Submitted:
10 January 2023
Posted:
12 January 2023
You are already at the latest version
Abstract
The FTF (Fusarium Transcription Factor) gene family is composed of two members (FTF1 and FTF2) with high sequence homology. Both genes encode Zn(II)2Cys6 binuclear zinc finger transcription factors involved in the modulation of virulence in the F. oxysporum species complex (FOSC). While FTF1 is a multicopy gene exclusive of highly virulent strains of FOSC and is located in the accessory genome, FTF2 is a single copy gene, located in the core genome, and well conserved in all filamentous ascomycete fungi, except yeast. The involvement of FTF1 in the colonization of the vascular system and regulation of the expression of SIX effectors has been stablished. To address the role of FTF2 we generated and characterized null FTF2 mutants in a F. oxysporum f. sp. phaseoli weakly virulent strain and analyzed them together with the equivalent mutants formerly obtained in a highly virulent strain. The results obtained highlight a role for FTF2 as a negative regulator of the production of macroconidia and demonstrate that it is required for full virulence and positive regulation of SIX effectors. In addition, gene expression analyses provide compelling evidence that FTF2 is involved in the regulation of hydrophobins likely required for plant colonization.
Keywords:
Fusarium oxysporum
; virulence
; FTF gene family
; plant colonization
; effector
1. Introduction
Fusarium oxysporum Schlechtend.:Fr. is an anamorphic species complex comprised of saprophytes and plant colonizers. The second group of strains may live inside the plant roots as endophytes or infect and cause disease in many important crops. However, the pathogenic individual isolates generally are able to infect only a single species, or a small group of them, which allows to classify them into host-specific forms known as formae speciales [1].
The wide pathogenic ability of F. oxysporum as species complex but restricted specificity at the strain level is surprising in comparison with other species of the genus Fusarium, such as Fusarium graminearum and Fusarium verticillioides. Recently, 106 formae speciales have been well document and 37 more have been reported but insufficiently documented [2]. Although F. oxysporum is mostly a plant colonizer its abilities are not restricted to the vegetal kingdom. There have been described isolates able to infect Caenorhabditis elegans [3], mice and humans [4]. The genetic basis responsible for this versatility probably rely in the particular architecture of the F. oxysporum genome. The core genome, very similar among all the formae speciales sequenced to date and also to the related species F. graminearum and F. verticillioides, encompasses 11 chromosomes that contain genes required for basic metabolism and differentiation. The accessory genome, which spands several lineage-specific chromosomes and some parts of the core genome chromosomes, contains sophisticated genomic elements linked to pathogenicity, virulence and host specificity [5,6]. The term “accessory” relates to the fact that the genetic elements in this genome are not required for basic saprophytic growth and development.
Horizontal transfer of whole or partial chromosomes may explain how an endophytic or saprophytic strain may become pathogenic and also the diversification of formae speciales [7,8]. However, it is not the only mechanism as pathogenicity also may evolve independently [7]. In this case, the most plausible scenario would imply mutation and functional diversification of preexisting genes in the core genome. To analyze this hypothesis a good starting point is the observation of how gene families have evolved and diversified.
Several gene families involved in pathogenicity and virulence have been described in F. oxysporum. The two most important ones are the SIX gene family of effectors [9] and the FTF gene family of transcription factors. Currenlty, 14 SIX genes have been identified in F. oxysporum f. sp. lycopersici. The corresponding proteins share the common feature of being “secreted in xylem”, but the coding genes are not phylogenetically related. On the contrary, the FTF gene family is composed of a unique core genome gene, FTF2, which is present in nonpathogenic strains and pathogenic strains of all formae speciales and encode a predicted polypeptide 1072 amino acids in length, and a variable number of FTF1 paralogs [10]. The number of paralogs range from a single one in formae speciales arabidopsis and conglutinans up to 10 in forma specialis lycopersici race 2. These paralogs may be putative functional genes showing some variability in the length of the encoded proteins (from 930 to 1070-1079 amino acids) or putative nonfunctional truncated pseudogenes that show combinations of small and large deletions and premature stop codons. In most cases they are linked to transposons, which suggests that duplication and translocation may be involved in their origin and diversification.
The FTF1 paralogs have been shown to be important virulence factors that are only expressed in planta during host colonization [11] and regulate the expression of some, if not all, the SIX effectors. The attenuation of FTF gene expression by means of gene silencing results in a dramatic reduction of virulence and reduced expression of several SIX genes. Gene replacement of the FTF2 gene in a highly virulent strain results in a less important reduction of virulence towards the host plant (common bean) [10].
In this work, we address the precise role of FTF2 in plant colonization, the nature of the genes regulated by this transcription factor and the type of defensive response that its expression induces in the plant host. We also propose a model to explain how the expression of the FTF gene family modulates virulence in F. oxysporum.
2. Materials and Methods
2.1. Fungal strains and culture conditions
The F. oxysporum f. sp. phaseoli strains FOP-SP1 (highly virulent) and FOP-SP4 (weakly virulent) [12,13] and strains FOP-SP1ΔFTF2 and SP1PgpdA::FTF1 [10] were used in this study. All strains were grown as previously described [12,14]. Fungal cultures were established from frozen mycelia stored on 25% glycerol v/v at -800C and incubated under controlled conditions (250C and continuous light) for 6 days (solid media) or 5 days at 120-180 rpm (liquid cultures).
2.2. Obtention of FTF2 null mutants in the weakly virulent strain FOP-SP4 of F. oxysporum f. sp. phaseoli
Plasmid pFTF2-KO [9] was used to obtain FOP-SP4 transformants with the FTF2 gene (locus FOXG_09390 in strain 4287 of F. oxysporum f. sp. lycopersici) inactivated by gene replacement with a selectable marker (the hygromicin resistance gene hph). Fungal transformations were performed following the Agrobacterium tumefaciens-mediated transformation procedure as previously described [11,15].
2.3. Sporulation, germination and saprophytic growth assays
To test the sporulation rate of the fungal strains, 106 fresh conidia were inoculated in 40 mL of PDB (Potato Dextrose Broth, Difco) and the cultures were incubated at 250C and 180 r.p.m. with continuous light. 2 mL aliquots were taken at 3, 5, 7 and 10 days post inoculation (dpi) and the concentration of spores was estimated with a Thoma cell counting chamber. Four independent cultures were analyzed in each biological experiment and experiments were repeated three times.
To test the germination rate of the fungal strains, a suspension of fresh microconidia prepared at a concentration of 106 spores/mL was used as initial culture. 100 µL of this suspension were placed in the center of an empty Petri dish. The plates were incubated at 25°C with continuous light and high humidity for a maximum of 10 hours. The concentration of germinated vs. non-germinated spores was estimated 0, 2, 4, 6, 8 and 10 hpi. A spore was considered as “germinated” when the length of the germinative tube was at least the same as the non-geminated spore. Three independent cultures per time point were analyzed in each biological experiment and experiments were repeated three times.
The saprophytic growth of the strains was tested on solid synthetic media. Minimal media [15] was amended either with 23.5 mM NaNO3 and different carbon sources (sucrose, mannose or xylose) at 3% or 0.3%, or with 3% sucrose and different nitrogen sources (NaNO3, NaNO2, ammonium tartrate or NH4NO3) at concentrations previously described [16]. The pH of minimal media amended with 3% sucrose and 23.5 mM NaNO3 [16] was adjusted to 4.0, 6.0 or 8.0 when analyzing the effect of pH on the fungal growth. Three plates per media and strain were inoculated and three independent biological experiments were analyzed. Plates were incubated at 250C under a 16/8 hours light/dark photoperiod. The diameter of the colony was measured 6 dpi.
2.4. Inoculation of common bean plants
Inoculation of P. vulgaris L. cv Blanca Riñón with conidia from F. oxysporum strains and transformants was carried out as previously described [12]. After inoculation, the plants were transferred to 50 ml Falcon® tubes filled with PGM (Plant Growth Medium) solution, covered with foil and incubated in hydroponic cultures as previously described [17]. Inoculation assays were repeated three times in a randomized design.
2.5. Confocal laser microscopy
Plants inoculated with FOP-SP1 wild type and FTF2 null mutant strains were maintained in hydroponic cultures and examined after infection as previously described with some modifications [17]. Longitudinal and cross sections were sliced from root system (1, 2, 3 dpi), root crown (5, 7 dpi) and hypocotyls (14, 21 dpi) and incubated overnight in 100% ethanol at 40C (ethanol was replaced if necessary). Next, sections were incubated in 10% KOH at 850C for 5 minutes. The samples were then washed 4 times with 1X PBS (Phosphate Buffered Saline) pH 7.4, and then the staining solution (10 µg/mL WGA-FITC -W11261, Invitrogen, ThermoFisher Scientific Inc.-; 20 µg/mL propidium iodide -P4170, Sigma-Aldrich-; 0.02% Tween 20 in 1X PBS pH 7.4) was added. The samples were vacuum infiltrated once for 10 minutes, followed by 10 times for two minutes each using a Savant DNA 120 SpeedVac Concentrator (ThermoFisher Scientific Inc., Waltham, MA, USA), de-stained washing 4 times with 1X PBS and stored overnight at 40C. Stained sections were visualized using a laser scanning spectral confocal microscope (TCS2-SP2, Leica Microsystems, Bensheim, Germany). The images were analyzed using the software LAS-Advanced Fluorescence Lite 1.8.2 (Leica Microsystems, Germany).
2.6. Nucleic acid manipulations
Samples of mycelium and plant for nucleic acid isolation were harvested or cut and immediately frozen at -800C. Genomic DNA was extracted from F. oxysporum mycelium according to the procedures previously described [11,12,18] and used in PCR reactions and Southern blot analysis. Southern blots were carried out as previously described [10,11]. Total DNA from inoculated plants was isolated using the “mini-prep” DNA extraction method [19]. Total RNA was extracted both from mycelium and plants using the SV Total RNA Isolation System Z3105 (Promega, Madison, WI, USA) according to the manufacturer’s recommendations. RNA was finally treated with TURBO DNA-freeTM Kit (AM1907, Invitrogen, ThermoFisher Scientific Inc., Waltham, MA, USA) to remove traces of DNA. The integrity of purified RNA was checked by running an aliquot in agarose gels and it was quantified using a Nanodrop Spectrophotometer (ThermoFisher Scientific Inc., Waltham, MA, USA).
2.7. Analysis of gene expression and fungal biomass quantification
The analysis of gene expression and the quantification of fungal burden was performed by means of Quantitative Reverse Transcription PCR (RT-qPCR) and qPCR, respectively. The qPCR reaction components and cycling conditions followed the recommendations of previously described protocols [17,20]. The efficiency of each pair of primers used in this work was verified prior to use them in the quantifications as it has been previously described [20]. All the reactions were performed in a StepOnePlusTM Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) according to manufacturer’s recommendations. The StepOneTM Software v2.3 (Applied Biosystems, Foster City, CA, USA) was used in the analysis of the data.
The Prime ScriptTM RT Reagent Kit (Takara Bio Europe, France) was used in the synthesis of cDNA. The conditions of the synthesis followed previously described indications [17]. The F. oxysporum EF1a and the common bean actin genes were used as endogenous reference genes. The 2-DDCt method [21] was selected for the calculations of relative expression levels of each gene. Samples from three different biological experiments and two independent cDNA preparations per biological experiment were obtained. Three replicas of each cDNA were analyzed to calculate the mean and standard deviation. All the primers used in RT-qPCR experiments are listed in Supplementary Table 1.
The quantification of the relative fungal DNA vs. plant DNA amount was performed according to previously described methods [20]. Three independent biological experiments were performed in a randomized design, six plants per time point and condition assayed in each experiment were collected for the isolation of DNA as it was described above, and three replicas of each DNA were analyzed. A total of 100 ng of each DNA extracted from inoculated plants were used as template in qPCR reactions. For the detection and quantification of fungal DNA primers SGE1-Fwd and SGE1-Rev were used to generate a fragment of the single-locus SGE1 gene (locus FOXG_10510 in strain 4287 of F. oxysporum f. sp. lycopersici) (Supplementary Table 1). Primers PR1-Fwd and PR1-Rev were designed to generate a fragment of the single-locus PR1 gene from common bean genome (Supplementary Table 1), which was used as an endogenous gene to normalize differences in DNA template amounts.
3. Results
3.1. Obtention of FTF2 null mutants by gene replacement
Gene replacement of the FTF2 allele in FOP-SP4 weakly virulent wild type was performed using plasmid pFTF2-KO essentially as described [10]. The plasmid and the strategy followed were designed to ensure the replacement of most of the FTF2 region leaving unaltered the FTF1 paralogues and the coding region of a putative locus adjacent to FTF2 (FOXG_09391). Several transformants were obtained and subjected to PCR and Southern analysis, which verified the deletion of FTF2, replaced by the hygromicin resistance (hph) maker gene (Fig. S1). Two transformants were selected for further analysis (FOP-SP4DFTF2-1 and FOP-SP4DFTF2-7), together with a confirmed ectopic transformant (FOP-SP4Ect-4). The lack of FTF2 transcript in the selected transformants was verified by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) (data not shown).
3.2. Functional domains of the FTF transcription factors
The FTF2 transcription factor and those transcription factors encoded by the FTF1 functional paralogues have two functional domains: a) the Zn(II)2Cys6 binuclear cluster DNA-binding motif exclusive of fungi and first characterized in the Saccharomyces cerevisiae GAL4 protein [22], and b) a “fungal specific transcription factor domain or “Middle Homology Region” (MHR) [23], also called “Major Homology Domain” (MHD) which is commonly associated to the Zn(II)2Cys6 domain[10,11].
A multiple alignment of the MHD region of 22 FTF1 paralogues representing 5 formae speciales (7 paralogues from F. oxysporum f. sp. lycopersici race 2 strain 4287, 5 from F. oxysporum f. sp. lycopersici race 3, 3 from F. oxysporum f. sp. melonis, 2 from F. oxysporum f. sp. raphani, 4 from F. oxysporum f. sp. vasinfectum and 1 from F. oxysporum f. sp. phaseoli) to the FTF2 equivalent region showed a high level of conservation of the amino acidic sequence, with some diversity among the FTF1 paralogues (Figure 1). The equivalent multiple alignment of the binuclear zing-finger domain showed almost a complete identity (data not shown).
In order to verify spatial differences in the domain structure, the Phyre2 web portal for protein modeling, prediction and analysis [24] was used to compare the predicted 3D structures of the MHD domain of the 22 paralogues and the FTF2 protein. No differences that could be correlated to polymorphisms in the amino acid sequence could be detected (data not shown). The 3D structures of the binuclear zing-finger domain were also analyzed, but no significative differences could be observed (data not shown).
Figure 1.
Multiple alignment of the predicted amino acid sequences of the MHD region of Gal4, FTF2 and FTF1 proteins. The corresponding locus code/name for the encoding gene of each protein and the species are shown in the first column. Deduced proteins from the following formae speciales were analyzed: FOP-SP1 (F. oxysporum f. sp. phaseoli); Fol 4287 (F. oxysporum f. sp. lycopersici race 2); Fol race 3 (F. oxysporum f. sp. lycopersici race 3); Fom (F. oxysporum f. sp. melonis); For (F. oxysporum f. sp. raphani) and Fov (F. oxysporum f. sp. vasinfectum).
Figure 1.
Multiple alignment of the predicted amino acid sequences of the MHD region of Gal4, FTF2 and FTF1 proteins. The corresponding locus code/name for the encoding gene of each protein and the species are shown in the first column. Deduced proteins from the following formae speciales were analyzed: FOP-SP1 (F. oxysporum f. sp. phaseoli); Fol 4287 (F. oxysporum f. sp. lycopersici race 2); Fol race 3 (F. oxysporum f. sp. lycopersici race 3); Fom (F. oxysporum f. sp. melonis); For (F. oxysporum f. sp. raphani) and Fov (F. oxysporum f. sp. vasinfectum).

3.3. Phenoypic characterization of FTF2 null mutants
FTF2 null mutants obtained by gene replacement of the native FTF2 locus (FOXG_09390 in the F. oxysporum f. sp. lycopersici genome) in the highly virulent wild type strain FOP-SP1 and the weakly virulent wild type strain FOP-SP4 were subjected to in vitro phenotypic characterization by analyzing the sporulation rate, the germination rate of conidia and the growth in synthetic media.
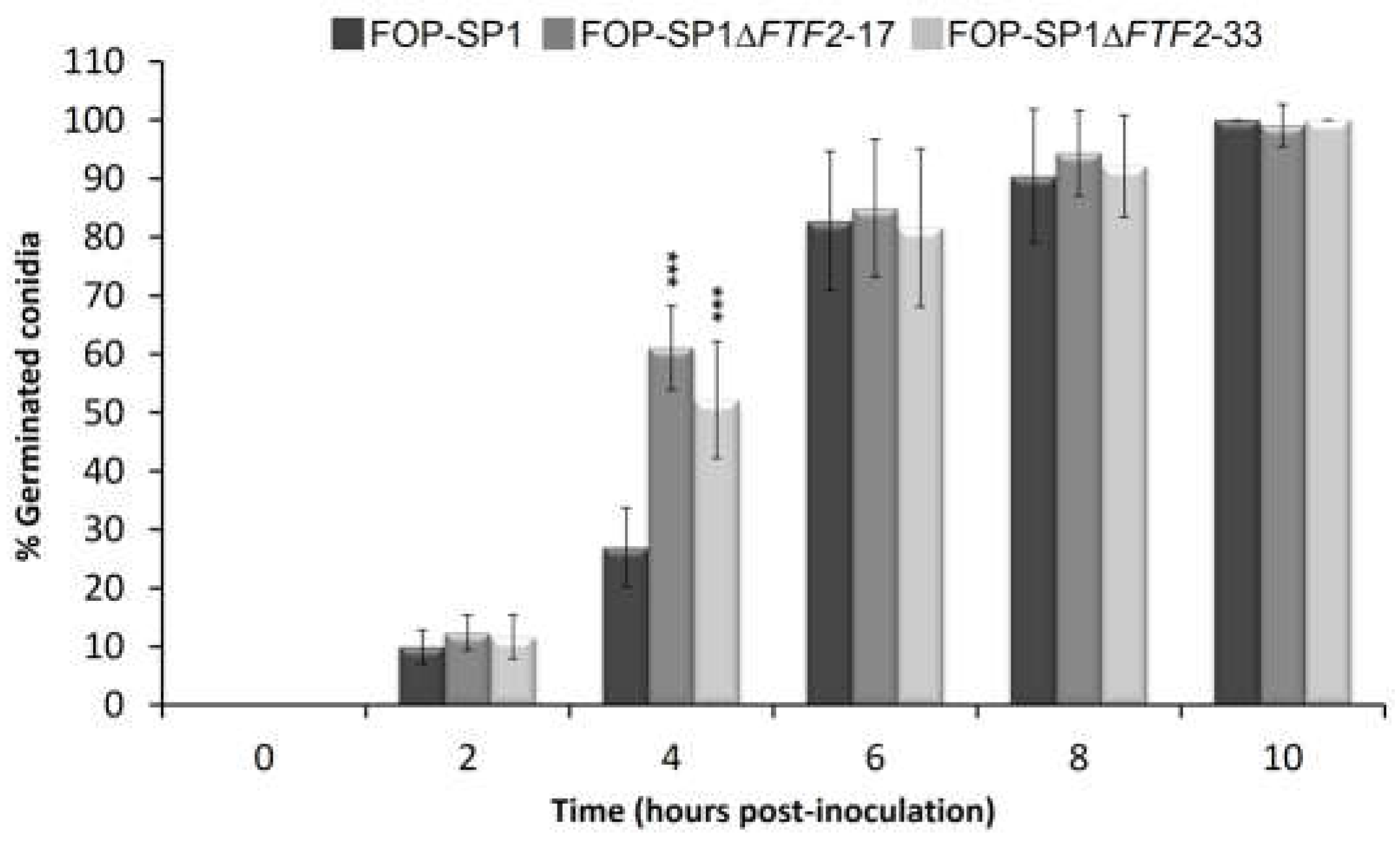
The sporulation rates of two independent mutants for each wild type genetic background were determined in liquid cultures for a maximum period of ten days after inoculation (Figure 2). The mutant strains showed a delay in the sporulation rates when compared to their respective wild types. However, by 7 to 10 days after inoculation sporulation rates were very similar. On the contrary, germination in the mutant strains started earlier than in the wild type strains, although the percentage of germinated spores was similar in both cases 6 h after inoculation (Figure 3; only the results of FOP-SP1DFTF2 and FOP-SP1 are shown).
In order to verify possible differences in radial growth rate or colony morphology between the mutant strains and the wild types, both groups of strains were grown on PDA and minimal medium amended either with sodium nitrate and different carbon sources at two concentrations (3% and 0.3%) or with sucrose 3% and different nitrogen sources. Also, growth was assayed on minimal medium amended with sucrose 3% and sodium nitrate, as the sole nitrogen source, adjusted to pH values 4.0, 6.0 and 8.0. No differences could be observed between the mutant strains and the original wild types (Supplementary Figure 2).
Figure 3.
Germination rates of FOP-SP1ΔFTF2 strains. Mutants and wild-type strains were incubated in PDB and germinated vs. non-germinated spores were analyzed for a maximum period of 10 hpi. Each bar represents the media ± standard deviation of three independent biological experiments. Significant differences were tested using an ANOVA analysis followed by a Dunnett’s test and are indicated by *** (P < 0.001). .
Figure 3.
Germination rates of FOP-SP1ΔFTF2 strains. Mutants and wild-type strains were incubated in PDB and germinated vs. non-germinated spores were analyzed for a maximum period of 10 hpi. Each bar represents the media ± standard deviation of three independent biological experiments. Significant differences were tested using an ANOVA analysis followed by a Dunnett’s test and are indicated by *** (P < 0.001). .
Sporulation rates were also determined for mycelia grown on solid media. Wild type strains do not produce macroconidia, neither when grown in liquid media (as shown before) nor when they are grown on synthetic solid media. However, the FTF2 null mutants obtained from FOP-SP1 (FOP-SP1DFTF2-17 and FOP-SP1DFTF2-33) showed a surprisingly high production of macroconidia (Figure 4 and Supplementary Table S2), either in PDA or in some amended minimal media (with glucose, xylose, glycerol, sucrose and mannose). Similar results were obtained for FOP-SP4DFTF2 mutants (results not shown).
3.4. Host plant colonization by FTF2 null mutants
One of the FTF2 gene replacement mutants obtained in the FOP-SP1 genetic background was used to analyze possible changes in the in planta colonization pattern. FOP-SP1 is a highly virulent strain, therefore the possible altered patterns of colonization determined by the absence of the FTF2 transcription factor should be more easily detected than in a weakly virulent strain. Longitudinal and transversal sections of the root system, root crown and hypocotyl of common bean plants were sampled at different times after inoculation. The characteristics and extent of fungal colonization were visualized by means of WGA-FITC and propidium iodide staining as shown in Figure 5.
The FTF2 mutant shows a reduced ability to colonize the root system, which is more evident three days after inoculation. Also, the ratio of colonized parenchymal tissue versus vascular tissue is increased at later stages, both in root crown and hypocotyl. This colonization pattern highly resembles that displayed by weakly virulent strains [20] and mutants attenuated in the expression of the FTF gene family [10].
The altered pattern of colonization and the drastic reduction of fungal hyphae inside the vascular system is not due to an overall reduction of the fungal burden. The amount of fungal biomass detected by means of RT-qPCR in plants inoculated with the wild type and the mutant strain was not significantly different (Supplementary Figure 3).
3.5. Plant defensive response
To evaluate the role of FTF2 in the induction of the plant defensive response, possible changes in the expression of selected plant genes were assessed in RT-qPCR experiments. To this point, the expression of genes involved in the salicylic acid response (PR1) and ethylene/jasmonic acid response (ERF1 and ERF2) was analyzed in the root system, root crown and hypocotyl of common bean plants inoculated with the wild type strain (FOP-SP1) or the FTF2 mutant strain (FOP-SP1DFTF2).
The results presented in Figure 6 show a significant increase in the expression of PR1 in roots colonized by the mutant strain 3 days post inoculation (dpi), although no differences in the expression of this gene could be detected in later stages of colonization. The expression of ERF1 and ERF2 in plants inoculated with the wild type was in line with the results previously obtained [20]. The lack of expression of FTF2 did not change this expression pattern.
3.6. Analysis of FTF2-responsive genes
To shed more light on the functional role of the FTF2 transcription factor we carried out an expression analysis of several putative FTF2-responsive genes. These genes were selected from a preliminary transcriptomic comparative analysis of 48 h old liquid cultures inoculated with FOP-SP4 and one of the FTF2 mutants obtained from this strain. As FOP-SP4 is a weakly virulent strain it lacks all the FTF1 paralogous genes, thus ensuring that the differentially expressed genes obtained from the transcriptomic analysis would be exclusively the result of FTF2 regulation.
We selected five genes: FOXG_14730 (putatively encoding a protein with SET domain), FOXG_02746 and FOXG_02748 (putatively encoding type II hydrophobins), FOXG_14731 (putatively encoding an oxygenase) and FTF2 itself, to analyze their expression in different genetic backgrounds, namely, both wild types (FOP-SP4 and FOP-SP1), the null mutants obtained by the FTF2 gene replacement in both strains (FOP-SP4DFTF2 and FOP-SP1DFTF2), a FOP-SP4 mutant containing an ectopic integration of the construction used to replace FTF2 (FOP-SP4-Ect-4), and, finally, a mutant of FOP-SP1 which constitutively express one of the functional FTF1 paralogs under the control of the gpdA promoter (SP1-PgpdA::FTF1) [10].
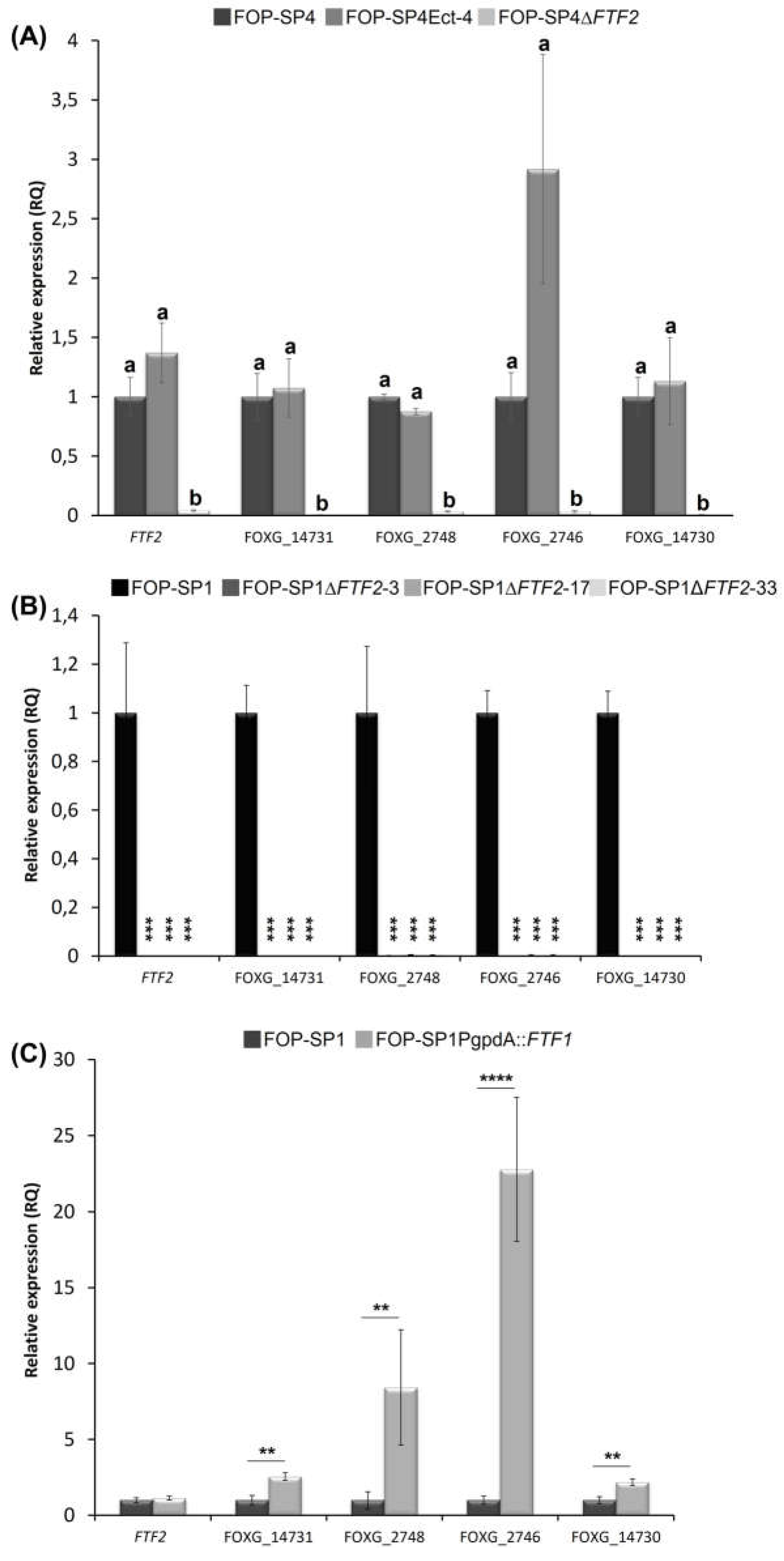
The RT-qPCR analysis of expression in mycelia grown in PDB medium is shown in Figure 7. As expected, expression of the five genes could not be detected in FOP-SP4DFTF2 (Figure 7A). Also, a complete lack of expression of the five genes could be observed in FOP-SP1DFTF2 mutants (Figure 7B). These results indicate that the genes analyzed are regulated by the FTF2 transcription factor. However, as there is no significant in vitro expression of the FTF1 paralogs, it could not be ruled out that FTF1 expression could activate the transcription of some or all of the selected genes.
To answer this question two approaches were followed. First, a transformant that expresses constitutively one of the FTF1 paralogs under the control of the gpdA promoter (SP1PgpdA::FTF1) [10] was analyzed (Figure 7C). All the genes, except FTF2, showed a significant upregulation when FTF1 was constitutively expressed. These results demonstrate that even though all the genes were selected on the basis of differential expression in the absence of FTF2, they also respond positively to the presence of FTF1. However, massive amounts of FTF1 transcripts, such as those produced by the strong gpdA promoter, are required for this activation.
Figure 7.
RT-qPCR analysis of expression of putative FTF2-responsive genes. A, Analysis performed in FOP-SP4ΔFTF2 strain. The value 1.0 was denoted for the transcript level of all genes in the wild-type strain FOP-SP4. Letters over each bar represent significant differences in the expression of the genes in the strains assayed after an ANOVA analysis followed by a Tukey’s HSD test (P < 0.05). B, Analysis in FOP-SP1ΔFTF2 strains. The value 1.0 was denoted for the transcript level of all genes in the wild-type strain FOP-SP1. Differences were tested using an ANOVA analysis followed by a Dunnett’s test and are indicated by *** (P < 0.001). C, Analysis of gene expression in SP1PgpdA::FTF1 strain. The value 1.0 was denoted for the transcript level of all genes in the wild-type strain FOP-SP1. Differences were tested using a t-test and significant differences are indicated by ** (P < 0.01) and **** (P < 0.0001). In all cases the expression ratios were normalized by using the EF1α gene as endogenous control and bars represent the media ± standard deviation of three independent biological experiments.
Figure 7.
RT-qPCR analysis of expression of putative FTF2-responsive genes. A, Analysis performed in FOP-SP4ΔFTF2 strain. The value 1.0 was denoted for the transcript level of all genes in the wild-type strain FOP-SP4. Letters over each bar represent significant differences in the expression of the genes in the strains assayed after an ANOVA analysis followed by a Tukey’s HSD test (P < 0.05). B, Analysis in FOP-SP1ΔFTF2 strains. The value 1.0 was denoted for the transcript level of all genes in the wild-type strain FOP-SP1. Differences were tested using an ANOVA analysis followed by a Dunnett’s test and are indicated by *** (P < 0.001). C, Analysis of gene expression in SP1PgpdA::FTF1 strain. The value 1.0 was denoted for the transcript level of all genes in the wild-type strain FOP-SP1. Differences were tested using a t-test and significant differences are indicated by ** (P < 0.01) and **** (P < 0.0001). In all cases the expression ratios were normalized by using the EF1α gene as endogenous control and bars represent the media ± standard deviation of three independent biological experiments.
Second, expression of the selected genes was analyzed in the course of plant colonization by the wild type and the corresponding FTF2 mutant strain. To this point, RT-qPCR experiments were performed with roots 3 days post inoculation (dpi), root crown 7 dpi and hypocotyls 21 dpi (Figure 8). None of the genes analyzed were expressed in a detectable way in plants inoculated with the FTF2 null mutant, thus showing that expression of FTF2, and not of FTF1, is required for their in planta expression. The results presented in Fig. 8A show a moderate induction of the expression of FTF2 which reaches a maximum in hypocotyl samples at 21 dpi. Transcripts of FOXG_02746 (the locus encoding a hydrophobin II) were detected in early stages of infection, while transcripts of FOXG_14730 (the locus encoding a protein with SET domain) and FOXG_14731 (the locus encoding an oxygenase) were only detected in root crown samples. By the time vascular wilt is at its maximum and the plants are dead (21 dpi) expression of none of the FTF2-responsive genes could be detected.
FTF2 overexpression mutants have been obtained in F. oxysporum f. sp. lycopersici, and it has been suggested that FTF2 regulates the expression of genes encoding the SIX effectors [25]. Although none of the SIX genes described to date were selected as putative FTF2-responsive genes, we analyzed SIX1 and SIX6 expression during colonization (Fig. 8B). A significant reduction in the accumulation of both transcripts was observed in root and root crown of plants colonized by FOP-SP1ΔFTF2, indicating that SIX1 and SIX6 are directly or indirectly regulated by FTF2. Curiously, the expression of SIX6 in hypocotyls at 21 dpi was significantly higher in plants colonized by FOP-SP1ΔFTF2 than in those colonized by the wild type FOP-SP1.
4. Discussion
It has been proposed that one of the main advantages of an accessory genome would be the acquisition of novel virulence factors that can expand host range [26] or modify the colonizing abilities. Novel or modified gene functions evolved from gene duplications and subsequent divergence might be well tested in the context of accessory chromosomes. It is not easy to explain how this mechanism may produce entirely new proteins able to perform new functions, except that already existing transcription factors may change to alternative forms with different regulatory abilities, thus reprograming the expression of whole sets of genes. The best known pathogenicity chromosome in F. oxysporum is chromosome 14 in the reference strain F. oxysporum f. sp. lycopersici 4287, which is part of the accessory genome. It contains 13 predicted transcription factor genes that cluster into nine families [25]. Interestingly, all these transcription factors, except TF3, have a homologue in the core genome, which strongly support the hypothesis that they have evolved by gene duplication of the core genome homologues. In the case of the FTF transcription factors gene family, the core genome homologue is FTF2 (chromosome 9 in the reference strain), and all the FTF1 paralogs are located in chromosomes pertaining to the accessory genome (chromosomes 3, 6, 14, 15 and the variable segment of chromosome 1) [10]. In the highly virulent strains of F. oxysporum f. sp. phaseoli, where this gene family was first described [8,11,14], all the FTF1 paralogs are located in a small chromosome, likely homologue to chromosome 14 [10,11,27].
To ascertain what advantages in terms of virulence may provide the FTF1 transcription factors a comparative analysis with FTF2 is required. In a former work we generated null FTF2 mutants from a highly virulent strain (FOP-SP1) to analyze FTF1 when FTF2 is inactive [10]. In the present work we describe and analyze null FTF2 mutants from a weakly virulent strain (FOP-SP4), which is devoid of all the FTF1 paralogs, with the aim to study the phenotypic effect of the complete absence of FTF genes.
First, multiple alignments and three-dimensional modelling were carried out to identify differences in the predicted spatial structure of the two main domains: the Zn(II)2Cys6 binuclear cluster and the middle homology region. No significative differences were found in the former one that might account for differences in DNA binding capabilities. The MHR domain is involved in the regulation of the activity of Zn(II)2Cys6 binuclear transcription factors [28]. The crystal structure of Cep3, a fungal transcription factor containing a MHR region has showed that this region contains eight motifs included in an all-alpha domain, called MHD (Middle Homology Domain) [29,30]. The multiple alignment of the amino acid sequences that encomprise MHD indicates that this domain shows some diversity, both in comparison to FTF2 and among the FTF1 proteins. However, the 3D analysis did not show any evidence that those differences might be correlated with modifications of the basic all-alpha domain. It cannot be ruled out that some of the polymorphisms observed may alter the function of the corresponding FTF1 proteins. However, both the alignment and the 3D modelling do not show changes in the all-alpha domain between FTF2 and all the FTF1 proteins that may suggest differences in the regulation abilities of both types of transcription factors.
Mutants altered in FTF2 showed slight differences with the wild types in sporulation and germination rates. However, the most striking difference was the drastic increase in the production of macroconidia when grown in solid media (either PDA or minimal medium supplemented with different carbon sources). Several transcription regulators essential for conidiation have been described in F. oxysporum. REN1 is required for normal development of micro and macronidia [31], while STUA is a positive regulator for the development of macroconidia and a negative regulator for the development of chlamydospores [32]. Also, several components of the Velvet complex are involved in conidiation. Disruption of veA, velB and velC causes a derepression of conidiation and an increase in the shape of microconidia [33]. The phenotype displayed in solid media by the FTF2 mutants is different to the above described and strongly suggests that FTF2 might be a negative regulator of the production of macroconidia.
FTF2 mutants show a slight reduction in virulence [10] but whether this is caused by an enhanced plant defensive response or a reduction in colonization capabilities has not been dilucidated. The results here reported strongly indicate that both causes contribute to the observed reduction. The plant colonization pattern exhibited by the FTF2 mutant here analyzed is characterized by first, a reduced ability to colonize the root system at early stages, and second, an increased ratio of parenchymal colonization versus vascular colonization at later stages. Both features are also distinctive of the colonization patterns exhibited by weakly virulent strains [20] and mutants attenuated in the expression of the FTF genes [10], although the FTF2 mutants are more virulent than the mutants attenuated in the FTF genes and as virulent as the weakly virulent strains. The reduced colonization of the root system is not a consequence of the reduction of fungal biomass, as it was also observed for the weakly virulent strains [20]. Our results strongly support the role of the FTF1 paralogs as critically required for massive colonization of the vascular system, which results in enhanced virulence towards the host plant. The higher rate of parenchymal colonization displayed by the FTF2 mutant correlates with an increased expression of PR1 by the host plant. This result agrees with former reports on the colonization pattern showed by weakly virulent strains [20].
Taking together all these observations, two basic plant colonization patterns by F. oxysporum may be depicted. Naturally occurring weakly virulent strains (such as FOP-SP4) or mutants with an altered expression of virulence factors (such as those defective in FTF2 or attenuated in the FTF genes) show an increase in the parenchymal/vascular colonization ratio that correlates with an induction of the host defensive response mediated by SA (as shown by the increased expression of PR1). On the opposite, highly virulent strains quickly enter the vascular cylinder and spread through the xylem vessels of the host plant. This rapid progression towards the vasculature of the plant requires avoidance of the SA mediated plant defensive response and high expression of all the members of the FTF gene family.
The structure of the functional domains in FTF2 and FTF1 do no suggest functional differences between both transcription factors. However, we show that, at least under different growing conditions, they regulate different genes. The genes encoding for a hydrophobin II, the protein with SET domain and the oxygenase do not show detectable expression during plant colonization by the FOP-SP1 mutant that lacks FTF2, although they respond to FTF1 overexpression during in culture growth. These results demonstrate that although FTF1 may potentially regulate these genes, their naturally occurring regulation, both in culture and in planta, correspond to FTF2.
Among the five genes selected to verify their possible regulation by FTF2, loci FOXG_02748 and FOXG_02746 are predicted to encode fungal hydrophobins. Hydrophobins are low molecular weight secreted proteins. They are characterized by moderate to high levels of hydrophobicity and the presence of eight conserved cysteines [34]. The stablished role for hydrophobins in fungi is to form a layer that allows fungal structures to breach the air-water interface or prevent water-logging [35]. They are also involved in rendering the conidial surface hydrophobic and resistant to water, thus facilitating dispersal of spores in the air [36]. Surface hydrophobins play a role in preventing immune recognition of airborne fungal spores [37]. Hydrophobins have been shown to play a role in fungal plant pathogenicity as mutants altered in their expression show reduced virulence towards the host plant [38,39,40]. This contribution to virulence has also been confirmed in F. graminearum, as mutants deficient in hydrophobins FgHyd2 and FgHyd3 show reduced growth and ability to penetrate through the water-air interface in the plant host. [41]. Regulation of the expression of hydrophobin encoding genes by FTF2 suggests the involvement of this transcription factor in conidia production and also in plant colonization, in accordance with the phenotype described for null mutants. These results are in line with those reported for another xylem colonizer such as Verticillium dahliae, which is able to produce hydrophobins in tomato xylem sap [42].
Transcriptomic analysis carried out in FTF2 overexpression mutants of F. oxysporum f. sp. lycopersici suggested that two SIX efectors (SIX1 and SIX6) are regulated by FTF2 [25]. Our results confirm this role, as both SIX1 and SIX6 are downregulated in FOP-SP1DFTF2 mutants during host colonization. Previously it had been shown that SIX1 and SIX6 are under positive control of the FTF1 paralogs [10]. The results here presented extend positive control of SIX effectors to the whole FTF gene family. Taking into account the differential expression of the members of the family in the time spatial frame of plant colonization, it is likely that a fine modulation of the expression of the effectors take place.
We propose the model depicted in Figure 9 to explain the role of the FTF family of transcription factors in plant colonization and the severity of disease produced by F. oxysporum [27]. Virulence and the degree of symptoms produced in the host would be the consequence of a cumulative expression of the active members of the FTF gene family and the repertoire of effectors present in the fungal strain. Lack of FTF1 paralogs and a reduced number of SIX effectors would result in mild symptoms, as those displayed by weakly virulent strains. On the other hand, an extended set of effectors and several active copies of the FTF1 paralogs would drastically increase the severity of disease (including death of the plant), as happens in the infections produced by highly virulent strains. This model predicts probable genetic features of F. oxysporum endophytes. As these strains are able to colonize plant parenchymal tissues with almost no growth inside the vasculature, they should lack FTF1 paralogs and show a reduced effector gene repertoire. A recent study conducted with F. oxysporum endophytes and more than 100 pathogenic strains confirms this prediction. It was found that a pathogenic lifestyle is associated with extended effector gene catalogs, while the endophytes analyzed clustered together based on the scarcity of effector candidates in their genomes [43]. Although no data on the presence of FTF1 is provided in the study, all the endophytic F. oxysporum strains we have analyzed to date are devoid of FTF1 copies, as it happens for the weakly virulent strains (data not shown). Moreover, the presence of FTF1 is proof of virulence [18].
The expression of the FTF gene family members to regulate virulence offers a good example of coordinated crosstalk between elements of the core genome and the accesory genome in F. oxysporum. It has been suggested the need of a spatial association between the gene expansion of transcription factors and their regulated genes [44]. However, there is no association between FTF2 (chromosome 9) and the target genes SIX1 and SIX6 (chromosome 14), thus showing that transcription factors in the core genome may regulate effector genes in the accessory genome.
5. Conclusions
We provide evidence that FTF2 is a negative regulator of macroconidia production, which, together with the predicted functions inferred for some of the genes it regulates, supports its role in basic metabolism and physiology, as it would be expected for a core genome gene. FTF2 is also required for full virulence, probably in a cumulative but not redundant manner with the FTF1 transcription factors, as it is described in the model proposed of virulence regulation by the FTF gene family. The appearance of multiple paralogs by gene duplication and diversification of FTF2 in the accessory genome would provide highly virulent strains with a selectable advantage in terms of quantitative positive regulation of effectors required for quick colonization of the xylem vessels. It remains to be clarified whether FTF2 is critical for plant colonization also in endophytic strains or only for virulence in a pathogenic lifestyle.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
JMDM, EPB and VCdC conceived the study, participated in the design of the experiments, the analysis of the results and final draft. VCdC performed the molecular lab work, infection assays and statiscal analysis. JMDM drafted the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants AGL2012-3987-CO2-01 from Ministerio de Economía y Competitividad (MINECO, Spain) and PID-2019-110605RB-100 from Ministerio de Ciencia e Innovación (MICINN, Spain). V. Casado-del Castillo was recipient of fellowship AP2010-2742 from Ministerio de Educación, Cultura y Deporte (MECD, Spain).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
References
- Armstrong, G.M.; Armstrong, J.K. Fusarium: diseases, biology and taxonomy; Pennsilvania State University Press: USA, 1981; pp. 391–400. [Google Scholar]
- Edel-Hermann, V.; Lecomte, C. Current Status of Fusarium oxysporum formae speciales and races. Phytopathology 2019, 109, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Nag, P.; Aggarwal, P.R.; Ghosh, S.; Narula, K.; Tayal, R.; Maheshwari, N.; Chakraborty, N.; Chakraborty, S. Interplay of neuronal and non-neuronal genes regulates intestinal DAF-16-mediated immune response during Fusarium infection of Caenorhabditis elegans. Cell Death Discov. 2017, 3, 17073. [Google Scholar] [CrossRef] [PubMed]
- Nag, P.; Paul, S.; Shriti, S.; Das, S. Defence response in plants and animals against a common fungal pathogen, Fusarium oxysporum. Curr. Res. Microb. Sciences 2022, 3, 100135. [Google Scholar] [CrossRef]
- Ma, L.-J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef]
- Yu, H.; Ayhan, D.H.; Martínez-Soto, D.; Cochavi, S.M.; Ma, L.-J. Accessory chromosomes of the Fusarium oxysporum species complex and their contribution to host niche adaptation. In Plant Relationships: Fungal-Plant Interactions; Scott, B., Mesarich, C., Eds.; Springer International Publishing: Cham, 2023; pp. 371–388. ISBN 978-3-031-16503-0. [Google Scholar]
- Ma, L.-J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.-J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef]
- Henry, P.M.; Pincot, D.D.A.; Jenner, B.N.; Borrero, C.; Aviles, M.; Nam, M.; Epstein, L.; Knapp, S.J.; Gordon, T.R. Horizontal chromosome transfer and independent evolution drive diversification in Fusarium oxysporum f. sp. fragariae. New Phytol. 2021, 230, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Houterman, P.M.; Cornelissen, B.J.C.; Rep, M. Suppression of plant resistance gene-based immunity by a fungal effector. PLoS Pathog. 2008, 4, e1000061–6. [Google Scholar] [CrossRef]
- Niño-Sánchez, J.; Casado-del Castillo, V.; Tello, V.; de Vega-Bartol, J.J.; Ramos, B.; Sukno, S.A.; Díaz-Mínguez, J.M. The FTF gene family regulates virulence and expression of SIX effectors in Fusarium oxysporum. Mol. Plant Pathol. 2016, 17, 1124–1139. [Google Scholar] [CrossRef]
- Ramos, B.; Alves-Santos, F.M.; García-Sánchez, M.A.; Martín-Rodrigues, N.; Eslava, A.P.; Díaz-Mínguez, J.M. The Gene coding for a new transcription factor (Ftf1) of Fusarium oxysporum Is only expressed during infection of common bean. Fungal Genet. Biol. 2007, 44, 864–876. [Google Scholar] [CrossRef]
- Alves-Santos, F.M.; Benito, E.P.; Eslava, A.P.; Diaz-Minguez, J.M. Genetic diversity of Fusarium oxysporum strains from common bean fields in Spain. Appl. Environ. Microbiol. 1999, 65, 3335–3340. [Google Scholar] [CrossRef]
- Alves-Santos, F.; Cordeiro-Rodrigues, L.; Sayagués, J.M.; Martín-Dominguez, R.; García-Benavides, P.; Crespo, M.C.; Díaz-Mínguez, J.M.; Eslava, A.P. Pathogenicity and race characterization of Fusarium oxysporum f. sp. phaseoli isolates from Spain and Greece. Plant Pathol. 2002, 51, 605–611. [Google Scholar] [CrossRef]
- de Vega-Bartol, J.J.; Martín-Dominguez, R.; Ramos, B.; García-Sánchez, M.-A.; Díaz-Mínguez, J.M. New virulence groups in Fusarium oxysporum f. sp. phaseoli: the expression of the gene coding for the transcription factor Ftf1 correlates with virulence. Phytopathology 2011, 101, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Mullins, E.; Chen, X.; Romaine, P.; Raina, R.; Geiser, D.; Kang, S. Agrobacterium-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathology 2001, 91, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Correll, J.C.; Klittich, C.J.R.; Leslie, J.F. Nitrate nonutilizing mutants of Fusarium oxysporum and their use in vegetative compatibility tests. Phytopathology 1987, 77, 1640–1646. [Google Scholar] [CrossRef]
- Castillo, V.C.-D.; Benito, E.P.; Díaz-Mínguez, J.M. In Planta Gene Expression analysis and colonization of Fusarium oxysporum. Methods Mol. Biol. 2022, 2391, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Alves-Santos, F.M.; Ramos, B.; García-Sánchez, M.A.; Eslava, A.P.; Díaz-Mínguez, J.M. A DNA-based procedure for in planta detection of Fusarium oxysporum f. sp. phaseoli. Phytopathology 2002, 92, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Afanador Kafury, Lucía; Haley, S.D.; Kelly, J.D. Adoption of a “mini-prep” DNA extraction method for RAPD marker analysis in common bean (Phaseolus vulgaris L.). Bean Improvement Cooperative. Annu. Report (USA) 1993, 36, 10–11. [Google Scholar]
- Niño-Sánchez, J.; Tello, V.; Casado-del Castillo, V.; Thon, M.R.; Benito, E.P.; Díaz-Mínguez, J.M. Gene expression patterns and dynamics of the colonization of common bean (Phaseolus vulgaris L.) by highly virulent and eeakly virulent strains of Fusarium oxysporum. Front. Microbiol. 2015, 6, 605–614. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pan, T.; Coleman, J. GAL4 transcription factor Is Not a "zinc finger" but forms a Zn(II)2 Cys6 binuclear cluster. Proc. Natl. Acad. Sci. USA 1990, 87, 2077–2081. [Google Scholar] [CrossRef]
- Schjerling, P.; Holmberg, S. Comparative amino acid sequence analysis of the C6 zinc cluster family of transcriptional regulators. Nucleic Acids Res. 1996, 24, 4599–4607. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- van der Does, H.C.; Fokkens, L.; Yang, A.; Schmidt, S.M.; Langereis, L.; Lukasiewicz, J.M.; Hughes, T.R.; Rep, M. Transcription factors encoded on core and accessory chromosomes of Fusarium oxysporum induce expression of effector genes. PLoS Genet. 2016, 12, e1006401-38. [Google Scholar] [CrossRef] [PubMed]
- Croll, D.; McDonald, B.A. The accessory genome as a cradle for adaptive evolution in pathogens. PLoS Pathog. 2012, 8, e1002608. [Google Scholar] [CrossRef]
- Casado-del Castillo, V. The roles of FTF2 and the pathogenicity chromosome of Fusarium oxysporum. PhD Thesis, University of Salamanca, Spain, 2017. [Google Scholar]
- Macpherson, S.; Larochelle, M.; Turcotte, B. A Fungal Family of Transcriptional Regulators: The zinc cluster proteins. Microbiol. Mol. Biol. Rev. 2006, 70, 583–604. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, J.J.; Sorger, P.K.; Harrison, S.C. Crystal structure of the yeast inner kinetochore subunit Cep3p. Structure 2007, 15, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Purvis, A.; Singleton, M.R. Insights into kinetochore–DNA interactions from the structure of Cep3Δ. EMBO Rep. 2008, 9, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.; Inoue, I.; Namiki, F.; Kunoh, H.; Tsuge, T. REN1 is required for development of microconidia and macroconidia, but not of chlamydospores, in the plant pathogenic fungus Fusarium oxysporum. Genetics 2004, 166, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.; Tsuge, T. FoSTUA, encoding a basic helix-loop-h protein, differentially regulates development of three kinds of asexual spores, macroconidia, microconidia, and chlamydospores, in the fungal plant pathogen Fusarium oxysporum. Euk. Cell 2004, 3, 1412–1422. [Google Scholar] [CrossRef]
- López-Berges, M.S.; Hera, C.; Sulyok, M.; Schäfer, K.; Capilla, J.; Guarro, J.; Di Pietro, A. The Velvet complex governs mycotoxin production and virulence of Fusarium oxysporum on plant and mammalian hosts. Mol. Microbiol. 2012, 87, 49–65. [Google Scholar] [CrossRef]
- Wessels, J.G.H. Hydrophobins, unique fungal proteins. Mycologist 2000, 14, 153–159. [Google Scholar] [CrossRef]
- Wang, X.; Shi, F.; Wösten, H.A.B.; Hektor, H.; Poolman, B.; Robillard, G.T. The SC3 hydrophobin self-assembles into a membrane with distinct mass transfer properties. Biophys. J. 2005, 88, 3434–3443. [Google Scholar] [CrossRef] [PubMed]
- Bayry, J.; Aimanianda, V.; Guijarro, J.I.; Sunde, M.; Latgé, J.-P. Hydrophobins—unique fungal proteins. PLoS Pathog. 2012, 8, e1002700. [Google Scholar] [CrossRef] [PubMed]
- Aimanianda, V.; Bayry, J.; Bozza, S.; Kniemeyer, O.; Perruccio, K.; Elluru, S.R.; Clavaud, C.; Paris, S.; Brakhage, A.A.; Kaveri, S.V. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 2009, 460, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.L.; Talbot, N.J. Surface attachment and pre-penetration stage development by plant pathogenic fungi. Annu. Rev. Phytopathol. 2001, 39, 385–417. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, J.R.; Spanu, P.D. Hydrophobins and the interactions between fungi and plants: Hydrophobins and fungus-plant interactions. Mol. Plant Pathol. 2002, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ahn, I.-P.; Rho, H.-S.; Lee, Y.-H. MHP1, a Magnaporthe grisea hydrophobin gene, is required for fungal development and plant colonization. Mol. Microbiol. 2005, 57, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Quarantin, A.; Hadeler, B.; Kröger, C.; Schäfer, W.; Favaron, F.; Sella, L.; Martínez-Rocha, A.L. Different hydrophobins of Fusarium graminearum are involved in hyphal growth, attachment, water-air interface penetration and plant infection. Front. Microbiol. 2019, 10, 751. [Google Scholar] [CrossRef]
- Maurus, I.; Leonard, M.; Nagel, A.; Starke, J.; Kronstad, J.W.; Harting, R.; Braus, G.H. Tomato xylem sap hydrophobins Vdh4 and Vdh5 are important for late stages of Verticillium dahliae plant infection. J. Fungi. 2022, 8, 1252. [Google Scholar] [CrossRef]
- Constantin, M.E.; Fokkens, L.; de Sain, M.; Takken, F.L.W.; Rep, M. Number of candidate effector genes in accessory genomes differentiates pathogenic from endophytic Fusarium oxysporum strains. Front. Plant Sci. 2021, 12, 761740. [Google Scholar] [CrossRef]
- Yang, H.; Yu, H.; Ma, L.-J. Accessory chromosomes in Fusarium oxysporum. Phytopathology 2020, 110, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Sporulation rates of FOP-SP1ΔFTF2 (A) and FOP-SP4ΔFTF2 (B) strains. Mutants and wild-type strains were cultured in PDB media for a maximum period of 10 days. Each bar represents the media ± standard deviation of three independent biological experiments. Significant differences were tested using an ANOVA analysis followed by a Dunnett´s test and are indicated by * (P < 0.05) and *** (P < 0.001).
Figure 2.
Sporulation rates of FOP-SP1ΔFTF2 (A) and FOP-SP4ΔFTF2 (B) strains. Mutants and wild-type strains were cultured in PDB media for a maximum period of 10 days. Each bar represents the media ± standard deviation of three independent biological experiments. Significant differences were tested using an ANOVA analysis followed by a Dunnett´s test and are indicated by * (P < 0.05) and *** (P < 0.001).
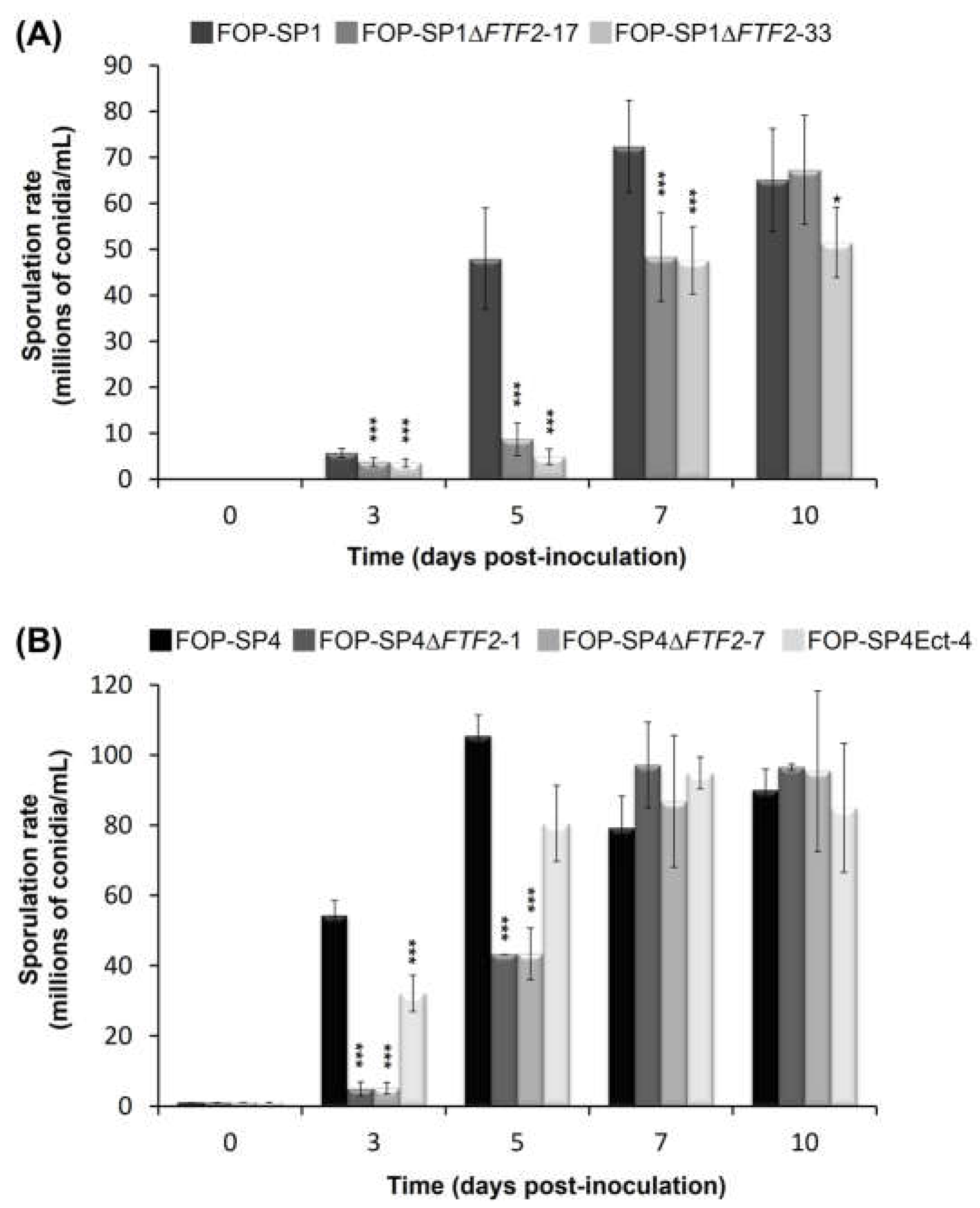
Figure 4.
Macroconidia production by FOP-SP1ΔFTF2 strains. Fungal strains were inoculated on synthetic solid media (PDA -A-, and minimal medium amended with NaNO3 as nitrogen source and glucose -B-, sucrose -C-, xylose -D-, mannose -E- or glycerol -F- as carbon source) under controlled conditions (25°C and a 16/8 h light/dark photoperiod). The conidia were harvested 6 days post-inoculation. Images of a suspension of harvested conidia were taken using a Leica DC300F camera adapted to the Leica DLMB microscope (Leica Microsystems, Bensheim, Germany).
Figure 4.
Macroconidia production by FOP-SP1ΔFTF2 strains. Fungal strains were inoculated on synthetic solid media (PDA -A-, and minimal medium amended with NaNO3 as nitrogen source and glucose -B-, sucrose -C-, xylose -D-, mannose -E- or glycerol -F- as carbon source) under controlled conditions (25°C and a 16/8 h light/dark photoperiod). The conidia were harvested 6 days post-inoculation. Images of a suspension of harvested conidia were taken using a Leica DC300F camera adapted to the Leica DLMB microscope (Leica Microsystems, Bensheim, Germany).
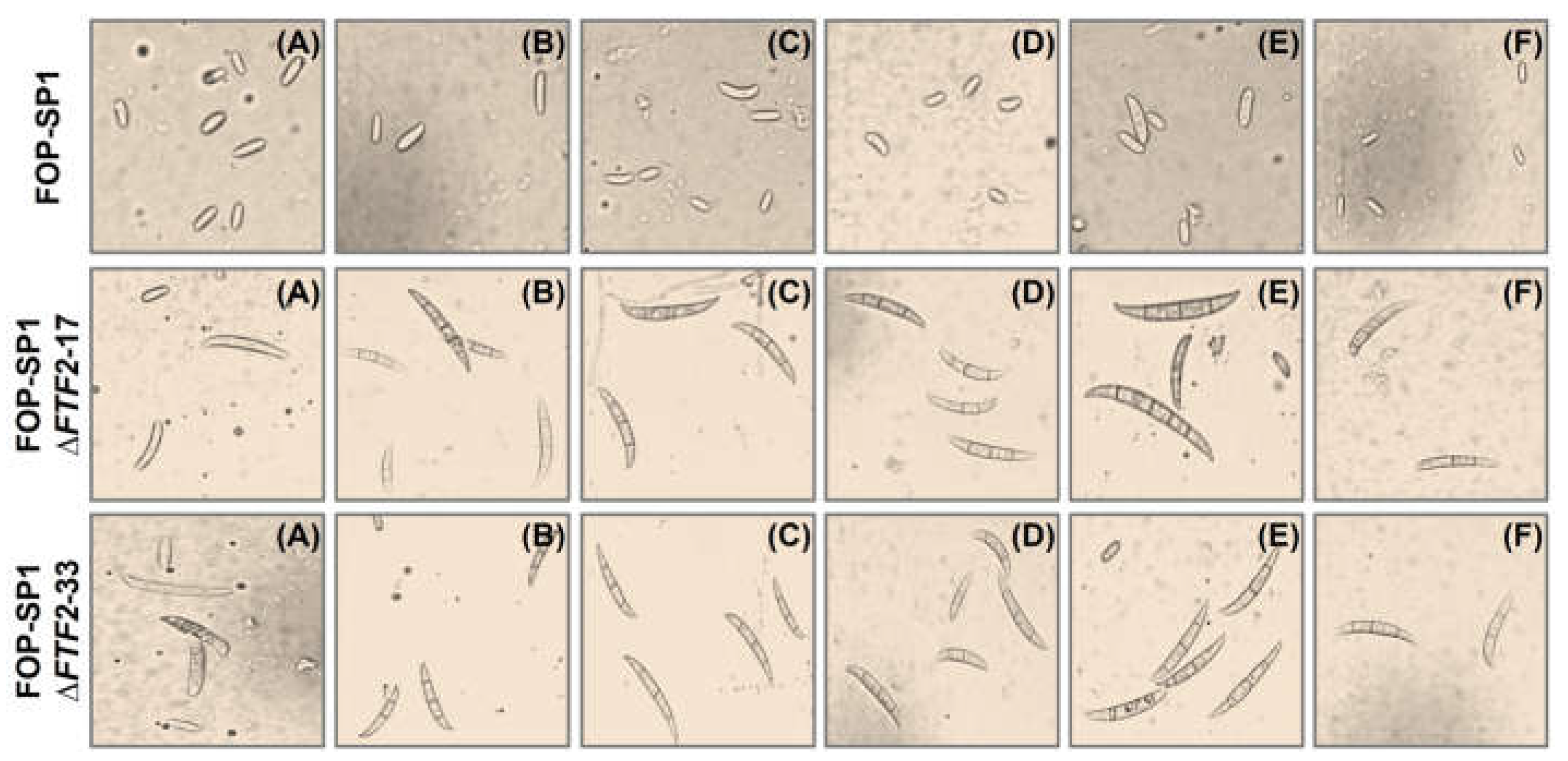
Figure 5.
Common bean colonization by FOP-SP1ΔFTF2 mutant. Sections of plants colonized by FOP-SP1 and a selected FOP-SP1ΔFTF2 mutant were double-stained with WGA Alexa FluorTM 488 and propidium iodide. Confocal laser microscopy was used to visualize the progress of in planta growth. Sections from the root system (A-F) were sliced at 1 (A, D), 2 (B, E) and 3 (C, F) dpi, from the root crown (G-J) at 5 (G, I) and 7 (H, J) dpi, and from hypocotyls (K-N) at 14 (K, M) and 21 (L, N) dpi. Upper images in each panel show plant tissues colonized by FOP-SP1, while bottom images correspond to the FOP-SP1ΔFTF2 mutant.
Figure 5.
Common bean colonization by FOP-SP1ΔFTF2 mutant. Sections of plants colonized by FOP-SP1 and a selected FOP-SP1ΔFTF2 mutant were double-stained with WGA Alexa FluorTM 488 and propidium iodide. Confocal laser microscopy was used to visualize the progress of in planta growth. Sections from the root system (A-F) were sliced at 1 (A, D), 2 (B, E) and 3 (C, F) dpi, from the root crown (G-J) at 5 (G, I) and 7 (H, J) dpi, and from hypocotyls (K-N) at 14 (K, M) and 21 (L, N) dpi. Upper images in each panel show plant tissues colonized by FOP-SP1, while bottom images correspond to the FOP-SP1ΔFTF2 mutant.

Figure 6.
RT-qPCR analysis of expression of common bean genes involved in the defense response (PR1, Pathogenesis Response; ERF, Ethylene Response Factors) in inoculated plants. The plant regions assayed and the time intervals after inoculation are indicated in the X axis (R3, root system 3 dpi; C7, root crown 7 dpi; H21, hypocotyls 21 dpi). The relative expression measurements in the Y axis are indicated in a logarithmic scale. Dark bars of each color indicate gene expression in plants colonized by FOP-SP1; light bars indicate gene expression in plants colonized by FOP-SP1ΔFTF2. The expression ratios were normalized by using the common bean actin gene as endogenous control. The value 1.0 was denoted for the transcript level of all genes in mock inoculated plants for each plant region (data not shown). The levels of expression for a pair of measurements.
Figure 6.
RT-qPCR analysis of expression of common bean genes involved in the defense response (PR1, Pathogenesis Response; ERF, Ethylene Response Factors) in inoculated plants. The plant regions assayed and the time intervals after inoculation are indicated in the X axis (R3, root system 3 dpi; C7, root crown 7 dpi; H21, hypocotyls 21 dpi). The relative expression measurements in the Y axis are indicated in a logarithmic scale. Dark bars of each color indicate gene expression in plants colonized by FOP-SP1; light bars indicate gene expression in plants colonized by FOP-SP1ΔFTF2. The expression ratios were normalized by using the common bean actin gene as endogenous control. The value 1.0 was denoted for the transcript level of all genes in mock inoculated plants for each plant region (data not shown). The levels of expression for a pair of measurements.
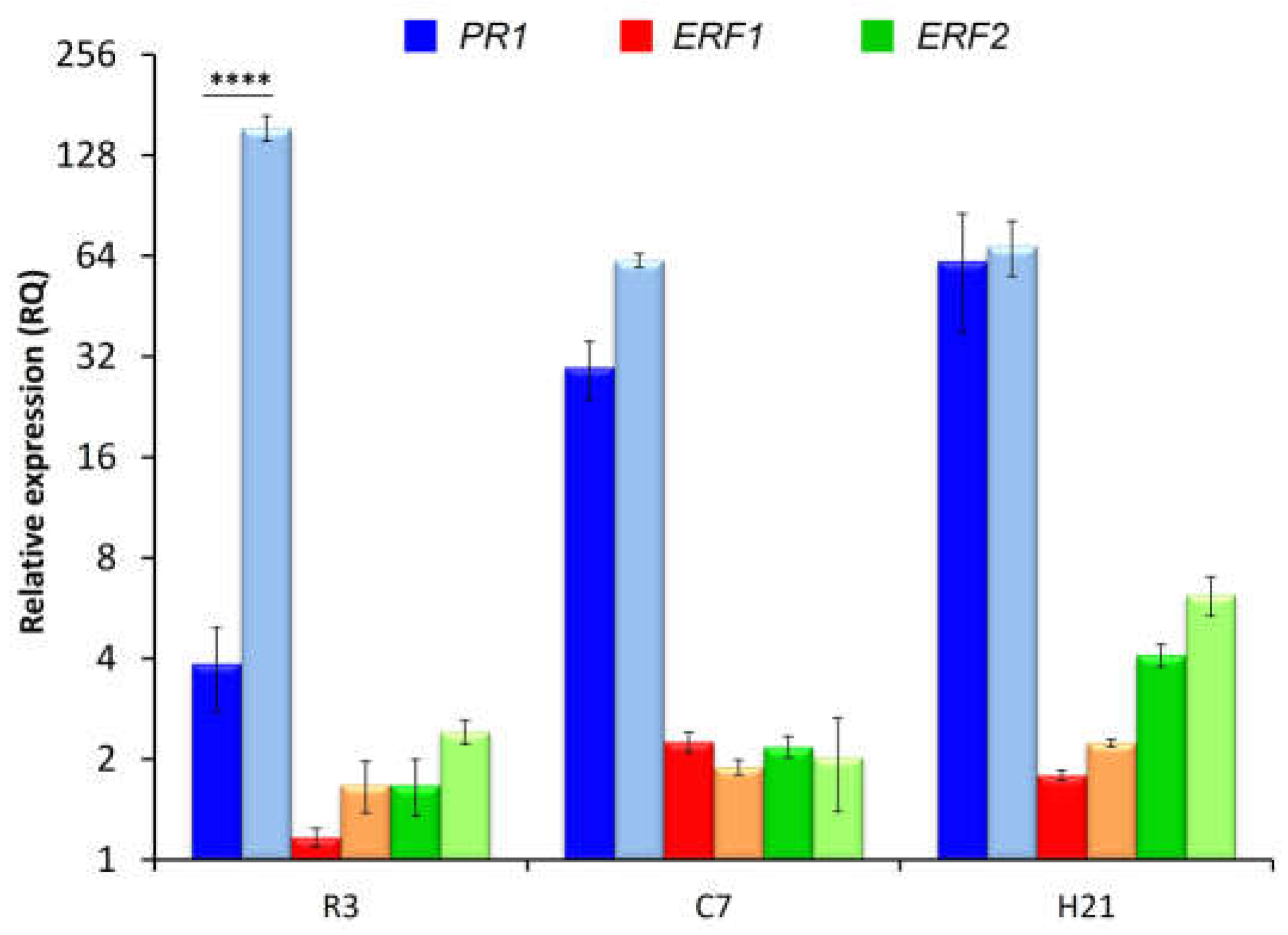
Figure 8.
RT-qPCR analysis of expression of putative FTF2-responsive genes (A) and two SIX effector genes (B) during common bean colonization by a FOP-SP1ΔFTF2 mutant. The plant regions assayed and the time intervals after inoculation are indicated in the X axis (R3, root system 3 dpi; C7, root crown 7 dpi; H21, hypocotyls 21 dpi). The relative expression measurements in the Y axis in (B) are indicated in a logarithmic scale. Dark bars indicate expression in plants colonized by FOP-SP1; light bars indicate expression in plants colonized by FOP-SP1ΔFTF2. The expression ratios were normalized by using the EF1α gene as endogenous control. The value 1.0 was denoted for the transcript level of genes FTF2 and FOXG_02746 in wild-type R3, of genes FOXG_14730 and FOXG_14731 in wild-type C7, and genes SIX1 and SIX6 in wild-type H21. The levels of expression for a pair of measurements were tested using a t-test and significant differences are indicated by * (P < 0.05), ** (P < 0.01), *** (P < 0.001) and **** (P < 0.0001).
Figure 8.
RT-qPCR analysis of expression of putative FTF2-responsive genes (A) and two SIX effector genes (B) during common bean colonization by a FOP-SP1ΔFTF2 mutant. The plant regions assayed and the time intervals after inoculation are indicated in the X axis (R3, root system 3 dpi; C7, root crown 7 dpi; H21, hypocotyls 21 dpi). The relative expression measurements in the Y axis in (B) are indicated in a logarithmic scale. Dark bars indicate expression in plants colonized by FOP-SP1; light bars indicate expression in plants colonized by FOP-SP1ΔFTF2. The expression ratios were normalized by using the EF1α gene as endogenous control. The value 1.0 was denoted for the transcript level of genes FTF2 and FOXG_02746 in wild-type R3, of genes FOXG_14730 and FOXG_14731 in wild-type C7, and genes SIX1 and SIX6 in wild-type H21. The levels of expression for a pair of measurements were tested using a t-test and significant differences are indicated by * (P < 0.05), ** (P < 0.01), *** (P < 0.001) and **** (P < 0.0001).
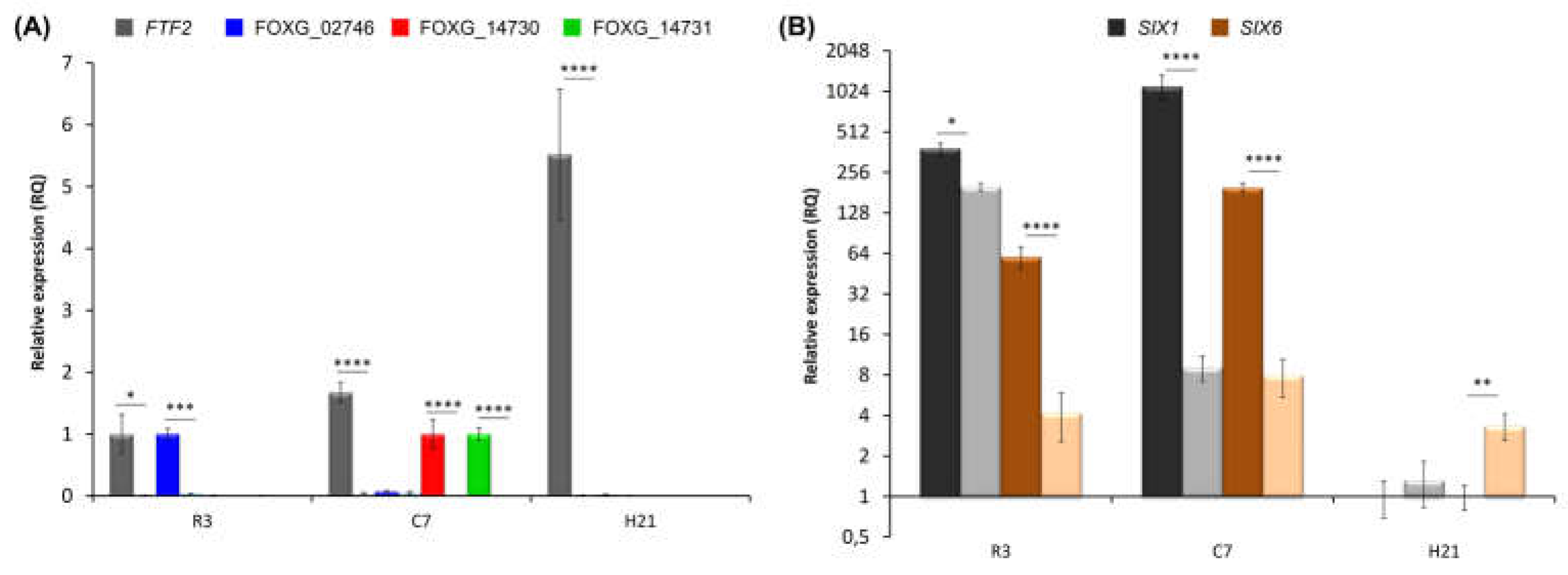
Figure 9.
Hypothetical model of regulation of vascular colonization effectors (VCE) and virulence during host plant colonization by F. oxysporum. VCE: Vascular Colonization Effectors. The yellow to red shade transition represents the buildup of effectors as the result of gene expression activation by the FTF family of transcription factors and the subsequent enhanced virulence.
Figure 9.
Hypothetical model of regulation of vascular colonization effectors (VCE) and virulence during host plant colonization by F. oxysporum. VCE: Vascular Colonization Effectors. The yellow to red shade transition represents the buildup of effectors as the result of gene expression activation by the FTF family of transcription factors and the subsequent enhanced virulence.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



