
Submitted:
03 January 2023
Posted:
04 January 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The novel coronavirus disease 19 (COVID-19) has resulted in an estimated 20 million excess deaths and the recent resurgence of COVID-19 in China is predicted to result in up to 1 million deaths over the next few months. With vaccines unable to halt transmission it is important to continue our quest for safe, effective, affordable drugs that will be available to all countries. Drug repurposing is one of the strategies being explored in this context. Recently, out of 7,817 approved drugs, 214 candidates were systematically down-selected using a combination of 11 filters including approval status, assay data against SARS-CoV-2, pharmacokinetic, pharmacodynamic and toxicity profiles. These drugs were subjected in this study to virtual screening against various targets of SARS-CoV-2 followed by molecular dynamic studies of the best scoring ligands against each target. The chosen molecular targets were Spike receptor binding domain, Nucleocapsid protein RNA binding domain, and key non-structural proteins 3, 5, 12, 13 and 14. Four drugs approved for other indications — alendronate, cromolyn, natamycin and treprostinil — look sufficiently promising from our in silicostudies to warrant further in vitro and in vivo investigations as appropriate to ascertain their extent of anti-viral activities.
Keywords:
COVID-19
; drug repurposing
; long COVID
; molecular docking
; molecular dynamics
; SARS-CoV-2
1. Introduction
The novel coronavirus disease 19 (COVID-19) and the post COVID-19 condition called long COVID have affected lives and livelihoods across the world. As of 1 January 2023, we had over 664 million confirmed COVID-19 cases and 6.62 million deaths recorded globally [1] and an estimated 20 million excess deaths obtained by extrapolating the findings published recently in The Lancet [2]. According to the WHO, circa 144 million people suffered from long COVID by the end of 2021 when the recorded cases stood at around 278 million [3, 4], therefore we are likely to have over 330 million long COVID patients worldwide already. The recent resurgence of COVID-19 in China is predicted to result in up to 1 million deaths over the next few months [5].
COVID-19 is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is part of the RNA virus family Coronaviridae and sub-family Coronavirinae. This sub-family includes four genera of viruses alpha coronaviruses, beta coronaviruses, gamma coronaviruses, and delta coronaviruses, which have been grouped primarily based on serology and phylogenetic clustering [6]. SARS-CoV-2 possesses four structural proteins (S, E, M, and N as defined below) and sixteen non-structural proteins (NSP1−16). The capsid formed from the nucleocapsid protein (N) is present outside the positive-sense, single-stranded RNA genome (+ssRNA) and the genome is further covered by an envelope that is mainly related to three structural proteins: membrane protein (M), spike protein (S), and envelope protein (E). Spike glycoproteins (S protein) form homotrimers that protrude from the host-derived viral envelope and furnish specificity for cellular entry receptors[7].
Vaccines have been useful to restore a level of normalcy but are unable to stop the transmission of the virus and its variants at population levels, therefore it is important to develop affordable therapies that are safe, effective and available, especially to low- and lower-middle income countries that have suffered from insufficient vaccination [8]. Various anti-viral drugs had been developed such as remdesivir, molnupiravir, favipiravir, nirmatrelvir, etc. but none of these meet all four requirements mentioned above. Remdesivir is a prodrug is available as a parenteral [9] while molnupiravir was not shown to have a good clinical outcome from the clinical trials data [10, 11]. Favipiravir was found to be not effective against disease progression [12] and nirmatrelvir, a medication developed by Pfizer is very potent in combination with ritonavir, but has a high price and low manufacturing capability to cater to global demand [13, 14].
Unfortunately, the mean and median capitalized research and development investment of getting a new drug to market were respectively US$1335.9 million and US$985.3 million for the period 2008-19 [15], therefore repurposing drugs approved for other indications to combat COVID-19 and long COVID is a worthwhile strategy [16]. This approach looks for new uses for approved or investigational drugs that are outside the scope of the original medical indication, and can dramatically shrink the costs and timeline involved. However, a major challenge is that there are 7,817 candidates in Compounds Australia open drug collection, so we recently developed a database to down-select the top candidates in a systematic manner [17, 18]. To validate our approach experimentally, 12 of the top 214 drugs, along with the current standards of care (remdesivir, molnupiravir and nirmatrelvir/ritonavir), were recently subjected to in vitro evaluation of anti-viral efficacy against SARS-CoV-2 Delta and Omicron variants of concern [19]. To decide which additional candidates from the list of 214 drugs should be evaluated next, this paper reports an in silico approach consisting of molecular docking studies followed by molecular dynamic studies.
2. Materials and Methodology
Jain et al. [18] have developed the open access, user-friendly ‘CoviRx’ platform to display each of the 7,817 drugs along with physical and chemical properties, original indication, available data from multiple assays, COVID-19 clinical trials, any red flags such as pregnancy concerns, contraindications, etc. and drugs that are similar to the query on the basis of the Tanimoto coefficient [20]. This work made use of CoviRx.org along with the pharmacological down-selection methodology described by MacRaild et al. [18] involving a combination of 11 filters such as approval status, assay data against SARS-CoV-2, pharmacokinetic, pharmacodynamic and toxicity profiles. In addition, the pregnancy and pan-assay interference compounds (PAINS) categories were included to rule out drugs unsafe for pregnant women and those likely to produce false positive results. In this study, the 214 drugs which passed all the filters were subjected to virtual screening against various targets of SARS-CoV-2 followed by molecular dynamic studies of the best scoring ligands against each target (work flow depicted in Figure 1) [21]. The molecular targets include Spike receptor binding domain (RBD), Nucleocapsid protein RNA binding domain (NPRBD), and selected NSPs such as RNA-dependent RNA polymerase (RdRp), Papain-like protease (PL pro), 3CL protease/main protease (3CL pro/ M pro), Helicase, and Exoribonuclease domain (Table 1) [7].
Protein Preparation
The virus-based targets with good resolution were downloaded from the protein data bank (PDB) (https://www.rcsb.org/). These proteins were prepared by adding hydrogens, refining the protein by optimization using PROPKA at pH 7.0, removing water molecules beyond 3.0Å, followed by structural minimization. PROPKA predicts the pKa values of ionizable groups in proteins and protein-ligand complexes based on the 3D structure. This process was performed using protein preparation wizard of Maestro 22 (Schrodinger, LLC, New York, NY, 2022) [22]. The details of virus-based protein targets and their respective PDB IDs are shown in Table 1.

Table 1.
Targets used for virtual screening.
| S. No | Target protein | PDB ID | Resolution |
|---|---|---|---|
| 1. | Spike Receptor Binding Domain | 6M0J | 2.45 Å |
| 2. | Nucleocapsid protein RNA Binding domain | 6VYO | 2.2 Å |
| 3. | Papain Like protease (NSP3) | 6W9C | 2.7 Å |
| 4. | Main protease (3CL protease) (NSP5) | 6W63 | 2.1 Å |
| 5. | RNA dependent RNA polymerase (NSP12) | 6M71 | 2.9 Å |
| 6. | Helicase (NSP13) | 7NNG | 2.38 Å |
| 7. | Exoribonuclease domain (NSP14) | 7R2V | 2.53 Å |
Ligand Preparation
The short-listed drugs (214) were downloaded from Pubchem database in “sdf” format (https://pubchem.ncbi.nlm.nih.gov/). These drugs were imported in Maestro, and LigPrep module was used to prepare the ligands by applying optimize potentials for liquid simulation (OPLS4) forcefield. The maximum ligand size was set to 500 atoms to remove larger molecules as they do not obey “Lipinski rule of five”. In addition, ionization was set to neutralize using the Epik module of Schrodinger suite.
Grid Generation
Receptor grid generation wizard was used to select the native position of the co-crystal ligands to validate the docking protocol. If a protein which do not possess co-crystal ligand, then an extensive literature search was conducted to look for the co-ordinates to be given as input for x, y and z co-ordinates to the grid wizard. On the other hand, for the spike protein (6M0J), as it does not possess co-crystal ligand, the key interacting residues with Angiotensin converting enzyme 2 (ACE2) were considered as the center of the grid.
Molecular Docking Studies
The ligands prepared using ligprep module, the minimized protein and the generated grid files were used for performing flexible ligand docking studies in extra precision (XP) mode by adding Epik state penalties. The number of poses to be included and to write were set to ten and one respectively. All other default parameters were retained.
Molecular Dynamics (MD)
The workstation used to perform the dynamic studies was of Tyrone, equipped with 120GB HDD and 12GB of NVIDIA graphic card. The solvent model used was of transferable intermolecular potential 3P (TIP3P) and the temperature was set to 310.15K. Firstly, the protein was preprocessed to identify the problems if any, and rectified by adding hydrogens and filling the missing residues using prime module of the software. Further, water molecules beyond 5 Å were removed and at pH of 7.0 ± 2.0, heteroatoms state was generated using the software’s Epik module [26]. Secondly, system builder wizard was run by choosing the TIP3P solvent model using OPLS4 force filed and generating an orthorhombic box with dimensions of 10x10x10 Å [27, 28]. The number of ions required to neutralize the system was calculated and added. In addition, salt with concentration of 0.15M was checked in. Thirdly, the output of system builder was used to carry out minimization step for the entire system of complex and solvent for 100ps. Lastly, MD simulation was run using isothermal-isobaric ensemble with constant number of particles (NPT) mode for 100ns.
3. Results and Discussion
Docking Validation
To validate the protein-ligand interaction, the co-crystal ligands from the targets were extracted and redocked in the same grid as that of the native ligand [29]. Among nine, only three targets (NSP5 (3CL pro), NSP13 (helicase), and NSP14 (exoribonuclease domain)) possess the co-crystalized ligands, therefore validation was carried to these three. Among these, for the protein corresponding to 3CL protease (PDB: 6W63) the random mean square deviation (RMSD) score of the co-crystal ligand was 1.6984 Å. For NSP13 (PDB: 7NNG) and NSP14 (PDB: 7R2V), the RMSD for corresponding co-crystals were 0.3814 Å and 1.035 Å respectively. As the RMSD was <2 Å, the docking methodology followed is within the acceptable limits (Figure 2).
Screening of Drugs and MD Studies
The shortlisted drugs among the FDA/TGA approved were subjected to docking studies against seven different virus-based targets. Few molecules that failed to bind to the targets in our docking studies were excluded. One of the 214 drug molecules, gramicidin, generated poses only for RdRp with high binding energy so it was also not considered further. From this screening process, the drug with the least binding energy against each target was subjected to MD studies. The complete summary of the drug hits corresponding to each target is further discussed in detailed in the following subsections.
Table 3.
Top results from docking studies against seven chosen targets.
| S. No | Target code | Drug name | Dock score (kcal/mol) |
Type of interactions | Interacting residues – Bond length (in Å) |
|---|---|---|---|---|---|
| 1. | Spike Receptor Binding Domain (6M0J) | Natamycin | -7.82 | H-bond | Tyr449: 1.97; Tyr453: 1.98; Gln498: 1.92; Asn501: 1.99, 2.46; Gly502: 2.15 |
| Alendronate | -7.69 | H-bond | Arg403: 1.95; Tyr453: 2.10; Ser494: 1.78, 1.9, 1.8; Gly496: 1.95 | ||
| 2. | N protein RNA Binding Domain (6VYO) | Cromolyn | -9.54 | H-bond | Ala154(A): 1.99; Asn55(D): 2.09; Ala55(D): 2.17; Arg92(D): 2.45; Ala173(D): 1.79; Arg149(D): 1.98 |
| Salt bridge | Arg107(D): 4.33; Arg149(D): 3.87 | ||||
| 3. | NSP3 (6W9C) |
Natamycin | -7.39 | H-bond | Asp(A) 108: 2.52; Lys157(C): 2.08; Leu162(C): 2.10; Gly163(C): 2.57; Glu167(C): 2.06 |
| 4. | NSP5 (6W63) |
Treprostinil | -10.76 | H-bond | Cys44: 1.63; Glu166: 1.97; Thr190: 1.87; Gln192: 1.97 |
| Co-crystal | -6.90 | H-bond | Gly143: 2.31; Asn142: 2.23; Hie163: 2.09; Glu166: 1.92 | ||
| Pi-pi stacking | Hie41: 5.14 | ||||
| 5. | NSP12 (6M71) |
Alendronate | -7.86 | H-bond | Asp452: 1.95; Arg553: 1.94; Arg555: 2.64; Thr556: 1.61, 1.7, 1.83; Asp623: 1.76, 1.76 |
| Remdesivir | -3.27 | H-bond | Asp618: 1.81, 1.98; Cys622: 2.66 | ||
| Pi-Pi stacking | Tyr455: 5.34 | ||||
| 6. | NSP13 (7NNG) |
Cromolyn | -5.87 | H-bond | Arg443: 2.14; Lys320: 2.23, 2.54; Gly538: 2.08 |
| Pi-cation | Arg443: 4.89, 4.81 | ||||
| Co-crystal | -4.06 | H-bond | Lys320: 2.15 | ||
| Salt bridge | Lys320: 2.88; Lys323: 2.96 | ||||
| 7. | NSP14 (7R2V) |
Cromolyn | -11.95 | H-bond | Leu366: 2.13; Tyr368: 1.99; Asn388: 1.85 |
| Pi-Pi stacking | Phe426: 4.2 | ||||
| Co-crystal | -11.747 | H-bond | Arg310: 1.83; Gly333: 1.99; Asp352: 1.78, 1.99; Ala353: 2.37; Tyr368: 2.01, 2.01; Trp385: 2.10 | ||
| Salt bridge | Arg310: 2.81 |
Spike Receptor Binding Domain (RBD)
Spike protein is a homotrimer with two subunits, namely S1 and S2 in each of the monomer. S1 contains the RBD which binds specifically to the host ACE2 to enable entry into the cells, and is prone to key mutations found in the different variants of concern [8]. On the other hand, S2 subunit arbitrates the viral-cell fusion followed by viral replication by infusing the viral genetic material [30, 31]. Therefore, blocking the S1 subunit will help prevent the fusion with ACE2 preventing the viral entry and its replication. When we carried out the docking studies of the shortlisted drugs with the viral spike protein (PDB: 6M0J) by giving key interacting residues with ACE2 as the active site grid, the drugs natamycin and alendronate had the least docking score of -7.82 kcal/mol and -7.69 kcal/mol respectively, making them promising candidates. The residues Tyr449, Tyr453, Ser494, Gln498, Asn501 and Gly502 have shown interactions with natamycin. Among these, most of the interactions are due to hydroxyl groups of the drug. Only two residues Ser494 and Tyr453 interacted with the 4-amino group of mycosamine sugar moiety (Figure 3a). Interestingly, these residues are among those that interacted with the ACE2 receptor during viral entry [32]. As, natamycin binds to the same residue, binding of the ACE2 receptor is not possible. Similar binding pattern is seen in case of alendronate, wherein the residue Arg403, Tyr453, Ser494 and Gly496 interacts with this drug. Therefore, due to better binding scores, there is a chance that these two drugs could prevent the entry of virus as well as inhibit the spike protein from viral replication.
To determine the behavior of ligand in a biological system, we used molecular dynamics simulation and found that natamycin was stable for the initial 20ns as the residue from the docked complex was retained during this period, but the ligand started losing few contacts after 20ns and moved slightly out of the active site pocket resulting in the increased RMSD and fluctuations until 60ns. After that, the ligand maintained interactions with Asn437, and to a lesser extent with Asn440, until the end of the simulation time. As shown in Figures 3b and 3c, the active site residue has low fluctuations in the RMSF plot; c.f. time frame analysis for pose view at different time frames of natamycin (Figure 3d). A different trend was observed in case of alendronate, wherein, the complex was stable for the initial 3ns and the contacts were same as that of docked complex. After 3ns, the molecule completely moved out of the assigned active site resulting in escalated RMSD to 60 Å and then declined to 32 Å at 5ns. During this, alendronate deviated from the active site and attached to the other side of the S1 protein by interacting with Asp427 and Asp428 (c.f. time frame analysis, Figure 3e). Multiple strong interactions were seen with Asp427 and 428 while few contacts were observed with Phe429 and Thr430. From 6ns, the ligand was stable and the interactions with Asp427 and Asp428 were retained until the end of the simulation with minor deviations (c.f. supplementary file 3.1). As natamycin stays in the pocket till the end of simulation, it is most likely to inhibit the spike protein, and in vitro studies could show promising results.
Nucleocapsid Protein RNA Binding Domain (NPRBD)
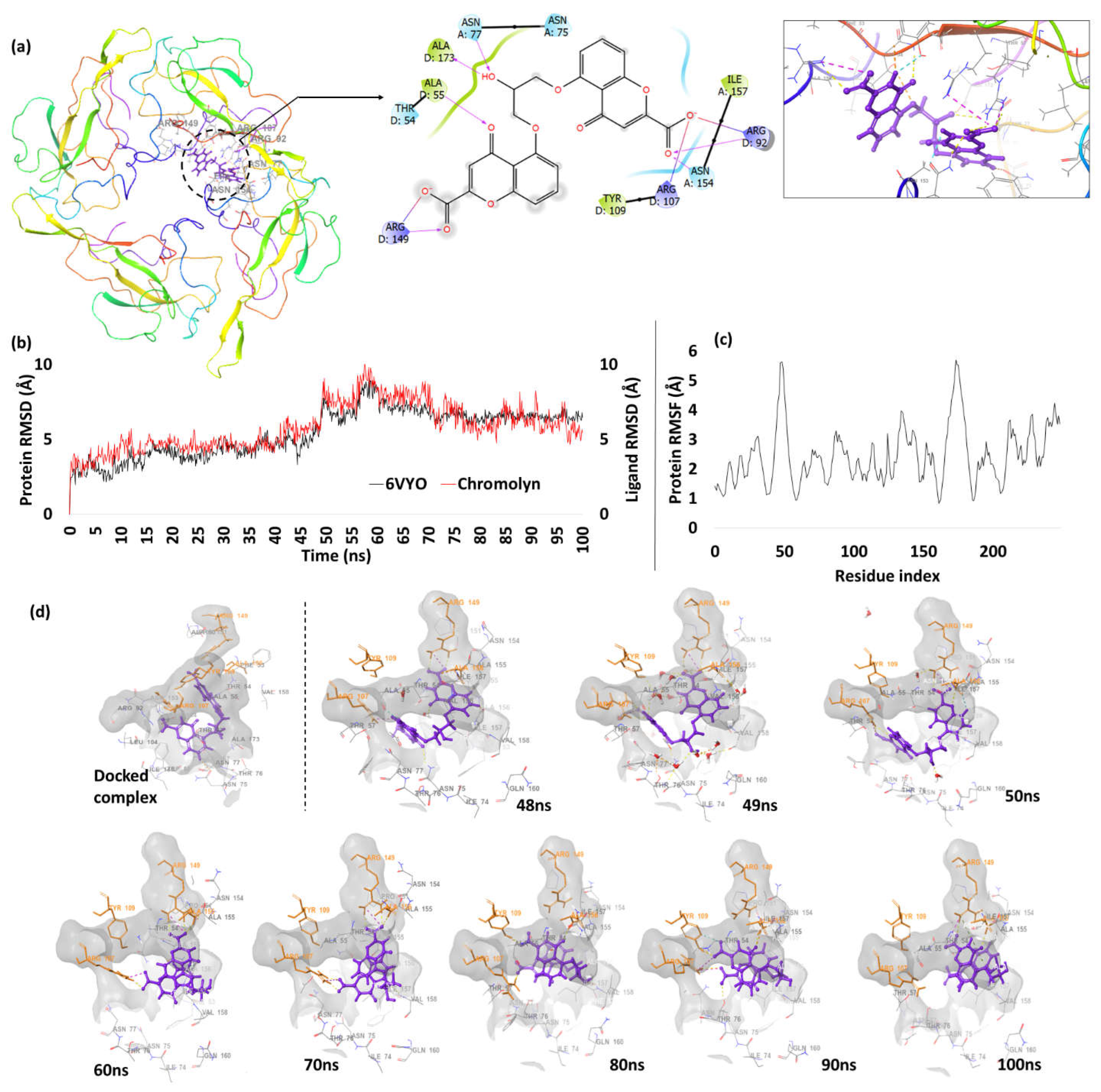
Nucleocapsid protein has a significant role in the viral structure as it binds to the virus’s genetic material (RNA) and facilitates the folding process of hammerhead ribozyme that catalyzes reversible cleavage and ligation reaction at specific RNA sites. This results in preventing the formation of unproductive conformations of the RNA and production of a helical ribonucleoprotein. Further, it also regulates various cellular processes such as, cell cycle progression, apoptosis and actin reorganization [33]. The nucleocapsid protein has two different domains which aid in different functions. The N-terminal domain (NTD) is very important in case of viral replication and transcription; while the C-terminal domain (CTD) has conserved dimerization mechanisms by forming hydrophilic and hydrophobic interactions [33]. Since, N-protein has a major role in viral replication, inhibition of this target can be beneficial for treating SARS-CoV-2 infection. Upon performing docking with NPRBD (PDB: 6VYO), the drug with the best score was found to be cromolyn, a mast cell stabilizer with the docking score of -9.54 kcal/mol. The residues that interacted with cromolyn include Ala55, Asn77, Arg92, Arg107, Arg149, Asn154 and Ala173. Except for Ala55, Asn77 and Ala173, all other residues interacted with the carboxyl terminals of the drug’s chromene moiety, while the other residues (Asn77 and Ala173) interacted with the hydroxyl at 2nd position of propane linker, and Ala55 interacted with the moiety’s keto group at the 4th position (Figure 4a).
MD analysis of cromolyn in complex with 6VYO shows good RMSD plot as consistent deviations were observed with the protein residues (Figure 4b). For the initial 48ns, the ligand has slight variations between RMSD of 3Å-5Å, during which the residues Trp52, Asn153, Asn154, Ala155 of chain A and Ala55, Thr57, Arg107, Tyr109, Arg149, Pro151, Ala156 and Asa173 of chain D shown strong interactions with the cromolyn molecule. After that, some of the interacting residues with the ligand were broken resulting in escalated RMSD between 6Å-10Å up to 75ns (Figure 4c).
Time frame analysis between 48ns-100ns shown in Figure 4d reveals that the ligand started uprooting from the binding site and was prevented by the strong interactions of Arg107 and Arg149 which held the cromolyn molecule within the site. The alkyl chain (hydroxyl at the 2nd position of propane) linker between two chromene moieties were likely responsible for these higher fluctuations. The protein had a similar fluctuating pattern as that of the ligand. Thr76 of chain A formed a new interaction while the residues Asn154, Ala155 of chain A and Arg107, Arg149, Ala156 of chain D retained their strong interactions with the cromolyn molecule until the end of the simulation. Further, from the ligand protein (LP) contacts, it is clear that the residues Arg107, Arg149 and Ala156 of chain D contributed to most of the interactions while the other residues only made moderate contributions along with water mediated interactions (c.f. supplementary file 3.2).
Figure 4.
(a) Docking pose of chromolyn representation in 3D (right) and 2D (left) (b) RMSD plot of cromolyn showing deviations during the complete simulation (c) RMSF plot of N protein RNA binding domain showing fluctuations of the residues during the simulation (d) time frame analysis of cromolyn showing poses at different time intervals.
Figure 4.
(a) Docking pose of chromolyn representation in 3D (right) and 2D (left) (b) RMSD plot of cromolyn showing deviations during the complete simulation (c) RMSF plot of N protein RNA binding domain showing fluctuations of the residues during the simulation (d) time frame analysis of cromolyn showing poses at different time intervals.
Papain Like Protease (PL pro)
PL proteases are multifunctional enzymes essential for replication of viruses. It is capable of recognizing and hydrolyzing ubiquitin and Interferon-stimulated gene 15 (ISG15; Ubiquitin-like protein) that covalently form an isopeptide bond with the target protein, thereby favouring the entry/replication of the viral cells within the host. Hence, inhibiting PL pro could be a good approach to prevent the viral replication in the host system [34]. From our studies, three drugs, natamycin, alendronate and metaproterenol, were found to have close docking scores of -7.39 kcal/mol, -7.16 kcal/mol and -7.13 kcal/mol respectively. The active site of PL pro is between two chains (A and C) that gives a proper shape for the ligand to fit (Figure 5a). Between these two chains, the residues that interacted with natamycin include Asp108 of chain A and Lys157, Leu162, Gly163 and Glu167 of chain C. The hydroxyl on the 25th carboxylic group of natamycin interacted with Lys157 and Leu162, while hydroxyl groups at 1st and 26th positions interacted with Gly163 and Asp108 respectively. Additionally, the hydroxyl at 6th position of the substituted sugar (mycosamine) interacted with Glu167.
Further MD analysis shows low fluctuations of the target residues and the ligand during the complete simulation as the RMSD was <3.2Å. After major fluctuations during 18-24ns and 44-56ns, the protein residues attained equilibrium for the rest of the simulation time. Change in Natamycin’s conformation from the docked pose resulted in the RMSD of 4Å, and we observed at 1ns that the residue Gln269 formed a contact with the drug while Asp108 and Asn109 formed water mediated interactions. The fluctuations were varying up to 7Å until 30ns and during 31ns-34ns and 40ns-48ns, the ligand had no contact with the protein resulting higher RMSD of 12Å. After 48ns, Tyr268 and Asn109 formed contact with natamycin resulting in decline of RMSD to 9Å and the ligand attained equilibrium as the fluctuations were not high towards the end (Figure 5b). RMSF analysis shows low fluctuations of the residues except those in the active site region due to drug interaction. The residues between 266-270 formed interactions with the ligand and resulted in higher RMSF due to fluctuations of the interacting atoms of the ligand (>4.0 Å) (Figure 5c). The residues that were in contact predominantly with natamycin were Trp106, Asp108, Asn109, Asn267 and Tyr268. Among these, Asp108 and Asn267 retained the interactions and prevented the ligand from moving outside the active site (c.f. Figure 5d). These two residues contributed to bulk of the simulation time (42% and 31% respectively), compared to Asn109, Thr265 and Tyr268 (10-15%) (c.f. supplementary file 3.3).
3. CL Protease/Main Protease
3CL protease, a C30 endopeptidase is found in almost all coronavirus variants and is involved in cleaving polyprotein at different conserved sites [35]. It possesses three major domains (D1, D2 and D3) and the active site remains between D1 and D2 [36]. From our docking analysis, a total of 14 drugs were found to have docking score close to the co-crystal ligand (-6.89 kcal/mol). Among these, two drugs were found to have docking score > -9.0 kcal/mol. These two drugs are treprostinil, a prostacyclin vasodilator, and ETC-1002 (bempedoic acid), an anti-hyperlipidemic agent, with docking scores of -10.76 kcal/mol and -9.65 kcal/mol, respectively. Since, treprostinil had the better score, we analyzed its binding pattern further. Treprostinil formed a total of four H-bonds each with Cys44 and Glu166, and two interactions each with Thr190 and Gln192. The hydroxyl group at 3rd position on the octyl side chain interacted with the carbonyl group of Thr190 and amino group of Gly192. In addition, the hydroxyl group at 2nd position of the cyclopentanaphthalene interacted with Glu166 and the carboxylic terminal of treprostinil formed H-bond with Cys44 (Figure 6a).
To understand the behavior of treprostinil, the docked pose of co-crystal ligand as well as treprostinil along with the protein was taken forward for molecular dynamics. During the simulation process, the fluctuations were low as the RMSD was below 3Å. The RMSD of the protein corresponding to treprostinil is relatively less compared to the co-crystal, indicating greater stability of the complex. Further, the ligand RMSD plot shows that co-crystal ligand had less RMSD than treprostinil until 50ns, however, huge deviations was observed until the end of the simulation. Treprostinil had RMSD ranging between 2Å-4Å until 35ns, beyond which the RMSD increased to 4Å from 37ns and attained equilibrium until the end of the study (Figure 6b). The residues that predominantly interacted with the ligand during the simulation process include Asp187, Gln189, Thr190 and Gln192. Additionally, these active site residues have fewer fluctuations during the simulation time as their random mean square fluctuation (RMSF) is below 2Å (Figure 6c). Among these residues, Asp187, Thr190 contributed for >40% of interaction while Glu166, Arg188, Gln189 and Gln192 contributed for interactions ranging between 10% and 20% (See supplementary file 3.4). The confirmations between 34ns-37ns were also captured as the ligand had slight increased RMSD during this interval, and it was observed that the drug retained within the active site. (Figure 6d). As treprostinil has far better docking score than the co-crystal ligand, and the complex is more stable, it can be considered a potential candidate to target main protease experimentally.
RNA Dependent RNA Polymerase (RdRp)
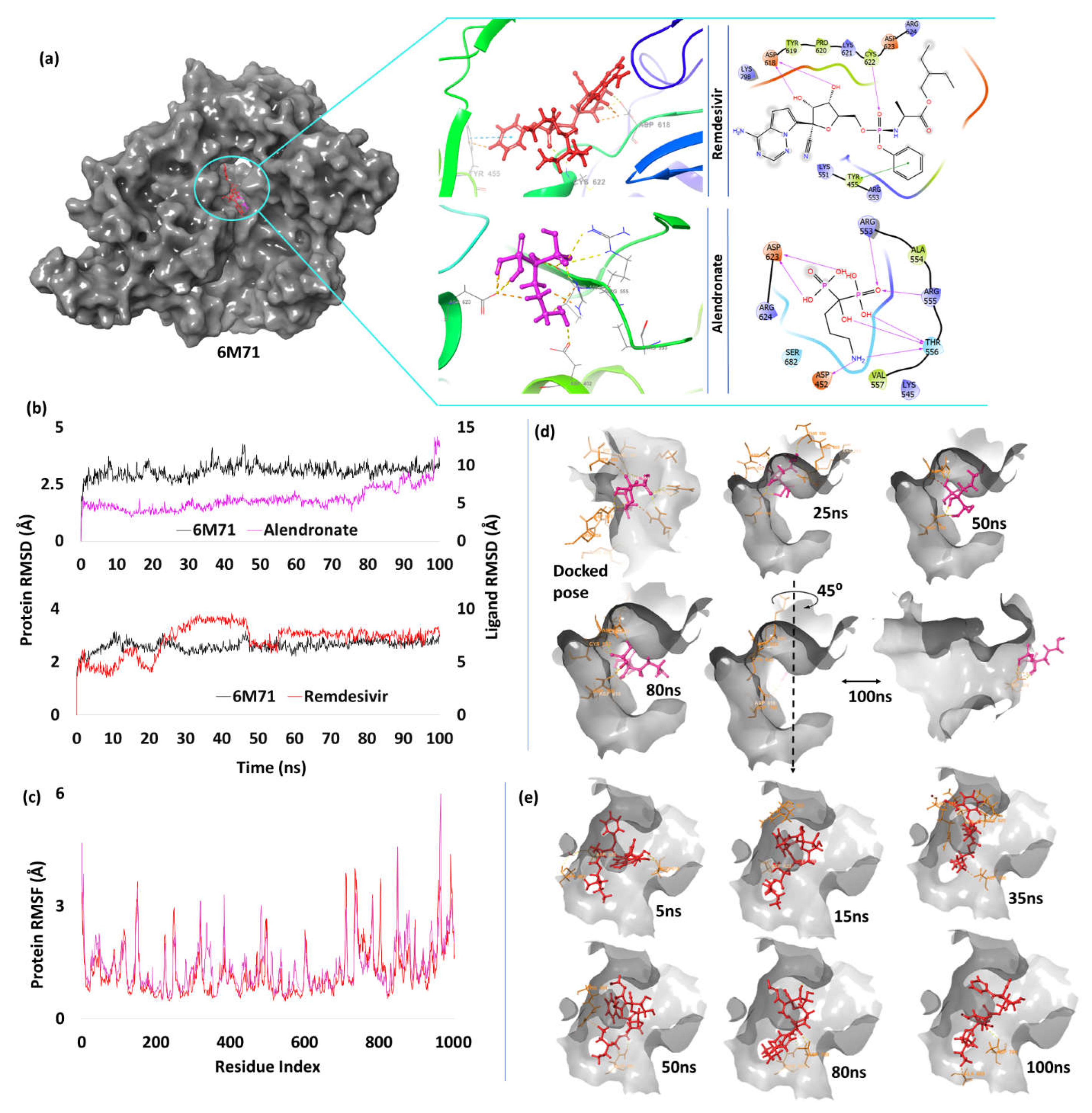
RNA dependent RNA polymerases are highly conserved proteins and catalyze the replication process of an RNA template. Therefore, inhibiting this target could help in preventing the replication of the viral genetic material, thereby reducing the viral load [37]. When we performed the docking studies with the shortlisted ligands, alendronate, a bisphosphonate drug, had the least binding score of -7.86 kcal/mol, better than the control drug remdesivir has lower score of -3.27 kcal/mol. Alendronate was able to form a total of eight interactions each with Asp452, Arg553, Arg555, two with Asp623, and three with Thr556. All the interactions are due to the functional groups present on the bisphosphonate molecule. On the other hand, remdesivir had only one H-bond interaction with each of Asp618, Pro620 and Cys622. Tyr455 also formed pi-pi stacking interaction with the drug due to aromaticity (Figure 7a).
MD studies of the alendronate complex with RdRp reveals a stable RMSD plot. Although it varied from its docking pose at 1ns, the interacting residue was still in contact with alendronate resulting in escalated RMSD to 6Å and greater stability with low 4Å - 6Å fluctuations until 80ns (Figure 7b). Asp623 and Asp760 had constant interactions with alendronate while Arg553, Arg555, Asp618, Lys621 and Cys622 had partial interactions. Beyond 80ns, the ligand further changed its conformation resulting in higher RMSD. The ligand for the last 20ns showed partial interactions with Arg553, Lys621, and Cyc622 while multiple strong interactions were seen with Asp618 and Asp623. The interactions with Asp760 slightly decreased during the end of the study. RMSF plot shows minor fluctuation in the active site residues (<2.5Å), indicating protein stability (Figure 7c). The LP contact plot shows Asp623 and Asp760 contributed 87% and 77% respectively, to the total interactions. Contributions of other residues such as Arg553 (36%), Arg555 (22%), Asp618 (21%), Lys621 (34%) and Cys622 (24%) were moderate (c.f. supplementary file 3.5, alendronate). The time frame analysis also shows the interaction of Asp623 and Asp760 during the fluctuating intervals (Figure 7d).
To compare our results with the control drug, MD analysis was carried out. In our analysis, remdesivir was stable for the initial 10ns with RMSD 4Å - 4.5Å, followed by fluctuation, especially at 20ns - 45ns up to 9Å, then a drop in RMSD of 6Å-7Å during 45ns-55ns, and equilibrium for the rest of the simulation (Figure 7b). The protein was also stable as the interacting residues were within the RMSF of 2.0Å (Figure 7c). The LP contact plot shows minor contributions of the residues during the simulation study, unlike alendronate complex (c.f. supplementary file 3.5, remdesivir). While the key interacting residues of RdRp were present in the MD study of remdesivir, the interactions with alendronate were much stronger. The residue that participated in the direct interactions with remdesivir include Ala550 (20%), Arg555 (11%), Asp623 (13%) and Asp760 (20%) with moderate to low contributions. Alendronate had multiple stronger interactions with the active site and the drug remained within the active site during the entire simulation.
Figure 7.
(a) Docking image of alendronate and remdesivir in the active site of RdRp (PDB_id: 6M71) (b) RMSD plot showing alendronate and remdesivir (c) RMSF plot showing fluctuations in protein residues (d) time frame analysis of alendronate showing its poses a at different intervals (e) time frame analysis of remdesivir showing its poses at different time intervals.
Figure 7.
(a) Docking image of alendronate and remdesivir in the active site of RdRp (PDB_id: 6M71) (b) RMSD plot showing alendronate and remdesivir (c) RMSF plot showing fluctuations in protein residues (d) time frame analysis of alendronate showing its poses a at different intervals (e) time frame analysis of remdesivir showing its poses at different time intervals.
Helicase
Helicases are the enzymes responsible for unwinding of helical structure of genetic material producing ssDNA/ssRNA that is essential for 3R’s (replication, recombination and repair) [38, 39]. The unwinding process occurs by breaking the H-bonds sequentially between two strands of the double helix and this whole process of bond breaking is catalyzed by ATPase that consumes a molecule of ATP and produces sufficient energy to break the bond [40]. Hence, preventing the bond breaking could halt the unwinding process and thus the 3R’s. As these enzymes are highly conserved across all coronaviruses, it is an interesting target to block the viral replication [41]. Upon docking of the shortlisted 214 FDA/TGA approved drugs, the molecule with best binding score was found to be cromolyn (-5.87 kcal/mol) which is slight better than the co-crystal ligand (-4.02 kcal/mol). The residue that participated in interactions with cromolyn were Lys320, Arg443 and Gly532, while only Lys320 was involved in interacting with the co-crystal ligand (Figure 8a).
RMSD plot from our MD studies reveal initial fluctuations followed by stability. Although Glu261, Ser289, Thr440, Arg442, and Glu540 were absent in the docked complex, they were found to interact with the ligand during the first 5ns of MD studies leading to RMSD of 6Å. Between 10ns -35ns the ligand fluctuated to 10Å after which it attained equilibrium (Figure 8b). During this, the protein active site residues had minor fluctuations (<2Å) as shown in the RMSF plot (Figure 8c). Beyond 35ns, Glu261 contributed significantly (64%) and other residues moderately (15%-20% for Asp260, Asn268, Phe291, Phe437, Thr440, Lys462 and His464) to ligand bonding. Two pi-cation interactions were also seen with His290 (10%) and His464 (11%) (see supplementary file 3.6, cromolyn). Despite fluctuations, the ligand never left the active site during the entire simulation (c.f. time frame analysis, Figure 8d). When we performed the MD studies to the co-crystal ligand, the RMSD trend was similar to that of cromolyn as RMSD escalated between 7Å-8Å for the first half of the simulation time and then declined to 6Å (Figure 8c). All the interacting residues with the co-crystal ligand were within the active site region and the major contributing residues include His290 (33%), Lys320 (76%), Tyr324 (30%), Arg443 (94%) and Lys569 (15%). Since cromolyn and the co-crystal ligand have similar behaviour during the simulation, both molecules could have similar potency.
Exoribonuclease Domain
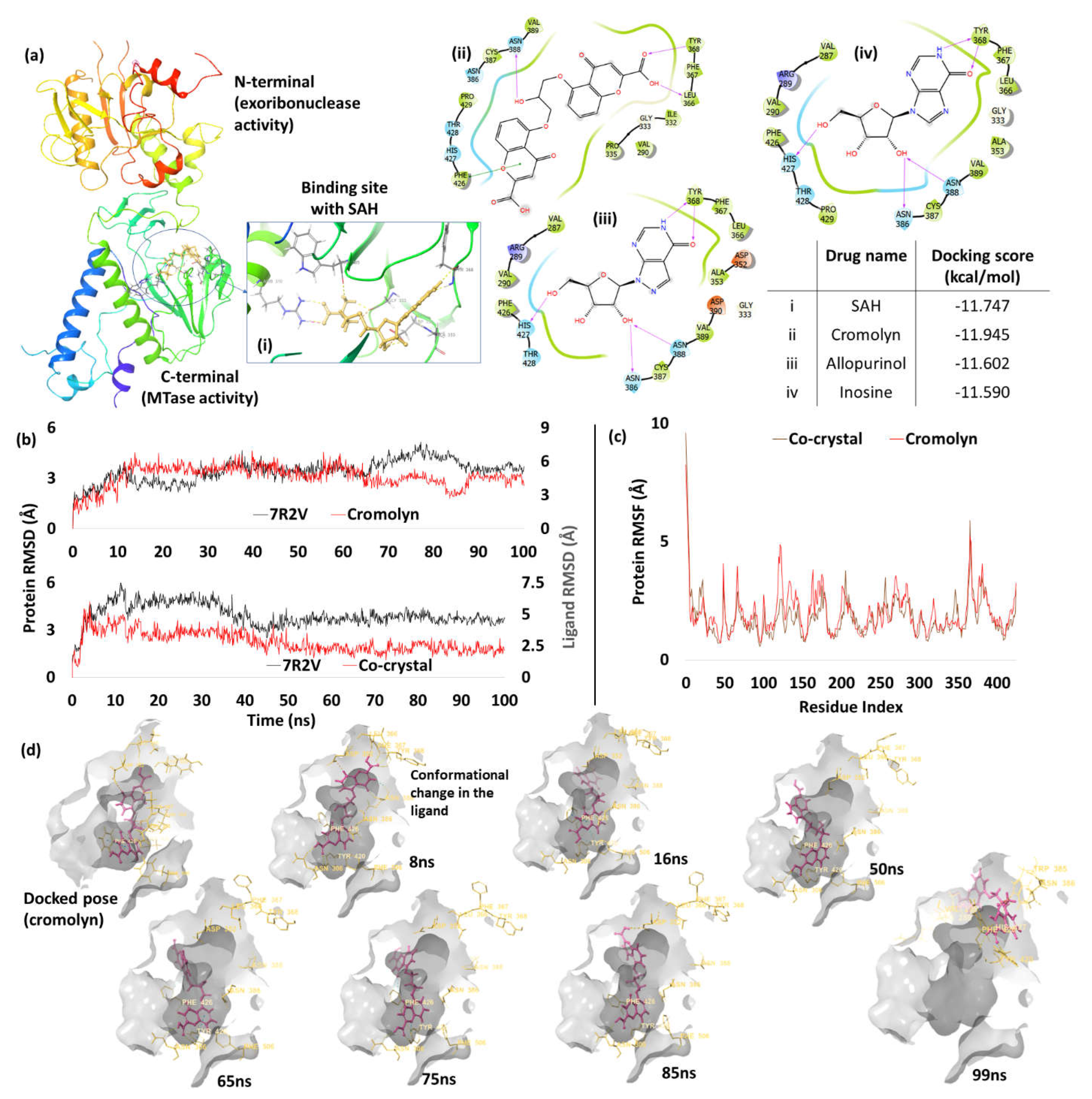
NSP14 has two terminals with different activities; the N-terminal has 3’-5’ exoribonuclease activity, while the C-terminal has N7-methyl transferase (MTase) activity [42]. While the C-terminal preserves the stability of the viral RNA, the N-terminal role is crucial for maintaining the RNA genome [43]. To exhibit MTase activity, NSP14 uses S-Adenosyl methionine (SAM) as a methyl donor that causes methylation on the Nitrogen at the 7th position of guanine (5’ end), which is necessary for mRNA stability as well as the translation process in human cells [44]. Therefore, by introducing a molecule with more affinity to the active site residues than the SAM, it is possible to block the activity of MTase. When we performed docking studies with this protein (PDB: 7R2V), three drugs had docking score closer to the co-crystal (-11.75 kcal/mol). These drugs were cromolyn (-11.95 kcal/mol), allopurinol (-11.60 kcal/mol) and inosine (-11.59 kcal/mol). Since cromolyn had a better score, we further analyzed it in terms of interactions. SAH has interactions with Arg310, Asp352, Ala353, Tyr368 and Trp385. Unlike the SAH, cromolyn had different interacting residues such Tyr368, Asn386 and Asn388. However, the binding site residues were the same in all three drugs (Figure 9a). As cromolyn had the best score, MD studies were conducted to analyze its behavior and compare its binding properties with that of the co-crystal ligand.
MD analysis of cromolyn shows RMSD ranging between 1.5Å - 6.4Å, and interactions with Asn306, Leu366, Tyr368, Asn388 and Phe426 until 12ns. After 16ns, water-mediated interactions were mainly observed and the RMSD was between 3Å-6Å. (Figure 9b). RMSF plot shows that active site residues in cromolyn complex had similar pattern to that of the co-crystal (Figure 9c). During the simulation, key contributing residues were Asn306 (62%), Trp385 (34%), Tyr420 (58%) and Phe426 (24%) (c.f. supplementary file 3.7, cromolyn). Time frame conformations of the ligand were captured to see the binding pose at different fluctuating intervals. From Figure 9d, it is observed that the change in the conformation was due to cromolyn’s rotatable bonds. When we compared the results with co-crystal ligand, the RMSD reveals fluctuations for the initial 5ns (RMSD 2.4Å - 5.6Å) which started to decline and attain equilibrium (fluctuations between 2.5Å-3Å) towards the end of the simulation (Figure 9b). The residue that contributed in the study include Gln313, Asp331, Ile332, Asp352, Tyr368, Trp385, Asn386 and Asn388. Among these, Asp352 (92%), Tyr368 (98%), Trp385 (74%) and Asn388 (41%) are especially important (c.f. supplementary file 3.7, co-crystal).
4. Conclusions and Future Studies
From the molecular docking and molecular dynamic studies, it is clear that the drugs, treprostinil, natamycin, cromolyn and alendronate have shown promising results against various targets of SARS-CoV-2. RMSD plots and time frame analysis have revealed the behaviour of these drugs during the simulation. In case of main protease, treprostinil had a better RMSD plot when compared to the co-crystal ligand, with limited fluctuations. On the other hand, the fluctuations of natamycin and the residues in papain like protease were similar, with interactions maintained until the end of the simulation. Cromolyn also shows interesting results with three different targets (N protein RNA binding domain, NSP13 and NSP14). It maintained constant interactions during the entire molecular dynamic simulation. Alendronate was able to form strong interactions with two targets (spike receptor binding domain and RNA dependent RNA polymerase). Its interaction with RdRp was far better than remdesivir, which is a current standard of care. From this study, we propose that these four drugs (treprostinil, alendronate, natamycin and cromolyn) could have anti-viral effects and could be a good candidate to treat SARS-CoV-2 infections. As treprostinil promotes vasodilation of pulmonary and systemic arterial vasculature, it might have beneficial effects for COVID-19 patients through improving oxygenation and nitric oxide levels in the blood vessels, inhibiting platelet aggregation, and its anti-inflammatory property [45-47]. Our study further shows that this antihypertensive drug has better binding pattern than the co-crystal ligand. Alendronate is an amino-bisphosphonate indicated for osteoporosis, and patients administered with this drug have reportedly low incidences of COVID-19 testing and hospitalisation [48, 49], which could be due to their immunomodulatory effects and active site interactions discerned in our study. Natamycin is an anti-fungal drug used in eye infections and we could find only one publication pertaining to COVID-19; an in silico study has suggested its use in combination with Baloxavir marboxil and RU85053 as a cocktail as they inhibit 3CLpro, PL pro and RdRp [50]. Our simulation study shows significant binding interactions with PL pro and better dynamic behaviour, thus strengthening its evidence base for further investigation. Our study shows that cromolyn, a mast cell stabilizer used as an asthma drug, is promising due to better binding score with multiple interactions and similar dynamic behaviour to that of the co-crystal; unsurprisingly, it is currently in phase III COVID-19 clinical trial [51, 52]. Further in vitro and in vivo studies must be carried out to verify our hypothesis and quantify the anti-viral activities of these four drugs. We are currently implementing the approach presented in this paper to our broader shortlist of 1029 repurposed drug candidates, and improving our filtering methodology with better alternatives to the Tanimoto coefficient [20, 53, 54]. We are also extending our approach to the key variants of SARS-CoV-2 [8].
Institutional Review Board Statement and Informed Consent
Not applicable as this study involved no human or animal subjects.
Data Availability Statement
Any underlying data not presented can be provided by the corresponding authors upon reasonable request.
Acknowledgements
Authors are grateful to their respective institutions for internal funding and infrastructure. The article reflects the views of the authors and does not represent the views or policies of any affiliating or funding agency. We thank our colleagues for their comments and support.
Conflict of Interest
The authors declare no conflict of interest.
Abbreviations
3CL protease – 3 Chymotrypsin like protease; ACE-2 – Angiotensin converting enzyme 2; ATP – Adenosine triphosphate; FDA – Food and drug administration; H-bond – Hydrogen bond; ISG15 - Interferon-stimulated gene 15; MD studies – Molecular dynamic studies; MTase: Methyl transferase; NSP – Non-structural protein; PAINS: Pan-assay interference compounds; OPLS4: Optimize potentials for liquid simulation; PDB: Protein data bank; PL pro: Papain like protease; RMSD – Root mean square deviation; SAM – S-Adenosyl methionine; SAH – S-Adenosyl homocysteine; sdf: structure data file; TGA – Therapeutic Goods Administration; TIP3P: Transferable intermolecular potential 3P; XP: extra precision
References
- COVID-19 Data Explorer - Our World in Data. (2022). Available online: https://ourworldindata.org/explorers/coronavirus-data-explorer (accessed on 23 November 2022).
- Wang, H.; Paulson, K.R.; Pease, S.A.; Watson, S.; Comfort, H.; et al. Estimating excess mortality due to the COVID-19 pandemic: a systematic analysis of COVID-19-related mortality, 2020-21. Lancet 2022, 399, 1513–1536. [Google Scholar] [CrossRef]
- World Health Organization. Monkeypox, COVID-19 & Other Global Health Issues Virtual Press Conference - 22 September 2022. (2022). Available online: https://www.who.int/publications/m/item/monkeypox--covid-19---other-global-health-issues-virtual-press-conference---22-september-2022 (accessed on 23 November 2022).
- World Health Organization. Weekly epidemiological update on COVID-19 - 28 December 2021. (2021). Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---28-december-2021 (accessed on 28 November 2022).
- Mallapaty, S. China COVID wave could kill one million people, models predict. Nature 19 December 2022. [CrossRef]
- Holmes, E.C.; Zhang, Y.Z. The evolution and emergence of hantaviruses. Curr Opin Virol. 2015, 10, 27–33. [Google Scholar] [CrossRef]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; et al. Role of Structural and Non-Structural Proteins and Therapeutic. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Van Vuren, P.J.; McAuley, A.J.; Kuiper, M.J.; Singanallur, N.B.; Bruce, M.P.; et al. Highly Thermotolerant SARS-CoV-2 Vaccine Elicits Neutralising Antibodies against Delta and Omicron in Mice. Viruses 2022, 14, 800. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health. Remdesivir. COVID-19 Treatment Guidelines. (2022). Available online: https://www.covid19treatmentguidelines.nih.gov/therapies/antiviral-therapy/remdesivir/ (accessed on 9 May 2022).
- Kozlov, M. Merck’s COVID pill loses its lustre: what that means for the pandemic. Nature 2021. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.C.; Hobbs, F.D.R.; Gbinigie, O.A.; Rahman, N.M.; Hayward, G.; et al. Molnupiravir plus usual care versus usual care alone as early treatment for adults with COVID-19 at increased risk of adverse outcomes (PANORAMIC): an open-label, platform-adaptive randomised controlled trial. Lancet 2022. [Google Scholar] [CrossRef]
- Szabo, B.G.; Lenart, K.S.; Petrik, B.; Gaspar, Z.; Kiss-Dala, N.; et al. Favipiravir treatment does not influence disease progression among adult patients hospitalized with moderate-to-severe COVID-19: a prospective, sequential cohort study from Hungary. Geroscience 2021, 43, 2205–2213. [Google Scholar] [CrossRef]
- Jimenez, D.; Paxlovid: what we know about Pfizer’s Covid-19 pill. (2022). Available online: https://www.pharmaceutical-technology.com/features/paxlovid-pfizer-covid-19-pill/ (accessed on 7 February 2022).
- National Institutes of Health. Ritonavir-Boosted Nirmatrelvir (Paxlovid). (2022). Available online: https://www.covid19treatmentguidelines.nih.gov/therapies/antiviral-therapy/ritonavir-boosted-nirmatrelvir--paxlovid-/ (accessed on 9 May 2022).
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Jain, H.A.; Agarwal, V.; Bansal, C.; Kumar, A.; Faheem, F.; et al. CoviRx: A User-Friendly Interface for Systematic Down-Selection of Repurposed Drug Candidates for COVID-19. Data 2022, 7, 164. [Google Scholar] [CrossRef]
- MacRaild, C.A.; Mohammed, M.U.R.; Faheem Murugesan, S.; Styles, I.K.; et al. Systematic Down-Selection of Repurposed Drug Candidates for COVID-19. Int J Mol Sci 2022, 23, 11851. [Google Scholar] [CrossRef]
- McAuley, A.J.; Jansen van Vuren, P.; Mohammed, M.-U.-R.; Faheem Goldie, S.; et al. Use of Human Lung Tissue Models for Screening of Drugs against SARS-CoV-2 Infection. Viruses 2022, 14, 2417. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, T.T. An Elementary Mathematical Theory of Classification and Prediction. In: Proc. IBM Internal Report. International Business Machines Corp. pp. 1–11.
- Kushwaha, P.P.; Singh, A.K.; Bansal, T.; Yadav, A.; Prajapati, K.S.; et al. Identification of Natural Inhibitors Against SARS-CoV-2 Drugable Targets Using Molecular Docking, Molecular Dynamics Simulation, and MM-PBSA Approach. Front Cell Infect Microbiol 2021, 728. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J Med Chem 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- El-Aziz, N.; Shehata, M.; Awad, O.; El-Sohaimy, S. Inhibition of COVID-19 RNA-Dependent RNA Polymerase by Natural Bioactive Compounds: Molecular Docking Analysis. Egypt. J. Chem 2021, 1989–2001. [Google Scholar] [CrossRef]
- Li, D.; Luan, J.; Zhang, L. Molecular docking of potential SARS-CoV-2 papain-like protease inhibitors. Biochem Biophys Res Commun 2021, 538, 72–79. [Google Scholar] [CrossRef]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm Sin B 2020, 10, 1228–1238. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; et al. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Nutt, D.R.; Smith, J.C. Molecular dynamics simulations of proteins: Can the explicit water model be varied? J Chem Theory Comput 2007, 3, 1550–1560. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; et al. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model 2009, 49, 444–460. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Cheng, Y.; Wu, Y. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol. Sin. 2020, 35, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J Chem Inf Model 2021, 61, 4425–4441. [Google Scholar] [CrossRef] [PubMed]
- Tatar, G.; Ozyurt, E.; Turhan, K. Computational drug repurposing study of the RNA binding domain of SARS-CoV-2 nucleocapsid protein with anti-viral agents. Biotechnol Prog 2021, 37, e3110. [Google Scholar] [CrossRef] [PubMed]
- Báez-Santos, Y.M.; St. John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed anti-viral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.; Batool, M.; Ain, Q.U.; Kim, M.S.; Choi, S. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int J Mol Sci 2021, 22, 9124. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Aftab, S.O.; Ghouri, M.Z.; Masood, M.U.; Haider, Z.; Khan, Z.; et al. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J Transl Med 2020, 18, 1–15. [Google Scholar] [CrossRef]
- Wu, Y. Unwinding and rewinding: Double faces of helicase? J. Nucleic Acids. 2012, 2012, 140601. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.M.; Frolov, M.V. The diverse roles of RNA helicases in RNAi. Cell Cycle 2009, 8, 3500–3505. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Marchand, B.; te Velthuis, A.J.W.; Snijder, E.J.; Weiss, S.; et al. Mechanism of nucleic acid unwinding by SARS-CoV helicase. PLoS ONE 2012, 7, e36521. [Google Scholar] [CrossRef]
- Pitsillou, E.; Liang, J.; Hung, A.; Karagiannis, T.C. The SARS-CoV-2 helicase as a target for anti-viral therapy: Identification of potential small molecule inhibitors by in silico modelling. J Mol Graph Model 2022, 114, 108193. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M. Coronavirus genomic nsp14-ExoN, structure, role, mechanism, and potential application as a drug target. J Med Virol. 2021, 93, 4258–4264. [Google Scholar] [CrossRef]
- Ogando, N.S.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Bredenbeek, P.J.; Posthuma, C.C.; et al. The Enzymatic Activity of the nsp14 Exoribonuclease Is Critical for Replication of MERS-CoV and SARS-CoV-2. J Virol 2020, 94, e01246–20. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; et al. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc Natl Acad Sci USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]
- Prasetya, E.; Mulia, B.; Luke, K. Inhaled prostacyclin analogues in COVID-19 associated acute respiratory distress syndrome: scientific rationale. Egypt Heart J. 2021, 73, 82. [Google Scholar] [CrossRef]
- Nasrullah, A.; Virk, S.; Shah, A.; Jacobs, M.; Hamza, A.; et al. Acute respiratory distress syndrome and the use of inhaled pulmonary vasodilators in the COVID-19 era: A narrative review. Life 2022, 12, 1766. [Google Scholar] [CrossRef]
- Rajpal, S.; Inpatient use of inhaled pulmonary vasodilator therapy in patients infected with COVID-19 - American College of Cardiology. (2020). Available online: https://www.acc.org/latest-in-cardiology/articles/2020/05/13/08/55/inpatient-use-of-inhaled-pulmonary-vasodilator-therapy-in-patients-infected-with-covid-19 (accessed on 2 January 2023).
- Degli Esposti, L.; Perrone, V.; Sangiorgi, D.; Andretta, M.; Bartolini, F.; et al. The use of oral amino-bisphosphonates and coronavirus disease 2019 (COVID-19) Outcomes. J. Bone Miner. Res. 2021, 36, 2177–2183. [Google Scholar] [CrossRef]
- Von Andrian, U.H.; Thompson, J.; Wang, Y.; Dreischulte, T.; Barreiro, O.; et al. Association between Bisphosphonate use and COVID-19 related outcomes: a retrospective cohort study. medRxiv 2022. [Google Scholar] [CrossRef]
- Murugan, N.A.; Kumar, S.; Jeyakanthan, J.; Srivastava, V. Searching for target-specific and multi-targeting organics for Covid-19 in the Drugbank database with a double scoring approach. Sci Rep 2020, 10, 19125. [Google Scholar] [CrossRef] [PubMed]
- Staten, M. New COVID-19 treatment being explored at TTUHSC El Paso. (2022) Available online:. Available online: https://www.krwg.org/regional/2022-01-19/new-covid-19-treatment-being-explored-at-ttuhsc-el-paso (accessed on 2 January 2023).
- Michelson, E.A.; Cromolyn sodium for treatment of COVID-19 pneumonia. ClinicalTrials.gov. (2021). Available online: https://clinicaltrials.gov/ct2/show/NCT05077917 (accessed on 2 January 2023).
- Miranda-Quintana, R.A.; Bajusz, D.; Rácz, A.; Héberger, K. Extended similarity indices: the benefits of comparing more than two objects simultaneously. Part 1: Theory and characteristics. J Cheminform; 2021, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Quintana, R.A.; Rácz, A.; Bajusz, D.; Héberger, K. Extended similarity indices: the benefits of comparing more than two objects simultaneously. Part 2: speed, consistency, diversity selection. J Cheminform 2021, 13, 33. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The shortlisting methodology is shown on the left and the targets used for the molecular docking study on the right.
Figure 1.
The shortlisting methodology is shown on the left and the targets used for the molecular docking study on the right.
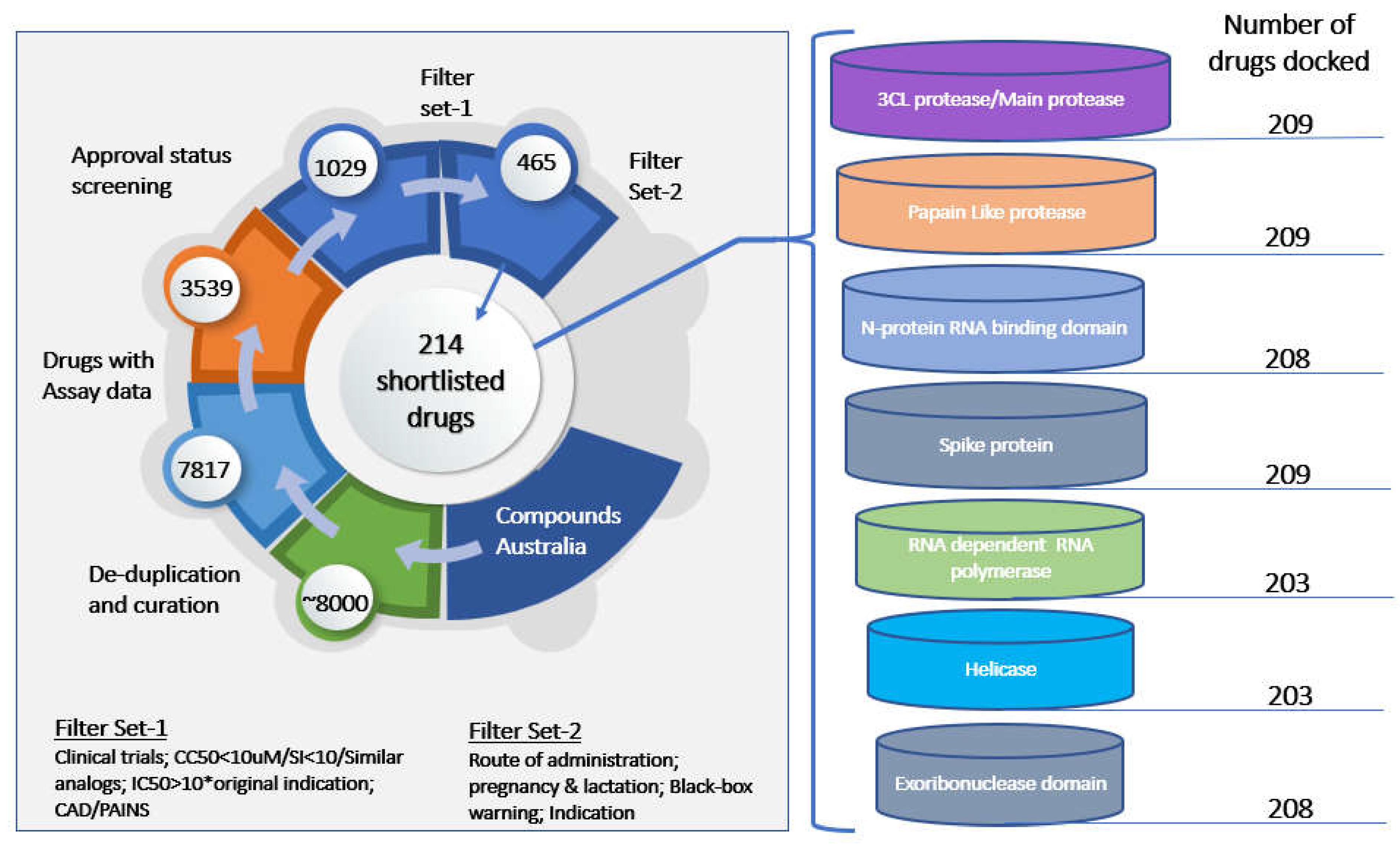
Figure 2.
Co-crystal RMSD of (a) 3CL protease (b) Helicase (c) Exoribonuclease domain; green color represents native ligand and blue represents redocked pose.
Figure 2.
Co-crystal RMSD of (a) 3CL protease (b) Helicase (c) Exoribonuclease domain; green color represents native ligand and blue represents redocked pose.
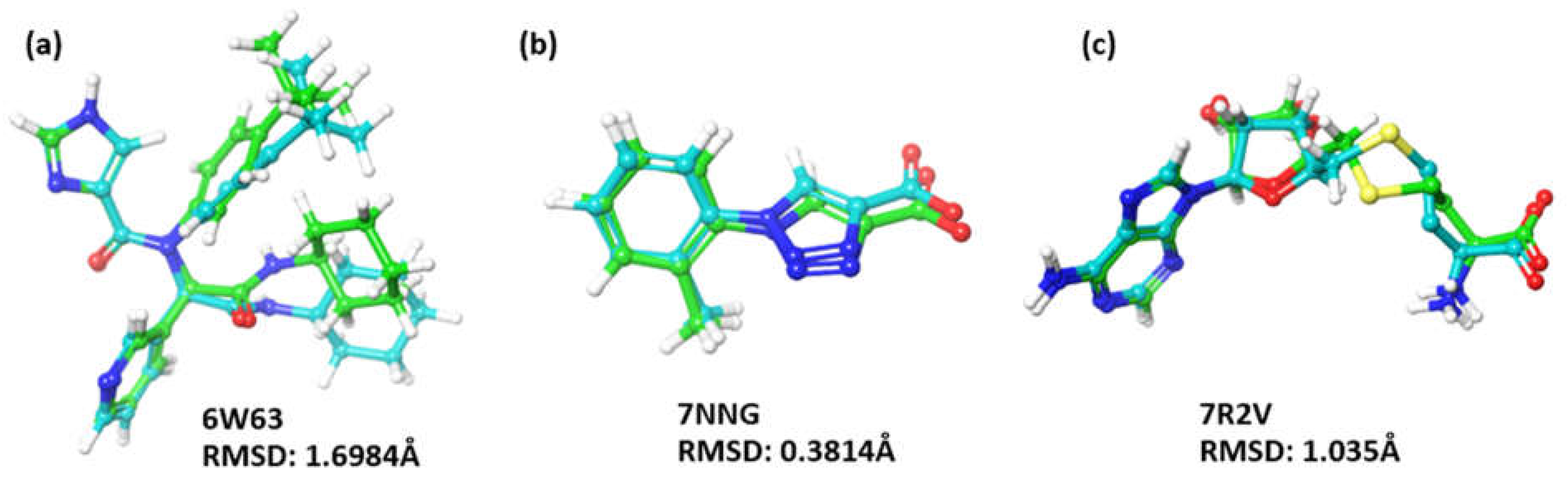
Figure 3.
(a) RMSD plot showing natamycin and alendronate (b) RMSF plot showing fluctuations in protein residues (c) time frame analysis of natamycin showing its poses at different intervals (d) time frame analysis of alendronate showing its poses at different time intervals.
Figure 3.
(a) RMSD plot showing natamycin and alendronate (b) RMSF plot showing fluctuations in protein residues (c) time frame analysis of natamycin showing its poses at different intervals (d) time frame analysis of alendronate showing its poses at different time intervals.
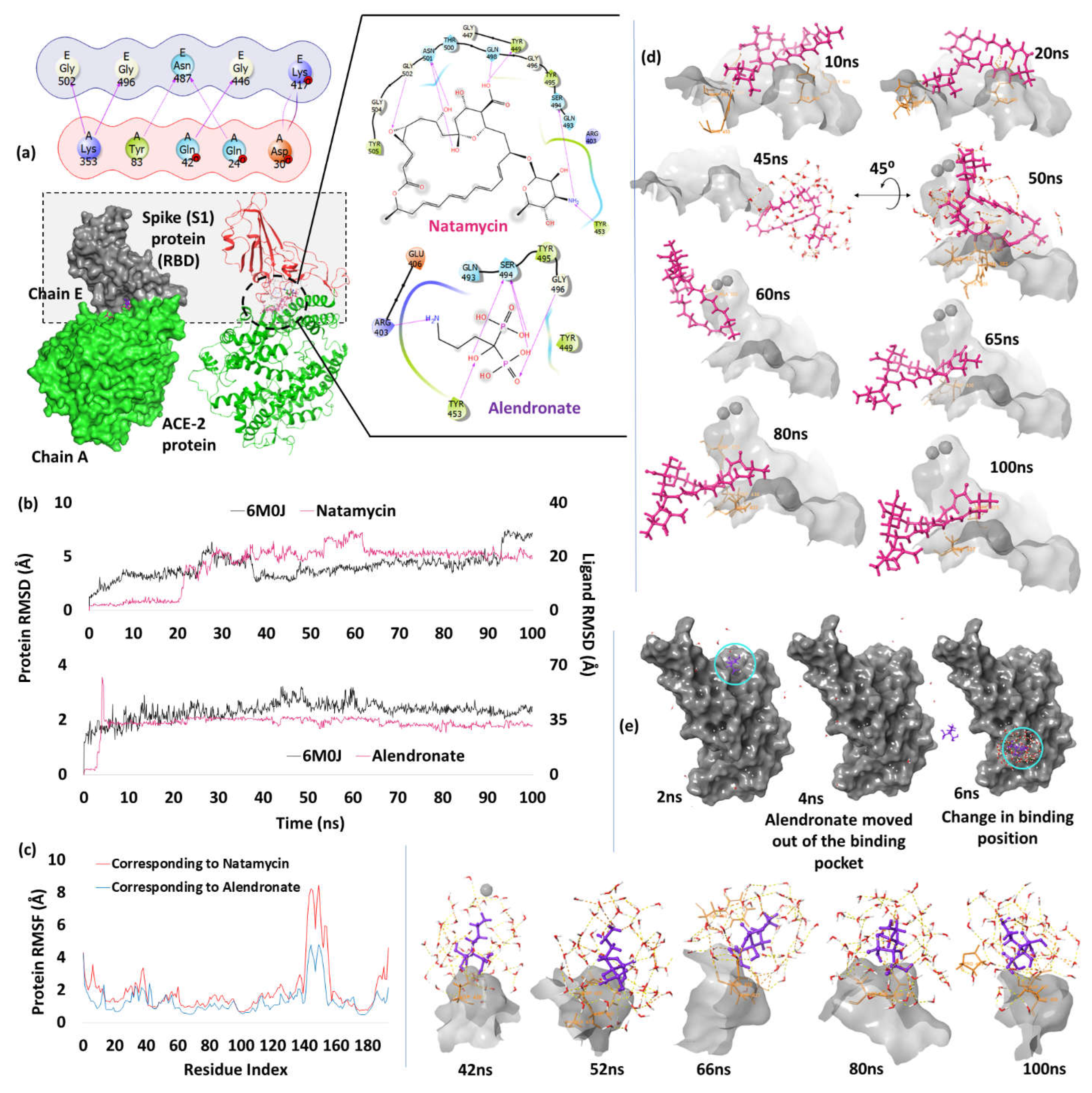
Figure 5.
(a) Docking pose representation in the active site between two chains A and C (b) RMSD deviation of natamycin and PL protease during the simulation time of 100ns (c) RMSF plot of the protein showing residual fluctuations (d) time frame analysis images of natamycin at intervals showing higher fluctuation.
Figure 5.
(a) Docking pose representation in the active site between two chains A and C (b) RMSD deviation of natamycin and PL protease during the simulation time of 100ns (c) RMSF plot of the protein showing residual fluctuations (d) time frame analysis images of natamycin at intervals showing higher fluctuation.
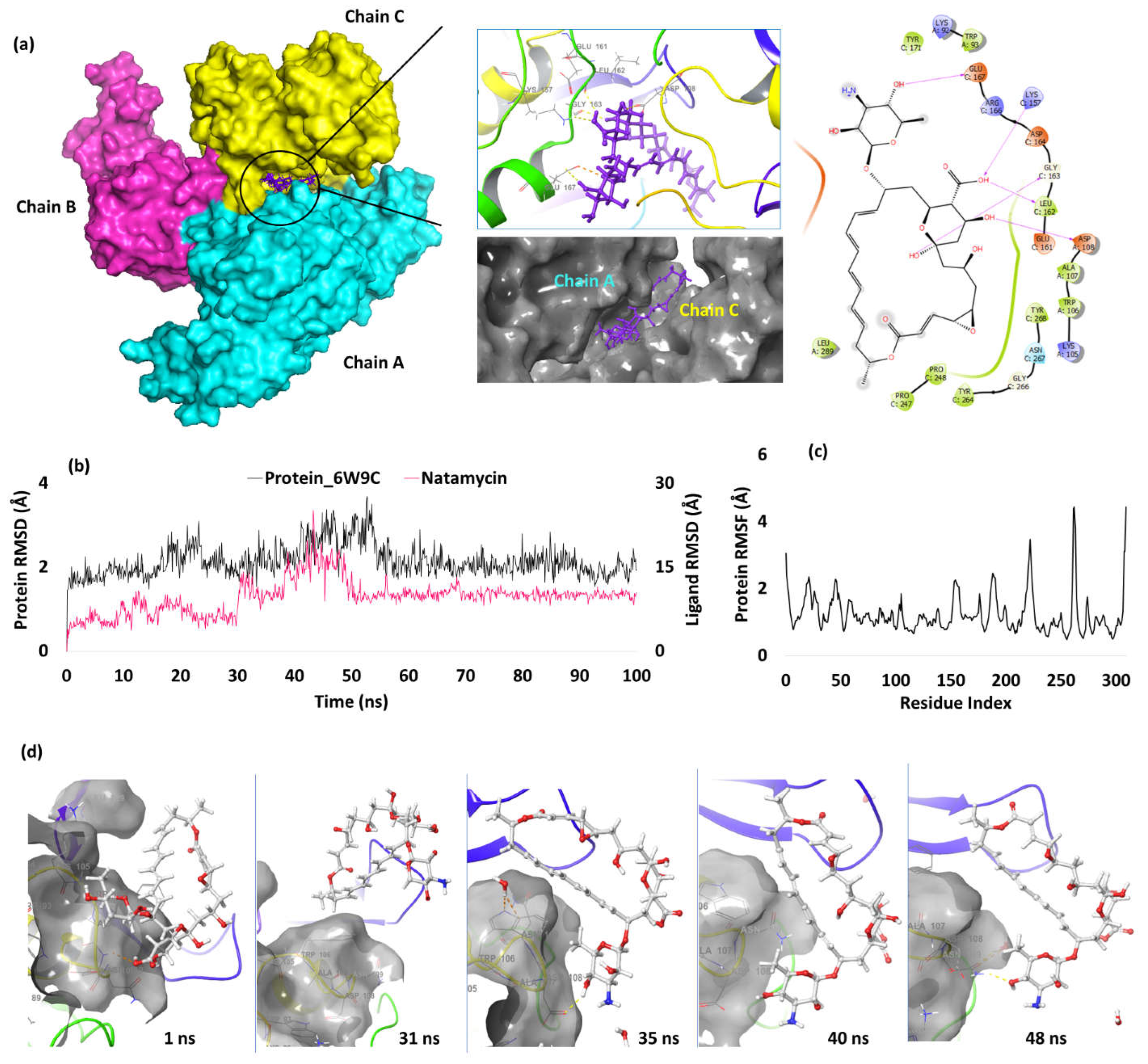
Figure 6.
(a) Docking results representation of trepreostinil in the active site between D1 and D2 (b) RMSD plot of 3CL protease (above) and ligands (below) (c) RMSF plot (d) time frame analysis of treprostinil showing the ligand poses at different time intervals.
Figure 6.
(a) Docking results representation of trepreostinil in the active site between D1 and D2 (b) RMSD plot of 3CL protease (above) and ligands (below) (c) RMSF plot (d) time frame analysis of treprostinil showing the ligand poses at different time intervals.
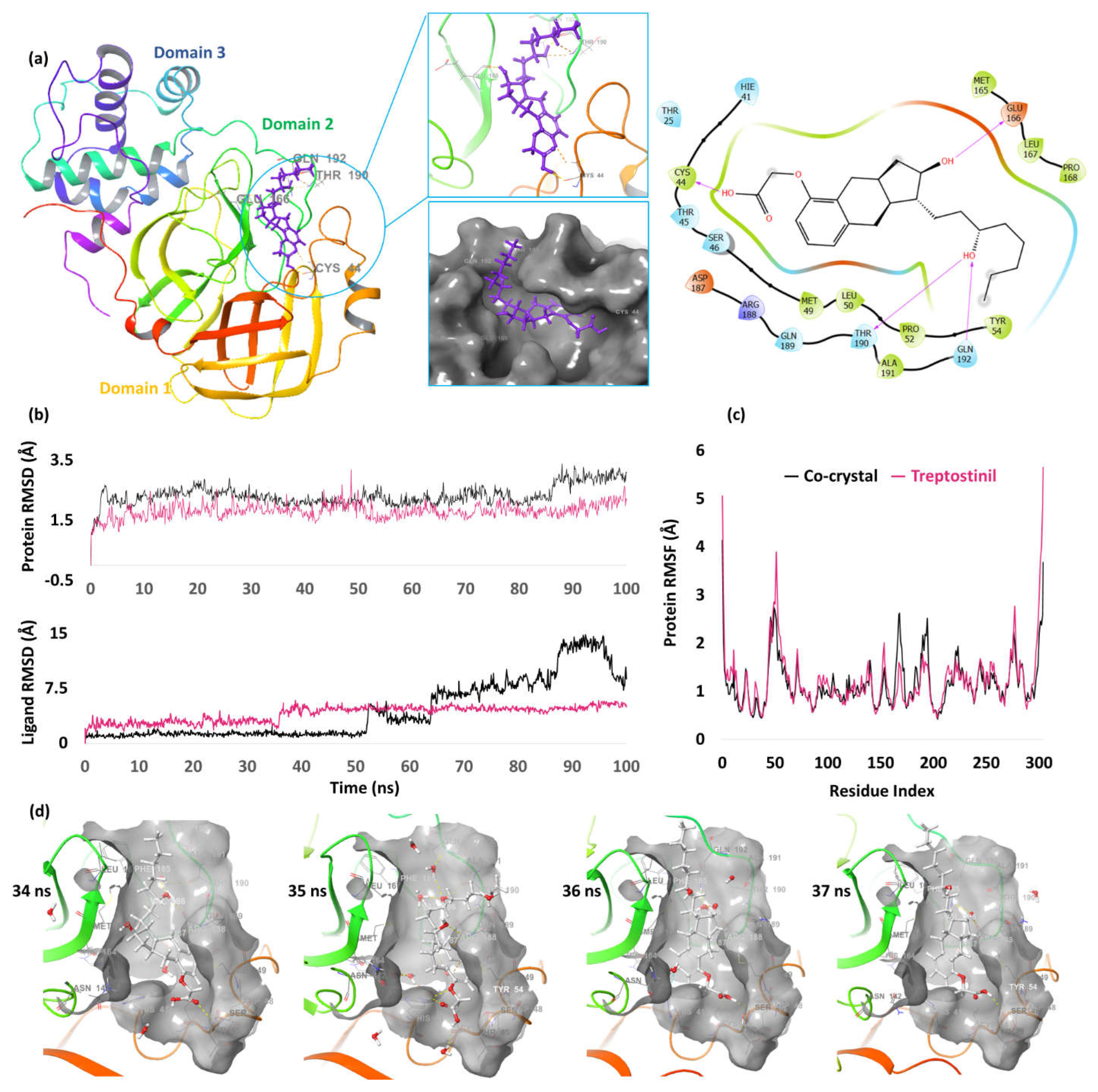
Figure 8.
Docking poses of cromolyn and co-crystal ligand in the active site between domains 1A and 2A (b) RMSD plot of cromolyn and co-crystal ligand (c) RMSF plot of helicase protein (7NNG) (d) time frame analysis of cromolyn representing various poses at different time intervals.
Figure 8.
Docking poses of cromolyn and co-crystal ligand in the active site between domains 1A and 2A (b) RMSD plot of cromolyn and co-crystal ligand (c) RMSF plot of helicase protein (7NNG) (d) time frame analysis of cromolyn representing various poses at different time intervals.
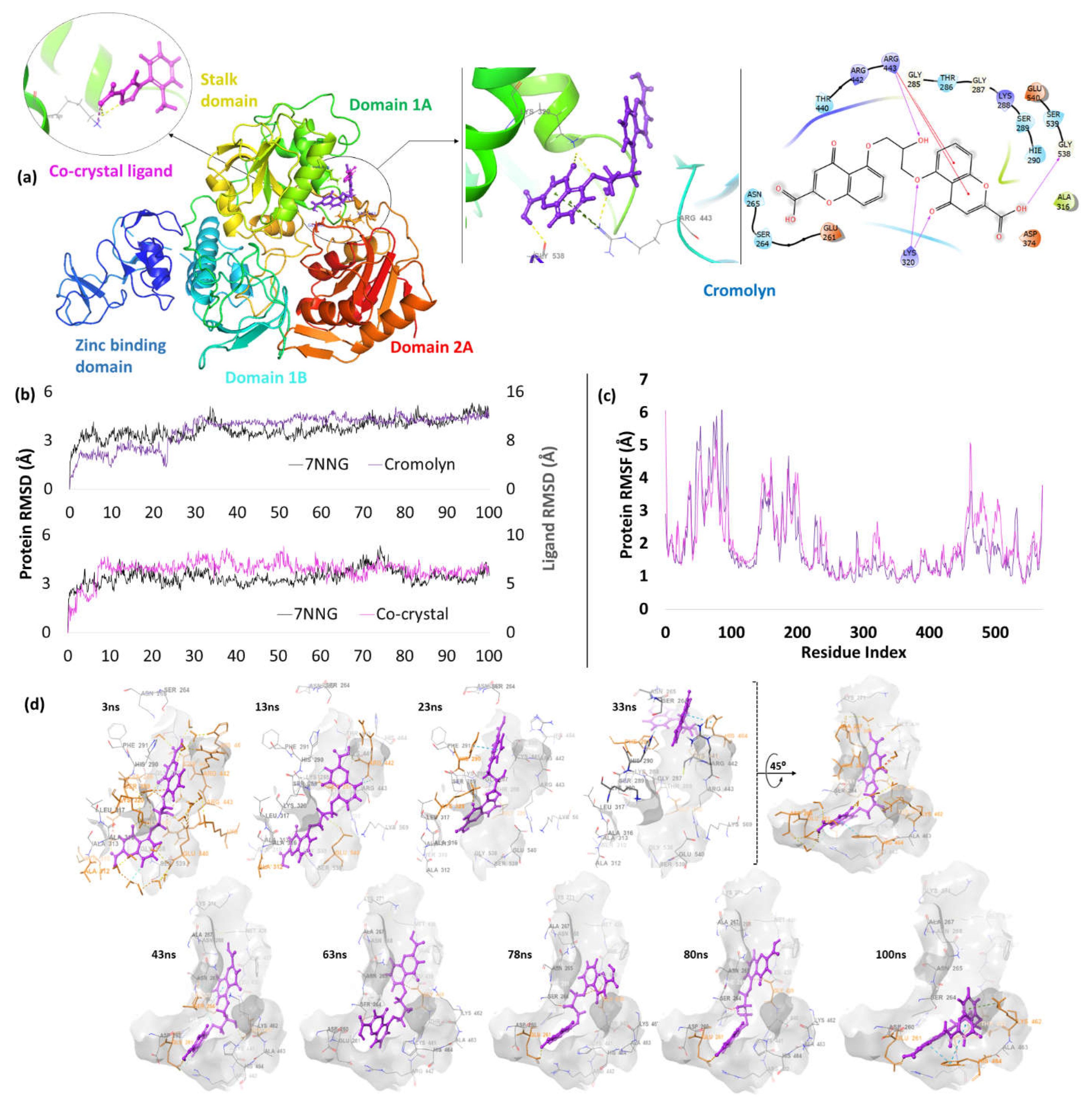
Figure 9.
Docking poses of cromolyn and co-crystal ligand in the C-terminal active site (b) RMSD plot of cromolyn and co-crystal ligand (c) RMSF plot of NSP14 (d) time frame analysis of cromolyn representing various poses at different time intervals.
Figure 9.
Docking poses of cromolyn and co-crystal ligand in the C-terminal active site (b) RMSD plot of cromolyn and co-crystal ligand (c) RMSF plot of NSP14 (d) time frame analysis of cromolyn representing various poses at different time intervals.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



