
Submitted:
21 January 2023
Posted:
24 January 2023
You are already at the latest version
Abstract
November 15th, 2022 was selected as the day where the human population reached the 8 billion mark [1]. This new reality will force the governments around the world to find ways to sustainably produce and secure food, water, and energy for their countries [2-4]. In terms of water, its quality and availability has been a matter of great concern in recent decades [5], especially with the emergence of new organic and inorganic pollutants [6]. Among organic contaminants, the detection of trace amounts of fluoroquinolone antibiotics, such as ciprofloxacin (CFX) and levofloxacin (LFX), in natural water bodies, has been of great concern in the scientific community [7,8]. Some of the side effects of the consumption of these antibiotics are nausea, diarrhea, abdominal pain, rash, low sugar levels, and antibiotic resistance to bacterial infections, among others [9,10]. It was estimated that in 2019 more than 1.27 million people died due to antibiotic-resistant bacterial infections [11], and this number is expected to rise to 10 million by 2050, if the trend continues [12]. Because of this, new ways to degrade antibiotics from water have been developed over the years. A method that has been implemented for some time is the use of photocatalysts for the degradation of these compounds in water [13]. Semiconductors such as titanium oxide (TiO2), zinc oxide (ZnO), zinc sulfide (ZnS), cadmium sulfide (CdS), strontium peroxide (SnO2), or tungsten trioxide (WO3), among others, are commonly used in photocatalytic processes [14-16]. Zinc oxide has been widely used due to its low cost and stability in aqueous solution, easy production, and because it is an environmentally friendly material [17,18]. It has been identified that some of the disadvantages of ZnO as photocatalyst are photocorrosion, recombination of electron-hole pairs, fast backward reactions, and inability to use visible light [18]. Multiple approaches have been implemented over the years to reduce these limitations. One of them is the use of noble metals such as platinum (Pt), gold (Au), or even silver (Ag) as cocatalysts [15-17,19]. These metals can increase the photocatalytic activity by reducing the recombination of electron-hole pairs, as well allowing the use of visible light [20]. For example, Quin and coworkers [21] prepared a bio-inspired hierarchical assembly of carbonized spinach leaves@Au/ZnO for the degradation of CFX under visible light. The results showed a degradation of 61% of the antibiotic in a period of 180 minutes. Chankhanittha et al. [22] developed different Ag@ZnO composites for the complete degradation of red dye and ofloxacin antibiotic in 25 and 80 minutes, respectively. The researchers attributed the improved photoactivity to the high electron-hole separation efficiency at the photocatalyst interface, as well as the creation of the Schottky barrier at the silver-zinc oxide interface.
Keywords:
Photodegradation
; Ciprofloxacin
; Levofloxacin
; Molybdenum disulfide
; Reduced Graphene Oxide
; Zin oxide
; Gold nanoparticles
1. Introduction
November 15th, 2022 was selected as the day where the human population reached the 8 billion mark [1]. This new reality will force the governments around the world to find ways to sustainably produce and secure food, water, and energy for their countries [2,3,4]. In terms of water, its quality and availability has been a matter of great concern in recent decades [5], especially with the emergence of new organic and inorganic pollutants [6]. Among organic contaminants, the detection of trace amounts of fluoroquinolone antibiotics, such as ciprofloxacin (CFX) and levofloxacin (LFX), in natural water bodies, has been of great concern in the scientific community [7,8]. Some of the side effects of the consumption of these antibiotics are nausea, diarrhea, abdominal pain, rash, low sugar levels, and antibiotic resistance to bacterial infections, among others [9,10]. It was estimated that in 2019 more than 1.27 million people died due to antibiotic-resistant bacterial infections [11], and this number is expected to rise to 10 million by 2050, if the trend continues [12]. Because of this, new ways to degrade antibiotics from water have been developed over the years. A method that has been implemented for some time is the use of photocatalysts for the degradation of these compounds in water [13]. Semiconductors such as titanium oxide (TiO2), zinc oxide (ZnO), zinc sulfide (ZnS), cadmium sulfide (CdS), strontium peroxide (SnO2), or tungsten trioxide (WO3), among others, are commonly used in photocatalytic processes [14,15,16]. Zinc oxide has been widely used due to its low cost and stability in aqueous solution, easy production, and because it is an environmentally friendly material [17,18]. It has been identified that some of the disadvantages of ZnO as photocatalyst are photocorrosion, recombination of electron-hole pairs, fast backward reactions, and inability to use visible light [18]. Multiple approaches have been implemented over the years to reduce these limitations. One of them is the use of noble metals such as platinum (Pt), gold (Au), or even silver (Ag) as cocatalysts [15,16,17,19]. These metals can increase the photocatalytic activity by reducing the recombination of electron-hole pairs, as well allowing the use of visible light [20]. For example, Quin and coworkers [21] prepared a bio-inspired hierarchical assembly of carbonized spinach leaves@Au/ZnO for the degradation of CFX under visible light. The results showed a degradation of 61% of the antibiotic in a period of 180 minutes. Chankhanittha et al. [22] developed different Ag@ZnO composites for the complete degradation of red dye and ofloxacin antibiotic in 25 and 80 minutes, respectively. The researchers attributed the improved photoactivity to the high electron-hole separation efficiency at the photocatalyst interface, as well as the creation of the Schottky barrier at the silver-zinc oxide interface.
Another material that has been gaining a lot of attention in recent years is molybdenum disulfide (MoS2). Some of the advantages of using MoS2 as cocatalyst are its high abundance, good stability, high catalytic activity, and low cost [23]. Studies using ZnO/MoS2 for the degradation of antibiotics in water were not found to the best of our knowledge, but different groups have implemented these composites for the degradation of other organic pollutants. Ahamad and coworkers [24] reported an 84.5% degradation of the pesticide dicotol in 90 minutes, using heterostructured nanocomposites containing MoS2/ZnO nanoparticles embedded in nitrogen/sulfur doped graphite carbon. They attributed the results to the formation of active photogenerated species such as OH•, •O2- and e-. Other heterostructured hybrid layered ZnO and MoS2 nanosheets composites have been synthesized by Benavente and coworkers [25], and tested in the degradation of methylene blue under direct solar light. The composites were able to degrade 75 % of the dye after 300 minutes of irradiation, and this behavior was justified on the basis that MoS2 played a key role in decreasing the bandgap of the heterostructure.
As noble metals and MoS2, reduced graphene oxide (rGO) has been implemented as cocatalyst for the degradation of organic pollutants from water. Properties such as large specific surface area, chemical stability, high electrical conductivity, and absorptivity make rGO an excellent functional material in different fields such as hydrogen production, optics, electronics and photocatalysis [26]. Arya and coworkers [27] prepared different heterostructures based on rGO-Bi2WO6 for the degradation of LFX under visible light at room temperature. The photocatalysts exhibited a degradation efficiency of 74.3% in 120 minutes. According to the researchers, the improvement in photocatalytic activity could be due to a reduction in the recombination rate of photoexcited charge carriers due to the introduction of graphene, which served as a charge carrier [27]. Another research group [28] developed ternary nanocomposites based on rGO-BiVO4-ZnO for CFX degradation using visible light radiation. In this case, a 98.4% degradation was observed after 60 min, attributing the results to charge transfer and excellent electron-hole separation by rGO doping [28].
In this research, and considering all the advantages of the mentioned materials, nine catalysts have been developed, based on heterostructures of Au nanoparticles on ZnO nanoparticles, and MoS2 and rGO nanosheets. These catalysts have been tested for the catalytic photodegradation of CFX and LFX in aqueous solution. The amounts of Au and MoS2 varied between 1% and 5% by weight, while keeping the amount of rGO constant at 1% w/w. The final composites were identified as follows: 1%Au@ZnONPs-1%MoS2-1%rGO, 3%Au@ZnONPs-1%MoS2-1%rGO, 5%Au@ZnONPs-1%MoS2-1%rGO, 1%Au@ZnONPs-3%MoS2-1%rGO, 3%Au@ZnONPs-3%MoS2-1%rGO, 5%Au@ZnONPS-3%MoS2-1%rGO, 1%Au@ZnONPS-5%MoS2-1%rGO, 3%Au@ZnONPs-5%MoS2-1%rGO, and 5%Au@ZnONPs-5%MoS2-1%rGO. The catalysts were characterized by Brunauer-Emmett-Teller (BET) surface area analysis, field emission scanning electron microscopy (FESEM), high resolution transmission electron microscopy (HRTEM), X-ray diffraction (XRD) analysis, Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), diffuse reflectance, and gas chromatography (GC-MS).
2. Results and Discussion
2.1. Characterization of the Photocatalysts
The as-synthesized photocatalysts were characterized and their photocatalytic activity was studied by the degradation of LFX and CFX in aqueous solution. The most efficient catalyst in the degradation of LFX, 5%Au@ZnONPs-3%MoS2-1%rGO, was selected to be fully characterized.
The BET surface area of the catalysts is shown in Table S1. As can be seen, the bare ZnONPs showed a surface area of 67 m2g-1, increasing with the incorporation of Au nanoparticles, and MoS2 and rGO nanosheets. This increase in surface area by the addition of other cocatalysts has been previously described [14,15,16,17]. The trend of the results shows that the higher percentages of Au and MoS2, the higher the surface area of the catalyst. The 5%Au@ZnONPs-5%MoS2-1%rGO composite showed the highest surface area, 151 m2g-1, which represents a difference of 84 m2g-1 compared to bare ZnONPs.
The precursors of the catalysts were characterized by field emission scanning electron microscopy, FESEM, (see Figure 1). ZnONPs at different magnifications (Figure 1a,b) show inhomogeneous particles with diameters ranging from ca. 15 to 20 nm. rGO (Figure 1c) consists of inhomogeneous particles formed by sheets that pack very close together, with different diameters (ca. 1 μm to 5 μm), similar to what has been observed in other works [29]. The MoS2 was previously delaminated by ultrasound treatment in aqueous solution (Figure 1d). As expected, delaminated MoS2 exhibits a layered structure with sizes ranging from ca. 1 µm to 6 µm [30].
Figure 2 shows the high-resolution transmission electron microscopy (HRTEM) images of the precursors. Figure 2a,b show the ZnONPs after the incorporation of AuNPs. As can be seen there, the ZnONPs are highly crystalline, showing distinct lattice fringes with an interplanar spacing of ca. 0.28 nm (inset of 2b), corresponding to the d-spacing of the [001] planes [14]. This is consistent with the results shown by the selected area electron diffraction (SAED) in the inset of Figure 2b, as well as by XRD analysis to be discussed later. The AuNPs also presented high crystallinity, with diameters of ca. 10 nm (Figure 2a,b ). Figure 2c corresponds to the rGO micrograph, clearly showing the lattice fringes with an interplanar distance of ca. 0.24 nm, which represent the d-spacing of the [002] planes [31]. The atomic structure of a highly exfoliated MoS2 is shown in Figure 2d. The SAED of MoS2 (inset of 2d) indicates a high crystalline material. It is possible that due to the ultrasound exfoliation process some structural defects may have been generated. The presence of these defects has not yet been verified, but it could influence the activity of the catalysts.
Figure 3 shows the Raman spectra of rGO, ZnONPs, MoS2, and the 5%Au@ZnONPs-3%MoS2-1%rGO catalyst. rGO (Figure 3a) shows two peaks at 1350 cm-1 and 1586 cm-1, corresponding at the D and G bands, respectively, and represent the presence of carbon atom lattice defects and in-plane stretching vibration from sp2 hybridization of carbon [32]. The ZnONPs (Figure 3b) shows different peaks at ca. 327 cm-1, 437 cm-1, 550 cm-1, and 1125 cm-1. The 327 cm-1 band is attributed to the second-order Raman spectrum [14], whereas the 437 cm-1 band was assigned to the E2 modes of Zn motion, corresponding to the band characteristic of the wurtzite phase [14]. The 550 cm-1 band is assigned to the E1 mode and usually originates from second-order Raman scattering [33]. The band at 1125 cm-1 was assigned to overtones and/or combination bands [14]. The Raman spectrum of MoS2 (see Figure 3c) shows two characteristic bands at ca. 383 cm-1 and 407 cm-1, which have been assigned to the E12g and A1g modes, respectively [30], being attributed to the exfoliation process and the formation MoS2 flakes with few layers [34,35]. The 5%Au@ZnONPs-3%MoS2-1%rGO catalyst (Figure 3d) only showed the G-band of rGO, possibly due to the low concentration of rGO (1% by weight) in the sample. The catalyst (Figure 3d) showed the four characteristic bands of ZnONPs, but with lower intensity and some small changes. These differences are attributed to the interaction with the other additives. The presence of the two main MoS2 bands was also evident in the catalyst. The presence of the most significant peaks of all the catalyst components confirmed the heterostructured nature of the composite.
Figure 4 shows the diffraction pattern of the catalyst 5%Au@ZnONPs-3%MoS2-1%rGO, along with that of ZnONPs, 5%Au@ZnONPs, MoS2 and rGO for comparison purposes. The diffraction peaks of ZnONPs (Figure 4a) can be unambiguously indexed to the ZnO phase of hexagonal wurtzite [36], whose reflections are dominant in the 5%Au@ZnONPs-3%MoS2-1%rGO catalyst, as observed in Figure 4e. Incorporation of 5%AuNPs (Figure 4b) does not reveal the appearance of a new peak at ca. 38.1°, corresponding to Au (111), possibly due to the high dispersion of the metal [37]. MoS2 (Figure 4c) shows several diffraction peaks at ca. 32°, 36°, 39°, 49°, and 58° that have been ascribed to (100), (102), (103), (105), and (110) crystalline planes of 2H-type MoS2 hexagonal phase (JCPDS # 75–1539), respectively [30,38,39]. rGO (Figure 4d) shows a peak at ca. 23.8°, assigned to the (002) crystal plane, indicating that in this reduced material most of the functional groups with oxygen, which are characteristic of graphene oxide, have been removed [40,41]. rGO shows a second peak close to 40°, at a slightly lower angle than expected (43°), which has been assigned to the (100) plane of the hexagonal carbon structure. Figure 4e shows the different diffraction peaks corresponding to the most active catalyst. To facilitate the identification of components, the same color code has been used. As can be seen, the most prominent components are shown in the catalyst although, as observed in 4b, no peak corresponding to gold is observed.
The representative elements of the most efficient catalyst (5%Au@ZnONPs-3%MoS2-1%rGO) were characterized by X-ray photoelectron spectroscopy (XPS). Zn2p (see Figure 5a) shows two components at 1044.2 eV and 1020.6 eV, with a characteristic spin-orbit splitting of 23.6 eV, that were ascribed to the Zn2p1/2 and Zn2p3/2 transitions of Zn2+, respectively [42,43]. Both transitions are very symmetrical, and the fitting to other possible states of Zn did not give results, so any additional contribution was ruled out. Figure 5b shows the transition corresponding to O1s. The transition is clearly asymmetric, and it has been possible to deconvolute into three components at ca. 530.1, 532.2 eV and 535.1 eV. The peak at ca. 530.1 eV, was assigned to O2- species in the ZnO network, and the one observed at 532.2 eV was assigned to O2- in oxygen-deficient regions, respectively [44]. Graphene oxide (rGO) should show a component below 530 eV, although this contribution should be masked by the peak at 530.1 eV. The component observed at the highest binding energy (535.1 eV) must correspond to species generated by the interaction of the ZnO nanoparticles with rGO. In fact, O1s components have been observed in rGO at BE above 535 eV, although their origin is not entirely clear [45]. Figure 5c shows the Au4f transition, with peaks at 83.9 eV and 87.6 eV that have been ascribed to Au4f7/2 and Au4f5/2, respectively. Both peaks, together with a characteristic spin-orbit splitting of 3.7 eV, evidence the presence of metallic gold [46]. The C1s spectrum (Figure 5d) is quite asymmetric and has been deconvolved into three components at 284.6, 287.2 and 289.2 eV, respectively. The most important contribution is observed at 284.6 eV, and has been ascribed to the sp2 carbon of rGO. The other two components observed at 287.2 and 289.2 eV can be attributed to C-OH and O=C-OH, respectively, and are possibly due to the presence of structural defects in rGO, produced during the graphite exfoliation process, and subsequent reduction of graphene [47,48]. Figure 5e shows the Mo3d and S2s transitions. Mo3d shows two well-defined and symmetrical peaks at 232.1 and 229.0 eV, which have been attributed to the Mo3d3/2 and Mo3d5/2 doublet, respectively, and assigned to the Mo4+ state in MoS2 [47,48]. The observed peak at ca. 226.4 eV corresponds to the contribution of S2s [48], characteristic of MoS2.
The different precursors and the most efficient catalyst (5%Au@ZnONPs-3%MoS2-1%rGO) were characterized by diffuse reflectance spectroscopy. From the reflectance in Kubelka-Munk units, the Tauc plots were obtained, which have allowed establishing the bandgaps of the different precursors and the most efficient catalyst (see Figure 6). The bandgap energy of the wurtzite crystalline phase of ZnO has been reported to be ca. 3.37 eV [49], while the synthesized ZnONPs (Figure 6a) showed a value of 3.24 eV. This slight difference could be due to the morphology and particle size of the semiconductor [49]. After the incorporation of 5 wt.% of AuNPs onto the bare ZnONPs, the bandgap energy decreased to 3.19 eV (Figure 6b). This reduction was expected and has been previously reported [13,14,15]. However, even when the bandgap is decreased, it is still in the ultraviolet region of the electromagnetic spectrum [15]. The MoS2 bandgap energy showed a value of 2.45 eV (Figure 6c). Depending on the degree of delamination, and the number of layers of the material, this value can vary widely from ca. 0.9 eV to values above 2.50 eV [50]. This effect has also been observed to depend on the degree of quantum confinement of the material along the c axis [50]. The rGO bandgap energy was 2.00 eV (Figure 6e), although, as previously described in other investigations, the level of reduction can greatly affect this value, with bandgaps ranging from ca. 0.20 eV to 2.00 eV [51]. Both MoS2 and rGO can absorb in the visible region, which undoubtedly improves the catalytic properties of the composite, promoting energy absorption in the visible range. The bandgap energy of the most efficient catalyst (5%Au@ZnONPs-3%MoS2-1%rGO) was 2.15 eV (Figure 6d), indicating that the catalyst can efficiently use visible light for catalytic processes, as confirmed in the degradation studies that will be discussed later.
2.2. Photodegradation of Levofloxacin and Ciprofloxacin
The ideals conditions in terms of antibiotic concentration, catalyst loading, and pH were determined for LFX, using the 5%Au@ZnONPs-1%MoS2-1%rGO, 5%Au@ZnONPs-3%MoS2-1%rGO, and 5%Au@ ZnONPs-5%MoS2-1%rGO catalysts, and the results can be found in the supplementary information section. In the case of CFX, these conditions have been previously determined in other studies by our research group [17,52].
It is reported [18] that the concentration of the antibiotic must be in a suitable range to improve the interaction with the active sites of the catalyst, avoiding the interaction between the LFX molecules, which could decrease the degradation rate. For that reason, the first parameter studied was the evaluation of the initial concentration of LFX (see Figure S1). To carry out the experiments, the catalyst loading, and pH of the solution was set to be 1.0 g/L and 7, respectively. As observed, the concentration of LFX varied between 2 μM – 50 μM, being 10 μM the concentration at which the three evaluated catalysts obtained the highest percentage of degradation. From 2 μM – 10 μM the photocatalytic activity increased, suggesting a good interaction between the catalyst and LFX. After 10 μM, the concentration began to decrease, and this was associated to the byproducts formed during the photodegradation. These byproducts can compete for the catalyst's active sites, reducing its efficiency. Also, at higher concentrations, the surface of the catalysts can become saturated leading to a decrease in the degradation rate [18]. The catalyst with the highest percentage of degradation at 10 μM was 5%Au@ZnONPs-3%MoS2-1%rGO (99.8%), followed by 5%Au@ZnONPs-5%MoS2-1%rGO (98%), and 5%Au@ ZnONPs-1%MoS2-1%rGO (95%), respectively.
The second parameter studied for LFX was the catalyst loading (see Figure S2). The LFX concentration used to carry out the experiment was 10 μM at pH=7. The amount of the catalysts varied between 0.2 g/L and 1.5 g/L, being 1.1 g/L the catalyst loading with the best results for the three composites tested. It is suggested that between 0.2 g/L and 1.1 g/L the interaction between LFX and the catalyst is enhanced, but at loadings over 1.1 g/L a decrease is observed. This might be associated to the poor interaction between LFX and the catalyst and/or the scattering of the irradiation due to the high amount of catalyst present in the solution [18]. The catalyst with the higher percentage of degradation was 5%Au@ZnONPs-3%MoS2-1%rGO (99.8%), followed by 5%Au@ZnONPs-1%MoS2-1%rGO (97.5%), and 5%Au@ ZnONPs-5%MoS2-1%rGO (94.8%), respectively.
The third parameter studied was pH (Figure S3). To carry out this experiment, only the highest efficiency catalyst (5%Au@ZnONPs-3%MoS2-1%rGO) was used, with a catalyst loading of 1.1 g/L and a concentration of 10 μM of LFX. As can be seen, the pH varied between 4 and 11, with pH=8 being the one with the highest percentage of degradation. At acidic pH (< 7), the photocatalytic process was not favorable and could be attributed to the repulsive forces between the positively charged ZnO surface and the LFX molecules [53]. At alkaline pH (>7), the photodegradation rate increased until pH=8, and then slowly decreased until reaching pH=9 and 11, suggesting attractive forces between the positively charged ZnO and the -OH ions present in the solution [53,54]. According to the experiments carried out, the ideal conditions for the catalytic photodegradation of LFX consisted of a 10 μM LFX concentration at pH=8 and with a catalyst loading of 1.1 g/L. For the CFX photodegradation reaction, the ideal conditions were 10 µM of CFX at pH 7 with a catalyst loading of 1.0 g/L.
Figure 7 shows the rate of catalytic photodegradation of CFX and LFX as a function of time. The photodegradation behavior of both antibiotics was very different depending on the catalyst. In the case of CFX (Figure 7a–c), it was observed that after 60 min the degradation varied from 70% to 96%. The three catalysts with the highest degradation percentages were 1%Au@ZnONPs-3%MoS2-1%rGO (96%), followed by 3%Au@ZnONPs-3%MoS2-1%rGO (90%), and 1 %Au@ZnONPs-1%MoS2-1%rGO (86%). The catalyst that produced less degradation (70%) was 5%Au@ZnONPs-5%MoS2-1%rGO. The observed results indicate that at higher percentages of AuNPs and MoS2, the degradation decreases. A possible explanation for this behavior could be that at higher loadings, AuNPs and MoS2 nanosheets can cause a slight scattering of radiation. On the other hand, the agglomeration of the particles could be improving the catalyst-catalyst contact instead of favoring the catalyst-CFX contact, which would imply a decrease in activity. [18]. In the case of LFX (Figure 7d–f), experiments were performed for 120 min instead of 60 min as done with CFX. This decision was made when the percentages of degradation of both antibiotics were compared at 60 minutes. In that time range, all catalysts could degrade 70-96% CFX, but only 65-80% LFX. This difference suggests that CFX degrades faster while LFX is more recalcitrant to degrade. Similar results have been reported before [17], and are attributed to factors such as pH and the chemical structure of the antibiotics. The catalyst with the highest percentage of LFX degradation during a reaction time of 120 min was 5%Au@ZnONPs-3%MoS2-1%rGO (99.8%), followed by 5%Au@ZnONPs-5%MoS2- 1%rGO (99%), and 3%Au@ZnONPs-3%MoS2-1%rGO (98%). The catalyst with the lowest percentage of degradation was 1%Au@ZnONPs-1%MoS2-1%rGO, (89%). Unlike what was observed in CFX, LFX degradation was favored with high percentages of both AuNPs and MoS2. It is possible that for LFX the high percentages of AuNPs and MoS2 improve the catalyst-LFX contact by creating new active sites for photocatalytic activity. Other research groups [3] have reported similar results with high percentages of other cocatalysts.
To study the effect and contribution of parameters such as catalyst, radiation and presence of oxygen, control experiments were carried out for the antibiotic LFX (Figure S4). For the anoxic experiments, the solution was purged with nitrogen gas (N2) for 180 min, whereas the photolytic experiments were performed without the presence of the catalyst (5%Au@ZnONPs-3%MoS2-1%rGO). For catalytic experiments, the radiation source was removed, and the solution was kept in the dark. As can be seen (Figure S4), LFX degradation is negligible when the oxygen source (anoxic), radiation source (photolysis) and catalyst (catalysis) are removed from the system. Without the oxygen source, radicals are not formed and oxidation and ultimately degradation of LFX does not occur. Without radiation sources, there is no activation of the catalyst, so electron-hole pairs (e-/h+) will not be generated, and degradation cannot continue. If the catalyst is removed, the degradation does not continue as the radiation source is not sufficient to degrade the LFX molecules. The stability of this fluoroquinolone in water is consistent with studies where CFX and LFX have been detected in surface waters [7,8].
To study the recyclability of the most efficient catalysts for the degradation of LFX (5%Au@ZnONPs-3%MoS2-1%rGO), and CFX (1%Au@ZnONPs-3%MoS2-1%rGO), 15 cycles were performed (Figure S5). The experiments consisted of recovering the catalyst after each cycle by centrifugation (3000 rpm for 20 min), followed by washing with deionized water and ethanol to remove any byproducts, and drying for 5 hours at 60 °C. After drying the catalyst, the same parameters were used for antibiotic degradation, as discussed above. As can be seen (Figure S5a), the degradation of LFX after 15 cycles experienced a low decrease in efficiency, until reaching a degradation percentage of 92.8 %, which represents a decrease in activity close to 7 %. In the case of CFX, the loss of efficiency is greater, reaching a degradation percentage close to 85.7 % after 15 cycles of use. This difference could eventually be associated with the different loading of gold in both catalysts. The most efficient catalyst for CFX photodegradation has only 1 % AuNPs, so a possible gold leaching could have a much greater effect (as observed in Figure S5b), compared to what happens with LFX, whose most efficient catalyst has 5%AuNPs (Figure S5a). The results obtained are certainly promising, taking into account that the catalysts are made up of three components whose synergistic behavior is maintained with few variations after each cycle of use. Furthermore, these results suggest that the catalysts could be used for longer cycles without excessively compromising degradation efficiency.
In semiconductor catalysis, electron-hole pairs (e-/h+) are formed when an electron leaves the valence band (VB) of the semiconductor and reaches the conduction band (CB) [13,14,15,16,17,18,19,20]. The hole (h+) that is formed acts as an oxidizing agent and can degrade substances that are prone to oxidation. If the electron that left the VB, for some reason, does not reach the CB and returns, recombination occurs. This recombination is one of the main disadvantages of semiconductor photocatalysis [14,15,16,17]. To reduce the probability of recombination, hole scavengers are often used [19]. The idea behind this is to incorporate a substance that is more susceptible to oxidation than the contaminant of interest. The substance will feel attraction to the h+ formed in the VB of the catalyst and will be oxidized, reducing the probability of recombination. The excited electrons that reach the CB of the catalyst can be gained by oxygen species, forming superoxide radicals (O2-) that can act as even stronger oxidizing agents than the valence band’s holes. These radicals then can degrade the antibiotics, leading to a higher degradation percentage. In this sense, and to evaluate the photodegradation mechanism, some scavengers were added to the reaction mixture: tert-butanol (t-butanol), 1,4-benzoquinone (1,4-BQ) and disodium salt of ethylenediaminetetraacetic acid, EDTA-Na2 (Figure S6). t-Butanol, 1,4-BQ and EDTA-Na2, were adopted as hydroxyl radical (•OH), superoxide radical (•O2−), and h+ scavenger, respectively. As can be seen, t-butanol hindered photoactivity noticeably, suggesting the main role of •OH reactive species in the photodegradation process. 1,4-BQ hindered the reaction, although clearly to a lesser extent, which supports the fact that ·O2− does not play as prominent a role as the hydroxyl radical in the degradation process. Finally, the presence of EDTA practically did not affect the reaction, thus it is evident that the holes generated during the catalytic process do not intervene in the photodegradation of LFX. Similar effects were observed for CFX (see Figure S6b), although in this case the effects of all scavengers were certainly greater.
The intermediates of the photodegradation of LFX and CFX by the catalysts that showed higher efficiency (5%Au@ZnONPs-3%MoS2-1%rGO and 1%Au@ZnONPs-3%MoS2-1%rGO, respectively), were characterized by GC-MS. Based on these results, a possible degradation pathway has been established (see Figure 8). For both contaminants, photodegradation was very fast. In the case of LFX (Figure 8a), three different and simultaneous degradation pathways have been suggested. Pathway 1 involves a first piperazin ring cleavage (m/z 308), followed by the loss of methyl groups (m/z 279) [56,57], and then mineralization. Pathway 2 shows a first degradation stage similar to 1 (m/z 279) [56,57], followed by decarboxylation and subsequent mineralization. In pathway 3, a depiperazinylation and defluorination is observed, for subsequent mineralization. In the case of CFX, photodegradation was even faster than in LFX, generating different compounds that have allowed establishing four possible pathways (Figure 8b). The first three pathways are characterized by simultaneous depiperazinylation and defluorination, leading to various intermediates (m/z: 284, 216 and 205) [58], with subsequent mineralization. Pathway 3 also experiences the loss of the cyclopropane ring. Pathway 4 shows the piperazin ring cleavage (m/z 220), followed by the cleavage of the moiety corresponding to the heterocycle with nitrogen and the cyclopropane ring (m/z 141), to continue with subsequent mineralization.
2.3. Mechanism for the Photodegradation of Levofloxacin and Ciprofloxacin
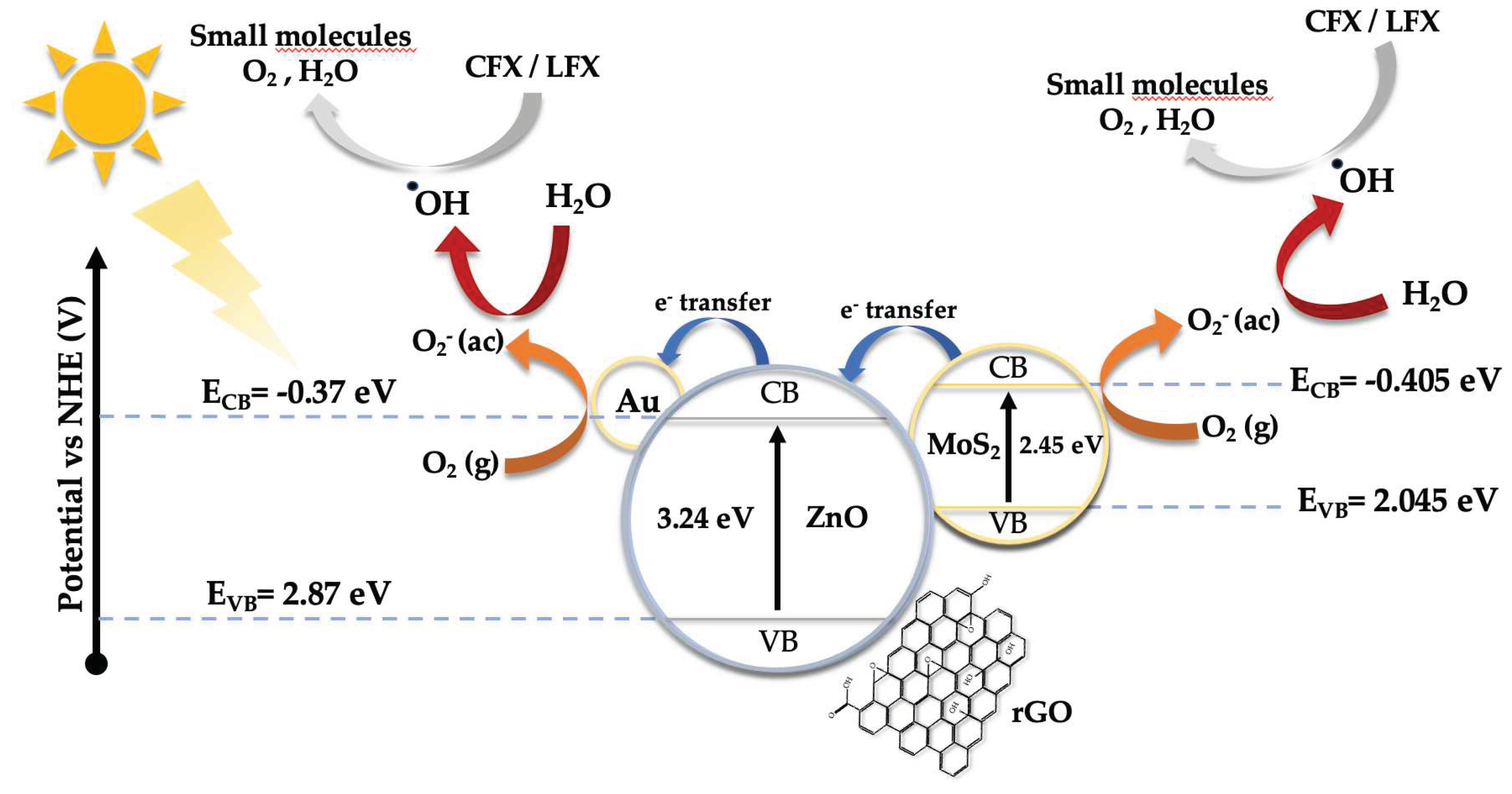
A possible mechanism of the catalytic photodegradation of LFX and CFX is shown in Figure 9. The band edge position and the migration direction of the photogenerated charge carriers were determined by the bandgap energies (Figure 6) and the Mulliken electronegativity theory [59], using the following equations:
ECB = X – EC – 0.5Eg
EVB = ECB + Eg
ECB is the edge potential of the conduction band, X is the absolute electronegativity, EC has a value of 4.50 eV and corresponds to the energy of free electrons on the hydrogen scale [60,61], Eg is the bandgap energy, and EVB is the edge potential of the valence band. The absolute electronegativity for ZnO and MoS2 are 5.75 eV and 5.32 eV, respectively, whereas the ECB and EVB edge potentials for Au@ZnO were -0.37 eV and 2.87 eV. In the case of MoS2, the edge potentials were -0.405 eV (ECB) and 2.045 eV (EVB). These values are similar to those reported by other research groups [17,62].
Under visible light, photons do not have enough energy to remove an electron from the VB of ZnO, so the route of degradation relies, mainly, on the other components of the system. In the case of MoS2, visible light is capable of removing electrons from the VB to the CB. Those electrons have enough energy to reduce oxygen (O2) molecules into superoxide radicals (•O2-), which in turn react with water to form hydroxyl radicals. Hydroxyl and superoxide radicals can oxidize and degrade CFX and LFX. Since MoS2 has a more negative ECB potential edge (-0.405 eV) than Au@ZnONPs (-0.37 eV), photoexcited electrons from the VB of MoS2 can be injected into the CB of Au@ZnONPs. Once there, O2 can gain those electrons by forming •O2- and hydroxyl radicals, which in turn can oxidize and degrade antibiotics. Multiples studies [63,64,65] have reported that the AuNPs can act as an electron sink, reducing the recombination of e-/h+ and providing active sites for the catalytic processes. Another advantage of using AuNPs is that under visible light (~580 nm depending on the AuNPs particle size) the phenomenon of surface plasmon resonance (SPR) occurs [63,64,65]. The SPR provides photoexcited electrons with enough energy to reduce the O2 molecules into •O2- radicals. Reduced graphene oxide (rGO) has a bandgap of 2.0 eV (Figure 6), allowing the use of visible light to form h+ and photoexcited electrons capable of producing superoxide and hydroxyl radicals. In addition, rGO has a high surface area which allows to create active sites for the photocatalytic process.
3. Materials and Methods
3.1. Materials
All the reactants were used as received and the solutions were prepared using deionized water (Milli-Q water; 18.2 MΩcm-1 at 25 oC). The synthesis of the ZnONPs required the use of zinc acetate (Zn(C2H3O2)2.2H2O; 98.99%) and sodium hydroxide (NaOH; 99.0%), both acquired by Sigma Aldrich (Milwaukee, WI, USA). For the incorporation of AuNPs onto the ZnONPs was used gold (III) chloride trihydrate (HAuCl4 .3H2O; ACS Reagent, 49.0+% Au basis) and sodium borohydride (NaBH4; 98.9%), provided by Sigma Aldrich (Milwaukee, WI, USA) and Acros Chemical (Newark, NJ, USA), respectively. The synthesis of the Au@ZnONPs-MoS2-rGO catalysts required the use of MoS2 (nanopowder, 90 nm diameter; 99% trace metals basis) and rGO (powder, carbon >75%, nitrogen <5%), also provided by Sigma Aldrich (Milwaukee, WI, USA). Levofloxacin (C18H20FN3O4; 98.0-102% anhydrous basis), ciprofloxacin (C17H18FN3O3; 98%), hydrogen peroxide (H2O2; 35% w/w), ethylenediaminetetraacetic acid (EDTA-Na2; ACS reagent, 99.4-100.6% powder), tert-butanol ((CH3)3COH); ACS reagent; >99.0%), 1,4 – benzoquinone (C6H4O2, reagent grade; >98%), and 0.45 μm syringe filters, were provided by Sigma Aldrich (Milwaukee, WI, USA).
3.2. Synthesis of the ZnONPs
The synthesis of ZnONPs has been described elsewhere [14], and consisted in mixing 25 mL of a 0.2 M Zn(C2H3O2)2 .2H2O solution with 50 mL of deionized water at a temperature of 60 oC. After the solution reached the desired temperature, 25 mL of a 4 M NaOH solution was added dropwise. The solution was kept at 60 oC under stirring for 2 hours. Then, the solution was allowed to cool, and the solid obtained was centrifuged and washed several times until neutral pH was reached in the washing waters. The final product was then collected and dried overnight at 60 °C.
3.3. Synthesis of the Au@ZnONPs
The incorporation of AuNPs onto ZnONPs has been previously described [14], and consisted in dispersing 500 mg of the ZnONPs in 250 mL of deionized water. The dispersion was stirred for 1 hour and after that, the desired amount of HAuCl4.3H2O was incorporated to the solution and stirred for 30 minutes. Then, a solution consisting of 10 mg of NaBH4 in 10 mL of deionized water, was added dropwise to the reaction mixture and stirred for 1 hour. The final product was collected, centrifuged, and washed several times with deionized water to remove any byproducts. The final product was dried overnight at 60 oC.
3.4. Synthesis of the Au@ZnONPs-MoS2-rGO Catalysts
The incorporation of MoS2 and rGO onto the Au@ZnONPs consisted in the dispersion of 500 mg of the Au@ZnONPs with the desired percentage of Au (1 wt.%, 3wt.%, or 5wt.%) into 250 mL of deionized water. Then, the desired amount of MoS2 was added to the dispersion. The MoS2 was previously exfoliated. The processes of the exfoliation consisted of mixing 4 g of the commercial MoS2 with 200 mL of deionized water. Then, the dispersion was sonicated using a Cole-Palmer Tip Sonicator (Cole-Parmer 750-Watt Ultrasonic Processor) for 6 hours in pulsed mode (40% amplitude, pulse on 5s, pulse off 10s). After that, the solution was kept static for 3 hours, and the supernatant was extracted and centrifuged for 30 min at 3000 rpm. Finally, the product was dried and sealed for use. The exfoliated MoS2 was added to the solution containing the dispersion of Au@ZnONPs, and 5 mg (1 wt.%) of rGO was incorporated to the reaction mixture. Subsequently, the solution was stirred for 1 hour, centrifuged, and washed several times with deionized water. The final product was dried at 60 oC overnight, and sealed until further use.
3.5. Characterization of the Photocatalysts
The BET surface area of the catalysts was analyzed using a Micrometrics ASAP 2020, with N2 adsorption isotherms at 77 K (Norcross, GA, USA). The morphology of the composites was characterized by field emission scanning electron microscopy (FESEM) using a FEI Verios 460L, equipped with a Quantax EDS Analyzer (Thermo Fisher Scientific, Hillsboro, Oregon, USA). Characterization by high-resolution transmission electron microscopy was carried out using a JEM 3000F microscope (JEOL, Peabody, MA, USA). The crystalline phase of the catalysts was studied by X-Rays diffraction (XRD) using a Bruker D8 Advance X-Ray diffractometer, operating at 40 kV and 40 mA (Billerica, MA, USA). Raman spectroscopy was carried out using a DXR Thermo Raman Microscope with a 532 nm laser source at 5mW power, and a resolution of 5 cm-1 (Waltham, MA, USA). X-Ray photoelectron spectroscopy (XPS), was carried out using a ESCALAB 220i-XL spectrometer with a non-monochromatic Mg Kα (1253.6 eV) radiation, operating at 20 mA and 12 kV (Waltham, MA, USA). The determination of the bandgap energies of the catalysts, along with the degradation of the antibiotics were carried in a Perkin Elmer Lambda 365 UV-Vis Spectrophotometer (Waltham, MA, USA). Analysis of photodegradation intermediates was carried out by gas chromatography/mass spectrometry (GC-MS), using a QP2020 Plus GC-MS (Shimadzu Corporation, Japan). Samples were separated using a 30 m × 0.25 mm i.d. capillary column. (Rtx-5MS, Restek Corporation, Bellefonte PA, USA), using helium (99.999%) as carrier gas.
3.6. Photocatalytic Experiments
Ideal conditions in terms of concentration (2μM - 50M), catalyst loading (0.5 g/L – 1.5 g/L), and pH (4 – 11) were determined for LFX before conducting the experiments. The photodegradation experiments consisted in preparing a solution of 10 μM of LFX and mixing it with 1.1 g/L of the desired catalyst. Then, the pH of the solution was adjusted to 8 by using sodium hydroxide (NaOH) or hydrochloric acid (HCl), and the solution was kept in the dark for 30 minutes under constant stirring. This was made to achieve the adsorption/desorption equilibrium between the catalyst and the solution. After that, 3 mL of a 0.005% H2O2 solution was added to the solution and the mixture was subjected to constant air bubbling to guarantee the presence of oxygen. Subsequently, the solution was surrounded with a solar simulator composed of two white light bulbs (60 watts and ca. 5200 lx). Then, the solar simulator was switched on and the reaction was conducted for 120 min at 22 oC, taking 5 mL aliquots every 10 min. The aliquots were filtered through 0.45 μm membrane filters to remove the catalyst, and were subsequently analyzed.
The ideal conditions of CPX were previously determined [17], and the experiments are similar to those described for LFX, with the difference that the catalyst loading was 1.0 g/L, the pH adjusted to 7, and the experiments performed for a maximum time of 60 min.
For the study of the intermediates of the photodegradation process, aliquots were obtained at different reaction times, which were filtered to eliminate the catalyst. These aliquots were diluted with 50 mL of deionized water and subjected to an extraction process with ethyl acetate by liquid-liquid extraction. The extract was evaporated to dryness using a rotary evaporator and then dissolved in 5 mL of methanol. GC-MS analysis was carried out by injecting 1 µL of sample into a gas chromatograph/mass spectrometer (GC-MS QP2020 Plus, Shimadzu, Japan). For both contaminants, a total of 4 injections were made, including a blank sample, using Helium as carrier gas.
4. Conclusions
The photocatalytic activity of nine catalysts containing zinc oxide, gold, molybdenum disulfide, and reduced graphene oxide was evaluated in the degradation of CFX and LFX. The results showed that LFX is more recalcitrant to degradation when compared to CFX and that the main route of degradation under visible light is the formation of hydroxyl radicals.
The CFX degradation ranged between 70% - 96%, with 1%Au@ZnONPs-3%MoS2-1%rGO being the catalyst with the highest percentage of degradation (96%) in 60 min. In the case of LFX, the degradation ranged from 85% - 99.8% in a maximum time of 120 min, and the catalyst with the highest percentage of degradation was 5%Au@ZnONPs-3%MoS2-1%rGO (99 .8%).
The use of t-butanol, 1,4-BQ and EDTA-Na2 as scavengers, together with the determination of bandgaps, allowed us to establish the possible mechanisms involved in the catalytic reaction of photodegradation of LFX and CFX.
The reaction intermediates were analyzed by GC-MS, which has made it possible to establish a possible photodegradation route for both antibiotics until reaching complete mineralization.
The recyclability tests showed that the most active catalyst in the case of LFX (5%Au@ZnONPs-3%MoS2-1%rGO) maintains ca. 93 % of its efficiency after 15 cycles. In the case of CFX, the most active catalyst was 1%Au@ZnONPs-3%MoS2-1%rGO, and after 15 cycles the efficiency was 85.7 %, lower than that observed in LFX. The results obtained suggest that the catalysts could be used for many more cycles and could also be evaluated to degrade other emerging organic pollutants in aqueous solution.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1. BET surface area of the synthesized catalysts; Figure S1: Evaluation of the initial concentration of LFX on the catalytic efficiency of 5%Au@ZnONPs-1%MoS2-1%rGO, 5%Au@ZnONPs-3%MoS2-1%rGO, and 5%Au@ ZnONPs-5%MoS2-1%rGO, in the photodegradation reaction; Figure S2: Evaluation of the catalyst loading of 5%Au@ZnONPs-1%MoS2-1%rGO, 5%Au@ZnONPs-3%MoS2-1%rGO, and 5%Au@ZnONPs-5%MoS2-1%rGO on the efficiency of the photodegradation reaction of LFX; Figure S3: Photocatalytic activity of 5%Au@ZnONPs-3%MoS2-1%rGO on the degradation of LFX under irradiation at different pH; Figure S4: Control experiments for 5%Au@ZnONPs-3%MoS2-1%rGO with LFX, under irradiation; Figure S5: Recyclability tests: 5%Au@ZnONPs-3%MoS2-1%rGO after 15 consecutive catalytic cycles of photodegradation of LFX (a); and 1%Au@ZnONPs-3%MoS2-1%rGO after 15 consecutive catalytic cycles of photodegradation of CFX (b); Figure S6: Photocatalytic activity in the presence of different scavengers under irradiation: 5%Au@ZnONPs-3%MoS2-1%rGO on the degradation of LFX at pH=8 (a); and 1%Au@ZnONPs-3%MoS2-1%rGO on the degradation of CFX at pH=7 (b).
Author Contributions
Conceptualization, A.M., and F.M.; methodology, A.M., F.M., and C.M.; formal analysis, A.M., and F.M.; investigation, A.M., L.S.-V., E.R., M.C., D.G., P.B.-R., D.O., K.F., and C.M..; resources, A.M., F.M.,M.C., C.M. and F.P.; writing—original draft preparation, A.M., and F.M.; writing—review and editing, A.M., and F.M.; supervision, A.M.,M.C., and F.M.; project administration, A.M. and F.M.; funding acquisition, A.M., F.M., E.R., C.M. and F.P. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support from NSF Center for the Advancement of Wearable Technologies-CAWT (Grant 1849243), from the Consortium of Hybrid Resilient Energy Systems CHRES (DE-NA0003982), and from the Spanish Ministry of Economy and Competitiveness, under NanoCat-Com Project (PID2021-124667OB-I00), are gratefully acknowledged.
Institutional Review Board Statement
Not applicable for studies not involving humans or animals.
Data Availability Statement
The data is contained in the article and is available from the corresponding authors on reasonable request.
Acknowledgments
The authors acknowledge Raúl S García for the development of part of the catalytic measurements. The facilities provided by the National Center for Electron Microscopy at Complutense University of Madrid (Spain), by “Instituto de Micro y Nanotecnología IMN-CNM, CSIC, CEI UAM + CSIC” and by the Materials Characterization Center at University of Puerto Rico are gratefully acknowledged. K.F. thanks PR NASA Space Grant Consortium for a graduate fellowship (#80NSSC20M0052). D.O. thanks Consortium of Hybrid Resilient Energy Systems (CHRES) for a graduate fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goujon, A. 8 Billion and Then What? Population Council, In The World at 8 Billion 2022, 16–17.
- Van Vliet, M.T.H.; Jones, E.R.; Florke, M.; Franssen, W.H.P.; Hanasaki, N.; Wada, Y.; Yearsley, J.R. Global water scarcity including surface water quality and expansions of clean water technologies. Environ. Res. Lett. 2021, 16, 024020. [Google Scholar] [CrossRef]
- Ricart, S.; Villar-Navascués, R.A.; Hernández-Hernández, M.; Rico-Amorós, A.M.; Olcina-Cantos, J.; Moltó-Mantero, E. Extending Natural Limits to Address Water Scarcity? The Role of Non-Conventional Water Fluxes in Climate Change Adaptation Capacity: A Review. Sustainability 2021, 13, 2473. [Google Scholar] [CrossRef]
- Ungureanu, N.; Vlăduț, V.; Voicu, G. Water Scarcity and Wastewater Reuse in Crop Irrigation. Sustainability 2020, 12, 9055. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Zhang, Q.; Liu, P.; Guo, R.; Jin, S.; Liu, J.; Chen, L.; Ma, Z.; Liu, Y. Evaluation and Analysis of Water Quality of Marine Aquaculture Area. Int. J. Environ. Res. Public Health 2020, 17, 1446. [Google Scholar] [CrossRef]
- De Baat, M.L.; Van der Oost, R.; Van der Lee, G.H.; Wieringa, N.; Hamers, T.; Verdonschot, P.F.M.; De Voogt, P.; Kraak, M.H.S. Advancements in effect-based surface water quality assessment. Water Research 2020, 183, 116017. [Google Scholar] [CrossRef]
- Serwecińska, L. Antimicrobials and Antibiotic-Resistant Bacteria: A Risk to the Environment and to Public Health. Water 2020, 12, 3313. [Google Scholar] [CrossRef]
- Zhuang, M.; Achmon, Y.; Cao, Y.; Liang, X.; Chen, L.; Wang, H.; Siame, B.A.; Leung, K.Y. Distribution of antibiotic resistance genes in the environment. Environ. Pollution 2021, 285, 117402. [Google Scholar] [CrossRef]
- Ghernaout, D.; Elboughdiri, N. Antibiotics Resistance in Water Mediums: Background, Facts, and Trends. Appl. Engine. 2020, 4, 1–6. [Google Scholar]
- Zheng, D.; Yin, G.; Liu, M.; Chen, C.; Jiang, Y.; Hou, L.; Zheng, Y. A systematic review of antibiotics and antibiotic resistance genes in estuarine and coastal environments. Sci. Total Environ. 2021, 777, 146009. [Google Scholar] [CrossRef]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Makabenta, J.M.V.; Nabawy, A.; Li, C.H.; Schmidt-Malan, S.; Patel, R.; Rotello, V.M. Nanomaterial-based therapeutics for antibiotic-resistant bacterial infections. Nat. Rev. Microbiol. 2021, 19, 23–36. [Google Scholar] [CrossRef]
- Pinilla, S.; Machín, A.; Park, S.-H.; Arango, J.C.; Nicolosi, V.; Márquez-Linares, F.; Morant, C. TiO2 -Based Nanomaterials for the Production of Hydrogen and the Development of Lithium-Ion Batteries. J. Phys. Chem. B 2018, 122, 972–983. [Google Scholar] [CrossRef]
- Machín, A.; Cotto, M.; Duconge, J.; Arango, J.C.; Morant, C.; Pinilla, S.; Soto-Vázquez, L.; Resto, E.; Márquez, F. Hydrogen Production via Water Splitting Using Different Au@ZnO Catalysts under UV–Vis Irradiation. J. Photochem. Photobiol. A: Chemistry 2018, 353, 385–394. [Google Scholar] [CrossRef]
- Machín, A.; Arango, J.C.; Fontánez, K.; Cotto, M.; Duconge, J.; Soto-Vázquez, L.; Resto, E.; Petrescu, F.I.T.; Morant, C.; Márquez, F. Biomimetic Catalysts Based on Au@ZnO–Graphene Composites for the Generation of Hydrogen by Water Splitting. Biomimetics 2020, 5, 39. [Google Scholar] [CrossRef]
- Machín, A.; Soto-Vázquez, L.; Colón-Cruz, C.; Valentín-Cruz, C.A.; Claudio-Serrano, G.J.; Fontánez, K.; Resto, E.; Petrescu, F.I.; Morant, C.; Márquez, F. Photocatalytic Activity of Silver-Based Biomimetics Composites. Biomimetics 2021, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Machín, A.; Fontánez, K.; Duconge, J.; Cotto, M.C.; Petrescu, F.I.; Morant, C.; Márquez, F. Photocatalytic Degradation of Fluoroquinolone Antibiotics in Solution by Au@ZnO-rGO-gC3N4 Composites. Catalysts 2022, 12, 166. [Google Scholar] [CrossRef]
- Soto-Vázquez, L.; Rolón-Delgado, F.; Rivera, K.; Cotto, M.C.; Ducongé, J.; Morant, C.; Pinilla, S.; Márquez-Linares, F.M. Catalytic Use of TiO2 Nanowires in the Photodegradation of Benzophenone-4 as an Active Ingredient in Sunscreens. J. Environ. Management 2019, 247, 822–828. [Google Scholar] [CrossRef]
- Martins, P.; Kappert, S.; Nga Le, H.; Sebastian, V.; Kühn, K.; Alves, M.; Pereira, L.; Cuniberti, G.; Melle-Franco, M.; Lanceros-Méndez, S. Enhanced Photocatalytic Activity of Au/TiO2 Nanoparticles against Ciprofloxacin. Catalysts 2020, 10, 234. [Google Scholar] [CrossRef]
- Kutuzova, A.; Dontsova, T.; Kwapinski, W. Application of TiO2-Based Photocatalysts to Antibiotics Degradation: Case of Sulfamethoxazole, Trimethoprim and Ciprofloxacin. Catalysts 2021, 11, 728. [Google Scholar] [CrossRef]
- Qin, Z.; Sun, H.; Tang, Y.; Chang, Z.; Yin, S.; Liu, Z. Bio-inspired hierarchical assembly of Au/ZnO decorated carbonized spinach leaves with enhanced photocatalysis performance. J. Alloys Comp. 2020, 829, 154393. [Google Scholar] [CrossRef]
- Chankhanittha, T.; Komchoo, N.; Senasu, T.; Piriyanon, J.; Youngme, S.; Hemavibool, K.; Nanan, S. Silver decorated ZnO photocatalyst for effective removal of reactive red azo dye and ofloxacin antibiotic under solar light irradiation. Colloids Surf. A Physicochem. Eng. Asp. 2021, 626, 127034. [Google Scholar] [CrossRef]
- Krishnan, U.; Kaur, M.; Kaur, G.; Singh, K.; Dogra, A.R.; Kumar, M.; Kumar, A. MoS2/ZnO nanocomposites for efficient photocatalytic degradation of industrial pollutants. Mater. Res. Bull. 2019, 111, 212–221. [Google Scholar] [CrossRef]
- Ahamad, T.; Naushad, M.; Al-Saeedi, S.I.; Almotairi, S.; Alshehri, S.M. Fabrication of MoS2/ZnS embedded in N/S doped carbon for the photocatalytic degradation of pesticide. Mater. Lett. 2020, 263, 127271. [Google Scholar] [CrossRef]
- Benavente, E.; Durán, F.; Sotomayor-Torres, C.; González, G. Heterostructured layered hybrid ZnO/MoS2 nanosheets with enhanced visible light photocatalytic activity. J. Phys. Chem. Solids 2018, 113, 119–124. [Google Scholar] [CrossRef]
- Govindaraj, T.; Mahendran, C.; Manikandan, V.S.; Archana, J.; Mohd Shkir, J.; Chandrasekaran, J. Fabrication of WO3 nanorods/RGO hybrid nanostructures for enhanced visible-light-driven photocatalytic degradation of Ciprofloxacin and Rhodamine B in an ecosystem. J. Alloys Comp. 2021, 868, 159091. [Google Scholar] [CrossRef]
- Arya, M.; Kaur, M.; Kaur, A.; Singh, S.; Devi, P.; Kansal, S.K. Hydrothermal synthesis of rGO-Bi2WO6 heterostructure for the photocatalytic degradation of levofloxacin. Op. Mater. 2020, 107, 110126. [Google Scholar] [CrossRef]
- Raja, A.; Rajasekaran, P.; Selvakumar, K.; Arunpandian, M.; Kaviyarasu, K.; Bahadur, A.; Swaminathan, M. Visible active reduced graphene oxide-BiVO4-ZnO ternary photocatalyst for efficient removal of ciprofloxacin. Sep. Purif. Technol. 2020, 233, 115996. [Google Scholar] [CrossRef]
- Arjun, N.; Uma, K.; Pan, GT.; Yang, T.C.K.; Sharmila, G. One-pot synthesis of covalently functionalized reduced graphene oxide–polyaniline nanocomposite for supercapacitor applications. Clean Techn. Environ. Policy 2018, 20, 2025–2035. [Google Scholar] [CrossRef]
- Fontánez, K.; García, D.; Ortiz, D.; Sampayo, P.; Hernández, L.; Cotto, M.; Ducongé, J.; Díaz, F.; Morant, C.; Petrescu, F.; Machín, A.; Márquez, F. Biomimetic Catalysts Based on Au@TiO2-MoS2-CeO2 Composites for the Production of Hydrogen by Water Splitting. Int. J. Mol. Sci. 2023, 24, 363. [Google Scholar] [CrossRef]
- Pushkareva, I.V.; Pushkarev, A.S.; Kalinichenko, V.N.; Chumakov, R.G.; Soloviev, M.A.; Liang, Y.; Millet, P.; Grigoriev, S.A. Reduced Graphene Oxide-Supported Pt-Based Catalysts for PEM Fuel Cells with Enhanced Activity and Stability. Catalysts 2021, 11, 256. [Google Scholar] [CrossRef]
- Usharani, B.; Murugadoss, G.; Rajesh Kumar, M.; Gouse Peera, S.; Manivannan, V. Reduced Graphene Oxide–Metal Oxide Nanocomposites (ZrO2 and Y2O3): Fabrication and Characterization for the Photocatalytic Degradation of Picric Acid. Catalysts 2022, 12, 1249. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, B.P.; Dhar, S.; Gondorf, A.; Spasova, M. Effect of surface groups on the luminescence property of ZnO nanoparticles synthesized by sol–gel route. Surf. Sci. 2012, 606, L13–L17. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Q.; Yap, C.C.R.; Tay, B.K.; Edwin, T.H.T.; Olivier, A.; Baillargeat, D. From Bulk to Monolayer MoS2: Evolution of Raman Scattering. Adv. Funct. Mater. 2012, 22, 1385–1390. [Google Scholar] [CrossRef]
- Castellanos-Gomez, A.; Quereda, J.; van der Meulen, H.P.; Agraït, N.; Rubio-Bollinger, G. Spatially Resolved Optical Absorption Spectroscopy of Single- and Few-Layer MoS 2 by Hyperspectral Imaging. Nanotechnol. 2016, 27, 115705. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Rehman, W.; Mansoob, M.; Tauseef, M.; Gul, A.; Haq, S.; Ullah, R.; Rab, A.; Menaa, F. Phytogenic Fabrication of ZnO and Gold Decorated ZnO Nanoparticles for Photocatalytic Degradation of Rhodamine B. J. Environ. Chem. Eng. 2021, 9, 104725. [Google Scholar] [CrossRef]
- Dediu, V.; Busila, M.; Tucureanu, V.; Bucur, F.I.; Iliescu, F.S.; Brincoveanu, O.; Iliescu, C. Synthesis of ZnO/Au Nanocomposite for Antibacterial Applications. Nanomater. 2022, 12, 3832. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Shen, W.; Li, L.; Wu, S.; Wang, W. 3D CoFe2O4 Nanorod/Flower-Like MoS2 Nanosheet Heterojunctions as Recyclable Visible Light-Driven Photocatalysts for the Degradation of Organic Dyes. Appl. Surf. Sci. 2018, 447, 711–723. [Google Scholar] [CrossRef]
- Ghasemipour, P.; Fattahi, M.; Rasekh, B.; Yazdian, F. Developing the Ternary ZnO Doped MoS2 Nanostructures Grafted on CNT and Reduced Graphene Oxide (rGO) for Photocatalytic Degradation of Aniline. Sci. Rep. 2020, 10, 4414. [Google Scholar] [CrossRef]
- Tarekegne, A.; Worku, D. Synthesis and Characterization of Reduced Graphene Oxide (rGO) Started from Graphene Oxide (GO) Using the Tour Method with Different Parameters. Adv. Mater. Sci. Eng. 2019, 14, 289–298. [Google Scholar]
- Jayachandiran, J.; Yesuraj, J.; Arivanandhan, M.; Raja, A.; Suthanthiraraj, A.; Jayavel, R.; Nedumaran, D. Synthesis and Electrochemical Studies of rGO/ZnO Nanocomposite for Supercapacitor Application. J. Inorg. Organomet. Polym. 2018, 28, 365–379. [Google Scholar] [CrossRef]
- Naseri, A.; Samadi, M.; Mahmoodi, N.M.; Pourjavadi, A.; Mehdipour, H.; Moshfegh, A.Z. Tuning Composition of Electrospun ZnO/CuO Nanofibers: Toward Controllable and Efficient Solar Photocatalytic Degradation of Organic Pollutants. J. Phys. Chem. C 2017, 121, 3327–3338. [Google Scholar] [CrossRef]
- Qiao, Y.; Li, J.; Li, H.; Fang, H.; Fan, D.; Wang, W. A Label-Free Photoelectrochemical Aptasensor for Bisphenol A Based on Surface Plasmon Resonance of Gold Nanoparticle-Sensitized ZnO Nanopencils. Biosens. Bioelectron. 2016, 86, 315–320. [Google Scholar] [CrossRef]
- Machín, A.; Arango, J.C.; Fontánez, K.; Cotto, M.; Duconge, J.; Soto-Vázquez, L.; Resto, E.; Petrescu, F.I.T.; Morant, C.; Márquez, F. Biomimetic Catalysts Based on Au@ZnO–Graphene Composites for the Generation of Hydrogen by Water Splitting. Biomimetics 2020, 5, 39. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, J.S.; Lee, K.Y.; Park, H.P.; Bae, J.S.; Mubarak, M.; Lee, H. Elucidation of an intrinsic parameter for evaluating the electrical quality of graphene flakes. Sci. Rep. 2019, 9, 557. [Google Scholar] [CrossRef]
- Briggs, D.; Seah, M. Practical Surface Analysis; Wiley: New York, NY, USA, 1994. [Google Scholar]
- Morimoto, N.; Kubo, T.; Nishina, Y. Tailoring the oxygen content of graphite and reduced graphene oxide for specific applications. Sci. Rep. 2016, 6, 21715. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Ambrosi, A.; Chua, C.K.; Pumera, M. Electron transfer properties of chemically reduced graphene materials with different oxygen contents. J. Mater. Chem. A 2014, 2, 10668–10675. [Google Scholar] [CrossRef]
- Zagorac, D.; Zagorac, J.; Pejić, M.; Matović, B.; Schön, J.C. Band Gap Engineering of Newly Discovered ZnO/ZnS Polytypic Nanomaterials. Nanomater. 2022, 12, 1595. [Google Scholar] [CrossRef]
- Rahman, I.A.; Purqon, A. First Principles Study of Molybdenum Disulfide Electronic Structure. J. Phys.: Conf. Ser. 2017, 877, 012026. [Google Scholar] [CrossRef]
- Shen, Y.; Yang, S.; Zhou, P.; Sun, Q.; Wang, P.; Wan, L.; Li, J.; Chen, L.; Wang, X.; Ding, S.; Zhang, D.W. Evolution of the band-gap and optical properties of graphene oxide with controllable reduction level. Carbon 2013, 62, 157–164. [Google Scholar] [CrossRef]
- Machín, A.; Fontánez, K.; García, D.; Sampayo, P.; Colón-Cruz, C.; Claudio-Serrano, G.J.; Soto-Vázquez, L.; Resto, E.; Petrescu, F.I.; Morant, C.; Márquez, F. Hydrogen Production and Degradation of Ciprofloxacin by Ag@TiO2-MoS2 Photocatalysts. Catalysts 2022, 12, 267. [Google Scholar] [CrossRef]
- Soto-Vázquez, L.; Cotto, M.; Ducongé, J.; Morant, C.; Márquez, F. Synthesis and photocatalytic activity of TiO2 nanowires in the degradation of p-aminobenzoic acid: A comparative study with a commercial catalyst. J. Environ. Manage. 2016, 167, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Prabavathi, S.L.; Saravanakumar, K.; Park, C.M.; Muthuraj, V. Photocatalytic degradation of levofloxacin by a novel Sm6WO12/g-C3N4 heterojunction: Performance, mechanism and degradation pathways. Sep. Purif. Technol. 2021, 257, 117985. [Google Scholar] [CrossRef]
- Jourshabani, M.; Shariatinia, Z.; Badiei, A. Synthesis and Characterization of Novel Sm2O3/S-Doped g-C3N4 Nanocomposites with Enhanced Photocatalytic Activities under Visible Light Irradiation. Appl. Surf. Sci. 2018, 427, 375–387. [Google Scholar] [CrossRef]
- Tsai, C.K.; Lee, Y.C.; Nguyen, T.T.; Horng, J.J. Levofloxacin Degradation under Visible-LED Photo-Catalyzing by a Novel Ternary Fe–ZnO/WO3 Nanocomposite. Chemosphere 2022, 298, 134285. [Google Scholar] [CrossRef]
- Xing, Z.; Wang, Z.; Chen, W.; Zhang, M.; Fu, X.; Gao, Y. Degradation of Levofloxacin in Wastewater by Photoelectric and Ultrasonic Synergy with TiO2/g-C3N4@AC Combined Electrode. J. Environ. Manage. 2023, 330, 117168. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Hu, X.; Peng, Q.; Zhou, L.; Tan, X.; Jiang, L.; Tang, C.; Wang, H.; Liu, S.; Wang, Y.; Ning, Z. Mechanisms Underlying the Photocatalytic Degradation Pathway of Ciprofloxacin with Heterogeneous TiO2. Chem. Eng. J. 2020, 380, 122366. [Google Scholar] [CrossRef]
- Prabavathi, S.L.; Saravanakumar, K.; Nkambule, T.T.I.; Muthuraj, V.; Mamba, G. Enhanced Photoactivity of Cerium TungstateModified Graphitic Carbon Nitride Heterojunction Photocatalyst for the Photodegradation of Moxifloxacin. J. Mater. Sci. Mater. Electron. 2020, 31, 11434–11447. [Google Scholar] [CrossRef]
- Cao, J.; Li, X.; Lin, H.; Chen, S.; Fu, X. In Situ Preparation of Novel p–n Junction Photocatalyst BiOI/(BiO)2CO3 with Enhanced Visible Light Photocatalytic Activity. J. Hazard. Mater. 2012, 239–240, 316–324. [Google Scholar] [CrossRef]
- Nethercot, A.H. Prediction of Fermi Energies and Photoelectric Thresholds Based on Electronegativity Concepts. Phys. Rev. Lett. 1974, 33, 1088–1091. [Google Scholar] [CrossRef]
- Chen, C.; Bi, W.; Xia, Z.; Yuan, W.; Li, L. Hydrothermal Synthesis of the CuWO4/ZnO Composites with Enhanced Photocatalytic Performance. ACS Omega 2020, 5, 13185–13195. [Google Scholar] [CrossRef]
- Badilescu, S.; Raju, D.; Bathini, S.; Packirisamy, M. Gold Nano-Island Platforms for Localized Surface Plasmon Resonance Sensing: A Short Review. Molecules 2020, 25, 4661. [Google Scholar] [CrossRef] [PubMed]
- Bereli, N.; Bakhshpour, M.; Topçu, A.A.; Denizli, A. Surface Plasmon Resonance-Based Immunosensor for Igm Detection with Gold Nanoparticles. Micromachines 2021, 12, 1092. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-W.; Pham, T.-T.-H.; Hsu, C.-C.; Lien, C.-H.; Phan, Q.-H. Single-Layer-Graphene-Coated and Gold-Film-Based Surface Plasmon Resonance Prism Coupler Sensor for Immunoglobulin G Detection. Sensors 2022, 22, 1362. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Field emission scanning electron microscopy (FESEM) images of the different components of the catalysts: ZnONPs at 50,000X (a) and 100,000X (b); rGO (c); and MoS2 (d).
Figure 1.
Field emission scanning electron microscopy (FESEM) images of the different components of the catalysts: ZnONPs at 50,000X (a) and 100,000X (b); rGO (c); and MoS2 (d).

Figure 2.
High resolution transmission electron microscopy (HRTEM) micrographs of the precursors: Au@ZnONPs at a magnification of 100,000X (a); Au@ZnONPs with two insets showing the selected area electron diffraction, SAED, and a micrograph at higher magnification showing the lattice fringes (b); rGO showing the lattice fringes (c); and the exfoliated MoS2 with an inset showing the SAED (d).
Figure 2.
High resolution transmission electron microscopy (HRTEM) micrographs of the precursors: Au@ZnONPs at a magnification of 100,000X (a); Au@ZnONPs with two insets showing the selected area electron diffraction, SAED, and a micrograph at higher magnification showing the lattice fringes (b); rGO showing the lattice fringes (c); and the exfoliated MoS2 with an inset showing the SAED (d).

Figure 3.
Raman spectra of rGO (a); ZnONPs (b); MoS2 (c); and 5%Au@ZnONPs-3%MoS2-1%rGO (d).
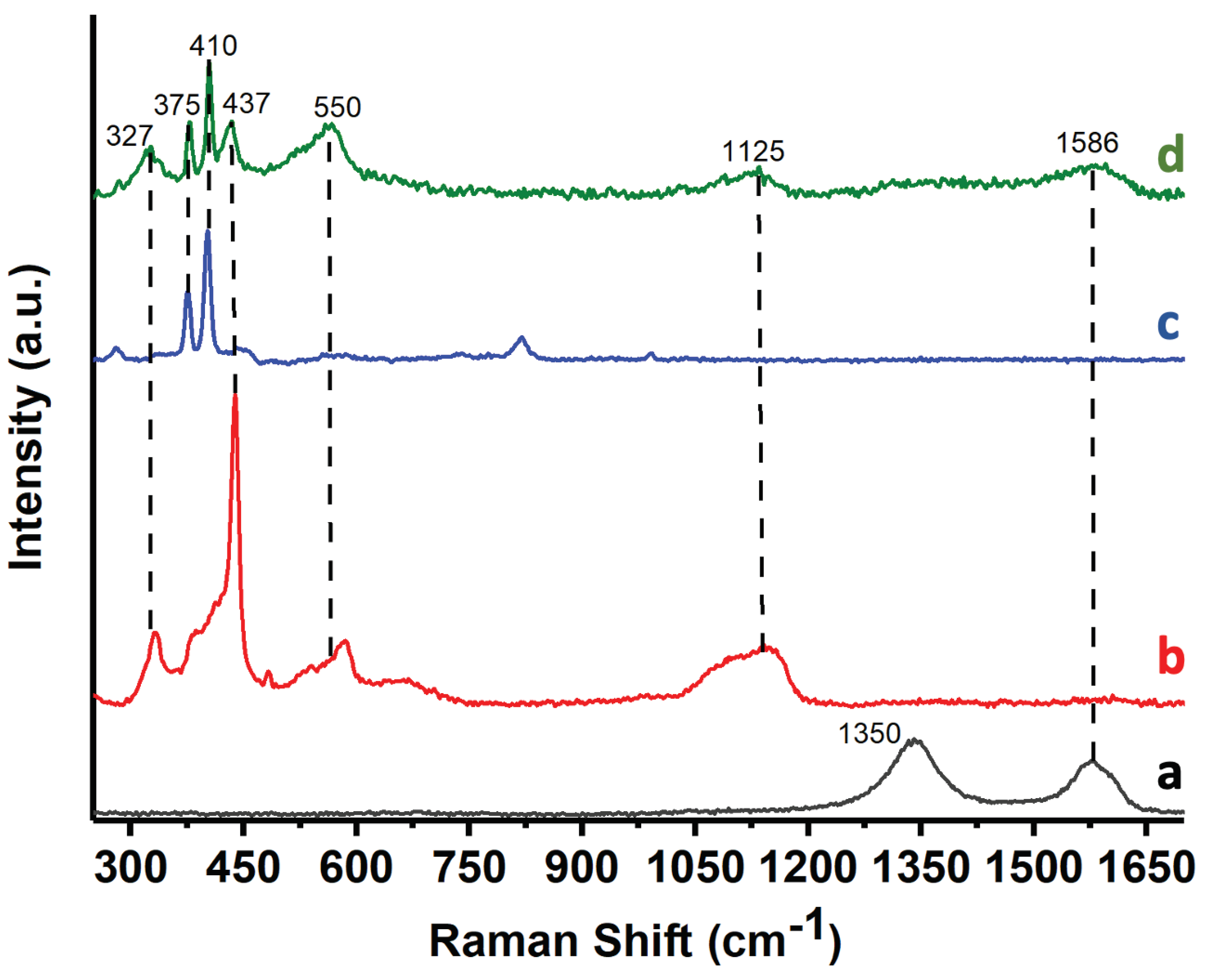
Figure 4.
XRD patterns of ZnONPs (a); 5%Au@ZnONPs (b); MoS2 (c); rGO (d); and 5%Au@ZnONPs-3%MoS2-1%rGO (d).
Figure 4.
XRD patterns of ZnONPs (a); 5%Au@ZnONPs (b); MoS2 (c); rGO (d); and 5%Au@ZnONPs-3%MoS2-1%rGO (d).
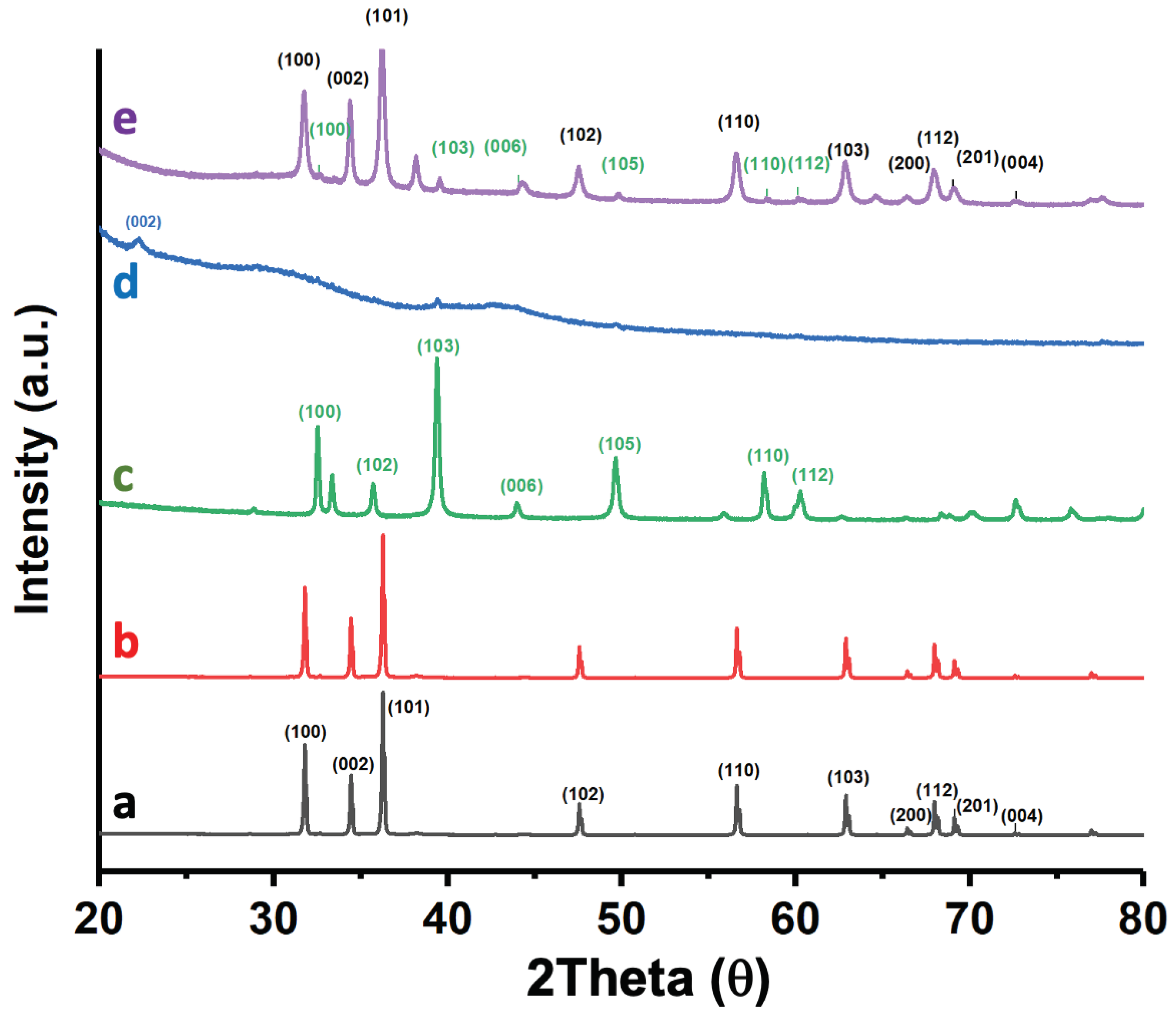
Figure 5.
XPS core level spectra for 5%Au@ZnONPs-3%MoS2-1%rGO: Zn2p (a); O1s (b); Au4f (c); C1s (d); and Mo3d-S2s (e).
Figure 5.
XPS core level spectra for 5%Au@ZnONPs-3%MoS2-1%rGO: Zn2p (a); O1s (b); Au4f (c); C1s (d); and Mo3d-S2s (e).
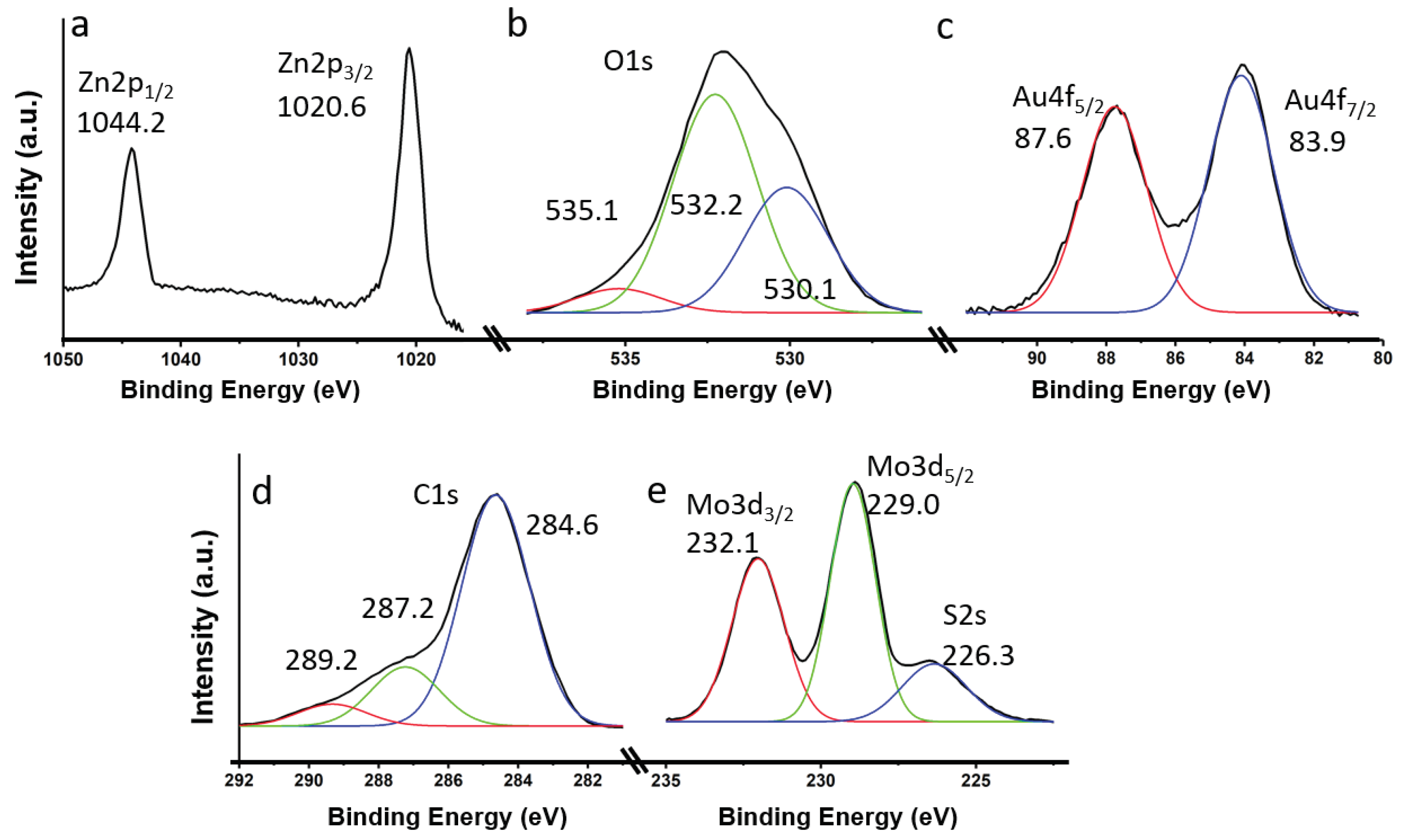
Figure 6.
Tauc plots of (αhn)2 versus energy (eV), and determination of the bandgap energy of ZnONPs (a); 5%Au@ZnONPs (b); MoS2 (c); 5%Au@ZnONPs-3%MoS2-1%rGO (d); and rGO (e).
Figure 6.
Tauc plots of (αhn)2 versus energy (eV), and determination of the bandgap energy of ZnONPs (a); 5%Au@ZnONPs (b); MoS2 (c); 5%Au@ZnONPs-3%MoS2-1%rGO (d); and rGO (e).
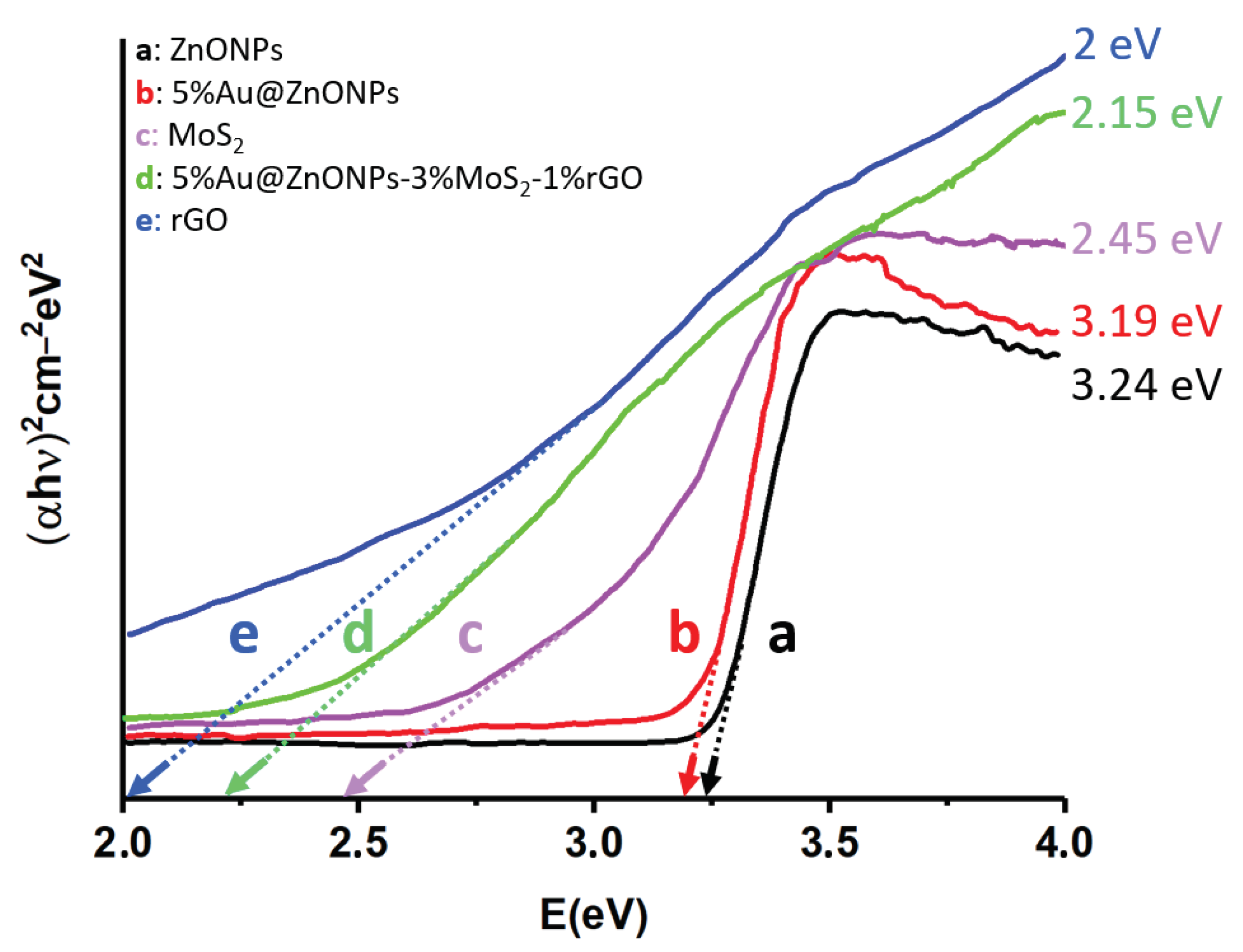
Figure 7.
Photodegradation rate of the antibiotics CFX (a,b,c) and LFX (d,e,f) as a function of time by the different synthesized catalysts.
Figure 7.
Photodegradation rate of the antibiotics CFX (a,b,c) and LFX (d,e,f) as a function of time by the different synthesized catalysts.
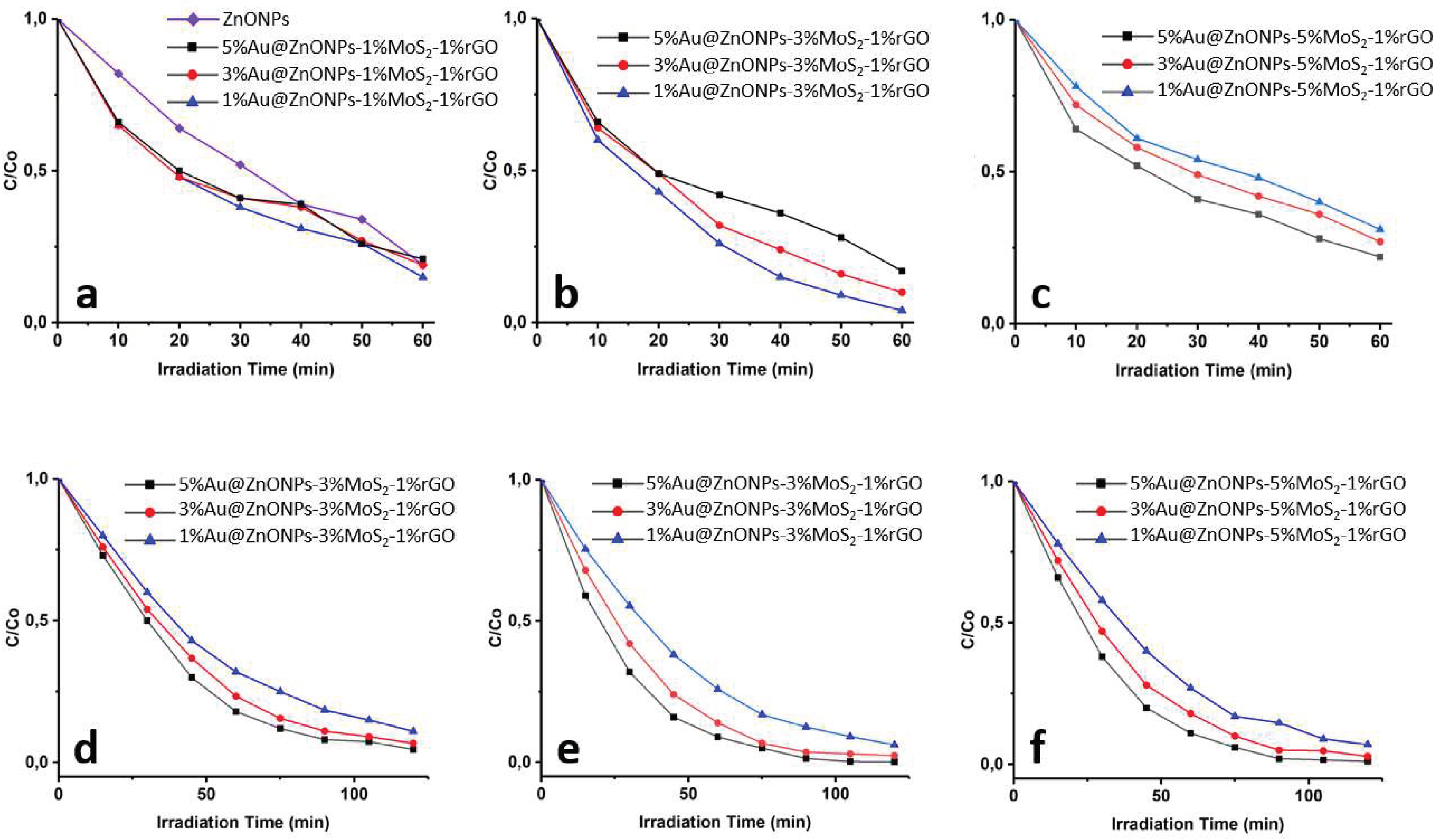
Figure 8.
Degradation pathway of LFX (a) and CFX (b) under the effect of the most active catalysts (5%Au@ZnONPs-3%MoS2-1%rGO and 1%Au@ZnONPs-3%MoS2-1% rGO, respectively).
Figure 8.
Degradation pathway of LFX (a) and CFX (b) under the effect of the most active catalysts (5%Au@ZnONPs-3%MoS2-1%rGO and 1%Au@ZnONPs-3%MoS2-1% rGO, respectively).
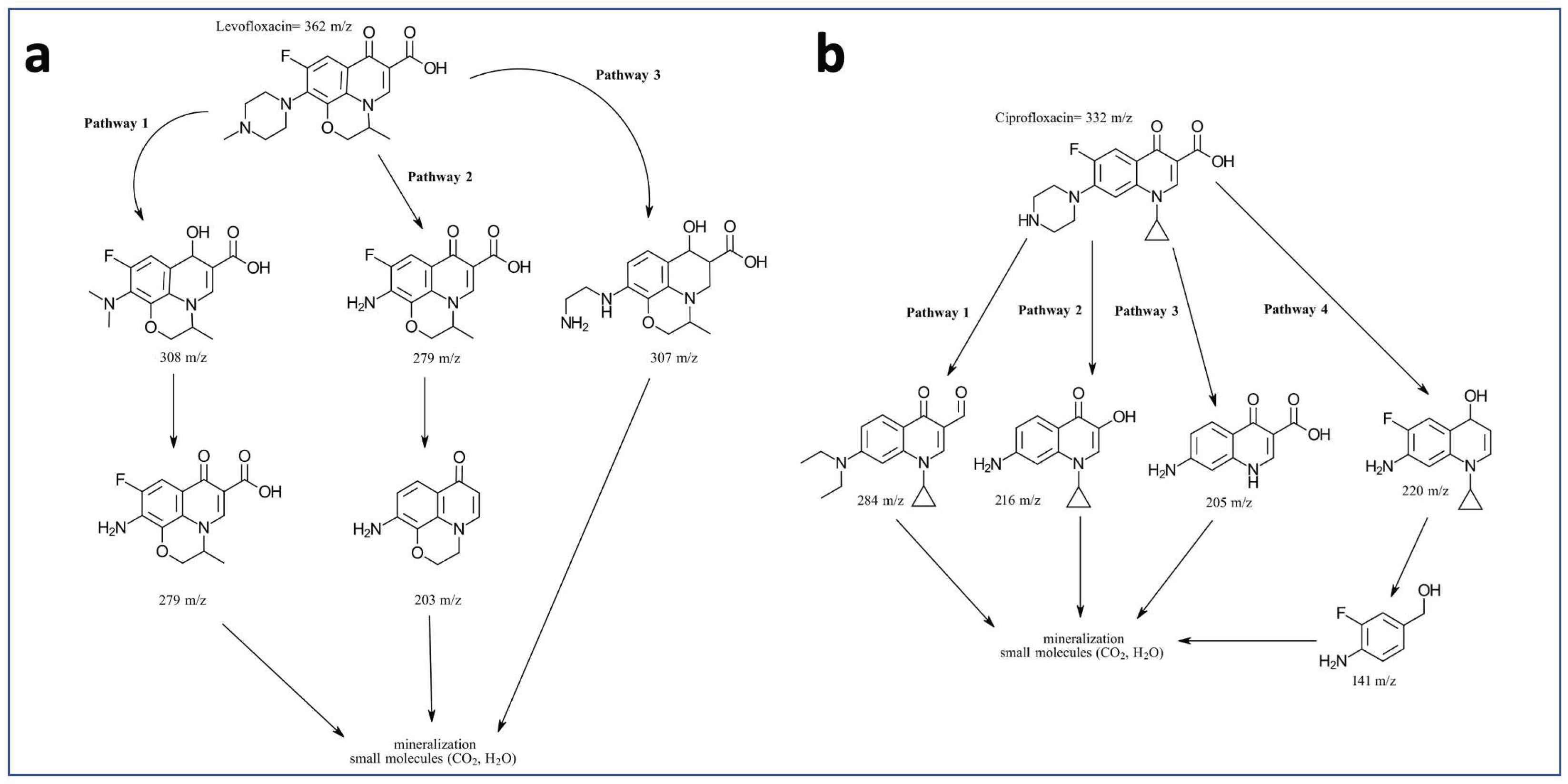
Figure 9.
Schematic diagram of the proposed mechanism for CFX and LFX photodegradation using Au@ZnONPs-MoS2-rGO catalysts under visible irradiation.
Figure 9.
Schematic diagram of the proposed mechanism for CFX and LFX photodegradation using Au@ZnONPs-MoS2-rGO catalysts under visible irradiation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



