
Submitted:
30 December 2022
Posted:
09 January 2023
You are already at the latest version
Abstract
Duchenne muscular dystrophy (DMD) is a debilitating and fatal genetic disease affecting 1/3500 boys globally, characterized by progressive muscle breakdown and eventual death with an average lifespan in the mid-late twenties. While no cure yet exists for DMD, gene and antisense therapies have been heavily explored in recent years to better treat this disease. Four antisense therapies have received conditional FDA approval, and many more exist in varying stages of clinical trials. These upcoming therapies often utilize novel drug chemistries to address limitations of existing therapies, and their development could herald the next generation of antisense therapy. This review article aims to summarize the current state of development for antisense-based therapies for the treatment of Duchenne muscular dystrophy, exploring candidates designed for both exon skipping and gene knockdown.
Keywords:
eteplirsen
; golodirsen
; viltolarsen
; casimersen
; WVE-N531
; SRP-5051
; DS-5141B
; NS-089/NCNP-02
; SCAAV9.U7.ACCA
; ATL1102
1. Introduction
Duchenne muscular dystrophy (DMD) is a fatal genetic disease stemming from mutations in the DMD gene located on the X-chromosome [1,2]. This gene is responsible for the production of dystrophin, a protein most notable for its role in maintaining muscular strength and stability. In the absence of dystrophin, muscle fibers are prone to progressive degradation and fibrosis.
The DMD gene itself is the largest gene in the human genome, and myriad mutations are possible across its 79 exons that can result in DMD. The most common causative mutations are whole-exon deletions or duplications that disrupt the open reading frame (ORF) of DMD and lead to a non-functional protein product, accounting for 60-70% of all DMD cases [3,4]. The remainder of cases arise from smaller nonsense, intronic, splice site, and UTR mutations. These mutations, particularly large exon deletions, tend to cluster in two mutational “hotspots” within DMD rather than occurring evenly across the gene. Approximately 73% of major deletions occur in the primary hotspot located between exons 43-55, with a smaller secondary hotspot encompassing 23% of major deletions between exons 2-22 [5].
Clinically, DMD affects approximately 1/3500 boys worldwide [6]. Patients typically present within the first few years of life with lower limb weakness and difficulty rising from the floor without the aid of their arms, a finding known as Gower’s sign [7]. By their early teenage years, most patients will experience a total loss of ambulatory ability and require the use of a wheelchair [8,9,10]. Between the ages of 18-20 patients typically require ventilatory assistance as the disease begins to affect the respiratory and cardiac muscles. This progression ultimately proves fatal for most patients, with DMD-related cardiomyopathy as the leading cause of death and a median lifespan of only 28 years [11]. No cure currently exists for DMD.
The current standard of care for DMD is usually corticosteroid treatment, which has been shown to delay the progression of disease and prolong ambulation [12]. However, this approach fails to address the underlying cause of DMD and is associated with numerous side effects including weight gain, bone weakness, and the potential for adrenal insufficiency with prolonged use [13].
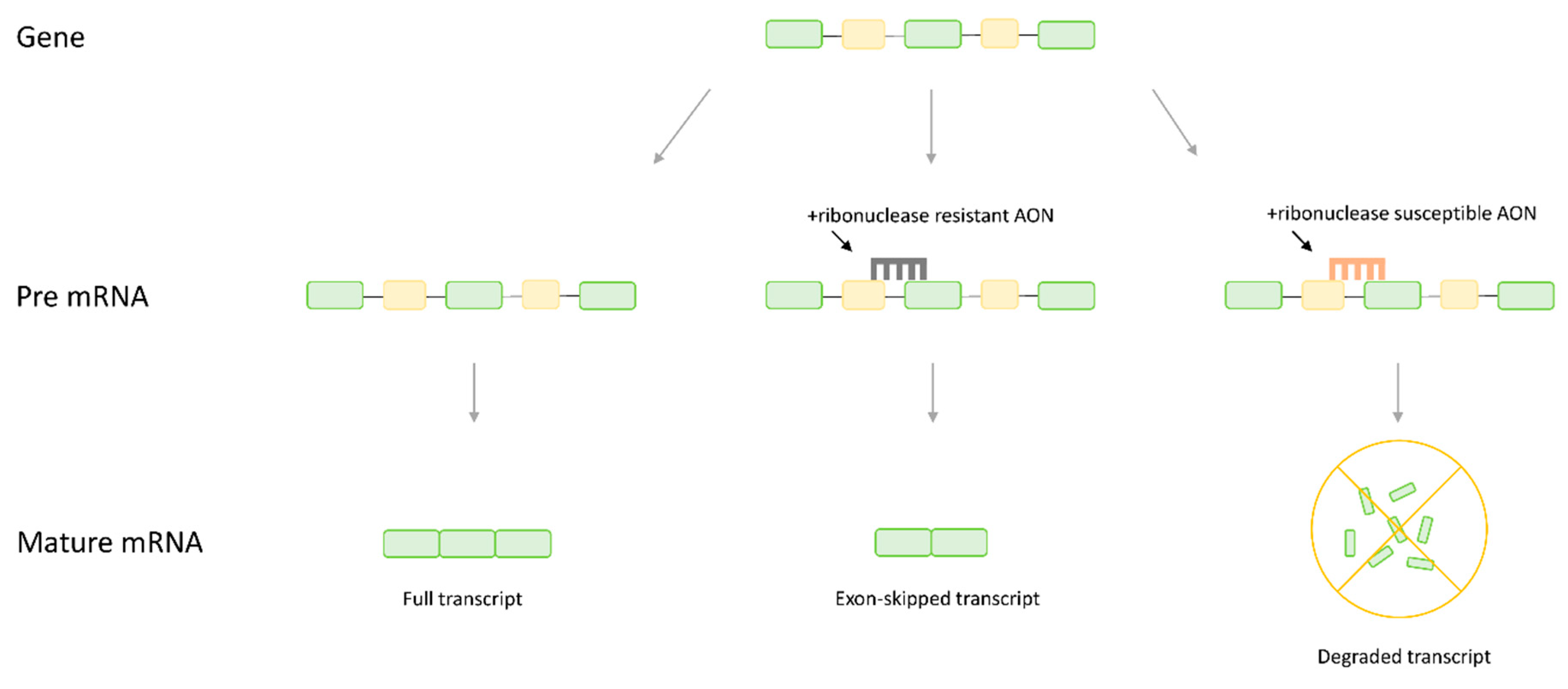
More recently, a novel treatment approach known as antisense therapy has gained traction, with four antisense-based drugs for DMD gaining FDA approval since 2016. Antisense therapy is a generalized term for therapeutics relying on small DNA-like molecules called antisense oligonucleotides (AONs). These AONs are designed to hybridize with targeted mRNA at which point therapeutic benefit can be conferred in one of two ways, either through splice-switching or gene knockdown, with the mechanism varying based on the specific chemistry of the AON and the target site [14,15] (Figure 1). If the AON is designed with oligonucleotides susceptible to ribonuclease H, such as endogenous DNA, ribonuclease is recruited to degrade the AON:mRNA complex, leading to a reduction in the abundance of mRNA transcripts and a corresponding reduction in protein product [16,17]. This approach is selected when knockdown of the targeted gene is desirable to alleviate a pathogenic phenotype. In contrast, spliceswitching involves using oligonucleotides with ribonuclease-resistant chemistry, such as phosphorodiamidate morpholinos, that target splice sites or splicing enhancers on the pre-mRNA transcript [15,18]. Rather than inducing degradation, these AONs sterically block the binding of splice machinery, manipulating splicing to favor inclusion or exclusion of a target region.
In the case of DMD, almost all antisense therapies use the splice switching approach, aiming to selectively skip DMD exons adjacent to a mutation in order to restore the reading frame of dystrophin [19,20]. The product of this skipped transcript is an internally deleted but partially functional dystrophin protein which has been demonstrated to impart benefit in both pre-clinical and clinical studies [19,21,22]. This approach, often referred to as exon skipping therapy (EST), has seen massive popularity for DMD in recent years. Four AON candidates have received conditional FDA approval for the treatment of DMD, and many more remain in clinical and pre-clinical trials. The purpose of this article is to provide an overview of these drugs, summarizing the current status of antisense therapy for the treatment of DMD.
2. Current AONs in Development for DMD
FDA Approval Obtained
At the time of writing, four AONs targeting three different exons have received conditional FDA approval for the treatment of DMD: eteplirsen (Sarepta, exon 51 skipping), viltolarsen (NS pharma, exon 53), golodirsen (Sarepta, exon 53), and casimersen (Sarepta, exon 45) [21,23,24,25]. All four drugs are phosphorodiamidate morpholino oligomers (PMOs), a class of synthetic AONs containing a six-sided morpholine ring and phosphorodiamidate backbone which is resistant to enzymatic degradation. While all four candidates have successfully completed initial phase III clinical trials, continued approval will be contingent upon long-term clinical benefit, which will be assessed via ongoing longitudinal phase III trials.
These longitudinal studies are expected to conclude in April 2024 for golodirsen and casimersen, November 2024 for eteplirsen, and December 2024 for viltolarsen [26,27,28]. As the most advanced antisense candidates for DMD, these studies will be pivotal for guiding the future of antisense therapy for DMD patients. The results from these studies will determine not only whether each currently approved AON will retain FDA approval, but also whether antisense and exon skipping therapy in general are suitable for the long-term treatment of DMD. Particularly for golodirsen, these results will aim to dispute previous results from a phase II extension study of golodirsen which found no significant improvement in clinical outcomes following 3 years of golodirsen treatment compared to natural history controls [29].
Phase II Clinical Trials
While no AON candidates other than the aforementioned PMOs with conditional FDA approval have entered Phase III trials, six different candidates are in varying stages of Phase II trials: ATL1102 (Antisense Therapeutics), SCAAV9.U7.ACCA (Astellas Pharma), SRP-5051 (Sarepta), NS-089/NCNP-02 (NS Pharma), WVE-N531 (Wave Life Sciences), and DS-5141B (Daiichi Sankyo).
ATL1102
ATL1102 is the most advanced of these candidates, having successfully completed Phase IIa trials in Australia [30]. Unlike other AONs for DMD which aim to restore dystrophin production, ATL1102 is instead focused on treating the inflammation associated with DMD. ATL1102 was designed with ribonuclease-susceptible 2′-O-(2-methoxyethyl) oligos targeted to CD49d mRNA, which encodes for a key subunit of the human very late antigen (VLA4) associated with inflammation [31]. By recruiting ribonucleases to degrade CD49d transcripts, ATL1102 thus aims to reduce inflammation and inflammation-mediated tissue damage in DMD [32].
Results from the completed Phase II trials found that weekly injection of ATL1102 was generally safe in patients, with no serious treatment-related adverse effects after 24 weeks of treatment [33] No significant changes were found in blood lymphocyte count, a marker of inflammation, throughout the study, although the authors did note a significant increase in CD3+CD49d+ T lymphocytes 4 weeks after stopping treatment. Further, no changes in grip strength or pinch strength were noted throughout the study [33] Based on the favorable safety data and apparent stabilizing effect of ATL1102, the authors expressed optimism in the continued development of this drug. However, no details regarding further trials are yet available.
SCAAV9.U7.ACCA
SCAAV9.U7.ACCA is another unique antisense candidate which aims to treat patients containing a duplication of DMD exon 2. This therapy strays away from direct AON treatment, instead opting for treatment with an adeno-associated virus (AAV) vector which allows for the endogenous production of non-coding U7 small nuclear RNAs (U7snRNAs) [34]. These U7snRNAs act in a similar fashion to AONs, inducing exon 2 skipping by targeting splice donor and acceptor sites on the pre-mRNA flanking exon 2 [35]. This leads to the production of normal, full-length dystrophin transcripts and proteins.
Pre-clinical studies in mice identified that SCAAV9.U7.ACCA was able to effectively skip one of the duplicate copies of exon 2, restoring the reading frame and enabling dystrophin production [35,36]. The researchers also found that both copies of exon 2 are occasionally skipped, resulting in an out-of-frame dystrophin transcript [37]. Interestingly, these transcripts still express functional dystrophin through an internal ribosomal entry site located on exon 5, so no correction was needed to account for this occurrence.
Phase I/II clinical trials are currently underway for SCAAV9.U7.ACCA, with an expected completion date of November 2025 [38]. This study will provide data regarding the safety of SCAAV9.U7.ACCA, as well as preliminary measures of efficacy and ability to restore dystrophin production. Notably, the safety of AAV-based therapies has been called into question recently following the tragic deaths of multiple patients in independent clinical trials assessing AAV-mediated drugs for DMD and X-linked myotubular myopathy [39,40,41]. Therefore, the safety findings from this study will be particularly important in determining whether the development of this therapy can proceed.
SRP-5051
A major limitation of existing PMO-based therapies is that PMOs exhibit poor cellular uptake and are rapidly cleared through the renal system [42]. This reduces the bioavailability and efficacy of PMO treatments, thus requiring a higher dose to confer therapeutic benefit. To address these limitations, recent research has explored the use of PMOs conjugated to cell-penetrating peptides, known as PPMOs [43,44]. By promoting cellular uptake, PPMOs are designed to be more effective than their unmodified PMO counterparts.
The most clinically advanced PPMO candidate is SRP-5051, a PPMO targeting exon 51 that is currently in phase II clinical trials [45]. SRP-5051, also known as vesleteplirsen, targets the same patient population as eteplirsen but with the added benefit of a proprietary cell-penetrating peptide moiety, and can therefore be thought of as a next-generation eteplirsen. As the first PPMO in clinical trials, validating the safety of SRP-5051 is of the utmost importance, and phase II trials were placed on clinical hold from June 2022 to September 2022 following an instance of treatment-related hypomagnesemia [45,46]. Despite this setback, phase II trials are expected to conclude in August 2024, and will provide information on the safety, pharmacokinetics, and preliminary efficacy of dystrophin restoration for SRP-5051.
WVE-N531
WVE-N531 is an AON targeting exon 53 designed using Wave Life Sciences’ proprietary phosphoryl guanidine-containing (PN) backbone [47]. Pre-clinical studies exploring this novel chemistry identified that compared to oligos with standard phosphorothioate or phosphodiester chemistries, PN oligos demonstrate improved efficacy, pharmacokinetics, and activity in difficult-to-reach brain tissues [48,49].
WVE-N531 is nearing the end of its phase I/II trials, with results expected in December 2022. These trials will primarily provide information regarding the safety and pharmacokinetics of WVE-N531, with secondary findings exploring preliminary efficacy as measured by dystrophin restoration [47]. While no formal results are available yet, an interim press release from Wave indicates that WVE-N531 demonstrated a positive safety profile across all tested doses after six weeks of treatment, and that despite low dystrophin levels effective exon skipping was observed at the mRNA level [50].
NS-089/NCNP-02
Developed by NS Pharma, NS-089/NCNP-02 is the first AON candidate aiming to induce skipping of exon 44 [51,52]. The chemistry used for this drug is unspecified, though it is reasonable to assume that it may use a similar PMO chemistry to NS Pharma’s previous AON candidate, viltolarsen. Unlike AONs in development for exons 45, 51, and 53 that must compete with the previously approved AONs for each exon, NS-089/NCNP-02 would become the only available treatment for mutations amenable to exon 44 skipping if approved.
NS-089/NCNP-02 is currently in a phase II extension study following the successful completion of phase I/II trials. A press release regarding the phase I/II study from NS Pharma identified that no serious adverse effects occurred during the trial which required discontinuation, and that dystrophin was effectively restored in a dose-dependent manner after treatment with NS-089/NCNP-02 [53]. Results from the extended phase II trial will provide further data regarding safety, dystrophin restoration, and clinical efficacy, and are expected in July 2023 [52].
DS-5141B
The final candidate in phase II development is DS-5141B, an AON designed to skip exon 45 constructed using 2'-O,4'-C-ethylene-bridged nucleic acid (ENA) oligos [54,55]. Compared to PMOs, ENAs were reported to demonstrate increased exon skipping efficiency in both skeletal and cardiac tissue in mouse studies, which is of particular importance given the high rate of cardiomyopathy-related mortality in DMD [54]. The authors hypothesize that this improvement is due to improved binding affinity with dystrophin pre-mRNA, however the mechanism is not well understood yet.
Following positive safety findings in phase I/II trials, DS-5141B is currently undergoing a phase II extension study which will further validate the safety of this AON, as well as provide preliminary clinical efficacy data assessing both skeletal muscle and cardiac phenotype [55,56,57]. The results from this study are expected in March 2023.
Pre-clinical/Phase I Clinical Trials
The majority of the newer DMD antisense therapies being developed are focused on AON conjugates, aiming to address the limitations associated with standard, unmodified AONs. Most of these conjugations involve proprietary technology designed to boost efficacy which will seek to disrupt the unmodified PMOs that have previously received FDA approval. However, given the early stage of development for many of these therapies, specific details are often unavailable.
PGN-EDO51
PGN-EDO51 (PepGen) is currently in the phase I stage of clinical testing. This oligo aims to skip DMD exon 51 and is constructed with PepGen’s Enhanced Delivery Oligonucleotide (EDO) technology, which is a proprietary cell-penetrating PPMO like Sarepta’s SRP-5051 [58]. While the available information surrounding PGN-EDO51 is minimal, a press release by PepGen claims that Phase I testing in healthy normal volunteers showed a favorable safety profile and high levels of exon 51 skipping [58]. Accordingly, PepGen will aim to initiate phase IIa trials in 2023.
ENTR-601-44
Another PPMO, ENTR-601-44, is also aiming to initiate clinical trials in the near future [59]. Created by Entrada Therapeutics using their Endosomal Escape Vehicle (EEV) platform, this PPMO is specifically designed to promote endosomal escape by inducing the collapse of endosomes which contain entrapped PPMO [60,61,62]. This approach improves the bioavailability of the conjugated AON, thus improving efficacy [63]. Entrada is in the process of submitting an Investigational New Drug (IND) application to the FDA, however a press release revealed that this may be delayed by a clinical hold imposed by the FDA in December 2022 [59]. Further details regarding this therapy may become available once ENTR-601-44 enters formalized clinical testing.
Taking a slightly different approach, both Dyne Therapeutics and Avidity Biosciences have recently unveiled antibody-conjugated AONS hybridized to transferrin receptor 1 (TfR1) antibodies [64,65]. Preclinical studies assessing this technology in mice have identified that TfR1-AONS demonstrate significantly increased dystrophin expression and functional improvement compared to unmodified PMO [64].
DYNE-251
Dyne’s candidate, DYNE-251, aims to use this technology to skip DMD exon 51. Phase I trials have recently begun with a planned extension to phase II, which will assess safety, dystrophin restoration, and clinical improvement after 24 weeks and 120 weeks of treatment [66]. This study is expected to conclude in November 2026.
AOC 1044
AOC 1044 is Avidity’s candidate and aims to treat patients with DMD amenable to exon 44 skipping. Avidity announced via press release that they were commencing clinical trials for AOC 1044 in October 2022, although at the time of writing no information is publicly available in the FDA database [67]. The trial is named EXPLORE44 and will assess the safety and preliminary efficacy of AOC 1044 treatment.

Table 1.
An overview of all antisense therapies currently in clinical development, organized by their therapeutic target.
Table 1.
An overview of all antisense therapies currently in clinical development, organized by their therapeutic target.
| Therapeutic Target | Name | AON Chemistry | Sponsor | Status |
|---|---|---|---|---|
| Exon 53 Skipping | viltolarsen | PMO | NS Pharma | Approved |
| golodirsen | PMO | Sarepta Therapeutics | Approved | |
| WVE-N531 | phosphoryl guanidine (PN) backbone | Wave Life Sciences | Phase I/II | |
| Exon 51 Skipping | eteplirsen | PMO | Sarepta Therapeutics | Approved |
| SRP-5051 | PPMO | Sarepta Therapeutics | Phase II | |
| PGN-EDO51 | PPMO | PepGen | Phase I | |
| DYNE-251 | Antibody-PMO | Dyne Therapeutics | Phase I | |
| Exon 45 Skipping | casimersen | PMO | Sarepta Therapeutics | Approved |
| DS-5141B | 2'-O,4'-C-ethylene-bridged nucleic acid (ENA) | Daiichi Sankyo | Phase II | |
| Exon 44 Skipping | NS-089/NCNP-02 | Unknown | NS Pharma | Phase II |
| AOC 1044 | Antibody-PMO | Avidity Biosciences | Phase I | |
| ENTR-601-44 | PPMO | Entrada Therapeutics | Preclinical | |
| Exon 2 Skipping | SCAAV9.U7.ACCA | AAV U7snRNA | Astellas Pharma | Phase I/II |
| CD49d Knockdown | ATL1102 | 2′-O-(2-methoxyethyl) | Antisense Therapeutics | Phase IIa |
Future Directions
Finally, although there are no candidates in the clinical developmental pipeline yet, multi-exon skipping is an approach that has the potential to transform the field of antisense therapy for the treatment of DMD [68]. Rather than the single-exon approach of current AONs, multi-exon skipping aims to skip multiple exons simultaneously with an AON “cocktail” [69]. This method improves the applicability of antisense therapy for DMD, and a cocktail skipping exons 45-55 has been validated in vitro and in vivo which could be applicable to nearly 50% of all DMD patients [69,70,71]. Furthermore, this research has demonstrated that spontaneous skipping can occur during multi-exon skipping which reduces the number of AONs required in the cocktail, reducing treatment complexity and cost [72]. At this time, the development of multi-exon skipping cocktails is limited by regulatory hurdles, as each AON in the cocktail must undergo separate clinical approval [24].
Although the focus of this article emphasized antisense therapy, numerous other therapeutic options are also under development. Microdystrophin therapy, which seeks to treat DMD by supplementing patients with an AAV vector encoding for a truncated but functional “microdystrophin” protein, has multiple candidates in phase III clinical trials and could soon join antisense therapies as FDA-approved DMD treatments [73,74,75,76]. Another approach which was shown some promise is readthrough therapy, which aims to treat DMD arising from premature termination codons [77,78]. Readthrough therapy has gained approval in Europe and seeks to also gain FDA approval with ongoing longitudinal phase III trials [79]. More recently, cell-based therapies have also been under development and have demonstrated notable benefit in cardiac tissue, which is a key weakness of current antisense therapies [80,81]. While further research is required to determine which therapies will prove most beneficial for patients, each approach – antisense or otherwise – helps to bring a functional treatment for DMD closer to the patients who need it most.
Author Contributions
Conceptualization, H.W.C and T.Y.; methodology, H.W.C and T.Y.; investigation, H.W.C and T.Y.; writing—original draft preparation, H.W.C.; writing—review and editing, H.W.C and T.Y.; supervision, T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
H.W.C is grateful for funding provided by the H Jean McDiarmaid Scholarship.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are grateful for the support provided by Muscular Dystrophy Canada, the Friends of Garrett Cumming Research Fund, the HM Toupin Neurological Science Research Fund, Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, and the Women and Children’s Health Research Institute (WCHRI).
Conflicts of Interest
H.W.C declares no conflicts of interest. T.Y. is a founder and shareholder of OligomicsTx, which aims to commercialize antisense oligonucleotide technology. The Yokota lab has previously been sponsored by Avidity Biosciences to aid with preclinical mouse studies, although H.W.C was uninvolved with the project.
References
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nature Reviews Disease Primers 2021 7:1 2021, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and Genetics of Dystrophin and Dystrophin-Related Proteins in Muscle. Physiol Rev 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.; Elshafey, A.; Al-balool, H.; Alaboud, H.; Ali, M.A. ben; Baqer, A.; Bastaki, L. Mutation Spectrum Analysis of Duchenne/Becker Muscular Dystrophy in 68 Families in Kuwait: The Era of Personalized Medicine. PLoS One 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Rossi, R.; Trabanelli, C.; Mauro, A.; Selvatici, R.; Falzarano, M.S.; Spedicato, N.; Margutti, A.; Rimessi, P.; Fortunato, F.; et al. The Genetic Landscape of Dystrophin Mutations in Italy: A Nationwide Study. Front Genet 2020, 0, 131. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Lim, K.R.Q.; Nakamura, A.; Yokota, T. Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges. J Pers Med 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifirò, G. Global Epidemiology of Duchenne Muscular Dystrophy: An Updated Systematic Review and Meta-Analysis. Orphanet Journal of Rare Diseases 2020 15:1 2020, 15, 1–20. [Google Scholar] [CrossRef]
- Chang, R.F.; Mubarak, S.J. Pathomechanics of Gowers’ Sign: A Video Analysis of a Spectrum of Gowers’ Maneuvers. Clin Orthop Relat Res 2012, 470, 1987. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef]
- Gardner-Medwin, D. Clinical Features and Classification of the Muscular Dystrophies. Br Med Bull 1980, 36, 109–116. [Google Scholar] [CrossRef]
- Nowak, K.J.; Davies, K.E. Duchenne Muscular Dystrophy and Dystrophin: Pathogenesis and Opportunities for Treatment: Third in Molecular Medicine Review Series. EMBO Rep 2004, 5, 872–876. [Google Scholar] [CrossRef]
- Broomfield, J.; Hill, M.; Guglieri, M.; Crowther, M.; Abrams, K. Life Expectancy in Duchenne Muscular Dystrophy. Neurology 2021, 97, e2304–e2314. [Google Scholar] [CrossRef] [PubMed]
- Gloss, D.; Moxley, R.T.; Ashwal, S.; Oskoui, M. Practice Guideline Update Summary: Corticosteroid Treatment of Duchenne Muscular Dystrophy - Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016, 86, 465–472. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-Term Effects of Glucocorticoids on Function, Quality of Life, and Survival in Patients with Duchenne Muscular Dystrophy: A Prospective Cohort Study. The Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C.A. Antisense Oligonucleotides: Basic Concepts and Mechanisms.
- Aartsma-Rus, A.; van Ommen, G.J.B. Antisense-Mediated Exon Skipping: A Versatile Tool with Therapeutic and Research Applications. RNA 2007, 13, 1609. [Google Scholar] [CrossRef] [PubMed]
- Pauli, A.; Montague, T.G.; Lennox, K.A.; Behlke, M.A.; Schier, A.F. Antisense Oligonucleotide-Mediated Transcript Knockdown in Zebrafish. PLoS One 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Molecular Therapy 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [PubMed]
- Niks, E.H.; Aartsma-Rus, A. Exon Skipping: A First in Class Strategy for Duchenne Muscular Dystrophy. Expert Opin Biol Ther 2017, 17, 225–236. [Google Scholar] [CrossRef]
- Kole, R.; Krieg, A.M. Exon Skipping Therapy for Duchenne Muscular Dystrophy. Adv Drug Deliv Rev 2015, 87, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Duddy, W.; Partridge, T. Optimizing Exon Skipping Therapies for DMD. In Proceedings of the Acta Myologica; Pacini Editore, December 2007; Vol. 26; pp. 179–184. [Google Scholar]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Echigoya, Y.; Nagata, T.; Kuraoka, M.; Kobayashi, M.; Aoki, Y.; Partridge, T.; Maruyama, R.; Takeda, S.; Yokota, T. Efficacy of Multi-Exon Skipping Treatment in Duchenne Muscular Dystrophy Dog Model Neonates. Molecular Therapy 2019, 27, 76–86. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the Treatment of Duchenne Muscular Dystrophy. Drug Des Devel Ther 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne Muscular Dystrophy. Drugs of Today 2020, 56, 491–504. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the Treatment of Duchenne Muscular Dystrophy. Drugs of Today 2019, 55, 627–639. [Google Scholar] [CrossRef]
- Study to Assess the Efficacy and Safety of Viltolarsen in Ambulant Boys With DMD (RACER53) - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04060199?term=NCT04060199&draw=2&rank=1 (accessed on 21 December 2022).
- A Study to Compare Safety and Efficacy of a High Dose of Eteplirsen in Participants With Duchenne Muscular Dystrophy (DMD) (MIS51ON) - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03992430?cond=NCT03992430&draw=2&rank=1 (accessed on 21 December 2022).
- Study of SRP-4045 (Casimersen) and SRP-4053 (Golodirsen) in Participants With Duchenne Muscular Dystrophy (DMD) - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02500381 (accessed on 21 December 2022).
- Servais, L.; Mercuri, E.; Straub, V.; Guglieri, M.; Seferian, A.M.; Scoto, M.; Leone, D.; Koenig, E.; Khan, N.; Dugar, A.; et al. Long-Term Safety and Efficacy Data of Golodirsen in Ambulatory Patients with Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A First-in-Human, Multicenter, Two-Part, Open-Label, Phase 1/2 Trial. Nucleic Acid Ther 2022, 32, 29–39. [Google Scholar] [CrossRef]
- ANZCTR – Registration. Available online: https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12618000970246 (accessed on 21 December 2022).
- Limmroth, V.; Barkhof, F.; Desem, N.; Diamond, M.P.; Tachas, G. CD49d Antisense Drug ATL1102 Reduces Disease Activity in Patients with Relapsing-Remitting MS. Neurology 2014, 83, 1780. [Google Scholar] [CrossRef]
- Pinto-Mariz, F.; Rodrigues Carvalho, L.; Prufer De Queiroz Campos Araujo, A.; de Mello, W.; Gonçalves Ribeiro, M.; Cunha, M.D.C.S.A.; Cabello, P.H.; Riederer, I.; Negroni, E.; Desguerre, I.; et al. CD49d Is a Disease Progression Biomarker and a Potential Target for Immunotherapy in Duchenne Muscular Dystrophy. Skelet Muscle 2015, 5. [Google Scholar] [CrossRef]
- Woodcock, I.; Tachas, G.; Desem, N.; Houweling, P.; Yiu, E.; Kean, M.; Emmanuel, J.; Kennedy, R.; Carroll, K.; Valle, K. de; et al. A Phase 2 Open-Label Study to Determine the Safety and Efficacy of Weekly Dosing of ATL1102 in Patients with Non-Ambulatory Duchenne Muscular Dystrophy. medRxiv 2226, 2022.01.16.22269029. [Google Scholar] [CrossRef]
- Gushchina, L. v.; Frair, E.C.; Rohan, N.; Bradley, A.J.; Simmons, T.R.; Chavan, H.D.; Chou, H.J.; Eggers, M.; Waldrop, M.A.; Wein, N.; et al. Lack of Toxicity in Nonhuman Primates Receiving Clinically Relevant Doses of an AAV9.U7snRNA Vector Designed to Induce DMD Exon 2 Skipping. Hum Gene Ther 2021, 32, 882–894. [Google Scholar] [CrossRef]
- Wein, N.; Vetter, T.A.; Vulin, A.; Simmons, T.R.; Frair, E.C.; Bradley, A.J.; Gushchina, L. v.; Almeida, C.F.; Huang, N.; Lesman, D.; et al. Systemic Delivery of an AAV9 Exon-Skipping Vector Significantly Improves or Prevents Features of Duchenne Muscular Dystrophy in the Dup2 Mouse. Mol Ther Methods Clin Dev 2022, 26, 279. [Google Scholar] [CrossRef]
- Wein, N.; Dunn, D.M.; Waldrop, M.A.; Gushchina, L. v.; Frair, E.C.; Weiss, R.B.; Flanigan, K.M. Absence of Significant Off-Target Splicing Variation with a U7snRNA Vector Targeting DMD Exon 2 Duplications. Hum Gene Ther 2021. [Google Scholar] [CrossRef]
- Wein, N.; Vulin, A.; Falzarano, M.S.; Szigyarto, C.A.K.; Maiti, B.; Findlay, A.; Heller, K.N.; Uhlén, M.; Bakthavachalu, B.; Messina, S.; et al. Translation from a DMD Exon 5 IRES Results in a Functional Dystrophin Isoform That Attenuates Dystrophinopathy in Humans and Mice. Nat Med 2014, 20, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- AAV9 U7snRNA Gene Therapy to Treat Boys With DMD Exon 2 Duplications. - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04240314 (accessed on 8 December 2021).
- Philippidis, A. After Third Death, Audentes’ AT132 Remains on Clinical Hold. https://home.liebertpub.com/hum 2020, 31, 908–910. [Google Scholar] [CrossRef] [PubMed]
- High-Dose AAV Gene Therapy Deaths. Nat Biotechnol 2020, 38, 910. [CrossRef]
- Pfizer Reports Patient Death in Early-Stage Duchenne Gene Therapy Trial, Halts Enrollment | FierceBiotech. Available online: https://www.fiercebiotech.com/biotech/pfizer-reports-death-patient-duchenne-trial-halts-enrolment (accessed on 26 December 2021).
- Shadid, M.; Badawi, M.; Abulrob, A. Antisense Oligonucleotides: Absorption, Distribution, Metabolism, and Excretion. Expert Opin Drug Metab Toxicol 2021, 17, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Moulton, J.D. Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy. Biochimica et Biophysica Acta (BBA) - Biomembranes 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-Conjugate Antisense Based Splice-Correction for Duchenne Muscular Dystrophy and Other Neuromuscular Diseases. EBioMedicine 2019, 45, 630. [Google Scholar] [CrossRef] [PubMed]
- Two-Part Study for Dose Determination of SRP-5051 (Vesleteplirsen) (Part A), Then Dose Expansion (Part B) in Participants With Duchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04004065?cond=NCT04004065&draw=2&rank=1 (accessed on 21 December 2022).
- Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Available online: https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-provides-update-srp-5051-treatment-duchenne (accessed on 21 December 2022).
- Open-Label Study of WVE-N531 in Patients With Duchenne Muscular Dystrophy - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04906460?term=53&cond=DMD&draw=2 (accessed on 21 December 2022).
- Kandasamy, P.; McClorey, G.; Shimizu, M.; Kothari, N.; Alam, R.; Iwamoto, N.; Kumarasamy, J.; Bommineni, G.R.; Bezigian, A.; Chivatakarn, O.; et al. Control of Backbone Chemistry and Chirality Boost Oligonucleotide Splice Switching Activity. Nucleic Acids Res 2022, 50, 5443–5466. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, P.; Liu, Y.; Aduda, V.; Akare, S.; Alam, R.; Andreucci, A.; Boulay, D.; Bowman, K.; Byrne, M.; Cannon, M.; et al. Impact of Guanidine-Containing Backbone Linkages on Stereopure Antisense Oligonucleotides in the CNS. Nucleic Acids Res 2022, 50, 5401–5423. [Google Scholar] [CrossRef] [PubMed]
- Wave Life Sciences Provides Positive Update, On. Available online: https://www.globenewswire.com/news-release/2022/12/19/2576214/0/en/Wave-Life-Sciences-Provides-Positive-Update-on-Proof-of-Concept-Study-for-WVE-N531-in-Duchenne-Muscular-Dystrophy.html (accessed on 21 December 2022).
- Exploratory Study of NS-089/NCNP-02 in DMD - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04129294 (accessed on 21 December 2022).
- Extension Study of NS-089/NCNP-02 in DMD - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05135663 (accessed on 21 December 2022).
- Study Shows the Efficacy of Antisense Oligonucleotide-Based Exon 44 Skipping Drug, NS-089/NCNP-02, for Patients with Duchenne Muscular Dystrophy (DMD) | 国立研究開発法人 国立精神・神経医療研究センター National Center of Neurology and Psychiatry. Available online: https://www.ncnp.go.jp/topics/2022/20220317e.html (accessed on 21 December 2022).
- Takaishi, K.; Kakuta, M.; Ito, K.; Kanda, A.; Takakusa, H.; Miida, H.; Masuda, T.; Nakamura, A.; Onishi, Y.; Onoda, T.; et al. Stunning Pharmacological Properties of DS-5141b, an Antisense Oligonucleotide Consisting of 2’-O,4’-C-Ethylene-Bridged Nucleic Acids and 2’-O-Methyl RNA, on Dystrophin MRNA Exon Skipping. Neuromuscular Disorders 2017, 27, S216. [Google Scholar] [CrossRef]
- Long-Term, Extension Study of DS-5141b in Patients With Duchenne Muscular Dystrophy - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04433234 (accessed on 21 December 2022).
- Daiichi Sankyo Announces the Results Summary of Phase 1/2 Clinical Trial in Japan for DS-5141 - Press Releases - Media - Daiichi Sankyo. Available online: https://www.daiichisankyo.com/media/press_release/detail/index_4112.html (accessed on 21 December 2022).
- Study of DS-5141b in Patients With Duchenne Muscular Dystrophy - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02667483 (accessed on 21 December 2022).
- PepGen Reports Positive Data from Phase 1 Trial of PGN-EDO51 for the Treatment of Duchenne Muscular Dystrophy | PepGen. Available online: https://investors.pepgen.com/news-releases/news-release-details/pepgen-reports-positive-data-phase-1-trial-pgn-edo51-treatment (accessed on 21 December 2022).
- Entrada Therapeutics Announces Clinical Hold on IND Application for ENTR-601-44 in Duchenne Muscular Dystrophy | BioSpace. Available online: https://www.biospace.com/article/releases/entrada-therapeutics-announces-clinical-hold-on-ind-application-for-entr-601-44-in-duchenne-muscular-dystrophy/ (accessed on 21 December 2022).
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding Cell Penetration of Cyclic Peptides. Chem Rev 2019, 119, 10241–10287. [Google Scholar] [CrossRef] [PubMed]
- Sahni, A.; Qian, Z.; Pei, D. Cell-Penetrating Peptides Escape the Endosome by Inducing Vesicle Budding and Collapse. ACS Chem Biol 2020, 15, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Larochelle, J.R.; Jiang, B.; Lian, W.; Hard, R.L.; Selner, N.G.; Luechapanichkul, R.; Barrios, A.M.; Pei, D. Early Endosomal Escape of a Cyclic Cell-Penetrating Peptide Allows Effective Cytosolic Cargo Delivery. Biochemistry 2014, 53, 4034–4046. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.A.; Yao, M.; Hall, J.; O’donnell, E.; Venkatesan, R.; Spring, S.; Wen, A.; Hsia, N.; Shen, P.; Russo, R.; et al. NAR Breakthrough Article Enhanced Exon Skipping and Prolonged Dystrophin Restoration Achieved by TfR1-Targeted Delivery of Antisense Oligonucleotide Using FORCE Conjugation in Mdx Mice. Nucleic Acids Res 2022. [Google Scholar] [CrossRef]
- Levin, A.A. Targeting Therapeutic Oligonucleotides. New England Journal of Medicine 2017, 376, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Safety, Tolerability, Pharmacodynamic, Efficacy, and Pharmacokinetic Study of DYNE-251 in Participants With Duchenne Muscular Dystrophy Amenable to Exon 51 Skipping - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05524883 (accessed on 21 December 2022).
- Avidity Biosciences Announces Phase 1/2 EXPLORE44TM Trial of AOC 1044 for Duchenne Muscular Dystrophy Mutations Amenable to Exon 44 Skipping. Available online: https://www.prnewswire.com/news-releases/avidity-biosciences-announces-phase-12-explore44-trial-of-aoc-1044-for-duchenne-muscular-dystrophy-mutations-amenable-to-exon-44-skipping-301646531.html (accessed on 21 December 2022).
- Aoki, Y.; Yokota, T.; Nagata, T.; Nakamura, A.; Tanihata, J.; Saito, T.; Duguez, S.M.R.; Nagaraju, K.; Hoffman, E.P.; Partridge, T.; et al. Bodywide Skipping of Exons 45-55 in Dystrophic Mdx52 Mice by Systemic Antisense Delivery. Proc Natl Acad Sci U S A 2012, 109, 13763–13768. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Echigoya, Y.; Duddy, W.; Saito, T.; Aoki, Y.; Takeda, S.; Yokota, T. Antisense PMO Cocktails Effectively Skip Dystrophin Exons 45-55 in Myotubes Transdifferentiated from DMD Patient Fibroblasts. PLoS One 2018, 13, e0197084. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Lim, K.R.Q.; Melo, D.; Bao, B.; Trieu, N.; Mizobe, Y.; Maruyama, R.; Mamchaoui, K.; Tanihata, J.; Aoki, Y.; et al. Exons 45-55 Skipping Using Mutation-Tailored Cocktails of Antisense Morpholinos in the DMD Gene. Mol Ther 2019, 27, 2005–2017. [Google Scholar] [CrossRef]
- Lee, J.J.A.; Saito, T.; Duddy, W.; Takeda, S.I.; Yokota, T. Direct Reprogramming of Human DMD Fibroblasts into Myotubes for In Vitro Evaluation of Antisense-Mediated Exon Skipping and Exons 45-55 Skipping Accompanied by Rescue of Dystrophin Expression. Methods Mol Biol 2018, 1828, 141–150. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Woo, S.; Melo, D.; Huang, Y.; Dzierlega, K.; Shah, M.N.A.; Aslesh, T.; Roshmi, R.R.; Echigoya, Y.; Maruyama, R.; et al. Development of DG9 Peptide-Conjugated Single- and Multi-Exon Skipping Therapies for the Treatment of Duchenne Muscular Dystrophy. Proc Natl Acad Sci U S A 2022, 119. [Google Scholar] [CrossRef]
- Shin, J.H.; Pan, X.; Hakim, C.H.; Yang, H.T.; Yue, Y.; Zhang, K.; Terjung, R.L.; Duan, D. Microdystrophin Ameliorates Muscular Dystrophy in the Canine Model of Duchenne Muscular Dystrophy. Molecular Therapy 2013, 21, 750. [Google Scholar] [CrossRef] [PubMed]
- le Guiner, C.; Servais, L.; Montus, M.; Larcher, T.; Fraysse, B.; Moullec, S.; Allais, M.; François, V.; Dutilleul, M.; Malerba, A.; et al. Long-Term Microdystrophin Gene Therapy Is Effective in a Canine Model of Duchenne Muscular Dystrophy. Nat Commun 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Microdystrophin Gene Transfer Study in Adolescents and Children With DMD - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03368742 (accessed on 8 December 2021).
- A Phase 3 Study to Evaluate the Safety and Efficacy of PF-06939926 for the Treatment of Duchenne Muscular Dystrophy - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04281485 (accessed on 8 December 2021).
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bönnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a Study of Ataluren-Mediated Dystrophin Production in Patients with Nonsense Mutation Duchenne Muscular Dystrophy. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F.; Osorio, A.N.; Tulinius, M.; Buccella, F.; Morgenroth, L.P.; Gordish-Dressman, H.; Jiang, J.; Trifillis, P.; Zhu, J.; et al. Safety and Effectiveness of Ataluren: Comparison of Results from the STRIDE Registry and CINRG DMD Natural History Study. J Comp Eff Res 2020, 9, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Long-Term Outcomes of Ataluren in Duchenne Muscular Dystrophy - Full Text View - ClinicalTrials.Gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03179631 (accessed on 18 August 2022).
- A Study of CAP-1002 in Ambulatory and Non-Ambulatory Patients With Duchenne Muscular Dystrophy (HOPE-3) - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05126758 (accessed on 8 August 2022).
- A Study of CAP-1002 in Ambulatory and Non-Ambulatory Patients With Duchenne Muscular Dystrophy - Full Text View - ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03406780 (accessed on 8 August 2022).
Figure 1.
A comparison of the effect of different antisense oligonucleotide (AON) chemistries on final mRNA transcript. Ribonuclease resistant AONs targeted to splice sites will induce splice switching, in this case leading to removal of an exon form the mature mRNA. Ribonuclease susceptible AONs will recruit ribonuclease H and induce hydrolysis, eliminating the transcript.
Figure 1.
A comparison of the effect of different antisense oligonucleotide (AON) chemistries on final mRNA transcript. Ribonuclease resistant AONs targeted to splice sites will induce splice switching, in this case leading to removal of an exon form the mature mRNA. Ribonuclease susceptible AONs will recruit ribonuclease H and induce hydrolysis, eliminating the transcript.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



