
Submitted:
28 December 2022
Posted:
03 January 2023
You are already at the latest version
Abstract
Background: High blood pressure (BP) has been implicated as a major risk factor for cardiovascular diseases in several global populations, including in individuals of African ancestry. Despite the elevated burden of high BP-induced cardiovascular diseases in Africa and other global populations with African ancestry, limited genetic studies have been carried out to explore the genetic machinery driving this phenomenon. Methods: We performed univariate and multivariate analyses using Genome-wide association studies (GWAS) and summary statistics data of 77,850 individuals of African ancestry for systolic (SBP) and diastolic blood pressure (DBP) traits. The six independent cohorts used included individuals derived from the African Partnership for Chronic Disease Research (APCDR), the UK Biobank, and the Million Veteran Program (MVP). Subsequently, we annotated, prioritized, visualized, and interpreted our meta-analyses results using FUMA, to gain further insight into the molecular mechanism(s) that contribute to the genetics of BP traits. Finally, loci attaining genome-wide significance, GWS (p<5x10-8) were also followed up with Bayesian fine-mapping to identify potential causal variants. Results: Our meta-analyses altogether identified 350 GWAS SNPs for SBP (166 SNPs) and DBP (184 SNPs, including two novel loci) whilst our multivariate GWAS method identified 166 SNPs (including three novel loci). Interestingly, in FUMA there was significant tissue enrichment of up-regulated differentially expressed genes (DEGs) in the sigmoid and transverse colon for SBP, as well as 10 significant gene sets from MAGMA gene set analyses, However, for DBP, no significant DEGs nor gene sets in MAGMA were found; instead, in DBP for gene property analysis for tissue specificity nine candidates were found to be significant and all nine were in different brain regions. Finally, Bayesian fine-mapping revealed that only 11 variants from the lead SNPs had >50% posterior probability (PP) of being causal and they included novel variant rs562545 (MOBP, PP = 77%) and 10 other previously published variants. Conclusion: Our results demonstrate the importance of performing GWAS in large sample sizes of global populations of African ancestry, including continental Africans; which yield novel insights, from novel loci to novel pathways/tissue expression candidates. Large-scale genomic datasets are required to enhance further discovery and fine-mapping of high-risk loci/variants in highly susceptible groups for cardiovascular disease and other related traits. Our study highlights the need for diversity in genetic research and the importance of expanding large GWASs to include ancestrally diverse populations.
Keywords:
systolic blood pressure
; diastolic blood pressure
; GWAS
; high blood pressure
; multivariate
; univariate
1. Introduction
Blood pressure (BP) is a quantitative trait that is affected by both multifactorial genetic and environmental factors [1,2,3]. The heritability of high blood pressure is estimated to be 30-50% [4]. Elevated blood pressure otherwise called hypertension is the leading risk factor for many cardiovascular diseases like stroke and coronary artery diseases [5,6]. The global prevalence of hypertension among adults aged 30-79 years increased significantly from 650 million in 1990 to 1.28 billion in 2019; with two-thirds of this burden coming from low-income countries (LMICs) [7]. When compared to other ethnic groups, African Americans and other African ancestry show a higher occurrence of high blood pressure [8,9,10,11].
Despite the global rise in the disease burden among individuals of African ancestry, limited genome-wide association studies (GWASs) of blood pressure traits have been conducted or included individuals of African ancestry [12,13]. For instance, the largest GWAS of blood pressure conducted to date in approximately a million individuals was predominantly consist of Europeans [15]. Additionally, only ~62% of all the genome-wide significant loci from this GWAS had the concordant direction of effects for individuals of African ancestry and moderate Pearson correlation coefficients with effect estimates in Europeans r2=0.37 in Africans, compared to the strong r2=0.78 for South Asians [15,16,17,18]. Another example is that majority of blood pressure GWASs conducted in African ancestry populations have small sample sizes [19,20,21,22,23] and they mostly use single trait approach without giving due consideration to the phenotypic relatedness and the relationship between the two traits (SBP and DBP), which is a possible link between risk-related clinical measures and arterial properties [24,25]. Thus, many novel insights into blood pressure traits in people of African ancestry remain to be discovered.
Furthermore, various GWAS reports have shown that the genetic determinants of blood pressure have small effect sizes and vary significantly between European and non-European populations [26]. Therefore, our study aim to extensively study the African population to better understand the genetic epidemiology underlying blood pressure traits in individuals of African descent. We also perform a multivariate GWAS in the hope that it will increase our study’s statistical power over the univariate approach and consequently increase the overall number of novel loci observed in our study.
We conducted the largest GWAS of blood pressure in over 77,850 people from the African Partnership for Chronic Disease and Research (APCDR), African ancestry people from the United Kingdom (UK-Biobank), and the Million Veteran Program in this study (MVP). Figure 1 depicts the overall study design; we used fixed effects meta-analysis across the cohorts. We then performed a multivariate analysis, fine-mapping, pathway and tissue enrichment test analysis, and pathway and tissue enrichment test analysis to highlight relevant biological processes and investigate causal relationships with disease traits.
2. Materials and Methods
2.1. Study Population
The full description of the study population can be found in the supplementary 1 cohort description while the study design can be found in Figure 1
2.2. Meta-analysis of BP Summary Statistics in African Ancestry Individuals
We aggregated BP association summary statistics across the three cohorts (UK-Biobank, APCDR-UGR, DSS, DCC, and AADM, and the MVP) and performed an inverse-variance-weighted meta-analysis implemented in GWAMA [37]. We used a total of seventy-seven thousand eight hundred and fifty sample sizes across the studied cohorts (Table 3). The resulting output was used for subsequent downstream analyses, and we then plotted the resulting p-value in a Manhattan plot.
2.3. Tissue Expression Enrichment Pathway analysis
We performed a gene-based analysis with the MAGMA 1.6 software (Multi-marker Analysis of Genomic Annotation) [38], which is available in FUMA [39]. Magma gene-based analysis is useful for analyzing and detecting multiple genetic markers of individuals with a weak effect, which is common in polygenic traits. The 1000 Genomes were used as a reference dataset to account for LD between SNPs, and the confounding effects of gene density and gene size were used as covariates. The pathway and tissue expression analyses were performed using the default parameters in FUMA, using the results obtained from the meta-analysis.
2.4. Functional mapping and annotation analysis
We used an online functional mapping and annotation tool (FUMA) [39] to annotate SNPs from the GWAMA meta-analysis with their biological functionality and then mapped them to genes using positional mapping and QTL association (blood eQTL) [40]. The independent SNPs were classified based on their P-values as genome-wide significant (P ≤ 5.0 × 10−8), independence from each other (r2 < 0.1) and LD threshold within a 1 Mb window. Furthermore, the independent SNPs were annotated for functional consequences on gene function using ANNOVAR [41]. For the positional mapping, genes were mapped to SNPs if the physical distance between them was < 10 kb. The eQTL mapping used data from the blood cis-eQTL, and SNPs were mapped to genes on the premise that the SNPs had a significant effect on the expression of the gene. And also, SNPs were filtered using a CADD score > 12.37, which is the threshold for deleterious scores (CADD scores are deleterious scores of genetic variants obtained by 63 functional annotations) [42]. Normalized gene expressions for 53 tissue types were obtained from GTEx. Other clumping parameters used were: reference panel to compute LD and MAF (minor allele frequencies)= 1000 genome project was used (AFR) [43]; minor allele frequency filter > 0.01; maximum distance between LD blocks to merge into a single locus for genomic risk loci= 250 kb, lead SNPs were classified as SNPs that were in LD with each other at r2 < 0.1.
2.5. Locus Definition
Lead SNPs from both univariate and multivariate analyses were defined based on positional mapping using 1Mb; SNPs that had reached the genome-wide significant threshold (p < 5 x 10-8) were considered to be associated with BP. Loci were defined by flanking distance mapping 500kb up and downstream of peak SNPs, and we retained SNPs with the lowest p-value from both the meta-analysis and multi-trait analysis.
2.6. Fine-mapping Analysis of Sentinel Variants
Following our result output from multi-trait analysis and meta-analysis, we performed Bayesian fine-mapping to identify possible causal variants for the locus ± 500kb of all the lead SNPs. We used a Bayesian approach [44] to fine-map the loci of the lead SNPs. The Z-scores for the SNPs were then used to compute the Bayes factor for each SNP denoted as , given by:
Where K is the number of studies). The posterior probability of driving the association for each SNP was computed by:
Where the summation in the denominator is over all SNPs at the locus.
Ninety-nine percent credible set sizes were calculated by sorting all SNPs at the locus according to their posterior probability from highest to lowest and then counting the number of SNPs required to achieve a cumulative posterior probability greater or equal to 0.99. High confidence was defined as index SNPs that account for more than 50% of the posterior probability of driving the BP association at a given signal.
2.7. Multivariate GWAS analysis
To further increase the statistical power for discovery, we employed a cross-phenotype approach implemented in the CPASSOC software [45]. The cross-phenotype association analysis accounts for the correlation of summary statistics data among traits and the participating cohorts and allows for both heterogeneity and homogeneity effects. The CPASSOC analysis generates two statistical tests: SHom and Shet, the latter of which is an extension of the former and improves statistical power when there is a difference in the genetic effect sizes across the traits. Meanwhile, the SHom test, which is similar to the fixed-effect meta-analysis approach, increases in power when the genetic effect sizes across the traits are the same.
3. Results
3.1. Results overview
We compiled GWAS summary statistics from three cohorts (Table 1), totaling 20,000,000 SNPs in 77,850 people of African descent. We used a univariate meta-analysis and a multivariate GWAS to find genetic variants linked to BP traits. At a genome-wide significant threshold of (P<5x10-8) for both known and novel loci, the meta-analysis and multivariate approaches identified both known and novel loci. We used FUMA and fine mapping to gain more insight into the likely causal variants and molecular mechanism(s) that contribute to the genetics of BP traits.
3.2. Univariate GWAS meta-analysis
Meta-analysis of all six cohorts (n=77,850) identified 166 significant variants for SBP (Supplementary Table 1) and 184 genome-wide significance variants (P<5x10-8) for DBP (Supplementary Table 2). The significant SNPs for both blood pressure traits were clumped at ±500Kb distance leaving 10 for SBP (Supplementary Table 3) and 9 lead SNPs for DBP (Supplementary Table 4). After clumping, out of the 19 SNPs identified across both traits, 2 were at least 1 Mbp away from any previously reported BP locus and therefore considered novel; an intergenic variant rs77534700 in AC074290.1 (p = 3.749e-08) and rs562545 an intronic variant at MOBP (p = 1.823e-09) and are both associated with DBP trait (Table 2, Figure 2A,B). Commonly known variants in CACNA1D, HTR4, SLC22A14, NPPA-AS1, C3orf73, KCNK3, RPL35P4, CASZ1, NPPA-AS1, CTC-436K13.2, KCNN3, RSPO3, ATP2B1, FGF5, ULK4, and NPPA-AS1 were associated with SBP and DBP (Supplementary Tables 3 and 4).
The CACNA1D gene is an intron variant that has previously been identified in other populations, including African Americans, and is thought to regulate the renin-aldosterone-angiotensin system. The previously observed associations of the genetic variant in the meta-analyses were predominantly from the MVP cohorts, which might be driven by the fact that the largest proportion of our sample size came from the MVP’s African American population. We plotted the resulting p-values from this association analysis on a Manhattan plot. (Figure 2).
3.3. Functional mapping and annotation analyses from FUMA from the meta-analysis
Using the default parameters on FUMA, we performed functional annotation on all SNPs in linkage disequilibrium (LD) to annotate and prioritize genes obtained from our meta-analysis. FUMA’s SNP2GENE function revealed that the functional consequences of SNPs on genes in SBP included 12 lead SNPs (Supplementary Table 5), 20 independent SNPs (Supplementary Table 6), and 10 genomic risk loci (Supplementary Table 7). Following that, in DBP, 13 lead SNPs (Supplementary Table 8), 27 independent SNPs (Supplementary Table 9), and 8 genomic risk loci (Supplementary Table 10) were identified. The majority of the markers in SBP were intergenic, followed by those in the intronic region. In DBP, the most significant proportion of the markers were intronic SNPs, followed by intergenic SNPs (Supplementary Figure 1). 19 genes were identified through positional and/or eQTL mapping in SBP (Supplementary Table 5), and 34 genes for DBP SNPs (Supplementary Table 6).
The MAGMA gene set, tissue expression, and pathway analyses were carried out as part of the FUMA workflow. According to the MAGMA gene set study, after Bonferroni correction, no DBP gene sets were significant, but 10 SBP gene sets were (Supplementary Table 7). SBP was specifically linked to gene sets involved in biological processes such as synapse assembly (including presynaptic membrane assembly and organization, postsynaptic density, and specialization assembly), cell-cell adhesion via plasma membrane adhesion molecules (i.e., the connection of one cell to another cell through the use of adhesion molecules that are at least partially embedded in the plasma membrane), and the Takada gastric cancer copy number (i.e. candidate genes in the regions of copy number loss in gastric cancer cell lines).
Based on MAGMA tissue expression analysis, SBP was not associated with any gene property analysis for tissue specificity. However, DBP was associated with nine tissue specificities, significantly associated with brain tissues: particularly the hippocampus, brain substantia nigra, brain amygdala, brain putamen basal ganglia, hypothalamus, cortex, anterior cingulate cortex BA24, caudate basal ganglia, and nucleus accumbens basal ganglia (Figure 3). Notably, the strongest enrichment was observed for genes expressed in the hippocampus, followed by putamen basal ganglia.
As part of the FUMA pipeline, we used GENE2FUNC to test differentially expressed genes (DEGs); DBP found no association with our GTEx v8 54 tissue types, but SBP found two significantly up-regulated DEGs in the sigmoid and transverse colon (Supplementary Figure 2). Finally, we tested the enrichment of input gene sets (adjusted p<0.05) and we found several gene sets previously associated with SBP, DBP, and correlated traits (Supplementary Figures S2–S8).
3.4. Fine-mapping of putatively causal variants
We performed Bayesian fine-mapping to pinpoint putative causal variants for distinct BP association signals using differences in the structure of LD between ancestry groups. Bayesian fine-mapping of the 19 distinct signals from the meta-analysis after clumping for DBP and SBP was undertaken in the region mapping 500kb up-and-down-stream, which together accounted for 99% posterior probability and was based on association summary statistics from the meta-analysis GWAS. Only 11 variants from the lead SNPs had >50% posterior probability (PP) of being causal, including the novel variant rs562545 (MOBP, PP = 77%) (Supplementary Table 14, Figure 4), and known variants; rs3821845 (CACNA1D, PP = 99%), rs12509595 (FGFR, PP = 99%), rs11129785 (SLC22A14, PP = 75%), rs12476527 (KCNK3, PP = 52%), rs5068 (NPPA-AS1, PP = 64%), rs73437338 (ATP2B1, PP = 52%), rs7720317 (CTC-436K13.2, PP = 59%), rs1984285 (KCNN3, PP = 99%). One of the lead SNPs, rs880315 (CASZ1, PP = 95%) was not the lead variant in the fine-mapping but was overlapped by other variants rs17035646 (PP = 99%) (Supplementary Table 8.
Multivariate GWAS analysis of blood pressure traits identifies additional novel loci
Using CPASSOC, we performed a multivariate analysis, this method identified 166 genome-wide significant loci associated with blood pressure (Supplementary Table 10 (p < 5 x 10-8). After clumping, we identified 21 independent significant SNPs, 3 novel SNPs, and 18 known SNPs (Supplementary Table 16). Interestingly, using the model assuming heterogeneity in CPASSOC, we identified 3 novel independent significant variants (Table 3), rs138493856 (DNAJC17P1/GLULP6, p = 6.132e-09), rs139235642 (RRM2, p = 2.798e-08) and rs72619992 (LOC105377644, p = 1.134e-08). The resulting p-values were then plotted and visualized in a Manhattan plot (Figure 5).

Table 3.
Novel variants of Blood pressure traits identified using multivariate methods.
| Nearest Gene | Lead SNPs | Chr | BP | Effect Allele | Other Allele | HET_Pvalue | Functional Consequence |
|---|---|---|---|---|---|---|---|
|
DNAJC17P1/ GLULP6 GLULP6GLULP6 GLULP6 |
rs138493856 | 2 | 194678067 | A | G | 6.1322e-09 | Intergenic variant |
| RRM2 | rs139235642 | 2 | 10278626 | T | C | 2.7981e-08 | intron variant NMD transcript variant |
| LOC105377644 | rs72619992 | 3 | 39407952 | A | C | 1.1339e-08 | Intron variant |
4. Discussion
This study describes the largest GWAS of blood pressure in African ancestry to date, involving a total of 77,850 individuals from the MVP, APCDR, and UK Biobank cohorts. The results of this analysis provide additional relevant information on the genetic and biological architecture of blood pressure traits in people of African ancestry.
According to our results, the multivariate GWAS approach had greater statistical power in identifying new variants than the univariate meta-analysis (Figure 6). Previous GWAS studies had shown the power of the multivariate approach, especially when dealing with traits that are highly correlated [27].
Five novel variants were discovered using both methods. The multivariate approach identified three variants: DNAJC17P1/GLULP6 (rs138493856), RRM2 (rs139235642), and LOC105377644 (rs72619992), while the univariate approach identified two variants: AC074290.1 (rs77534700) and MOBP (rs562545). The DNAJC17P1/GLULP6 gene, which is located in the intergenic region, is known to be associated with susceptibility to infectious disease measurement [28] as well as educational attainment [29]. The RRM2 is a protein-coding gene that encodes one of two non-identical subunits for ribonucleotide reductase and is highly expressed in the bone marrow (28.1) and lymph node (20.5), along with other tissues [30]. The high expression of this gene can lead to the abnormal proliferation of histiocytes and can also be used as a marker for malignant changes in ovarian endometriosis [31]. The rs72619992 variant in LOC105377644 is an uncharacterized RNA gene that belongs to the ncRNA class and does not code for any protein. In AC074290.1, our univariate method identified an uncharacterized pseudogene. According to the GWAS catalog, the MOBP gene, which is a myelin-associated oligodendrocyte-associated protein, is linked to Alzheimer’s disease [32], cognitive performance, and other brain-related disorders [33]. The MOBP gene is thought to be involved in both frontotemporal dementia and nervous system development. We used the largest BP summary statistics from European ancestry individuals to look up our lead SNPs, while some of the lead SNPs were found to be replicated at replicated at P-value 0.05. None of the SNPs identified as being novel replicated (Supplementary 13).
In the meta-analysis results, our in silico functional mapping and annotation analyses from FUMA revealed several biologically relevant signals. SBP gene sets, for example, were significantly associated with associated biological systems such as several synapse assembly components (such as components correlated to nervous system development/neurons and chemical or electrical synapses), candidate genes in regions of copy number loss in gastric cancer cell lines, cell-cell adhesion via plasma membrane adhesion molecules (possibly, part of action potentials generated by the movement of ions through transmembranous channels), and cell-cell adhesion via plasma membrane adhesion.
In addition, the SBP meta-analysis tissue enrichment analysis was associated with significantly up-regulated DEGs in the sigmoid and transverse colon (Supplementary Figure 1); which may suggest that gut microbiota may play a role in the regulation of gastro-renal axis and blood pressure [34]. Furthermore, the most interesting enrichment of input genes in gene sets significant in the Reactome was in the cardiac conduction and muscle contraction pathways for the SBP meta-analysis, which are the mechanisms and pathways that elicit rapid changes in the heart rate, blood pressure, and respond to changes in autonomic tone. On the other hand, our DBP MAGMA tissue expression analysis highlighted nine brain tissue types associated with DBP. For instance, the putamen, caudate, and nucleus accumbens basal ganglia are input nuclei as well as part of the corpus striatum, and the substantia nigra is a basal ganglia function-related nuclei, which are all involved in processing movement-related information. Dysfunction in this region is known to be associated with movement disorders like Huntington’s, as correlated by GWAS catalog genes highlighted. In addition, the GWAS catalog genes included in gene sets included blood pressure traits and their interactions with alcohol and cigarette smoking, hence, these may be interesting environmental risk factors that should be investigated for their impact on BP traits in populations of African descent. Further investigation is needed to understand this, as different regions have different drinking and smoking habits.
Furthermore, our tissue expression analysis shows that DBP gene expression is enriched in the brain hippocampus (Figure 3), a brain region that is essential for learning and memory [35]. According to one study, hypertension is linked to decreased functional hippocampus connectivity and impaired memory [36]. As a result, more research is needed to understand our findings from in silico functional mapping and annotation analyses, as well as their mechanism.
Our current study has several strengths. First, our study is the largest SBP and DBP GWAS meta-analysis of an African population; thus, it has allowed us to find novel loci and replicate prior findings. Secondly, our functional mapping and annotation found several biologically relevant regions, that support our genetic findings, and these regions, tissues, and pathways are good candidates to explore further to elucidate the pathogenesis of blood pressure-related disorders like hypertension and prevent or treat them better. Finally, fine-mapping recommended target candidate loci to test in vivo and in vitro to improve our understanding of the regulators and genetic factors that affect blood pressure traits. The CPASSOC used for our multivariate GWAS increased statistical power and reflected the nature of the multivariate effect of traits on the genetic factor.
One of our limitations is that the "black" participants in our study are primarily from admixed regions with a variety of characteristics. Thus, our study used a small sample size from the continental African population, and this may be the reason why most of our variants were identified from the MVP dataset, as this data had the largest sample size (Table 3). Although our study is the largest study of SBP and DBP genetics, the overall sample size was small compared to contemporary GWASs for other traits. Thus, future studies will need to include more continental Africans to make sure our genetic risk factors can be used to make genetic risk scores that are inclusive of all or most African populations and their full range of diversity. Due to the diversity in African genome, latent sub-structuring could inflate the results, but this effect was minimized by adjusting for principal components in the GWAS model by the contributing cohorts. Second, the paucity of functional genomics information specific to African people makes it challenging to evaluate the functional relevance of the relationships found. Thirdly, regional environmental factors, including dietary variations, variances in the prevalence of TB and HIV, and other non-communicable disease factors, could potentially have an impact on BP outcomes; however, there isn’t enough research on these aspects in our target group. Afrocentric GWAS data are grossly limited, hence we used Blood pressure GWAS data from individual of African ancestry available and accessible to the authors.
In conclusion, we have conducted the largest GWAS of blood pressure in African ancestry, which has significantly enabled an in-depth understanding of the genetic component. Our analysis emphasizes the relevance of applying fine-mapping and multivariate methods to correlated trait and their increase in statistical power toward the discovery of causal variants. These strategies offer a reliable approach to better understanding the genetic epidemiology of blood pressure disease in African ancestry and treatment development strategy. Lastly, to better understand the implication of these results, future studies could replicate the result on the European population.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
OS is supported by the Africa Research Excellence Fund (AREF-325-SORE-F- C0904). SF is supported by the Wellcome Trust grant (220740/Z/20/Z) at the MRC/UVRI and LSHTM. TC is an international training fellow supported by the Wellcome Trust grant (214205/Z/18/Z).
Authors’ contribution
SF conceptualize the idea, BU and OS contributed to the preliminary analyses and manuscript drafting, OS contributed to the Meta-analysis and Multivariate analyses, TM contributed to the functional analysis, SF contributed to the supervision of the manuscript, TC and AK contributed to the overviewing of the manuscript.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of respective data used in this study. The DDS was approved by the Biomedical Research Ethics Committee at the University of KwaZulu-Natal (reference: BF030/12) and the UK National Research Ethics Service (reference: 14/WM/1061). This UGR -GPC was approved by the Science and Ethics Committee of the UVRI, the Ugandan National Council for Science and Technology and the East of England-Cambridge South NHS Research Ethics Committee United Kingdom. The DCC was approved by the Biomedical Research Ethics Committee at the University of KwaZulu-Natal (reference: BF078/08) and the UK National Research Ethics Service (reference: 11/H0305/6). The AADM study obtained ethical approval from the Institutional Review Boards (IRB) of all participating institutions.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Muñoz, M.; Pong-Wong, R.; Canela-Xandri, O.; Rawlik, K.; Haley, C.S.; Tenesa, A. Evaluating the contribution of genetics and familial shared environment to common disease using the UK Biobank. Nat. Genet. 2016, 48, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Feinleib, M.; et al. The NHLBI Twin Study of Cardiovascular Disease Risk Factors: Methodology Summary of Results. Am. J. Epidemiol. 1977, 106, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Poulter, N.R.; Prabhakaran, D.; Caulfield, M. Hypertension. The Lancet 2015, 386, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; et al. Framingham Heart Study 100K Project: genome-wide associations for blood pressure and arterial stiffness. BMC Med. Genet. 2007, 8 (Suppl. S1), S3. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Bayraktutan, U. Risk factors for ischaemic stroke. Int. J. Stroke Off. J. Int. Stroke Soc. 2008, 3, 105–116. [Google Scholar] [CrossRef]
- European Stroke Organisation (ESO) Executive Committee and ESO Writing Committee. Guidelines for management of ischaemic stroke and transient ischaemic attack 2008. Cerebrovasc. Dis. Basel Switz. 2008, 25, 457–507. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet Lond. Engl. 2021, 398, 957–980. [Google Scholar] [CrossRef]
- Carson, A.P.; Howard, G.; Burke, G.L.; Shea, S.; Levitan, E.B.; Muntner, P. Ethnic differences in hypertension incidence among middle-aged and older adults: the multi-ethnic study of atherosclerosis. Hypertens. Dallas Tex 1979 2011, 57, 1101–1107. [Google Scholar] [CrossRef]
- Berenson, G.S.; Wattigney, W.A.; Webber, L.S. Epidemiology of hypertension from childhood to young adulthood in black, white, and Hispanic population samples. Public Health Rep. Wash. DC 1974 1996, 111 (Suppl. S2), 3–6. [Google Scholar]
- Writing Group Members; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef]
- Chor, D.; et al. Prevalence, Awareness, Treatment and Influence of Socioeconomic Variables on Control of High Blood Pressure: Results of the ELSA-Brasil Study. PloS One 2015, 10, e0127382. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; et al. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009, 41, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Newton-Cheh, C.; et al. Eight blood pressure loci identified by genome-wide association study of 34,433 people of European ancestry. Nat. Genet. 2009, 41, 666–676. [Google Scholar] [CrossRef] [PubMed]
- “Genetic Variants in Novel Pathways Influence Blood Pressure and Cardiovascular Disease Risk. Nature 2011, 478, 103–109. [CrossRef]
- Evangelou, E.; et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018, 50, 1412–1425. [Google Scholar] [CrossRef] [PubMed]
- Ehret, G.B.; et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011, 478, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat. Genet. 2011, 43, 531–538. [Google Scholar] [CrossRef]
- Warren, H.R.; et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat. Genet. 2017, 49, 403–415. [Google Scholar] [CrossRef]
- Adeyemo, A.; et al. A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet. 2009, 5, e1000564. [Google Scholar] [CrossRef]
- Franceschini, N.; et al. Genome-wide Association Analysis of Blood-Pressure Traits in African-Ancestry Individuals Reveals Common Associated Genes in African and Non-African Populations. Am. J. Hum. Genet. 2013, 93, 545–554. [Google Scholar] [CrossRef]
- Giri, A.; et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat. Genet. 2019, 51, 51–62. [Google Scholar] [CrossRef]
- Liang, J.; et al. Single-trait and multi-trait genome-wide association analyses identify novel loci for blood pressure in African-ancestry populations. PLOS Genet. 2017, 13, e1006728. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.R.; et al. Association of genetic variation with systolic and diastolic blood pressure among African Americans: the Candidate Gene Association Resource study. Hum. Mol. Genet. 2011, 20, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, N.; et al. Genome-wide Association Analysis of Blood-Pressure Traits in African-Ancestry Individuals Reveals Common Associated Genes in African and Non-African Populations. Am J Hum Genet 2013, 93, 545–54. [Google Scholar] [CrossRef] [PubMed]
- Hendry, L.M.; Sahibdeen, V.; Choudhury, A.; Norris, S.A.; Ramsay, M.; Lombard, Z. Insights into the genetics of blood pressure in black South African individuals: the Birth to Twenty cohort. BMC Med. Genomics 2018, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- He, J.; et al. Genome-wide association study identifies 8 novel loci associated with blood pressure responses to interventions in Han Chinese. Circ. Cardiovasc. Genet. 2013, 6, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Fatumo, S.; Carstensen, T.; Nashiru, O.; Gurdasani, D.; Sandhu, M.; Kaleebu, P. Complimentary Methods for Multivariate Genome-Wide Association Study Identify New Susceptibility Genes for Blood Cell Traits. Front. Genet. 2022, 10. Available online: https://www.frontiersin.org/article/10.3389/fgene.2019.00334 (accessed on 2 June 2022). [CrossRef]
- Nudel, R.; et al. A large-scale genomic investigation of susceptibility to infection and its association with mental disorders in the Danish population. Transl. Psychiatry 2019, 9, 283. [Google Scholar] [CrossRef]
- Okbay, A.; et al. Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nat. Genet. 2022, 54, 437–449. [Google Scholar] [CrossRef]
- Fagerberg, L.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics MCP 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Yang, B.; Wang, T.; Li, N.; Zhang, W.; Hu, Y. The High Expression of RRM2 Can Predict the Malignant Transformation of Endometriosis. Adv. Ther. 2021, 38, 5178–5190. [Google Scholar] [CrossRef] [PubMed]
- Sherva, R.; et al. Genome-wide association study of the rate of cognitive decline in Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2014, 10, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Need, A.C.; et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Hum. Mol. Genet. 2009, 18, 4650–4661. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jose, P.A.; Zeng, C. Gastrointestinal–Renal Axis: Role in the Regulation of Blood Pressure. J. Am. Heart Assoc. 2017, 6, e005536. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.S.; Dhikav, V. Hippocampus in health and disease: An overview. Ann. Indian Acad. Neurol. 2012, 15, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Rolls, E.T.; Cheng, W.; Feng, J. Hypertension is associated with reduced hippocampal connectivity and impaired memory. EBioMedicine 2020, 61, 103082. [Google Scholar] [CrossRef]
- Mägi, R.; Morris, A. GWAMA: Software for genome-wide association meta-analysis. BMC Bioinformatics 2010, 11, 288. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Watanabe, K.; Taskesen, E.; Van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Westra, H.-J.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Hutchinson, A.; Watson, H.; Wallace, C. Improving the coverage of credible sets in Bayesian genetic fine-mapping. PLOS Comput. Biol. 2020, 16, e1007829. [Google Scholar] [CrossRef]
- Li, X.; Zhu, X. Cross-Phenotype Association Analysis Using Summary Statistics from GWAS. Methods Mol. Biol. Clifton NJ 2017, 1666, 455–467. [Google Scholar] [CrossRef]
Figure 1.
Study design schematic for discovery and validation of loci. APCDR; African Partnership for Control of Disease Research, UKB; United Kingdom Biobank, MVP; Million Veteran Program.
Figure 1.
Study design schematic for discovery and validation of loci. APCDR; African Partnership for Control of Disease Research, UKB; United Kingdom Biobank, MVP; Million Veteran Program.
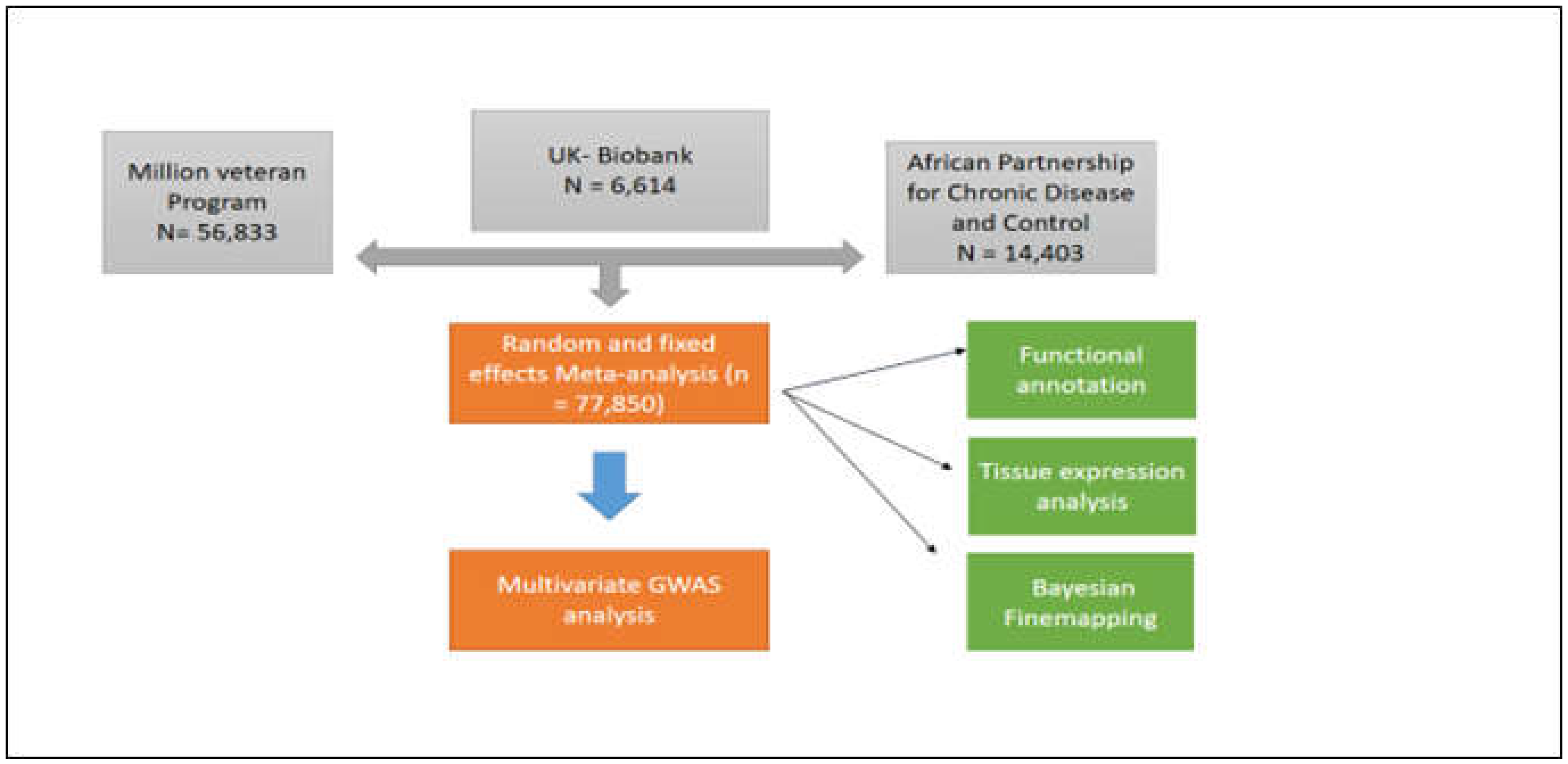
Figure 2.
Manhattan plots showing the minimum P-value for the association across (A) DBP and (B) SBP blood pressure traits, computed using inverse-variance fixed-effect meta-analysis from 75,850 individuals. Each point on the Manhattan plots denotes a variant, with the X-axis representing the genomic position and the Y-axis representing the association level -log 10 (P-value). The horizontal red line shows the genome-wide significant threshold p-value = 5 X10–8.
Figure 2.
Manhattan plots showing the minimum P-value for the association across (A) DBP and (B) SBP blood pressure traits, computed using inverse-variance fixed-effect meta-analysis from 75,850 individuals. Each point on the Manhattan plots denotes a variant, with the X-axis representing the genomic position and the Y-axis representing the association level -log 10 (P-value). The horizontal red line shows the genome-wide significant threshold p-value = 5 X10–8.
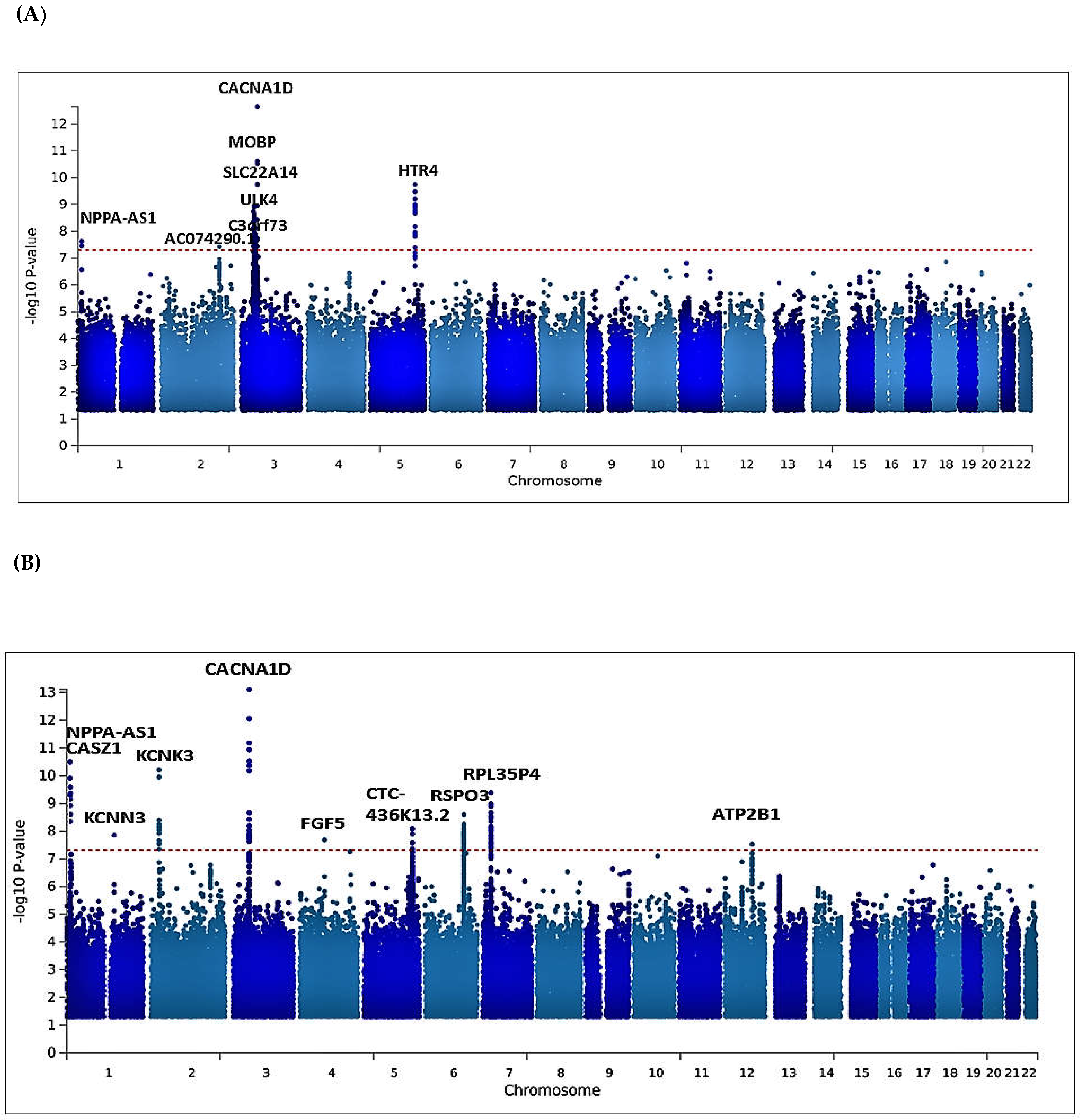
Figure 3.
MAGMA tissue expression analysis using gene expression per tissue based on GTEx RNAseq data for 53 specific tissue types. Significant tissue is shown in red.
Figure 3.
MAGMA tissue expression analysis using gene expression per tissue based on GTEx RNAseq data for 53 specific tissue types. Significant tissue is shown in red.
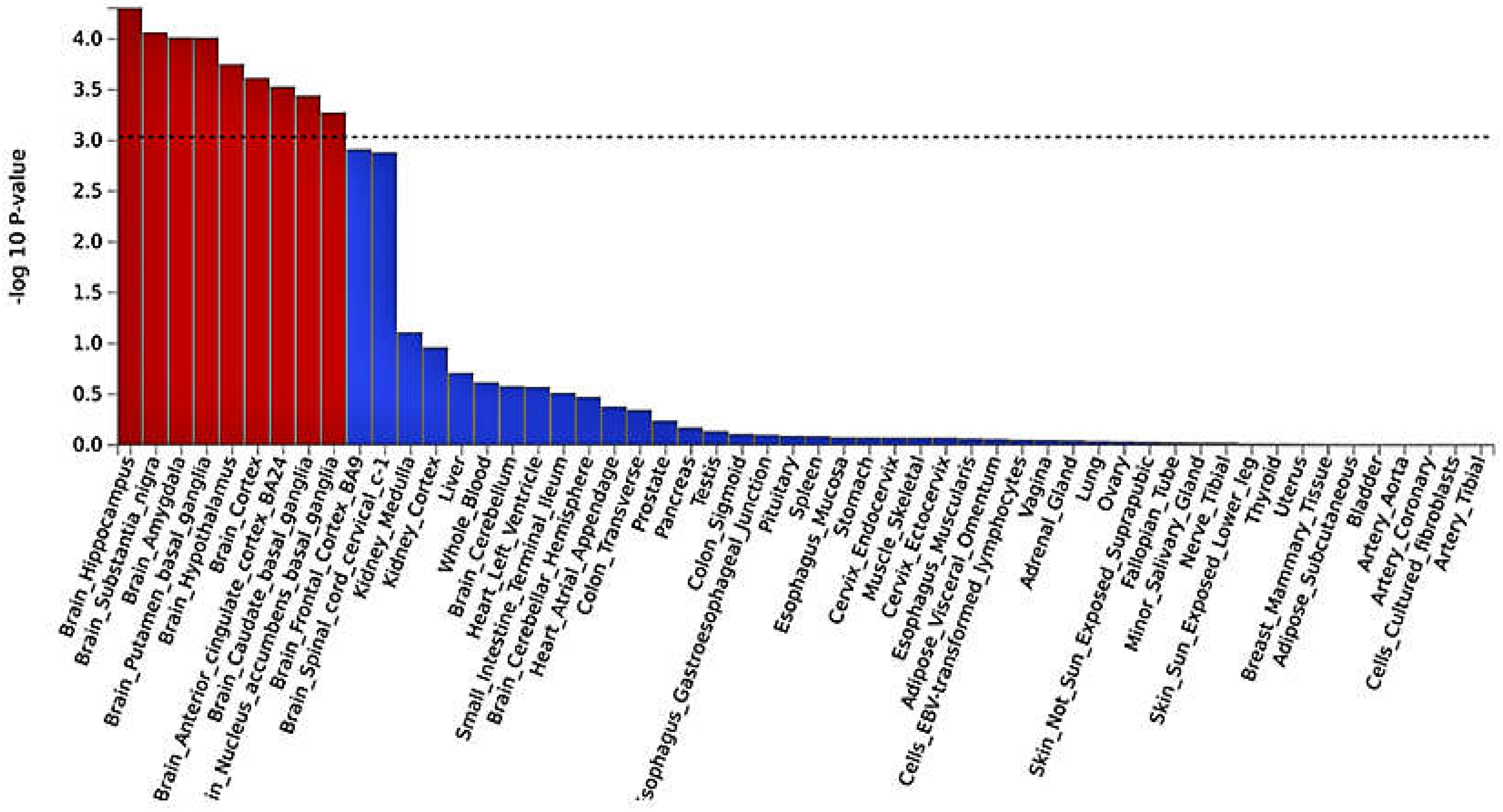
Figure 4.
Regional visualization of the GWAS of –log10 of the P-value of genomic location MOBP (rs562545 in purple) (A), AC074290.1 (rs77534700 in purple)(B), with each dot representing SNP on the corresponding genes at the bottom.
Figure 4.
Regional visualization of the GWAS of –log10 of the P-value of genomic location MOBP (rs562545 in purple) (A), AC074290.1 (rs77534700 in purple)(B), with each dot representing SNP on the corresponding genes at the bottom.
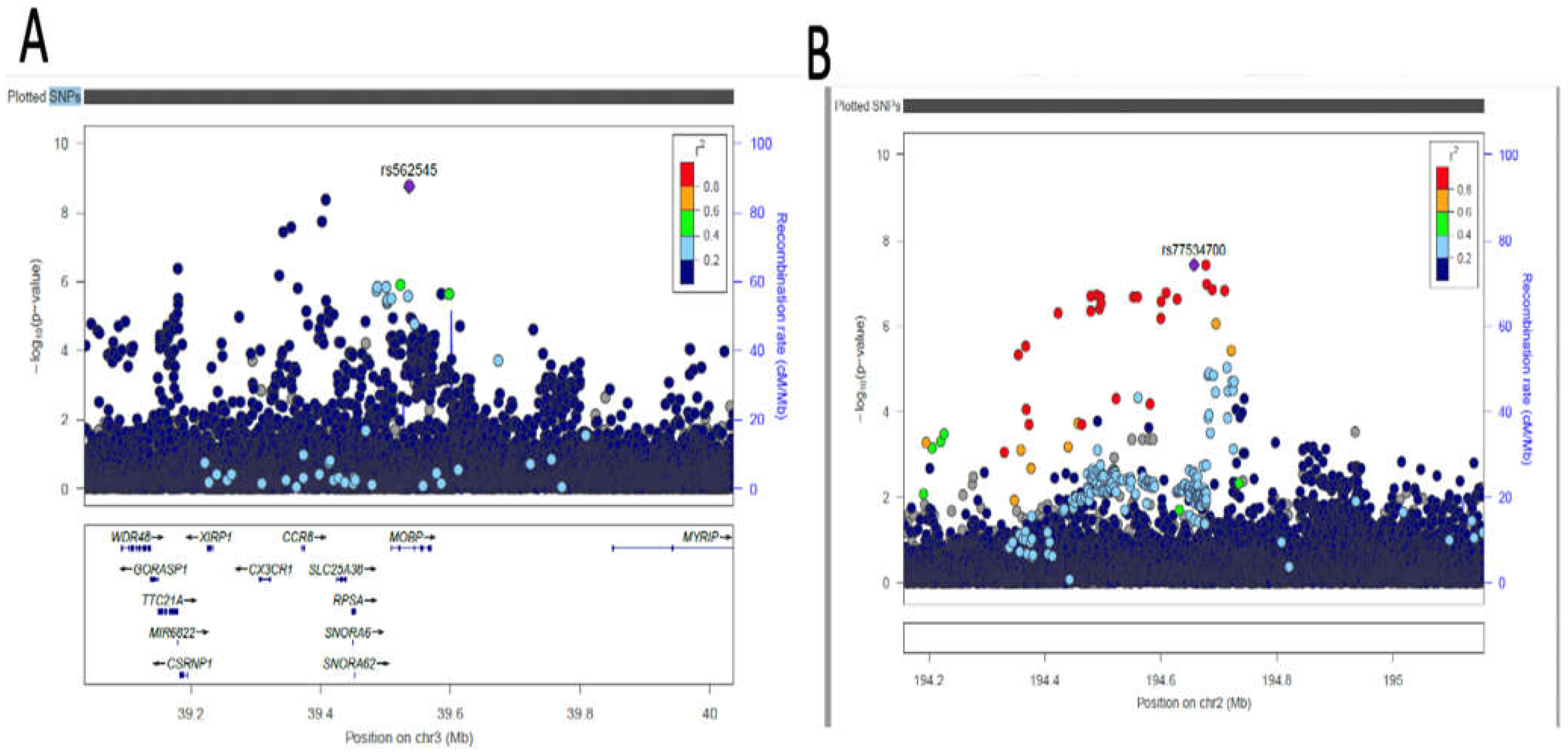
Figure 5.
Manhattan plots showing P-value for the association computed from CPASSOC results output. Each point on the Manhattan plots denotes a variant, with the X-axis representing the genomic position and the Y-axis representing the association level -log 10 (P-value). The horizontal red line shows the genome-wide significant threshold p-value = 5 X10–8
Figure 5.
Manhattan plots showing P-value for the association computed from CPASSOC results output. Each point on the Manhattan plots denotes a variant, with the X-axis representing the genomic position and the Y-axis representing the association level -log 10 (P-value). The horizontal red line shows the genome-wide significant threshold p-value = 5 X10–8
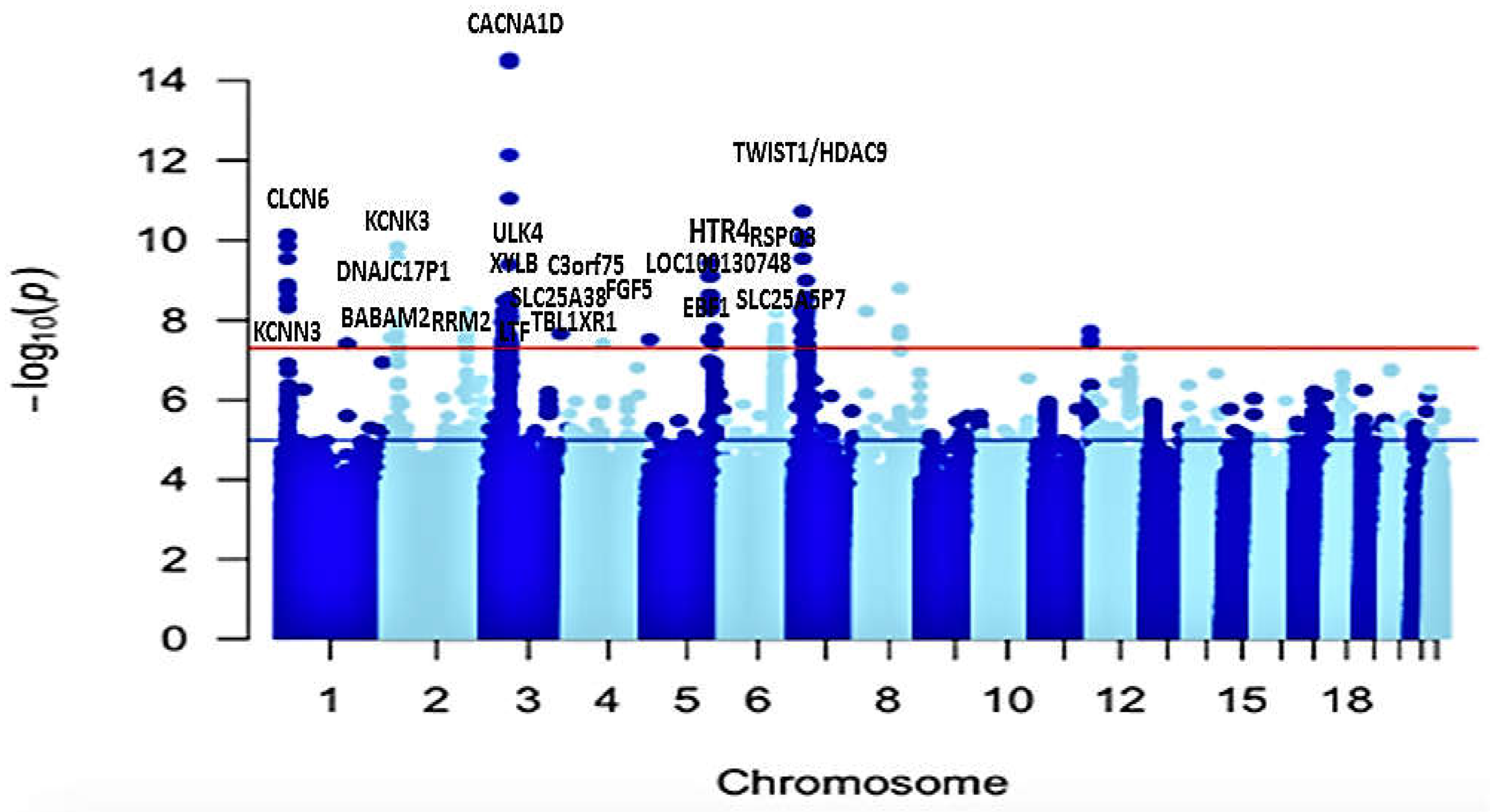
Figure 6.
Venn diagram comparing the loci found by Meta-analysis and multivariate GWAS analysis. Both methods identified both known, the novel loci are indicated with an asterisk.
Figure 6.
Venn diagram comparing the loci found by Meta-analysis and multivariate GWAS analysis. Both methods identified both known, the novel loci are indicated with an asterisk.
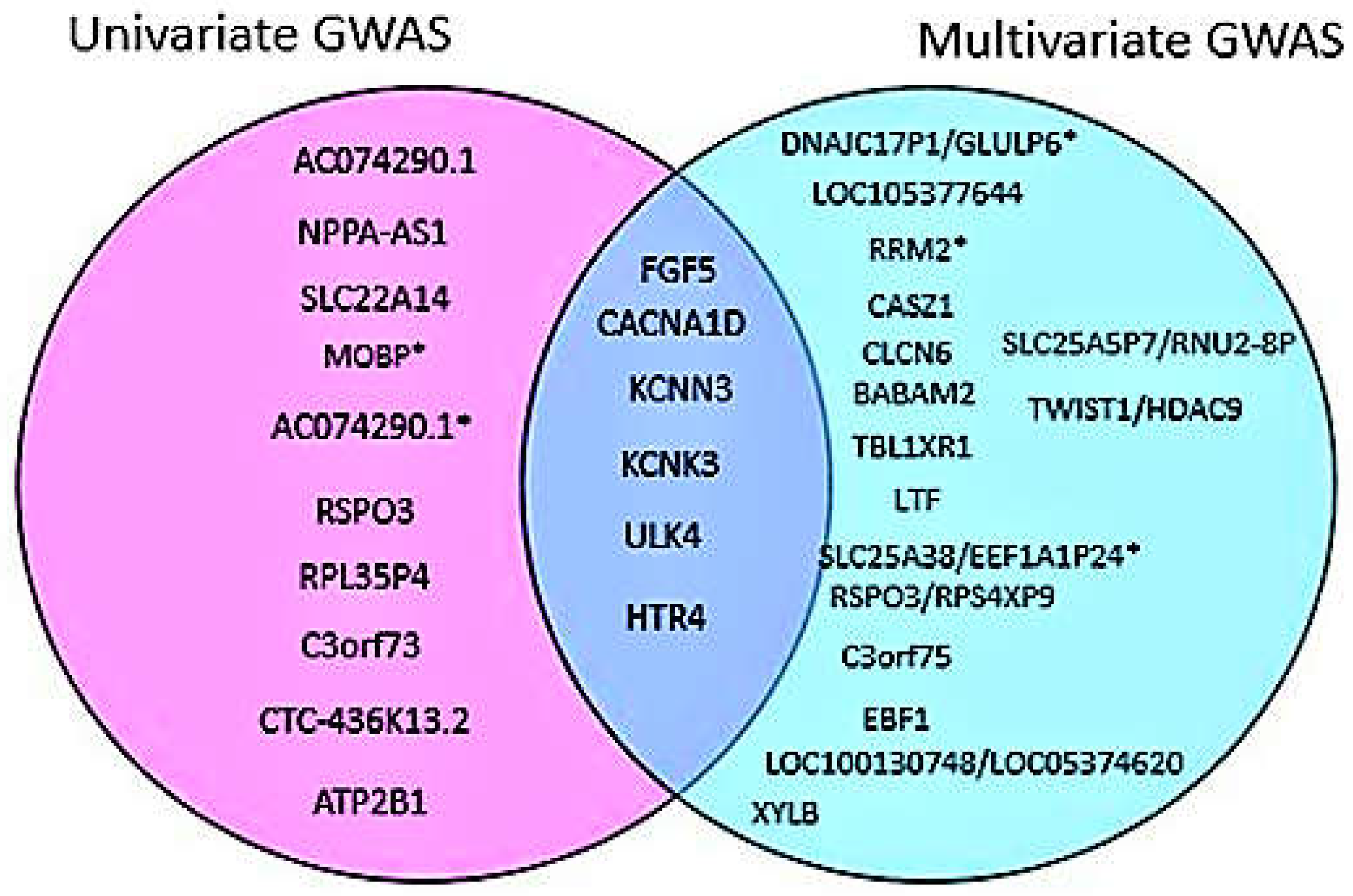
Table 1.
Description of study cohort used in this study.
| Cohort | Continent | Country | Sample Size (N) | Phenotype | Type of Cohorts | Imputation Panel and Genome Build |
|---|---|---|---|---|---|---|
| APCDR-UGR (Gurdasani et.al.,2020) |
Africa | Uganda | 6,407 | DBP SBP |
Observational | Africa genome panel, hg19 |
| APCDR-DCC (Gurdasani et.al.2020) |
Africa | South-Africa | 1,600 | DBP SBP |
Observational | Africa genome panel, hg19 |
| APCDR-DDS (Gurdasani et.al.,2020) |
Africa | South-Africa | 1,165 | DBP SBP |
Case-control | Africa genome panel, hg19 |
| APCDR-AADM (Gurdasani et.al.2020) |
Africa | Nigeria Ghana Kenya |
5,231 | DBP SBP |
Case-control | Africa genome panel, hg19 |
| MVP – AFR | America | USA | 56,833 | DBP SBP |
Observational | 1000 Genome, hg19 |
| UKB – AFR (Sudlow C,et.al.2015) |
Europe | UK | 6,614 | DBP SBP |
Observational | 1000 Genome, hg19 |
Table 2.
Novel distinct variant identified using a meta-analysis approach.
| Nearest Gene | Lead SNPs | Chr | BP | Effect Allele | Other Allele | Trait | Beta | SE | MAF | P-value | Functional Consequence |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AC074290.1 | rs77534700 | 2 | 194678067 | A | G | DBP | -0.0967 | 0.0176 | 0.0836 | 3.749e-08 | Intergenic variant |
| MOBP | rs562545 | 3 | 39536524 | A | G | DBP | 0.0593 | 0.0099 | 0.8973 | 1.823e-09 | Intron variant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



