
Submitted:
12 April 2023
Posted:
13 April 2023
You are already at the latest version
Abstract
Following the central dogma of molecular biology, gene expression heterogeneity can aid in predicting and explaining the wide variety of protein products, functions, and, ultimately, heterogeneity in phenotypes. There is currently overlapping terminology used to describe the types of diversity in gene expression profiles, and overlooking these nuances can misrepresent important biological information. Here, we describe transcriptome diversity as a measure of the heterogeneity in 1) the expression of all genes within a sample or a single gene across samples in a population (gene-level diversity) or 2) the isoform-specific expression of a given gene (isoform-level diversity). We first overview modulators and quantification of transcriptome diversity at the gene level. Then, we discuss the role alternative splicing plays in driving transcript isoform-level diversity and how it can be quantified. Additionally, we overview computational resources for calculating gene-level and isoform-level diversity for high-throughput sequencing data. Finally, we discuss future applications of transcriptome diversity. This review provides a comprehensive overview of how gene expression diversity arises, and how measuring it determines a more complete picture of heterogeneity across proteins, cells, tissues, organisms, and species.
Keywords:
Gene expression
; transcriptome diversity
; transcriptional variation
; transcript diversity
; isoform-level diversity
; gene-level diversity
Introduction
Following the central dogma of molecular biology, gene expression heterogeneity can aid in predicting and explaining the wide variety of protein products, functions, and, ultimately, heterogeneity in phenotypes. Over the past few decades, transcriptomic expression profiles have been assayed in many ways, with the two most common approaches being microarray-based and sequencing-based [1]. More recently, microarrays have been surpassed by Next-Generation Sequencing (NGS), also known as second-generation or short-read sequencing [2,3]. To assay gene expression with NGS technology (also known as RNA sequencing, RNA-Seq), RNA is first reverse-transcribed into cDNA, fragmented, and then constructed into an NGS library that is then read by the sequencer and then computationally mapped to the transcriptome for quantification [3]. The ability to barcode cells in combination with low-input protocols has also enabled single-cell/nuclei RNA-Seq (sc/snRNA-Seq) to measure the transcriptomic profiles of individual cells [4,5]. Most recently, third-generation or long-read sequencing popularized by Pacific Biosciences [6] (PacBio) and Oxford Nanopore Technologies [7,8] (ONT) has allowed the sequencing of contiguous reads of up to 2.3 million bases, considerably longer than the longest human messenger RNA (mRNA) transcripts [9]. These long reads are enabled by single-molecule real-time (SMRT) and ionic nanopore technology innovations by PacBio and ONT, respectively. In addition, long-read RNA-Seq (lrRNA-Seq) is capable of directly sequencing RNA and detecting its modifications, which was previously not possible with short-read technologies that require reverse transcription into cDNA [10]. Additionally, more technologies are being developed for the rapidly growing field of long-read sequencing at the single-cell level [11,12,13].
As both these and newer technologies continue to evolve with the goal of measuring transcriptomic profiles more precisely, the need to quantify and interpret those profiles continues to grow [11,12,13]. Dating back to microarray experiments, differential expression (DE) is the most common analysis of gene expression, and it is frequently used in both bulk RNA-Seq and scRNA-Seq. Generally, a basic mean DE analysis determines whether individual genes are up- or down-regulated (i.e., more highly or lowly expressed, respectively) between conditions, for example, across tissues [14,15] or disease states [16,17]. There are two main kinds of differential analysis, differential variability [18] and differential mean, with the differential mean being the most common [19]. Popular R packages for differential mean expression include DESeq2 [20], EdgeR [21], and limma [22]. DE analysis is typically done at the gene level, collapsing all counts from a sequencing library that map to a single gene unit even though many genes undergo alternative splicing (AS) to produce several different mRNA molecules or isoforms. Alternatively, DE can also be examined at the isoform level by comparing reads mapped to each specific transcript as independent entities with the aforementioned [20,21,22] or other DE packages. Some caveats of DE are that this analysis alone may miss biological complexity and information [23], that DE genes are often not causal (i.e., are disease-induced [24]), or that the function of causal genes changes but mRNA expression levels remain unchanged [25], and that interpretation must also account for biases associated with the expression level or abundance of reads for a particular gene [25]. These caveats are also important considerations when performing DE analysis in scRNA-Seq as well [26].
An additional consideration for analyzing transcriptomic profiles is the need to quantify the complexity of biological systems because nothing in biology acts in isolation. To illustrate, a perturbed gene will likely have perturbed interactions with other genes and proteins in that biological system, which in turn may also contribute to phenotypic differences. There are different ways to assess these coordinated patterns of gene expression, such as grouping genes together by pathway or function in pathway analyses (e.g., KEGG [27] and GO [28]) to determine which pathways are up- or down-regulated between conditions. While this analytical approach considers genes in aggregate by function and interactions, it does not include all of their known or predicted interactions and genes can be part of multiple pathways, which confounds the analysis. Representing gene expression profiles as biological networks presents an alternative or complementary approach to differential gene expression analysis, and such networks have been shown to be dysregulated in disease [29]. These network biology approaches allow researchers to incorporate additional information to combine multiple information types and improve in silico-predicted interactions within a condition [30].
However, the above approaches fail to describe how expression patterns change between conditions. Transcriptome diversity quantifies gene expression changes at 1) the gene level: the total expression of all genes within a sample or a gene across samples in a population, and 2) the isoform level: the isoform-specific expression of a given gene (Figure 1). For example, such approaches have identified unique disease-related genes across 16 human disease data sets compared to differential expression alone [23]. Here we review the causes and measurement of transcriptome diversity across samples, genes, and isoforms in protein-coding genes. While diversity also occurs in non-coding transcripts, that expression does not lie within the scope of this review. Previous literature has primarily focused either on diversity at the gene level [31] or the isoform level [32]. Here, we review both kinds of diversity found in transcriptomes and delineate some boundaries for how to describe and quantify this diversity. Therefore, we first overview modulators and quantifications of transcriptome diversity at the gene level. Then, we discuss the role alternative splicing plays in driving transcript isoform-level diversity and how it can be quantified. Additionally, we overview computational resources for calculating gene-level and isoform-level diversity for high-throughput sequencing data. Finally, we discuss future applications of transcriptome diversity.
I. Gene-level Diversity in Gene Expression Profiles
A. Biological Processes that Lead to Gene-level Diversity
As gene expression analyses have become a critical tool for furthering phenotypic, mechanistic, and evolutionary interpretation, it is vital to understand the forces guiding gene expression heterogeneity [33]. Like other biological processes, inherent biological noise in gene expression has been observed ubiquitously across species [34]. This stochasticity is driven by many processes within the cell, including transcription/translation initiation and mRNA/protein degradation [34]. In extreme cases, this stochastic gene expression noise has been shown to reduce fitness in yeast cells [35]. However, previous studies have noted that genetic and environmental factors are the two main drivers of biological heterogeneity in gene expression [36]. However, additional intrinsic factors like cell cycle [37], circadian rhythm [38], and aging [39,40] (which are also influenced by genetic and environmental factors) also contribute to gene expression heterogeneity.
Promoters, enhancers, and transcription factors are key genetic features contributing to gene expression heterogeneity observed across species, tissues, and cell types [41,42]. The heavily studied RNA polymerase II core promoter directly regulates gene expression [43,44], and natural variations in promoter regions are linked directly to both gene expression and phenotype heterogeneity [45]. By regulating transcription levels distally, enhancers also influence gene expression heterogeneity within specific cell types, tissues, and even species [46,47]. Similar to promoters, alteration in an enhancer region can lead to phenotypic changes by impacting gene expression [48]. Transcription factors are essential regulatory proteins that drive gene expression by interacting with DNA sequences like promoters and enhancers to control transcriptional processes [49,50]. Studies like the Encyclopedia of DNA Elements (ENCODE) project, which integrated over 450 experiments of 119 transcription factors, have demonstrated that transcription factors have dynamic regulatory networks that lead to measurable heterogeneity in homeostatic gene expression [51].
Additionally, epigenetic processes including DNA methylation, histone modifications, and other environmental or stress responses, can also drive gene expression heterogeneity. DNA methylation, notably mammalian m5C (methyl groups at the 5’ cytosine of a C-G dinucleotide) [52], regulates gene expression in multiple ways [53], including through transcription factor binding, the functionality of enhancers, insulator elements, and promoters, and by altering chromatin conformation [54]. Various studies have noted correlations between gene expression and DNA methylation, further supporting its role as a possible driver for gene expression heterogeneity. Post-translational histone modifications (e.g., acetylation, methylation, phosphorylation, or ubiquitination) are also known to be correlated with gene expression [55,56,57] and can even be used to predict gene expression [56]. Environmental and stress-related effects, like hypoxia, can also impact the heterogeneity of gene expression. Many organismal studies have observed the impact of stress on producing a biological response and subsequent regulation of various genes to alleviate environmental damages (e.g., in oxidative stress) [58,59,60].
B. Methods for Quantifying Gene-level Transcriptome Diversity
Researchers have applied different approaches to empirically calculate gene expression heterogeneity for both bulk and single-cell/nuclei transcriptome profiles, including coefficient of variation (CV) [23], variance [61], and others [62]. While the gene expression terms variation and diversity both describe changes in gene expression across samples, variation/variability are more frequently associated with measures of dispersion (e.g., CV, variance), and diversity is more commonly associated with these probability-based measures, particularly Shannon or information entropy (Figure 2A). In fact, the application of CV and variance to gene expression profiling analysis is sometimes known as expression variance (EV) [62]. Originally described by Alemu et al., EV showed tissue-specific variation across gene expression profiles [63] and was later used to show expression variation associated with aging and methylation [64].
Standard deviation describes the dispersion of the data in relation to its mean. Building on standard deviation, CV considers the standard deviation of the gene expression sample divided by its mean and thus is a standardized measure [23]. Therefore CV can be used to compare across conditions or datasets to identify disease-associated genes that are not identified by DE alone [23]. Additionally, other studies have applied both technical CV and biological CV (BCV) to describe RNA-Seq gene expression variation associated with technical or biological variables, respectively, as well as [21] normalized CV to examine gene expression variation, for example, across neurological diseases [61]. Recent studies have also used CV to understand how gene expression variability among therapeutic targets determines drug effectiveness and safety, thus improving therapeutic development methodologies [65]. Another empirical measurement of gene expression is variance. In the Mar et al. study, variance measures the significance of the mean difference between groups by using a t-test or ANOVA [61], but the term has also been used synonymously with gene expression variability [61,64,66,67]. For example, Bachtiary et al. applied variance (here defined as standard deviation squared) to measure the variation of expression between and within cervical cancer patient samples [68]. Gene expression variance has also been studied in human populations, where functional connections between low-variance genes and fundamental cell processes and high-variance genes with immune processes suggest that variance is biologically meaningful and not merely reflective of stochastic noise [69].
Though CV and variance are some of the most common methods for empirically calculating variation, there are a few other ways of describing variation across gene expression. For example, differential variability analysis can also be performed with Bartlett's, Levene's, median absolute deviation (MAD), or Fligner–Killeen tests, yet the R package MDSeq based on reparameterization of the real-valued negative binomial, which was shown to outperform these methods [19]. On the other hand, the range of gene expression observed is one of the simplest measures of variability. Though generally not used in its simplest form (i.e., maximum value minus minimum value), a modified version of range has been used. For example, dynamic range, the log10 ratio between the maximum and minimum normalized gene expression counts, has been used to compare the expression of orthologous genes between humans and mice to determine genes constrained throughout early vertebrate evolution [70] as well as to describe gene expression variation patterns across organs and tissues [71]. Additionally, researchers have developed a metric based on a ratio of the percentage of reads covering a proportion of the genome to quantify gene expression variation [72]. When a large percentage of reads covers a smaller number of total genes in the genome, it indicates lower variability in that condition than when the percentage of reads spans over a larger set of genes in another condition. However, these metrics are biased towards longer genes if gene size is not properly accounted for during analysis.
In 1948, Shannon defined entropy as the probability of uncertainty of an outcome or the amount of choice in the outcome based on how much information [73]. The basis of Information Theory, Shannon entropy, is the log of the event probability so that an event with full certainty or a probability of one would have no surprise. Over the years, Shannon entropy has been applied to numerous biological processes, including gene expression [74]. When using Shannon entropy in this context, gene expression measurements for a specific gene are the information used to measure uncertainty, or as we describe it, diversity [75] (Figure 2). Previous studies have employed Shannon entropy to study diversity in drug targets [76], tissue-specificity [77], species-specificity [75], and even intraspecies genomic DNA information [78]. When used to compare gene expression in RNA-Seq data, differential Shannon entropy, compared to differential CV and DE, identified genes overlapping with CV-identified genes but also included unique disease-associated genes [23], underlining that Shannon entropy can identify biological signals that CV and DE do not. Shannon entropy has also been used in combination with WGCNA analyses by calculating entropy from the betweenness of networks [79]. Additionally, studies using adaptations of Shannon entropy, such as Tsallis entropy (also known as HCDT entropy), have divided gene-level diversity into two categories: alpha and beta diversity [80], where alpha diversity represents the diversity of a single profile, and beta diversity represents diversity between samples within a group. This particular example of Tsallis entropy allows a researcher to able to manipulate a parameter (q) that can adjust the weight of highly-expressed genes [80], therefore giving a higher degree of control and leaving room for interpreting more biologically- relevant information at different levels of q. The introduction of alpha and beta diversity nomenclature is an eloquent way to describe the diversity shown in Figure 1A, with alpha diversity representing diversity across genes or transcripts within a sample and beta diversity being the two-dimensional diversity across all samples in a group or population, though this nomenclature is not yet widely used. Example analytical packages that apply entropy and variation in the context of gene expression diversity are described in Table 1, although many of these analyses are performed without specialized software.
Altogether, the aforementioned gene expression studies demonstrate not only the importance of further understanding the drivers of this gene expression diversity but also the importance of developing new and comprehensive ways to quantify this diversity through various methodologies. Quantifying gene-level transcriptome diversity is a salient part of ascertaining how biological processes lead to phenotypic manifestations, including in a disease context. Therefore, it is imperative to examine other sources of diversity, such as heterogeneity in mRNA transcripts due to AS.
II. Isoform-level Diversity in Gene Expression Profiles
A. Biological Processes that Lead to Isoform-level Diversity
Before the start of the human genome project, the human genome was expected to have approximately 100,000 genes [84] based on the approximated number of protein products. However, after completing the project, the human genome actually had between 20,000 and 25,000 genes, much less than projected [85]. While humans may not have more genes than all other organisms, their splicing patterns are more specific and complex [86]. Compared to other eukaryotic organisms, humans have the highest relative splicing abundance, and this abundance steadily decreases for species with a larger evolutionary divergence from humans [86]. Because 94% of human genes undergo AS [87], most genes have a variety of transcript isoforms that, in many cases, result in proteins with unique functions, therefore increasing protein diversity. There are between six and eight types of AS events depending on the classification used, and their abundance varies by species, with exon skipping being the most common in animals and intron retention more common in plants and fungi [88]. In addition to RNA splicing, differences in transcript usage in organisms can also be driven by alternative promoter usage (e.g., producing different transcripts of the brain-derived neurotrophic factor (BDNF) gene [89]) or 3′ end usage [90]. In this section, we will focus on the heterogeneity driven by RNA splicing.
RNA splicing occurs as part of the process to produce mature mRNA from pre-RNA and was first described in 1977 [91]. RNA splicing happens in virtually all multi-exonic genes through either constitutive splicing (splicing out an intron that is always excluded from a final transcript) or AS (variably splicing out alternative exons and/or introns resulting in diverse mRNA sequences). Exons that are always incorporated in the final or mature transcript are described as constitutive exons, while alternatively-spliced exons are those that vary in usage from transcript to transcript [92]. This AS process is performed by the spliceosome, which contains approximately 170 proteins [93], including many RNA-protein complexes called small nuclear ribonucleoproteins (snRNPs), and recognizes splice sites to facilitate the transesterification reactions that lead to intron removal (further reviewed in [94,95], [32]). There is currently a debate on how much of this splicing is functional or controlled because not all alternative transcripts are protein-coding, AS changes can be driven stochastically [96], and AS transcripts can be sensitive to nonsense-mediated decay. While not all AS transcripts are functional, ribosomal profiling experiments indicate that over 75% of medium-to highly expressed AS transcripts are bound to ribosomes and translated into proteins [97] so it is still highly likely that many of these are made into proteins, as further underscoring AS is biologically relevant.
A key property of AS is its high specialization to a given biological condition. For example, AS is species-specific [86], and as organisms gain more evolutionarily complexity, their AS patterns become more similar to humans. AS is also sex-specific and can lead to sex-specific traits. For example, in fruit flies (Drosophila), the sex splicing gene doublesex (dsx) controls sexual differentiation [98] and is regulated by AS. In addition to more extensive sex-specific splicing in fruit flies [99,100,101], sex-specific splicing has also been shown in fish [102], birds [103], non-human primates [104], and humans [105,106]. Moreover, AS is critical in developmental changes, particularly as coordinated AS changes help define tissue identity [32]. In fact, AS is tissue-specific [107], driven at least in part by tissue-specific splicing factors [108]. These tissue-specific splicing factors govern complex splicing regulatory networks [109] that can influence protein interaction networks and thereby increase the functional diversity of proteins [110]. Further, AS is also highly cell-type specific [111], and the recent increase in single-cell studies has highlighted an increasing number of cases of AS that are cell-type and even cell-subtype specific [112]. This has been particularly well-documented in the brain during neuronal differentiation [113] (e.g., the cerebral cortex [111]) and in immune cells [114].
Changes in AS are also associated with many diseases [115,116]. Currently, an estimated 15% of human disease-causing point mutations result in an AS defect [117], and many diseases and disorders are associated with disrupted splicing patterns, like spinal muscular atrophy, cancer, and autism spectrum disorder (ASD) [118,119]. Because of all of these known changes in AS across numerous biological conditions, some pathogenic and others benign, it is critical for genomic researchers to quantify changes in AS using RNA-Seq. Numerous methodological approaches with software implementations for analyzing and quantifying isoform-level heterogeneity (including specialized approaches for single-cell or long-read data) are discussed in further detail in the next section.
B. Methods for Quantifying Isoform-level Diversity
Measuring alternatively-spliced transcript expression diversity requires first identifying and then quantifying transcripts from RNA-Seq data. One way to quantify alternatively spliced transcripts from gene expression data relies on identifying reads that cover splice junctions, the genomic loci where two exons have been spliced together [120]. This process varies depending on the transcript quantification tool, as some tools only count if an exon is included at all (i.e., if any reads map to that exon), while others search for junctions to determine if an exon is spliced in, because reads mapping to a free exon (i.e., not including a junction) cannot resolve where in the transcript that exon has been spliced. A major limitation is that there must be sufficient read depth to detect all splice junctions from short-read data [121]. In some cases, junctions are specific to unique transcripts, so a read mapping to a unique junction could indicate that that transcript is being expressed without having any continuous reads capturing the entire transcript. One of these splice-junction-based methods is Splice Expression Variation Analysis (SEVA) [122], which compares the variability of the multivariate distribution of splice junction expression profiles between conditions. On the other hand, because short reads with few junctions usually do not match a unique transcript, probabilistic methods can be used to estimate exon inclusion [123] (also known as percent spliced in, PSI) [124], greatly reducing the precision of transcript quantification.
There are different terminologies for the number of concepts and analyses that fall under the umbrella of isoform-level diversity. Differential transcript (or isoform) expression (DTE) is a data analysis method similar to differential gene expression as it identifies up- or down-expression of specific transcripts in one condition versus another. However, differential transcript usage (DTU), sometimes referred to as differential isoform usage, isoform switching, or differential splicing [125], can determine differential proportions of transcript expression within a gene across conditions [126]. Soneson et al. describe three methods for DTU: assembly-based, type of AS-based, and differential exon usage [126]. Due to limitations with short reads not covering entire transcripts that overlap, differential exon usage (DEU) can also be used in a similar way to measure shifts in functional unit expression (i.e., bins) across conditions, usually comparing PSI values [126]. Table 2 includes analysis packages comparing transcript expression across conditions and used for DTU, DEU (PSI), or other analyses.
However, short-read sequencing technology often fails to adequately resolve transcripts because the typical mRNA is over 1kb, whereas most short-read RNA-Seq data is only 100-200 bases in length [3], i.e., only long enough to cover an exon or less. The advent of long-read technologies has created an opportunity to capture more detailed gene expression profiles, especially for resolving transcript expression, but also for accurate sequencing and subsequent mapping of repetitive, hard-to-map, and/or duplicated gene regions [127]. Additionally, as lrRNA-Seq approaches can sequence full-length novel transcript isoforms, they are continuing to identify novel transcripts, including those that are lowly expressed [128]. While most of the short-read tools for transcript quantification can be used on long-read data, there are transcript quantification tools that are specialized for long-read sequencing data, like FLAIR [129] and BAMBU [130], which include steps for correcting misalignments that can result from less accurate reads. Applying variance and entropy-based diversity quantification approaches in combination with these lrRNA-Seq technologies, therefore captures transcriptomic changes across biological conditions and phenotypes.
Additionally, isoform-level diversity can be described by enumerating the total number of isoforms [131,132], herein called isoform number diversity. In contrast to counting the number of transcripts, the distribution of isoform expression for a gene, such as in DTU, can also be considered isoform-level diversity or isoform usage. Similarly to the gene expression level, variance and Shannon entropy can be used to describe this isoform-level diversity (Figure 2) [133]. One way to measure isoform-level diversity is the Fano factor, or the squared variance over the mean [134], which describes the distribution of alternatively spliced transcripts while adjusting for the mean expression of that gene. Another method is by Shannon entropy, where a gene with many isoforms could be less diverse than a gene with few isoforms if the latter has a more even distribution of expression and perhaps equal usage of those gene products. Figure 2B provides an example of isoform-level diversity quantified with Shannon entropy.
The first instance of using Shannon entropy to describe diversity in three types of alternative transcription (AS, polyadenylation, and transcription initiation) using targeted microarray expression data was by Ritchie et al. in 2008 [133]. Ritchie et al.'s rationale was that Shannon entropy could capture aberrant transcription seen in cancer and was therefore used to compare patient cancerous tissue transcriptomic profiles with non-cancerous tissue transcriptomic profiles. The authors found that out of the three types of transcription studied, only AS had increased diversity in cancer tissues. They concluded that these changes in entropy are unlikely to reflect changes in gene function because they found it unlikely that, in a cancer context, shifts in isoform expression are functional or controlled. This general approach to measuring entropy has also been used to compare transcript diversity across conditions in the brain [127] and epithelial cells [135].
Several software approaches have been developed for comparing isoform-level diversity with Shannon entropy, including Cuffdiff (from Cufflinks) [136,137,138], Whippet [139], and SplicingFactory [140]. Cuffdiff uses Jensen-Shannon divergence, which, like Shannon entropy, relies on probability to compare the distribution of transcript expression across conditions [138]. Whippet [139] applies Shannon entropy to define the entropy of individual AS events instead of at the gene level, meaning that each alternatively spliced exon is given a value based on PSI. SplicingFactory [140] is unique because it also includes multiple other methods for assaying diversity across isoforms of the same gene, like the Gini Index (originally developed for describing wealth inequalities) and the Simpson Index (originally developed to measure ecological diversity). The aforementioned Tsallis entropy could also potentially be used in an isoform context to describe biological heterogeneity since it has been shown to provide more information than Shannon entropy or Simpson Index alone though it has not yet been applied to study alternatively-spliced isoform distributions. While some approaches have proved more popular than others, there is no standout or one best method to examine variability and diversity across isoforms as of yet. This means that researchers still need to think deeply about what analyses they do want to perform based on their questions of interest.

Table 2.
Software packages that detect isoform-level diversity and variability in exon and isoform usage. This table includes the name of the software package, the year published, the type of data it can be used with, and the analysis type. Note: As terminology used by authors to describe a particular method varies, the analysis type listed in the table is standardized according to the defined terminology in this review. Entries are sorted from most to least recent. DEU - differential exon usage, PSI - percent spliced in, DTU - differential transcript usage. Citation counts are from Pubmed, March, 2023.
Table 2.
Software packages that detect isoform-level diversity and variability in exon and isoform usage. This table includes the name of the software package, the year published, the type of data it can be used with, and the analysis type. Note: As terminology used by authors to describe a particular method varies, the analysis type listed in the table is standardized according to the defined terminology in this review. Entries are sorted from most to least recent. DEU - differential exon usage, PSI - percent spliced in, DTU - differential transcript usage. Citation counts are from Pubmed, March, 2023.
| Package Name | Year | Bulk or Single Cell | Analysis Type: Exon/Transcript or Other | Citation count | Language |
| Insplico [141] | 2023 | Both | Other - Splicing Order | 0 | Perl |
| acorde [142] | 2022 | Single-cell | DTU and coDTU | 2 | R |
| SpliZ [143] | 2022 | Single-cell | DEU (PSI) | 4 | Python |
| DTUrtle [144] | 2021 | Both | DTU | 3 | R |
| NanoCount [145] | 2021 | Bulk | DTU | 11 | R |
| SplicingFactory [140] | 2021 | Bulk | Other - Diversity | 0 | R |
| scisorseqr [146] | 2021 | Single-cell | DTU (modified) | 39 | R |
| satuRn [147] | 2021 | Both | DTU | 0 | R |
| ASCOT [112] | 2020 | Single-cell | DEU (PSI) | 24 | Python |
| BANDITS [148] | 2020 | Bulk | DTU | 10 | R |
| Sierra [149] | 2020 | Single-cell | DTU | 28 | R |
| RATs [150] | 2019 | Bulk | DTU | 10 | R |
| SUPPA2 [151] | 2018 | Bulk | DEU (PSI) | 193 | Python |
| LeafCutter [152] | 2018 | Bulk | Other - Intron Excision | 246 | R/Python |
| Whippet [139] | 2018 | Bulk | DTU | 61 | Julia |
| GSReg/SEVA [122] | 2018 | Bulk | Other - Variability | 6 | R |
| IsoformSwitchAnalyzeR [153] | 2017 | Bulk | DTU | 104 | R |
| Census/Monocle [154] | 2017 | Single-cell | DEU (PSI) | 610 | R |
| BRIE [155] | 2017 | Single-cell | DEU (PSI) | 50 | Python |
| DRIM-Seq [156] | 2016 | Bulk | DTU | 49 | R |
| JunctionSeq [157] | 2016 | Bulk | DEU (PSI) | 81 | R |
| MAJIQ [158] | 2016 | Bulk | DEU (PSI) | 188 | Python/C++ |
| SGSeq [159] | 2016 | Bulk | DEU (PSI) | 63 | R |
| SingleSplice [160] | 2016 | Single-cell | DTU | 36 | R/Perl |
| Limma (diffSplice) [22] | 2015 | Bulk | DEU (PSI) | 15473 | R |
| VAST-TOOLS [161] | 2014 | Bulk | DTU | 339 | R/Perl |
| rMATS [162] | 2014 | Bulk | DEU (PSI) | 982 | Python/C++ |
| CuffDiff2 [163] | 2013 | Bulk | DEU (PSI) | 2341 | C++ |
| SplicingCompass [164] | 2013 | Bulk | DTU | 39 | R |
| DEXSeq [165] | 2012 | Bulk | DEU (PSI) | 874 | R |
| SpliceTrap [123] | 2011 | Bulk | DEU (PSI) | 59 | C++/Perl |
| MISO [166] | 2010 | Bulk | DEU (PSI) | 876 | Python/C |
To summarize, AS contributes to the gene expression diversity observed by increasing the number of products that can be produced by a single gene. Quantifying the isoform-level diversity of a given gene will identify not only which isoforms are highly expressed but how isoform expression shifts between conditions. With the increasing popularity of more accurate sequencing and long-read transcriptomics, more attention should be spent on DTU and looking for the functional relevance of these differentially used or diverse transcripts.
Conclusion
The types of data most compatible with the analysis discussed in this review are primarily high-throughput RNA-Seq, long-read RNA-Seq, and scRNA-Seq. However, not all measured gene expression profile heterogeneity is due to the condition being studied but may be due to technical (i.e., sample processing and sequencing preparation, such as sequencing depth) or biological (i.e., gene differing RNA synthesis and degradation rates) artifacts. Sequencing depth is particularly important because sequencing depth directly impacts the ability to confidently detect more lowly expressed genes or isoforms. At the isoform level, if a researcher is interested in all the isoforms of a lowly-expressed gene, they would want to sequence an experiment at a higher depth to ensure they are capturing as many different isoforms as possible. Likewise, sequencing length impacts confidence in full-length isoform detection, and full-length scRNA-Seq protocols allow for the detection of splice variants. Therefore, data normalization is critical. For example, normalization approaches based on sequencing depth (such as TPM) are widely used for comparing across groups in bulk expression data, because without normalization there may be scenarios where a gene is expressed in equal proportions, but the condition with more reads overall may appear to have higher gene expression than the other condition. While normalization is a critical step, there are many nuances to comparing sequencing results across conditions and samples because there are many factors that can influence TPM values such as protocol, tissue type, RNA strandedness, and RNA compartmentalization [167]. For more information on these limitations, we refer readers to the following references [167,168].
Additional considerations are needed for sc/snRNA-Seq data because of its well-documented data sparsity [169], though the degree of sparsity varies by platform. Compared to bulk RNA-Seq approaches, TPM is typically not used for most scRNA-Seq libraries due to scRNA-Seq specific variation (e.g., technical dropouts), and methods such as counts per million (CPM), high-count filtering CPM, and others (e.g., scran [170]) are alternatively used, for more information please refer to [171]. When measuring diversity, taking into account different cell type population proportions is key because larger cell clusters may appear to have greater transcriptomic diversity based on more sequencing reads. Also, when measuring variability across sample populations technical batch effects should always be examined and potentially corrected or minimized, to focus on biologically relevant signals for the study. When designing scRNA-Seq data generation experiments, it is ideal to plan for the kinds of analyses being performed beforehand and ensure the data generated is powered for specific hypotheses. One example of this is enriching for rare cell types to ensure there are enough of those cells for analyses like differential expression. Further, due to data sparsity in scRNA-Seq data, isoform-resolution gene expression requires increased read depth to capture isoforms of interest. Moreover, most scRNA-Seq protocols exhibit preparation-specific read bias that should be accounted for when integrating across protocols because this bias can make it difficult to unambiguously align reads and distinguish between isoforms [172].
In conclusion, measuring transcriptome diversity when analyzing gene expression profiles is an integral analysis step to capturing biological information in tandem with the differential expression of individual genes and this literature review underscores the primary axes of heterogeneity that exist and can be measured in transcriptomic data. Having terminology clarified to better navigate this heterogeneity will enable researchers to simultaneously explore transcriptome diversity at the gene and isoform level, and to better interpret and articulate their findings. Gene expression is a critical facet of complex living systems, and research can extract additional information with current computational methods by investigating transcriptome diversity. Critically, omitting gene-level or isoform-level diversity from gene expression analysis could miss biological information since phenotypic diversity is partially driven by gene expression fluctuations and AS [88]. Combining both applications of diversity to better understand high-dimensional gene expression data can provide insight into the transcriptional differences between contexts and reveal gene expression differences that traditional DE analyses cannot like novel therapeutic targets [23]. Therefore, we recommend examining and comparing variability, diversity, and differential expression at the gene and isoform level, when possible, to capture more of this complexity.
Because of the massive amounts of transcriptomic data being generated, there is an unprecedented opportunity for discovery. Increased sequencing depth, longer reads, and more cells/samples will further facilitate transcriptomic diversity studies. Here, we provided an overview of the drivers of gene expression variability and diversity and described how it has been quantified across genes and at the isoform level. Additionally, we summarize resources for calculating diversity and variability (Table 1 and Table 2). Applying additional variation and diversity measurements in transcriptomic analysis has the potential to capture additional gene expression profile changes between conditions and, in the future, could be adapted to additional gene expression profile analyses, such as in spatial transcriptomics, network biology, personalized medicine, and drug repositioning. Incorporation of these transcriptomic data with other data modalities (such as epigenetics, metabolomics, or proteomics) may further increase the ability to make functional inferences. Clinically, these multimodal meta-analyses may have the potential to also elucidate new therapeutic targets for hard-to-treat disorders, further underscoring the importance of transcriptome diversity.
Acknowledgments
We thank Tabea Soelter for her thoughtful feedback on this manuscript. All figures were created using BioRender.com.
References
- Mantione KJ, Kream RM, Kuzelova H, et al. Comparing bioinformatic gene expression profiling methods: microarray and RNA-Seq. Med. Sci. Monit. Basic Res. 2014, 20, 138–142. [CrossRef]
- Niedringhaus TP, Milanova D, Kerby MB, et al. Landscape of next-generation sequencing technologies. Anal. Chem. 2011, 83, 4327–4341. [CrossRef] [PubMed]
- Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [CrossRef] [PubMed]
- Haque A, Engel J, Teichmann SA, et al. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75.
- Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [CrossRef]
- Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [CrossRef] [PubMed]
- Branton D, Deamer DW, Marziali A, et al. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 2008, 26, 1146–1153. [CrossRef]
- Garalde DR, Snell EA, Jachimowicz D, et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [CrossRef]
- Amarasinghe SL, Su S, Dong X, et al. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30.
- Parker MT, Knop K, Sherwood AV, et al. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. Elife 2020, 9.
- Tian L, Jabbari JS, Thijssen R, et al. Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing. Genome Biol. 2021, 22, 1–24.
- Singh M, Al-Eryani G, Carswell S, et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat. Commun. 2019, 10, 3120. [CrossRef] [PubMed]
- Hardwick SA, Hu W, Joglekar A, et al. Single-nuclei isoform RNA sequencing unlocks barcoded exon connectivity in frozen brain tissue. Nat. Biotechnol. 2022, 1–11.
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [CrossRef] [PubMed]
- Melé M, Ferreira PG, Reverter F, et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [CrossRef]
- Manczak M, Park BS, Jung Y, et al. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004, 5, 147–162. [CrossRef] [PubMed]
- Brown J 3rd, Theisler C, Silberman S, et al. Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J. Biol. Chem. 2004, 279, 34674–34681. [CrossRef]
- Ho JWK, Stefani M, dos Remedios CG, et al. Differential variability analysis of gene expression and its application to human diseases. Bioinformatics 2008, 24, i390–8. [CrossRef]
- Ran D, Daye ZJ. Gene expression variability and the analysis of large-scale RNA-seq studies with the MDSeq. Nucleic Acids Res. 2017, 45, e127. [CrossRef]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550.
- McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [CrossRef]
- Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015, 43, e47. [CrossRef] [PubMed]
- Wang K, Phillips CA, Rogers GL, et al. Differential Shannon entropy and differential coefficient of variation: alternatives and augmentations to differential expression in the search for disease-related genes. Int. J. Comput. Biol. Drug Des. 2014, 7, 183–194. [CrossRef] [PubMed]
- Porcu E, Sadler MC, Lepik K, et al. Differentially expressed genes reflect disease-induced rather than disease-causing changes in the transcriptome. Nat. Commun. 2021, 12, 5647. [CrossRef] [PubMed]
- Hudson NJ, Dalrymple BP, Reverter A. Beyond differential expression: the quest for causal mutations and effector molecules. BMC Genomics 2012, 13, 356.
- Squair JW, Gautier M, Kathe C, et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021, 12, 5692. [CrossRef]
- Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–62. [CrossRef]
- Gene Ontology Consortium. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 2021, 49, D325–D334. [CrossRef] [PubMed]
- de la Fuente, A. From ‘differential expression’ to ‘differential networking’ – identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010, 26, 326–333. [Google Scholar] [CrossRef]
- Glass K, Huttenhower C, Quackenbush J, et al. Passing messages between biological networks to refine predicted interactions. PLoS One 2013, 8, e64832.
- de Jong TV, Moshkin YM, Guryev V. Gene expression variability: the other dimension in transcriptome analysis. Physiol. Genomics 2019, 51, 145–158. [CrossRef] [PubMed]
- Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [CrossRef]
- Storey JD, Madeoy J, Strout JL, et al. Gene-expression variation within and among human populations. Am. J. Hum. Genet. 2007, 80, 502–509. [CrossRef] [PubMed]
- Raser JM, O’Shea EK. Noise in gene expression: origins, consequences, and control. Science 2005, 309, 2010–2013. [CrossRef] [PubMed]
- Wang Z, Zhang J. Impact of gene expression noise on organismal fitness and the efficacy of natural selection. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, E67–76.
- Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007, 3, 1724–1735.
- Whitfield ML, Sherlock G, Saldanha AJ, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 2002, 13, 1977–2000. [CrossRef]
- Zhang R, Lahens NF, Ballance HI, et al. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 16219–16224. [CrossRef]
- Viñuela A, Snoek LB, Riksen JAG, et al. Genome-wide gene expression regulation as a function of genotype and age in C. elegans. Genome Res. 2010, 20, 929–937. [CrossRef]
- Viñuela A, Brown AA, Buil A, et al. Age-dependent changes in mean and variance of gene expression across tissues in a twin cohort. Hum. Mol. Genet. 2018, 27, 732–741. [CrossRef]
- Schoenfelder S, Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [CrossRef]
- Nott A, Holtman IR, Coufal NG, et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 2019, 366, 1134–1139. [CrossRef]
- Butler JEF, Kadonaga JT. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev. 2002, 16, 2583–2592. [CrossRef]
- Danino YM, Even D, Ideses D, et al. The core promoter: At the heart of gene expression. Biochim. Biophys. Acta 2015, 1849, 1116–1131. [CrossRef]
- Duan P, Xu J, Zeng D, et al. Natural Variation in the Promoter of GSE5 Contributes to Grain Size Diversity in Rice. Mol. Plant 2017, 10, 685–694. [CrossRef]
- Ong C-T, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011, 12, 283–293. [CrossRef]
- Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 1981, 27, 299–308. [CrossRef]
- Gallagher MD, Chen-Plotkin AS. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [CrossRef]
- Lambert SA, Jolma A, Campitelli LF, et al. The Human Transcription Factors. Cell 2018, 172, 650–665.
- Papavassiliou AG. Molecular medicine. Transcription factors. N. Engl. J. Med. 1995, 332, 45–47. [CrossRef]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef]
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [CrossRef]
- Jones MJ, Goodman SJ, Kobor MS. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [CrossRef]
- Jones, PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Karlić R, Chung H-R, Lasserre J, et al. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2926–2931. [CrossRef]
- Cheng C, Yan K-K, Yip KY, et al. A statistical framework for modeling gene expression using chromatin features and application to modENCODE datasets. Genome Biol. 2011, 12, R15.
- Araki Y, Wang Z, Zang C, et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 2009, 30, 912–925. [CrossRef]
- Feidantsis K, Giantsis IA, Vratsistas A, et al. Correlation between intermediary metabolism, Hsp gene expression, and oxidative stress-related proteins in long-term thermal-stressed Mytilus galloprovincialis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 319, R264–R281. [CrossRef]
- Hasthanasombut, Paisarnwipatpong. Expression of OsBADH1 gene in Indica rice (Oryza sativa L.) in correlation with salt, plasmolysis, temperature and light stresses. Plant Omics.
- Zhang TY, Labonté B, Wen XL, et al. Epigenetic mechanisms for the early environmental regulation of hippocampal glucocorticoid receptor gene expression in rodents and humans. Neuropsychopharmacology 2013, 38, 111–123. [CrossRef]
- Mar JC, Matigian NA, Mackay-Sim A, et al. Variance of gene expression identifies altered network constraints in neurological disease. PLoS Genet. 2011, 7, e1002207.
- Komurov K, Ram PT. Patterns of human gene expression variance show strong associations with signaling network hierarchy. BMC Syst. Biol. 2010, 4, 154.
- Alemu EY, Carl JW Jr, Corrada Bravo H, et al. Determinants of expression variability. Nucleic Acids Res. 2014, 42, 3503–3514.
- Bashkeel N, Perkins TJ, Kærn M, et al. Human gene expression variability and its dependence on methylation and aging. BMC Genomics 2019, 20, 941.
- Simonovsky E, Schuster R, Yeger-Lotem E. Large-scale analysis of human gene expression variability associates highly variable drug targets with lower drug effectiveness and safety. Bioinformatics 2019, 35, 3028–3037. [CrossRef]
- Igolkina AA, Armoskus C, Newman JRB, et al. Analysis of Gene Expression Variance in Schizophrenia Using Structural Equation Modeling. Front. Mol. Neurosci. 2018, 11, 192. [CrossRef]
- Sturm G, List M, Zhang JD. Tissue heterogeneity is prevalent in gene expression studies. NAR Genom Bioinform 2021, 3, lqab077.
- Bachtiary B, Boutros PC, Pintilie M, et al. Gene expression profiling in cervical cancer: an exploration of intratumor heterogeneity. Clin. Cancer Res. 2006, 12, 5632–5640. [CrossRef]
- Wolf S, Melo D, Garske KM, et al. Characterizing the landscape of gene expression variance in humans. bioRxiv 2023. 2022.11.15.516646.
- Pervouchine DD, Djebali S, Breschi A, et al. Enhanced transcriptome maps from multiple mouse tissues reveal evolutionary constraint in gene expression. Nat. Commun. 2015, 6, 5903. [CrossRef]
- Breschi A, Djebali S, Gillis J, et al. Gene-specific patterns of expression variation across organs and species. Genome Biol. 2016, 17, 151.
- Chen Y, Davidson NM, Wan YK, et al. A systematic benchmark of Nanopore long read RNA sequencing for transcript level analysis in human cell lines. bioRxiv 2021. 2021.04.21.440736.
- Shannon, CE. A Mathematical Theory of Communication. Bell System Technical Journal 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Martínez O, Reyes-Valdés MH. Defining diversity, specialization, and gene specificity in transcriptomes through information theory. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 9709–9714. [CrossRef]
- Ameri AJ, Lewis ZA. Shannon entropy as a metric for conditional gene expression in Neurospora crassa. G3 2021, 11.
- Fuhrman S, Cunningham MJ, Wen X, et al. The application of shannon entropy in the identification of putative drug targets. Biosystems. 2000, 55, 5–14. [CrossRef]
- Schug J, Schuller W-P, Kappen C, et al. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 2005, 6, R33.
- Machado T, J. A. Shannon Entropy Analysis of the Genome Code. Math. Probl. Eng. 2012, 2012. [Google Scholar]
- Monaco A, Amoroso N, Bellantuono L, et al. Shannon entropy approach reveals relevant genes in Alzheimer’s disease. PLoS One 2019, 14, e0226190.
- Dérian N, Pham H-P, Nehar-Belaid D, et al. The Tsallis generalized entropy enhances the interpretation of transcriptomics datasets. PLoS One 2022, 17, e0266618.
- Kim MC, Gate R, Lee DS, et al. memento: Generalized differential expression analysis of single-cell RNA-seq with method of moments estimation and efficient resampling. bioRxiv 2022. 2022.11.09.515836.
- Zhang JD, Hatje K, Sturm G, et al. Correction to: Detect tissue heterogeneity in gene expression data with BioQC. BMC Genomics 2018, 19, 558.
- Wang K, Phillips CA, Saxton AM, et al. EntropyExplorer: an R package for computing and comparing differential Shannon entropy, differential coefficient of variation and differential expression. BMC Res. Notes 2015, 8, 832.
- Salzberg, SL. Open questions: How many genes do we have? BMC Biol. 2018, 16, 94. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [CrossRef]
- Barbosa-Morais NL, Irimia M, Pan Q, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [CrossRef]
- Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [CrossRef]
- Wright CJ, Smith CWJ, Jiggins CD. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 2022.
- Ayoubi TA, Van De Ven WJ. Regulation of gene expression by alternative promoters. FASEB J. 1996, 10, 453–460. [CrossRef]
- Alasoo K, Rodrigues J, Danesh J, et al. Genetic effects on promoter usage are highly context-specific and contribute to complex traits. Elife 2019, 8.
- Berget SM, Moore C, Sharp PA. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA*. Proceedings of the National Academy of Sciences 1977, 74, 3171–3175. [CrossRef]
- Patrick E, Buckley M, Yang YH. Estimation of data-specific constitutive exons with RNA-Seq data. BMC Bioinformatics 2013, 14, 31.
- Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [CrossRef] [PubMed]
- Graveley, BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Smith CW, Valcárcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem. Sci. 2000, 25, 381–388. [CrossRef]
- Wan Y, Larson DR. Splicing heterogeneity: separating signal from noise. Genome Biol. 2018, 19, 86.
- Weatheritt RJ, Sterne-Weiler T, Blencowe BJ. The ribosome-engaged landscape of alternative splicing. Nat. Struct. Mol. Biol. 2016, 23, 1117–1123. [CrossRef]
- Burtis KC, Baker BS. Drosophila doublesex gene controls somatic sexual differentiation by producing alternatively spliced mRNAs encoding related sex-specific polypeptides. Cell 1989, 56, 997–1010. [CrossRef]
- McIntyre LM, Bono LM, Genissel A, et al. Sex-specific expression of alternative transcripts in Drosophila. Genome Biol. 2006, 7, R79.
- Brown JB, Boley N, Eisman R, et al. Diversity and dynamics of the Drosophila transcriptome. Nature 2014, 512, 393–399. [CrossRef]
- Gibilisco L, Zhou Q, Mahajan S, et al. Alternative Splicing within and between Drosophila Species, Sexes, Tissues, and Developmental Stages. PLoS Genet. 2016, 12, e1006464.
- Naftaly AS, Pau S, White MA. Long-read RNA sequencing reveals widespread sex-specific alternative splicing in threespine stickleback fish. Genome Res. 2021, 31, 1486–1497. [CrossRef] [PubMed]
- Rogers TF, Palmer DH, Wright AE. Sex-Specific Selection Drives the Evolution of Alternative Splicing in Birds. Mol. Biol. Evol. 2021, 38, 519–530. [CrossRef] [PubMed]
- Blekhman R, Marioni JC, Zumbo P, et al. Sex-specific and lineage-specific alternative splicing in primates. Genome Res. 2010, 20, 180–189. [CrossRef] [PubMed]
- Trabzuni D, Ramasamy A, Imran S, et al. Widespread sex differences in gene expression and splicing in the adult human brain. Nat. Commun. 2013, 4, 2771. [CrossRef] [PubMed]
- Karlebach G, Veiga DFT, Mays AD, et al. The impact of biological sex on alternative splicing. bioRxiv 2020, 490904.
- Xu Q, Modrek B, Lee C. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res. 2002, 30, 3754–3766. [CrossRef]
- Grosso AR, Gomes AQ, Barbosa-Morais NL, et al. Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res. 2008, 36, 4823–4832. [CrossRef]
- Zhang C, Zhang Z, Castle J, et al. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008, 22, 2550–2563. [CrossRef]
- Buljan M, Chalancon G, Eustermann S, et al. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 2012, 46, 871–883. [CrossRef]
- Zhang X, Chen MH, Wu X, et al. Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 2016, 166, 1147–1162.e15. [CrossRef]
- Ling JP, Wilks C, Charles R, et al. ASCOT identifies key regulators of neuronal subtype-specific splicing. Nat. Commun. 2020, 11, 137. [CrossRef]
- Song Y, Botvinnik OB, Lovci MT, et al. Single-Cell Alternative Splicing Analysis with Expedition Reveals Splicing Dynamics during Neuron Differentiation. Mol. Cell 2017, 67, 148–161.e5. [CrossRef] [PubMed]
- Shalek AK, Satija R, Adiconis X, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [CrossRef] [PubMed]
- Kim HK, Pham MHC, Ko KS, et al. Alternative splicing isoforms in health and disease. Pflugers Arch. 2018, 470, 995–1016. [CrossRef]
- Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [CrossRef] [PubMed]
- Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 1992, 90, 41–54.
- Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [CrossRef]
- Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [CrossRef]
- Pan Q, Shai O, Lee LJ, et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [CrossRef]
- Steijger T, Abril JF, Engström PG, et al. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 2013, 10, 1177–1184. [CrossRef]
- Afsari B, Guo T, Considine M, et al. Splice Expression Variation Analysis (SEVA) for inter-tumor heterogeneity of gene isoform usage in cancer. Bioinformatics 2018, 34, 1859–1867. [CrossRef]
- Wu J, Akerman M, Sun S, et al. SpliceTrap: a method to quantify alternative splicing under single cellular conditions. Bioinformatics 2011, 27, 3010–3016. [CrossRef]
- Venables JP, Klinck R, Bramard A, et al. Identification of alternative splicing markers for breast cancer. Cancer Res. 2008, 68, 9525–9531. [CrossRef]
- Merino GA, Conesa A, Fernández EA. A benchmarking of workflows for detecting differential splicing and differential expression at isoform level in human RNA-seq studies. Brief. Bioinform. 2019, 20, 471–481. [CrossRef]
- Soneson C, Matthes KL, Nowicka M, et al. Isoform prefiltering improves performance of count-based methods for analysis of differential transcript usage. Genome Biol. 2016, 17, 12.
- Dougherty ML, Underwood JG, Nelson BJ, et al. Transcriptional fates of human-specific segmental duplications in brain. Genome Res. 2018, 28, 1566–1576. [CrossRef] [PubMed]
- Sharon D, Tilgner H, Grubert F, et al. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [CrossRef] [PubMed]
- ang AD, Soulette CM, van Baren MJ, et al. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020, 11, 1438. [CrossRef]
- Chen Y, Sim A, Wan Y, Goeke J. bambu: Reference-guided isoform reconstruction and quantification for long read RNA-Seq data. 2022.
- Leung SK, Jeffries AR, Castanho I, et al. Full-length transcript sequencing of human and mouse cerebral cortex identifies widespread isoform diversity and alternative splicing. Cell Rep. 2021, 37, 110022. [CrossRef]
- Palmer CR, Liu CS, Romanow WJ, et al. Altered cell and RNA isoform diversity in aging Down syndrome brains. Proc. Natl. Acad. Sci. U. S. A. 2021, 118.
- Ritchie W, Granjeaud S, Puthier D, et al. Entropy measures quantify global splicing disorders in cancer. PLoS Comput. Biol. 2008, 4, e1000011.
- Oguchi Y, Ozaki Y, Abdelmoez MN, et al. NanoSINC-seq dissects the isoform diversity in subcellular compartments of single cells. Sci Adv 2021, 7.
- Padonou F, Gonzalez V, Provin N, et al. Aire-dependent transcripts escape Raver2-induced splice-event inclusion in the thymic epithelium. EMBO Rep. 2022, 23, e53576. [CrossRef] [PubMed]
- Roberts A, Trapnell C, Donaghey J, et al. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [CrossRef] [PubMed]
- Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [CrossRef] [PubMed]
- Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [CrossRef] [PubMed]
- Sterne-Weiler T, Weatheritt RJ, Best AJ, et al. Efficient and Accurate Quantitative Profiling of Alternative Splicing Patterns of Any Complexity on a Laptop. Mol. Cell 2018, 72, 187–200.e6. [CrossRef]
- Dankó B, Szikora P, Pór T, et al. SplicingFactory-splicing diversity analysis for transcriptome data. Bioinformatics 2021.
- Gohr A, Iñiguez LP, Torres-Méndez A, et al. Insplico: effective computational tool for studying splicing order of adjacent introns genome-wide with short and long RNA-seq reads. Nucleic Acids Res. 2023.
- Arzalluz-Luque A, Salguero P, Tarazona S, et al. acorde unravels functionally interpretable networks of isoform co-usage from single cell data. Nat. Commun. 2022, 13, 1828. [CrossRef] [PubMed]
- Olivieri JE, Dehghannasiri R, Salzman J. The SpliZ generalizes ‘percent spliced in’ to reveal regulated splicing at single-cell resolution. Nat. Methods 2022, 19, 307–310. [CrossRef]
- Tekath T, Dugas M. Differential transcript usage analysis of bulk and single-cell RNA-seq data with DTUrtle. Bioinformatics 2021.
- Gleeson J, Leger A, Prawer YDJ, et al. Accurate expression quantification from nanopore direct RNA sequencing with NanoCount. Nucleic Acids Res. 2022, 50, e19. [CrossRef] [PubMed]
- Joglekar A, Prjibelski A, Mahfouz A, et al. A spatially resolved brain region- and cell type-specific isoform atlas of the postnatal mouse brain. Nat. Commun. 2021, 12, 463. [CrossRef]
- Gilis J, Vitting-Seerup K, Van den Berge K, et al. satuRn: Scalable analysis of differential transcript usage for bulk and single-cell RNA-sequencing applications. F1000Res. 2021, 10, 374. [CrossRef]
- Tiberi S, Robinson MD. BANDITS: Bayesian differential splicing accounting for sample-to-sample variability and mapping uncertainty. Genome Biol. 2020, 21, 69.
- Patrick R, Humphreys DT, Janbandhu V, et al. Sierra: discovery of differential transcript usage from polyA-captured single-cell RNA-seq data. Genome Biol. 2020, 21, 167.
- Froussios K, Mourão K, Simpson G, et al. Relative Abundance of Transcripts ( RATs): Identifying differential isoform abundance from RNA-seq. F1000Res. 2019, 8, 213. [CrossRef]
- Trincado JL, Entizne JC, Hysenaj G, et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40.
- Li YI, Knowles DA, Humphrey J, et al. Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 2018, 50, 151–158. [CrossRef]
- Vitting-Seerup K, Sandelin A. The Landscape of Isoform Switches in Human Cancers. Mol. Cancer Res. 2017, 15, 1206–1220. [CrossRef] [PubMed]
- Qiu X, Hill A, Packer J, et al. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 2017, 14, 309–315. [CrossRef] [PubMed]
- Huang Y, Sanguinetti G. BRIE: transcriptome-wide splicing quantification in single cells. Genome Biol. 2017, 18, 123.
- Nowicka M, Robinson MD. DRIMSeq: a Dirichlet-multinomial framework for multivariate count outcomes in genomics. F1000Res. 2016, 5, 1356. [CrossRef] [PubMed]
- Hartley SW, Mullikin JC. Detection and visualization of differential splicing in RNA-Seq data with JunctionSeq. Nucleic Acids Res. 2016, 44, e127.
- Vaquero-Garcia J, Barrera A, Gazzara MR, et al. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 2016, 5, e11752. [CrossRef] [PubMed]
- Goldstein LD, Cao Y, Pau G, et al. Prediction and Quantification of Splice Events from RNA-Seq Data. PLoS One 2016, 11, e0156132. [CrossRef]
- Welch JD, Hu Y, Prins JF. Robust detection of alternative splicing in a population of single cells. Nucleic Acids Res. 2016, 44, e73. [CrossRef]
- Irimia M, Weatheritt RJ, Ellis JD, et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 2014, 159, 1511–1523. [CrossRef]
- Shen S, Park JW, Lu Z-X, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E5593–601.
- Trapnell C, Hendrickson DG, Sauvageau M, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [CrossRef]
- Aschoff M, Hotz-Wagenblatt A, Glatting K-H, et al. SplicingCompass: differential splicing detection using RNA-seq data. Bioinformatics 2013, 29, 1141–1148. [CrossRef]
- Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [CrossRef]
- Katz Y, Wang ET, Airoldi EM, et al. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [CrossRef]
- Zhao S, Ye Z, Stanton R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA 2020, 26, 903–909. [CrossRef] [PubMed]
- Van den Berge K, Hembach KM, Soneson C, et al. RNA Sequencing Data: Hitchhiker’s Guide to Expression Analysis. Annu. Rev. Biomed. Data Sci. 2019, 2, 139–173. [CrossRef]
- Jiang R, Sun T, Song D, et al. Statistics or biology: the zero-inflation controversy about scRNA-seq data. Genome Biol. 2022, 23, 31.
- Lun ATL, Bach K, Marioni JC. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016, 17, 75.
- Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 2019, 15, e8746. [CrossRef] [PubMed]
- Archer N, Walsh MD, Shahrezaei V, et al. Modeling Enzyme Processivity Reveals that RNA-Seq Libraries Are Biased in Characteristic and Correctable Ways. Cell Syst 2016, 3, 467–479.e12. [CrossRef] [PubMed]
Figure 1.
Types of transcriptome diversity from gene expression profiles. Gene-level transcriptome diversity can be measured across samples in a population or as the diversity of expression across genes within a condition. Isoform-level transcriptome diversity can be measured as the number of isoforms or the distribution of isoform expression.
Figure 1.
Types of transcriptome diversity from gene expression profiles. Gene-level transcriptome diversity can be measured across samples in a population or as the diversity of expression across genes within a condition. Isoform-level transcriptome diversity can be measured as the number of isoforms or the distribution of isoform expression.
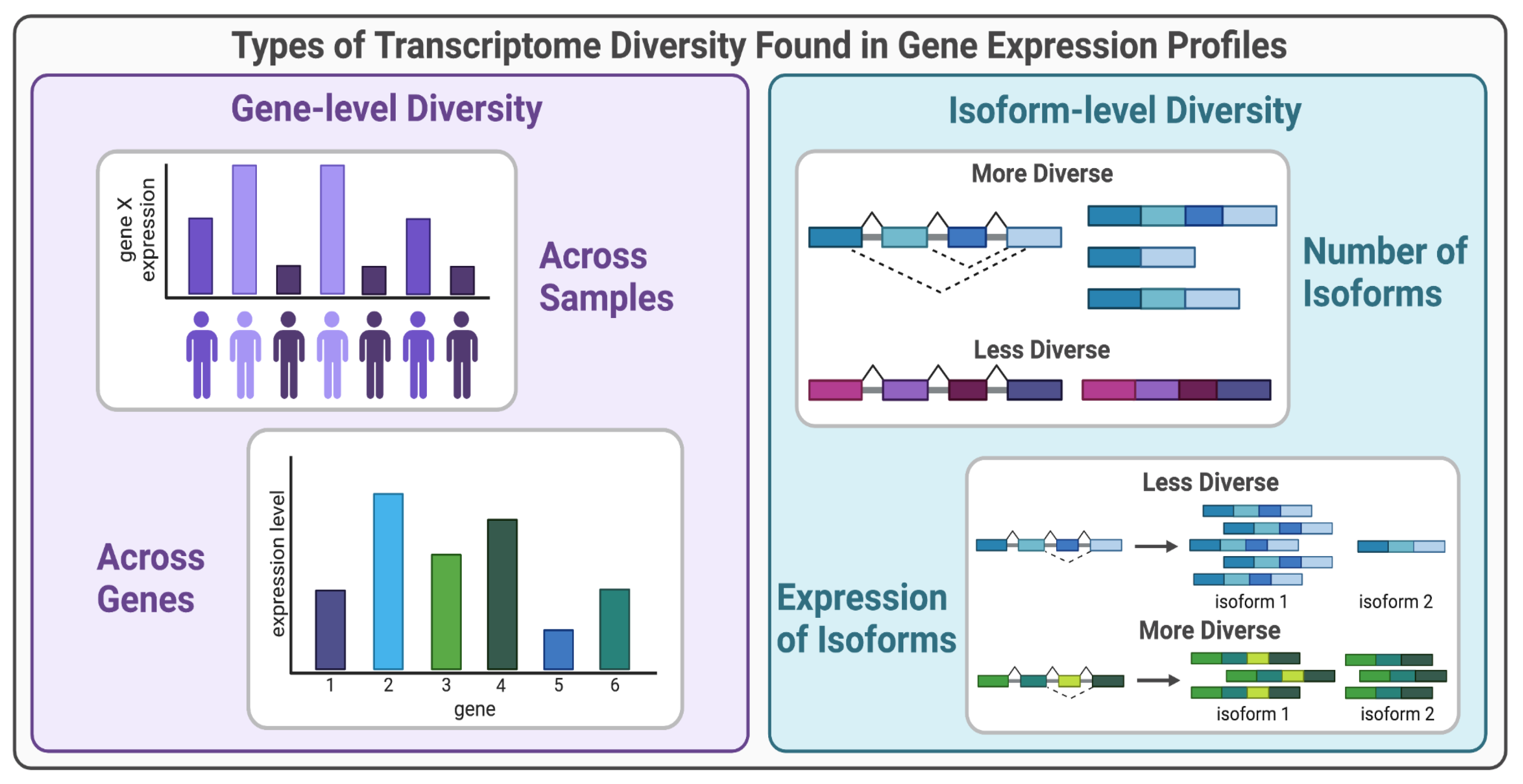
Figure 2.
Shannon entropy can be used to quantify transcriptome diversity. 2A) A toy example showing the principle of Shannon entropy when describing gene-level diversity. When there is a uniform distribution of gene expression values, the Shannon entropy is higher than when gene expression is concentrated on a smaller number of genes. 2B) A toy example of the principle of Shannon entropy when used to describe isoform-level diversity. Even when there are varying numbers of isoforms, the entropy or diversity is at its maximum when the distribution is most uniform or flat.
Figure 2.
Shannon entropy can be used to quantify transcriptome diversity. 2A) A toy example showing the principle of Shannon entropy when describing gene-level diversity. When there is a uniform distribution of gene expression values, the Shannon entropy is higher than when gene expression is concentrated on a smaller number of genes. 2B) A toy example of the principle of Shannon entropy when used to describe isoform-level diversity. Even when there are varying numbers of isoforms, the entropy or diversity is at its maximum when the distribution is most uniform or flat.
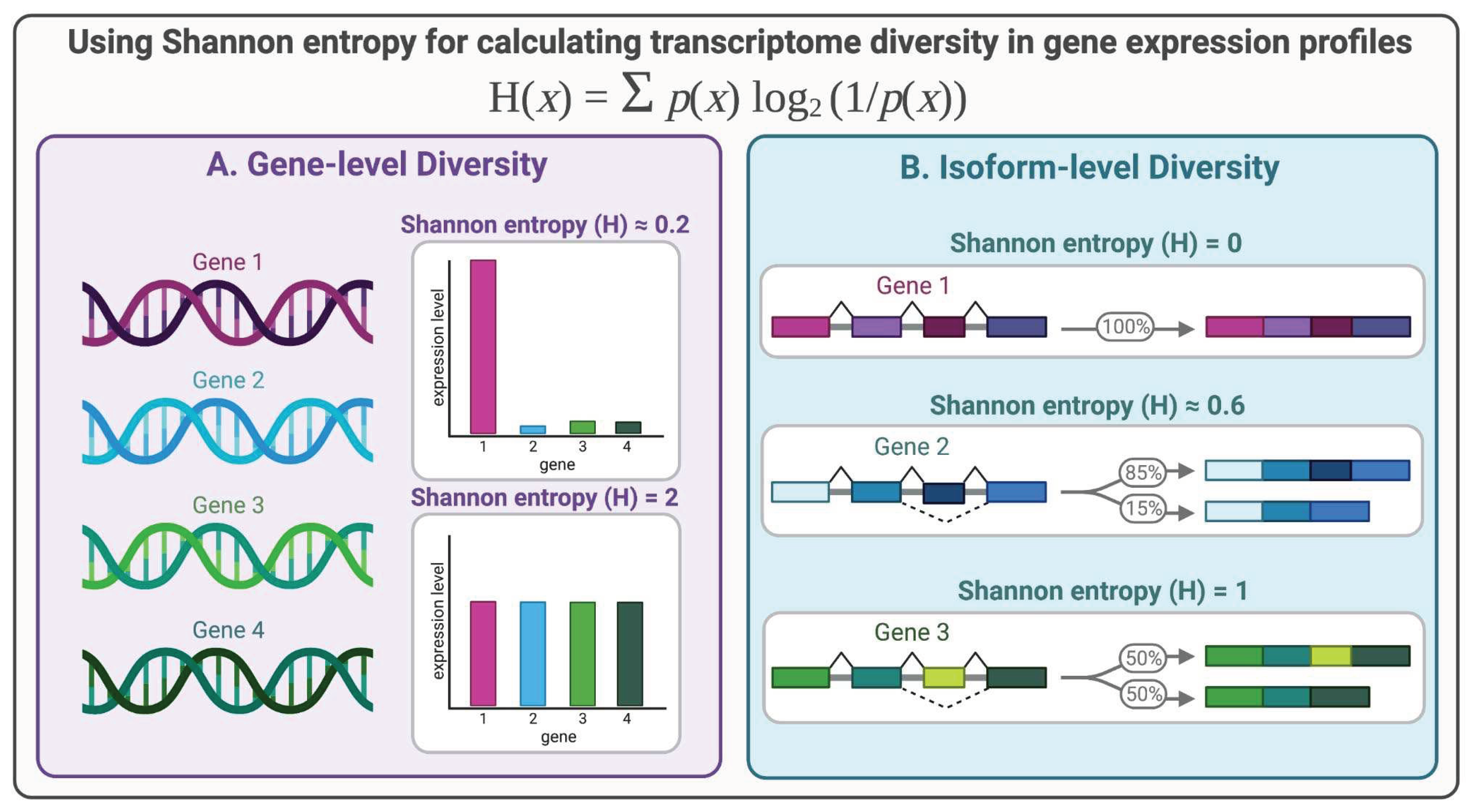
Table 1.
Software Packages that Detect Gene-level Transcriptome Diversity. This table includes the name of the software package, the year published, gene expression data it can be used with, and the transcriptome diversity metric. Entries are sorted from most to least recent. Citation counts are from Pubmed, March, 2023.
Table 1.
Software Packages that Detect Gene-level Transcriptome Diversity. This table includes the name of the software package, the year published, gene expression data it can be used with, and the transcriptome diversity metric. Entries are sorted from most to least recent. Citation counts are from Pubmed, March, 2023.
| Name of Package | Year | Bulk or Single Cell | Gene-level Transcriptome Diversity Metrics | Citation Count | Language |
| memento [81] | 2022 | Single-cell | Variation | 0 | Python |
| BioQC [82] | 2017 | Bulk | Shannon Entropy | 26 | R |
| MDSeq [19] | 2017 | Bulk | Variation | 15 | R |
| EntropyExplorer [83] | 2015 | Bulk | Differential Shannon Entropy | 9 | R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



