
Submitted:
27 January 2023
Posted:
30 January 2023
You are already at the latest version
Abstract
The gap junction-coupled astroglial network plays a central role in the regulation of neuronal activity and synchronization, but its involvement in the pathogenesis of neuronal diseases is not yet understood. Here we present the current state of knowledge about the impact of impaired glial coupling in the development and progression of epilepsy and discuss whether astrocytes represent alternative therapeutic targets. We focus mainly on temporal lobe epilepsy (TLE), which is the most common form of epilepsy in adults, characterized by high therapy resistance. Functional data from TLE patients and corresponding experimental models point to a complete loss of astrocytic coupling, but preservation of the gap junction forming proteins connexin43 (Cx43) and connexin30 (Cx30) in hippocampal sclerosis. Several studies further indicate that astrocyte uncoupling represents a causal event in the initiation of TLE, as it occurs very early in epileptogenesis, clearly preceding dysfunctional changes in neurons. However, more research is needed to fully understand the role of gap junction channels in epilepsy and to develop safe and effective therapeutic strategies targeting astrocytes.
Keywords:
astrocyte
; gap junction coupling
; connexin 43
; connexin 30
; hippocampus
; epilepsy
; ep-ileptogenesis
; temporal lobe epilepsy
1. Introduction
Epilepsy is a disease of the brain that affects about 1% of the world's population and is characterised by the occurrence of recurrent unprovoked epileptic seizures [1,2]. Although more than twenty antiepileptic drugs (AEDs) are currently on the market, seizures cannot be fully controlled in about-one third of patients. Furthermore, available AEDs merely provide symptomatic treatment but are not able to prevent or reverse the development of the disorder [3]. These drugs act primarily on neurons by modulating voltage-activated ion channels, enhancing GABAergic inhibition or attenuating excitatory neurotransmission [3,4]. However, as the efficacy and tolerability of AEDs have not substantially improved in recent decades and drugs with disease-modifying (antiepileptogenic) effects are still not available, this neurocentric strategy in the development of AEDs is increasingly being challenged. Indeed, it seems plausible that not only defective neurons but also a disruption of the control mechanisms that regulate neuronal excitability and synaptic transmission can cause epilepsy [5,6]. Astrocytes not only have a crucial homeostatic and supportive functions, such as supply of energy substrates to neurons, control of extracellular ion and water homeostasis, clearance of neurotransmitters from the synapse or regulation of the blood-brain barrier (BBB) permeability, but also actively modulate synaptic transmission and neuronal plasticity through the release of gliotransmitter [5,7]. Important prerequisite for the implementation and/or efficiency of most of these regulatory functions is the fact that astrocytes are extensively electrically and metabolically interconnected via low-resistance aqueous channels termed gap junctions (GJs). This leads to the formation of large syncytium-like functional networks that overlap with neuronal synaptic networks and enable their coordinated regulation and synchronisation [8,9]. Astrocytic gap junctions consist mainly of the channel proteins connexin43 (Cx43) and connexin30 (Cx30), whose relative expression levels vary considerably across developmental stages and brain regions [9,10,11]. In the adult mouse and human hippocampus astrocyte coupling appears to be predominately mediated by Cx43 [10,12,13], while mouse thalamic astrocytes couple mainly through Cx30 [11]. The two astrocytic Cx isoforms possess different biophysical properties (unitary conductances, selectivity, transjunctional voltage sensitivity, and gating properties) and are differentially regulated [14,15]. The gating properties of Cx43-based channels as well as the trafficking, assembly and degradation of Cx43 proteins are thightly regulated by phosphorylation [16]. In contrast, there are no reports on Cx30 phosphorylation. Astrocytic Cxs can form in addition to homotypic GJ channels also functional heterotypic channels with each other and with oligodendrocytic Cxs [11,17,18]. In addition, under certain (primarily pathological) conditions, undocked Cx hemichannels (HCs) might also function as transmembrane channels, allowing the bidirectional exchange of ions and small molecules between the cytoplasm and the extracellular space [8,19]. Finally, there is growing evidence that Cx proteins play an important role in several other fundamental processes, including adhesion, migration, cellular life cycle, neurogenesis, cell death and signal transduction [8,20,21].
2. Role of glial Cx channels in epilepsy
In view of the essential role of the astrocyte network for neuronal function and brain homeostasis, it is not surprising that various neurological disorders including stroke, migraine, gliomas, Alzheimer’s disease, and epilepsy have been linked to alterations in astrocyte coupling [8]. The role of astrocytic network disturbances in the development and progression of epilepsy is not yet conclusively understood, but there is growing evidence that they are fundamentally involved [5,22,23,24,25]. In principle, disruption of GJ coupling between astrocytes can affect neuronal excitability and synchronisation in many ways (Figure 1). An important epilepsy-promoting functional consequence of such a disruption would be the reduced astrocytic capacity to buffer elevated extracellular K+ concentrations ([K+]o) during high neuronal activity. In the process known as spatial K+ buffering, the excess neuronally released [K+]o is passively taken up by astrocytes via Kir4.1 channels, distributed in the GJ-coupled network and released at sites of lower [K+]o [26]. This process, which is driven by the difference between the local K+ equilibrium potential and the more negative membrane potential of the glial syncytium, cannot be sustained in the absence of coupling, resulting in stronger [K+]o increases and consequently in a more pronounced neuronal depolarization and enhanced neuronal excitability [26,27,28,29,30]. In addition to K+, the redistribution of Na+ ions through the GJ-coupled astrocytic network might also play an important role in preventing neuronal hyperexcitability. In fact, astrocytic glutamate transporters are mainly responsible for the removal of excess glutamate during synaptic activity, a process driven by the Na+ gradient and involving massive co-uptake of the ion [31,32,33]. Reduced GJ coupling could promote accumulation of cytosolic Na+ in astrocytes, resulting in a reduced driving force for uptake of synaptically released glutamate and thus increased excitatory neurotransmission [34]. Furthermore, such increased cytosolic Na+ can trigger the reversal of the Na+/Ca2+ exchanger (NCX), resulting in effective astrocytic Ca2+ uptake and therefore defective Ca2+-dependent processes such as aberrant gliotransmitter release, which in turn also affects network excitability [31,33,35,36]. Finally, the cytosolic K+ and Na+ accumulation enhances water influx and thus the extent of activity-dependent astrocytic swelling. The resulting reduction in extracellular space volume further increase the concentration of extracellular ions and neurotransmitters, exacerbating the seizure promoting effect of impaired spatial K+ and glutamate buffering [22,37]. Experimental confirmation for these theoretical considerations is provided by experiments on brain slices from transgenic mice with coupling-deficient astrocytes or after pharmacological disruption of GJ communication. Uncoupling in fact resulted in impaired spatial K+ and Na+ buffering [29,30,32,38,39], reduced astrocytic glutamate clearance [37] and hypertrophic astrocytes [37,40]. Together, these findings point to an anti-epileptic function of the astroglial network. On the other hand, it was shown that the astrocytic network is crucial for the effective supply of energetic metabolites from blood vessels to sites of high neuronal activity [41,42]. It can be assumed that this function of the network has less effect on the initiation than on the maintenance of seizure activity. Thus, an acute reduction of astrocytic GJ coupling may include rapid seizure-promoting consequences due to reduced K+ and glutamate buffering, but delayed seizure-suppressing effects due to insufficient energy supply [22,23,31]. Reduced coupling may also restrict the propagation of Na+ and Ca2+ through the astrocyte network and thus its ability to synchronise large populations of neurons. This effect may counteract hypersynchronous neuronal firing that characterises epilepsy [5,22,31,43].
In addition to intercellular GJ channels, functional Cx HCs have also been reported to play an important role in the pathology of various diseases, including epilepsy, although it is unclear how cells can maintain their integrity upon opening of such large non-specific pores [8,44]. In the healthy brain, Cx HC do not appear to have a significant open probability, but this increases in response to various stress conditions such as metabolic inhibition, hypoxia/ischemia, inflammation, strong depolarisation or altered intra- and extracellular Ca2+ concentrations [8,19]. In epilepsy, opened HCs were proposed to release neuroactive molecules such as glutamate, ATP or D-serine which in turn enhance neuronal excitability and synchronization. Accordingly, Cx HC activation has been proposed to have pro-epileptic effects [8,22,45,46,47].
Finally, non-channels functions might also be involved in pathogenesis of epilepsy. For example, Cx43 was shown to alter the expression and dose-response curve of glial purinergic P2Y1 receptors even in the absence of functional channels [8,48]. Due to its involvement in glial Ca2+ mobilisation and the propagation of astrocytic Ca2+ waves, the metabotropic P2Y1 receptor has been attributed an important role in epilepsy [49,50]. Another example of an epilepsy-relevant non-channel function of Cx proteins was provided by Pannasch and colleagues, who showed that Cx30 proteins control the intrusion of astroglial processes into the synaptic cleft, thereby indirectly modulating the efficiency of synaptic glutamate clearance and thus the strength of excitatory synaptic transmission [51].
3. Connexin expression and GJ coupling in human epilepsy
The most relevant information about astrocytic changes in human epilepsy has been gained from epileptogenic brain tissue specimens surgically resected from patients with drug-resistant temporal lobe epilepsy (TLE). Comparative studies of epilepsy-related changes in Cx expression have been described in several publications, in which either surgical specimens from TLE patients with and without hippocampal sclerosis (HS), or epileptic tissue samples from epilepsy patients with specimens from nonepileptic patients were compared [12,52,53,54,55,56,57,58]. Except one study [56], all found upregulation of Cx43 transcripts or protein in epileptic or sclerotic tissue. A detailed analysis was carried out by Deshpande and co-workers who compared not only whole cell protein levels of the two astrocytic Cx isoforms but also the plasma membrane-associated protein fractions, subcellular distribution and phosphorylation status of Cx43 between sclerotic and non-sclerotic human tissue [12]. Neither total nor membrane-associated Cx30 protein levels nor subcellular distribution of Cx30 differed between both conditions. Interestingly, in HS increased Cx43 protein levels were found only in the total protein fraction, but not in the plasma membrane fraction. Importantly, the authors also reported a pronounced subcellular redistribution towards the perivascular endfeet as well as an altered phosphorylation pattern of Cx43 in HS, a finding suggestive of altered astrocytic coupling [12]. Information on functional astrocytic changes in human epileptic tissue is sparse, due to the limited availability of live tissue, the technical difficulties of recording from them and the lack of healthy control tissue. First evidence of a disturbance in the ability of astrocytes to spatially buffer K+ in human TLE with HS has already been gained more than 2 decades ago, although it remained unclear whether changes in GJ coupling contributed to the phenomenon [59,60,61,62]. Characterization of functional GJ coupling between human astrocytes in the sclerotic and non-sclerotic hippocampus has been performed several years later by tracer diffusion analysis [24]. Intriguingly, in non-HS control-like tissue (obtained from patients with lesion-associated TLE, [62,63]) the extent of astrocyte coupling was in the same range as in the adult rodent hippocampus, indicating that GJ coupling has no influence on the pathogenesis of this type of epilepsy [13,24]. In contrast, complete loss of bona fide astrocytes and GJ coupling was found in hippocampal slices from TLE-HS [24]. Together, these data suggest that Cx proteins are abundantly expressed in the sclerotic hippocampus from TLE patients, but do not form functional GJ channels, most likely due to subcellular redistribution, morphological alterations and/or abnormal phosphorylation of Cx43.
4. Connexin expression, communication and regulation in experimental epilepsy
The loss of astrocytic coupling observed in human tissue raises the question of whether this is causally involved in the genesis of epilepsy or whether it represents merely a consequence of the disease. To answer this question, information is required about the early pathophysiological changes that ultimately culminate in chronic epilepsy. As human tissue specimens are only available from the late chronic state of the disorder, animal models that closely mimic the human situation are needed. Most experimental TLE models involve the chemical or electrical induction of status epilepticus (SE) that triggers chronic epilepsy after a latent period of days to weeks [64,65,66]. Astrocyte changes occurring during SE or the latency period can be considered as possible causative factors in epileptogenesis. In accordance with the human Cx expression studies, Cx43 and Cx30 mRNA and/or protein were found either unchanged or upregulated in TLE models [12,67,68,69,70,71,72,73,74,75]. Intriguingly, in the unilateral intracortical kainate injection model of TLE-HS [24] the changes in astrocytic Cx expression, subcellular distribution and phosphorylation closely resembled those described in human tissues. Consistently, of the two astrocytic Cxs, only total protein levels of Cx43 were upregulated in the sclerotic hippocampus, while the membrane-associated levels were unaltered. Also in accordance with the situation in human tissue, Cx43 exhibited subcellular redistribution towards perivascular endfeet and altered phosphorylation pattern in HS [12]. Interestingly, the model also recapitulated the complete loss of GJ coupling between astrocytes in chronic human TLE and revealed that uncoupling and the associated disruption of K+ buffering started already at a very early stage of epileptogenesis, even before the onset of neurodegeneration and chronic seizures activity [24]. This finding implies that uncoupling of astrocytes plays a causal role in the initiation of TLE. A similar reduction of GJ coupling was also observed 5 days after hyperthermia-induced febrile seizures [76], a finding that might provide a mechanistic explanation for the increased risk of developing human TLE following prolonged juvenile febrile seizures [77]. In contrast, Takahashi and colleagues reported increased coupling between hippocampal astrocytes one week after systemic injection of kainate into rats [74]. Differences between models (e.g. in the time course of histopathological and functional changes associated with epileptogenesis) may account for these contradictory findings.
The signaling pathway underlying loss of astrocyte coupling in TLE remains unknown, but there are good reasons to assume that increased levels of pro-inflammatory cytokines are involved. Indeed, inflammation is a hallmark of both human and experimental epilepsy, and there is evidence that the process is not only a consequence but also a causal factor in the etiology of the disease [78]. Initial evidence for the regulation of astrocytic GJ coupling by inflammatory mediators came from cell culture studies, which showed that the pro-inflammatory cytokines IL-1β and TNFα released from microglia inhibit coupling between cultured astrocytes [79,80,81]. Interestingly, the cytokines exerted an opposite effect on the Cx HC, which were activated in their presence [80]. In later work, inhibition of astrocytic coupling could also be induced in situ after incubation of acute hippocampal slices from mice with IL-1β and TNFα, as well as in vivo by systemic injection of the inflammation-inducing molecule lipopolysaccharide [24]. Direct demonstration of the involvement of microglia-derived cytokines in astrocytic uncoupling in epileptic tissue was recently provided by Henning and co-workers, who showed that seizure activity triggered by intracortical kainate injections decreased GJ coupling between hippocampal astrocytes only in acute slices from wild-type mice, but not in slices from transgenic mice with microglia-specific TNFα deletion or from mice with depleted microglia [82]. These data suggest that at this early time point (4 h after kainate injection), astrocytic coupling is inhibited by microglial TNFα. The cytokine probably exerts its effects by binding to astrocytic TNF receptors, which in turn might mediate the observed changes in Cx expression and/or phosphorylation [12,76], or even trigger necroptotic astrocytic cell death as recently described in the same model [83].
Another pathological mechanism that may be involved in GJ regulation arises from the fact that epilepsy is associated with a breakdown of the BBB, allowing serum proteins such as albumin to enter the brain parenchyma [84]. Extravasated serum albumin was proposed to be endocytosed by astrocytes via binding to transforming growth factor beta (TGFβ) receptors to reduce Cx expression and GJ coupling [85,86,87]. However, in a recent study no major astrocytic albumin uptake could be detected 4 h and 1 d after intracortical kainate injection, and inhibition of TGFβR1 kinase inhibition did not prevent seizure-induced GJ uncoupling, arguing against an involvement of this mechanism in the regulation of GJs during early epileptogenesis [88]. Nevertheless, as uncoupling and albumin extravasation was observed at the chronic stage in this TLE model [12,24], a later contribution of the albumin-TGFβR1signalling pathway to astrocytic dysfunction cannot be ruled out.
5. Impact of glial Cx gene knockout on neuronal excitability and epileptogenesis
Important insights into the role of astrocytic GJ channels in the regulation of neuronal excitability and susceptibility to seizures have been gained from transgenic mice with coupling-deficient astrocytes, which were generated by Wallraff and colleagues by crossing astrocyte-specific Cx43-deficient mice (Cx43fl/fl:hGFAP-Cre mice [89]) with mice globally lacking Cx30 (Cx30-/- [30,90]). The resulting double knockout (dKO) mice showed not only a complete disruption of astrocytic coupling and, as mentioned above, impaired K+ and glutamate buffering, but also spontaneous epileptiform activity in acute hippocampal slices [30]. In a subsequent study, dKO mice exhibited a higher number but less severe PTZ-induced acute seizures in vivo during short term (30 min) EEG/video recording, a finding the authors attributed to an increased neuronal excitability but decreased neuronal release probability and synchronization [91]. Recently, dKO mice were subjected to the intracortical kainate injection model of TLE-HS to assess not only consequences of Cx deficiency on seizure susceptibility but also on the whole process of epileptogenesis [92]. Remarkably, dKO mice showed substantially increased seizure and interictal spike activity during the chronic phase, but less pronounced HS, indicating that the interruption of astrocytic GJ communication promotes chronic seizures but attenuates seizure-induced histopathological changes [92]. However, neither the constitutive dKO mice nor the recently generated inducible, astrocyte-specific dKO mice (Cx30fl/fl:Cx43fl/fl:GLASTCreERT2; [40]) showed spontaneous seizures or abnormal EEG activity in vivo [40,91]. Of course, one must bear in mind that in dKO mice not only intercellular coupling but also the Cx proteins are missing, which does not correspond to the situation in human and experimental epilepsy where the proteins are even upregulated [12]. Hence, potential pro-epileptic non-channel and HC functions of Cxs are not considered in these mice. Moreover, astrocyte uncoupling in human and experimental TLE is spatially restricted to the sclerotic area and the epileptic foci, while Cx deletion in dKO mice concerns the entire brain. Finally, compensatory developmental changes in the constitutive mouse line and residual coupling due to incomplete Cre recombination in the inducible mouse line limit the conclusions that can be drawn from these experiments.
Mice lacking only Cx30 developed less severe behavioural convulsions during the first 2 h after systemic kainate injection, a phenomenon that was dependent only on the presence of the protein but not on functional channels [72]. The authors attributed this to more effective astrocytic glutamate clearance in the absence of Cx30, which is consistent with the above-mentioned non-channel function of the protein in controlling the invasion of astrocytic processes into the synaptic cleft [51,72].
6. Therapeutic potential of targeting glial Cx channels in epilepsy
The low efficacy and tolerability of currently available AEDs and the lack of antiepileptogenic, disease-modifying drugs underscore the urgent need for new cellular and molecular targets and therapeutic strategies in drug development. The evidence from the literature described above suggests that the Cx channels, especially those composed of Cx43, play an important role in epilepsy and may therefore constitute promising targets for new AEDs. Interestingly, it has been shown that impaired GJ coupling in cytokine- or LPS-incubated astroglia/microglia co-cultures or in acute hippocampal slices from LPS-injected mice could be normalized by treatment with the commonly used AED levetiracetam, leading to the assumption that the antiepileptogenic properties of the drug are at least partly attributable to its effect on coupling [24,81]. The suitability of substances that directly inhibit or increase GJ coupling as antiepileptic drugs is questionable, as Cxs are ubiquitously expressed in almost all tissues and cell types [93] and therefore systemic administration likely has side effects. For example, Cx43 is the most abundant GJ protein in the heart where it plays a key role in the coordinated spread of electrical activity. Thus, its inhibition or activation in the heart would have deleterious effects on heart function. Another problem is the lack of specificity of substances that modulate GJs. For instance, carbenoxolone (CBX), the most commonly used GJ blocker, is not only unable to distinguish between different Cx isoforms or between GJ channels and HCs, but was also shown to inhibit voltage-gated Ca2+ channels, purinergic P2X7 receptors and tonic GABAA receptor currents [94,95,96]. Accordingly, several studies have shown that CBX inhibits synaptic transmission and neuronal network excitability in a GJ–independent manner, questioning its suitability for assessing the role of GJs in epilepsy [94,97,98,99,100]. This should be kept in mind when drawing conclusions from the extensive literature on antiepileptic effects of CBX treatment in epilepsy models (for review see [101]). Similarly, no substances are known to specifically increase astrocytic coupling. Regarding the systemic side effects, it would probably make more sense not to search for more specific GJ blockers/activators but for substances that prevent/reverse the molecular mechanisms or signaling pathways that lead to impaired coupling under pathological conditions. For example, it was recently reported that epileptiform activity inhibits astrocytic coupling via Na+/HCO3- cotransporter-mediated intracellular alkalization, and that blocker of this cotransporter have antiepileptic effects [102]. As data from human and experimental TLE indicate that GJ inhibition is mediated by phosphorylation-mediated closure of Cx43 [12,76], compounds that disrupt aberrant Cx phosphorylation may have therapeutic potentials in the treatment of epilepsy. In this context, antiarrhythmic peptides such as the Cx43-targeting peptide danegaptide (ZP1609), developed for the treatment of reperfusion injury after acute myocardial infarction [103], were discussed [8,46]. Indeed, danegaptide was found to normalize pathological phosphorylation-induced closure of Cx43 not only in the heart but also between astrocytes in a mouse brain ischemia/reperfusion model [104,105]. However, the peptide exhibited no effect on astrocyte uncoupling induced by intracortical kainate injections (unpublished data from our group), indicating that the mechanism of Cx43 inhibition differs between epilepsy and ischemia.
In addition to GJ channels, specific Cx HC blockade has also been proposed as a promising strategy for treating epilepsy [45,47]. In accordance with the proposed seizure-promoting consequences of HC opening, inhibition of Cx43 HCs with the Cx mimetic peptide TAT-Gap19 or the small molecule D4, which were described to block HC activity without affecting GJ coupling, yielded anticonvulsant effects in mice and rats in different models of seizures and epilepsy [45,47]. The advantage of this approach is that HC activation occurs only under pathological conditions, so that systemic administration of HC blockers should not have major side effects [45,47]. However, more research is needed to understand the biophysical properties and specific role of these channels in the healthy and diseased brain before the therapeutic potential of Cx HC blockers can be established.
7. Conclusions
Evidence is accumulating showing that astrocyte Cx channels are critically implicated in epilepsy, but the underlying mechanisms and therapeutic importance are still poorly understood. In the sclerotic hippocampus of TLE patients, astrocytic GJ coupling is completely lost, despite the fact that expression of the two astrocytic Cx isoforms does not decrease. This might simply due to loss of physical contact of glial processes as a result of the dramatic morphological alterations astrocytes undergo in sclerosis (Hinterkeuser et al., 2000; Bedner et al., 2015). However, it can also not be excluded that besides altered intercellular GJ coupling, increased Cx HC activity or non-channel properties of Cxs affect neuronal excitability and synchronization. Maybe, only a combination of several of these factors is sufficient to generate neuronal hyperexcitability, which would explain why mice with genetic deletion of astrocytic Cxs do not exhibit spontaneous behavioral convulsions. Identifying the exact mechanisms underlying generation and progression of this complex disease remains a challenge. Nevertheless, since astrocytic uncoupling is one of the earliest detectable cellular changes in experimental TLE that precedes the onset of neurodegeneration and chronic seizures, it can be considered as a causative factor. To validate the causality of the dysfunction, it would be necessary to proof that its suppression prevents the genesis of the disease. However, this requires detailed knowledge of the mechanism of uncoupling and the identification of substances that specifically inhibit it. The growing recognition of the importance of the astrocytic network in epilepsy, together with the increasingly sophisticated technical tools and methods available today, give rise to optimism that the mechanisms governing epilepsy will soon be better understood. This opens new avenues for future research and therapeutic interventions.
Author Contributions
Both authors wrote the manuscript and agreed to its final version.
Funding
This research was funded by BMBF, grant numbers 01DN20001 and 16GW0182; and EU, grant number H2020 MSCA-ITN project 722053.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE Official Report: A Practical Clinical Definition of Epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Kwon, C.-S.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L.; Jetté, N. Prevalence and Incidence of Epilepsy: A Systematic Review and Meta-Analysis of International Studies. Neurology 2017, 88, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol Rev 2020, 72, 606–638. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E. Mechanisms of Action of Antiepileptic Drugs: The Search for Synergy. Current Opinion in Neurology 2010, 23, 157–163. [Google Scholar] [CrossRef]
- Boison, D.; Steinhäuser, C. Epilepsy and Astrocyte Energy Metabolism. Glia 2018, 66, 1235–1243. [Google Scholar] [CrossRef]
- Binder, D.K.; Steinhäuser, C. Astrocytes and Epilepsy. Neurochem Res 2021, 46, 2687–2695. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol Rev 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Giaume, C.; Naus, C.C.; Sáez, J.C.; Leybaert, L. Glial Connexins and Pannexins in the Healthy and Diseased Brain. Physiological Reviews 2020, 101, 93–145. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Koulakoff, A.; Roux, L.; Holcman, D.; Rouach, N. Astroglial Networks: A Step Further in Neuroglial and Gliovascular Interactions. Nat Rev Neurosci 2010, 11, 87–99. [Google Scholar] [CrossRef]
- Gosejacob, D.; Dublin, P.; Bedner, P.; Hüttmann, K.; Zhang, J.; Tress, O.; Willecke, K.; Pfrieger, F.; Steinhäuser, C.; Theis, M. Role of Astroglial Connexin30 in Hippocampal Gap Junction Coupling. Glia 2011, 59, 511–519. [Google Scholar] [CrossRef]
- Griemsmann, S.; Höft, S.P.; Bedner, P.; Zhang, J.; von Staden, E.; Beinhauer, A.; Degen, J.; Dublin, P.; Cope, D.W.; Richter, N.; et al. Characterization of Panglial Gap Junction Networks in the Thalamus, Neocortex, and Hippocampus Reveals a Unique Population of Glial Cells. Cereb Cortex 2015, 25, 3420–3433. [Google Scholar] [CrossRef]
- Deshpande, T.; Li, T.; Herde, M.K.; Becker, A.; Vatter, H.; Schwarz, M.K.; Henneberger, C.; Steinhäuser, C.; Bedner, P. Subcellular Reorganization and Altered Phosphorylation of the Astrocytic Gap Junction Protein Connexin43 in Human and Experimental Temporal Lobe Epilepsy. Glia 2017, 65, 1809–1820. [Google Scholar] [CrossRef]
- Bedner, P.; Jabs, R.; Steinhäuser, C. Properties of Human Astrocytes and NG2 Glia. Glia 2020, 68, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Valiunas, V.; Manthey, D.; Vogel, R.; Willecke, K.; Weingart, R. Biophysical Properties of Mouse Connexin30 Gap Junction Channels Studied in Transfected Human HeLa Cells. The Journal of Physiology 1999, 519, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Valiunas, V.; Weingart, R.; Brink, P.R. Formation of Heterotypic Gap Junction Channels by Connexins 40 and 43. Circ Res 2000, 86, E42–49. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Connexin43 Phosphorylation: Structural Changes and Biological Effects. Biochemical Journal 2009, 419, 261–272. [Google Scholar] [CrossRef]
- Manthey, D.; Banach, K.; Desplantez, T.; Lee, C.G.; Kozak, C.A.; Traub, O.; Weingart, R.; Willecke, K. Intracellular Domains of Mouse Connexin26 and -30 Affect Diffusional and Electrical Properties of Gap Junction Channels. J. Membrane Biol. 2001, 181, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Magnotti, L.M.; Goodenough, D.A.; Paul, D.L. Functional Heterotypic Interactions between Astrocyte and Oligodendrocyte Connexins. Glia 2011, 59, 26–34. [Google Scholar] [CrossRef]
- Giaume, C.; Leybaert, L.; Naus, C.; Sáez, J.-C. Connexin and Pannexin Hemichannels in Brain Glial Cells: Properties, Pharmacology, and Roles. Front. Pharmacol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Decrock, E.; Leybaert, L.; Bultynck, G.; Himpens, B.; Vanhaecke, T.; Rogiers, V. Non-Channel Functions of Connexins in Cell Growth and Cell Death. Biochimica et Biophysica Acta (BBA) - Biomembranes 2012, 1818, 2002–2008. [Google Scholar] [CrossRef]
- Kunze, A.; Congreso, M.R.; Hartmann, C.; Wallraff-Beck, A.; Hüttmann, K.; Bedner, P.; Requardt, R.; Seifert, G.; Redecker, C.; Willecke, K.; et al. Connexin Expression by Radial Glia-like Cells Is Required for Neurogenesis in the Adult Dentate Gyrus. Proc Natl Acad Sci U S A 2009, 106, 11336–11341. [Google Scholar] [CrossRef] [PubMed]
- Steinhäuser, C.; Seifert, G.; Bedner, P. Astrocyte Dysfunction in Temporal Lobe Epilepsy: K+ Channels and Gap Junction Coupling. Glia 2012, 60, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Bedner, P.; Steinhäuser, C. Altered Kir and Gap Junction Channels in Temporal Lobe Epilepsy. Neurochemistry International 2013, 63, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Bedner, P.; Dupper, A.; Hüttmann, K.; Müller, J.; Herde, M.K.; Dublin, P.; Deshpande, T.; Schramm, J.; Häussler, U.; Haas, C.A.; et al. Astrocyte Uncoupling as a Cause of Human Temporal Lobe Epilepsy. Brain 2015, 138, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Henning, L.; Unichenko, P.; Bedner, P.; Steinhäuser, C.; Henneberger, C. Overview Article Astrocytes as Initiators of Epilepsy. Neurochem Res 2022. [Google Scholar] [CrossRef] [PubMed]
- Orkand, R.K. Introductory Remarks: Glial-Interstitial Fluid Exchange. Annals of the New York Academy of Sciences 1986, 481, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Walz, W. Role of Astrocytes in the Clearance of Excess Extracellular Potassium. Neurochemistry International 2000, 36, 291–300. [Google Scholar] [CrossRef]
- Kofuji, P.; Newman, E.A. Potassium Buffering in the Central Nervous System. Neuroscience 2004, 129, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Breithausen, B.; Kautzmann, S.; Boehlen, A.; Steinhäuser, C.; Henneberger, C. Limited Contribution of Astroglial Gap Junction Coupling to Buffering of Extracellular K+ in CA1 Stratum Radiatum. Glia 2020, 68, 918–931. [Google Scholar] [CrossRef]
- Wallraff, A.; Köhling, R.; Heinemann, U.; Theis, M.; Willecke, K.; Steinhäuser, C. The Impact of Astrocytic Gap Junctional Coupling on Potassium Buffering in the Hippocampus. J. Neurosci. 2006, 26, 5438–5447. [Google Scholar] [CrossRef]
- Henneberger, C. Does Rapid and Physiological Astrocyte–Neuron Signalling Amplify Epileptic Activity? The Journal of Physiology 2017, 595, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Langer, J.; Stephan, J.; Theis, M.; Rose, C.R. Gap Junctions Mediate Intercellular Spread of Sodium between Hippocampal Astrocytes in Situ. Glia 2012, 60, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Kirischuk, S.; Parpura, V.; Verkhratsky, A. Sodium Dynamics: Another Key to Astroglial Excitability? Trends in Neurosciences 2012, 35, 497–506. [Google Scholar] [CrossRef]
- Rose, C.R.; Felix, L.; Zeug, A.; Dietrich, D.; Reiner, A.; Henneberger, C. Astroglial Glutamate Signaling and Uptake in the Hippocampus. Front Mol Neurosci 2017, 10, 451. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, J.; Wellmann, M.; Sotomayor-Zárate, R.; Bonansco, C. Gliotransmission: A Novel Target for the Development of Antiseizure Drugs. Neuroscientist 2020, 26, 293–309. [Google Scholar] [CrossRef]
- Müller, J.; Timmermann, A.; Henning, L.; Müller, H.; Steinhäuser, C.; Bedner, P. Astrocytic GABA Accumulation in Experimental Temporal Lobe Epilepsy. Front Neurol 2020, 11, 614923. [Google Scholar] [CrossRef] [PubMed]
- Pannasch, U.; Vargová, L.; Reingruber, J.; Ezan, P.; Holcman, D.; Giaume, C.; Syková, E.; Rouach, N. Astroglial Networks Scale Synaptic Activity and Plasticity. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 8467–8472. [Google Scholar] [CrossRef] [PubMed]
- Bazzigaluppi, P.; Weisspapir, I.; Stefanovic, B.; Leybaert, L.; Carlen, P.L. Astrocytic Gap Junction Blockade Markedly Increases Extracellular Potassium without Causing Seizures in the Mouse Neocortex. Neurobiology of Disease 2017, 101, 1–7. [Google Scholar] [CrossRef]
- EbrahimAmini, A.; Bazzigaluppi, P.; Aquilino, M.S.; Stefanovic, B.; Carlen, P.L. Neocortical in Vivo Focal and Spreading Potassium Responses and the Influence of Astrocytic Gap Junctional Coupling. Neurobiology of Disease 2021, 147, 105160. [Google Scholar] [CrossRef]
- Hösli, L.; Binini, N.; Ferrari, K.D.; Thieren, L.; Looser, Z.J.; Zuend, M.; Zanker, H.S.; Berry, S.; Holub, M.; Möbius, W.; et al. Decoupling Astrocytes in Adult Mice Impairs Synaptic Plasticity and Spatial Learning. Cell Rep 2022, 38, 110484. [Google Scholar] [CrossRef]
- Rouach, N.; Koulakoff, A.; Abudara, V.; Willecke, K.; Giaume, C. Astroglial Metabolic Networks Sustain Hippocampal Synaptic Transmission. Science 2008, 322, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Philippot, C.; Griemsmann, S.; Jabs, R.; Seifert, G.; Kettenmann, H.; Steinhäuser, C. Astrocytes and Oligodendrocytes in the Thalamus Jointly Maintain Synaptic Activity by Supplying Metabolites. Cell Rep 2021, 34, 108642. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gonzalo, M.; Losi, G.; Chiavegato, A.; Zonta, M.; Cammarota, M.; Brondi, M.; Vetri, F.; Uva, L.; Pozzan, T.; de Curtis, M.; et al. An Excitatory Loop with Astrocytes Contributes to Drive Neurons to Seizure Threshold. PLoS Biol. 2010, 8, e1000352. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, B.S.; Hansen, D.B.; Ransom, B.R.; Nielsen, M.S.; MacAulay, N. Connexin Hemichannels in Astrocytes: An Assessment of Controversies Regarding Their Functional Characteristics. Neurochem Res 2017, 42, 2537–2550. [Google Scholar] [CrossRef] [PubMed]
- Walrave, L.; Pierre, A.; Albertini, G.; Aourz, N.; Bundel, D.D.; Eeckhaut, A.V.; Vinken, M.; Giaume, C.; Leybaert, L.; Smolders, I. Inhibition of Astroglial Connexin43 Hemichannels with TAT-Gap19 Exerts Anticonvulsant Effects in Rodents. Glia 2018, 66, 1788–1804. [Google Scholar] [CrossRef] [PubMed]
- Walrave, L.; Vinken, M.; Leybaert, L.; Smolders, I. Astrocytic Connexin43 Channels as Candidate Targets in Epilepsy Treatment. Biomolecules 2020, 10, 1578. [Google Scholar] [CrossRef]
- Guo, A.; Zhang, H.; Li, H.; Chiu, A.; García-Rodríguez, C.; Lagos, C.F.; Sáez, J.C.; Lau, C.G. Inhibition of Connexin Hemichannels Alleviates Neuroinflammation and Hyperexcitability in Temporal Lobe Epilepsy. Proceedings of the National Academy of Sciences 2022, 119, e2213162119. [Google Scholar] [CrossRef] [PubMed]
- Scemes, E. Modulation of Astrocyte P2Y1 Receptors by the Carboxyl Terminal Domain of the Gap Junction Protein Cx43. Glia 2008, 56, 145–153. [Google Scholar] [CrossRef]
- Nikolic, L.; Nobili, P.; Shen, W.; Audinat, E. Role of Astrocyte Purinergic Signaling in Epilepsy. Glia 2020, 68, 1677–1691. [Google Scholar] [CrossRef]
- Vezzani, A.; Ravizza, T.; Bedner, P.; Aronica, E.; Steinhäuser, C.; Boison, D. Astrocytes in the Initiation and Progression of Epilepsy. Nat Rev Neurol 2022, 18, 707–722. [Google Scholar] [CrossRef]
- Pannasch, U.; Freche, D.; Dallérac, G.; Ghézali, G.; Escartin, C.; Ezan, P.; Cohen-Salmon, M.; Benchenane, K.; Abudara, V.; Dufour, A.; et al. Connexin 30 Sets Synaptic Strength by Controlling Astroglial Synapse Invasion. Nature Neuroscience 2014, 17, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Gorter, J.A.; Jansen, G.H.; Leenstra, S.; Yankaya, B.; Troost, D. Expression of Connexin 43 and Connexin 32 Gap-Junction Proteins in Epilepsy-Associated Brain Tumors and in the Perilesional Epileptic Cortex. Acta Neuropathol 2001, 101, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Collignon, F.; Wetjen, N.M.; Cohen-Gadol, A.A.; Cascino, G.D.; Parisi, J.; Meyer, F.B.; Marsh, W.R.; Roche, P.; Weigand, S.D. Altered Expression of Connexin Subtypes in Mesial Temporal Lobe Epilepsy in Humans. Journal of Neurosurgery 2006, 105, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.G.; Bechberger, J.F.; Paul, D.L. Gap Junction Gene Expression in Human Seizure Disorder. Experimental Neurology 1991, 111, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Wallace, G.C.; Holmes, C.; McDowell, M.L.; Smith, J.A.; Marshall, J.D.; Bonilha, L.; Edwards, J.C.; Glazier, S.S.; Ray, S.K.; et al. Hippocampal Tissue of Patients with Refractory Temporal Lobe Epilepsy Is Associated with Astrocyte Activation, Inflammation, and Altered Expression of Channels and Receptors. Neuroscience 2012, 220, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Elisevich, K.; Rempel, S.A.; Smith, B.J.; Edvardsen, K. Hippocampal Connexin 43 Expression in Human Complex Partial Seizure Disorder. Experimental Neurology 1997, 145, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, C.G.; Green, C.R.; Nicholson, L.F.B. Upregulation in Astrocytic Connexin 43 Gap Junction Levels May Exacerbate Generalized Seizures in Mesial Temporal Lobe Epilepsy. Brain Research 2002, 929, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Garbelli, R.; Frassoni, C.; Condorelli, D.F.; Salinaro, A.T.; Musso, N.; Medici, V.; Tassi, L.; Bentivoglio, M.; Spreafico, R. Expression of Connexin 43 in the Human Epileptic and Drug-Resistant Cerebral Cortex. Neurology 2011, 76, 895–902. [Google Scholar] [CrossRef]
- Heinemann, U.; Gabriel, S.; Jauch, R.; Schulze, K.; Kivi, A.; Eilers, A.; Kovacs, R.; Lehmann, T.-N. Alterations of Glial Cell Function in Temporal Lobe Epilepsy. Epilepsia 2000, 41, S185–S189. [Google Scholar] [CrossRef]
- Jauch, R.; Windmüller, O.; Lehmann, T.-N.; Heinemann, U.; Gabriel, S. Effects of Barium, Furosemide, Ouabaine and 4,4′-Diisothiocyanatostilbene-2,2′-Disulfonic Acid (DIDS) on Ionophoretically-Induced Changes in Extracellular Potassium Concentration in Hippocampal Slices from Rats and from Patients with Epilepsy. Brain Research 2002, 925, 18–27. [Google Scholar] [CrossRef]
- Kivi, A.; Lehmann, T.-N.; Kovács, R.; Eilers, A.; Jauch, R.; Meencke, H.J.; Deimling, A.V.; Heinemann, U.; Gabriel, S. Effects of Barium on Stimulus-Induced Rises of [K+]o in Human Epileptic Non-Sclerotic and Sclerotic Hippocampal Area CA1. European Journal of Neuroscience 2000, 12, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Hinterkeuser, S.; Schröder, W.; Hager, G.; Seifert, G.; Blümcke, I.; Elger, C.E.; Schramm, J.; Steinhäuser, C. Astrocytes in the Hippocampus of Patients with Temporal Lobe Epilepsy Display Changes in Potassium Conductances. European Journal of Neuroscience 2000, 12, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Blümcke, I.; Beck, H.; Lie, A.A.; Wiestler, O.D. Molecular Neuropathology of Human Mesial Temporal Lobe Epilepsy. Epilepsy Research 1999, 36, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Schmidt, D. Modern Antiepileptic Drug Development Has Failed to Deliver: Ways out of the Current Dilemma. Epilepsia 2011, 52, 657–678. [Google Scholar] [CrossRef]
- Jefferys, J.; Steinhäuser, C.; Bedner, P. Chemically-Induced TLE Models: Topical Application. Journal of Neuroscience Methods 2016, 260, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Rusina, E.; Bernard, C.; Williamson, A. The Kainic Acid Models of Temporal Lobe Epilepsy. eNeuro 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, D.F.; Mudò, G.; Trovato-Salinaro, A.; Mirone, M.B.; Amato, G.; Belluardo, N. Connexin-30 MRNA Is Up-Regulated in Astrocytes and Expressed in Apoptotic Neuronal Cells of Rat Brain Following Kainate-Induced Seizures. Molecular and Cellular Neuroscience 2002, 21, 94–113. [Google Scholar] [CrossRef]
- Condorelli, D.F.; Trovato-Salinaro, A.; Mudò, G.; Mirone, M.B.; Belluardo, N. Cellular Expression of Connexins in the Rat Brain: Neuronal Localization, Effects of Kainate-Induced Seizures and Expression in Apoptotic Neuronal Cells. European Journal of Neuroscience 2003, 18, 1807–1827. [Google Scholar] [CrossRef]
- Hussein, A.M.; Abbas, K.M.; Abulseoud, O.A.; El-Hussainy, E.-H.M.A. Effects of Ferulic Acid on Oxidative Stress, Heat Shock Protein 70, Connexin 43, and Monoamines in the Hippocampus of Pentylenetetrazole-Kindled Rats. Can J Physiol Pharmacol 2017, 95, 732–742. [Google Scholar] [CrossRef]
- Kinjo, E.R.; Higa, G.S.V.; Morya, E.; Valle, A.C.; Kihara, A.H.; Britto, L.R.G. Reciprocal Regulation of Epileptiform Neuronal Oscillations and Electrical Synapses in the Rat Hippocampus. PLOS ONE 2014, 9, e109149. [Google Scholar] [CrossRef]
- McCracken, C.B.; Roberts, D.C.S. A Single Evoked Afterdischarge Produces Rapid Time-Dependent Changes in Connexin36 Protein Expression in Adult Rat Dorsal Hippocampus. Neurosci Lett 2006, 405, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Pannasch, U.; Dossi, E.; Ezan, P.; Rouach, N. Astroglial Cx30 Sustains Neuronal Population Bursts Independently of Gap-Junction Mediated Biochemical Coupling. Glia 2019, 67, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Güldenagel, M.; Beck, H.; Teubner, B.; Traub, O.; Gutiérrez, R.; Heinemann, U.; Willecke, K. Expression of Connexin Genes in Hippocampus of Kainate-Treated and Kindled Rats under Conditions of Experimental Epilepsy. Molecular Brain Research 2000, 83, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.K.; Vargas, J.R.; Wilcox, K.S. Increased Coupling and Altered Glutamate Transport Currents in Astrocytes Following Kainic-Acid-Induced Status Epilepticus. Neurobiology of Disease 2010, 40, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Tang, Y.C.; Lu, Q.Y.; Xiao, X.L.; Song, T.B.; Tang, F.R. Astrocytic Cx 43 and Cx 40 in the Mouse Hippocampus during and after Pilocarpine-Induced Status Epilepticus. Exp Brain Res 2015, 233, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Dupper, A.; Deshpande, T.; Graan, P.N.E.D.; Steinhäuser, C.; Bedner, P. Experimental Febrile Seizures Impair Interastrocytic Gap Junction Coupling in Juvenile Mice. Journal of Neuroscience Research 2016, 94, 804–813. [Google Scholar] [CrossRef]
- Choy, M.; Dubé, C.M.; Ehrengruber, M.; Baram, T.Z. Inflammatory Processes, Febrile Seizures, and Subsequent Epileptogenesis. Epilepsy Curr 2014, 14, 15–22. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory Pathways as Treatment Targets and Biomarkers in Epilepsy. Nat Rev Neurol 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Même, W.; Calvo, C.-F.; Froger, N.; Ezan, P.; Amigou, E.; Koulakoff, A.; Giaume, C. Proinflammatory Cytokines Released from Microglia Inhibit Gap Junctions in Astrocytes: Potentiation by β-Amyloid. The FASEB Journal 2006, 20, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Sáez, P.J.; Sáez, J.C.; Giaume, C. Cx43 Hemichannels and Gap Junction Channels in Astrocytes Are Regulated Oppositely by Proinflammatory Cytokines Released from Activated Microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Haghikia, A.; Ladage, K.; Hinkerohe, D.; Vollmar, P.; Heupel, K.; Dermietzel, R.; Faustmann, P.M. Implications of Antiinflammatory Properties of the Anticonvulsant Drug Levetiracetam in Astrocytes. Journal of Neuroscience Research 2008, 86, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Henning, L.; Antony, H.; Breuer, A.; Müller, J.; Seifert, G.; Audinat, E.; Singh, P.; Brosseron, F.; Heneka, M.T.; Steinhäuser, C.; et al. Reactive Microglia Are the Major Source of Tumor Necrosis Factor Alpha and Contribute to Astrocyte Dysfunction and Acute Seizures in Experimental Temporal Lobe Epilepsy. Glia 2023, 71, 168–186. [Google Scholar] [CrossRef]
- Wu, Z.; Deshpande, T.; Henning, L.; Bedner, P.; Seifert, G.; Steinhäuser, C. Cell Death of Hippocampal CA1 Astrocytes during Early Epileptogenesis. Epilepsia 2021, 62, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Friedman, A. Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? International Journal of Molecular Sciences 2020, 21, 591. [Google Scholar] [CrossRef] [PubMed]
- David, Y.; Cacheaux, L.P.; Ivens, S.; Lapilover, E.; Heinemann, U.; Kaufer, D.; Friedman, A. Astrocytic Dysfunction in Epileptogenesis: Consequence of Altered Potassium and Glutamate Homeostasis? J. Neurosci. 2009, 29, 10588–10599. [Google Scholar] [CrossRef] [PubMed]
- Cacheaux, L.P.; Ivens, S.; David, Y.; Lakhter, A.J.; Bar-Klein, G.; Shapira, M.; Heinemann, U.; Friedman, A.; Kaufer, D. Transcriptome Profiling Reveals TGF-β Signaling Involvement in Epileptogenesis. J. Neurosci. 2009, 29, 8927–8935. [Google Scholar] [CrossRef] [PubMed]
- Braganza, O.; Bedner, P.; Hüttmann, K.; von Staden, E.; Friedman, A.; Seifert, G.; Steinhäuser, C. Albumin Is Taken up by Hippocampal NG2 Cells and Astrocytes and Decreases Gap Junction Coupling. Epilepsia 2012, 53, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Henning, L.; Steinhäuser, C.; Bedner, P. Initiation of Experimental Temporal Lobe Epilepsy by Early Astrocyte Uncoupling Is Independent of TGFβR1/ALK5 Signaling. Front Neurol 2021, 12, 660591. [Google Scholar] [CrossRef] [PubMed]
- Theis, M.; Jauch, R.; Zhuo, L.; Speidel, D.; Wallraff, A.; Döring, B.; Frisch, C.; Söhl, G.; Teubner, B.; Euwens, C.; et al. Accelerated Hippocampal Spreading Depression and Enhanced Locomotory Activity in Mice with Astrocyte-Directed Inactivation of Connexin43. J. Neurosci. 2003, 23, 766–776. [Google Scholar] [CrossRef]
- Teubner, B.; Michel, V.; Pesch, J.; Lautermann, J.; Cohen-Salmon, M.; Söhl, G.; Jahnke, K.; Winterhager, E.; Herberhold, C.; Hardelin, J.-P.; et al. Connexin30 (Gjb6)-Deficiency Causes Severe Hearing Impairment and Lack of Endocochlear Potential. Human Molecular Genetics 2003, 12, 13–21. [Google Scholar] [CrossRef]
- Chever, O.; Dossi, E.; Pannasch, U.; Derangeon, M.; Rouach, N. Astroglial Networks Promote Neuronal Coordination. Sci. Signal. 2016, 9, ra6–ra6. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, T.; Li, T.; Henning, L.; Wu, Z.; Müller, J.; Seifert, G.; Steinhäuser, C.; Bedner, P. Constitutive Deletion of Astrocytic Connexins Aggravates Kainate-Induced Epilepsy. Glia 2020, 68, 2136–2147. [Google Scholar] [CrossRef] [PubMed]
- Bedner, P.; Steinhäuser, C.; Theis, M. Functional Redundancy and Compensation among Members of Gap Junction Protein Families? Biochimica et Biophysica Acta (BBA) - Biomembranes 2012, 1818, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Vessey, J.P.; Lalonde, M.R.; Mizan, H.A.; Welch, N.C.; Kelly, M.E.M.; Barnes, S. Carbenoxolone Inhibition of Voltage-Gated Ca Channels and Synaptic Transmission in the Retina. Journal of Neurophysiology 2004, 92, 1252–1256. [Google Scholar] [CrossRef] [PubMed]
- Suadicani, S.O.; Pina-Benabou, M.H.D.; Urban-Maldonado, M.; Spray, D.C.; Scemes, E. Acute Downregulation of Cx43 Alters P2Y Receptor Expression Levels in Mouse Spinal Cord Astrocytes. Glia 2003, 42, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Ransom, C.B.; Ye, Z.; Spain, W.J.; Richerson, G.B. Modulation of Tonic GABA Currents by Anion Channel and Connexin Hemichannel Antagonists. Neurochem Res 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rekling, J.C.; Shao, X.M.; Feldman, J.L. Electrical Coupling and Excitatory Synaptic Transmission between Rhythmogenic Respiratory Neurons in the PreBötzinger Complex. J. Neurosci. 2000, 20, RC113–RC113. [Google Scholar] [CrossRef] [PubMed]
- Rouach, N.; Segal, M.; Koulakoff, A.; Giaume, C.; Avignone, E. Carbenoxolone Blockade of Neuronal Network Activity in Culture Is Not Mediated by an Action on Gap Junctions. The Journal of Physiology 2003, 553, 729–745. [Google Scholar] [CrossRef]
- Chepkova, A.N.; Sergeeva, O.A.; Haas, H.L. Carbenoxolone Impairs LTP and Blocks NMDA Receptors in Murine Hippocampus. Neuropharmacology 2008, 55, 139–147. [Google Scholar] [CrossRef]
- Tovar, K.R.; Maher, B.J.; Westbrook, G.L. Direct Actions of Carbenoxolone on Synaptic Transmission and Neuronal Membrane Properties. Journal of Neurophysiology 2009, 102, 974–978. [Google Scholar] [CrossRef]
- Volnova, A.; Tsytsarev, V.; Ganina, O.; Vélez-Crespo, G.E.; Alves, J.M.; Ignashchenkova, A.; Inyushin, M. The Anti-Epileptic Effects of Carbenoxolone In Vitro and In Vivo. International Journal of Molecular Sciences 2022, 23, 663. [Google Scholar] [CrossRef] [PubMed]
- Onodera, M.; Meyer, J.; Furukawa, K.; Hiraoka, Y.; Aida, T.; Tanaka, K.; Tanaka, K.F.; Rose, C.R.; Matsui, K. Exacerbation of Epilepsy by Astrocyte Alkalization and Gap Junction Uncoupling. J. Neurosci. 2021, 41, 2106–2118. [Google Scholar] [CrossRef] [PubMed]
- Engstrøm, T.; Nepper-Christensen, L.; Helqvist, S.; Kløvgaard, L.; Holmvang, L.; Jørgensen, E.; Pedersen, F.; Saunamaki, K.; Tilsted, H.-H.; Steensberg, A.; et al. Danegaptide for Primary Percutaneous Coronary Intervention in Acute Myocardial Infarction Patients: A Phase 2 Randomised Clinical Trial. Heart 2018, 104, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Dhein, S.; Hagen, A.; Jozwiak, J.; Dietze, A.; Garbade, J.; Barten, M.; Kostelka, M.; Mohr, F.-W. Improving Cardiac Gap Junction Communication as a New Antiarrhythmic Mechanism: The Action of Antiarrhythmic Peptides. Naunyn-Schmied Arch Pharmacol 2010, 381, 221–234. [Google Scholar] [CrossRef]
- Freitas-Andrade, M.; Bechberger, J.; Wang, J.; Yeung, K.K.C.; Whitehead, S.N.; Shultz Hansen, R.; Naus, C.C. Danegaptide Enhances Astrocyte Gap Junctional Coupling and Reduces Ischemic Reperfusion Brain Injury in Mice. Biomolecules 2020, 10, 353. [Google Scholar] [CrossRef]
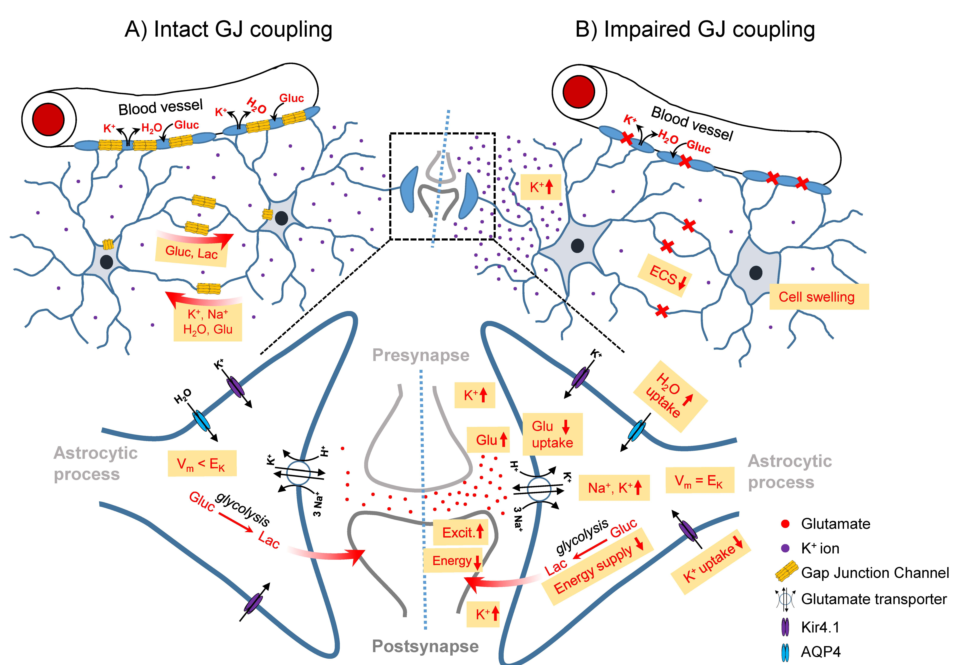
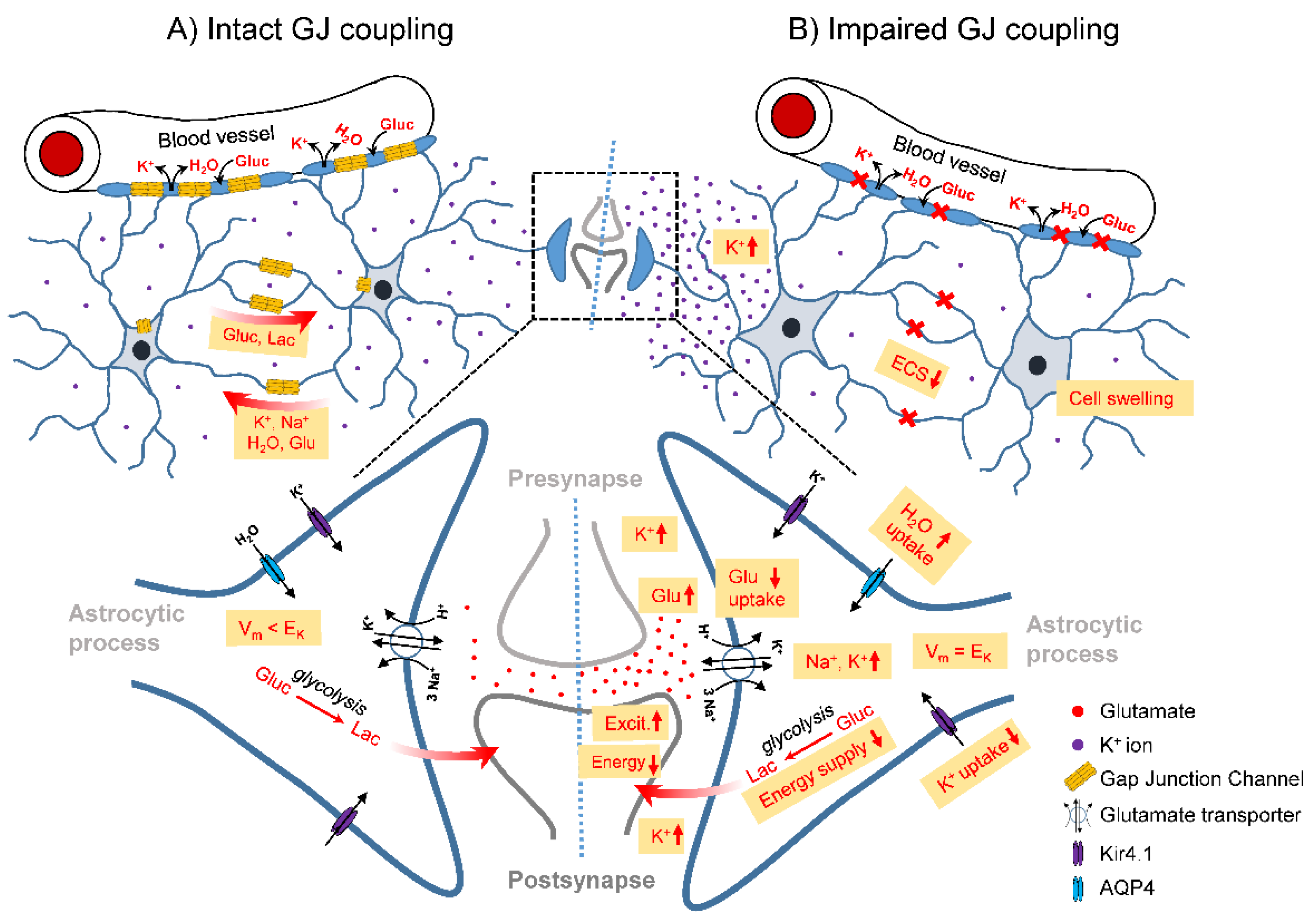
Figure 1.
Schematic illustration of the role of the GJ-coupled astrocytic network in the healthy brain (left) and consequences of its disconnection (right) on astrocyte homeostatic functions and neuronal activity. A) In the intact astrocytic network, excessive extracellular K+ released during high neuronal activity is taken up by astrocytes, redistributed through the astrocytic network and released at regions of lower extracellular K+ concentrations. This process is energy-independent, as uptake and release are driven by the difference between the local astrocyte membrane potential (Vm) and the K+ equilibrium potential (EK). Astrocytic synaptic glutamate (Glu) buffering leads to massive Na+ co-uptake, which is distributed in the astrocytic network. Astrocytic GJ coupling mediates activity-dependent trafficking of glucose (Gluc) and its metabolic product lactate (Lac) from blood vessels to sites of high energy demand. B) In the absence of GJ coupling, am increased extracellular K+ concentration leads to depolarisation of nearby astrocytes (Vm = Ek), resulting in cessation of K+ uptake and re-distribution through spatial buffering. This leads to increased/prolonged extracellular K+ transients and increased/prolonged neuronal depolarisation, lowering the threshold for seizure generation. Loss of coupling also leads to intracellular accumulation of Na+ taken up together with glutamate. Na+ accumulation and depolarisation triggered by elevated extracellular K+ reduce the driving force for glutamate uptake, resulting in a seizure-promoting prolongation of excitatory synapse activation. In addition, intracellular accumulation of K+ and Na+ trigger astrocytic water uptake and a reduction of the extracellular space (ECS) volume due to astrocytic swelling, which in turn further increases the concentration of extracellular ions and neurotransmitters, exacerbating the seizure-promoting effect of impaired K+ and glutamate buffering. On the other hand, uncoupled astrocytes are less effective at providing energy substrates, which counteracts neuronal hyperactivity.
Figure 1.
Schematic illustration of the role of the GJ-coupled astrocytic network in the healthy brain (left) and consequences of its disconnection (right) on astrocyte homeostatic functions and neuronal activity. A) In the intact astrocytic network, excessive extracellular K+ released during high neuronal activity is taken up by astrocytes, redistributed through the astrocytic network and released at regions of lower extracellular K+ concentrations. This process is energy-independent, as uptake and release are driven by the difference between the local astrocyte membrane potential (Vm) and the K+ equilibrium potential (EK). Astrocytic synaptic glutamate (Glu) buffering leads to massive Na+ co-uptake, which is distributed in the astrocytic network. Astrocytic GJ coupling mediates activity-dependent trafficking of glucose (Gluc) and its metabolic product lactate (Lac) from blood vessels to sites of high energy demand. B) In the absence of GJ coupling, am increased extracellular K+ concentration leads to depolarisation of nearby astrocytes (Vm = Ek), resulting in cessation of K+ uptake and re-distribution through spatial buffering. This leads to increased/prolonged extracellular K+ transients and increased/prolonged neuronal depolarisation, lowering the threshold for seizure generation. Loss of coupling also leads to intracellular accumulation of Na+ taken up together with glutamate. Na+ accumulation and depolarisation triggered by elevated extracellular K+ reduce the driving force for glutamate uptake, resulting in a seizure-promoting prolongation of excitatory synapse activation. In addition, intracellular accumulation of K+ and Na+ trigger astrocytic water uptake and a reduction of the extracellular space (ECS) volume due to astrocytic swelling, which in turn further increases the concentration of extracellular ions and neurotransmitters, exacerbating the seizure-promoting effect of impaired K+ and glutamate buffering. On the other hand, uncoupled astrocytes are less effective at providing energy substrates, which counteracts neuronal hyperactivity.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



