
Submitted:
04 January 2023
Posted:
06 January 2023
You are already at the latest version
Abstract
Plazomicin is a recently U.S. Food and Drug Administration (FDA)-approved semisynthetic aminoglycoside. Its structure consists of a sisomicin scaffold modified by adding a 2(S)-hydroxy aminobutyryl group at the N1 position and a hydroxyethyl substituent at the 6′ position. These substitutions produced a molecule refractory to most aminoglycoside-modifying enzymes. The main enzyme within this group that recognizes plazomicin as substrate is the aminoglycoside 2′-N-acetyltransferase type Ia [AAC(2′)-Ia], which reduces the antibiotic’s potency. Designing formulations that combine an antimicrobial with an inhibitor of resistance is a recognized strategy to extend the useful life of existing antibiotics. We have recently found that several metal ions inhibit acetylation of numerous aminoglycosides catalyzed by the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib]. In particular, Ag1+, which also enhances the effect of aminoglycosides by other mechanisms, is very effective in interfering with AAC(6′)-Ib-mediated resistance to amikacin. Here we report that silver acetate is a potent inhibitor of AAC(2′)-Ia-mediated acetylation of plazomicin in vitro, and it reduces resistance levels of Escherichia coli carrying aac(2′)-Ia. The resistance reversion assays produced equivalent results when the structural gene was expressed under the control of the natural or the blaTEM-1 promoters. The antibiotic effect of plazomicin in combination with silver was bactericidal, and the mix did not show significant toxicity to human embryonic kidney 293 (HEK293) cells.
Keywords:
AAC(2′)-Ia
; aminoglycoside 2′-N-acetyltransferase type Ia
; aminoglycoside
; multidrug resistance
; metal ions
; plazomicin
; adjuvant
1. Introduction
Nosocomial- and community-acquired pathogens have become resistant to many different antibiotics, and in some cases, they are virtually untreatable [1]. Numerous Enterobacterales clinical isolates possess genes coding for aminoglycoside modifying enzymes, extended-spectrum β-lactamases, and carbapenemases [2,3]. Colistin remains an option for treating life-threatening multidrug-resistant infections caused by some of these bacteria [4]. However, resistant variants have already been found in several geographical regions and may soon become prevalent [5,6]. Other novel options include cefiderocol and new β-lactams/β-lactamase inhibitors [7,8,9]. Aminoglycosides are excellent tools for treating a wide variety of Gram-negative and Gram-positive infections. Unfortunately, the rise and dissemination of aminoglycoside modifying enzymes the major mechanism of resistance to these antibiotics in the clinical setting, have reduced their effectiveness [10,11,12]. Therefore, developing new antibiotics or therapeutic strategies is necessary to generate viable treatment options [13]. Numerous analogs to natural aminoglycosides have been designed to resist the action of resistance enzymes. Many of these compounds, known as semisynthetic aminoglycosides, are refractory to the action of most aminoglycoside modifying enzymes and have been successfully introduced in the clinical setting to treat resistant infections [14,15]. Plazomicin is a next-generation semisynthetic aminoglycoside designed modifying sisomicin by the addition of a 2(S)-hydroxy aminobutyryl group at the N1 position and a hydroxyethyl substituent at the 6′ position [16,17]. These modifications result in a molecular structure refractory to most aminoglycoside modifying enzymes [10,18,19,20,21]. Plazomicin was approved in 2018 by the FDA to be used in patients with limited or no options for alternative treatment. It is active against multidrug resistant Enterobacterales, including strains producing carbapenemases and extended-spectrum β-lactamases, while showing tolerable levels of nephrotoxicity and ototoxicity [19,22,23]. Unfortunately, despite the substitutions that make plazomicin a non-substrate for most aminoglycosides modifying enzymes, the AAC(2′)-Ia enzyme identified in the chromosome of Providencia stuartii, can catalyze the inactivation of the antibiotic molecule through the transfer of an acetyl group from acetyl-CoA to the 2’-N-position [24,25]. Although this enzyme is not usually found in clinical isolates, it is a matter of time before it disseminates and becomes prevalent if the use of plazomicin increases. An obvious path to deal with the rise and dissemination of aminoglycoside modifying enzymes is the continuous design of semisynthetic aminoglycosides. However, design of new generations of semisynthetic aminoglycosides has proven costly and time consuming. These stumbling blocks warrant exploring alternative strategies such as the development of inhibitors of the enzymatic inactivation that together with the aminoglycoside form a combination therapy effective against resistant pathogens [13]. The recent finding that metal ions, some of them in complex with ionophores, inhibit the AAC(6′)-Ib-catalyzed acetylation of aminoglycosides and induce a reduction in the minimal inhibitory concentration (MIC) of amikacin to susceptibility levels proved this concept and increased expectations that multidrug resistant infections could be treated by these combination therapies [26,27,28,29,30,31]. In particular, Ag1+ inhibits the acetylation of amikacin by AAC(6′)-Ib and reverses resistance in bacteria in culture at low concentrations without needing an ionophore [31]. This article describes the inhibition of AAC(2′)-Ia-mediated plazomicin-resistance by Ag1+ in Escherichia coli harboring a recombinant clone containing the aac(2′)-Ia gene.
2. Results
2.1. Effect of Ag1+ on AAC(2')-Ia-mediated acetylation of plazomicin
Ag1+ drastically interfered with the acetylation of plazomicin (Figure 1). The presence of silver acetate in the reaction mixture produced significant inhibition while the addition of sodium acetate did not produce any changes in acetylation levels. These results, taking together with previous research showing that metal ions have the capability to inhibit the enzymatic acetylation of aminoglycosides, are an encouraging indication that Ag1+ could serve as an adjuvant to plazomicin if AAC(2’)-Ia or a similar enzyme disseminates among bacterial pathogens.
2.2. Effect of Ag1+ on AAC(2')-Ia-mediated resistance to plazomicin
The E. coli TOP10(pUC57AAC2Ia) MIC of plazomicin, as determined using commercial E-strips, was 12 μg/ml. To assess the effect of Ag1+ on the resistance to plazomicin of growing cells, E. coli TOP10(pUC57AAC2Ia) was cultured in the presence of silver acetate in addition to plazomicin. A reduction in the level of growth was observed when only plazomicin was present (Table 1). However, the cells grew to an OD600 consistent with heavy growth, confirming that AAC(2')-Ia confers substantial resistance to the antibiotic. Addition of silver acetate at 4 μM was sufficient to completely inhibit growth in the presence of plazomicin at a sub-MIC concentration (4 μg/ml) (Table 1). Furthermore, when the concentration of plazomicin was 8 μg/ml, still a sub-MIC value, 2 μM silver acetate was enough to inhibit growth (Table 1). A control experiment adding sodium acetate showed no differences in growth in the presence or absence of the addition (Table 1). The results described in this section unequivocally indicated that Ag1+ interferes with resistance to amikacin mediated by AAC(6')-Ib.
Studies on the expression of the aac(2')-Ia gene suggest that in its natural location, the P. stuartii chromosome, is subjected to regulation [32]. To discard any regulatory role in the action of Ag1+, the AAC(2')-Ia open reading frame was placed downstream of the blaTEM-1 promoter and cloned using pUC57 as cloning vector to generate the recombinant plasmid pUC57PBLAAAC2Ia. The MIC of plazomicin of E. coli TOP10(pUC57PBLAAAC2Ia) was 12/16 μg/ml. Table 2 shows that the results of this experiment were similar to those observed with the gene under the control of the natural promoter. The resistance levels in liquid medium were slightly lower with the gene that carries the blaTEM-1 promoter. However, inhibition of resistance by Ag1+ was identical with both promoters.
2.3. Bactericidal effect
Plazomicin showed bactericidal activity in previous studies [33]. Time-kill assays were used to evaluate if the phenotypic conversion to susceptibility observed when E. coli TOP10(pUC57AAC2Ia) was cultured in the presence of silver acetate in addition to plazomicin was due to a bactericidal effect. Figure 2 shows that adding silver acetate and plazomicin at a sub-MIC concentration had a robust bactericidal effect. Conversely, healthy growth was observed when one of the components was omitted. As in a previous report [33], regrowth was observed after 10 h incubation. It is worth noting that in the past study, regrowth in time-kill assays was observed with concentrations of up to 2x or 4x MIC values depending on the strain assayed. The bases for regrowth remain to be elucidated. Possible causes are the emergence of resistance or presence of tolerant variants in the culture. The results of the experiments described in this section demonstrated that plazomicin, in the presence of Ag1+ ions, exerted bactericidal action on E. coli cells in which resistance is caused by the presence of the AAC(2')-Ia enzyme. The bactericidal effect of plazomicin plus Ag1+ ions on E. coli resistant cells was similar to that of plazomicin alone on susceptible E. coli cells.
2.3. Cytotoxicity of the mix plazomicin/silver acetate
An essential factor for the viability of combination therapies is that they show low toxicity to the host. A preliminary analysis of the cytotoxicity of the mix investigated in this work was carried out using HEK293 cells. Figure 3 shows that the exposure of the cells to the combination or the individual components at the active concentrations did not cause significant mortality. While these experiments are a preliminary step toward understanding the toxicity of the combination plazomicin/silver acetate, the results warrant further development towards overcoming the action of the AAC(2')-Ia enzyme.
3. Discussion
Infectious diseases are a leading cause of death, compromised health, and disability [34]. Outbreaks of bacterial infection, usually associated with multidrug resistance, are increasingly reported and may soon be responsible for millions of deaths per year [35,36,37]. Furthermore, the increase in hard-to-treat or untreatable bacteria also threatens medical procedures like surgery, cancer treatment and other chronic diseases, organ transplants, dental work, and care for premature infants [13,38,39,40]. The impact of the drug resistance crisis is such that it was listed as one of the top ten global health threats [41]. Compounding the problem, unlike in the past when new antibiotics were available if existing ones became ineffective, the number of new antibiotics in development is dangerously low [1]. It is necessary to devise methodologies that extend the life of antibiotics currently in use. The development of inhibitors of mechanisms of resistance that can be administered in combination with the cognate antibiotic can be a viable strategy to treat resistant bacteria [11,13]. Although there are no inhibitors of resistance to aminoglycosides in clinical use, this course of action has already been proven successful for β-lactamase-mediated resistance to β-lactams [42].
While plazomicin is a new aminoglycoside antibiotic, there are already enzymes that can inactivate it. AAC(2')-Ia catalyzes the inactivation of plazomicin by acetylation and reduces its potency [18]. Following the steps of previous work indicating that selected metal ions interfere with the acetylation reaction [26,27,28,29,31,43,44], we tested the effect of Ag1+. We chose this ion because, unlike other metals, ionophores were not necessary to observe reversion of resistance in growing bacterial cells when tested as an inhibitor of resistance to aminoglycosides mediated by AAC(6')-Ib [31]. Plazomicin was readily acetylated in vitro in a soluble extract of cells containing a recombinant clone harboring aac(2')-Ia. The AAC(2')-Ia activity results in resistance to plazomicin as determined by measuring MIC values for E. coli cells carrying recombinant clones that include the gene expressed under the control of the natural or the constitutive blaTEM promoters. Since both recombinant plasmids were generated using the same plasmid vector, the gene dosage in both strains must be identical. Therefore, the similarity of the MIC values showed by both strains suggests that the described transcriptional regulation of expression of aac(2')-Ia [45] does not impact resistance levels in the conditions used in our assays. The effect of silver acetate was also identical in the strains harboring the structural gene expressed under the control of both promoters. When plazomicin was present at 4 or 8 μg/ml, the concentrations needed to overcome resistance were as low as 1 and 2 μM, respectively. It is worth noting that it has been described that silver ions potentiate the effect of aminoglycoside and other antibiotics by mechanisms still in discussion. There is consensus that silver ions increase membrane permeability, which can enhance the effect of antibiotics against Gram-negative bacteria [46,47,48]. It has also been suggested that silver-mediated increased production of reactive oxygen species plays a role in potentiating the action of aminoglycosides [47]. However, this latter possibility has not been confirmed. Our prior [31] and present results indicate that silver ions also enhance the action of aminoglycosides by interfering with enzymatic inactivation, at least in the case of two aminoglycoside modifying enzymes (AAC(6')-Ib and AAC(2')-Ia). We conclude that silver ions potentiate aminoglycosides by multiple mechanisms that result in the observed phenotypic conversion to susceptibility. These facts, taken together with the confirmation that plazomicin retains its bactericidal action when acting in concert with Ag+1 to inhibit growth of resistant bacteria, and that the combination plazomicin/silver acetate at the active concentrations does not exhibit cytotoxicity, make these mixes excellent candidates to extend the useful life of plazomicin and other aminoglycosides.
4. Materials and Methods
4.1. Bacterial strains and plasmids
Escherichia coli TOP10 F- mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL(StrR) endA1 nupG was transformed with the plasmid pUC57AAC2Ia and used for all assays. The plasmid pUC57AAC2Ia was constructed inserting the P. stuartii aac(2′)-Ia gene (accession number L06156, nucleotides 12-820)[45] into the BamHI/HindIII sites of pUC57. The plasmid pUC57PBLAAAC2Ia was generated fusing the blaTEM promoter and Shine-Dalgarno sequences, fragment encompassing nucleotides 4154-4225 (reverse complement, accession number J01749) [49] to the aac(2′)-Ia open reading frame (fragment encompassed by nucleotides 264-810, accession number L06156) [45]. Transformation of E. coli TOP10 with pUC57AAC2Ia was carried out as described by Cohen et al [50].
4.2. Bacterial growth
Bacteria were cultured in Lennox L broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl) with the addition of 2% agar in the case of solid medium. Plazomicin resistance levels were determined in cation-adjusted Mueller-Hinton broth at 37°C with shaking. Growth was assessed measuring the optical density at 600 nm (OD600) of the cultures containing the specified additions. Plazomicin was generously supplied by Cipla Therapeutics.
4.3. MIC determination
MIC values were determined using plazomicin commercial E-strips (Liofilchem S.r.l., Roseto degli Abruzzi, Italy) following the recommendations of the supplier on Mueller-Hinton agar plates.
4.4. Time-kill assays
Bacterial cells were cultured in cation-adjusted Mueller-Hinton broth until they reached 107 CFU/ml. At this time the cultures were divided in four aliquots, one of them was left intact and the others were supplemented with either 8 μg/ml plazomicin, 4 μM silver acetate, or both. Incubation was continued at 37°C with shaking and the CFUs were determined after the indicated times [29].
4.5. Acetyltransferase assays
Total soluble proteins (enzymatic extracts) were prepared as before [31]. Briefly, cells were recovered from cultures by centrifugation, resuspended in a 0.5 mM MgCl2 solution, and sonicated with a Heat Systems Ultra- sonic, Inc., Model No. H-IA (Plainview, NY) cell disrupter. The soluble proteins were then separated by centrifugation in a microfuge for 10 min at 4°C. The protein content of the extracts was determined using a commercial reagent (Bio-Rad). Acetyltransferase activity was assessed using the phosphocellulose paper binding assay [51]. Soluble extract (120 μg protein) obtained from E. coli TOP10(pUC57AAC2Ia) cells was added to the reaction mixture (200 mM Tris HCl pH 7.6 buffer, 0.25 mM MgCl2, 330 μM plazomicin, the indicated concentrations of sodium acetate or silver acetate, and 0.05 μCi of [acetyl-1-14C]-acetyl-coenzyme A (specific activity 60 μCi/μmol) in a final volume of 30 μl. After incubating the reaction mixture at 37 °C for 30 minutes, 20 μl were spotted on phosphocellulose paper strips. The unreacted radioactive substrate was washed once by submersion in 80 °C water followed by two washes with room temperature water. The phosphocellulose paper strips were then dried and the radioactivity corresponding to acetylated plazomicin was determined.
4.6. Cytotoxicity assays
The cytotoxicity of the combination silver acetate/plazomicin on HEK293 cells [52] was assessed as previously described [53]. One thousand cells per well were cultured on flat-bottom 96-well, black microtiter plates for 12 h before addition of the compounds to be tested. Incubation was then continued for 24 h before the cells were washed with sterile D-PBS, resuspended in the LIVE/DEAD reagent (2 μM ethidium homodimer 1 and 1 μM calcein-AM) (Molecular Probes), and incubated for 30 min at 37°C. At this moment the fluorescence levels at 645 nm (dead cells) and 530 nm (live cells) were measured. The percentage of dead cells was calculated relative to the untreated control cells. Control of maximum toxicity was calculated on cells treated with 70% methanol for 20 min. Experiments were conducted in triplicate. The results were expressed as mean ± SD of three independent experiments.
5. Conclusions
There is an urgent need for novel strategies to extend the useful life of antibiotics currently in use. Latest-generation antibiotics such as plazomicin can quickly be overcome by resistance mechanisms developed by bacteria. The ability of Ag1+ to interfere with the action of AAC(2′)-Ia, an aminoglycoside modifying enzyme that mediates the inactivation of plazomicin, makes it an excellent candidate as a plazomicin adjuvant to eliminate this enzyme as a threat to the effectivity to this antibiotic. The low cytotoxicity observed at the active concentrations makes the combination plazomicin/Ag1+ a viable option for treating multidrug resistant infections.
Author Contributions
Conceptualization, D.N. and M.E.T.; methodology, D.N., A.M., T.T., K.P., V.J., M.S.R., and M.E.T.; formal analysis, D.N., A.M., T.T., V.J., M.S.R., and M.E.T.; investigation, D.N., A.M., T.T., J.S., V.J., M.S.R., and M.E.T.; resources; writing—original draft preparation, M.E.T.; writing—review and editing, D.N., A.M., J.S., B. E., K. P., V.J., M.S.R., and M.E.T.; supervision, M.E.T.; funding acquisition, M.E.T.; project administration, M.E.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Public Health Service Grants 2R15AI047115 (MET) and SC3GM125556 (MSR) from the National Institute of Health. T.T. was partially supported by grant LA Basin Minority Health and Health Disparities Research Training Program (MHRT) T37MD001368 from the National Institute on Minority Health and Health Disparities, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
Plazomicin was generously supplied by Cipla Therapeutics. We thank Carlos Tolmasky for help with data analysis.
References
- Boucher, H. W. Bad bugs, no drugs 2002-2020: progress, challenges, and call to action. Trans Am Clin Climatol Assoc 2020, 131, 65–71. [Google Scholar] [PubMed]
- Adler, A.; Friedman, N. D.; Marchaim, D. Multidrug-resistant Gram-negative bacilli: infection control implications. Infect Dis Clin North Am 2016, 30, 967–997. [Google Scholar] [CrossRef]
- Bassetti, M.; Garau, J. Current and future perspectives in the treatment of multidrug-resistant Gram-negative infections. J Antimicrob Chemother 2021, 76, iv23–iv37. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; El Chakhtoura, N. G.; Yasmin, M.; Bonomo, R. A. Polymyxins: to combine or not to Combine? Antibiotics (Basel) 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Machuca, I.; Gutierrez-Gutierrez, B.; Rivera-Espinar, F.; Cano, A.; Gracia-Ahufinger, I.; Guzman-Puche, J.; Marfil-Perez, E.; Perez-Nadales, E.; Caston, J. J.; Bonomo, R. A.; Carmeli, Y.; Paterson, D.; Pascual, A.; Martinez-Martinez, L.; Rodriguez-Bano, J.; Torre-Cisneros, J.; Group, R. E. I. External validation of the INCREMENT-CPE mortality score in a carbapenem-resistant Klebsiella pneumoniae bacteraemia cohort: the prognostic significance of colistin resistance. Int J Antimicrob Agents 2019, 54, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Santiago, J.; Cornejo-Juarez, P.; Silva-Sanchez, J.; Garza-Ramos, U. Polymyxin resistance in Enterobacterales: overview and epidemiology in the Americas. Int J Antimicrob Agents 2021, 58, 106426. [Google Scholar] [CrossRef] [PubMed]
- Burillo, A.; Bouza, E. Controversies over the management of infections caused by Amp-C- and ESBL-producing Enterobacterales: what questions remain for future studies? Curr Opin Infect Dis 2022, 35, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yamawaki, K. Cefiderocol: discovery, chemistry, and In vivo profiles of a novel siderophore cephalosporin. Clin Infect Dis 2019, 69, S538–S543. [Google Scholar] [CrossRef]
- Mezcord, V.; Wong, O.; Pasteran, F.; Corso, A.; Tolmasky, M. E.; Bonomo, R. A.; Ramirez, M. S. Role of beta-lactamase inhibitors on cefiderocol activity against carbapenem-resistant Acinetobacter species. Int J Antimicrob Agents 2022, 106700. [Google Scholar]
- Ramirez, M. S.; Tolmasky, M. E. Aminoglycoside modifying enzymes. Drug Resist Updat 2010, 13, 151–171. [Google Scholar] [CrossRef]
- Labby, K. J.; Garneau-Tsodikova, S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med Chem 2013, 5, 1285–1309. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M. S.; Nikolaidis, N.; Tolmasky, M. E. Rise and dissemination of aminoglycoside resistance: the aac(6')-Ib paradigm. Front Microbiol 2013, 4, 121. [Google Scholar] [CrossRef]
- Tolmasky, M. E. Strategies to prolong the useful life of existing antibiotics and help overcoming the antibiotic resistance crisis. In Frontiers in Clinical Drug Research-Anti Infectives; Atta-ur-Rhaman, Ed.; Bentham Books: Sharjah, UAE, 2017; Volume 1, pp. 1–27. [Google Scholar]
- Ramirez, M. S.; Tolmasky, M. E. Amikacin: uses, resistance, and prospects for inhibition. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Serio, A. W.; Keepers, T.; Andrews, L.; Krause, K. M. Aminoglycoside revival: review of a historically important class of antimicrobials undergoing rejuvenation. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef]
- Aggen, J. B.; Armstrong, E. S.; Goldblum, A. A.; Dozzo, P.; Linsell, M. S.; Gliedt, M. J.; Hildebrandt, D. J.; Feeney, L. A.; Kubo, A.; Matias, R. D.; Lopez, S.; Gomez, M.; Wlasichuk, K. B.; Diokno, R.; Miller, G. H.; Moser, H. E. Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob Agents Chemother 2010, 54, 4636–4642. [Google Scholar] [CrossRef] [PubMed]
- Endimiani, A.; Hujer, K. M.; Hujer, A. M.; Armstrong, E. S.; Choudhary, Y.; Aggen, J. B.; Bonomo, R. A. ACHN-490, a neoglycoside with potent in vitro activity against multidrug-resistant Klebsiella pneumoniae isolates. Antimicrob Agents Chemother 2009, 53, 4504–4507. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; Ejim, L.; Stogios, P. J.; Koteva, K.; Bordeleau, E.; Evdokimova, E.; Sieron, A. O.; Savchenko, A.; Serio, A. W.; Krause, K. M.; Wright, G. D. Plazomicin retains antibiotic activity against most aminoglycoside modifying enzymes. ACS Infect Dis 2018, 4, 980–987. [Google Scholar] [CrossRef]
- Saravolatz, L. D.; Stein, G. E. Plazomicin: a new aminoglycoside. Clin Infect Dis 2020, 70, 704–709. [Google Scholar] [CrossRef]
- Lin, J.; Nishino, K.; Roberts, M. C.; Tolmasky, M.; Aminov, R. I.; Zhang, L. Mechanisms of antibiotic resistance. Front Microbiol 2015, 6, 34. [Google Scholar] [CrossRef]
- Clark, J. A.; Burgess, D. S. Plazomicin: a new aminoglycoside in the fight against antimicrobial resistance. Ther Adv Infect Dis 2020, 7, 2049936120952604. [Google Scholar] [CrossRef]
- Tang, H. J.; Lai, C. C. Plazomicin-associated Nephrotoxicity. Clin Infect Dis 2020, 71, 1130–1131. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, A.; Di Franco, S.; Donatiello, V.; Maffei, V.; Fittipaldi, C.; Fiore, M.; Coppolino, F.; Sansone, P.; Pace, M. C.; Passavanti, M. B. Plazomicin against multidrug-resistant bacteria: a scoping review. Life (Basel) 2022, 12. [Google Scholar] [CrossRef]
- Macinga, D. R.; Rather, P. N. The chromosomal 2'-N-acetyltransferase of Providencia stuartii: physiological functions and genetic regulation. Front Biosci 1999, 4, D132–140. [Google Scholar] [CrossRef]
- Bassenden, A. V.; Dumalo, L.; Park, J.; Blanchet, J.; Maiti, K.; Arya, D. P.; Berghuis, A. M. Structural and phylogenetic analyses of resistance to next-generation aminoglycosides conferred by AAC(2') enzymes. Sci Rep 2021, 11, 11614. [Google Scholar] [CrossRef] [PubMed]
- Lin, D. L.; Tran, T.; Alam, J. Y.; Herron, S. R.; Ramirez, M. S.; Tolmasky, M. E. Inhibition of aminoglycoside 6'-N-acetyltransferase type Ib by zinc: reversal of amikacin resistance in Acinetobacter baumannii and Escherichia coli by a zinc ionophore. Antimicrob Agents Chemother 2014, 58, 4238–4241. [Google Scholar] [CrossRef] [PubMed]
- Chiem, K.; Fuentes, B. A.; Lin, D. L.; Tran, T.; Jackson, A.; Ramirez, M. S.; Tolmasky, M. E. Inhibition of aminoglycoside 6'-N-acetyltransferase type Ib-mediated amikacin resistance in Klebsiella pneumoniae by zinc and copper pyrithione. Antimicrob Agents Chemother 2015, 59, 5851–5853. [Google Scholar] [CrossRef] [PubMed]
- Chiem, K.; Hue, F.; Magallon, J.; Tolmasky, M. E. Inhibition of aminoglycoside 6'-N-acetyltransferase type Ib-mediated amikacin resistance by zinc complexed with clioquinol, an ionophore active against tumors and neurodegenerative diseases. Int J Antimicrob Agents 2018, 51, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Magallon, J.; Chiem, K.; Tran, T.; Ramirez, M. S.; Jimenez, V.; Tolmasky, M. E. Restoration of susceptibility to amikacin by 8-hydroxyquinoline analogs complexed to zinc. PLoS One 2019, 14, e0217602. [Google Scholar] [CrossRef] [PubMed]
- Magallon, J.; Vu, P.; Reeves, C.; Kwan, S.; Phan, K.; Oakley-Havens, C.; Ramirez, M. S.; Tolmasky, M. E. Amikacin in combination with zinc pyrithione prevents growth of a carbapenem-resistant/multidrug-resistant Klebsiella pneumoniae isolate. Int J Antimicrob Agents 2021, 58, 106442. [Google Scholar] [CrossRef]
- Reeves, C. M.; Magallon, J.; Rocha, K.; Tran, T.; Phan, K.; Vu, P.; Yi, Y.; Oakley-Havens, C. L.; Cedano, J.; Jimenez, V.; Ramirez, M. S.; Tolmasky, M. E. Aminoglycoside 6'-N-acetyltransferase type Ib [AAC(6')-Ib]-mediated aminoglycoside resistance: phenotypic conversion to susceptibility by silver ions. Antibiotics (Basel) 2020, 10. [Google Scholar] [CrossRef]
- Ding, X.; Baca-DeLancey, R. R.; Rather, P. N. Role of SspA in the density-dependent expression of the transcriptional activator AarP in Providencia stuartii. FEMS Microbiol Lett 2001, 196, 25–29. [Google Scholar] [CrossRef]
- Thwaites, M.; Hall, D.; Shinabarger, D.; Serio, A. W.; Krause, K. M.; Marra, A.; Pillar, C. Evaluation of the bactericidal activity of plazomicin and comparators against multidrug-resistant Enterobacteriaceae. Antimicrob Agents Chemother 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A. S. Emerging and reemerging infectious diseases: the perpetual challenge. Acad Med 2005, 80, 1079–1085. [Google Scholar] [CrossRef]
- Sprenger, M.; Fukuda, K. Antimicrobial resistance. New mechanisms, new worries. Science 2016, 351, 1263–1264. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E. Rise in the prevalence of resistance to extended-spectrum cephalosporins in the USA, nursing homes and antibiotic prescribing in outpatient and inpatient settings. J Antimicrob Chemother 2021, 76, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar]
- Pierce, G. N.; Resch, C.; Mourin, M.; Dibrov, P.; Dibrov, E.; Ravandi, A. Bacteria and the growing threat of multidrug resistance for invasive cardiac interventions. Rev Cardiovasc Med 2022, 23, 15. [Google Scholar] [CrossRef] [PubMed]
- Teillant, A.; Gandra, S.; Barter, D.; Morgan, D. J.; Laxminarayan, R. Potential burden of antibiotic resistance on surgery and cancer chemotherapy antibiotic prophylaxis in the USA: a literature review and modelling study. Lancet Infect Dis 2015, 15, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, A. K.; Boucher, H. W.; Fowler, V. G., Jr.; Jezek, A.; Outterson, K.; Greenberg, D. E. Antibiotic resistance in the patient with cancer: escalating challenges and paths forward. CA Cancer J Clin 2021, 71, 488–504. [Google Scholar] [CrossRef]
- WHO Ten threats to global health in 2019. Available online: https://www.who.int/news-room/spotlight/ten-threats-to-global-health-in-2019 (accessed on 16 December 2022).
- Bush, K.; Bradford, P. A. Interplay between beta-lactamases and new beta-lactamase inhibitors. Nat Rev Microbiol 2019, 17, 295–306. [Google Scholar] [CrossRef]
- Li, Y.; Green, K. D.; Johnson, B. R.; Garneau-Tsodikova, S. Inhibition of aminoglycoside acetyltransferase resistance enzymes by metal salts. Antimicrob Agents Chemother 2015, 59, 4148–4156. [Google Scholar] [CrossRef]
- Bohlmann, L.; De Oliveira, D. M. P.; El-Deeb, I. M.; Brazel, E. B.; Harbison-Price, N.; Ong, C. Y.; Rivera-Hernandez, T.; Ferguson, S. A.; Cork, A. J.; Phan, M. D.; Soderholm, A. T.; Davies, M. R.; Nimmo, G. R.; Dougan, G.; Schembri, M. A.; Cook, G. M.; McEwan, A. G.; von Itzstein, M.; McDevitt, C. A.; Walker, M. J. Chemical synergy between ionophore PBT2 and zinc reverses antibiotic resistance. mBio 2018, 9. [Google Scholar] [CrossRef]
- Rather, P. N.; Orosz, E.; Shaw, K. J.; Hare, R.; Miller, G. Characterization and transcriptional regulation of the 2'-N-acetyltransferase gene from Providencia stuartii. J Bacteriol 1993, 175, 6492–6498. [Google Scholar] [CrossRef]
- Herisse, M.; Duverger, Y.; Martin-Verstraete, I.; Barras, F.; Ezraty, B. Silver potentiates aminoglycoside toxicity by enhancing their uptake. Mol Microbiol 2017, 105, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Morones-Ramirez, J. R.; Winkler, J. A.; Spina, C. S.; Collins, J. J. Silver enhances antibiotic activity against gram-negative bacteria. Sci Transl Med 2013, 5, 190ra181. [Google Scholar] [CrossRef]
- Barras, F.; Aussel, L.; Ezraty, B. Silver and antibiotic, new facts to an old story. Antibiotics (Basel) 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J. G. Nucleotide sequence of the ampicillin resistance gene of Escherichia coli plasmid pBR322. Proc Natl Acad Sci U S A 1978, 75, 3737–3741. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S. N.; Chang, A. C.; Hsu, L. Nonchromosomal antibiotic resistance in bacteria: genetic transformation of Escherichia coli by R-factor DNA. Proc Natl Acad Sci U S A 1972, 69, 2110–2114. [Google Scholar] [CrossRef]
- Haas, M. J.; Dowding, J. E. Aminoglycoside-modifying enzymes. Methods Enzymol 1975, 43, 611–628. [Google Scholar]
- Graham, F. L.; Smiley, J.; Russell, W. C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Tran, T.; Chiem, K.; Jani, S.; Arivett, B. A.; Lin, D. L.; Lad, R.; Jimenez, V.; Farone, M. B.; Debevec, G.; Santos, R.; Giulianotti, M.; Pinilla, C.; Tolmasky, M. E. Identification of a small molecule inhibitor of the aminoglycoside 6'-N-acetyltransferase type Ib [AAC(6')-Ib] using mixture-based combinatorial libraries. Int J Antimicrob Agents 2018, 51, 752–761. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Effect of Ag1+ on (2’)-Ia activity. Assays were performed in triplicate by the phosphocellulose paper binding method with soluble extracts obtained from E. coli TOP10(pUC57AAC2Ia) cells, the reaction mixture containing 200 mM Tris HCl pH 7.6 buffer, 0.25 mM MgCl2, 330 μM plazomicin, the indicated concentrations of silver acetate (Ag1+) or sodium acetate (Na1+), and 0.05 μCi of [acetyl-1-14C]-acetyl-coenzyme A (specific activity 60 μCi/μmol) in a final volume of 30 μl.
Figure 1.
Effect of Ag1+ on (2’)-Ia activity. Assays were performed in triplicate by the phosphocellulose paper binding method with soluble extracts obtained from E. coli TOP10(pUC57AAC2Ia) cells, the reaction mixture containing 200 mM Tris HCl pH 7.6 buffer, 0.25 mM MgCl2, 330 μM plazomicin, the indicated concentrations of silver acetate (Ag1+) or sodium acetate (Na1+), and 0.05 μCi of [acetyl-1-14C]-acetyl-coenzyme A (specific activity 60 μCi/μmol) in a final volume of 30 μl.
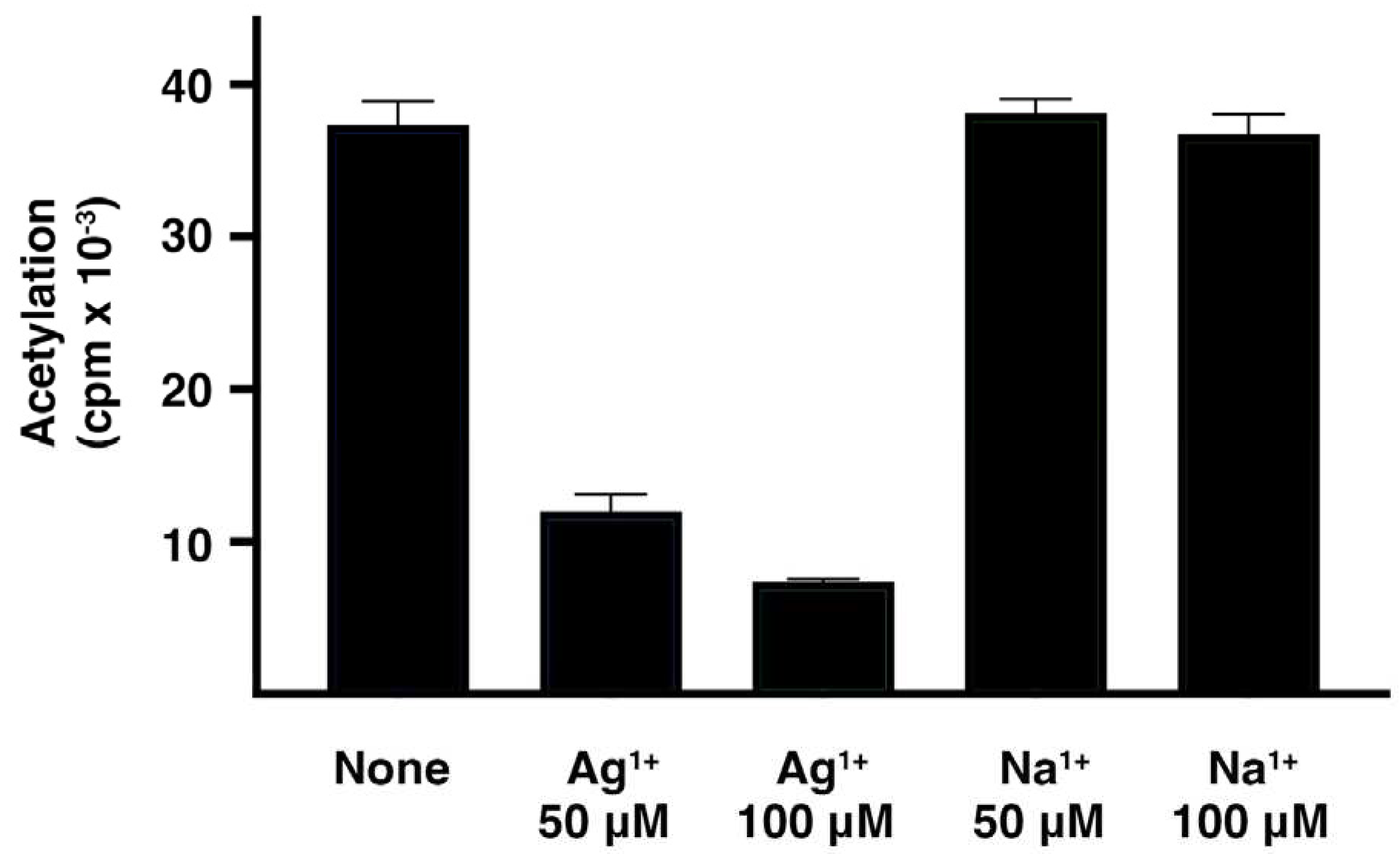
Figure 2.
Time-kill assays for plazomicin in the presence of silver acetate. E. coli TOP10(pUC57AAC2Ia) was incubated at 37 °C until the cell concentration reached the CFU/mL indicated in the figure at time zero. At this moment, the culture was divided in four aliquots and each one was supplemented with the compounds and concentrations indicated. The cultures were incubated at 37 °C, and CFU/mL values were measured at different intervals. AgAc, silver acetate. Assays were done in duplicate, and the values are mean ± SD of two independent experiments.
Figure 2.
Time-kill assays for plazomicin in the presence of silver acetate. E. coli TOP10(pUC57AAC2Ia) was incubated at 37 °C until the cell concentration reached the CFU/mL indicated in the figure at time zero. At this moment, the culture was divided in four aliquots and each one was supplemented with the compounds and concentrations indicated. The cultures were incubated at 37 °C, and CFU/mL values were measured at different intervals. AgAc, silver acetate. Assays were done in duplicate, and the values are mean ± SD of two independent experiments.
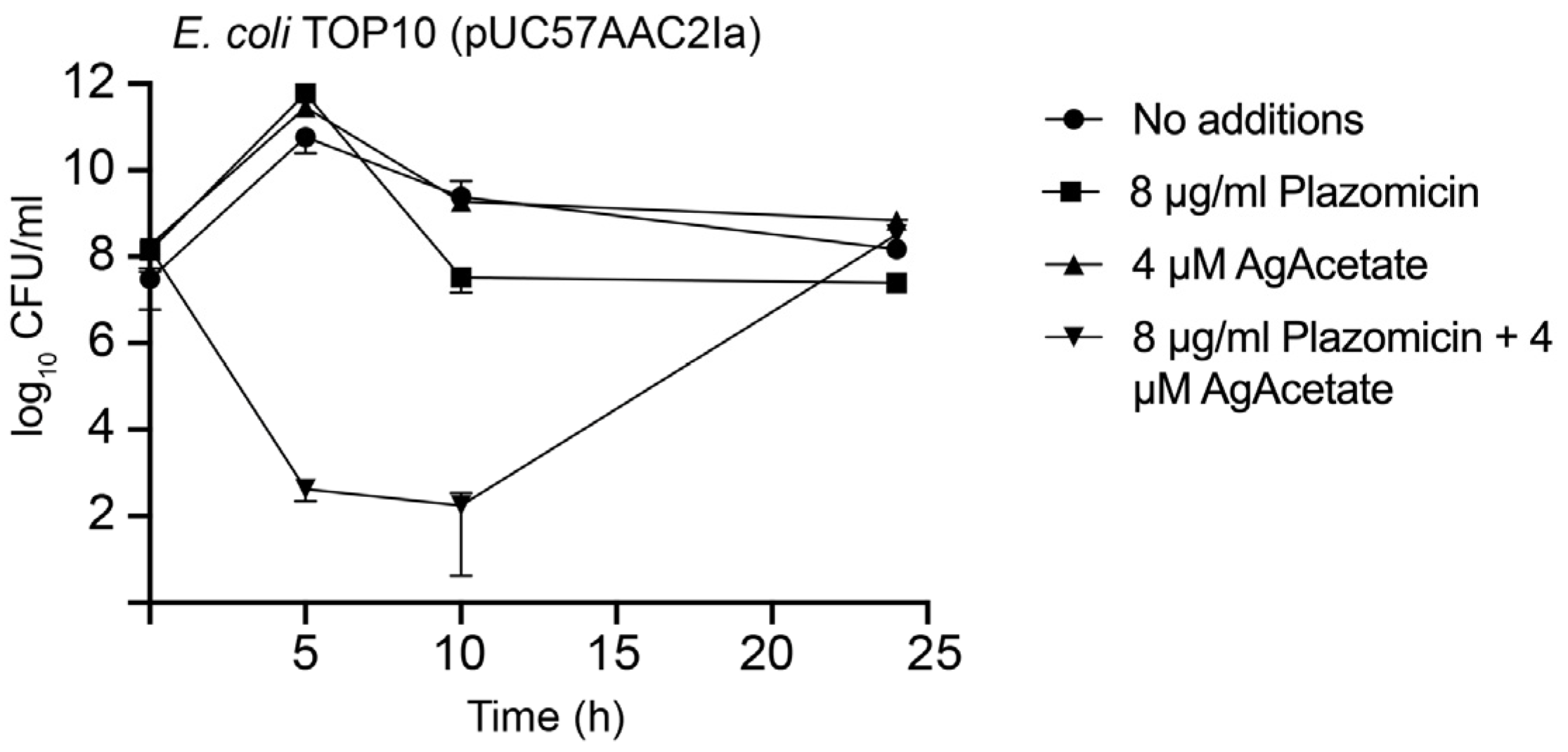
Figure 3.
Cytotoxicity of silver acetate and plazomacin. Cytotoxicity on HEK293 cells was assayed using a LIVE/DEAD kit as described in the Materials and Methods section. The percentage of surviving cells was calculated relative to cells untreated (striped bar). Control of maximum toxicity was determined by incubating the cells in 70% methanol. Assays were done in triplicate and the values are mean ± SD of three independent experiments.
Figure 3.
Cytotoxicity of silver acetate and plazomacin. Cytotoxicity on HEK293 cells was assayed using a LIVE/DEAD kit as described in the Materials and Methods section. The percentage of surviving cells was calculated relative to cells untreated (striped bar). Control of maximum toxicity was determined by incubating the cells in 70% methanol. Assays were done in triplicate and the values are mean ± SD of three independent experiments.
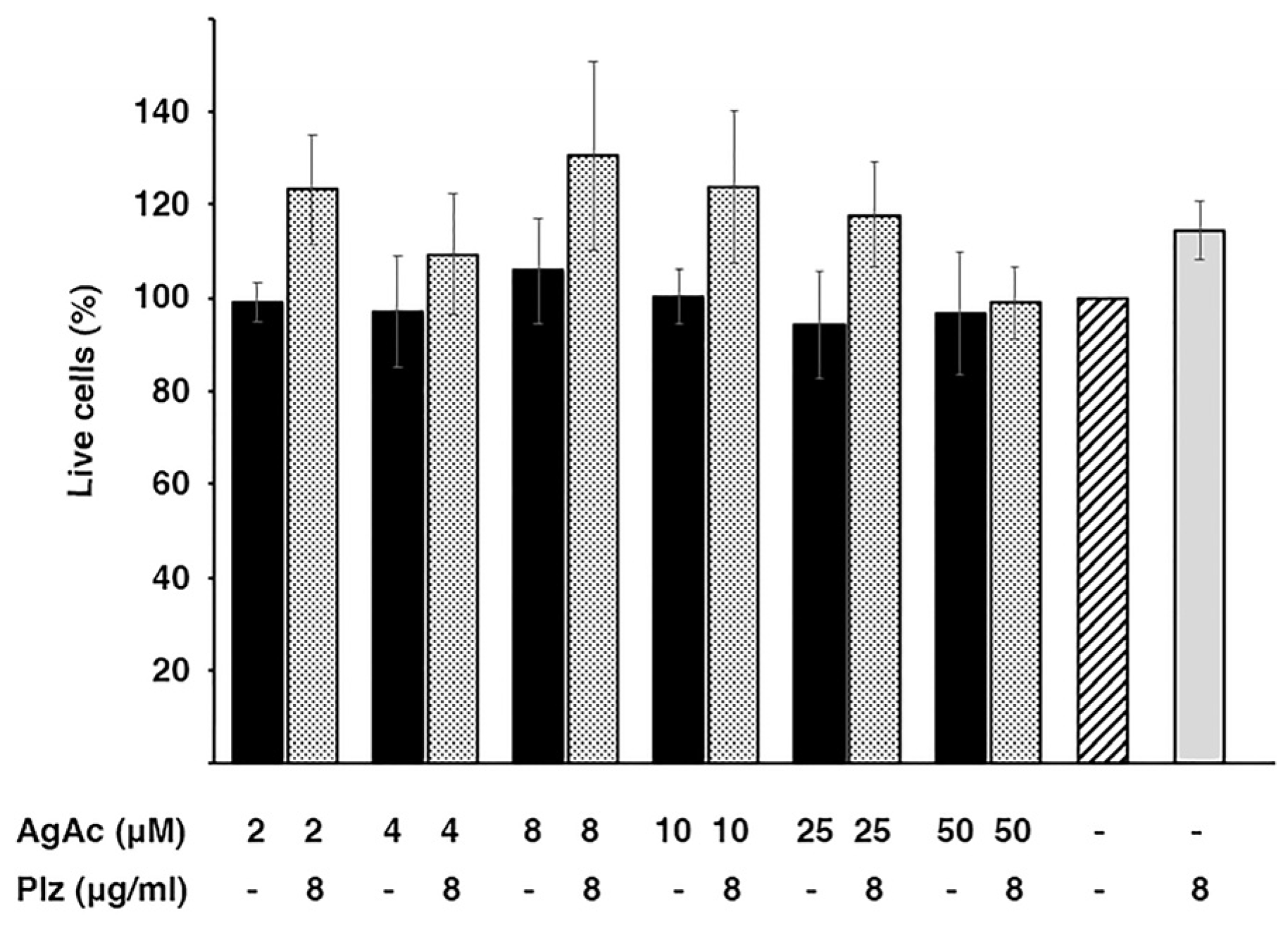

Table 1.
Growth in the presence of plazomicin and silver acetate.
| Plazomicin (μg/ml) | Silver Acetate (μM) OD600 |
Sodium Acetate (μM) OD600 |
||||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 4 | 0 | 8 | |
| 0 | 3.21 ± 0.02 | 3.09 ± 0.01 | 3.09 ± 0.02 | 3.18 ± 0.11 | 3.04 ± 0.06 | 3.12 ± 0.08 |
| 4 | 3.05 ± 0.08 | 2.86 ± 0.11 | 1.95 ± 0.01 | 0.12 ± 0.01 | 1.39 ± 0.03 | 1.34 ± 0.06 |
| 8 | 1.21 ± 0.01 | 1.26 ± 0.01 | 0.10 ± 0.03 | 0.02 ± 0 | 1.13 ± 0.02 | 1.08 ± 0.04 |
Cultures of E. coli TOP10(pUC57AAC2Ia) were performed in cation-adjusted Mueller Hinton with the indicated additions.
Table 2.
Growth in the presence of plazomicin and silver acetate.
| Plazomicin (μg/ml) | Silver Acetate (μM) OD600 |
||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 4 | ||
| 0 | 3.54 ± 0.01 | 3.48 ± 0.02 | 3.50 ± 0.07 | 3.35 ± 0.10 | |
| 4 | 1.71 ± 0.02 | 1.66 ± 0.09 | 0.95 ± 0.09 | 0.09 ± 0.07 | |
| 8 | 0.68 ± 0.06 | 0.24 ± 0.02 | 0.14 ± 0.06 | 0.03 ± 0 | |
Cultures of E. coli TOP10(pUC57PBLAAAC2Ia) were performed in cation-adjusted Mueller Hinton with the indicated additions.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



