 2013;12(3):342–353. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



