
Submitted:
23 January 2023
Posted:
24 January 2023
You are already at the latest version
Abstract
Our understanding of how microbiome signatures are modulated in wild fish populations remains poorly known and has, until now, mostly been inferred from studies in commercial and farmed fish populations. Here, we have studied for the first time changes in the skin and blood microbiomes of the Salmo trutta population of the volcanic Kerguelen archipelago located at the northern limit of the Antarctic Ocean. Kerguelen is a natural framework of population expansion and a likely situation under further climate change in distribution areas. Our results showed that S. trutta of Kerguelen has a microbiome signature distinct from those of salmonids of the Northern Hemisphere. Our study also revealed that the skin and blood microbiomes differ between sedentary and migratory S. trutta. While 18 phyla were shared between both groups of trout, independent of the compartment, six phyla were unique to migratory trout. Further analyses showed that microbiome signatures undergo significant site-specific variations that correlate, in some cases, to the peculiarity of specific ecosystems. Our study also revealed the presence of potential pathogens at particular sites and the impact of abiotic factors on the microbiome, most notably due to the volcanic nature of the environment. This study contributes to a better understanding of the factors that modulate the microbiome signatures of migratory and sedentary fish populations. It will also help better monitor climate change's impacts on the colonization process in the sub-Antarctic region.
Keywords:
Blood microbiome
; skin microbiome
; migration
; Kerguelen Islands
; 16S rRNA
1. Introduction
The microbiota is now recognized as a key player in health and disease progression. Defining the microbiome signature, which includes nucleic acid originating from microorganisms, has thus become an essential component of the biomarker schematic in medicine and ecology. In clinical settings, for example, specific signatures will determine if a cancer patient will respond to particular therapies [1,2]. This progress has been greatly facilitated by the development of next-generation sequencing (NGS) technologies for the analysis of 16S ribosomal RNA (rRNA) gene amplicons, which allows rapid evaluation of microbial biodiversity while overcoming the limitations of cultivation-based methods. Despite these technical developments, our understanding of the microbiome in wild animals is only in its infancy. This is particularly true in fish populations, most notably those inhabiting environments sensitive to pollution and climate change. Moreover, until now, studies of the fish microbiome have mostly focused on the gut microbiota and, to a lesser extent, on the skin (mucosal microbiota). In fish, in particular, the skin mucus represents the first barrier defense against infectious stressors in the environment. The nonpathogenic skin microbial flora helps the immune system protect itself against pathogens [3,4]. Its composition depends on several factors, most notably host genetics and their surrounding environment. Captivity, pollution, and habitat transition are other factors known to affect the microbiome signature of animals [5,6,7]. However, most microbiome studies in fish have been conducted in fish farms or experimental settings simulating freshwater-to-seawater transitions [8,9]. We still need to learn more about how the skin microbiome differs between migratory and sedentary wild fish populations. Considering the importance of the microbiome as a biomarker and the need to adopt less costly and invasive procedures, the concept of “circulating microbiome DNA” (cmDNA) has been developed as an alternative to studying the microbiome in clinical settings [10]. Since the middle of the 20th century, we have known that DNA fragments are present in the blood [11], but it is only in recent years that the advent of NGS methods has allowed us to better understand the origin of these circulating DNA fragments. We now know that blood contains a vast array of self- and nonself DNA fragments originating from multiple tissues, including infected tissues, and that a significant percentage of the DNA within the blood originates from microorganisms [12,13,14]. Combined with the 16S rRNA approach, cmDNA analysis has thus become a central tool for studying an individual's health status and measuring its ability to respond to treatment or environmental stressors [15,16,17,18,19]. Several recent studies have applied this concept to study the hemolymphatic microbiome of bivalves and its changes in response to environmental stressors [20,21,22]. The existence of a blood microbiome is a concept that is now widely accepted in humans and animals (including pigs, cows, goats, rodents, camels, and dogs [21,23,24,25,26,27,28,29]. A blood microbiome has also been reported in fish and discussed in reviews [5,30,31,32]. The existence of a circulating microbiome has also been reported in invertebrates. In many cases, this has been applied to facilitate the detection of pathogens. The Kerguelen Islands (also known as the Desolation Islands) are located in the Southern Ocean just north of the polar front. Long-term monitoring studies have shown that this archipelago, which comprises more than 300 islands that cover more than 7000 km2 and almost 3 000 km of shoreline, is particularly sensitive to global warming [33,34]. Previously devoid of freshwater fish, the Kerguelen Islands were populated at multiple sites with various stocks of fish over three decades (1962–1993). Overall, 22 successful imports were made with eggs collected from 8 salmonid species in the Northern Hemisphere [35]. Salmo trutta (S. trutta) is among the different species that successfully colonized the largest number of watersheds following the early occurrence of alternative anadromous (e.g., migratory) to pursue its life cycle in seawater [36]. Given its remoteness and limited anthropogenic activities, Kerguelen’s salmonid populations thus provide a unique subpolar environment to study the impact of the environment and migratory activity on microbiome compartments and to understand better how climate change can impact these populations [37]. In the present work, we studied the skin (mucus) and circulating (blood) microbiomes of migratory and sedentary S. trutta populations collected at different sites in Kerguelen. We first assessed the overall composition of both the skin and blood microbiota in both resident and migrating trout. We then proceeded to a comparison for both skin and blood samples and resident and migratory traits. We also analyzed site-specific variations to establish whether the local environment could trigger substantial contrasts in microbiota. Our study also revealed the presence of potential pathogens at certain sites and the impact of abiotic factors on the microbiome, most notably due to the volcanic nature of the environment. Finally, we further showed that FTA® Cards-based sampling is perfectly adapted for establishing mucosal and circulating 16S rRNA microbiome signatures, an interesting avenue for long-term monitoring programs in remote and sensitive polar environments.
2. Materials and Methods
2.1. Sample Collection
A total of 83 skin and blood microbiomes collected from 52 trout samples were analyzed (Table S1). Sedentary trout were captured by electrofishing in freshwater rivers, and migratory trout were captured by trolling (fly fishing). Immediately after capture, the fish were anesthetized, as described by Marandel et al. (2018). Blood was immediately withdrawn from the caudal vein using sterile nonheparinized syringes and spotted on Whatman 903™ Flinders Technology Associates filter paper (FTA® Cards) (Sigma–Aldrich, Oakville, ON, Canada). To avoid cross-contamination between samples, all samples were allowed to air dry and kept individually in a plastic bag with a desiccant, as previously described [39]. Mucus samples were collected using a sterile scalpel blade and scraping along the fish's lateral line and immediately spotted to cover an entire disc of the FTA cards. The same preservation method was applied as above. The care and use of field-sampled animals complied with the Government of France's animal welfare laws, guidelines and policies (Comité d'Ethique) as approved by the Terres Australes et Antarctiques Françaises administration. All methods are reported under ARRIVE guidelines.
2.2. DNA Extraction, Preprocessing and Sequencing
All procedures were conducted in a white room where pressure, temperature, and humidity were controlled to eliminate contaminants. Individual discs were cut from the FTA® Cards using a sterile 5.0 mm single round hole punch. Total DNA was isolated using the QIAamp DNA Investigator Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. DNA was quantified in duplicate using a Quant-iT™ PicoGreen® dsDNA detection kit (Molecular Probes, Eugene OR). Amplification of the 16S ribosomal RNA (rRNA) genes and 16S gene amplicon sequencing for all DNA samples were performed at the Centre d'Expertise et de Services Génome Québec (Montréal, QC, Canada) using the universal primers 341F (5’- CCTACGGGNGGCWGCAG-3’) and 805R (5’-GACTACHVGGGTATCTAATCC- 3’) [40]. Sequence libraries were prepared by Génome Québec with the TruSeq® DNA Library Prep Kit (Illumina, San Diego, CA, USA) and quantified using the KAPA Library Quantification Kit for Illumina platforms (Kapa Biosystems). Paired-end sequences were generated on a MiSeq platform PE300 (Illumina Corporation, San Diego, CA, USA) with the MiSeq Reagent Kit v3 600 cycles (Illumina, San Diego, CA, USA). The raw data files are publicly available on the NCBI Sequence Read Archive (PRJNA772886).
2.3. 16S rRNA Data Processing
Illumina sequence data (FASTQ files) were trimmed using Cutadapt (version 2.8). The 16S rRNA (V3-V4) amplicon sequence variants (ASVs) were generated with the DADA2 pipeline (version 1.16.0) [41] and subsequently analyzed within the R environment (R version 4.0.3) [42]. RDP (Ribosomal Database Project) 16 classifier database was used for ASV taxonomy assignment [43]. RDP contains 16S rRNA sequences available from the International Nucleotide Sequence Database Collaboration (INSDC) databases. Some studies have also compared RDP and SILVA [44,45]. Archaea and unclassified ASVs at the phylum level (representing 4.7% of the total ASVs) were removed. The phyloseq, microbiomeSeq, microbiomeMarker and vegan software packages were used to characterize the microbial communities [46,47,48,49].
2.4. Statistical Analysis
The alpha diversity index, including Shannon, Pielou’s evenness and richness, between the different groups was calculated using the phyloseq R package [46]. The comparisons of the average values of each group were performed using Wilcoxon rank sum tests. Microbiota composition differences among groups were determined using multivariate analysis of variance with permutation (PERMANOVA) with 9999 permutations followed by pairwise permutation tests. Heatmaps were generated based on the relative abundance and constructed with the 30 most abundant genera. Linear discriminant analysis (LDA) identified the effect size (LEfSe) that differentiates the samples among these taxa. The threshold on the logarithmic score of the LDA analysis was set to 3.0. Differences in the overall bacterial community composition among specimens were determined based on the UniFrac distance and visualized by principal coordinates analysis (PCoA). Permutation multivariate analysis of dispersion (PERMDISP) was also conducted with the betadisper function to test for the homogeneity of multivariate dispersions (i.e., deviations from centroids) among the specimens.
3. Results
3.1. General View of the Mucosal and Circulating Bacterial Microbiome
A total of 83 samples were collected from the blood (n = 45) and skin mucus (n = 38) of sedentary and migratory trout captured at different sites of Kerguelen (Figure 1). Each sample was immediately stored on-site on FTA cards (Table S1).
For each sample, the cmDNA was determined by sequencing the V3-V4 hypervariable region of the 16S rRNA gene amplicons. A total of 1 976 043 paired-end sequences passed quality filtering (23 808 ± 14 925 per sample). Amplicon sequence variants (ASVs) were generated from 12 985 high-quality reads, and 89.2% of samples contained more than 10 000 reads. We first examined the overall skin and blood microbiomes at the phylum level, focusing on phyla representing at least 2% of all ASVs. Our results showed that the microbiomes of S. trutta were dominated by Proteobacteria (~45-75%) and Bacteroidetes (~5-11%) and, to a lesser extent, Parcubacteria (~0.1-20%), Firmicutes (~1-9%), Actinobacteria (~3-6%), and Planctomycetes (~2-5%) (Figure 2A). Parcubacteria, which has been described in the seawater of polar regions [50], was more abundant in the blood (17.1%) than in the mucus (0.3%) (p = 0.014 (LefSE)). Firmicutes and Chloroflexi were also significantly more abundant in the blood of the migratory trout compared to the sedentary population (p < 0.05 linear discriminant analysis effect size (LEfSE)). At the genus level, we found a dominance of genera under Proteobacteria, the most abundant being the Aliivibrio genus, and to a lesser extent, bacterial DNA derived from Sphingomonas, Pseudoalteromonas, Yersinia and Aquabacterium, among others (Figure 2B). Concerning differences between the skin and blood microbiomes, we found that the Corynebacterium, Streptococcus, Acidovorax and Acinetobacter genera were only found in the blood but not in the skin mucus. In contrast, Psychrobacter, Yersinia, and Aeromonas were only found in the circulating microbiome. Discriminatory genera between the blood and mucus are shown in Figure S1 (LDA score). Aliivibrio had the highest LDA score in the mucus, and Flavobacterium dominated the blood microbiome. Overall, these results indicate that the bacterial microbiome signature differs between the skin and blood microbiomes.
3.2. Comparative Analysis between Sedentary and Migratory S. trutta
We next examined whether the skin and blood microbiome signatures differ between sedentary and migratory trout. We first compared the alpha diversity between migratory and sedentary populations using three diversity indices: richness, Shannon index, and Pielou’s evenness. Globally, we found no significant differences in the alpha diversity between the two populations for either the skin or blood microbiome (Figure 3). The alpha diversity was also not dependent on sex differences (Figure S2). Further analyses revealed, however, significant differences at the phylum and genus levels in the composition of the microbiomes between the sedentary and migratory trout. The high abundance of rare ASVs in the blood of migratory trout was reflected by the Chao1 index (Figure 4A).
Focusing on unique and shared phyla, we found that 18 were shared between sedentary and migratory trout, independent of the compartment. Seven phyla (BRC1, Chlorobi, Elusimicrobia, Ignavibacteriae, Lentisphaera, Poribacteria, and Tenericutes) were unique to migratory trout (Figure 4B). Another two phyla (Spirochaetes and SR1) were unique to the mucus of both populations, and one phylum (Chlorobi) was uniquely found in the blood but not in the mucus. A clear dominance of unique bacteria in the mucosal microbiome of migratory trout was also found at the genus level (Figure 4C). Overall, migratory trout had more than six times the number of unique genera in their compartment compared to sedentary trout (233 versus 39). This result is consistent with a multivariate analysis (PERMANOVA) showing that the global composition of the microbiome was significantly different between the two populations (UniFrac PERMANOVA, F(1, 82) = 1.44, p =0.02).
To further study the differences between migratory and sedentary trout, we examined the distribution of phyla and genera. At the phylum level, we found that the skin microbiome of sedentary trout had significantly (p < 0.05) higher DNA fragments of Cyanobacteria/chloroplasts and Verrucomicrobia than migratory trout (Figure 5). In contrast, the latter had higher levels of Firmicutes in both the skin and blood microbiomes. At the genus level, sedentary trout had a higher abundance of Aquabacterium (p < 0.05), Flavobacterium (p < 0.01), Luteolibacteria (p < 0.01), and Sphingomonas (p < 0.05) than the skin microbiome of migratory trout (Figure 6). In contrast, the skin microbiome of migratory trout had higher levels of Aeromonas (p < 0.05) and Yersinia (p < 0.05) (Figure 6). Other differences in the skin microbiome that approached conventional levels of significance (p < 0.10) were also observed in the case of Methylobacterium and Pseudoalteromonas. Taken together, these results indicate that the skin microbiome signatures differed between migratory and sedentary trout.
3.3. Site-Specific Variations
A general view of individual site-specific variations for phyla that constituted at least 1.5% of the total phylum is shown in Figure 7. Independent of the sampling site and the compartment, Proteobacteria was the dominant phylum in the majority of samples, especially in the skin microbiome. This abundance of Proteobacteria was almost absolute in samples collected at Rivière-du-Nord, independent of the population and the compartment. The presence of Parcubacteria was also omnipresent at all sites except at Rivière-du-Nord. Focusing on the 30 most abundant genera at the genus level, we found that Aliivibrio was abundant in Rivière-du-Nord, in both sedentary and migratory trout, and in the skin microbiome of migratory trout in Val-Travers (Figure S3). At Rivière-du-Nord and other sites, the skin microbiome was particularly rich in psychrophilic genera in migratory trout (Figure S4) compared to other sites. We also paid special attention to bacterial genera associated with the degradation of hydrocarbons, such as polycyclic aromatic hydrocarbons (PAHs). We found that migratory trout sampled at Acaena were particularly rich in this regard compared to other sites (Figures S5 and S6) [51]. Migratory trout from this site also had skin and blood microbiome signatures that were abundant in potentially pathogenic genera compared to other migratory or sedentary trout (Figure S7). Taken together, these results showed that both skin and blood microbiome signatures are site-dependent and may reveal specific environmental conditions.
3.4. Functional Analysis of the cmDNA
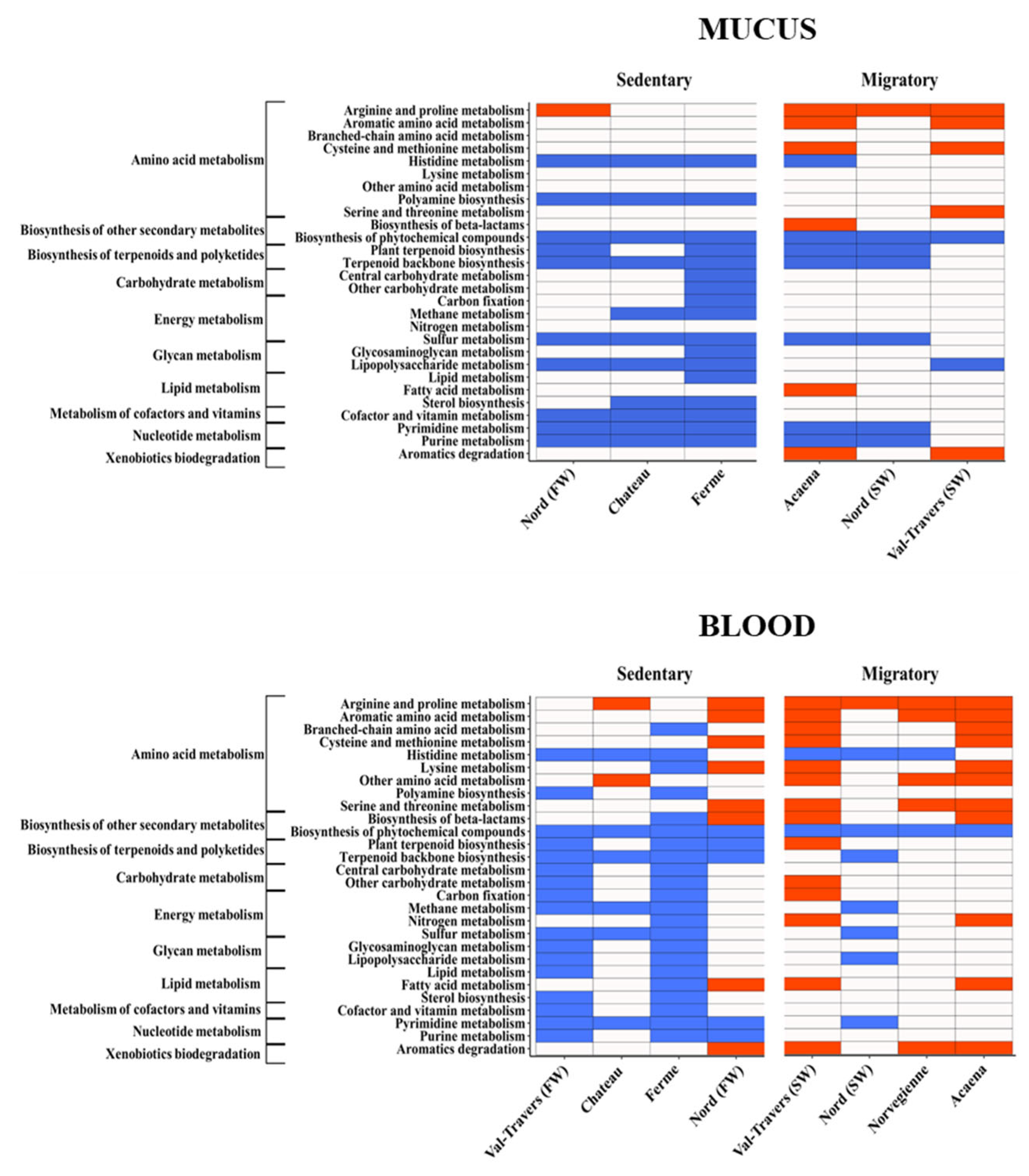
Predictive functional analysis from 16S rRNA profiling of cmDNA is routinely used to link the abundance of specific taxa with metabolic profiles [52,53,54]. To further explore the distinctive traits between sedentary and migratory S. trutta, we studied the functional content of the blood microbiome predicted from the KEGG database using the Piphillin tool (http://secondgenome.com/Piphillin) [53]. This analysis revealed two major differences. The first is the distinct difference in the metabolic profiles of migratory versus sedentary trout (Figure 8). We found a clear shift in lipid metabolism, consistent with previous studies in the salmonids [55]. A similar shift has recently been observed in lipid metabolism in the skin microbiome of S. salar during experimental seawater acclimatization [8]. Among the other potential differences between the sedentary and migratory microbial communities in terms of metabolism, we found a generally higher amino metabolism and biosynthesis of beta-lactams in the latter form, consistent with previous findings by Dehler and colleagues [8].
4. Discussion
In the present work, we compared the skin and blood microbiomes of sedentary and migratory S. trutta of the Kerguelen Islands. We have shown that the skin and blood microbiome signatures 1) differ at both the phylum and genus levels, 2) between migratory and sedentary trout, and 3) are site-dependent. This is the first study comparing the skin and blood microbiomes of wild sedentary and migratory salmonids and the first characterization of the circulating blood microbiome in a fish species. Finally, from a methodological perspective, our logistically simple and minimally invasive sampling platform offers an alternative approach for the long-term monitoring of fish populations in sensitive and remotely polar ecosystems.
Studies on the microbiome composition of wild fish populations remain relatively scarce and have, until now, mostly focused on the gut- and skin-associated microbiomes in fish farms. In the case of salmonids, Lokesh and Kiron (2016) have shown that the skin-associated microbiome of Atlantic salmon (S. salar) during the transition from freshwater to seawater in Norway was dominated by Proteobacteria, Bacteroidetes and Firmicutes [56]. Our data also revealed an abundance of Proteobacteria and Bacteroidetes in the skin-associated microbiome of both migratory and sedentary S. trutta of Kerguelen. This dominance was also found in the blood microbiome. In our study, however, the dominance of Proteobacteria was not related to the abundance of the Oleispira genus, as reported for S. salar. We did not find a dominance of either Oleispira (or detected, for that matter) in the skin mucus (or in the blood) of migratory trout, as opposed to what is observed for S. salar [8]. Another difference from the study reported for S. salar is that we did not find a dominance of Firmicutes in the skin-associated microbiome of sedentary trout [8]. Instead, we found that Firmicutes were more abundant in migratory S. trutta. This was true for both mucosal and blood microbiomes. This increase in Firmicutes in migratory S. trutta was driven by the presence of Aerococcus, Bacillus, Hathewaya and Clostridium_sensu_stricto genera, which was different from the Firmicutes found in the sedentary trout, which mainly included Staphylococcus and Lactobacillus genera. We also found an increased abundance of Verrucomicrobia and Cyanobacteria in the skin mucus of sedentary trout. This shift was not found in the blood microbiome. Other bacteria previously found in the skin mucus of S. salar, such as Thalassomonas, Psychromonas, Agarivorans, Pseudoalteromonas, Marinomonas, Arcobacter, Perlucidibaca and Octadecabacter, were also absent in S. trutta. However, a clear difference at the phylum level was the abundance of Actinobacteria and Parcubacteria in the blood microbiome, but not in the mucus, of both migratory and sedentary trout. This signature is, in fact, similar to a recent metagenomic study showing that bacteria enriched in seawater in polar regions were mostly Proteobacteria, Actinobacteria, Bacteroidetes and Parcubacteria [50]. Interestingly, we also found a similar signature in the hemolymphatic (blood-like) microbiome of mussel species (Figure S8) that inhabit the coastal marine ecosystems of Kerguelen [57]. These results suggest the existence of a possible “Kerguelen signature,” at least at the phylum level, driven by the environmental conditions of Kerguelen. Metagenomic profiling using the 16S rRNA microbiome signature is a cost-effective and rapid means to screen for candidate pathogens associated with infectious and noninfectious diseases in a given population [16,58,59,60,61]. Here, we paid particular attention to this aspect, given the history of salmonids in Kerguelen and their isolation from other salmonid populations. Our data revealed the presence of Aliivibrio and Pseudomonas within the skin mucus and blood microbiomes of all trout from Kerguelen. These genera include several pathogenic strains, such as Aliivibrio salmonicida, a common pathogen found in fish farms [62]. Our study also revealed, in the skin microbiome of migratory trout at Acaena, the presence of Renibacterium. This genus includes Renibacterium salmoninarum, the causative agent of bacterial kidney disease, a deadly disease affecting wild and cultured salmonids worldwide [63]. This pathogen was introduced in 1987 following the importation from the United States of Chinook salmon into the Armor basin. This was one of the reasons why the Aquasaumon Sea ranching project at Armor was abandoned [64]. There is a possibility that the bacteria have spread since infected juveniles escaped from Armor in 1987. Moreover, Artic chars, kept in the Armor Hatchery, were released into the nearby Lac des Fougères in 1991 [35,65]. As of 2012, no signs of the disease have been observed [35]. Among the other sites that were distinguishable was Rivière-du-Nord. The microbiome of either sedentary or migratory trout showed a unique signature. This was generally apparent in the blood microbiome of sedentary trout, which were dominated by two genera: Aliivibrio and Photobacterium. The dominance of Aliivibrio was also found in the skin microbiome. Whether such dominance of Aliivibrio reveals the presence of Aliivibrio salmonicida, a common pathogen known to cause cold-water vibriosis in salmonids [66], is certainly an issue that warrants further investigation. This pathogen, mostly found in estuaries, is usually found in high amounts in the blood of moribund fish. ASVs corresponding to Aliivibrio salmonicida were only found at Rivière-du-Nord, while all ASVs from other sites corresponded to Aliivibrio logei, a commonly found genus in the skin and gut microbiota [67]. Interestingly, the sedentary trout sampled at Rivière-du-Nord harboured an almost identical metabolic profile to migratory trout in general [68]. It is important to note, however, that the presence of blood DNA fragments of bacterial pathogens does not always reflect the onset of a disease. Rather, it provides a rapid, ethical and sensitive means to detect dysbiosis and to alert to their presence and whether they do indeed express disease-associated genes. It is important to note, however, that despite all precautions taken during sampling and laboratory procedures, and even though we used blank FTA cards, it is important to recognize that one cannot completely rule out contaminations, especially when studying wild populations in a remote marine ecosystem. Future studies are thus needed to confirm the presence of these pathogenic strains and the potential existence of site-specific reservoirs. Overall, our study may help to evaluate better the impact of specific microbial structures on the fitness-related traits of specific populations, including their dispersal and reproductive abilities. Indeed, their presence in pathogenic strains would imply a higher energy expenditure on the immune system and a possible eco-evolutionary effect on MHC-related genes. For instance, MHC genes are highly homozygous in the Val Travers population, which is quite unusual compared to the available literature. This could be partly related to inbreeding or relaxation of selective pressures of pathogens [69].
Our data further revealed that another type of information that can be obtained from microbiome-based signatures is related to the physico-chemical characteristics of the ecosystem. A clear example is the abundance of hydrocarbon degrader genera within the microbiomes of migratory trout sampled at Acaena. We hypothesize that their presence at the mouth of the Acaena River is possibly due to the nearby presence of multiple lignite deposits, also called “brown coal,” at the Ravin du Charbon and the Ravin Jaune near the Acaena River [70].
To our knowledge, this is one of the rare studies of the blood microbiome of fish. Historically, most studies on the microbiome composition were carried out on the gut microbiome with emphasis on their role in nutrition and related diseases, including inflammatory diseases. However, advances in sequencing NGS technologies have shown that plasma DNA fragments originate from multiple organs, providing an opportunity to obtain a systemic view of tissue damage. It is now possible to determine the exact tissue origin of plasma DNA fragments by examining footprints of consensus sequences specific for transcription factors [71]. These findings have contributed to the emerging concept of the circulating microbiome and subsequent studies showing that the blood contains a rich source of microbial DNA to assess the host’s health status [12,13,72]. As the blood microbiome originates from multiple tissues [73,74], the possibility of using a single drop of blood for monitoring a much broader microbial diversity within a host opens the door for easier monitoring and understanding of factors that contribute to shaping the microbiome in aquatic species (and terrestrial for that matter), most notably in response to environmental stressors, including climate change.
Finally, we would like to discuss the sampling approach used in our study briefly. This study and our previous work in bivalves indicate that FTA® card-based sampling is perfectly adapted for establishing skin and blood (or hemolymphatic) 16S rRNA microbiome signatures. The efficacy of FTA® cards as a stable means to preserve DNA samples, even at room temperature, has been well documented [39]. Such a minimally invasive and ethical (nonlethal) sampling procedure is particularly well adapted for long-term monitoring programs in remote areas and for limiting the impact of large cohort studies on a given population inhabiting, for example, natural reserves, such as the Kerguelen Islands, for endangered species, or for storage and transport for fieldwork in areas where proper conditions for RNA preservation are challenging to achieve [75]. Sampling using FTA® cards is gaining momentum as it is compatible with basic nucleic acid-based detection methods. It has been used, for example, for molecular diagnosis, detecting viruses, etc. [76,77]. It is particularly useful for the safe transportation of infectious material, which is rapidly inactivated upon binding the nucleic acid to the chemically modified paper [78]. This low-cost method is logistically simple (without the need to maintain a cold chain for sample integrity) and is ideally adapted for biobanking.
5. Conclusions
In conclusion, we have shown that migratory and sedentary trout of Kerguelen have unique microbiome signatures compared to other salmonid populations reported in the Northern Hemisphere. It will be interesting to study the impacts of these microbial shifts on host physiology and how these signatures will change in response to climate change and the colonization process of salmonids in this unique ecosystem.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
F.C: Conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing - original draft, Writing - review & editing; S. F.: Conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing - original draft, writing - review & editing; R. V.: Conceptualization, research, methodology, validation, writing -review & editing; J. L.: Conceptualization, data curation, investigation, methodology, resources, validation, writing - review & editing; Y. S.-P.: Conceptualization, funding acquisition, analysis, methodology, project administration, resources, supervision, validation, visualization, writing -original draft, Writing - review & editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science and Engineering Research Council of Canada and the Institut Polaire Paul-Émile Victor. This study is part of the Zone Atelier et Terres Australes LTSER.
Institutional Review Board Statement
The care and use of experimental animals complied with the French environmental and animal welfare laws, guidelines, and policies as approved by the ethical committee for birds and fishes in the French region Nouvelle Aquitaine (CEEA073, authorization APAFIS#16249-201807241223324v3, project name “Ecologie évolutive de la colonisation des Îles Kerguelen par les salmonidés”).
Data Availability Statement
The raw data files used in this study are publicly available on the NCBI Sequence Read Archive (PRJNA772886).
Acknowledgments
This research was performed at Kerguelen with the logistic support of the French Polar Institute Paul-Emile-Victor (IPEV). The authors would like to thank all the personnel from the IPEV and the Terres Australes et Antarctiques Françaises (TAAF) for their help and hospitality during our stay. The authors would also like to thank Jean-Pierre Féral and Thomas Saucède and the PROTEKER team for their expert advice and technical support and Dr. Jep Lokesh for helpful discussions on the manuscript. The author would also like to thank Marlène Fortier for her excellent technical help.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Chen, H.; Ma, Y.; Liu, Z.; Li, J.; Li, X.; Yang, F.; et al. Circulating microbiome DNA: An emerging paradigm for cancer liquid biopsy. Cancer Lett. 2021, 521, 82–87. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar]
- Cabillon, N.A.R.; Lazado, C.C. Mucosal barrier functions of fish under changing environmental conditions. Fishes 2019, 4, 2. [Google Scholar] [CrossRef]
- Lowrey, L.; Woodhams, D.C.; Tacchi, L.; Salinas, I. Topographical mapping of the rainbow trout (Oncorhynchus mykiss) microbiome reveals a diverse bacterial community with antifungal properties in the skin. Appl. Environ. Microbiol. 2015, 81, 6915–6925. [Google Scholar] [CrossRef]
- Tarnecki, A.M.; Brennan, N.P.; Schloesser, R.W.; Rhody, N.R. Shifts in the skin-associated microbiota of hatchery-reared common snook Centropomus undecimalis during acclimation to the wild. Microb. Ecol. 2019, 77, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Uren Webster, T.M.; Consuegra, S.; Hitchings, M.; Garcia de Leaniz, C. Interpopulation variation in the Atlantic Salmon microbiome reflects environmental and genetic diversity. Appl. Environ. Microbiol. 2018, 84, e00691–18. [Google Scholar] [CrossRef] [PubMed]
- Uren Webster, T.M.; Rodriguez-Barreto, D.; Castaldo, G.; Gough, P.; Consuegra, S.; de Leaniz, C.G. Environmental plasticity and colonisation history in the Atlantic salmon microbiome: a translocation experiment. Mol. Ecol. 2020, 29, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Dehler, C.E.; Secombes, C.J.; Martin, S.A. Seawater transfer alters the intestinal microbiota profiles of Atlantic salmon (Salmo salar L.). Sci. Rep. 2017, 7, 13877. [Google Scholar]
- Rudi, K.; Angell, I.L.; Pope, P.B.; Vik, J.O.; Sandve, S.R.; Snipen, L.G. Stable core gut microbiota across the freshwater-to-saltwater transition for farmed Atlantic salmon. Appl. Environ. Microbiol. 2018, 84, e01974–17. [Google Scholar] [CrossRef]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-method characterization of the human circulating microbiome. Front. Microbiol. 2019, 9, 3266. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Blauwkamp, T.A.; Thair, S.; Rosen, M.J.; Blair, L.; Lindner, M.S.; Vilfan, I.D.; et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat. Microbiol. 2019, 4, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.K.; Blauwkamp, T.A.; Kertesz, M.; Bercovici, S.; Truong, C.; Banaei, N. Liquid biopsy for infectious diseases: sequencing of cell-free plasma to detect pathogen DNA in patients with invasive fungal disease. Diagn. Microbiol. Infect. Dis. 2018, 92, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Kowarsky, M.; Camunas-Soler, J.; Kertesz, M.; De Vlaminck, I.; Koh, W.; Pan, W.; et al. Numerous uncharacterized and highly divergent microbes which colonize humans are revealed by circulating cell-free DNA. Proc. Natl. Acad. Sci. USA. 2017, 114, 9623–9628. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: the D.E.S.I.R. study. Plos One. 2013, 8, e54461. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.J.; Leem, S.; Kim, S.A.; Yang, J.; Lee, Y.B.; Kim, S.S.; et al. Circulating microbiota-based metagenomic signature for detection of hepatocellular carcinoma. Sci. Rep. 2019, 9, 7536. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Du, M.; Wang, L. Bioinformatics analysis for circulating cell-free DNA in cancer. Cancers 2019, 11, 805. [Google Scholar] [CrossRef]
- Paisse, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; et al. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef]
- Potgieter, M.; Bester, J.; Kell, D.B.; Pretorius, E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol. Rev. 2015, 39, 567–591. [Google Scholar] [CrossRef]
- Auguste, M.; Lasa, A.; Pallavicini, A.; Gualdi, S.; Vezzulli, L.; Canesi, L. Exposure to TiO2 nanoparticles induces shifts in the microbiota composition of Mytilus galloprovincialis hemolymph. Sci. Total Environ. 2019, 670, 129–137. [Google Scholar] [CrossRef]
- Lokmer, A.; Wegner, M.K. Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 2015, 9, 670–682. [Google Scholar] [CrossRef]
- Wilkins, L.G.E.; Leray, M.; O'Dea, A.; Yuen, B.; Peixoto, R.S.; Pereira, T.J.; et al. Host-associated microbiomes drive structure and function of marine ecosystems. PLoS Biol. 2019, 17, e3000533. [Google Scholar] [CrossRef]
- Rynkiewicz, E.C.; Hemmerich, C.; Rusch, D.B.; Fuqua, C.; Clay, K. Concordance of 605 bacterial communities of two tick species and blood of their shared rodent host. Mol. Ecol. 2015, 24, 2566–2579. [Google Scholar] [CrossRef]
- Jeon, S.J.; Cunha, F.; Vieira-Neto, A.; Bicalho, R.C.; Lima, S.; Bicalho, M.L.; et al. Blood as a route of transmission of uterine pathogens from the gut to the uterus in cows. Microbiome. 2017, 5, 109. [Google Scholar] [CrossRef] [PubMed]
- Scarsella, E.; Sandri, M.; Monego, S.D.; Licastro, D.; Stefanon, B. Blood Microbiome: A New Marker of Gut Microbial Population in Dogs? Vet. Sci. 2020, 7, 198. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.; Lee, M.S.; Park, I.; Ko, H.S.; Yun, S.; Jang, D.H.; et al. Analysis of Porcine Model of Fecal-Induced Peritonitis Reveals the Tropism of Blood Microbiome. Front. Cell. Infect. Microbiol. 2021, 11, 676650. [Google Scholar] [CrossRef] [PubMed]
- Scarsella, E.; Zecconi, A.; Cintio, M.; Stefanon, B. Characterization of microbiome on feces, blood and milk in dairy cows with different milk leucocyte pattern. Animals 2021, 11, 1463. [Google Scholar] [CrossRef] [PubMed]
- Vientós-Plotts, A.I.; Ericsson, A.C.; Rindt, H.; Reinero, C.R. Blood cultures and blood microbiota analysis as surrogates for bronchoalveolar lavage fluid analysis in dogs with bacterial pneumonia. BMC Vet Res. 2021, 17, 129. [Google Scholar] [CrossRef]
- Tilahun, Y.; Pinango, J.Q.; Johnson, F.; Lett, C.; Smith, K.; Gipson, T.; et al. Transcript and blood-microbiome analysis towards a blood diagnostic tool for goats affected by Haemonchus contortus. Sci. Rep. 2022, 12, 5362. [Google Scholar]
- Tarnecki, A.M.; Patterson, W.F.; Arias, C.R. Microbiota of wild-caught red snapper Lutjanus campechanus. BMC Microbiol. 2016, 16, 245. [Google Scholar] [CrossRef]
- Kelly, C.; Salinas, I. Under pressure: interactions between commensal microbiota and the teleost immune system. Front. Immunol. 2017, 8, 559. [Google Scholar] [CrossRef] [PubMed]
- Pratte, Z.A.; Besson, M.; Hollman, R.D.; Stewart, F.J. The gills of reef fish support a distinct microbiome influenced by host-specific factors. Appl. Environ. Microbiol. 2018, 84, e00063–18. [Google Scholar] [CrossRef] [PubMed]
- Féral, J.-P.; Saucède, T.; Poulin, E.; Marschal, C.; Marty, G.; Roca, J.C.; et al. PROTEKER: implementation of a submarine observatory at the Kerguelen islands (Southern Ocean). Underw. Technol. 2016, 34, 3–10. [Google Scholar]
- Meredith, M.; Sommerkorn, M.; Cassotta, S.; Derksen, C.; Ekaykin, A.; Hollowed, A.; et al. Polar Regions. In IPCC Special Report on the Ocean and Cryosphere in a Changing Climate; Pörtner, H.-O., Roberts, D.C., Masson-Delmotte, V., Zhai, P., Tignor, M., Poloczanska, E., Mintenbeck, K., Alegría, A., Nicolai, M., Okem, A., Petzold, J., Rama, P.B., Weyer, N.M., Eds.; Cambridge University Press: Cambridge, UK and New York, NY, USA, 2019; pp. 203–320. [Google Scholar]
- Lecomte, F.; Beall, E.; Chat, J.; Davaine, P.; Gaudin, P. The complete history of salmonid introductions in the Kerguelen Islands, Southern Ocean. Polar Biol. 2013, 36, 457–475. [Google Scholar] [CrossRef]
- Dodson, J.J.; Aubin-Horth, N.; Thériault, V.; Páez, D.J. The evolutionary ecology of alternative migratory tactics in salmonid fishes. Biol. Rev. 2013, 88, 602–625. [Google Scholar] [CrossRef] [PubMed]
- Labonne, J.; Vignon, M.; Prévost, E.; Lecomte, F.; Dodson, J.J. Invasion Dynamics of a Fish-Free Landscape by Brown Trout (Salmo trutta). Plos One. 2013, 8, e71052. [Google Scholar] [CrossRef]
- Marandel, L.; Gaudin, P.; Guéraud, F.; Glise, S.; Herman, A.; Plagues-Juan, E.; et al. A reassessment of the carnivorous status of salmonids: Hepatic glucokinase is expressed in wild fish in Kerguelen Islands. Sci. Total Environ. 2018, 612, 276–285. [Google Scholar] [CrossRef]
- Caza, F.; Joly de Boissel, P.G.; Villemur, R.; Betoulle, S.; St-Pierre, Y. Liquid biopsies for omics-based analysis in sentinel mussels. PLoS One. 2019, 14, e0223525. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Wang, L.-Y.; Ke, W.J.; Sun, X.-B.; Liu, J.-F.; Gu, J.-D.; Mu, B.-Z. Comparison of bacterial community in aqueous and oil phases of water-flooded petroleum reservoirs using pyrosequencing and clone library approaches. Appl. Microbiol. Biotechnol. 2014, 98, 4209–4221. [Google Scholar] [CrossRef]
- Vilo, C.; Dong, Q. Evaluation of the RDP classifier accuracy using 16S rRNA gene variable regions. Metagenomics. 2012, 1, 104303. [Google Scholar] [CrossRef]
- Balvočiūtė, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT— how do these taxonomies compare? BMC Genomics. 2017, 18, 114. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; microbiomeMarker: microbiome biomarker analysis. R package version 0.0. 1.9000. Available online: https://github.com/yiluheihei/microbiomeMarker (accessed on 23 October 2022).
- Ssekagiri, A.; Sloan, W.; Ijaz, U.Z.; microbiomeSeq: An R package for analysis of microbial communities in an environmental context. ISCB Africa ASBCB Conference. Available online: https://github.com/umerijaz/microbiomeSeq (accessed on 22 February 2019).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; et al. Vegan: Community Ecology Package. R package version 2.5-7. Available online: https://cran.r-project.org/package=vegan (accessed on 11 October 2022).
- Cao, S.; Zhang, W.; Ding, W.; Wang, M.; Fan, S.; Yang, B.; et al. Structure and function of the Arctic and Antarctic marine microbiota as revealed by metagenomics. Microbiome 2020, 8, 47. [Google Scholar] [CrossRef]
- Hedaoo, M.; Gore, D.; Fadnavis, S.; Dange, M.; Soni, M.A.; Kopulwar, A.P. Bioinformatics approach in speciation of oil degrading uncultured bacterium and its frequency recording. J. Pharm. Res. 2018, 12, 628–635. [Google Scholar]
- Koner, S.; Chen, J.S.; Hsu, B.M.; Tan, C.W.; Fan, C.W.; Chen, T.H.; et al. Assessment of Carbon Substrate Catabolism Pattern and Functional Metabolic Pathway for Microbiota of Limestone Caves. Microorganisms. 2021, 9, 1789. [Google Scholar] [CrossRef]
- Narayan, N.R.; Weinmaier, T.; Laserna-Mendieta, E.J.; Claesson, M.J.; Shanahan, F.; Dabbagh, K.; et al. Piphillin predicts metagenomic composition and dynamics from DADA2-corrected 16S rDNA sequences. BMC Genom. 2020, 21, 56. [Google Scholar] [CrossRef]
- Peng, W.; Huang, J.; Yang, J.; Zhang, Z.; Yu, R.; Fayyaz, S.; et al. Integrated 16S rRNA sequencing, metagenomics, and metabolomics to characterize gut microbial composition, function, and fecal metabolic phenotype in non-obese type 2 diabetic Goto-Kakizaki rats. Front. Microbiol. 2020, 10, 3141. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, M.A. Alterations in lipid metabolism accompanying smoltification and seawater adaptation of salmonid fish. Aquaculture. 1989, 82, 191–203. [Google Scholar] [CrossRef]
- Lokesh, J.; Kiron, V. Transition from freshwater to seawater reshapes the skin-associated microbiota of Atlantic salmon. Sci. Rep. 2016, 6, 1–10. [Google Scholar]
- Ferchiou, S.; Caza, F.; Villemur, R.; Betoulle, S.; St-Pierre, Y. Species- and site-specific circulating bacterial DNA in Subantarctic sentinel mussels Aulacomya atra and Mytilus platensis. Sci. Rep. 2022, 12, 9547. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.-P.; Yu, A.L. Application of cell-free DNA sequencing in characterization of bloodborne microbes and the study of microbe-disease interactions. PeerJ. 2019, 7, e7426. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, Y.; Guo, S.; Mei, Z.; Liao, H.; Dong, H.; et al. Tumor microbiome contributes to an aggressive phenotype in the basal-like subtype of pancreatic cancer. Commun. Biol. 2021, 4, 1019. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, G.; Dinakaran, V.; Rajendhran, J.; Swaminathan, K. Blood microbiota and circulating microbial metabolites in diabetes and cardiovascular disease. Trends Endocrinol. Metab. 2020, 31, 835–847. [Google Scholar] [CrossRef]
- Onarheim, A.M.; Wiik, R.; Burghardt, J.; Stackebrandt, E. Characterization and identification of two Vibrio species indigenous to the intestine of fish in cold sea water; description of Vibrio iliopiscarius sp. nov. Syst. Appl. Microbiol. 1994, 17, 370–379. [Google Scholar] [CrossRef]
- Delghandi, M.R.; El-Matbouli, M.; Menanteau-Ledouble, S. Renibacterium salmoninarum ─ The causative agent of bacterial kidney disease in salmonid fish. Pathogens. 2020, 9, 845. [Google Scholar] [CrossRef] [PubMed]
- Caraguel, J.M.; Guerri, O.; Davaine, P. Pacage marin du saumon à Kerguelen, In: Rapport de campagne ARMOR 1992, INRA (1993).
- Eldøy, S.H.; Davidsen, J.; Vignon, M.; Power, M. The biology and feeding ecology of Arctic charr in the Kerguelen Islands. J. Fish Biol. 2020, 98, 526–536. [Google Scholar] [CrossRef]
- Austin, B.; Austin, D.A. Vibrionaceae representatives. In Bacterial Fish Pathogens; Austin, B., Austin, D.A., Eds.; Springer: Dordrecht, NL, 2012; pp. 357–411. [Google Scholar]
- Klemetsen, T.; Karlsen, C.R.; Willassen, N.P. Phylogenetic revision of the genus Aliivibrio: intra-and inter-species variance among clusters suggest a wider diversity of species. Front. Microbiol. 2021, 12, 626759. [Google Scholar] [CrossRef]
- Davidsen, J.G.; Bordeleau, X.; Eldøy, S.H.; Whoriskey, F.; Power, M.; Crossin, G.T.; et al. Marine habitat use and feeding ecology of introduced anadromous brown trout at the colonization front of the sub-Antarctic Kerguelen archipelago. Sci. Rep. 2021, 11, 11917. [Google Scholar] [CrossRef]
- Labonne, J.; Kaeuffer, R.; Gueraud, F.; Zhou, M.; Manicki, A.; Hendry, A.P. From the bare minimum : genetics and selection in populations founded by only a few parents. Evol. Ecol. Res. 2016, 17, 21–34. [Google Scholar]
- Frey, F.A.; Weis, D.; Yang, H.J.; Nicolaysen, K.; Leyrit, H.; Giret, A. Temporal geochemical trends in Kerguelen Archipelago basalts: evidence of decreasing magma supply from the Kerguelen plume. Chem. Geol. 2000, 164, 61–80. [Google Scholar] [CrossRef]
- Snyder, M.W.; M., K.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissue origin. Cell. 2016, 164, 57–68. [CrossRef]
- Abril, M.K.; Barnett, A.S.; Wegermann, K.; Fountain, E.; Strand, A.; Heyman, B.M.; et al. Diagnosis of Capnocytophaga canimorsus sepsis by next generation whole-genome sequencing. Open Forum Infect. Dis. 2016, 3, ofw144. [Google Scholar] [CrossRef]
- Miller, R.R.; Montoya, V.; Gardy, J.L.; Patrick, D.M.; Tang, P. Metagenomics for pathogen detection in public health. Genome Med. 2013, 5, 81. [Google Scholar] [CrossRef]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Cardona-Ospina, J.A.; Villalba-Miranda, M.F.; Palechor-Ocampo, L.A.; Mancilla, L.I.; Sepúlveda-Arias, J.C. A systematic review of FTA cards® as a tool for viral RNA preservation in fieldwork: Are they safe and effective? Prev. Vet. Med. 2019, 172, 104772. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.L.; Montiel, E.R.; Gimeno, I.M. Validation of Marek’s disease diagnosis and monitoring of Marek’s disease vaccines from samples collected in FTA® Cards. Avian Dis. 2009, 54, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Moscoso, H.; Raybon, E.O.; Thayer, S.G.; Hofacre, C.L. Molecular detection and serotyping of infectious bronchitis virus from FTA® filter paper. Avian Dis. 2005, 49, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Jóźwiak, M.; Wyrostek, K.; Domańska-Blicharz, K.; Olszewska-Tomczyk, M.; Śmietanka, K.; MInta, Z. Application of FTA® Cards for detection and storage of avian influenza virus. J. Vet. Res. 2016, 60, 1–6. [Google Scholar] [CrossRef]
- Poli, A.; Finore, I.; Romano, I.; Gioiello, A.; Lama, L.; Nicolaus, B. Microbial diversity in extreme marine habitats and their biomolecules. Microorganisms. 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Kuchi, N.; Khandeparker, L.; C. , A.A. Response of the bacterial metagenome in port environments to changing environmental conditions. Mar. Pollut. Bull. 2021, 172, 112869. [Google Scholar] [CrossRef] [PubMed]
- Austin, B.; Austin, D.; Sutherland, R.; Thompson, F.; Swings, J. Pathogenicity of vibrios to rainbow trout (Oncorhynchus mykiss, Walbaum) and Artemia nauplii. Environ. Microbiol. 2005, 7, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.-H.; Jensen, I.; Mikkelsen, H.; Bjørn, P.-A.; Jansen, P.A.; Bergh, Ø. Disease interaction and pathogens exchange between wild and farmed fish populations with special reference to Norway. Aquaculture. 2011, 315, 167–186. [Google Scholar] [CrossRef]
Figure 1.
Map of the French subantarctic Kerguelen Islands showing the location of the rivers. Sedentary trout were collected from freshwater (FW), and migratory trout were collected from the mouth of the river (seawater (SW)).
Figure 1.
Map of the French subantarctic Kerguelen Islands showing the location of the rivers. Sedentary trout were collected from freshwater (FW), and migratory trout were collected from the mouth of the river (seawater (SW)).
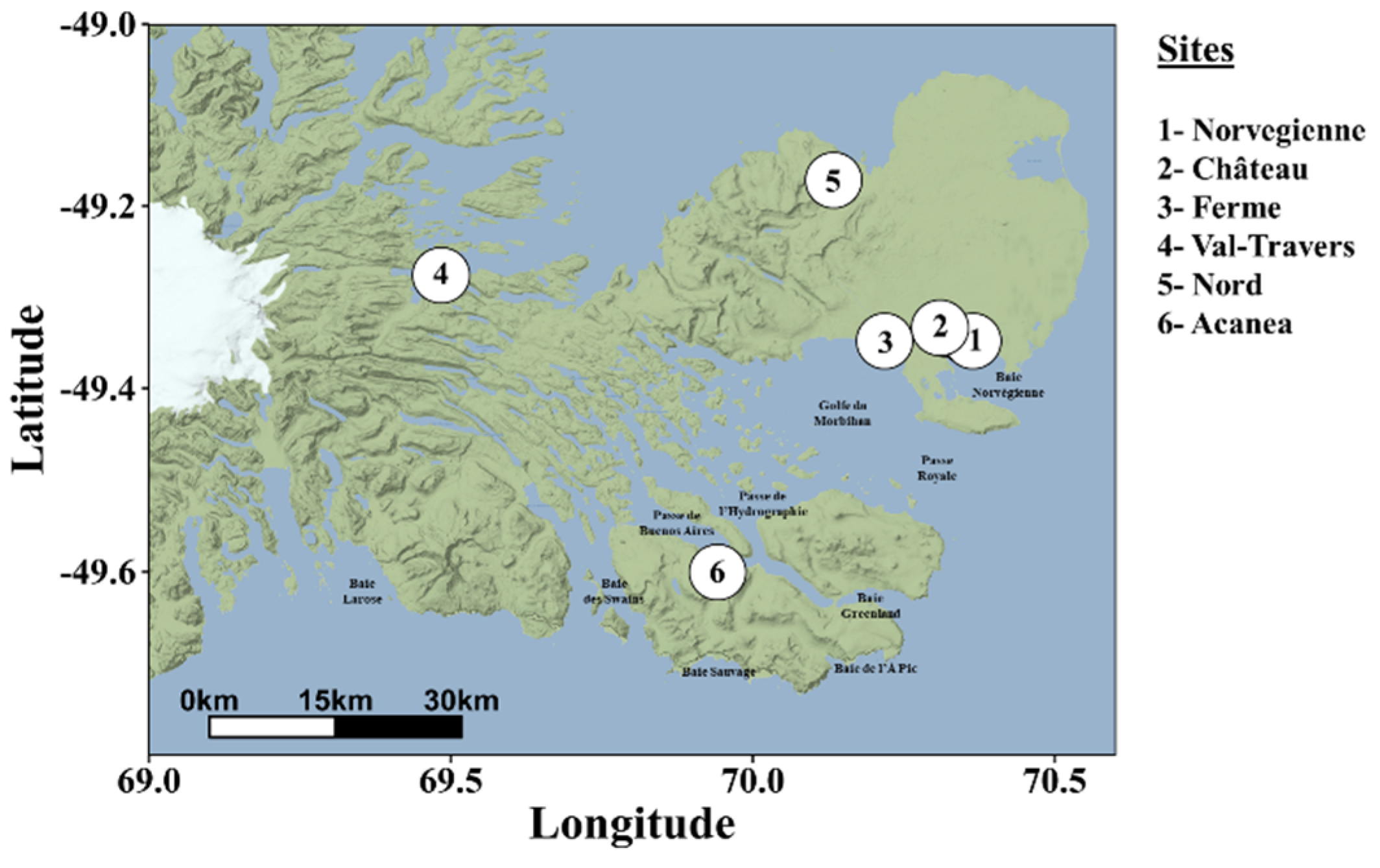
Figure 2.
Phylum and genus level analysis of circulating and mucosal bacterial DNA. (A) Phylum and (B) genus levels were analyzed for all samples (left) and mucosal and circulating compartments (right) in brown trout (S. trutta). Phylum and genus with a relative abundance of ≤ 1.5% are represented as "Other.".
Figure 2.
Phylum and genus level analysis of circulating and mucosal bacterial DNA. (A) Phylum and (B) genus levels were analyzed for all samples (left) and mucosal and circulating compartments (right) in brown trout (S. trutta). Phylum and genus with a relative abundance of ≤ 1.5% are represented as "Other.".
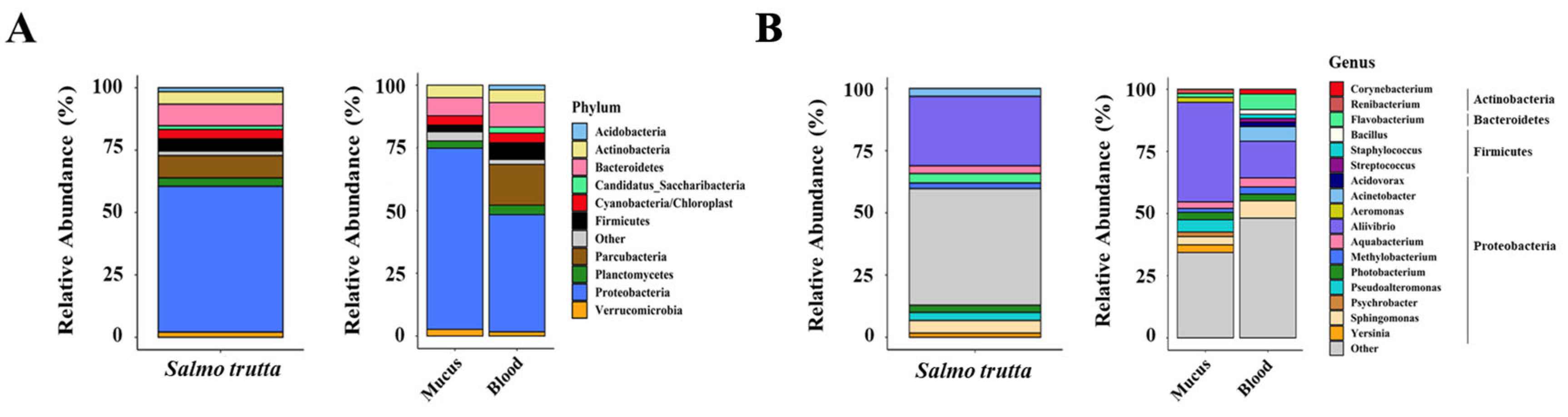
Figure 3.
Alpha diversity analysis of both S. trutta phenotypes for circulating and mucosal microbiomes. Species evenness, observed richness and Shannon diversity indexes were calculated for each group. No significant differences were observed between migratory and sedentary specimens for the same compartment.
Figure 3.
Alpha diversity analysis of both S. trutta phenotypes for circulating and mucosal microbiomes. Species evenness, observed richness and Shannon diversity indexes were calculated for each group. No significant differences were observed between migratory and sedentary specimens for the same compartment.
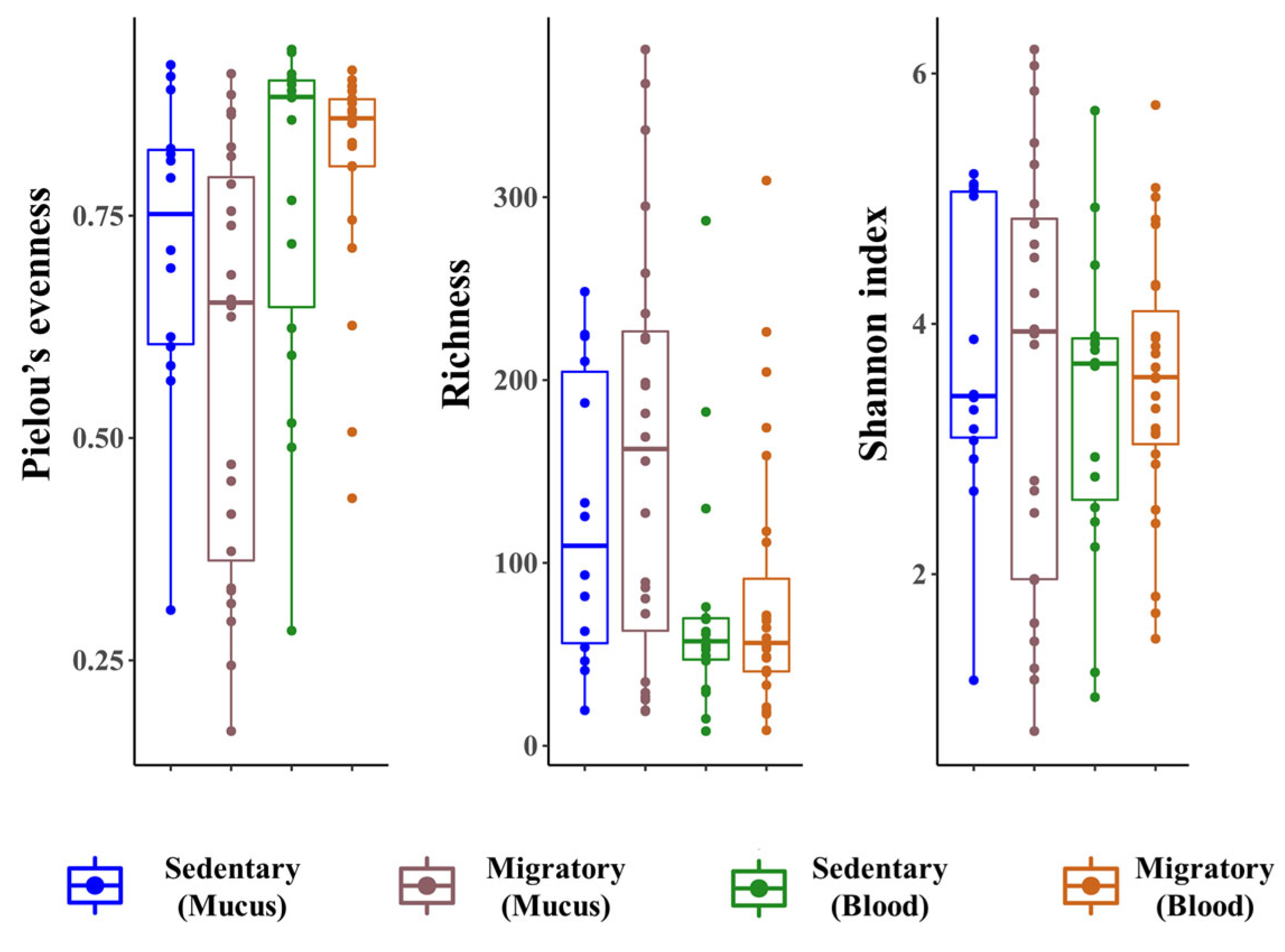
Figure 4.
Comparisons between migratory and sedentary trout in both compartments. (A) The Chao1 index was calculated for each group. Venn diagrams showing the number of unique and shared (B) bacterial phyla and (C) genera in mucosal and circulating compartments in both sedentary and migratory brown trout (S. trutta). Note: * 0.01 < p ≤ 0.05, ** 0.001 < p ≤ 0.01, *** p ≤ 0.001.
Figure 4.
Comparisons between migratory and sedentary trout in both compartments. (A) The Chao1 index was calculated for each group. Venn diagrams showing the number of unique and shared (B) bacterial phyla and (C) genera in mucosal and circulating compartments in both sedentary and migratory brown trout (S. trutta). Note: * 0.01 < p ≤ 0.05, ** 0.001 < p ≤ 0.01, *** p ≤ 0.001.
Figure 5.
Relative abundance (%) of the top 10 bacterial phyla in circulating and mucosal microbiomes in S. trutta. Bar graph analysis displays the logarithm (base 10) of the relative abundance (mean ± SE). White and black colors represent migratory and sedentary brown trout, respectively.
Figure 5.
Relative abundance (%) of the top 10 bacterial phyla in circulating and mucosal microbiomes in S. trutta. Bar graph analysis displays the logarithm (base 10) of the relative abundance (mean ± SE). White and black colors represent migratory and sedentary brown trout, respectively.
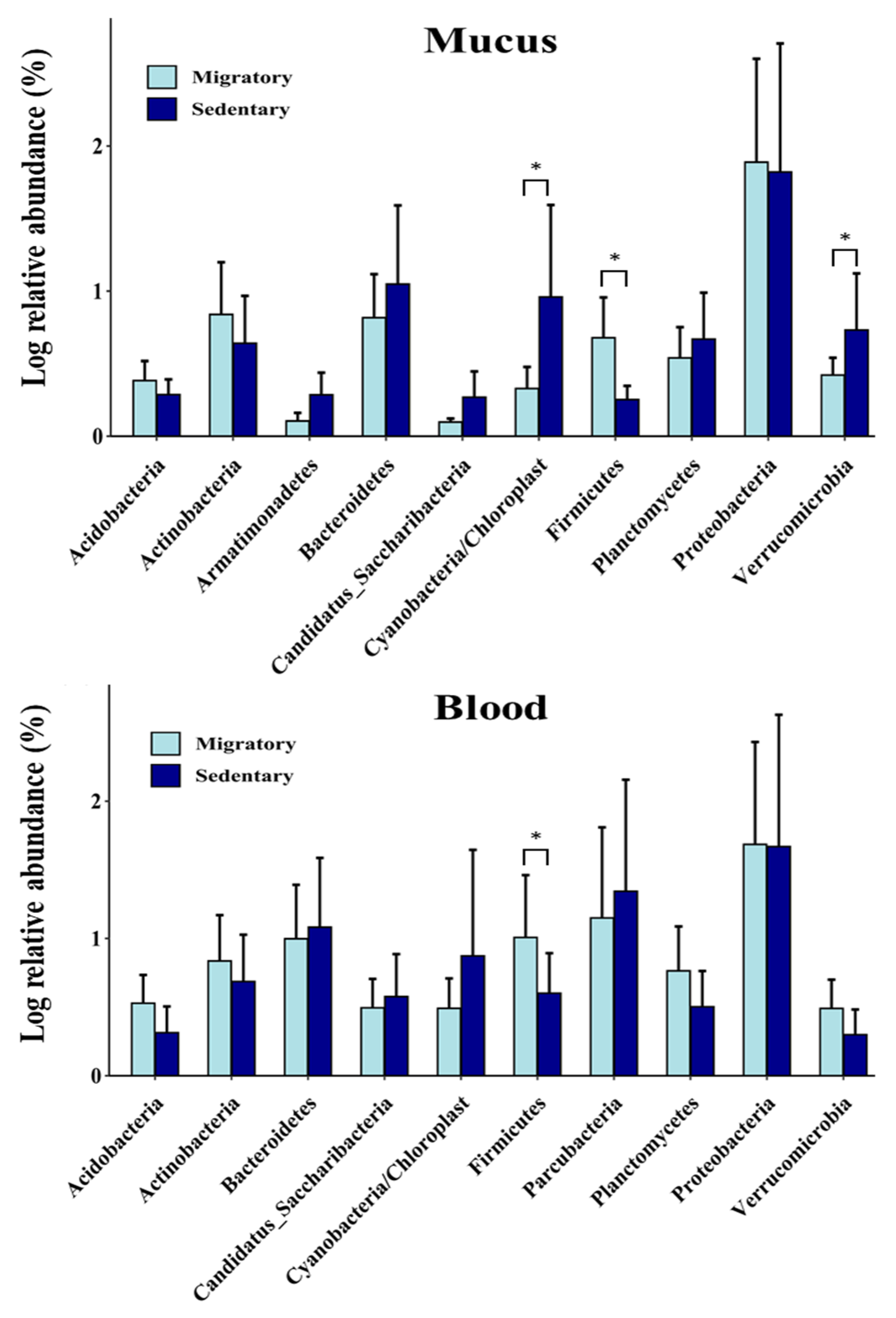
Figure 6.
Relative abundance (%) of the top 10 bacterial genera in circulating and mucosal microbiomes in S. trutta. Bar graph analysis displays the logarithm (base 10) of the relative abundance (mean ± SE). White and black colors represent migratory and sedentary brown trout, respectively.
Figure 6.
Relative abundance (%) of the top 10 bacterial genera in circulating and mucosal microbiomes in S. trutta. Bar graph analysis displays the logarithm (base 10) of the relative abundance (mean ± SE). White and black colors represent migratory and sedentary brown trout, respectively.
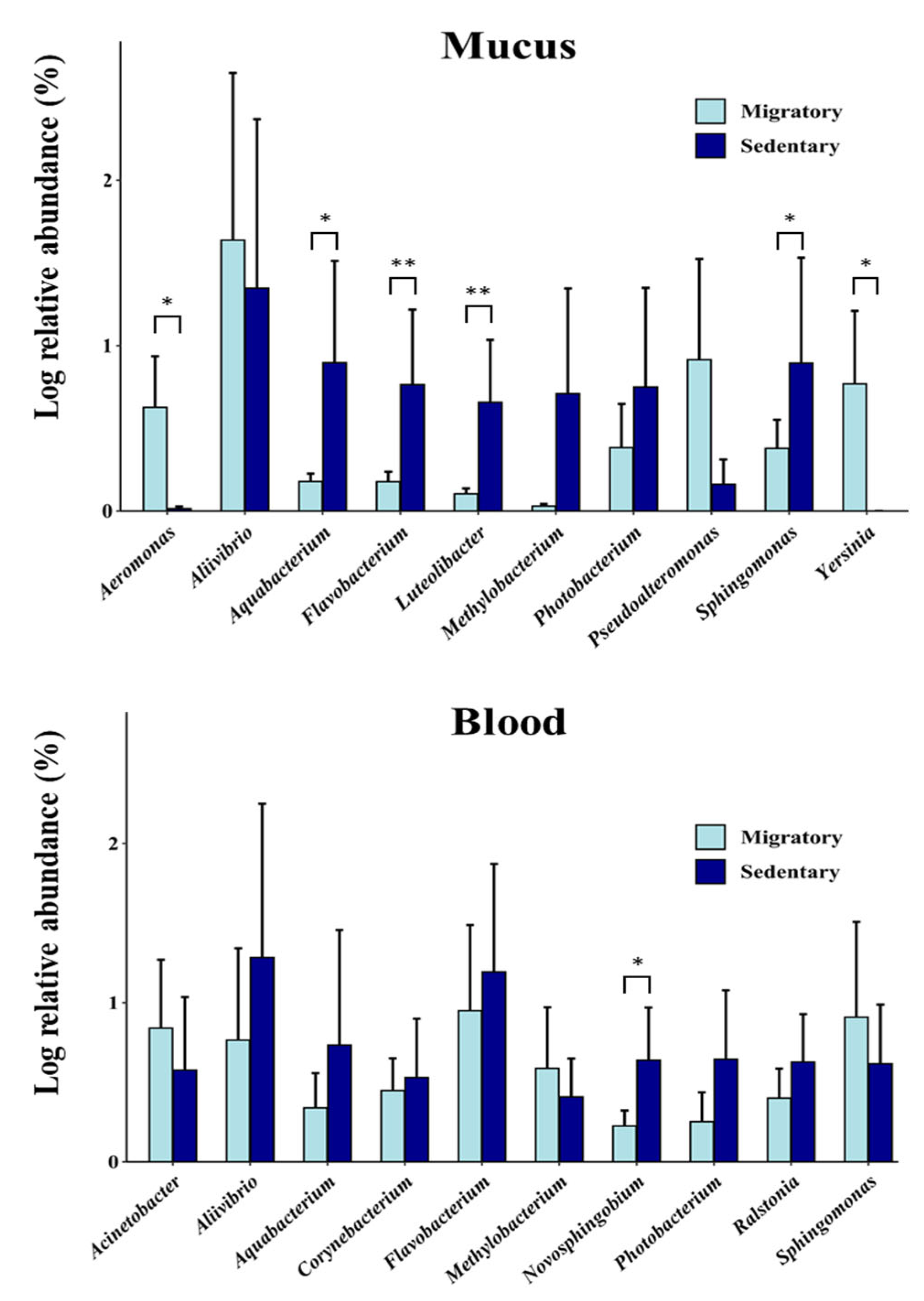
Figure 7.
Inter-individual variability of circulating and mucosal microbiomes at the phylum level. Bar plots display the major phyla between sedentary (left) and migratory (right) brown trout (S. trutta) collected at different sites. Phyla with a relative abundance of ≤ 1.5% are represented as "Other.".
Figure 7.
Inter-individual variability of circulating and mucosal microbiomes at the phylum level. Bar plots display the major phyla between sedentary (left) and migratory (right) brown trout (S. trutta) collected at different sites. Phyla with a relative abundance of ≤ 1.5% are represented as "Other.".
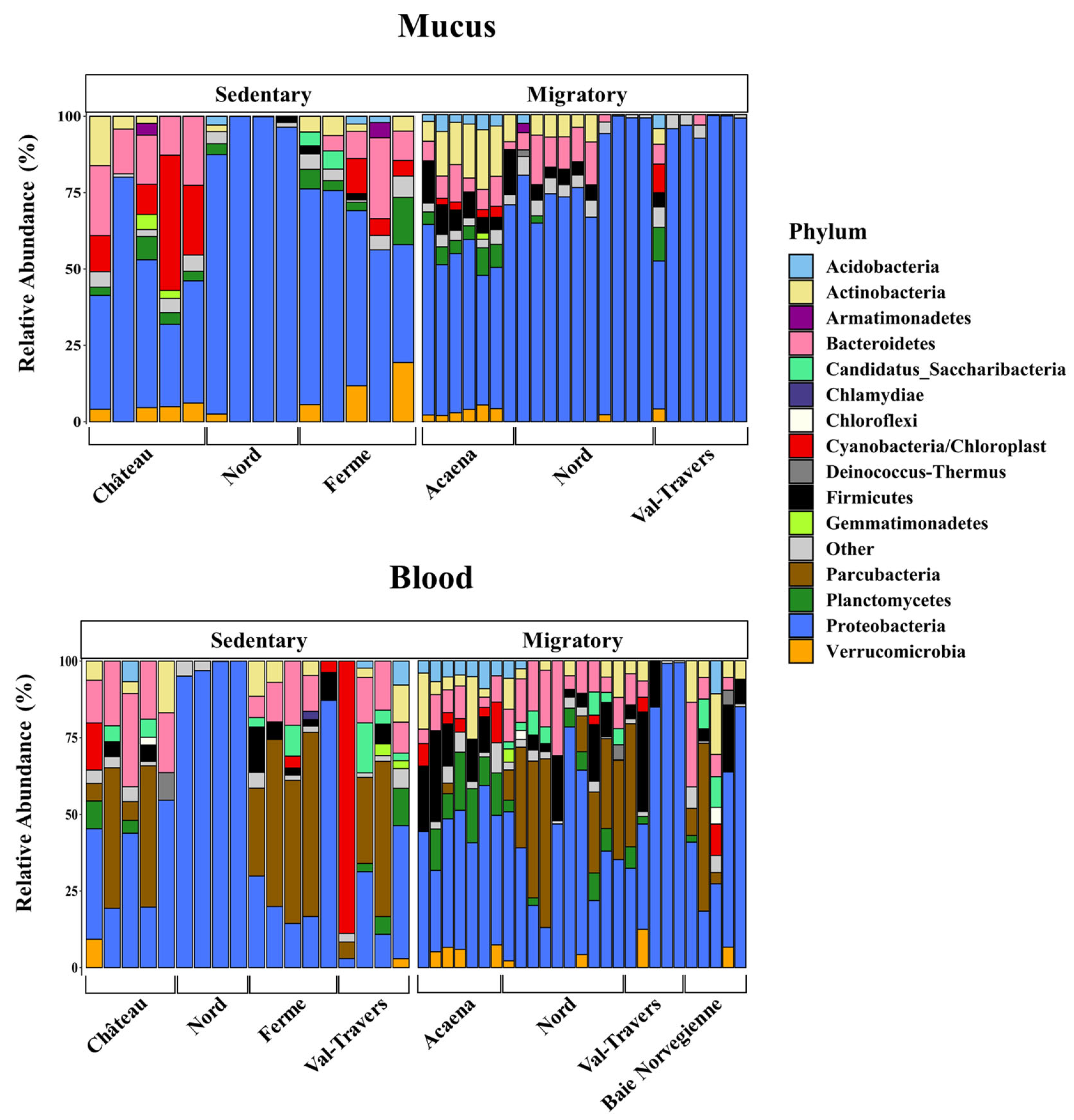
Figure 8.
Heatmaps show the relative abundance (natural logarithm base) of metabolic pathways based on 16S rRNA data in migratory and resident trout collected in different sites.
Figure 8.
Heatmaps show the relative abundance (natural logarithm base) of metabolic pathways based on 16S rRNA data in migratory and resident trout collected in different sites.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



