
Submitted:
30 January 2023
Posted:
01 February 2023
You are already at the latest version
Abstract
A separate group consists of compounds that are able to simultaneously interact with both orthosteric and allosteric sites, which are classified as bitopic GPCR ligands [74, 76-81]. They have two pharmacophores, one of which binds with high affinity to the orthosteric site, while the other, with lower affinity, binds to the allosteric site. If these sites are spatially separated in the receptor, then the pharmacophores in the bitopic ligand must be connected with a flexible linker, the length of which exactly corresponds to the distance between the orthosteric and allosteric sites. At the same time, it is important that the linker does not significantly affect the conformational rearrangements in the receptor caused by its activation by orthosteric and (or) allosteric agonists [74, 79].

Keywords:
G-protein-coupled receptor
; allosteric site
; allosteric modulator
; pepducin
; heterotrimeric G-protein
; autoantibody
; thyroid-stimulating hormone receptor
; luteinizing hormone receptor
; proteinase-activated receptor
; chemokine receptor
1. Introduction
G protein-coupled receptors (GPCRs), located in the plasma membrane, are the most representative and widespread superfamily of receptor proteins in all representatives of multicellular eukaryotes. GPCRs have been found in fungi [1,2], plants [3], and in all studied invertebrates and vertebrates [4,5,6,7]. Prototypes of the structural domains of both GPCRs and the adapter and regulatory proteins that interact with them appeared at the earliest stages of evolution, already at the level of prokaryotes and unicellular eukaryotes [2,8,9]. Through GPCRs, various extracellular signals, including photons, hydrogen ions, hormones, neurotransmitters, growth factors, nutrients, metabolites, and odorants, exert their regulatory effects on target cells. The result of the interaction of GPCR with signaling molecules is its transition to an active conformation and triggering of intracellular signaling cascades, which, through genomic and non-genomic mechanisms, regulate fundamental cellular processes, such as growth, metabolism, differentiation, apoptosis, and autophagy. The fact that the therapeutic effect of about a third of pharmacological drugs used in medicine is due to their influence on GPCRs and their signaling pathways [10,11] is the basis for the great importance of studying the molecular mechanisms of GPCR regulation.
Over the past two decades, many paradigms regarding the functioning of GPCRs and the transmission of hormonal signals through them have undergone significant revision. In the 1990s, it was generally accepted that hormone-activated GPCRs mediated signal transduction to intracellular targets almost exclusively via the heterotrimeric G-proteins, hence the name “G protein-coupled receptors”. However, subsequently, numerous evidences were obtained that other regulatory proteins, including β-arrestins, can function as a transducer component for GPCR, and moreover, the signaling through GPCR can be independent of G proteins, including, for example, transactivation of other groups of receptors [12,13,14,15,16,17]. Initially, it was thought that in GPCR-mediated signaling, β-arrestins were mainly responsible for GPCR desensitization, inducing dissociation of the G-protein α-subunit from the activated receptor and mediating endocytosis of the ligand-receptor complex into the cell, resulting in its degradation in the proteasome or recycling of the free receptor and its incorporation into the plasma membrane. At the turn of 1990-2000, the involvement of β-arrestins in GPCR-mediated regulation of the mitogen-activated protein kinase (MAPK) and other effector proteins and transcription factors was demonstrated and then studied [12,13,17,18,19,20,21,22,23,24]. The dual role of β-arrestins in signal transduction is largely due to their ability to stay in distinct conformations. Being in the “tail” conformation, β-arrestins preferentially interact with the intracellular C-terminal domain of GPCR, which is phosphorylated by specific G protein-coupled receptor kinases (GRKs), while in the “core” conformation they are capable of interact with the transmembrane domain (TMD) of the receptor [25,26,27,28]. Along with their signaling functions, β-arrestins are able to modulate the interaction of GPCRs with different types of G-proteins, as was shown for type 1 parathyroid hormone receptor (PTH1R) coupled to both Gs- and Gq/11-proteins [29]. It is important to point out that the functions of different types of arrestins, β1 and β2, in parathyroid hormone-induced signaling differ significantly, which is due to differences in their conformations when binding to an activated PTH1R [29].
The signaling functions of β-arrestins are determined by the pattern of GRK-mediated phosphorylation of intracellular loops (ICLs) of the receptor, which, together with a set of β-arrestin forms, predetermines a large number of variants for β-arrestin-dependent effects [30,31,32]. Phosphorylation of the ICLs by GRK2 and GRK3 usually leads to β1-arrestin-dependent desensitization of GPCR and its trafficking into the cell, while GRK5- and GRK6-mediated phosphorylation induces β2-arrestin-dependent signaling, although this not a general rule [33,34]. The signaling functions of β-arrestins also depend on the spatial positioning of phosphorylation sites, the so-called "phosphocode". In one variant of the location of phosphorylation sites in the V2-vasopressin receptor and AT1a-angiotensin II receptor, β1-arrestin stimulates the MAPK cascade, while in another location in the B2-bradykinin receptor, β1-arrestin inhibits ERK1/2 activity, and a change in the “phosphocode” in the mutant receptor leads to a change in β1-arrestin effect on the MAPK cascade [35]. The recruitment of β-arrestins and the dynamics of the formation of their complex with GPCR largely depend on the content and pattern of phosphoinositides in the plasma membrane [36]. After GRK phosphorylation, some types of GPCRs do not require phosphoinositides for the formation of GPCR–β-arrestin complexes, and such complexes are stable during endocytosis and continue to perform signaling functions in endosomes. For other GPCRs, phosphoinositides, including phosphatidylinositol-4,5-bisphosphate (PIP2), are required to form such complexes. A decrease in their content in the membrane during endocytosis leads to dissociation of the GPCR–β-arrestin complexes and recycling of the free receptor into the plasma membrane [36,37]. It is important that the stability of GPCR–β-arrestin complexes in endosomes is the basis for the implementation of “intracellular” signaling through β-arrestins and various types of G proteins in endosomes and the Golgi apparatus [38,39,40]. Thus, the paradigm of GPCR-mediated signal transduction and generation of second messengers only in the plasma membrane has been revised. Moreover, there is now evidence that for a number of GPCRs, it is “intracellular” signaling that makes the main contribution to the production of second messengers and the regulation of intracellular effector systems and GPCR-dependent gene transcription [39,41].
At an early stage of GPCR studies, it was believed that after high-affinity binding to an orthosteric agonist, the receptor is transformed into a single active conformation, in which it becomes capable of stimulating a certain type of G-protein and its coupled enzyme, which generate second messengers. As a result, GPCRs have been subdivided into families of Gs-, Gq/11-, Gi/o- and G12/13-coupled receptors based on the type of G protein that is preferentially activated. As is known, the activation of the Gs-protein leads to the stimulation of adenylate cyclase (AC) and an increase in the intracellular cAMP level, and the activation of Gq/11-proteins causes the stimulation of phosphoinositide-specific phospholipase Cβ (PLCβ) and the generation of second messengers, such as intracellular calcium and diacylglycerol. The activation of Gi/o-proteins, the main donors of Gβγ-subunits, affects the activity of Gβγ-dimer-sensitive isoforms of AC and G-protein-regulated ion channels, modulates the activity of PLCβ and inhibits (through the Gαi-subunit) hormone-stimulated AC activity. However, over the past 20 years, there has been extensive evidence that an agonist-coupled GPCR is able to simultaneously, albeit with varying efficiency, activate several types of G-proteins, as well as at least two types of β-arrestins, triggering several intracellular signaling cascades (see more details, see [7,23]. This is due to the ability of GPCR to be in several active conformations at once, which are quite close in energy characteristics, but each of which mediates the activation of a certain type of G-protein or β-arrestin [42,43,44]. The time during which an agonist-bound receptor can be in each such conformation largely determines its ability to activate a certain transducer protein. The first data on the possibility of GPCR being in several active conformations, which depends, among other things, on the nature of the agonist, were obtained in the late 1990s [45,46,47], and confirmed the concept of selective signaling agonism, also referred to as “agonist-specific trafficking of receptor signaling”, formulated by Terry Kenakin back in 1995 [48].
The multiplicity of active conformations of the agonist-bound GPCR, as well as the various scenarios of its signaling and traffic that depend on this, suggest the existence of mechanisms that affect the stability, dynamics, and ratio of these conformations and, thus, determine the formation of a functionally active complex with a certain transducer and(or) regulatory protein. Along with the nature of an orthosteric agonist, the physicochemical properties and lipid composition of the plasma membrane, ionic strength, composition and acidity (pH) of the extra- and intracellular medium, the redox potential, the concentration and ratio of amino acids, lipids, oligo- and polypeptides, and nutrients are of great importance. These physicochemical factors and substances, including the simplest ions, can affect the stability of the ligand-bound GPCR and its biased activity towards intracellular effectors, thereby predetermining the intensity and complexity of the cell response to an external stimulus. All this formed the concept of allosteric regulation of GPCRs and its decisive role in the biased regulation of intracellular signaling pathways, and changed another paradigm, according to which ligands of the orthosteric site were considered the key, if not the only, regulators of GPCRs (for more details, see [49]).
Unlike GPCR allosteric regulators, orthosteric agonists bind with high affinity to a single orthosteric site in the receptor, which can be localized either in extracellular loops (ECLs) or within the ТМD near the extracellular entrance to the transmembrane tunnel. Such binding, as a rule, leads to a significant stimulatory effect, inducing the transition of the receptor to the active state. The interaction of ligands with allosteric sites is characterized by a significantly lower affinity and more moderately and selectively affects the basal and (or) orthosteric agonist-stimulated GPCR activity. The GPCR contains not one, but several allosteric sites that differ in localization, configuration, and functional activity. These sites also differ in their influence on the conformation and accessibility of the orthosteric site, the ability of the receptor to form homo- and heterooligomeric complexes, and the efficiency of the interaction with transducer and regulatory proteins [49,50,51,52,53,54].
Allosteric sites can be topologically isolated or partially overlap with each other, as well as with an orthosteric site, which suggests the existence of both relatively simple and more complex reciprocal relationships between them. In most cases, the orthosteric site is highly conserved among GPCRs that are activated by a particular ligand, while allosteric sites are characterized by significant structural variability, although this is not a general rule for them [55,56,57,58]. As a result, the action of allosteric regulators specific for a certain type of GPCR is highly selective and often biased towards intracellular targets. There are few examples of how allosteric regulators specific to one GPCR are able to bind to allosteric sites of another receptor. This has been shown for allosteric sites located at the interface between the TMD and ICLs in structurally related types of receptors, such as chemokine receptors [59,60], β1- and β2-adrenergic receptors (β1/β2-AR) [61,62], and glucagon and glucagon-like peptide-1 receptors [63].
The currently developed paradigm of the decisive role of allosterism in the functioning of GPCRs is in good agreement with the rethinking of the principles of organization of receptor complexes. In the recent years, there has been much evidence that GPCRs function as di- and oligomeric complexes, and these complexes can be both homo- and heteromeric, including different types and subtypes of receptors, often implementing different functions. The formation of complexes not only significantly changes the affinity of GPCRs for orthosteric agonists, but also determines their specificity and effectiveness for various types of G-proteins and β-arrestins, and also affects the endocytosis of ligand-receptor complexes and their further fate in the cell [64,65]. Each GPCR-protomer in the receptor complex functions as an allosteric regulator with respect to other protomers of this complex. The properties of allosteric regulators are also inherent in G-proteins, β-arrestins, and receptor-activity-modifying proteins (RAMPs) [66,67,68,69,70]. Their effect on the affinity of the receptor for an orthosteric agonist largely depends on the functional state and set of these proteins, and in the case of G-proteins, the subunit composition and stability of the heterotrimeric complex are of great importance [23]. According to modern concepts, GPCRs and signaling proteins interacting with them form multicomponent complexes that are stabilized by anchoring on scaffold proteins, multifunctional regulators of intracellular signaling, which can also include β-arrestins [7,21,71]. Such complexes also exhibit a wide range of allosteric effects, which can affect not only the GPCR affinity for orthosteric and allosteric ligands, but also the signaling bias and features of intracellular receptor traffic.
This review is devoted to the problem of allosteric regulation of various classes of GPCRs, and also focuses on the multiplicity of allosteric sites, their location and functional role, as well as on various regulators and modulators that specifically interact with these sites. Considerable attention is paid to the diversity of endogenous and synthetic allosteric regulators that act on the receptor both from outside and inside the cell, as well as by changing the contact between the membrane lipids and the receptor TMD.
2. Classification of Allosteric GPCR Regulators
According to the ability to influence basal and orthosteric/allosteric agonist-stimulated activity, the ligands of GPCR allosteric sites can be divided into allosteric modulators that have no intrinsic activity and allosteric regulators that affect GPCR activity in the absence of orthosteric agonists [50]. In the case of allosteric modulators, the ligand, by binding to the allosteric site, changes or retains unchanged the affinity of the orthosteric agonist for GPCR binding and/or its ability to activate the receptor, which is assessed by its maximum stimulating effect. An allosteric ligand that increases the affinity and/or efficacy of an orthosteric agonist is classified as a positive allosteric modulator (PAM), while an allosteric ligand that reduces the affinity and/or efficacy of an orthosteric agonist is classified as a negative allosteric modulator (NAM). When an allosteric ligand does not significantly affect the affinity and efficacy of an orthosteric agonist, but is able to change some other characteristics of its interaction with GPCR, for example, selectivity for various types of G-proteins and β-arrestins and the specificity of activation of intracellular target proteins, it belongs to the silent allosteric modulators (SAM). Allosteric ligands that have their intrinsic activity can function as full agonists, inverse agonists and neutral antagonists, and their action is independent of orthosteric site occupancy. Such independence of the action of allosteric ligands can be realized only when the orthosteric and allosteric sites do not overlap and do not interact through ligand-induced conformational rearrangements [50]. When an allosteric ligand functions as a full agonist and at the same time enhances the affinity and/or efficacy of an orthosteric agonist, it is classified as ago-PAM. If an allosteric ligand, being a full agonist, reduces the affinity and/or efficacy of an orthosteric agonist, it is classified as ago-NAM. In the case when it reduces the effectiveness of an orthosteric agonist, acting as an antagonist, but at the same time increases the affinity of an orthosteric agonist to the receptor, functioning as a PAM, it is classified as a PAM-antagonist [72].
Regulatory influences caused by the specific binding of ligands to the orthosteric and allosteric sites of the GPCRs are reciprocal. Just as binding of a ligand to an allosteric site can change the binding characteristics and activation pattern of an orthosteric site, binding of a ligand to an orthosteric site can change the accessibility and regulatory properties of one or more allosteric sites. Given the multiplicity of allosteric sites, the mechanisms of such relationships between receptor sites can be quite complex. Moreover, depending on the nature and binding characteristics of orthosteric ligands, the same allosteric regulator can influence the affinity and efficiency of these ligands in different ways, functioning as PAM, NAM, or SAM [50,73,74]. Thus, the assignment of a ligand to one or another group of allosteric regulators or modulators is relative. In each specific case, the pharmacological profile of the allosteric ligand largely depends on the nature of the receptor-bound orthosteric agonist, the structural and functional characteristics of the receptor (phosphorylation, N-glycosylation, and other post-translational modifications), the formation of homo- or hetero-oligomeric complexes, the microenvironment (multicomponent complexes with transducer, adapter and regulatory proteins), as well as on the physicochemical properties of the plasma membrane, ionic composition and acidity of the extracellular and intracellular environment.
Given the variety of regulatory influences of allosteric ligands on various parameters of the interaction of orthosteric agonists with the receptor, in recent years a model has been widely used that describes a ternary complex that includes GPCR and receptor-bound allosteric and orthosteric ligands [75]. The effect of an allosteric ligand on the affinity of an orthosteric agonist is described by the factor α, while its effect on the efficacy (maximum regulatory effect) of the orthosteric agonist is described by the factor β. When the allosteric ligand increases the affinity and efficacy of the orthosteric agonist, the factors α and β are above zero (PAM). If the influence of the allosteric ligand is the opposite, then these factors have negative values (NAM). In the absence of a significant effect, the factors α and β are equal to zero (SAM). To assess the intrinsic activity of allosteric ligands, the factor τ is used, which for full agonists has values above zero (full allosteric agonist, ago-PAM, ago-NAM), and for an inverse agonist or neutral antagonist it has negative values. For “pure” allosteric modulators (PAM, NAM, SAM, PAM-antagonist), the factor τ is equal to zero. However, for each specific case and for each specific “allosteric ligand–orthosteric ligand” pair, the factors α, β, and τ can vary in magnitude and change their sign.
A separate group consists of compounds that are able to simultaneously interact with both orthosteric and allosteric sites, which are classified as bitopic GPCR ligands [74,76,77,78,79,80,81]. They have two pharmacophores, one of which binds with high affinity to the orthosteric site, while the other, with lower affinity, binds to the allosteric site. If these sites are spatially separated in the receptor, then the pharmacophores in the bitopic ligand must be connected with a flexible linker, the length of which exactly corresponds to the distance between the orthosteric and allosteric sites. At the same time, it is important that the linker does not significantly affect the conformational rearrangements in the receptor caused by its activation by orthosteric and (or) allosteric agonists [74,79].
3. Localization and Number of Allosteric Sites in GPCRs
Allosteric sites can be localized in all structural domains of the GPCRs: (1) in the ECL, including the external entrance (vestibule) to the transmembrane tunnel, (2) within the TMD, where usually an orthosteric site is located, both in the upper (extracellular) and lower (intracellular) portions of heptahelical transmembrane (TM7) bundle, (3) in the ICL and in their interfaces with transmembrane regions (TMs) responsible for interaction with G-proteins, β-arrestins and GRKs, as well as (4) on the outer-lateral surface of the TMD, where the receptor contacts the lipid matrix of the plasma membrane. Unlike a single orthosteric site, the number of allosteric sites in GPCR can be very significant. Estimation of the number and localization of allosteric sites in each receptor is a very difficult task, although significant progress in this direction has been made due to the development of new approaches for the identification of GPCR allosteric sites [53,54,82,83].
The FTMap (http://ftmap.bu.edu/) and FTSite (http://ftsite.bu.edu/) programs are widely used to search for allosteric sites in GPCRs. The FTMap has been successfully tested to search for allosteric sites in β2-AR, A2A-adenosine, and M2-muscarinic receptors [84,85,86]. In 2019, using the FTMap and its modified version FTSite, a large-scale study was carried out to identify allosteric sites in 17 GPCRs belonging to their various families. It is important that for all these receptors there was information on their allosteric regulation and X-ray diffraction data, as a result of which the results obtained were compared with the available experimental evidence on the localization of such sites in the studied receptors [82]. For allosteric sites located within the TMD, both near the entrance to the transmembrane tunnel and in the internal cavity of the TMD, the predictive ability of the FTMap and FTSite was 80 and 88%, respectively. For sites located in hydrophilic loops and on the outer-lateral surface of the TM7 bundle, the predictive ability was significantly lower. As a result, the overall predictive ability of the FTMap and FTSite programs for all allosteric sites was 69 and 76%, respectively [82]. In the case of hydrophilic loops, the limitations of the approaches used are due to the high mobility of these loops and, in most cases, the lack of reliable X-ray diffraction data, while for sites located on the outer-lateral surfaces of the TMD, such limitations are due to the features of FTMap and FTSite, adapted mainly to hydrophilic and globular proteins without taking into account their interaction with the hydrophobic phase.
Molecular dynamics methods can also be used to search for allosteric sites, and the most interesting here is the identification and study of allosteric sites located on the outer-lateral surface of the TM7 bundle and, therefore, available for interaction with membrane lipids, as was recently shown for β2-AR, glucagon receptor, M2-muscarinic receptor, P2Y1 purinergic receptor, D2-dopamine receptor, type 2 proteinase-activated receptor (PAR2), C5a anaphylatoxin chemotactic 1 receptor [83,87,88]. Since these sites are involved in the formation and stabilization of receptor complexes (homo- and hetero-oligomeric), they mediate the allosteric effects of receptor di- and oligomerization on their functional activity. Along with this, these sites are targets for plasma membrane lipids, which, therefore, can function as allosteric modulators, as demonstrated for cholesterol and phospholipids [89,90,91].
Recently, a comprehensive analysis of the topology of allosteric sites in GPCRs, which also makes it possible to predict “orphan” allosteric sites, was performed using the in-silico docking of small molecules probes, during which 557 GPCR structures for 113 receptors were studied [53]. As a result, a “pocketome” of allosteric sites was constructed for various classes of GPCRs (A, B1, B2, C, D1, and F). In total, for receptors of the largest class A, in the cavities between the hydrophobic helical regions forming the 7TM bundle, up to 11 allosteric sites were identified, the existence of which has experimental evidence, and up to 8 sites that can be considered as potential allosteric sites. Along with this, Interhelical Binding Site 1 (IBS1) and adjacent secondary binding sites IBS2 and IBS3 were identified in the central part of the TMD [53]. In GPCRs of classes A and B, IBS1 functions as an orthosteric site, and in GPCRs of class B, which are activated by peptide hormones, in addition to IBS1, the segments of the hydrophilic ECL and the outer vestibule of the transmembrane tunnel also take part in the formation of the orthosteric site. In GPCRs of class C, which have a significant ectodomain containing an orthosteric site, the IBS1 site does not interact with the orthosteric ligand, usually a large glycoprotein hormone, and functions as an allosteric site. Of considerable interest are the results of the structural and functional analysis of two “orphan” allosteric sites OS5 and OS9, which are located at the lower portion of the 7TM bundle. The OS5 site is located in the cavity between the TM5 and TM6, while the OS9 is in the cavity formed by the intracellularly oriented ends of TM1 and TM7 and the H8 helix. The cavity that forms the OS5 is highly conserved among GPCRs of classes A and B1, while the OS9 cavity is structurally similar among all known GPCRs, except class F. Mutations of amino acid residues that form the OS5 and OS9 sites in β2-AR and M3-muscarinic receptor had a strong effect on both the activation of G-proteins and the recruitment and activation of β-arrestins, and these effects were significantly different, which may indicate a bias of structural changes in the OS5 and OS9 sites in relation to the intracellular signaling pathways [53]. Mutations in the locus corresponding to the OS9 in the AT1-angiotensin receptor led to changes in the activity of Gq/11-protein- and β-arrestin-dependent cascades [92], which confirms the functional importance of this site in allosteric regulation of class A GPCRs. Thus, new tools have been obtained for the search and validation of allosteric sites in GPCRs, and this may soon allow mapping each of the classes of these receptors by the location and functional activity of allosteric sites, which is important for an allosteric ligand-based drug discovery and design [53,54].
However, it should be noted that the identification of allosteric sites in hydrophilic loops, and in particular, in large extracellular and cytoplasmic domains, is a very complex and far from being solved problem, especially since these sites, as a rule, are targets for a large number of proteins involved in GPCR-signaling, as well as for autoantibodies to the extracellular regions of the receptors.
4. Diversity of Endogenous Allosteric Regulators of GPCRs
The multiplicity of GPCR allosteric sites, which differ both in topology and environment, provides for the presence of allosteric regulators of different chemical nature, both extracellular and intracellular. At present, such regulators have been found in almost all classes of compounds, from simple ions to large protein complexes, including those involved in GPCR-dependent signaling pathways. Most of them function as allosteric modulators and have no intrinsic activity [68,69,91,93,94,95,96,97,98,99,100].
The most important and characteristic group of allosteric GPCR regulators are signal proteins, such as G-proteins, β-arrestins, and RAMPs, which are capable of forming a functionally active complex with the receptor and are responsible for the transduction of the hormonal signal into the cell. As is known, as a result of interaction with a ligand-activated receptor, a heterotrimeric G-protein dissociates into a GTP-bound Gα-subunit and a Gβγ-dimer, which become available for binding to effector proteins (AC, PLCβ, phosphatidylinositol-3-kinase, G-protein-activated ion channels, and others). The GTP-bound Gα-subunit and Gβγ-dimer allosterically affect the conformation of the orthosteric site and reduce its affinity for agonists, downregulating signal transduction. The first evidence of this was obtained almost half a century ago, in experiments on the effect of GTP and its non-hydrolysable analogs on the binding of agonists to β-AR [101,102], and later were confirmed for other receptors [73]. At the same time, GTP-free G-protein, which is a stable αβγ-heterotrimeric complex, increases the affinity of the receptor for an orthosteric agonist, and this is due to the stabilization of the “closed” conformation of the GPCR, which is characterized by restricted access to and egress from the hormone-binding site [68]. Due to the difficulty in dissociating the agonist from the “closed” conformation of the receptor, the stability of the GPCR-orthosteric ligand complex is increased, resulting in an increase in the affinity of the receptor for agonists. It is important to note that the interaction of the constitutively activated GPCR with the G-protein also results in the stabilization of the “closed” conformation, which prevents the binding of orthosteric agonists with receptor and makes GPCR signaling independent of them [68].
Along with GTP-free G-proteins, antibodies that mimic them also stabilize the “closed” conformation of GPCR, as was shown for nanobody Nb80 produced in response to agonist-activated β2-AR [93]. Antibodies have been developed that act in the opposite way, stabilizing the inactive GPCR conformation, which is characterized by low affinity for the ligand, as demonstrated for nanobody Nb60. This proves the existence of both positive and negative allosteric mechanisms of the influence of both G-proteins and antibodies mimicking them on the binding and efficiency of the orthosteric agonist [69]. The receptor allosteric site responsible for interaction with the G-protein is located at the interface between the cytoplasmic ends of the TMs and the proximal regions of the ICL2 and ICL3, and it interacts specifically with the Gα-subunit C-terminal segment and other GPCR-interacting regions of the G-protein [103]. As in the case of the agonist–GPCR–G-protein ternary complex, allosteric interactions were demonstrated for the agonist–GPCR–β-arrestin ternary complex, the existence of which was postulated back in 1997 by Gurevich and colleagues [104]. Binding of β-arrestins to GPCR increases the affinity of the receptor for orthosteric agonists [66,69], especially those that are biased towards β-arrestin signaling [105].
The preference for the formation of a certain ternary complex for receptors capable of interacting with several transducer proteins depends on a number of factors, but their role and influence on the stability of these complexes is not well understood. There is no doubt that the allosteric mechanisms that regulate the formation of a functionally active ternary complex play a decisive role in biased agonism, leading to selective activation of a certain type of G-protein or β-arrestin by an orthosteric agonist [49]. At the same time, it is not completely clear what is the trigger for the formation of a biased ternary complex. Such triggers can be binding to the receptor of a biased orthosteric agonist, which recruits a certain type of transducer protein into the complex, or the formation of a pre-activation complex between the agonist-free receptor and an inactive G-protein or β-arrestin, which creates suitable conditions for effective binding of a biased agonist and triggering signal transduction [106,107,108].
In favor of the first model, the so-called “ligand” mechanism, evidence suggests that agonist binding to the receptor is able to recruit inactive, GDP-bound, G-proteins with the formation of a ternary complex and, thereby, trigger signal transduction [109]. At the same time, the limited availability of a certain G-protein and the competition between the different types of G-proteins for binding to the ligand-activated receptor are not consistent with the data on the high rate and high efficiency of the formation of the ternary complex and the transduction of the hormonal signal into the cell. The “ligand” mechanism is unable to explain the fact that, in the absence of an orthosteric ligand, constitutively activated GPCR recruit and activate G-proteins [110,111]. Moreover, such receptors selectively activate certain types of G proteins or β-arrestins [112,113]. The alternative mechanism involves a different sequence of events. At the first stage, a pre-activation complex is formed that includes a ligand-free receptor and an inactive form of a certain G-protein, and this predetermines not only the binding of a certain orthosteric agonist, but also biased agonism [108]. Currently, the formation of such pre-activation complexes has been postulated for a number of GPCRs [114,115,116,117,118,119,120,121,122,123]. In the pre-activation complex, the interaction between TM3 and TM6 is weakened, and their conformational mobility increases. Although this is not enough for full activation of the receptor, the weakening of the interaction between TM3 and TM6 facilitates the agonist-induced shift of TM6 outward, which changes the location of the α5-helix of the Gα-subunit in the intracellular vestibule of the GPCR transmembrane tunnel, induces a decrease in the affinity of the Gα-subunit for GDP and provides an effective interaction of activated G-protein with receptor ICLs [108]. Instead of G-proteins, the pre-activation complex may contain β-arrestins, which stabilize the conformation of the orthosteric site, which has a high affinity for β-arrestin-biased agonists [106,124].
Along with highly specialized components of GPCR signaling, endogenous allosteric regulators of GPCR include some classes of antibodies produced against extracellular regions of receptors, small proteins, oligopeptides, steroids, fatty acids, amino acids, and even simple ions [91,94,95,96,97,98,99,100]. Some allosteric regulators specifically affect a particular receptor or closely related GPCRs, while others are non-specific and affect the activity of a large number of GPCRs belonging to different families. Such receptor-nonspecific activity has been demonstrated for sodium ions, which are NAMs for a large number of GPCRs, stabilizing their inactive state and reducing their stimulation by full agonists [97,125,126]. Sodium ions bind to an allosteric site located within the TMD, which includes several highly conserved amino acid residues, the most important of which is aspartic acid in TM2. This residue is critical for the translocation of sodium ions to the allosteric site and for their retention in this site by ionic bonds, and the replacement of aspartic acid with alanine blocks the NAM activity of these ions [125,127]. The effects of sodium ions on GPCR activity are carried out at their physiological concentrations, which indicates the involvement of Na+ in the regulation of GPCR signaling in real biological systems. Sodium ions make a significant contribution to the selectivity of activation of intracellular cascades by agonists, as shown for opioid receptors [128,129]. Using site-directed mutagenesis and molecular docking, a sodium ion-binding site has been identified in all classes of GPCRs. It is located in the middle of the transmembrane tunnel and includes amino acid residues from TM1, TM2, TM3, TM6 and TM7 [53]. Zinc and magnesium ions, being allosteric modulators for a large number of GPCRs, can function both as PAM and NAM [95,97,130]. It should be noted that a certain contribution to the effects of magnesium ions can be made by their ability to stimulate the GDP/GTP exchange and GTPase activity of G-proteins [131].
Among lipids, cholesterol is the most important allosteric regulator of GPCRs, and its effects on receptor activity are highly dependent on the content of cholesterol in the membrane, the type of receptor, and the nature of the orthosteric ligand [91,132,133]. A significant number of GPCRs contain consensus motifs for cholesterol binding (Cholesterol Recognition/Interaction Amino Acid Consensus motif, CRAC), which were first identified in β2-AR [134] and then found in other GPCRs, although often in modified form [91,135,136]. Along with cholesterol, other lipids are involved in the allosteric regulation of GPCRs, including phosphatidylinositol-4,5-bisphosphate, phosphoinositols [90] and phosphoserines [89]. Different lipids act on receptors through binding to different allosteric sites and, accordingly, affect their activity in different ways. For the 5-HT1A-serotonin receptor, it has been shown that cholesterol directly affects the binding characteristics of the orthosteric site, while phospholipid phosphatidylinositol 4-phosphate interacts with the intracellular site and improves the functional coupling between the agonist-activated receptor and the Gi/o-protein [137]. More complex allosteric effects of lipids on GPCR activity are possible, due to different mechanisms of their action on receptors and their complexes with other signal proteins. In addition to direct interaction with receptor allosteric sites, lipids, affecting the physicochemical properties of the plasma membrane (fluidity, tension, mechanical stability), are able to change the conformational characteristics of the GPCRs and the kinetics of its activated states, as shown for photosensitive rhodopsin [138]. Lipids can affect subcellular compartmentalization and stability of complexes between GPCRs and different regulatory and adapter proteins, as well as the formation of caveolae and lipid rafts, which make a significant contribution to the functional activity of GPCRs [139,140]. This is especially relevant in connection with the concept of “spatial bias” in signaling from the plasma membrane to intracellular compartments such as endosomes, the Golgi apparatus, and nuclear membranes [38,141,142]. All of these mechanisms can work together and, thus, affect GPCR activity in an unpredictable way, making it difficult to assess the contribution of each specific lipid and each specific mechanism to receptor-mediated signaling.
Unexpected was the discovery of the fact that the activity of receptors can be voltage-dependent, and this is due to allosteric mechanisms. As a result, in the recent years, the membrane potential has been considered as a “physicochemical” allosteric regulator [143]. Depolarization can affect both the activity of receptors and the efficiency of their stimulation by various ligands in different ways [144]. Substitutions of tryptophan residues (Trp99 in TM3 and Trp422 in TM7) for alanine in the transmembrane allosteric site of the M2-muscarinic receptor abolished the voltage dependence of agonist affinity, although they did not affect the conformational changes induced by depolarization [145]. All this indicates complex relationships between clusters of amino acid residues in the receptor TMD, which function as voltage sensors, and the activity of allosteric and orthosteric sites [143]. It is possible that depolarization can affect the binding of mono- and divalent ions, in particular sodium ions, to the receptor [146], although experimental data do not yet confirm this [147,148].
5. GPCR-Complexes and Allosteric Regulation
Significant allosteric effects on GPCR activity are exerted by their formation of homo- and heteromeric complexes, since the affinity of orthosteric and allosteric ligands for receptors, their efficiency, and bias towards intracellular cascades largely depend on the degree of oligomerization and the subunit composition of such complexes [65]. In vitro experiments with allosteric modulators of glutamate receptors showed that Ro 64-5229 (mGlu2-specific NAM), biphenylindanone A (mGlu2-specific PAM), and VU0361737 and PHCCC (mGlu4-specific PAMs) did not affect affinity and efficacy of orthosteric agonists for heterodimeric mGlu2/mGlu4-glutamate receptors. At the same time, these modulators were effective in the case of homodimeric glutamate receptors, such as mGlu2/mGlu2 (Ro 64-5229, biphenylindanone A) and mGlu4/mGlu4 (VU0361737, PHCCC) [149]. Surprisingly, the simultaneous addition of mGlu2- and mGlu4-specific PAMs did not affect the binding characteristics of the heterodimer receptors, indicating a more complex nature of the PAM effects on receptor activity. Under the in vivo conditions, the regulation of mGlu2- and mGlu4-glutamate receptors by these modulators differed from that in the in vitro conditions, which was due to the existence of various populations of homo- and heterooligomeric glutamate receptors in the target tissues [150].
Another model of heterodimer-specific allosterism is due to the effect of transactivation or transinhibition of one subunit of the heterodimeric GPCR complex upon binding of the allosteric modulator to its other subunit. In the complex formed by subtypes 2 and 3 of the type 1 taste receptor (T1R2, T1R3), the orthosteric agonist binds only to the T1R2 subunit, while the allosteric modulators cyclamate (PAM) and lactisol (NAM) regulate the activity of the heterodimeric T1R2/T1R3 by specific binding to T1R3 subunit [151,152]. The functional activity of the metabotropic γ-aminobutyric acid receptor (GABAB) is due to the formation of a heterodimeric complex formed by the GABAB1- and GABAB2-subunits, in which the GABAB1 binds to the orthosteric agonist, while the GABAB2 increases the affinity of GABAB1 to the orthosteric agonist and provides signal transduction to G protein [153]. The compound CGP7930 with PAM activity binds to the TMD of GABAB2-subunit, which leads to increased stimulation of the GABAB1-subunit by a specific agonist [154]. At the same time, the compound COR758, a recently developed NAM for GABAB-receptors, binds to an allosteric site localized in the GABAB1-subunit [155], indicating the coexistence of regulatory mechanisms, both dependent and independent of heterodimerization [156].
The reciprocal relationships between complex formation and the activity of allosteric regulators are determined by the localization of allosteric sites and their involvement in the formation of contacts between receptor subunits in the di- and oligomeric complex. This allows allosteric regulators to stabilize such complexes, thereby controlling the binding characteristics of the receptor and its ability to activate transducer proteins, or, conversely, prevent the formation of complexes. Binding of the mGlu5-glutamate receptor to PAM (Nb43 antibody, low molecular weight compound CDPPB) leads to the formation of a more compact homodimeric complex due to closer contact between TM6 of both protomers, and this increases the functional response of the receptor to the orthosteric agonist [157]. The compound MPEP with NAM activity for the mGlu5-glutamate receptor has the opposite effect, destabilizing such a complex [157].
The convergence of protomers through increased interaction between their TM6 has also been demonstrated for other active forms of receptors, including the GABAB-receptor and the calcium-sensing receptor [158]. At the same time, dimeric complexes with close TM3 and TM4 in the mGlu2-glutamate receptor [159] and close TM3 and TM5 in the GABAB-receptor [160] correspond to the inactive state of the receptors. There is every reason to believe that the leading role in this also belongs to allosteric sites located in the receptor regions involved in di- and oligomerization. For example, in TM6, according to molecular docking data, several allosteric sites are located: KS7 in the central part of the TM7 bundle, KS8 in its lower portion, and KS9 at the border of the TMD and ICLs [53], and all of them are targets for PAMs [157,161]. At the same time, in the TM3/TM4 and TM3/TM5 contacts, there are a significantly smaller number of such sites: in the first case, KS2 site located in the upper portion of the TM7 bundle, and in the second case, KS5 site located in its lower portion [53].
It cannot be ruled out that allosteric sites that are located outside the molecular determinants responsible for GPCR di- and oligomerization are involved in the formation of receptor complexes. In turn, there are reasons to believe that the formation of complexes significantly affects not only the conformation and binding characteristics of the orthosteric site, but also the corresponding characteristics of allosteric sites, including those that are topologically distant from the protomer contact surface in the GPCR complex. Perhaps this explains the fine targeted regulation of biased agonism through the formation of di- and olgomeric complexes, since in this case the conformation of allosteric sites that are located in the lower part of the TM7 bundle and in the ICLs and interact with G-proteins and β-arrestins can change significantly and specifically.
All of the above indicates a variety of allosteric effects on GPCR signaling, which are realized with the involvement of a large number of allosteric sites located in different domains and subdomains of the receptors. The following sections will discuss the localization of allosteric sites, the mechanisms of allosteric regulation, and some endogenous and synthetic allosteric regulators for several groups of receptors, including various types of chemokine receptors, PARs, the receptors of thyroid-stimulating (TSHR) and luteinizing hormones (LHR), and β-AR. The choice of these receptors is due to rather extensive information about their allosteric sites and the diversity of their endogenous and synthetic allosteric regulators, including autoantibodies and synthetic pepducins. At the same time, for a number of other GPCRs, there is also a lot of data on allosteric regulation and their analysis is presented in a number of recent comprehensive reviews and analytical articles: for muscarinic acetylcholine receptors [162,163,164,165], for metabotropic glutamate receptors [162,166,167], for 5-hydroxytryptamine (serotonine) receptors [168,169], for dopamine receptors [168,170], for opioid receptors [169,171,172], for cannabinoid receptors [167,173,174,175], for adenosine receptors [176], for neuropeptide Y receptors [177], for melanocortin receptors [177], for angiotensin receptors [178], for glucagon-like peptide-1 receptors [167,179], and for free fatty acid receptors [50].
6. Allosteric Regulators of Chemokine Receptors
A feature of chemokine receptors is a large number of specific endogenous ligands and many signaling pathways regulated through them. Thus, the CCR1 chemokine receptor is a target for at least 11 chemokines, some of which activate predominantly G-proteins, while others activate β-arrestins [180]. Accordingly, allosteric modulators and regulators are very important for biased agonism in the case of chemokine-dependent signaling cascades [181].
In terms of the development of allosteric regulators, the chemokine receptors CXCR1, CXCR2, CXCR3, CXCR4, CCR1, CCR2, CCR5, CCR7, CCR9 are the most studied [181,182]. Structurally related CXCR1 and CXCR2 receptors are activated by chemokines of the CXCR1/2 family, which causes increased proliferation of endothelial cells, stimulates their migration, and makes a significant contribution to survival, proliferation, and angiogenesis of vascular endothelial cells [183]. The CXCR4 receptor and its endogenous ligand, stromal cell-derived factor-1 (CXCL12/SDF-1), are involved in the regulation of leukocyte transport, B-cell lymphopoiesis, bone marrow myelopoiesis, and are involved in the regulation of survival and proliferation of hematopoietic stem cells [184]. CXCR4 is highly expressed in many tumors and also stimulate tumor angiogenesis and metastasis [185,186,187,188]. CXCR3, which is expressed on the surface of activated T cells, B cells and natural killer cells, and its ligands (CXCL9, CXCL10, CXCL11) has been implicated in the development of infectious, autoimmune, and neoplastic diseases [189]. CCR2, CCR7, and CCR9 are involved in the immune response and pathogenesis of numerous inflammatory diseases [190,191,192], as well as in tumor metastasis [188,193,194]. CCR5 functions as a co-receptor upon HIV entry into the cell, making it a valuable target for HIV therapy [195], and is also involved in acute graft-versus-host disease after allogeneic hematopoietic cell transplantation [196].
Some of the currently developed allosteric regulators of chemokine receptors bind to sites located inside the transmembrane tunnel, but most of them, both of low molecular weight and peptide nature (pepducins), interact with allosteric sites located in ICLs and their interfaces with TMs (Figure 1). The pharmacological profile of allosteric agonists varies greatly in terms of receptor activation by different endogenous agonists, as well as in the signaling cascades they regulate, indicating their inherent biased allosterism [197]. As noted above, Na+ and ions of divalent metals, binding to the receptor transmembrane allosteric site, function as their allosteric modulators. As a result, the compounds that form a complex with them can significantly affect the GPCR activity. Chelators such as phenanthroline and bipyridine complexed with zinc or copper cations, when penetrated within the transmembrane allosteric site of the CCR1 receptor, enhanced specific binding of the chemokine CCL3 to the receptor, acting as PAM. At the same time, they inhibited the binding of another chemokine, CCL5, to the receptor, acting as NAM, which indicates their biased allosterism [198].
There are data on the allosteric regulation of a significant number of chemokine receptors, including CXCR1, CXCR2, CXCR3, CXCR4, CCR2, CCR5, CCR7, and CCR9. Allosteric sites in them can be located at different loci of the TMD: in the upper (outwardly oriented) portion of the transmembrane tunnel (interfaces including the membrane-proximal segments of ECLs and extracellular segments of TMs, as well as the outer vestibule of the transmembrane channel) (designated as AS-1), in the central part of the 7TM bundle (AS-2), and in the lower (oriented to the cytoplasm) portion of the transmembrane tunnel (interfaces, including the membrane-proximal segments of ICLs and the cytoplasmic endings of TMs) (AS-3). In each of these loci, there are several cavities in which allosteric sites can be located, both overlapping and spatially separated. Along with this, allosteric sites can be located in hydrophilic loops, being targets for autoantibodies and synthetic pepducins. Endogenous allosteric regulators of chemokine receptors are sodium (NAM) and zinc (PAM) ions, as well as cholesterol and membrane phospholipids (mainly with PAM activity). Synthetic small allosteric regulators interact with allosteric sites located at all three loci, AS-1, AS-2, and AS-3, while pepducins, lipidated derivatives of peptides corresponding to ICLs, are transported across the plasma membrane and interact with intracellular sites. The Figure shows the following small regulators interacting with the AS-1 locus: Maraviroc (CCR5-selective NAM), Reparixin, Ladarixin and their analogs (CXCR1/CXCR2-selective NAMs and/or non-competitive allosteric antagonists), Plerixafor (allosteric CXCR4-inhibitor), FAUC1036 (biased allosteric CXCR3-agonist), T140 and its analog TN14003 (biased allosteric CXCR4-antagonists), and the peptide X4-2-6 (TM2/ECL1 of CXCR4; allosteric CXCR4-antagonist). The following ligands bind specifically to the AS-2 locus: RAMX3 and its biased analogs 8-azaquinazolinone (8-AQ) derivative 1b, BD064 and BD103 (CXCR3-selective NAMs), DF2755A (noncompetitive allosteric CXCR1/CXCR2-antagonist), and to the AS-3 locus: Vercirnon and its analogs (allosteric CCR9-antagonists), Navarixin (allosteric antagonist of CXCR1, CXCR2 and CCR7), CCR2-RA-[R] and Cmp2105 (allosteric CCR2-antagonists and/or NAMs). Pepducins, such as N-palmitoylated peptides X1/2pal-i3 (ICL3 of CXCR1/CXCR2; CXCR1/CXCR2-selective NAM), PZ-218 (ICL1 of CXCR4; CXCR4-selective NAM), and PZ-210 (ICL3 of CXCR4; CXCR4-selective NAM), as well as lithocholic acid-modified X1/2LCA-i1 (ICL1 CXCR1/CXCR2; allosteric CXCR1/CXCR2-inhibitor and/or NAM) and ATI-2341 (ICL1 of CXCR4; CXCR4-selective PAM) interact with intracellular sites involved in functional coupling with G-proteins and β-arrestins. Antibodies against extracellular regions of chemokine receptors are able to interact with ECLs and the extracellular N-terminal domain, but their effects remain poorly understood. Allosteric regulators that increase receptor activity (full agonists) are placed in red squares, while allosteric regulators that decrease receptor activity (antagonists, NAM) are placed in green squares.
One of the first allosteric regulators of chemokine receptors used in medicine was Maraviroc, developed in 2007. It has demonstrated NAM activity for CCR5 and is being used to treat HIV patients [199]. The anti-HIV activity of Maraviroc is due to its ability to prevent the entry of the virus into the cell by inhibiting CCR5 [200]. Maraviroc penetrates the transmembrane tunnel of CCR5, where it binds to an allosteric site located just below the outer vestibule, which is formed by amino acid residues from all TMs, except TM4 [200,201].
Of considerable interest among the allosteric regulators of chemokine receptors are Reparixin, Ladarixin (DF 2156A) and a number of their functional analogs that interact with the transmembrane allosteric site of the CXCR1 and CXCR2 receptors (Figure 1). This site is formed by TM1, TM3, TM6, and TM7. Currently, Reparixin and its analogues are considered as drugs that can reduce or prevent inflammatory and autoimmune processes, as well as suppress the development and metastasis of tumors [197,202]. Ladarixin, a potent allosteric inhibitor of CXCR1 and CXCR2, is highly effective (IC50 of about 0.1 nM) in preventing damage to pancreatic islets caused by proinflammatory factors and autoimmune reactions, indicating its potential antidiabetic activity [203,204]. Ladarixin binds to an allosteric site located in the upper portion of the transmembrane tunnel, and one of the key residues interacting with it are Lys126 TM3, Asp293 TM7 and Arg289 in ECL2 [205]. Reparixin, a non-competitive allosteric inhibitor of CXCR1 and CXCR2, suppresses the stimulatory effects of interleukin-8 (IL-8) on these receptors, to the greatest extent in the case of CXCR1, but does not affect their affinity for this agonist [206,207]. Reparixin has now been shown to be clinically successful as an anti-inflammatory drug, including in the treatment of patients with severe pneumonia caused by COVID19, and Reparixin treatment has not been associated with an increased risk of secondary infections [202].
Studies of the CXCR3 receptor made it possible to identify two partially overlapping allosteric sites, one of which was located in the outer vestibule of the transmembrane tunnel, and the other was located deep in this tunnel, in its middle [208]. For the first allosteric site, the biased allosteric agonist FAUC1036, and for the second site, the nonselective NAM RAMX3 were developed [209]. Later based on the RAMX3 structure, the biased NAMs, 8-azaquinazolinone derivative 1b [210] and the compounds BD064 and BD103 were created [211]. It is assumed that the recently developed compound DF2755A with the activity of a noncompetitive allosteric antagonist of CXCR1 and CXCR2 interacts with the same site [212]. DF2755A suppressed the stimulatory effects of orthosteric agonists of these receptors on neutrophil chemotaxis, and under the in vivo conditions reduced inflammatory and post-operative pain [212,213].
The allosteric inhibitor Plerixafor (AMD3100), which interacts with an allosteric site located in the outer vestibule of the transmembrane tunnel of the CXCR4 receptor, suppresses the stimulatory effects of the CXCL12 chemokine on the activity of this receptor, but does not affect its activation by the CXCL12 chemokine fragment, which also has the activity of a full CXCR4 agonist, but differs from the full-length chemokine in the receptor-binding site [214]. Plerixafor is not selective for intracellular signaling cascades, inhibiting both G-protein-regulated signaling and β-arrestin-mediated endocytosis, and the consequence of this is an increase in the expression of CXCR4 and the development of tolerance to this drug during its long-term use [215], which limits its clinical use for mobilization of stem cells from the bone marrow during transplantation. As an alternative, a peptide allosteric antagonist X4-2-6 was developed, structurally corresponding to TM2 and ECL1 of CXCR4, which formed a ternary complex with the receptor and the chemokine CXCL12 [216]. Being in this complex, the chemokine CXCL12 remained capable of stimulating G-protein-dependent pathways, but did not affect the recruitment of β-arrestins, as a result of which there was no compensatory accumulation of CXCR4 on the surface of target cells and no tolerance was developed to the peptide antagonist. Simultaneously with the peptide antagonist X4-2-6, biased small allosteric antagonists of T140 and its more stable and less toxic analog TN14003 were developed, which, in the case of inhibition of β-arrestin-mediated pathways, had the IC50 values 100 times higher than for Gi/o-protein-dependent pathways [217]. Compound TN14003 showed high activity in preventing degeneration of articular cartilage in osteoarthritis in guinea pigs [218]. It is important that in CXCR4, both small allosteric antagonists and the X4-2-6 peptide bind to similarly located allosteric sites [216,217].
Based on a comparative analysis of the structures of the chemoattractant receptor C5aR and the chemokine receptors CXCR1 and CXCR2, as well as taking into account the data of site-directed mutagenesis, an allosteric site was identified in the region of the external entry into the TMD of the C5aR receptor, which did not coincide with its orthosteric site. This allowed the development of DF2593A, which reduced agonist-stimulated C5aR activity and suppressed pain syndromes caused by acute and chronic inflammation [219].
Currently, a number of small allosteric GPCR antagonists have been developed, which, like pepducins, interact with intracellular allosteric sites and prevent the effective interaction of receptors with G-proteins and (or) β-arrestins (Figure 1). Among chemokine receptors, such antagonists have been found for three of their types, CCR2, CCR7, and CCR9. Among the allosteric CCR9 antagonists, the most interesting are Vercirnon, which has a very high selectivity for binding to CCR9 [59], and its analogs (including 4-aminopyrimidine derivatives), which may be useful in the treatment of Crohn's disease [220]. They interact with the intracellular allosteric site of CCR9, which is formed by the cytoplasmic ends of TM1, TM2, TM3, and TM6, and also includes both charged and hydrophobic residues localized in TM7, H8, and ICL1 [220]. A similar environment has intracellular allosteric sites in the CCR2 and CCR7 receptors, which are similar in their architecture to CCR9. This is supported by data on the 3D structure of complexes of CCR2 and CCR7 receptors with low molecular weight allosteric antagonists CCR2-RA-[R] [60] and Cmp2105 [221], respectively. In all three types of receptors, the main molecular determinants responsible for the binding of allosteric inhibitors are a tyrosine residue located at the cytoplasmic end of TM7 and a relatively variable Gly-Glu/Val-Lys/Arg-Phe-XX-Asp/Tyr sequence located in the H8 helix. The hydroxypyrrolinone group of CCR2-RA-[R] (CCR2), the thiadiazole-dioxide group of Cmp2105 (CCR7), and the sulfonamide group of Vercirnon (CCR9) interact with this sequence [221]. Navarixin, which is similar in pharmacological profile and efficacy to Cmp2105 and is an antagonist of CXCR1, CXCR2, and CCR7 [222], also interacts with the allosteric site formed by TM7 and H8. However, in this case, the cyclobutene-dione group of Navarixin is less suitable, which reduces the effectiveness of the interaction with CCR7, although it does not prevent high efficiency of Navarixin as an inhibitor of CXCR1 and CXCR2, which have a structurally different intracellular allosteric site [221]. The occupancy of intracellular allosteric sites of chemokine receptors by the antagonists prevents their interaction with the C-terminal segment of the Gαi/o-subunit and β-arrestin, since these sites, although to varying degrees, overlap with G-protein and β-arrestin-binding surfaces of the GPCR [223,224]. The consequence of this is the stabilization of the inactive state of the receptor and its low affinity for the agonist, since, as noted above, the formation of a complex between the GPCR and the inactive forms of G-protein or β-arrestin is a necessary condition for the transition of the receptor to a high-affinity state with its further activation by the agonist.
Pepducins as Regulators of Chemokine Receptors
Of considerable interest are pepducins, derivatives of ICLs of chemokine receptors, which, interacting with cytoplasmically oriented allosteric sites, modulate the activity of receptors and their coupling with G-proteins and β-arrestins [225,226,227,228,229] (Figure 1). Given their ability to multicenter binding, their regulatory effects are much broader and more diverse than those of small ligands of intracellular allosteric sites.
N-palmitoylated peptide Palm-RTLFKAHMGQKHRAMR (X1/2pal-i3), corresponding to ICL3 of CXCR1 and CXCR2, demonstrated the properties of NAM for CXCR1/CXCR2, inhibiting endothelial cell proliferation induced by endogenous CXCR1/CXCR2 ligands, IL-8 and the CXCL1/GRO chemokine, and also reduced the proliferation of human umbilical vein endothelial cells induced by metalloproteinase MMP1, a stimulator of chemokine secretion with angiogenic activity. Pepducin X1/2pal-i3 and peptide YSRVGRSVTD (X1/2LCA-i1) modified with lithocholic acid at the N-terminus and structurally corresponding to the ICL1 of CXCR1 and CXCR2 inhibited IL-8- and CXCL1/GRO-induced vascular tube formation, and their efficacy was comparable to the anticancer drug bevacizumab. Three-week treatment of mice with X1/2pal-i3 decreased the intensity of angiogenesis by 5 times and significantly reduced tumor growth in ovarian cancer xenografts [183]. A truncated analog of pepducin X1/2pal-i3, Palm-RTLFKAHMGQKHR, reduced the spontaneous formation of intestinal adenomas in Apc–/+ mice [230].
N-palmitoylated peptides PZ-218 (Palm-MGYQKKLRSMTD) and PZ-210 (Palm-SKLSHSKGHQKRKALK), corresponding to ICL1 (63–74) and ICL3 (224–239) of the CXCR4, inhibited CXCL12/SDF-1-induced responses in human neutrophils and mice, and also blocked CXCL12/SDF-1-mediated migration of lymphoma and lymphocytic leukemia cells, while palmitate-free analogs were inactive [226,231]. Both CXCR4-pepducins, including those in combination with the anticancer drug rituximab, suppressed the survival and metastasis of disseminated lymphoma xenografts. The effect of rituximab was blocked by treatment with the chemokine CXCL12/SDF-1, while this did not occur with the combined use of pepducins and rituximab, indicating synergism of their inhibitory effect on CXCR4 and different localization of binding sites [232].
Along with pepducins with NAM activity for CXCR4, CXCR4-derived pepducins with allosteric agonist and/or PAM activity have been developed, including pepducin ATI-2341 corresponding to the ICL1 of CXCR4 [225]. Pepducin ATI-2341 regulates chemotaxis in various cell cultures and is involved in the mobilization and activation of polymorphonuclear neutrophils and hematopoietic stem cells [225,233]. Unlike CXCL12/SDF-1 used in the clinic, which promotes the mobilization of not only polymorphonuclear neutrophils and hematopoietic stem cells, but also lymphocytes, ATI-2341 does not have a noticeable effect on the mobilization of lymphocytes, which indicates its selectivity. This is due to the fact that the targets of ATI-2341 are various isoforms of Gi/o-proteins, while CXCL12/SDF-1, along with Gi/o-proteins, stimulates β-arrestins and G12/13-proteins. ATI-2341 does not induce GRK-mediated phosphorylation of the CXCR4 receptor, which prevents its desensitization and maintains cell sensitivity to CXCR4-agonists [228]. In addition, ATI2341 protects brain endothelial cells from radiation damage, which indicates the prospect of its use for the repair of tissues damaged during chemotherapy or radiation therapy, through increasing the survival of vascular endothelial cells [234].
These data suggest that the ICL3 and ICL1 in the CXCR1, CXCR2 and CXCR4 receptors are either themselves involved in the formation of cytoplasmically oriented allosteric sites or interact with complementary receptor regions that form such sites [226,232]. Moreover, as in the case of small allosteric antagonists [59,60,220,221], pepducin-induced decrease in the accessibility of the vestibule leading to a transmembrane tunnel of chemokine receptors on the cytoplasmic side, or a change in its conformation, can prevent the formation of the GPCR–G-protein/β-arrestin complex and, thereby, inhibit signal transduction.
Autoantibodies to Chemokine Receptors CXCR3 and CXCR4
It is generally accepted that GPCR autoantibodies are produced against the extracellular regions of the receptor. Depending on the epitope to which they specifically bind, and on their type (monovalent, bivalent), autoantibodies are able to either stimulate or suppress the basal or agonist-stimulated activity of a recognized receptor and, thereby, be involved in the pathogenesis of various diseases [99,235,236,237]. In this regard, it is interesting that in the case of CXCR3 and CXCR4, patterns of autoantibodies have been identified that are specific not only to extracellular regions, but also to their ICLs. It has been shown that in the blood of patients with systemic sclerosis, a severe autoimmune disease, autoantibodies to the ICL2, ICL3 and the membrane-proximal region of the cytoplasmic C-terminal domain of CXCR3 predominate, while in healthy subjects the proportion of autoantibodies to extracellular regions of this receptor was significantly higher [238]. Importantly, autoantibodies against the extracellular N-terminal region of CXCR3 were associated with a better prognosis for systemic sclerosis [239]. This indicates a significant therapeutic potential of autoantibodies to extracellular regions of CXCR3 and CXCR4, but requires further study of the allosteric regulation of these receptors.
7. Allosteric Regulators of Proteinase-Activated Receptors
A significant progress has now been made in the development of allosteric regulators of the PAR family of GPCRs. This family includes 4 types of PARs (PAR1, PAR2, PAR3, and PAR4), which are critical for the control of hemostasis and platelet function, are involved in the regulation of embryonic development, inflammation, wound healing, as well as tumorigenesis and metastasis [240,241,242,243]. Thrombin-induced platelet activation is enhanced in the case of erosion and rupture of atherosclerotic plaques and in percutaneous coronary intervention, causing arterial thrombosis, the main cause of myocardial infarction and ischemic stroke. Thrombin exerts its effects on platelets through PAR1 and PAR4, coupled to different types of G-proteins (Gi/o, Gq/11 and G12/13), as well as through β-arrestin pathways, and the activating effect of thrombin on platelet aggregation is realized mainly through Gq/11-proteins [241,243,244,245]. After Gq/11-mediated activation of PLCβ in platelets, the level of intracellular calcium increases and diacylglycerol-sensitive isoforms of protein kinase C is activated, which leads to activation of αIIb/β3-integrins, their translocation to the plasma membrane, and increased platelet aggregation. Along with this, the release of ADP from platelets, an agonist of P2Y12-purinergic receptors, is enhanced, which stimulates the synthesis of thromboxane A2, a mediator of platelet aggregation and degranulation. It has been established that PAR1 expression is increased in tumor cells, and this is accompanied by an increase in the activity of matrix metalloproteinase-1 (MMP1), which functions as a PAR1 agonist [246,247]. Accordingly, PAR1 agonists enhance oncogenesis, invasion, and metastasis, while PAR1 antagonists, on the contrary, suppress them, preventing the tumor progression and improving the cancer prognosis [246,248,249]. Agonist-induced activation of PAR1 has a strong anti-inflammatory effect and protects endothelial cells from destruction during sepsis [250]. However, prolonged PAR1 activation may have the opposite effect, impairing endothelial cell function and exacerbating sepsis [251,252].
Regulatory effects mediated through PAR1 and PAR4 are determined by the ability of these receptors to form heterodimeric complexes with PAR2 and PAR3 [241,253]. In the case of PAR1, the formation of a heterodimeric complex with PAR2 provides PAR1-induced activation of PAR2 and stimulation of thrombin-mediated β-arrestin signaling, which significantly contributes to PAR-dependent effects on blood coagulation and inflammation [254]. As a result, allosteric regulators of the PARs with antagonistic or NAM activity can be used to prevent thrombosis, attenuate endothelial dysfunctions in sepsis, and also as anticancer drugs. In turn, allosteric agonists and PAMs may be useful for increasing cell survival and wound healing [243,255,256].
Small allosteric ligands of PARs
A relatively large group of PAR1 allosteric regulators are parmodulins, which are small molecules that act by binding to a cytoplasmically oriented allosteric site [257,258,259,260] (Figure 2). Parmodulins have a biased activity because they act on some types of PAR1-coupled G-proteins, but have a little effect on the activity of other types of G-proteins. They have been shown to inhibit Gq/11-mediated granule secretion caused by the mobilization of calcium ions from intracellular depots, but do not affect signaling pathways through G12/13-proteins, thereby normalizing platelet activity but not affecting platelet shape. Unlike the orthosteric PAR1 agonist, parmodulins inhibit PAR1 moderately and reversibly, which is consistent with their pharmacological profile as NAMs [257,259,260]. All this indicates the prospects for the development of parmodulin-based antithrombotic agents [257,259]. In animal models, the therapeutic effects of parmodulins are due to the attenuation of inflammatory reactions and their protective effect on endothelial cells [259,261]. Parmodulin ML161 (PM2) significantly reduced the size of myocardial infarction in mice with myocardial ischemia-reperfusion injury [262].
The study of the binding of parmodulins to PAR1 made it possible to identify the segments involved in the formation of the intracellular allosteric site that interacts with these regulators. It includes the cytoplasmic portion of the TM7 bundle, ICL1, and the H8 helix, since, as was shown for parmodulin JF5, palmitoylation of the C-terminal domain of the receptor, which stabilizes the H8 helix, is critical for JF5-induced activity [257]. Accordingly, the localization of the allosteric site in the area of contact between PAR1 and transducer proteins (G-proteins, β arrestins) can mediate such distinct, selective effects of parmodulins in relation to certain signaling cascades.
Along with allosteric modulators of PAR1, the compound GB83 with PAM activity was recently developed, which potentiated the stimulatory effects of thrombin and peptidic PAR1-agonists on PAR1 activity, but did not affect its basal activity [263]. Using molecular docking, GB83 was shown to bind to a site located near or overlapping with a transmembrane orthosteric site to which the PAR1-antagonist vorapaxar specifically binds (Figure 2). GB83 significantly enhanced PAR1-mediated cell survival and migration, and accelerated skin wound healing in the in vivo experiments [263].
There is now evidence of allosteric regulation and localization of allosteric sites for PAR1, PAR2 and PAR4. As in the case of chemokine receptors (see Figure 1), allosteric sites in PARs can be located at different TMD loci (AS-1, AS-2, AS-3), in the ECLs where they are available for interaction with autoantibodies, as well as in the ICLs where they are targets for pepducins. The small compounds, such as GB83 (PAR1-selective PAM), AZ3451 (allosteric PAR2-inhibitor), I-191 (PAR2-selective NAM) and cross-linked heterobivalent allosteric PAR1/PAR2 inhibitor, RWJ-58259/imidazopyridazine (IZP) derivative), interact with allosteric sites located in the upper and central parts of the TM7 bundle. The low molecular weight parmodulins, including the PAR1-allosteric inhibitors ML161 (PM2) and JF5, as well as pepducins interact with intracellular sites, including the AS-3 locus. The following pepducins have been obtained that have specific activity for PAR1, PAR2 and PAR4: Palm-SGRRYGHALR (P4pal-10) (ICL3 of PAR4, allosteric PAR4-antagonist and NAM, and allosteric antagonist for the non-cognate receptors PAR1, FPR2 and FFAR2), Palm-ATGAPRLPST (P4pal-i1) (ICL1 of PAR4, allosteric PAR4-inhibitor), Palm-RCLSSSAVANRS (P1pal-12), Palm-RSLSSSAVANRS (P1pal-12S) and their analog P1pal-7 (PZ-128) (PAR1 of ICL3, allosteric PAR1-antagonists), Pal-RCLSSSAVANRSKKSRALF (P1pal-19) and Palm-AVANRSKKSRALF (P1pal-13) (PAR1 of ICL3, full allosteric PAR1-agonists), Palm-RSSAMDENSEKKRKSAIK270-287 (P2pal-18S) and its analog PZ-235 (ICL3 of PAR2, allosteric PAR1/2-antagonists). Data on autoantibodies to PARs are fragmentary, but antibodies to extracellular regions of PAR1 in patients with systemic sclerosis have been shown to function as allosteric full agonists and/or PAM for PAR1. Many of the effects of allosteric regulators are due to their effect on the stability of heterodimeric PAR1/PAR2 complexes, as shown for the heterobivalent PAR1/PAR2-inhibitor and pepducins P1pal-12S and P2pal-18S. Allosteric regulators that increase receptor activity (full agonists, PAM) are placed in red squares, while allosteric regulators that decrease receptor activity (antagonists, NAM) are placed in green squares.
Among the small allosteric modulators of PAR2, the compound AZ3451 has been the most studied [264,265,266]. It binds to the allosteric site of PAR2 formed by the TM2, TM3, and TM4, and at nanomolar concentrations suppresses the stimulation of Gq/11-proteins, ERK1/2 and β-arrestin2-mediated signaling, induced by PAR2-agonists, including the small orthosteric agonist AZ2429 [266]. Since the allosteric site, the target for AZ3451, overlaps with the orthosteric site with which the tethered ligand interacts, the conformation of this site should change significantly after activation of the PARs, which makes a significant contribution to PAR-mediated signal transduction. In the absence of PAR2 activation by the agonist, AZ3451 had no effect on the basal activity of the receptor, which allows it to be considered as a NAM [266]. A similar pattern of activity is demonstrated by heterobivalent PAR1/PAR2 ligands, which are constructed based on PAR1- (RWJ-58259) and PAR2-antagonists (imidazopyridazine derivatives) crosslinked with polyethylene glycol [267]. Such constructs should affect the conformation of the PAR1 and PAR2 allosteric sites, which leads to the inhibition of Gq/11-mediated calcium mobilization in endothelial and cancer cells induced by PAR1- and PAR2-agonists. A possible range of therapeutic applications for AZ3451 include the treatment of postoperative osteoarthritis, as indicated by its ability to prevent postoperative cartilage degradation in rats [265].
Recently, a non-competitive allosteric inhibitor of I-191, an imidazopyridazine derivative, has been developed that suppresses PAR2 activation by different agonists (trypsin, peptide, non-peptidic PAR2-agonists) [268]. Compound I-191 reduced agonist-stimulated PAR2-effects, including Gq/11-protein-mediated Ca2+ release, Gi-protein-mediated inhibition of forskolin-stimulated AC, stimulation of ERK1/2 activity, and activation of small G-proteins of the Ras family (RhoA) [268]. Inhibiting the effects of PAR2-agonists at nanomolar concentrations, I-191 showed no activity in their absence, which allows us to classify it as a PAR2-specific NAM. Currently, several weaker PAR2-antagonists have been developed, including those selective for certain intracellular cascades, which are active at micromolar concentrations, which is typical for allosteric regulators, and are potentially capable of binding to the transmembrane allosteric site of PAR2. However, no convincing evidence has yet been obtained to attribute them to NAMs [264,269,270].
Pepducins as Allosteric Regulators of PARs
The greatest success in the creation of allosteric regulators of PAR1, PAR2, and PAR4 is due to the development of pepducins, which are lipidated derivatives of their ICLs [242,271,272,273,274,275,276,277] (Figure 2).
Pepducin Palm-ATGAPRLPST (P4pal-i1), corresponding to the ICL1 of PAR4, inhibited the signal transduction generated by thrombin through its cognate receptor [272]. P4pal-i1 blocked PAR4-mediated chemotaxis and prevented platelet aggregation induced by the PAR4-agonist AYPGKF-amide without affecting PAR1-mediated effects. The combined use of P4pal-i1 and PAR1-antagonist RWJ-56110 led to the complete disappearance of the effects of thrombin, indicating the effectiveness of simultaneous blockade of PAR1 and PAR4 [272]. The combined use of P4pal-i1 and the thrombolytic bivalirudin, which specifically binds to thrombin, completely prevented platelet aggregation induced by a submaximal concentration of α-thrombin [272,278]. P4pal-i1 also prevented the development of arterial thrombosis in guinea pigs with carotid artery occlusion, and the combined use of P4pal-i1 and bivalirudin was more effective than monotherapy [272]. Palm-SGRRYGHALR (P4pal-10), corresponding to PAR4 ICL3, was less specific than P4pal-i1 and had both PAR4-antagonist and inverse PAR1-agonist activity [271,273]. It reduced platelet aggregation induced by the PAR4-agonist AYPGKF-amide by 45–70% but had a little effect on platelet aggregation induced by the PAR1-agonist SFLLRN-amide [271].
At micromolar concentrations, pepducin Palm-RCLSSSAVANRS (P1pal-12) and its truncated analog P1pal-7, corresponding to PAR1 ICL3, inhibited PAR1 agonist-induced PLCβ stimulation [271,279,280]. Pepducin P1pal-12 also inhibited rat aortic vasorelaxation induced by the PAR1-agonist TFLLR-amide without affecting the corresponding effect of the PAR2-agonist [281]. Pepducin PZ-128, which was developed from the structure of P1pal-7, inhibited PAR1-mediated platelet aggregation and arterial thrombosis in guinea pigs and monkeys, and its effect was enhanced in the presence of the antiplatelet drug clopidogrel [275]. Further studies have shown that PZ-128 has no significant side effects both in experimental animals and in patients with ischemic heart disease or with multiple risk factors for coronary artery disease [242,275,282,283]. PZ-128 had a dose-dependent inhibitory effect on platelet aggregation stimulated by the PAR1-agonist SFLLRN-amide, but did not affect the corresponding effects of PAR4-agonists. PZ-128 did not affect bleeding, coagulation, biochemical and metabolic parameters, electrocardiogram parameters, and the drug was well tolerated when administered intravenously at a dose of 0.5 mg/kg. Currently, PZ-128 has successfully completed clinical trials (phase 2) in patients with cardiovascular disease, demonstrating safety and efficacy in the suppression of myonecrosis and arterial thrombosis [277].
The longer pepducin Pal-RCLSSSAVANRSKKSRALF (P1pal-19), in contrast to P1pal-12 and P1pal-7, demonstrated full PAR1-agonist activity and stimulated Gq/11- and Gi/o-proteins, similar to selective PAR1-agonists [271,281]. This indicates that the modification of the structure of PAR-pepducins not only affects the efficiency and selectivity of their action, but also changes their pharmacological profile.
A significant progress has been made in the development of PAR1-pepducins with antitumor activity [242,276]. The first of these was P1pal-7 (PZ-128), which inhibited tumor growth, suppressed metastasis, and reduced cell survival in breast, ovarian, and lung cancers [242,246,247,275,282,284]. P1pal-7 and its extended analog P1pal-12 suppressed PAR1-dependent migration of OVCAR-4, IGROV-1, and SKOV-3 ovarian carcinoma cells into ascitic fluid obtained from patients with ovarian cancer [246]. P1pal-7 enhanced the inhibitory effect of the anticancer drug Taxotere on tumor angiogenesis in ovarian cancer [246]. The antitumor effect of P1pal-7 was based on the inhibition of Akt kinase, a key antiapoptotic protein [284]. P1pal-7 was also effective in the treatment of lung cancer, reducing lung tumor growth by 75%, and in this it was comparable to bevacizumab (Avastin), a potent inhibitor of tumor angiogenesis [285]. In this case, the antitumor effect of P1pal-7 was due to the inhibition of PAR1-mediated activation of ERK1/2, an effector component of the MAPK cascade. P1pal-7 also suppressed the growth and invasive activity of Lewis lung carcinoma cells [247].
Pepducin Palm-RSLSSSAVANRS (P1pal-12S), with activity of allosteric PAR1-antagonist, had an anti-inflammatory effect in a mouse model of bacterial peritonitis, significantly increasing animal survival. However, its injection 4 h after the induction of peritonitis did not affect the mortality of mice. Pepducin Palm-AVANRSKKSRALF (P1pal-13), which is also a derivative of PAR1 ICL3, with the activity of an allosteric PAR1-agonist, was not effective when administered immediately after inflammation induction, but increased the survival rate of mice when administered 4 h later under the conditions of developed sepsis. In Par1 knockout mice, none of the studied pepducins affected sepsis [250]. These data indicate that at the early and late stages of sepsis the optimization of the use of PAR1-pepducins with agonistic and antagonistic activity can lead to a pronounced therapeutic effect. Pepducin P1pal-13 also had a proangiogenic effect in mice with carotid ligation [286]. Its effect required the presence of both PAR1 and PAR2, which indicates that the target of pepducin is the PAR1–PAR2 complex. This complex is also a target for Palm-RSSAMDENSEKKRKSAIK270-287 (P2pal-18S), an ICL3 derivative of PAR2 that inhibits migration of neutrophils and cholangiocarcinoma cells stimulated by both trypsin (PAR1-agonist) and selective PAR2-agonists [286,287].
Pepducin PZ-235, corresponding to PAR2 ICL3, with activity of allosteric antagonist showed potent anti-inflammatory effect in both the chronic inflammation and liver fibrosis model [288] and the atopic dermatitis model [289]. Treatment of mice with CCl4-induced liver fibrosis with PZ-235 suppressed the manifestations of fibrosis, reduced collagen deposits, reduced levels of pro-inflammatory cytokines and other biochemical markers of non-alcoholic fatty liver disease, attenuated the severity of steatosis, reduced triglyceride levels and transaminase activity, and inhibited by 50-100% stellate cell proliferation [288]. PZ-235 had a protective effect in the case of hepatocellular necrosis by attenuating PAR2-mediated production of reactive oxygen species in hepatocytes. When interacting with the PAR2 allosteric site, pepducin PZ-235 forms a well-structured amphipathic α-helix, structurally similar to the corresponding ICL3 region and the neighboring TM6 segment of this receptor [288]. In the in vitro experiments, PZ-235 significantly reduced PAR2-mediated expression of inflammatory factors, including nuclear factor-κB (NF-κB), thymic stromal lymphopoietin (TSLP), tumor necrosis factor-α (TNF-α) and differentiation marker K10 in human keratinocytes, as well as ked to a decrease in the expression of IL-4 and IL-13 in human mast cells [289]. In mice with oxazolone- and DNFB-induced dermatitis, PZ-235 reduced skin thickening (by 43–100%) and leukocyte crusting (by 57%), and also inhibited leukocyte chemotaxis to PAR2-agonists in the ex vivo [289]. PZ-235 treatment of filaggrin-deficient mice exposed to dust mite allergens for 8 weeks reduced total leukocyte and T-cell infiltration, epidermal thickness, and incidence of skin thickening, scaling, excoriation, and total lesion severity score [289].
In some cases, PAR-derived pepducins are able to influence the activity of receptors that are not homologous to PARs, as was shown for pepducin P4Pal10 corresponding to the ICL3 of PAR4 [290]. This pepducin not only functions as an antagonist and NAM for PAR4, but is also able to modulate the functional activity of a number of other receptors that do not belong to the PAR family [290,291]. P4Pal10 reduces the stimulation of Gi/o-proteins via the type 2 formyl peptide receptor (FPR2) and is a weak agonist for the type 2 short chain free fatty acid receptor (FFAR2) [290]. Therefore, structural similarity with the ICLs of the target receptor is not a necessary condition for the effect of pepducin on it. Possible explanations for the “non-specificity” of the PAR4-derived pepducin P4Pal10 on other receptors may be the existence of a common configuration of allosteric sites even in unrelated receptors. At the same time, which is more likely, pepducin can interact with heterooligomeric complexes, which, in addition to PAR4, also include non-homologous receptors (FPR2, FFAR2).
Autoantibodies to PAR1
Endogenous allosteric regulators of PAR1 are autoantibodies produced against its extracellular domains [237,292,293,294]. A significant proportion of patients with systemic sclerosis have autoantibodies to extracellular regions of PAR1, which are able to activate the receptor, like thrombin, thus acting as allosteric agonists [293]. Incubation of human dermal microvascular endothelial cells (HMECs) with IgG isolated from the blood of patients with systemic sclerosis resulted in increased release of IL-6 and phosphorylation of a number of target protein kinases (Akt, p70S6K, ERK1/2), the targets of PAR1-agonists, and these effects of autoantibodies were specifically blocked by PAR1-antagonists [293]. Interestingly, patients with severe forms of COVID-19 also have high titers of anti-PAR1 autoantibodies that stimulate PAR1 in endothelial cells and platelets, and thus contribute to the pathogenesis of microthrombosis. A positive correlation was shown between the level of autoantibodies to PAR1 and the content of the thrombosis marker D-dimer [294].
8. Allosteric Regulators of Pituitary Glycoprotein Hormone Receptors
The family of pituitary glycoprotein hormone receptors includes the receptors of thyroid-stimulating hormone (TSH) and the receptors of luteinizing (LH) and follicle-stimulating hormones (FSH) [295,296]. A feature of this family is a significant extracellular domain (ectodomain), where the orthosteric site is located, to which αβ-heterodimeric hormones containing the same α-subunit and variable β-subunits bind with high affinity.
8.1. Thyroid-Stimulating Hormone Receptor
The TSH receptor (TSHR) is localized predominantly on the surface of thyrocytes, but is sometimes found on the surface of adipocytes, fibroblasts, and other cells [297]. In the active state, the TSHR forms a dimeric or oligomeric complex [298,299,300]. The binding of TSHR to the hormone stimulates the synthesis and release of thyroid hormones by thyrocytes, and also regulates the growth and differentiation of these cells. In TSHR, the ectodomain contains a subdomain with 11 leucine-rich repeats (LRRs) and a cysteine-rich hinge region that functions as a spacer and is located between the LRR subdomain and TMD [301]. Endogenous regulators of TSHR are TSH, specific autoantibodies to TSHR, and thyrostimulin, which bind to the orthosteric site of TSHR, as well as to allosteric sites formed by the LRR subdomain, the hinge region, and the ECLs of TMD [302,303].
The main function of the hinge region is its inhibitory effect on the activity of TSHR, as well as on the specificity of its interaction with TSH and other endogenous regulators [304,305,306,307,308]. A relatively short region of Asp403–Asn406, located at the C-terminus of the hinge region, is considered as an “intrinsic” allosteric TSHR agonist [306,309,310]. Upon binding to the hormone, the conformation of TSHR changes, which allows this region to interact with the receptor ECLs and TMD and activate G-proteins. The peptide containing this region has specific TSHR agonist activity, which is due to its interaction with TSHR ECL1 [305,307,309]. The existence of an internal allosteric agonist is unique for GPCR and indicates the complex nature of the regulation of the interaction between the ectodomain and the TMD in TSHR [308]. It should be noted that th extracellular N-terminal regions of a number of other GPCRs also function as “intrinsic” allosteric regulators and are involved in biased allosterism, as shown for GPR35 [311].
After the hormone binds to the TSHR ectodomain, the cavity within the TMD remains free, although it is involved in the stabilization of the TSHR–G-protein preactivation complex and in the transmission of conformational changes from the ligand-bound ectodomain to the ICLs [312,313]. Allosteric sites are located in this cavity, and there is every reason to believe that their activity can be regulated by simple ions and (or) lipophilic molecules interacting with TSHR hydrophobic helices. At the same time, these sites are targets for synthetic low molecular weight compounds, which, through allosteric mechanisms, can regulate the intrinsic and hormone-stimulated activity of TSHR (Figure 3). At present, significant progress has been made both in the study of the configuration of TSHR transmembrane allosteric sites and in the development of small compounds that can bind to them and demonstrate the activity of allosteric regulators and modulators [314,315,316,317,318,319,320,321,322,323,324,325].
The first allosteric regulators of TSHR were developed back in the 2000s based on thieno[2,3-d]-pyrimidine structure with the activity of allosteric agonists of LH receptor (LHR), including the most active LHR-agonists Org41841 and Org43553 [326,327]. By analyzing their binding to TSHR, the 3D structure of the transmembrane allosteric site of TSHR was proposed [328,329,330], which was similar to that of LHR, and this subsequently allowed the development of a large number of its small ligands with different pharmacological profiles [315,317,318,319,320,331,332,333,334]. It was found that this site is formed by amino acid residues located in TM3, TM4, TM5, TM6, and TM7, and is covered from above by ECL2 segments. Unlike LHR, the entrance to the allosteric site of TSHR is narrower and is formed mainly by hydrophobic and large amino acid residues. These residues are located at the interfaces between ECL2 and neighboring TM4 and TM5 segments, as well as at the interface formed by ECL3 and TM6 [330].
The TSHR belongs to a family of class C GPCRs having a large extracellular domain in which an orthosteric site is located and which is a target for autoantibodies. Most of the small regulators of TSHR bind to allosteric sites located in the cavity of the AS-2 locus or slightly higher, in the lower part of the AS-1 locus. The following low molecular weight compounds specifically interact with allosteric sites in the AS-2 locus: compound C2, NCGC00168126-01 and its analog NCGC00165237-01 (full allosteric TSHR-agonists), MS437, MS438 (biased allosteric TSHR-agonists, did not affect Gi-proteins), TPY3m (biased allosteric TSHR-agonist, mainly activates Gs-proteins), MSq1 (biased allosteric TSHR-agonist, mainly activates Gq/11-proteins), NCGC00379308 (biased PAM, predominantly activates β-arrestins), TPY1, NIDDK/CEB-52 and its analogs NCGC00242595 and NCGC00242364 (allosteric TSHR-antagonists), NCGC00161856, NCGC00229600, compound S37a, TP48 (allosteric inverse TSHR-agonists). Pepducin 612–627(Palm) (ICL3 of TSHR) interact with intracellular allosteric site and functions as allosteric TSHR-agonist. Stimulating TSHR autoantibodies (TSAb) interact with the LRR subdomain, hinge region, and ECLs, while blocking TSHR autoantibodies (TSBAb) mainly interact the N-terminal and central portions of the LRR subdomain. Allosteric regulators that increase receptor activity (full agonists, PAMs) are placed in red squares, allosteric regulators with neutral antagonist activity are placed in green squares, while allosteric regulators with inverse agonist activity are placed in blue squares.
Among the large number of full allosteric agonists for TSHR, the compound NCGC00168126-01 and its analogue NCGC00165237-01 were characterized by high specific activity [315,335]. Under the in vitro conditions, they stimulated the AC activity in cells with expressed TSHR and increased the expression of thyroglobulin and other TSH-dependent genes in human thyrocytes, and their effects were comparable to those of TSH [315,335]. When administered to mice by various routes, NCGC00165237–01 increased the thyroxine level and increased the uptake of radioactive iodine by the thyroid gland, which was due to an increase in the expression of thyroglobulin, a precursor of thyroid hormones, and thyroperoxidase, which catalyzes the binding of iodine to tyrosine residues of thyroglobulin [335]. Based on the structure of thieno[2,3-d]-pyrimidine, we developed the compound TPY3m, which stimulated the production of thyroxine when it was administered intraperitoneally to rats, and also stimulated the expression of thyroid-specific genes in the cell culture of thyrocytes FRTL-5 and in the thyroid of rats [336]. Quite unexpectedly, TPY3m did not cause a decrease in the expression of the Tshr gene, and when combined with TSH (cultured FRTL-5 cells) or thyroliberin (rat thyroid), it partially restored it. This may be one of the mechanisms for maintaining the stimulating effects of TSH (in vitro, FRTL-5) and thyroliberin (in vivo, rats) on the production of thyroid hormones under conditions of their combined use with TPY3m [336].
Small agonists are able to activate TSHR mutants that are insensitive to TSH [337]. In a culture of HEK-EM293 cells with expressed mutant TSHR (Cys41Ser or Leu252Pro substitutions in the ectodomain), the low molecular weight agonist C2 increased the intracellular cAMP level, while TSH was inactive. At the same time, the Leu467Pro substitution in the allosteric site, which prevented TSHR binding to C2, blocked its effect on AC activity [337]. Thus, allosteric TSHR agonists may be effective in subclinical hypothyroidism associated with thyroid resistance to TSH due to mutations in the TSHR ectodomain.
Since TSHR is functionally coupled to several types of G-proteins, including Gi/o-proteins, the main donors of the Gβγ-dimer, an important task was to create allosteric TSHR agonists biased against certain types of G-proteins. In 2015, Rauf Latif and colleagues made the first attempt to create biased agonists and developed compounds MS437 and MS438, which at nanomolar concentrations were able to activate Gs-, Gq/11- and G12-proteins, but did not affect Gi-proteins [338]. Moreover, MS438 showed high efficiency in relation to the activation of the Gs-proteins and AC. Both compounds specifically bound to an allosteric site located in the TMD, but despite a similar regulatory pattern, there were some differences in interaction with the amino acid residues that form this site. When interacting with TM3, compound MS437 formed close contacts with Thr501, while MS438 formed close contacts with Ser505 and Glu506 [338]. Further studies led to the creation of a biased allosteric MSq1 agonist, which in CHO cells with expressed TSHR at nanomolar concentrations stimulated the Gq/11-protein and increased the activity of calcium-dependent isoforms of protein kinase C, with almost no effect on Gs- and G12/13-proteins and MAPK cascade [339]. Using molecular docking, it was found that MSq1 binds to a site that is formed by TM1, TM2, TM3, and TM7 and is covered from above by segments of the ECL1 and ECL2. Comparison of the binding of the Gq/11-selective agonist MSq1 with the compound MS438, which preferentially activates the Gs-protein, revealed significant differences in the pattern of interaction with the amino acid residues that form the allosteric site, which is the “key” to the further development of biased allosteric TSHR agonists [339], including those focused on the biased regulation of β-arrestin pathways [340].
The need to develop PAMs for the regulation of TSHR is due to the prospects for their use to prevent osteoporosis, since TSH plays an important role in the formation of bone tissue, preventing its loss and stimulating its formation [341]. The stimulating effect of TSH on osteoblasts and bone tissue formation is realized mainly through β1-arrestin-regulated cascades [342], and therefore the search for PAMs for TSHR was carried out among ligands biased for β-arrestins. As a result, the compound NCGC00379308 with PAM activity was developed, which potentiated TSH-induced translocation of β1-arrestin to TSHR, but did not affect the Gs- and Gq/11-dependent cascades, demonstrating specificity for β-arrestin signaling [318]. NCGC00379308, when treated with human U2OS cells, did not affect the expression of TSH-dependent genes OPN and ALPL, encoding osteopontin and alkaline phosphatase, and the secretion of osteopontin, but significantly potentiated their stimulation by TSH. Thus, NCGC00379308 can increase the efficiency of endogenous TSH on the growth and differentiation of osteoblast progenitors through β-arrestin-dependent mechanisms [318].
The development of allosteric inverse agonists and neutral antagonists for TSHR is necessary for the treatment of autoimmune hyperthyroidism (Graves' disease) and associated ophthalmopathy, as well as for the treatment of thyroid cancer caused by activating mutations in TSHR [315,322,325]. In 2008, the compound NIDDK/CEB-52 was developed with the activity of a neutral TSHR antagonist, in the phenyl ring of which a methoxypropylene group was introduced to facilitate penetration into the transmembrane allosteric site of the receptor [343]. Incubation of HEK-EM 293 cells with NIDDK/CEB-52 significantly reduced AC stimulation by both TSH and TSHR-stimulating antibodies, and incubation of human thyrocytes with NIDDK/CEB-52 caused a three-fold decrease in TSH-stimulated thyroperoxidase gene expression [343]. Since NIDDK/CEB-52 suppressed LHR activity to a small extent, more selective neutral TSHR antagonists, such as NCGC00242595 and NCGC00242364, were subsequently developed, which in vitro reduced the AC stimulation by TSH and TSHR-stimulating antibodies without affecting the basal activity of the enzyme [317,344]. Treatment of mice with NCGC00242364 reduced the thyroliberin-stimulated thyroxine levels by 44% and the expression of genes encoding Na+/I--symporter and thyroperoxidase by 75 and 83%. NCGC00242364 also reduced thyroxine levels and thyroid gene expression in mice treated with TSHR-stimulating antibodies. These data indicate the high efficacy of NCGC00242364 and its analogues in the treatment of Graves' disease. Since these compounds do not affect the basal activity of TSHR, this avoids the development of hypothyroidism, one of the undesirable effects when using allosteric inverse agonists of TSHR [315,317].
Among inverse TSHR agonists, compounds NCGC00161856 [345] and NCGC00229600 [346] had significant activity. They inhibited not only stimulated TSH or antibodies, but also basal TSHR activity. NCGC00229600 also prevented TSH- and antibody-dependent AC stimulation in primary culture of fibroblasts obtained from the retroorbital region of patients with Graves' disease and characterized by increased TSHR expression [347]. The ability of NCGC00229600 to suppress TSHR activity in retroorbital fibroblasts is important in terms of preventing ophthalmopathy, the most severe symptom of Graves' disease [348].
We have developed two thieno[2,3-d]-pyrimidine derivatives, TPY1 and TP48, with antagonistic activity against TSHR [349,350]. When administered to rats, TPY1 reduced thyroliberin-stimulated production of thyroid hormones and the expression of thyrogloubin, thyroperoxidase, and Na+/I--symporter genes in the thyroid, but did not affect basal levels of thyroid hormones, indicating its activity as a neutral allosteric TSHR antagonist [350]. In turn, TP48 not only suppressed thyroid hormones levels and expression of thyroid-specific genes stimulated by thyroliberin, but 3.5 hours after administration to rats, it reduced their basal levels of thyroid hormones and the expression of the Nis gene encoding Na+/I--symporter [349,350]. This indicates that the TP48 functions as a mild inverse TSHR agonist [350].
In 2019, the compound S37a with seven centers of chirality was developed, one of the stereoisomers of which, at micromolar concentrations, suppressed the AC activity and the cAMP accumulation in HEK293 cells with expressed TSHR when they were treated with TSH and stimulatory monoclonal antibodies TSAb M22 and KSAb1, oligoclonal stimulatory antibodies TSAb, characteristic of patients with Graves' disease, as well as a small C2 agonist [322]. S37a, when administered orally to mice, had a high bioavailability (53%) and was not toxic [322], which indicates the prospects for its clinical trials, especially since there are currently effective and safe approaches for the treatment of Graves' disease and Graves' ophthalmopathy not developed [351,352].
In addition to allosteric sites localized in the ECLs and TMD, such sites are also located in the ICLs of TSHR, as evidenced by our results on the stimulating effect on TSHR activity of pepducin, which corresponds to region 612–627 of TSHR ICL3 [353,354] (Figure 3). Pepducin 612–627(Palm) modified with palmitate at the C-terminus stimulated the AC activity in rat thyroid membranes, and its action was specific for TSHR and was not detected in membranes where TSHR was absent [353]. When administered intranasally to rats, 612-627(Palm) increased the level of thyroid hormones in them, and this effect was dose-dependent [354]. The obtained data suggest that 612-627(Palm) is an intracellular allosteric TSHR agonist, and hydrophobic palmitate is critical for its activity, providing the transport of pepducin into the cell, since the non-palmitoylated analog was inactive [353,354].
Autoantibodies to the TSH Receptor
Autoimmune thyroid diseases are found in an average of 5% of the population, the most common among which is diffuse toxic goiter (Graves' disease), which develops as a result of an autoimmune attack on TSHR [355]. For almost 40 years, the presence of antibodies to TSHR has been used to differentiate Graves' disease [356], and this approach is constantly being improved [357]. Assessment of the titer and pattern of autoantibodies to TSHR is widely used to differentiate Graves' disease from toxic multinodular goitre type A [358] and other forms of thyrotoxicosis [359].
Autoantibodies to TSHR have the activity of full agonists, inverse agonists and antagonists, and in some cases do not have their intrinsic activity, but allosterically affect the activity of TSHR stimulated by TSH or stimulatory autoantibodies [360,361,362,363,364]. Stimulating autoantibodies bind to TSHR, which is in an inactive conformation, activate the receptor, and trigger signaling cascades through Gs- and Gq/11-proteins, which leads to increased synthesis and secretion of thyroid hormones [365,366]. At the same time, they can compete with TSH for the binding site in the receptor ectodomain or allosterically affect its conformation. Binding of stimulatory autoantibodies is carried out with the receptor ectodomain, but can also include the hinge region connecting the ectodomain with the TMD, and regions of ECLs connecting TMs [362,366,367,368] (Figure 3). Neutral antibodies interact with the receptor in a similar way, with the only difference that for them the main molecular determinants are localized not in the ectodomain, but in the hinge region, which includes the sequence 316-366 [366,369]. Monoclonal antibodies to the TSHR hinge region, isolated from the blood serum of animals with experimental autoimmune thyroiditis, stimulated TSHR, increased the level of cAMP in thyrocytes, activated the 3-phosphoinositide pathway, the MAPK cascade, and different isoforms of protein kinase C, and also increased the activity of the pro-inflammatory factor NF-kB [367]. These data indicate a functional similarity between TSH and TSHR-stimulating antibodies and demonstrate that the hormone and stimulating antibodies interact with highly overlapping sites located in the receptor ectodomain, although these sites are still not completely identical [368]. The effect of stimulatory autoantibodies on TSHR is an increase in thyroid hormone production, leading to autoimmune hyperthyroidism (Graves' disease) as well as Graves' ophthalmopathy, and more than half of patients with Graves' disease are positive for such antibodies [370,371].
Blocking autoantibodies inhibit the binding of TSHR to TSH and stimulatory TSHR antibodies, preventing activation of the receptor and thereby reducing the production of thyroid hormones by thyrocytes. Blocking antibodies bind to linear and “conformational” epitopes located in the LRR subdomain, which includes a significant part of the TSHR ectodomain [366,368,372]. However, unlike stimulating antibodies, for which the “center” of binding to the LRR subdomain is shifted to its C-terminus, in the case of blocking TSHR autoantibodies, such a “center” is shifted to its N-terminus and, thus, only partially overlaps with the binding sites for TSH and stimulating autoantibodies [366,368]. This is supported by the fact that the Trp265 located in the tenth LRR, corresponding to the C-terminal portion of the LRR subdomain, effectively interacts with the light chain of stimulating autoantibody M22, but is not involved in binding with inhibitory antibody K1-70 [368]. The ability of blocking autoantibodies to prevent TSHR hyperactivation can be used to treat Graves' disease and Graves' orbitopathy [340,368,373]. Currently used antithyroid drugs have many side effects, and even after five years of treatment of patients with Graves' disease with these drugs, 21% of them remain positive for TSHR autoantibodies [374]. However, it should be taken into account that, along with the suppression of thyroid hormone production, some blocking and neutral autoantibodies can affect Gq/11-dependent signaling pathways, as well as enhance cell proliferation by activating the MAPK cascade [365,366], which indicates their varying degrees of bias in relation to TSHR-dependent signaling pathways.
8.2. Luteinizing hormone receptor
LHR has a significant similarity with TSHR both in topology in the membrane and in activation mechanisms, since in this case gonadotropin, LH or human chorionic gonadotropin (hCG), binds with high affinity to the ectodomain, which leads to conformational changes in the TMD and provides a functional interaction of LHR ICLs with G-proteins and β-arrestins [375,376]. In the stimulation of LHR by gonadotropin, the cavity of the transmembrane tunnel, in which allosteric sites are located, remains ligand-free. It is filled with water molecules that form hydrogen bonds with the polar groups of amino acid residues located in the inner cavity of this site. In LHR, as well as in the FSH receptor, along with the main allosteric site, there is an additional (minor) allosteric site, binding to which modulates the activity of the main site [377,378]. The main allosteric site is formed by TM3, TM4, TM5, and TM6 [378,379,380], while the additional allosteric site is formed by TM1, TM2, TM3, and TM7 [377]. Both sites are deepened to varying degrees into the TMD and overlap, resulting in a wide variety of pharmacological profiles of their ligands, including allosteric agonists and antagonists, allosteric modulators (PAM, NAM) and ago-PAM [333,381] (Figure 4).
Like TSHR, LHR has a large extracellular domain in which an orthosteric site is located, and the small LHR regulators bind to allosteric sites located in the cavity of the AS-2 locus. The following low molecular weight compounds specifically interact with allosteric sites in the AS-2 locus: thieno[2,3-d]-pyrimidine derivatives Org41841, Org43553, TP03, TP04 and their analogs (allosteric LHR-agonists or ago-PAMs, predominantly activate Gs-proteins), the derivatives of 1,3,5-pyrazole (compound 1) and terphenyl (LUF5771) (allosteric LHR-agonists), and thieno[2,3-d]-pyrimidine derivative TP31 (allosteric LHR-antagonists). Pepducin NKDTKIAKK-Nle-A(562-572)-K(Palm)A (ICL3 of LHR) interact with intracellular allosteric site and functions as allosteric LHR-agonist. Autoantibodies to the extracellular regions of LHR have been suggested, but have not been characterized to date. Allosteric regulators that increase receptor activity (full agonists, ago-PAM) are placed in red squares, while allosteric regulators with neutral antagonist activity are placed in green squares.
The need to create allosteric regulators is due to both the low selectivity of LH and hCG in relation to intracellular cascades, and the peculiarities of pharmacological preparations of gonadotropins, since their urinary forms (hCG) have a significant number of bioactive impurities and are variable in specific activity, while recombinant forms (LH, hCG) significantly differ from natural gonadotropins in the pattern of N-glycosylation and regulatory properties. Along with this, gonadotropins are not very effective in the case of mutant forms of LHR, which have a reduced ligand-binding ability or impaired translocation to the membrane, even when binding to gonadotropin is preserved [382,383,384]. In this regard, it should be noted that some of the developed small allosteric LHR agonists have the properties of chaperones, which stabilize the structure of LHR and facilitate their translocation to the plasma membrane, which increases their sensitivity to endogenous LH, as was experimentally shown for thieno[2,3-d]-pyrimidine derivative Org42599 [385,386,387].
The first allosteric regulators of LHR with agonist activity were obtained in 2002, the most effective of which were the thieno[2,3-d]-pyrimidine derivatives Org41841 and Org43553, which act on LHR at nanomolar concentrations [326]. They specifically bound to LHR without interfering with the activation of the receptor by gonadotropins, and were characterized by a bias towards Gs-proteins and cAMP-dependent cascades, and very weakly stimulated Gq/11-proteins and phosphoinositide metabolism, as well as β-arrestins [388,389,390]. By studying the binding of Org41841 and Org43553 to LHR with mutations in the TMD, it was found that the Tуг570, Phe585 and Tyr643 are important for the formation of the allosteric site, and this site is structurally similar to that in TSHR [329]. Negatively charged Glu506 localized in TM3 was key for the interaction with thieno[2,3-d]-pyrimidines, since its replacement with alanine blocked the binding of Org41841 to mutant LHR. This is due to the formation of a salt bridge between the carboxyl group of glutamic acid and the positively charged amino group of the allosteric ligand [327,329]. In vivo, Org43553 was effective not only when administered intraperitoneally and subcutaneously, but also when administered orally, due to its good absorption and stability in the gastrointestinal tract [390,391,392,393]. Oral administration of Org 43553 stimulated ovulation in immature mice and adult rats. The resulting eggs were of good quality, had high fertility, and when implanted with a high yield, they gave viable embryos. Rats did not show signs of ovarian hyperstimulation, which is often observed with gonadotropin treatment, and this was due to a relatively mild stimulation of steroidogenesis and a lower half-life of the drug (two times lower than that of hCG) [390]. In addition, Org 43553 had little effect on the production of vascular endothelial growth factor, the signaling pathways of which are involved in increased vascular permeability and provoke ovarian hyperstimulation syndrome [392]. Oral administration of Org 43553 at the same dose to male rats stimulated testicular steroidogenesis and increased blood testosterone levels [390]. When taken orally by women of reproductive age, Org 43553 (300 mg) caused ovulation in 83% of them, without significant adverse effects, including without provoking ovarian hyperstimulation syndrome [393].
We have developed a series of thieno[2,3-d]-pyrimidine derivatives with the properties of allosteric agonists and (or) ago-PAM and allosteric antagonists of LHR [323,324,394,395,396,397,398,399,400,401,402]. Compounds TP03 and TP04, the most active of the full agonists, stimulated the AC activity in testicular membranes isolated from rat testes, increased testosterone production by cultured Leydig cells, and also stimulated testicular steroidogenesis and increased testosterone levels when administered intraperitoneally, subcutaneously, and orally to both healthy male rats and animals with androgen deficiency caused by types 1 and 2 diabetes mellitus and aging [395,400,402,403,404]. Using molecular docking, it was found that the efficiency and selectivity of the studied thieno[2,3-d]-pyrimidines correlate with the characteristics of their binding to the transmembrane allosteric site of LHR, and the key parameter of such binding was the intensity of hydrophobic contact, while the Coulomb interactions and hydrogen bonds were less significant [400]. When co-administered with low doses of gonadotropin, thieno[2,3-d]-pyrimidines potentiated its effect, and potentiation was more pronounced in rats with type 1 diabetes mellitus [405]. As with hCG, TP03 and TP04 improved spermatogenesis in diabetic and aging rats [400,402,406,407]. When TP03 was administered to metformin-treated diabetic rats, a significant increase in its steroidogenic effect was shown upon short-term (but not long-term) TP03 administration [402,406]. In contrast to hCG, long-term administration of TP03 and TP04 to male rats did not suppress the expression of the Lhr gene in the testes, and their steroidogenic effect was preserved and even increased, which indicates the possibility of their long-term use to stimulate testicular steroidogenesis in the conditions of androgen deficiency [400,401]. TP03 has been shown to stimulate ovarian steroidogenesis in proestrus adult female rats pretreated with a GnRH antagonist [408]. These results indicate the promise of using TP03 and TP04 for correcting androgen status and improving spermatogenesis in metabolic disorders and aging, as well as for controlled ovulation induction.
Along with thieno[2,3-d]-pyrimidines, 1,3,5-pyrazole and terphenyl derivatives can also function as allosteric LHR ligands with full and inverse agonist activity [378,409,410]. Among terphenyl derivatives, LUF5771 inhibited LHR stimulation by gonadotropins and Org 43553 [378,410]. Compound 1, the most active pyrazole derivative with activity of full agonist, stimulated the AC activity and increased testosterone production by Leydig cells both in cell culture and when administered intraperitoneally to male rats [409]. Like Org43553, the pyrazole and terphenyl derivatives did not compete with hCG for binding with LHR, which indicates in favor of their interaction with the transmembrane allosteric site of the receptor [378,409].
Based on the thieno[2,3-d]-pyrimidine structure, we created an allosteric TP31 antagonist, which, in the in vitro experiments, reduced hCG- and TP03-stimulated AC activity in testicular membranes, and when administered intraperitoneally to male rats, it slightly reduced the baseline testosterone and largely inhibited the stimulatory effect of hCG on testosterone production [397,398] (Figure 4). Since pre-treatment with TP31 suppressed TP03-induced receptor activation in a dose-dependent manner, this indicates a common binding site. Compared to thieno[2,3-d]-pyrimidines with pronounced agonistic activity, TP31 has an additional ethylamine group in the second position of the heterocyclic ring of nicotinic acid, which changes the volume and charge of the variable substituent in 5-amino-4-(3-aminophenyl)-N-(tert-butyl)-2-(methylsulfanyl)thieno[2,3-d]pyrimidine-6-carboxamide, a framework molecule for the synthesis of LHR allosteric regulators [398]. Small ligands with inverse agonist or antagonist activity can be used to suppress steroidogenic function in the treatment of prostate cancer and in the treatment of gonadotropin-dependent tumors.
The importance of intracellular allosteric sites in the regulation of LHR activity is supported by our data on the effect of peptides derived from ICL3 of this receptor [411,412,413]. It should be mentioned that ICL3, both in LHR and in many other GPCRs, includes the key determinants responsible for functional coupling with G-proteins. ICL3-derived peptide NKDTKIAKK-Nle-A(562-572)-K(Palm)A, corresponding to LHR region 562–572, responsible for interaction with Gs-proteins, increased the basal AC activity in testicular membranes, inhibiting hCG-induced stimulation of the enzyme [412] (Figure 4). When administered intratesticularly to male rats, NKDTKIAKK-Nle-A(562-572)-K(Palm)A increased testosterone levels, exerting a stimulating effect on testicular steroidogenesis, but when administered intraperitoneally, its effect was weak, which may indicate its degradation in the bloodstream [412]. Thus, pepducins corresponding to LHR ICL3 may be effective as intracellular allosteric regulators of LHR, but further studies are required to improve their stability and bioavailability.
Since LHR is similar in structure and regulatory mechanisms to TSHR, it cannot be ruled out that it can also be allosterically regulated by autoantibodies against the LHR ectodomain. However, at present, reliable data on the possibility of such regulation, as well as on the association between the presence of autoantibodies to LHR and reproductive dysfunctions, have not been obtained. There is a clinical study in which an attempt was made to trace the relationship between the presence of LHR-stimulating autoantibodies in the blood of women with polycystic ovary syndrome (PCOS) and the severity of their hyperandrogenemia [414]. However, the data obtained indicate a low occurrence of anti-LHR autoantibodies in both the control and PCOS groups, and also demonstrate the absence of significant differences in the occurrence of these antibodies between groups. At the same time, there is evidence that autoantibodies to the FSH receptor may be involved in the pathogenesis of FSH-resistant PCOS [415]. As a result, further studies are needed to study LHR autoantibodies, primarily in patients resistant to LH.
9. Allosteric Regulators of β-Adrenergic Receptors
As in the structure of most GPCRs, allosteric sites in β-AR are located in all major subdomains, which is confirmed by the influence of autoantibodies to ECLs, synthetic ICL-derived pepducins, as well as the endogenous and synthetic small compounds capable of interacting with sites located on at various loci of the TMD and at its interfaces with ICLs. The multiplicity of these sites was demonstrated using the ligand competitive saturation (SILCS) computational method, which made it possible not only to predict the possible localization and functional activity of allosteric sites in the β2-AR, but also to suggest potential candidates as regulators of these sites [416]. Below, we will briefly discuss some synthetic small molecular weight allosteric regulators, since they, like small endogenous regulators (cholesterol metal inos, etc.), are described in detail in a number of reviews and experimental works [416,417,418,419,420,421], and we will pay more attention to autoantibodies and pepducins.
In 2017, based on the DNA-encoded small-molecule library, compound 15 (Cmpd-15) with NAM activity for β2-AR was developed [422]. Cmpd-15 is able to enter the cell and bind specifically to the intracellular allosteric site formed by the cytoplasmic ends of TM1, TM2, TM3 and TM7, as well as the ICL1 and the H8 helix of β2-AR [62]. The result of this is the stabilization of the receptor in an inactive state, a decrease in the affinity of β2-AR for orthosteric agonists and inhibition of its binding to G-proteins and β-arrestins [62,420]. As shown for the polyethylene glycol-carboxylic acid derivative Cmpd-15PA, which has the same pharmacological characteristics as Cmpd-15, the polar groups of this derivative bind to the allosteric site through interactions with polar amino acid residues (Thr2746.36, Ser3298.47, Asp3318.49, Asn692.40, Arg63ICL1), while the cyclohexyl and phenyl rings of Cmpd-15PA interacted with the hydrophobic package formed by the residues Val541.53 and Ile581.57 at the end of TM1, Leu64ICL1 (ICL1), Ile722.43 (TM2), Leu2756.37 (TM6), as well as Tyr3267.53 from the conserved NPxxY sequence located in TM7, and Phe3328.50 located in the H8 helix [62]. Subsequently, analogs of Cmpd-15 with NAM activity for β2-AR were synthesized, and the study of their structure and molecular docking confirmed the decisive importance of their interaction with hydrophobic residues that form the allosteric site of β2-AR [423].
In 2018, the compound 6 (Cmpd-6) with PAM activity for β2-AR was developed, which also interacted with an allosteric site located in the region of the intracellular vestibule of the transmembrane tunnel (ICL2, cytoplasmic ends of TM3 and TM4), stabilizing the active conformation of the receptor [422,424]. This compound interfered with the effects of the biased β-AR regulator carvedilol. Cmpd-6, on the one hand, increased the affinity of carvedilol for β2-AR and thereby enhanced its inhibitory effect on β2-agonist-induced stimulation of Gs-proteins and cAMP signaling, and, on the other hand, enhanced β-arrestin pathways activated by carvedilol, which led to stimulation of ERK1/2, increased receptor endocytosis and its trafficking into lysosomes [425]. Surprisingly, Cmpd-6 facilitated carvedilol-mediated activation of the β-arrestin pathways in the case of β1-AR as well, indicating its bias in the regulation of these pathways when binding to different types of β-AR [426]. Activation of the β-arrestin pathways mediates the cardioprotective effect of carvedilol, including suppression of ischemia/reperfusion injury-induced apoptosis, whereby the development of Cmpd-6 and its analogs acting as β-arrestin-biased PAMs opens up new prospects for the development of cardioprotective agents [426].
Allosteric sites localized on the cytoplasmic side of the β-AR TMD are targets not only for low molecular weight compounds, but also for pepducins corresponding to the ICLs and ICL/TM interfaces of β-AR. In 2014, the group of Jeffrey Benovic synthesized pepducins corresponding to different regions of ICL1, ICL2, and ICL3 of β2-AR [427]. As a result, three groups of pepducins were identified that differ in the mechanisms of action on β2-AR, Gs-proteins and β-arrestins. The first group included pepducins, which activated Gs-proteins in a receptor-independent manner. Pepducins of the second group had a biased activity towards the β-arrestin pathways, causing activation of MAPK, desensitization of β2-AR, and, as a result, reducing or blocking the cell response to β2-agonists. Pepducins of the third group receptor-dependently stimulated Gs-proteins and cAMP-signaling pathways, stabilizing the active conformation of β2-AR, and these effects were not accompanied by site-specific phosphorylation of the receptor by GRK-family kinases, which prevented β-arrestin-mediated internalization of β2-ARs and maintained or even increased their sensitivity to β2-agonists [427].
During congestive heart failure, the level of catecholamines increases and their stimulating effect on β-AR increases, which in cardiomyocytes leads to chronic activation of Gs-dependent β1-AR and Gi/o-dependent β2-AR. As a result, apoptotic processes leading to the death of cardiomyocytes are enhanced, the expression of β1-AR is reduced, and the inotropic reserve of the myocardium is weakened. Blockers of the β-AR orthosteric site, as a rule, have low selectivity for transducer proteins, and this negatively affects the activity of G-protein-independent β-arrestin pathways responsible for transient β-AR desensitization and cardiomyocyte survival. Like carvedilol, in cultured cardiomyocytes, pepducin ICL1–9, corresponding to β2-AR ICL1, stimulated β2-AR phosphorylation, induced β-arrestin recruitment and subsequent β2-AR internalization, and activated β-arrestin signaling cascades, functioning as β-arrestin-biased allosteric agonist [428]. Unlike carvedilol, pepducin ICL1-9 induced calcium-independent, but dependent on β2-AR and β-arrestin, cardiomyocyte contractility. The study of the mechanisms of action of ICL1-9 showed that, along with the activation of ERK1/2, it increases the activity of the epidermal growth factor receptor, which indicates its powerful growth-stimulating and regenerative potential [428]. Intramyocardial injection of pepducin ICL1-9 into mice reduced the size of myocardial infarction induced by ischemia/reperfusion, reduced the death of cardiomyocytes, improved heart function in acute and prolonged remodeling of myocardial fibrosis, indicating its pronounced cardioprotective effect in acute ischemic myocardial injury [429]. The antiapoptotic effect of ICL1-9 was dependent on the functional state of β2-AR, as well as on the presence of β-arrestins, since this effect was not detected in mutant mice lacking β2-AR and β-arrestins. A key role in the anti-apoptotic effect of ICL1-9 belongs to the signaling pathway, including RhoA, ROCK, and protein kinase D, which can be regulated both through β-arrestins and Gi-proteins that can functionally couple with β2-AR [429,430]. Thus, pepducins that function as ligands for intracellular allosteric sites of β2-AR and have a wide range of pharmacological activity are good candidates for the development of new drugs for the treatment of cardiovascular diseases [229,428,429,430].
Currently, a large number of autoantibodies to ECLs of β1- and β2-AR are known, which are closely associated with cardiovascular diseases [431,432,433]. The most extensive and clinically important group are autoantibodies to β1-AR ECL2, the presence of certain types of which is the root cause of dilated and ischemic cardiomyopathy [433,434,435,436,437]. Antibodies to ECL2, depending on the epitope for which they are produced, upon binding to β1-AR, induce conformational changes that stabilize both the active and inactive conformation of the receptor. In the case of stabilization of the active conformation, an increase in the affinity of β1-AR for the agonist and an increase in cAMP production are observed, which leads to cardiovascular pathology [435,438]. Among the pattern of autoantibodies to β1-AR isolated from the blood of patients with dilated cardiomyopathy, antibodies predominate that suppress the internalization of receptors, cause a β1-AR-mediated increase in intracellular cAMP levels in the absence of an agonist, and inhibit the stimulating effect of isoproterenol on AC activity, and this may indicate on their inherent activity of ago-NAMs, which are biased towards cAMP-dependent signaling pathways [438]. The fact that the decrease in β1-AR internalization caused by autoantibodies does not lead to an increase in agonist-induced AC activity can be explained as the fact that recycling of desensitized receptors is impaired. A certain contribution may be made by the negative influence of autoantibodies on the binding characteristics and accessibility of the orthosteric site. It is important that autoantibodies to β1-AR were detected in a significant proportion of healthy subjects without any signs of cardiovascular pathology, but these autoantibodies, unlike those in patients with dilated cardiomyopathy, did not have a stimulating effect on β1-AR and in some cases even had weak antagonistic activity [436].
To identify ECL epitopes that are most significant for β-AR activity, autoantibodies were studied by immunoprecipitation with peptides and their constructs derived from different ECL regions of β1- and β2-AR, as well as by immunization of experimental animals with peptides structurally corresponding to these regions [432,439]. When analyzing the immunoprecipitation of autoantibodies to β1-AR in patients with different forms of cardiomyopathy, it was found that immunogenic epitopes in all cases were localized in ECL2 (195–225) of β1-AR. In cardiomyopathy in patients with Chagas disease, the epitope included the region 201-205, in postpartum cardiomyopathy the epitope corresponded to the region 200-210, and in dilated cardiomyopathy, the epitopes were located in overlapping regions 183-208, 197-202, 206-212 and 213-218, including TM4 segments adjacent to ECL2 [439,440,441]. More recently, to establish a more precise localization of the epitope responsible for the production of cardiomyopathy-inducing autoantibodies, a cyclic peptide corresponding to β1-AR ECL2 was used, which made it possible to identify the key molecular determinants of this epitope, including the NDPK211–214 segment and the C209↔C215 cysteine bridge. Segment 211-214 was located at the C-terminal end of the backward-oriented α-helix constituting ECL2 in β1-AR [432]. In this case, the trigger of allosteric activation of β1-AR is most likely the Asp212, the negative charge of which is neutralized upon interaction with the autoantibody. This assumption is based on the fact that substitution of Asp212 for asparagine leads to paradoxical activation of the mutant receptor by antagonists, broxaterol and terbutaline [442]. In rodent experiments, a cyclic peptide corresponding to β1-AR ECL2 prevented the development of chronic heart failure caused by autoantibodies to β1-AR [432,443]. A cardioprotective effect on myocardial injury in mice was also demonstrated for another cyclic peptide, RD808, also corresponding to ECL2 of β1-AR [444].
Monovalent Fab-fragments of antibodies that were produced against the ECL2 region 173-180 of β2-AR bound to β2-AR and inhibited the stimulatory effects of β2-agonists on AC activity in cultured cardiomyocytes, while bivalent antibodies stimulated β2-AR-mediated signaling pathways and caused contraction of cardiomyocytes, and these effects of bivalent antibodies were due to their two-center binding to receptor molecules, which promotes β2-AR dimerization [445]. Subsequently, it was found that the stimulatory effects of these autoantibodies are specific for cAMP-dependent cascades mediated through Gs-proteins, which indicates their bias towards β2-AR-coupled transducer proteins [446]. These data indicate that the pharmacological profile of autoantibodies to β2-AR depends on the number and localization of binding sites in the extracellular domains of the receptors.
10. Conclusion
Allosteric regulation plays a decisive role in the functioning of all known representatives of GPCRs, since allosteric mechanisms are involved in the regulation of their basal activity and all the main stages of GPCR-mediated signal transduction, including orthosteric ligand binding, and are also responsible for receptor desensitization, endocytosis of ligand-receptor complexes (including intracellular signaling) and their recycling or degradation. Since ligands of allosteric sites affect various functions of the receptor, including biased agonism, and are characterized by varying degrees of selectivity for GPCRs, this predetermines their diversity in chemical structure, pharmacological profile, and localization and configuration of the sites with which they interact. Low-specific allosteric regulators include simple ions (protons, Na+, Zn2+, Mg2+, Mn2+, and some others), which penetrate into the inner cavity of the transmembrane tunnel of most GPCRs, and, being in complex with the amino acid residues that form the allosteric site, modulate their functional activity. This determines the influence of the ionic composition and pH in the extra- and intracellular environments on the activity of GPCRs, and may mediate the effect of the electrochemical properties of the membrane on GPCR signaling. Ion chelators, including peptides and proteins, affect the concentration and ratio of ions available for interaction with allosteric sites, and thus are able to regulate the GPCRs. This greatly expands the range of potential allosteric regulators.
An important group of allosteric modulators is membrane lipids (cholesterol, phospholipids, gangliosides, and others) that interact with allosteric sites of GPCRs located on the outer membrane-contacting surface of the TMD, as well as in interfaces formed by TMs and hydrophilic loops. These interactions are involved in the regulation of receptor activity, since they affect the conformational mobility and configuration of the transmembrane tunnel, and are critical for the ability of receptors to form GPCR-complexes and oligomeric complexes with other components of GPCR signaling. The influence of the physicochemical properties and composition of membranes on the activity of GPCRs is carried out largely through allosteric lipid-receptor interactions, and this occurs not only in the plasma membrane, but also in the membranes of endosomes, the Golgi apparatus, and the nucleus, where intracellular GPCR signaling occurs. It is not entirely clear to what extent the effects of lipids are specific to a particular receptor. Some lipids, such as cholesterol, target various groups of GPCRs, while other lipids, such as PIP2, may be receptor selective and biased for intracellular signaling.
Endogenous allosteric GPCR regulators include heterotrimeric G-proteins and β-arrestins, which are part of pre-activation complexes and mediate signal transduction after receptor activation by an orthosteric or allosteric agonist. Unlike small endogenous regulators, the interaction of transducer proteins with cytoplasmic subdomains and the intracellular vestibule of the receptor's transmembrane tunnel is usually multicentric and often involves molecular determinants that form various intracellular allosteric sites. In addition, in the functionally active state, GPCRs form homo- and heterodimeric complexes in which protomers interact allosterically, and this is another allosteric mechanism that is responsible for the stability and mutual transitions of active and inactive receptor conformations, the ability of the receptor to be activated by an orthosteric agonist, and also for biased agonism. The formation of homo- and heterodimeric GPCR-complexes involves multiple allosteric sites located in hydrophilic loops and on the outer surfaces of their TMD. Each protomer in the complex is subject to multiple allosteric influences. As a result, in a complex that includes several protomers, including those from different types of receptors, the allosteric regulation becomes much more complicated.
Highly specific allosteric regulators of GPCRs are autoantibodies developed against antigenic determinants located in their ECLs. They can stimulate or, conversely, inhibit GPCRs, as well as modulate their activity, including biased regulation of intracellular cascades. Considering the fact that the number of identified and studied autoantibodies to GPCRs is steadily increasing, there is no doubt that for many receptors there is a wide range of such autoantibodies with a different profile of functional activity, and these autoantibodies can be the root causes of many diseases. At the same time, autoantibodies to GPCRs are found in healthy subjects, although they differ from autoantibodies in patients with autoimmune diseases. This indicates that, even under normal conditions, autoantibodies to GPCRs may be involved in the allosteric regulation of GPCR-signaling.
The complexity of allosteric effects on GPCRs caused by numerous regulatory molecules differing in structure, availability, and mechanisms of action predetermines the multiplicity and different topology of allosteric sites in GPCRs. Allosteric regulators can interact with the following loci in GPCRs: 1) with ECLs, which affects the binding of orthosteric agonists to class B and C GPCRs and the formation of homo- and heteromeric receptor complexes; 2) with the internal cavity of the transmembrane tunnel, which affects the binding of orthosteric agonists to GPCRs of classes A and B and the formation of pre-activation complexes with G-proteins and β-arrestins, and provides biased agonism; 3) with ICLs, which affects the interaction of the receptor with transducer and adapter proteins and the stability of active and inactive states of the receptor, and also has a key role in the bias of intracellular signaling and in the further fate of the ligand-receptor complex; 4) with the lateral, membrane-contacting surface of the TMD, which affects the interaction of the receptor with membrane lipids and the stability of di- and oligomeric GPCR-complexes, and also determines the configuration of the internal cavities of the TMD, modulating the binding of orthosteric and allosteric ligands. Along with this, allosteric effects can be mediated by the interaction of various molecules, "indirect" allosteric regulators, with signal proteins that form functionally active complexes with GPCRs and are "direct" allosteric regulators (G-proteins, β-arrestins, RAMPs, protomers of other receptors, etc.). In such cases, one can speak of stepwise, both temporal and spatial, allosteric regulation of GPCR-containing signaling complexes. As a result, the search and development of artificial (synthetic) allosteric ligands should take into account the topological diversity and functional complexity of the endogenous allosteric regulation of GPCRs. It is important that synthetic allosteric ligands are balanced and harmoniously included in the mechanisms of allosteric endogenous regulation, and supplement and modify it in the necessary way.
In recent years, for many GPCRs, impressive progress has been made in the development of a large number of allosteric regulators with various pharmacological activities, and many of them are not only selective for a certain type of receptor, but also characterized by a biased activity towards intracellular cascades, which is very valuable and in high demand in medicine. Using molecular docking and molecular biological approaches, new populations of allosteric sites in GPCRs have been identified, which creates prerequisites for the search for their endogenous regulators, as well as for the creation of their artificial ligands. At the same time, it should be noted that in the process of translating the results of studies on the efficacy and safety of allosteric regulators obtained in vitro and in animal models, many intractable problems appear in the clinic, and this is one of the reasons for the very limited arsenal of pharmacological drugs based on allosteric GPCR-regulators. More intensive studies are needed on the allosteric mechanisms of regulation of GPCRs in real biological systems, at the organism level, where receptors are under the influence of many factors. At the same time, despite the difficulties and limitations, there is no doubt that, given the high specificity of action, including biased agonism, and also taking into account moderate activity, which excludes blocking or hyperactivation of receptors, as is often observed for orthosteric ligands, allosteric GPCR-regulators GPCRs will occupy a significant niche in the pharmacological industry in the coming years.
Funding
This research was funded by the Russian Science Foundation (project No. 19-75-20122).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AC | adenylate cyclase |
| β-AR | β-adrenergic receptor |
| ECL | extracellular loop |
| FFAR2 | type 2 short chain free fatty acid receptor |
| FPR2 | type 2 formyl peptide receptor |
| FSH | follicle-stimulating hormone |
| GPCR | G protein-coupled receptors |
| GRK | G protein-coupled receptor kinase |
| hCG | human chorionic gonadotropin |
| ICL | intracellular loop |
| LH | luteinizing hormone |
| LHR | luteinizing hormone receptor |
| MAPK | mitogen-activated protein kinase |
| NAM | negative allosteric modulator |
| NF-κB | nuclear factor-κB |
| PAM | positive allosteric modulator |
| PAR | proteinase-activated receptor |
| PIP2 | phosphatidylinositol-4,5-bisphosphate |
| PLCβ | phosphoinositide-specific phospholipase Cβ |
| RAMP | receptor-activity-modifying protein |
| SAM | silent allosteric modulator |
| SDF-1 | stromal cell-derived factor-1 (CXCL12/SDF-1) |
| TM | transmembrane region |
| TM7 bundle | heptahelical transmembrane bundle |
| TMD | transmembrane domain |
| TNF-α | tumor necrosis factor-α |
| TSH | thyroid-stimulating hormone |
| TSHR | thyroid-stimulating hormone receptor |
References
- Brown, N.A.; Schrevens, S.; van Dijck, P.; Goldman, G.H. Fungal G-Protein-Coupled Receptors: Mediators of Pathogenesis and Targets for Disease Control. Nat. MicroBiol. 2018, 3, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Shpakov, A.O.; Pertseva, M.N. Signaling Systems of Lower Eukaryotes and Their Evolution. Int. Rev. Cell Mol. Biol. 2008, 269, 151–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Dong, D.; Guo, L.; Dong, X.; Leng, J.; Zhao, B.; Guo, Y.-D.; Zhang, N. Research Advances in Heterotrimeric G-Protein α Subunits and Uncanonical G-Protein Coupled Receptors in Plants. Int. J. Mol. Sci. 2021, 22, 8678. [Google Scholar] [CrossRef] [PubMed]
- Reboul, J.; Ewbank, J.J. GPCRs in Invertebrate Innate Immunity. Biochem. Pharmacol. 2016, 114, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Singh, V. GPCR Signaling in C. Elegans and Its Implications in Immune Response. Adv. Immunol. 2017, 136, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, Y.; Li, T.; Feng, X. G-Protein Coupled Receptors (GPCRs): Signaling Pathways, Characterization, and Functions in Insect Physiology and Toxicology. Int. J. Mol. Sci. 2021, 22, 5260. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Zhao, T.; Yun, Y.; Xie, X. Recent Progress in Assays for GPCR Drug Discovery. Am J. Physiol. Cell Physiol. 2022, 323, C583–C594. [Google Scholar] [CrossRef] [PubMed]
- Martín-Mora, D.; Fernández, M.; Velando, F.; Ortega, Á.; Gavira, J.A.; Matilla, M.A.; Krell, T. Functional Annotation of Bacterial Signal Transduction Systems: Progress and Challenges. Int. J. Mol. Sci. 2018, 19, 3755. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sun, D.; Zhu, J.; Liu, W. Two-Component Signal Transduction Systems: A Major Strategy for Connecting Input Stimuli to Biofilm Formation. Front. MicroBiol. 2018, 9, 3279. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; Liu, Q.; Zhao, S.; Shui, W.; Jiang, Y.; Wang, M.W. G Protein-Coupled Receptors: Structure- and Function-Based Drug Discovery. Signal. Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Lefkowitz, R.J. The Role of Beta-Arrestins in the Termination and Transduction of G-Protein-Coupled Receptor Signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of Receptor Signals by Beta-Arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, A.C.; Dunn, H.; Ferguson, S.S. Regulation of GPCR Activity, Trafficking and Localization by GPCR-Interacting Proteins. Br. J. Pharmacol. 2012, 165, 1717–1736. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Guerra, G.; Parisi, M.; de Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-Surface Receptors Transactivation Mediated by g Protein-Coupled Receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.E.; Blaxall, B.C. G Protein Coupled Receptor-Mediated Transactivation of Extracellular Proteases. J. Cardiovasc. Pharmacol. 2017, 70, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Han, Y.; Duan, L.; Chung, K.Y. Scaffolding of Mitogen-Activated Protein Kinase Signaling by β-Arrestins. Int. J. Mol. Sci. 2022, 23, 1000. [Google Scholar] [CrossRef] [PubMed]
- Daaka, Y.; Luttrell, L.M.; Ahn, S.; della Rocca, G.J.; Ferguson, S.S.; Caron, M.G.; Lefkowitz, R.J. Essential Role for G Protein-Coupled Receptor Endocytosis in the Activation of Mitogen-Activated Protein Kinase. J. Biol. Chem. 1998, 273, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Pei, G. Beta-Arrestin Signaling and Regulation of Transcription. J. Cell Sci. 2007, 120, 213–218. [Google Scholar] [CrossRef]
- Tian, X.; Kang, D.S.; Benovic, J.L. β-Arrestins and G Protein-Coupled Receptor Trafficking. Handb. Exp. Pharmacol. 2014, 219, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Seyedabadi, M.; Ghahremani, M.H.; Albert, P.R. Biased Signaling of G Protein Coupled Receptors (GPCRs): Molecular Determinants of GPCR/Transducer Selectivity and Therapeutic Potential. Pharmacol. Ther. 2019, 200, 148–178. [Google Scholar] [CrossRef] [PubMed]
- Seyedabadi, M.; Gharghabi, M.; Gurevich, E.V.; Gurevich, V.V. Structural Basis of GPCR Coupling to Distinct Signal Transducers: Implications for Biased Signaling. Trends Biochem. Sci. 2022, 47, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.-L.; Zhou, Y.; Zhang, C.-Y.; Gao, Z.-A.; Duan, J.-X. The Role and Mechanism of β-Arrestin2 in Signal Transduction. Life Sci. 2021, 275, 119364. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Galtes, D.; Wang, J.; Rockman, H.A. G Protein-Coupled Receptor Signaling: Transducers and Effectors. Am J. Physiol. Cell Physiol. 2022, 323, C731–C748. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Westfield, G.H.; Xiao, K.; Reis, R.I.; Huang, L.-Y.; Tripathi-Shukla, P.; Qian, J.; Li, S.; Blanc, A.; Oleskie, A.N.; Dosey, A.M.; Su, M.; Liang, C.R.; Gu, L.L.; Shan, J.M; Chen, X.; Hanna, R.; Choi, M.; Yao, X.J; Klink, B.U.; Kahsai, A.W.; Sidhu, S.S.; Koide, S.; Penczek, P.A.; Kossiakoff, A.A.; Woods, V.L. Jr.; Kobilka, B.K.; Skiniotis, G.; Lefkowitz, R.J. Visualization of Arrestin Recruitment by a G-Protein-Coupled Receptor. Nature 2014, 512, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Wang, J.K.; Bauer, B.; Townshend, R.J.L.; Hollingsworth, S.A.; Olivieri, J.E.; Xu, H.E.; Sommer, M.E.; Dror, R.O. Molecular Mechanism of GPCR-Mediated Arrestin Activation. Nature 2018, 557, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Wingler, L.M.; Choi, M.; Pani, B.; Manglik, A.; Kruse, A.C.; Lefkowitz, R.J. Sortase Ligation Enables Homogeneous GPCR Phosphorylation to Reveal Diversity in β-Arrestin Coupling. Proc. Natl. Acad. Sci. USA 2018, 115, 3834–3839. [Google Scholar] [CrossRef]
- Asher, W.B.; Terry, D.S.; Gregorio, G.G.A.; Kahsai, A.W.; Borgia, A.; Xie, B.; Modak, A.; Zhu, Y.; Jang, W.; Govindaraju, A.; Huang, L.Y.; Inoue, A.; Lambert, N.A.; Gurevich, V.V.; Shi, L.; Lefkowitz, R.J.; Blanchard, S.C.; Javitch, J.A. GPCR-Mediated β-Arrestin Activation Deconvoluted with Single-Molecule Precision. Cell 2022, 185, 1661–1675.e16. [Google Scholar] [CrossRef]
- Haider, R.S.; Matthees, E.S.F.; Drube, J.; Reichel, M.; Zabel, U.; Inoue, A.; Chevigné, A.; Krasel, C.; Deupi, X.; Hoffmann, C. β-Arrestin1 and 2 Exhibit Distinct Phosphorylation-Dependent Conformations When Coupling to the Same GPCR in Living Cells. Nat. Commun. 2022, 13, 5638. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular Mechanism of β-Arrestin-Biased Agonism at Seven-Transmembrane Receptors. Annu. Rev. Pharmacol. Toxicol 2012, 52, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Masureel, M.; Hollingsworth, S.A.; Heydenreich, F.M.; Suomivuori, C.-M.; Brinton, C.; Townshend, R.J.L.; Bouvier, M.; Kobilka, B.K.; Dror, R.O. How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell 2020, 183, 1813–1825.e18. [Google Scholar] [CrossRef]
- Premont, R.T. Keys to the Kingdom: GPCR Phosphorylation Patterns Direct β-Arrestin. EMBO Rep. 2020, 21, e51249. [Google Scholar] [CrossRef]
- Kim, J.; Ahn, S.; Ren, X.-R.; Whalen, E.J.; Reiter, E.; Wei, H.; Lefkowitz, R.J. Functional Antagonism of Different G Protein-Coupled Receptor Kinases for Beta-Arrestin-Mediated Angiotensin II Receptor Signaling. Proc Natl. Acad. Sci. USA 2005, 102, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, F.; Zhang, D.; Liu, Z.; Lin, A.; Liu, C.; Xiao, P.; Yu, X.; Sun, J.-P. Phosphorylation of G Protein-Coupled Receptors: From the Barcode Hypothesis to the Flute Model. Mol. Pharmacol. 2017, 92, 201–210. [Google Scholar] [CrossRef]
- Baidya, M.; Kumari, P.; Dwivedi-Agnihotri, H.; Pandey, S.; Chaturvedi, M.; Stepniewski, T.M.; Kawakami, K.; Cao, Y.; Laporte, S.A.; Selent, J.; Inoue, A.; Shukla, A.K. Key Phosphorylation Sites in GPCRs Orchestrate the Contribution of β-Arrestin 1 in ERK1/2 Activation. EMBO Rep. 2020, 21, e49886. [Google Scholar] [CrossRef] [PubMed]
- Janetzko, J.; Kise, R.; Barsi-Rhyne, B.; Siepe, D.H.; Heydenreich, F.M.; Kawakami, K.; Masureel, M.; Maeda, S.; Garcia, K.C.; von Zastrow, M.; Inoue, A.; Kobilka, B.K. Membrane Phosphoinositides Regulate GPCR-β-Arrestin Complex Assembly and Dynamics. Cell 2022, 185, 4560–4573.e19. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; Kobilka, B.K. Structure of the Neurotensin Receptor 1 in Complex with β-Arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Eichel, K.; von Zastrow, M. Subcellular Organization of GPCR Signaling. Trends Pharmacol. Sci. 2018, 39, 200–208. [Google Scholar] [CrossRef]
- Sutkeviciute, I.; Vilardaga, J.-P. Structural Insights into Emergent Signaling Modes of G Protein-Coupled Receptors. J. Biol. Chem. 2020, 295, 11626–11642. [Google Scholar] [CrossRef] [PubMed]
- Peña, K.A. Endosomal Parathyroid Hormone Receptor Signaling. Am J. Physiol. Cell Physiol. 2022, 323, C783–C790. [Google Scholar] [CrossRef] [PubMed]
- Liccardo, F.; Luini, A.; di Martino, R. Endomembrane-Based Signaling by GPCRs and G-Proteins. Cells 2022, 11, 528. [Google Scholar] [CrossRef]
- Maudsley, S.; Martin, B.; Luttrell, L.M. G Protein-Coupled Receptor Signaling Complexity in Neuronal Tissue: Implications for Novel Therapeutics. Curr. Alzheimer. Res. 2007, 4, 3–19. [Google Scholar] [CrossRef]
- Maudsley, S.; Patel, S.A.; Park, S.-S.; Luttrell, L.M.; Martin, B. Functional Signaling Biases in G Protein-Coupled Receptors: Game Theory and Receptor Dynamics. Mini Rev. Med. Chem. 2012, 12, 831–840. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of Signalling and Biased Agonism in G Protein-Coupled Receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- Perez, D.M.; Hwa, J.; Gaivin, R.; Mathur, M.; Brown, F.; Graham, R.M. Constitutive Activation of a Single Effector Pathway: Evidence for Multiple Activation States of a G Protein-Coupled Receptor. Mol. Pharmacol. 1996, 49, 112–122. [Google Scholar]
- Seifert, R.; Gether, U.; Wenzel-Seifert, K.; Kobilka, B.K. Effects of Guanine, Inosine, and Xanthine Nucleotides on Beta(2)-Adrenergic Receptor/G(s) Interactions: Evidence for Multiple Receptor Conformations. Mol. Pharmacol. 1999, 56, 348–358. [Google Scholar] [CrossRef]
- Ghanouni, P.; Gryczynski, Z.; Steenhuis, J.J.; Lee, T.W.; Farrens, D.L.; Lakowicz, J.R.; Kobilka, B.K. Functionally Different Agonists Induce Distinct Conformations in the G Protein Coupling Domain of the Beta 2 Adrenergic Receptor. J. Biol. Chem. 2001, 276, 24433–24436. [Google Scholar] [CrossRef]
- Kenakin, T. Agonist-Receptor Efficacy. I: Mechanisms of Efficacy and Receptor Promiscuity. Trends Pharmacol. Sci. 1995, 16, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Bock, A.; Bermudez, M. Allosteric Coupling and Biased Agonism in G Protein-Coupled Receptors. FEBS J. 2021, 288, 2513–2528. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Bender, E.; Schamberger, J.; Eitner, F. Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators. Int. J. Mol. Sci. 2021, 22, 1763. [Google Scholar] [CrossRef] [PubMed]
- Jakubík, J.; El-Fakahany, E.E. Allosteric Modulation of GPCRs of Class A by Cholesterol. Int. J. Mol. Sci. 2021, 22, 1953. [Google Scholar] [CrossRef]
- Liu, L.; Fan, Z.; Rovira, X.; Xue, L.; Roux, S.; Brabet, I.; Xin, M.; Pin, J.-P.; Rondard, P.; Liu, J. Allosteric Ligands Control the Activation of a Class C GPCR Heterodimer by Acting at the Transmembrane Interface. Elife 2021, 10, e70188. [Google Scholar] [CrossRef]
- Hedderich, J.B.; Persechino, M.; Becker, K.; Heydenreich, F.M.; Gutermuth, T.; Bouvier, M.; Bünemann, M.; Kolb, P. The Pocketome of G-Protein-Coupled Receptors Reveals Previously Untargeted Allosteric Sites. Nat. Commun. 2022, 13, 2567. [Google Scholar] [CrossRef]
- Persechino, M.; Hedderich, J.B.; Kolb, P.; Hilger, D. Allosteric Modulation of GPCRs: From Structural Insights to in Silico Drug Discovery. Pharmacol. Ther. 2022, 237, 108242. [Google Scholar] [CrossRef]
- Christopoulos, A. Advances in G Protein-Coupled Receptor Allostery: From Function to Structure. Mol. Pharmacol. 2014, 86, 463–478. [Google Scholar] [CrossRef]
- Congreve, M.; Oswald, C.; Marshall, F.H. Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs. Trends Pharmacol. Sci. 2017, 38, 837–847. [Google Scholar] [CrossRef]
- Lu, S.; Shen, Q.; Zhang, J. Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms. Acc. Chem. Res. 2019, 52, 492–500. [Google Scholar] [CrossRef]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural Insights into G-Protein-Coupled Receptor Allostery. Nature 2018, 559, 45–53. [Google Scholar] [CrossRef]
- Oswald, C.; Rappas, M.; Kean, J.; Doré, A.S.; Errey, J.C.; Bennett, K.; Deflorian, F.; Christopher, J.A.; Jazayeri, A.; Mason, J.S.; Congreve, M.; Cooke, R.M.; Marshall, F.H. Intracellular Allosteric Antagonism of the CCR9 Receptor. Nature 2016, 540, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qin, L.; Zacarías, N.V.O.; de Vries, H.; Han, G.W.; Gustavsson, M.; Dabros, M.; Zhao, C.; Cherney, R.J.; Carter, P.; Stamos, D.; Abagyan, R.; Cherezov, V.; Stevens, R.C.; IJzerman, A.P.; Heitman, L.H.; Tebben, A.; Kufareva, I.; Handel, T.M. Structure of CC Chemokine Receptor 2 with Orthosteric and Allosteric Antagonists. Nature 2016, 540, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Kahsai, A.W.; Pani, B.; Wang, Q.-T.; Zhao, S.; Wall, A.L.; Strachan, R.T.; Staus, D.P.; Wingler, L.M.; Sun, L.D.; Sinnaeve, J.; Choi, M.; Cho, T.; Xu, T.T.; Hansen, G.M.; Burnett, M.B.; Lamerdin, J.E.; Bassoni, D.L.; Gavino, B.J.; Husemoen, G.; Olsen, E.K.; Franch, T.; Costanzi, S.; Chen, X.; Lefkowitz, R.J. Allosteric “Beta-Blocker” Isolated from a DNA-Encoded Small Molecule Library. Proc. Natl. Acad. Sci. USA 2017, 114, 1708–1713. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ahn, S.; Kahsai, A.W.; Meng, K.-C.; Latorraca, N.R.; Pani, B.; Venkatakrishnan, A.J.; Masoudi, A.; Weis, W.I.; Dror, R.O.; Chen, X.; Lefkowitz, R.J.; Kobilka, B.K. Mechanism of Intracellular Allosteric Β2AR Antagonist Revealed by X-Ray Crystal Structure. Nature 2017, 548, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Yang, D.; Wang, Y.; de Graaf, C.; Zhou, Q.; Jiang, S.; Liu, K.; Cai, X.; Dai, A.; Lin, G.; Liu, D.; Wu, F.; Wu, Y.; Zhao, S.; Ye, L.; Han, G.W.; Lau, J.; Wu, B.; Hanson, M.A.; Liu, Z.J.; Wang, M.W.; Stevens, R.C. Human GLP-1 Receptor Transmembrane Domain Structure in Complex with Allosteric Modulators. Nature 2017, 546, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Erol, I.; Salmas, R.E.; Aksoydan, B.; Kantarcioglu, I. Oligomerization and Cooperativity in GPCRs from the Perspective of the Angiotensin AT1 and Dopamine D2 Receptors. Neurosci. Lett. 2019, 700, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yuan, Y.; Chen, Y.; Yu, J.; Wang, J.; Chen, J.; Guo, Y.; Pu, X. Biased Activation Mechanism Induced by GPCR Heterodimerization: Observations from ΜOR/ΔOR Dimers. J. Chem. Inf. Model. 2022, 62, 5581–5600. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Dion, S.B.; Onorato, J.J.; Ptasienski, J.; Kim, C.M.; Sterne-Marr, R.; Hosey, M.M.; Benovic, J.L. Arrestin Interactions with G Protein-Coupled Receptors. Direct Binding Studies of Wild Type and Mutant Arrestins with Rhodopsin, Beta2-Adrenergic, and M2 Muscarinic Cholinergic Receptors. J. Biol. Chem. 1995, 270, 720–731. [Google Scholar] [CrossRef]
- Christopoulos, A.; Christopoulos, G.; Morfis, M.; Udawela, M.; Laburthe, M.; Couvineau, A.; Kuwasako, K.; Tilakaratne, N.; Sexton, P.M. Novel Receptor Partners and Function of Receptor Activity-Modifying Proteins. J. Biol. Chem. 2003, 278, 3293–3297. [Google Scholar] [CrossRef]
- DeVree, B.T.; Mahoney, J.P.; Vélez-Ruiz, G.A.; Rasmussen, S.G.F.; Kuszak, A.J.; Edwald, E.; Fung, J.-J.; Manglik, A.; Masureel, M.; Du, Y.; Matt, R.A.; Pardon, E.; Steyaert, J.; Kobilka, B.K.; Sunahara, R.K. Allosteric Coupling from G Protein to the Agonist-Binding Pocket in GPCRs. Nature 2016, 535, 182–186. [Google Scholar] [CrossRef]
- Staus, D.P.; Strachan, R.T.; Manglik, A.; Pani, B.; Kahsai, A.W.; Kim, T.H.; Wingler, L.M.; Ahn, S.; Chatterjee, A.; Masoudi, A.; Kruse, A.C.; Pardon, E.; Steyaert, J.; Weis, W.I; Prosser, R.S.; Kobilka, B.K.; Costa, T.; Lefkowitz, R.J. Allosteric Nanobodies Reveal the Dynamic Range and Diverse Mechanisms of G-Protein-Coupled Receptor Activation. Nature 2016, 535, 448–452. [Google Scholar] [CrossRef]
- Pioszak, A.A.; Hay, D.L. RAMPs as Allosteric Modulators of the Calcitonin and Calcitonin-like Class B G Protein-Coupled Receptors. Adv. Pharmacol. 2020, 88, 115–141. [Google Scholar] [CrossRef]
- Edward Zhou, X.; Melcher, K.; Eric Xu, H. Structural Biology of G Protein-Coupled Receptor Signaling Complexes. Protein Sci. 2019, 28, 487–501. [Google Scholar] [CrossRef]
- Kenakin, T.; Strachan, R.T. PAM-Antagonists: A Better Way to Block Pathological Receptor Signaling? Trends Pharmacol. Sci. 2018, 39, 748–765. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A.; Kenakin, T. G Protein-Coupled Receptor Allosterism and Complexing. Pharmacol. Rev. 2002, 54, 323–374. [Google Scholar] [CrossRef] [PubMed]
- Reinecke, B.A.; Wang, H.; Zhang, Y. Recent Advances in the Drug Discovery and Development of Dualsteric/ Bitopic Activators of G Protein-Coupled Receptors. Curr. Top. Med. Chem. 2019, 19, 2378–2392. [Google Scholar] [CrossRef] [PubMed]
- Jakubík, J.; Randáková, A.; Chetverikov, N.; El-Fakahany, E.E.; Doležal, V. The Operational Model of Allosteric Modulation of Pharmacological Agonism. Sci. Rep. 2020, 10, 14421. [Google Scholar] [CrossRef] [PubMed]
- Valant, C.; Sexton, P.M.; Christopoulos, A. Orthosteric/Allosteric Bitopic Ligands: Going Hybrid at GPCRs. Mol. Interv. 2009, 9, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Valant, C.; Robert Lane, J.; Sexton, P.M.; Christopoulos, A. The Best of Both Worlds? Bitopic Orthosteric/Allosteric Ligands of g Protein-Coupled Receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; Sexton, P.M.; Christopoulos, A. Bridging the Gap: Bitopic Ligands of G-Protein-Coupled Receptors. Trends Pharmacol. Sci. 2013, 34, 59–66. [Google Scholar] [CrossRef]
- Fronik, P.; Gaiser, B.I.; Sejer Pedersen, D. Bitopic Ligands and Metastable Binding Sites: Opportunities for G Protein-Coupled Receptor (GPCR) Medicinal Chemistry. J. Med. Chem. 2017, 60, 4126–4134. [Google Scholar] [CrossRef] [PubMed]
- Egyed, A.; Kiss, D.J.; Keserű, G.M. The Impact of the Secondary Binding Pocket on the Pharmacology of Class A GPCRs. Front. Pharmacol. 2022, 13, 847788. [Google Scholar] [CrossRef]
- Ferrisi, R.; Gado, F.; Polini, B.; Ricardi, C.; Mohamed, K.A.; Stevenson, L.A.; Ortore, G.; Rapposelli, S.; Saccomanni, G.; Pertwee, R.G.; Laprairie, R.B.; Manera, C.; Chiellini, G. Design, Synthesis and Biological Evaluation of Novel Orthosteric-Allosteric Ligands of the Cannabinoid Receptor Type 2 (CB2R). Front. Chem. 2022, 10, 984069. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, A.E.; Mason, J.S.; Vajda, S.; Keserű, G.M. Analysis of Tractable Allosteric Sites in G Protein-Coupled Receptors. Sci. Rep. 2019, 9, 6180. [Google Scholar] [CrossRef] [PubMed]
- Renault, P.; Giraldo, J. Dynamical Correlations Reveal Allosteric Sites in G Protein-Coupled Receptors. Int. J. Mol. Sci. 2020, 22, 187. [Google Scholar] [CrossRef] [PubMed]
- Ivetac, A.; McCammon, J.A. Mapping the Druggable Allosteric Space of G-Protein Coupled Receptors: A Fragment-Based Molecular Dynamics Approach. Chem. Biol. Drug Des. 2010, 76, 201–217. [Google Scholar] [CrossRef]
- Miao, Y.; Nichols, S.E.; McCammon, J.A. Mapping of Allosteric Druggable Sites in Activation-Associated Conformers of the M2 Muscarinic Receptor. Chem. Biol. Drug Des. 2014, 83, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Caliman, A.D.; Miao, Y.; McCammon, J.A. Mapping the Allosteric Sites of the A2A Adenosine Receptor. Chem. Biol. Drug Des. 2018, 91, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Ciancetta, A.; Gill, A.K.; Ding, T.; Karlov, D.S.; Chalhoub, G.; McCormick, P.J.; Tikhonova, I.G. Probe Confined Dynamic Mapping for G Protein-Coupled Receptor Allosteric Site Prediction. ACS Cent Sci. 2021, 7, 1847–1862. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Karlov, D.S.; Pino-Angeles, A.; Tikhonova, I.G. Intermolecular Interactions in G Protein-Coupled Receptor Allosteric Sites at the Membrane Interface from Molecular Dynamics Simulations and Quantum Chemical Calculations. J. Chem. Inf. Model. 2022, 62, 4736–4747. [Google Scholar] [CrossRef]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Masureel, M.; van Antwerpen, P.; Kobilka, B.K.; Govaerts, C. Allosteric Regulation of G Protein-Coupled Receptor Activity by Phospholipids. Nat. Chem. Biol. 2016, 12, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.-Y.; Hoi, K.K.; Liko, I.; Hedger, G.; Horrell, M.R.; Song, W.; Wu, D.; Heine, P.; Warne, T.; Lee, Y.; Carpenter, B.; Plückthun, A.; Tate, C.G.; Sansom, M.S.P.; Robinson, C.V. PtdIns(4,5)P2 Stabilizes Active States of GPCRs and Enhances Selectivity of G-Protein Coupling. Nature 2018, 559, 423–427. [Google Scholar] [CrossRef]
- Jafurulla, M.; Aditya Kumar, G.; Rao, B.D.; Chattopadhyay, A. A Critical Analysis of Molecular Mechanisms Underlying Membrane Cholesterol Sensitivity of GPCRs. Adv. Exp. Med. Biol. 2019, 1115, 21–52. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; He, X.; Yang, Z.; Chai, Z.; Zhou, S.; Wang, J.; Rehman, A.U.; Ni, D.; Pu, J.; Sun, J.; Zhang, J. Activation Pathway of a G Protein-Coupled Receptor Uncovers Conformational Intermediates as Targets for Allosteric Drug Design. Nat. Commun. 2021, 12, 4721. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; Choi, H.-J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; Schnapp, A.; Konetzki, I.; Sunahara, R.K.; Gellman, S.H.; Pautsch, A.; Steyaert, J.; Weis, W.I.; Kobilka, B.K. Structure of a Nanobody-Stabilized Active State of the β(2) Adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Wittmann, H.-J.; Schneider, E.H.; Seifert, R. Modulation of GPCRs by Monovalent Cations and Anions. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 363–380. [Google Scholar] [CrossRef] [PubMed]
- van der Westhuizen, E.T.; Valant, C.; Sexton, P.M.; Christopoulos, A. Endogenous Allosteric Modulators of G Protein-Coupled Receptors. J. Pharmacol. Exp. Ther. 2015, 353, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Kessler, P.; Marchot, P.; Silva, M.; Servent, D. The Three-Finger Toxin Fold: A Multifunctional Structural Scaffold Able to Modulate Cholinergic Functions. J. NeuroChem. 2017, 142 Suppl 2, 7–18. [Google Scholar] [CrossRef]
- Zarzycka, B.; Zaidi, S.A.; Roth, B.L.; Katritch, V. Harnessing Ion-Binding Sites for GPCR Pharmacology. Pharmacol. Rev. 2019, 71, 571–595. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.M.; Traynor, J.R.; Alt, A. Allosteric Modulator Leads Hiding in Plain Site: Developing Peptide and Peptidomimetics as GPCR Allosteric Modulators. Front. Chem. 2021, 9, 671483. [Google Scholar] [CrossRef] [PubMed]
- Skiba, M.A.; Kruse, A.C. Autoantibodies as Endogenous Modulators of GPCR Signaling. Trends Pharmacol. Sci. 2021, 42, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.M.; Feld, A.P. Nanobodies as Probes and Modulators of Cardiovascular G Protein-Coupled Receptors. J. Cardiovasc. Pharmacol. 2022, 80, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Mullikin, D.; Caron, M.G. Regulation of Beta-Adrenergic Receptors by Guanyl-5’-Yl Imidodiphosphate and Other Purine Nucleotides. J. Biol. Chem. 1976, 251, 4686–4692. [Google Scholar] [CrossRef] [PubMed]
- Maguire, M.E.; van Arsdale, P.M.; Gilman, A.G. An Agonist-Specific Effect of Guanine Nucleotides on Binding to the Beta Adrenergic Receptor. Mol. Pharmacol. 1976, 12, 335–339. [Google Scholar] [PubMed]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-Activated Structure of Β2-Adrenoceptor Stabilized by an Engineered Nanobody. Nature 2013, 502, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Pals-Rylaarsdam, R.; Benovic, J.L.; Hosey, M.M.; Onorato, J.J. Agonist-Receptor-Arrestin, an Alternative Ternary Complex with High Agonist Affinity. J. Biol. Chem. 1997, 272, 28849–28852. [Google Scholar] [CrossRef] [PubMed]
- Strachan, R.T.; Sun, J.; Rominger, D.H.; Violin, J.D.; Ahn, S.; Rojas Bie Thomsen, A.; Zhu, X.; Kleist, A.; Costa, T.; Lefkowitz, R.J. Divergent Transducer-Specific Molecular Efficacies Generate Biased Agonism at a G Protein-Coupled Receptor (GPCR). J. Biol. Chem. 2014, 289, 14211–14224. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pani, B.; Gokhan, I.; Xiong, X.; Kahsai, A.W.; Jiang, H.; Ahn, S.; Lefkowitz, R.J.; Rockman, H.A. β-Arrestin-Biased Allosteric Modulator Potentiates Carvedilol-Stimulated β Adrenergic Receptor Cardioprotection. Mol. Pharmacol. 2021, 100, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.; Chung, K.Y. The Conformational Dynamics of Heterotrimeric G Proteins During GPCR-Mediated Activation. Subcell. Biochem. 2022, 99, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Mafi, A.; Kim, S.-K.; Goddard, W.A. The Mechanism for Ligand Activation of the GPCR-G Protein Complex. Proc. Natl. Acad. Sci. USA 2022, 119, e2110085119. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R. How Receptors Talk to Trimeric G Proteins. Curr. Opin. Cell Biol. 1997, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Leurs, R.; Smit, M.J.; Alewijnse, A.E.; Timmerman, H. Agonist-Independent Regulation of Constitutively Active G-Protein-Coupled Receptors. Trends Biochem. Sci. 1998, 23, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Wenzel-Seifert, K. Constitutive Activity of G-Protein-Coupled Receptors: Cause of Disease and Common Property of Wild-Type Receptors. Naunyn. Schmiedebergs. Arch. Pharmacol. 2002, 366, 381–416. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Hall, D.A.; Giraldo, J. Can Adding Constitutive Receptor Activity Redefine Biased Signaling Quantification? Trends Pharmacol. Sci. 2019, 40, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Ceraudo, E.; Horioka, M.; Mattheisen, J.M.; Hitchman, T.D.; Moore, A.R.; Kazmi, M.A.; Chi, P.; Chen, Y.; Sakmar, T.P.; Huber, T. Direct Evidence That the GPCR CysLTR2 Mutant Causative of Uveal Melanoma Is Constitutively Active with Highly Biased Signaling. J. Biol. Chem. 2021, 296, 100163. [Google Scholar] [CrossRef] [PubMed]
- Nobles, M.; Benians, A.; Tinker, A. Heterotrimeric G Proteins Precouple with G Protein-Coupled Receptors in Living Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 18706–18711. [Google Scholar] [CrossRef] [PubMed]
- Galés, C.; van Durm, J.J.J.; Schaak, S.; Pontier, S.; Percherancier, Y.; Audet, M.; Paris, H.; Bouvier, M. Probing the Activation-Promoted Structural Rearrangements in Preassembled Receptor-G Protein Complexes. Nat. Struct. Mol. Biol. 2006, 13, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, M.A.; Maurel, D.; Binet, V.; Fink, M.; Prézeau, L.; Ansanay, H.; Pin, J.-P. Real-Time Analysis of Agonist-Induced Activation of Protease-Activated Receptor 1/Galphai1 Protein Complex Measured by Bioluminescence Resonance Energy Transfer in Living Cells. Mol. Pharmacol. 2007, 71, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Dong, C.; Wu, G.; Lambert, N.A. Inactive-State Preassembly of G(q)-Coupled Receptors and G(q) Heterotrimers. Nat. Chem. Biol. 2011, 7, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Kilander, M.B.C.; Petersen, J.; Andressen, K.W.; Ganji, R.S.; Levy, F.O.; Schuster, J.; Dahl, N.; Bryja, V.; Schulte, G. Disheveled Regulates Precoupling of Heterotrimeric G Proteins to Frizzled 6. FASEB J. 2014, 28, 2293–2305. [Google Scholar] [CrossRef] [PubMed]
- Andressen, K.W.; Ulsund, A.H.; Krobert, K.A.; Lohse, M.J.; Bünemann, M.; Levy, F.O. Related GPCRs Couple Differently to Gs: Preassociation between G Protein and 5-HT7 Serotonin Receptor Reveals Movement of Gαs upon Receptor Activation. FASEB J. 2018, 32, 1059–1069. [Google Scholar] [CrossRef]
- Ulsund, A.H.; Dahl, M.; Frimurer, T.M.; Manfra, O.; Schwartz, T.W.; Levy, F.O.; Andressen, K.W. Preassociation between the 5-HT7 Serotonin Receptor and G Protein Gs: Molecular Determinants and Association with Low Potency Activation of Adenylyl Cyclase. FASEB J. 2019, 33, 3870–3886. [Google Scholar] [CrossRef]
- Jang, W.; Adams, C.E.; Liu, H.; Zhang, C.; Levy, F.O.; Andressen, K.W.; Lambert, N.A. An Inactive Receptor-G Protein Complex Maintains the Dynamic Range of Agonist-Induced Signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 30755–30762. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Teng, X.; Zheng, S. Molecular Basis for the Selective G Protein Signaling of Somatostatin Receptors. Nat. Chem. Biol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-H.; Lin, J.; Ji, S.-Y.; Zhou, X.E.; Mao, C.; Shen, D.-D.; He, X.; Xiao, P.; Sun, J.; Melcher, K.; Zhang, Y.; Yu, X.; Xu, H.E. Structure Insights into Selective Coupling of G Protein Subtypes by a Class B G Protein-Coupled Receptor. Nat. Commun. 2022, 13, 6670. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; Tate, C.G. Molecular Basis of β-Arrestin Coupling to Formoterol-Bound Β1-Adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Neale, C.; Sljoka, A.; Lyda, B.; Pichugin, D.; Tsuchimura, N.; Larda, S.T.; Pomès, R.; García, A.E.; Ernst, O.P.; Sunahara, R.K.; Prosser, R.S. Mechanistic Insights into Allosteric Regulation of the A2A Adenosine G Protein-Coupled Receptor by Physiological Cations. Nat. Commun. 2018, 9, 1372. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Eddy, M.T.; Gao, Z.-G.; Han, G.W.; Lian, T.; Deary, A.; Patel, N.; Jacobson, K.A.; Katritch, V.; Stevens, R.C. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 2018, 26, 259–269.e5. [Google Scholar] [CrossRef] [PubMed]
- Massink, A.; Gutiérrez-de-Terán, H.; Lenselink, E.B.; Ortiz Zacarías, N.V.; Xia, L.; Heitman, L.H.; Katritch, V.; Stevens, R.C.; IJzerman, A.P. Sodium Ion Binding Pocket Mutations and Adenosine A2A Receptor Function. Mol. Pharmacol. 2015, 87, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.-P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular Control of δ-Opioid Receptor Signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Livingston, K.E.; Traynor, J.R. Disruption of the Na+ Ion Binding Site as a Mechanism for Positive Allosteric Modulation of the Mu-Opioid Receptor. Proc. Natl. Acad. Sci. USA 2014, 111, 18369–18374. [Google Scholar] [CrossRef]
- Sato, S.; Huang, X.-P.; Kroeze, W.K.; Roth, B.L. Discovery and Characterization of Novel GPR39 Agonists Allosterically Modulated by Zinc. Mol. Pharmacol. 2016, 90, 726–737. [Google Scholar] [CrossRef]
- Birnbaumer, L.; Zurita, A.R. On the Roles of Mg in the Activation of G Proteins. J. Recept. Signal. Transduct. Res. 2010, 30, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Levitt, E.S.; Clark, M.J.; Jenkins, P.M.; Martens, J.R.; Traynor, J.R. Differential Effect of Membrane Cholesterol Removal on Mu- and Delta-Opioid Receptors: A Parallel Comparison of Acute and Chronic Signaling to Adenylyl Cyclase. J. Biol. Chem. 2009, 284, 22108–22122. [Google Scholar] [CrossRef] [PubMed]
- Potter, R.M.; Harikumar, K.G.; Wu, S.V.; Miller, L.J. Differential Sensitivity of Types 1 and 2 Cholecystokinin Receptors to Membrane Cholesterol. J. Lipid Res. 2012, 53, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.-P.; Chien, E.Y.T.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A Specific Cholesterol Binding Site Is Established by the 2.8 A Structure of the Human Beta2-Adrenergic Receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [PubMed]
- van Aalst, E.; Konieleeri, J.; Wylie, B.J. In Silico Identification of Cholesterol Binding Motifs in the Chemokine Receptor CCR3. Membranes (Basel) 2021, 11, 570. [Google Scholar] [CrossRef] [PubMed]
- McGraw, C.; Koretz, K.S.; Oseid, D.; Lyman, E.; Robinson, A.S. Cholesterol Dependent Activity of the Adenosine A2A Receptor Is Modulated via the Cholesterol Consensus Motif. Molecules 2022, 27, 3529. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Huang, S.; Zhang, H.; Mao, C.; Zhou, X.E.; Cheng, X.; Simon, I.A.; Shen, D.-D.; Yen, H.-Y.; Robinson, C.V.; Harpsøe, K.; Svensson, B.; Guo, J.; Jiang, H.; Gloriam, D.E.; Melcher, K.; Jiang, Y.; Zhang, Y.; Xu, H.E. Structural Insights into the Lipid and Ligand Regulation of Serotonin Receptors. Nature 2021, 592, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Soubias, O.; Gawrisch, K. The Role of the Lipid Matrix for Structure and Function of the GPCR Rhodopsin. Biochim. Biophys. Acta 2012, 1818, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.H.; Murray, F.; Insel, P.A. G-Protein-Coupled Receptor-Signaling Components in Membrane Raft and Caveolae Microdomains. Handb. Exp. Pharmacol. 2008, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Żuk, J.; Bartuzi, D.; Miszta, P.; Kaczor, A.A. The Role of Lipids in Allosteric Modulation of Dopamine D2 Receptor-In Silico Study. Molecules 2022, 27, 1335. [Google Scholar] [CrossRef] [PubMed]
- Hanyaloglu, A.C. Advances in Membrane Trafficking and Endosomal Signaling of G Protein-Coupled Receptors. Int. Rev. Cell Mol. Biol. 2018, 339, 93–131. [Google Scholar] [CrossRef] [PubMed]
- Crilly, S.E.; Puthenveedu, M.A. Compartmentalized GPCR Signaling from Intracellular Membranes. J. Membr. Biol. 2021, 254, 259–271. [Google Scholar] [CrossRef] [PubMed]
- David, D.; Bentulila, Z.; Tauber, M.; Ben-Chaim, Y. G Protein-Coupled Receptors Regulated by Membrane Potential. Int. J. Mol. Sci. 2022, 23, 13988. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, A.; Marti-Solano, M.; Drabek, M.; Bünemann, M.; Kolb, P.; Rinne, A. The Allosteric Site Regulates the Voltage Sensitivity of Muscarinic Receptors. Cell Signal. 2018, 42, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Dekel, N.; Priest, M.F.; Parnas, H.; Parnas, I.; Bezanilla, F. Depolarization Induces a Conformational Change in the Binding Site Region of the M2 Muscarinic Receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Vickery, O.N.; Machtens, J.-P.; Tamburrino, G.; Seeliger, D.; Zachariae, U. Structural Mechanisms of Voltage Sensing in G Protein-Coupled Receptors. Structure 2016, 24, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Ågren, R.; Sahlholm, K.; Nilsson, J.; Århem, P. Point Mutation of a Conserved Aspartate, D69, in the Muscarinic M2 Receptor Does Not Modify Voltage-Sensitive Agonist Potency. Biochem. Biophys. Res. Commun. 2018, 496, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Tauber, M.; ben Chaim, Y. The Activity of the Serotonergic 5-HT1A Receptor Is Modulated by Voltage and Sodium Levels. J. Biol. Chem. 2022, 298, 101978. [Google Scholar] [CrossRef] [PubMed]
- Kammermeier, P.J. Functional and Pharmacological Characteristics of Metabotropic Glutamate Receptors 2/4 Heterodimers. Mol. Pharmacol. 2012, 82, 438–447. [Google Scholar] [CrossRef]
- Yin, S.; Noetzel, M.J.; Johnson, K.A.; Zamorano, R.; Jalan-Sakrikar, N.; Gregory, K.J.; Conn, P.J.; Niswender, C.M. Selective Actions of Novel Allosteric Modulators Reveal Functional Heteromers of Metabotropic Glutamate Receptors in the CNS. J. Neurosci. 2014, 34, 79–94. [Google Scholar] [CrossRef]
- Jiang, P.; Cui, M.; Zhao, B.; Snyder, L.A.; Benard, L.M.J.; Osman, R.; Max, M.; Margolskee, R.F. Identification of the Cyclamate Interaction Site within the Transmembrane Domain of the Human Sweet Taste Receptor Subunit T1R3. J. Biol. Chem. 2005, 280, 34296–34305. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Jiang, P.; Maillet, E.; Max, M.; Margolskee, R.F.; Osman, R. The Heterodimeric Sweet Taste Receptor Has Multiple Potential Ligand Binding Sites. Curr. Pharm. Des. 2006, 12, 4591–4600. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, D. Keeping the Balance: GABAB Receptors in the Developing Brain and Beyond. Brain Sci. 2022, 12, 419. [Google Scholar] [CrossRef] [PubMed]
- Binet, V.; Brajon, C.; le Corre, L.; Acher, F.; Pin, J.-P.; Prézeau, L. The Heptahelical Domain of GABA(B2) Is Activated Directly by CGP7930, a Positive Allosteric Modulator of the GABA(B) Receptor. J. Biol. Chem. 2004, 279, 29085–29091. [Google Scholar] [CrossRef] [PubMed]
- Porcu, A.; Mostallino, R.; Serra, V.; Melis, M.; Sogos, V.; Beggiato, S.; Ferraro, L.; Manetti, F.; Gianibbi, B.; Bettler, B.; Corelli, F.; Mugnaini, C.; Castelli, M.P. COR758, a Negative Allosteric Modulator of GABAB Receptors. Neuropharmacology 2021, 189, 108537. [Google Scholar] [CrossRef] [PubMed]
- Nieto, A.; Bailey, T.; Kaczanowska, K.; McDonald, P. GABAB Receptor Chemistry and Pharmacology: Agonists, Antagonists, and Allosteric Modulators. Curr. Top. Behav. Neurosci. 2022, 52, 81–118. [Google Scholar] [CrossRef] [PubMed]
- Koehl, A.; Hu, H.; Feng, D.; Sun, B.; Zhang, Y.; Robertson, M.J.; Chu, M.; Kobilka, T.S.; Laeremans, T.; Steyaert, J.; Tarrasch, J.; Dutta, S.; Fonseca, R.; Weis, W.I.; Mathiesen, J.M.; Skiniotis, G.; Kobilka, B.K. Structural Insights into the Activation of Metabotropic Glutamate Receptors. Nature 2019, 566, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Mosyak, L.; Kurinov, I.; Zuo, H.; Sturchler, E.; Cheng, T.C.; Subramanyam, P.; Brown, A.P.; Brennan, S.C.; Mun, H.-C.; Bush, M.; Chen, Y.; Nguyen, T.X.; Cao, B.; Chang, D.D.; Quick, M.; Conigrave, A.D.; Colecraft, H.M.; McDonald, P.; Fan, Q.R. Structural Mechanism of Ligand Activation in Human Calcium-Sensing Receptor. Elife 2016, 5, e13662. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, D.; Fan, H.; Xu, C.; Tai, L.; Lin, S.; Han, S.; Tan, Q.; Wang, X.; Xu, T.; Zhang, H.; Chu, X.; Yi, C.; Liu, P.; Wang, X.; Zhou, Y.; Pin, J.P.; Rondard, P.; Liu, H.; Liu, J.; Sun, F.; Wu, B.; Zhao, Q. Structures of Human MGlu2 and MGlu7 Homo- and Heterodimers. Nature 2021, 594, 589–593. [Google Scholar] [CrossRef]
- Papasergi-Scott, M.M.; Robertson, M.J.; Seven, A.B.; Panova, O.; Mathiesen, J.M.; Skiniotis, G. Structures of Metabotropic GABAB Receptor. Nature 2020, 584, 310–314. [Google Scholar] [CrossRef]
- Shaye, H.; Ishchenko, A.; Lam, J.H.; Han, G.W.; Xue, L.; Rondard, P.; Pin, J.-P.; Katritch, V.; Gati, C.; Cherezov, V. Structural Basis of the Activation of a Metabotropic GABA Receptor. Nature 2020, 584, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Nickols, H.H.; Conn, P.J. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol. Dis. 2014, 61, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Jakubik, J.; El-Fakahany, E.E. Current Advances in Allosteric Modulation of Muscarinic Receptors. Biomolecules 2020, 10, 325. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Lawrence, A.J. Allosteric modulation of muscarinic receptors in alcohol and substance use disorders. Adv. Pharmacol. 2020, 88, 233–275. [Google Scholar] [CrossRef] [PubMed]
- Szczurowska, E.; Szánti-Pintér, E.; Randáková, A.; Jakubík, J.; Kudova, E. Allosteric Modulation of Muscarinic Receptors by Cholesterol, Neurosteroids and Neuroactive Steroids. Int. J. Mol. Sci. 2022, 23, 13075. [Google Scholar] [CrossRef] [PubMed]
- Gregory, K.J.; Noetzel, M.J.; Niswender, C.M. Pharmacology of metabotropic glutamate receptor allosteric modulators: structural basis and therapeutic potential for CNS disorders. Prog. Mol. Biol. Transl. Sci. 2013, 115, 61–121. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Jun, J.J.; Cuyler, J.; Xie, X.Q. Covalent allosteric modulation: An emerging strategy for GPCRs drug discovery. Eur. J. Med. Chem. 2020, 206, 112690. [Google Scholar] [CrossRef] [PubMed]
- Fasciani, I.; Petragnano, F.; Aloisi, G.; Marampon, F.; Carli, M.; Scarselli, M.; Maggio, R.; Rossi, M. Allosteric Modulators of G Protein-Coupled Dopamine and Serotonin Receptors: A New Class of Atypical Antipsychotics. Pharmaceuticals (Basel) 2020, 13, 388. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Alvi, S.; Valant, C.; Christopoulos, A.; Carbone, S.E.; Poole, D.P. Therapeutic potential of allosteric modulators for the treatment of gastrointestinal motility disorders. Br. J. Pharmacol. 2022. [CrossRef] [PubMed]
- Kaczor, A.A.; Wróbel, T.M.; Bartuzi, D. Allosteric Modulators of Dopamine D2 Receptors for Fine-Tuning of Dopaminergic Neurotransmission in CNS Diseases: Overview, Pharmacology, Structural Aspects and Synthesis. Molecules 2022, 28, 178. [Google Scholar] [CrossRef] [PubMed]
- Obeng, S.; Hiranita, T.; León, F.; McMahon, L.R.; McCurdy, C.R. Novel Approaches, Drug Candidates, and Targets in Pain Drug Discovery. J. Med. Chem. 2021, 64, 6523–6548. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R. Biased, Bitopic, Opioid-Adrenergic Tethered Compounds May Improve Specificity, Lower Dosage and Enhance Agonist or Antagonist Function with Reduced Risk of Tolerance and Addiction. Pharmaceuticals (Basel) 2022, 15, 214. [Google Scholar] [CrossRef] [PubMed]
- Dopart, R.; Lu, D.; Lichtman, A.H.; Kendall, D.A. Allosteric modulators of cannabinoid receptor 1: developing compounds for improved specificity. Drug Metab. Rev. 2018, 50, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Gado, F.; Meini, S.; Bertini, S.; Digiacomo, M.; Macchia, M.; Manera, C. Allosteric modulators targeting cannabinoid cb1 and cb2 receptors: implications for drug discovery. Future Med. Chem. 2019, 11, 2019–2037. [Google Scholar] [CrossRef] [PubMed]
- Leo, L.M.; Abood, M.E. CB1 Cannabinoid Receptor Signaling and Biased Signaling. Molecules 2021, 26, 5413. [Google Scholar] [CrossRef] [PubMed]
- Korkutata, M.; Agrawal, L.; Lazarus, M. Allosteric Modulation of Adenosine A2A Receptors as a New Therapeutic Avenue. Int. J. Mol. Sci. 2022, 23, 2101. [Google Scholar] [CrossRef] [PubMed]
- Blough, B.; Namjoshi, O. Small Molecule Neuropeptide S and Melanocortin 4 Receptor Ligands as Potential Treatments for Substance Use Disorders. Handb. Exp. Pharmacol. 2020, 258, 61–87. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.D.; Karnik, S.S. Structural perspectives on the mechanism of signal activation, ligand selectivity and allosteric modulation in angiotensin receptors: IUPHAR Review 34. Br. J. Pharmacol. 2022, 179, 4461–4472. [Google Scholar] [CrossRef] [PubMed]
- Malik, F.; Li, Z. Non-peptide agonists and positive allosteric modulators of glucagon-like peptide-1 receptors: Alternative approaches for treatment of Type 2 diabetes. Br. J. Pharmacol. 2022, 179, 511–525. [Google Scholar] [CrossRef]
- Rajagopal, S.; Bassoni, D.L.; Campbell, J.J.; Gerard, N.P.; Gerard, C.; Wehrman, T.S. Biased agonism as a mechanism for differential signaling by chemokine receptors. J. Biol. Chem. 2013, 288, 35039–35048. [Google Scholar] [CrossRef]
- Miao, M.; de Clercq, E.; Li, G. Clinical Significance of Chemokine Receptor Antagonists. Expert. Opin. Drug Metab. Toxicol. 2020, 16, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Eiger, D.S.; Boldizsar, N.; Honeycutt, C.C.; Gardner, J.; Rajagopal, S. Biased Agonism at Chemokine Receptors. Cell. Signal. 2021, 78, 109862. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Tressel, S.L.; Kaimal, R.; Balla, M.; Lam, F.H.; Covic, L.; Kuliopulos, A. Identification of a Metalloprotease-Chemokine Signaling System in the Ovarian Cancer Microenvironment: Implications for Antiangiogenic Therapy. Cancer Res. 2010, 70, 5880–5890. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T.; Nakajima, T.; Tachibana, K.; Iizasa, H.; Bleul, C.C.; Yoshie, O.; Matsushima, K.; Yoshida, N.; Springer, T.A.; Kishimoto, T. Molecular cloning and characterization of a murine pre-B-cell growth-stimulating factor/stromal cell-derived factor 1 receptor, a murine homolog of the human immunodeficiency virus 1 entry coreceptor fusin. Proc. Natl. Acad. Sci. USA 1996, 93, 14726–14729. [Google Scholar] [CrossRef] [PubMed]
- Scala, S.; Ottaiano, A.; Ascierto, P.A.; Cavalli, M.; Simeone, E.; Giuliano, P.; Napolitano, M.; Franco, R.; Botti, G.; Castello, G. Expression of CXCR4 predicts poor prognosis in patients with malignant melanoma. Clin. Cancer Res. 2005, 11, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- Konoplev, S.; Rassidakis, G.Z.; Estey, E.; Kantarjian, H.; Liakou, C.I.; Huang, X.; Xiao, L.; Andreeff, M.; Konopleva, M.; Medeiros, L.J. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer 2007, 109, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Tavor, S. Role of CXCR4 in the pathogenesis of acute myeloid leukemia. Theranostics 2013, 3, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Mishan, M.A.; Ahmadiankia, N.; Bahrami, A.R. CXCR4 and CCR7: Two Eligible Targets in Targeted Cancer Therapy. Cell Biol. Int. 2016, 40, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Wang, S.; Ni, H.; Zhao, P.; Chen, G.; Xu, B.; Yuan, L. The Role of CXCR3 and Its Ligands in Cancer. Front. Oncol. 2022, 12, 1022688. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Borsig, L.; Heikenwalder, M. CCL2-CCR2 Signaling in Disease Pathogenesis. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Moschovakis, G.L.; Bubke, A.; Friedrichsen, M.; Ristenpart, J.; Back, J.W.; Falk, C.S.; Kremmer, E.; Förster, R. The Chemokine Receptor CCR7 Is a Promising Target for Rheumatoid Arthritis Therapy. Cell Mol. Immunol. 2019, 16, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Sun, M.; Yang, Z.; Lu, C.; Wang, Q.; Wang, H.; Deng, C.; Liu, Y.; Yang, Y. The Roles of CCR9/CCL25 in Inflammation and Inflammation-Associated Diseases. Front. Cell Dev. Biol. 2021, 9, 686548. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M. Chemokine Receptor CCR5: Insights into Structure, Function, and Regulation. Cell Signal. 2004, 16, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R. The Chemokine System and Cancer. J. Pathol. 2012, 226, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; Holmes, M.C.; Gregory, P.D.; Ando, D.G.; Kalos, M.; Collman, R.G.; Binder-Scholl, G.; Plesa, G.; Hwang, W.T.; Levine, B.L.; June, C.H. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ren, H.-Y. C-C Chemokine Receptor 5 and Acute Graft-versus-Host Disease. Immun. Inflamm. Dis. 2022, 10, e687. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, M.; Cesta, M.C.; Locati, M. Allosteric Modulation of Chemoattractant Receptors. Front. Immunol. 2016, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.C.; Thiele, S.; Ulven, T.; Schwartz, T.W.; Rosenkilde, M.M. Positive versus Negative Modulation of Different Endogenous Chemokines for CC-Chemokine Receptor 1 by Small Molecule Agonists through Allosteric versus Orthosteric Binding. J. Biol. Chem. 2008, 283, 23121–23128. [Google Scholar] [CrossRef] [PubMed]
- Lagane, B.; Garcia-Perez, J.; Kellenberger, E. Modeling the Allosteric Modulation of CCR5 Function by Maraviroc. Drug Discov. Today Technol. 2013, 10, e297–305. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; Zhang, W.; Xie, X.; Yang, H.; Jiang, H.; Cherezov, V.; Liu, H.; Stevens, R.C.; Zhao, Q.; Wu, B. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef]
- Swinney, D.C.; Beavis, P.; Chuang, K.-T.; Zheng, Y.; Lee, I.; Gee, P.; Deval, J.; Rotstein, D.M.; Dioszegi, M.; Ravendran, P.; Zhang, J.; Sankuratri, S.; Kondru, R.; Vauquelin, G. A Study of the Molecular Mechanism of Binding Kinetics and Long Residence Times of Human CCR5 Receptor Small Molecule Allosteric Ligands. Br. J. Pharmacol. 2014, 171, 3364–3375. [Google Scholar] [CrossRef] [PubMed]
- Landoni, G.; Zangrillo, A.; Piersanti, G.; Scquizzato, T.; Piemonti, L. The Effect of Reparixin on Survival in Patients at High Risk for In-Hospital Mortality: A Meta-Analysis of Randomized Trials. Front. Immunol. 2022, 13, 932251. [Google Scholar] [CrossRef] [PubMed]
- Citro, A.; Valle, A.; Cantarelli, E.; Mercalli, A.; Pellegrini, S.; Liberati, D.; Daffonchio, L.; Kastsiuchenka, O.; Ruffini, P.A.; Battaglia, M.; Allegretti, M.; Piemonti, L. CXCR1/2 Inhibition Blocks and Reverses Type 1 Diabetes in Mice. Diabetes 2015, 64, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Piemonti, L.; Keymeulen, B.; Gillard, P.; Linn, T.; Bosi, E.; Rose, L.; Pozzilli, P.; Giorgino, F.; Cossu, E.; Daffonchio, L.; Goisis, G.; Ruffini, P.A.; Maurizi, A.R.; Mantelli, F.; Allegretti, M. Ladarixin, an inhibitor of the interleukin-8 receptors CXCR1 and CXCR2, in new-onset type 1 diabetes: A multicentre, randomized, double-blind, placebo-controlled trial. Diabetes Obes. Metab. 2022, 24, 1840–1849. [Google Scholar] [CrossRef] [PubMed]
- Bertini, R.; Barcelos, L.S.; Beccari, A.R.; Cavalieri, B.; Moriconi, A.; Bizzarri, C.; di Benedetto, P.; di Giacinto, C.; Gloaguen, I.; Galliera, E.; Corsi, M.M.; Russo, R.C.; Andrade, S.P.; Cesta, M.C.; Nano, G.; Aramini, A.; Cutrin, J.C.; Locati, M.; Allegretti, M.; Teixeira, M.M. Receptor Binding Mode and Pharmacological Characterization of a Potent and Selective Dual CXCR1/CXCR2 Non-Competitive Allosteric Inhibitor. Br. J. Pharmacol. 2012, 165, 436–454. [Google Scholar] [CrossRef] [PubMed]
- Bertini, R.; Allegretti, M.; Bizzarri, C.; Moriconi, A.; Locati, M.; Zampella, G.; Cervellera, M.N.; di Cioccio, V.; Cesta, M.C.; Galliera, E.; Martinez, F.O.; Di Bitondo, R.; Troiani, G.; Sabbatini, V.; D'Anniballe, G.; Anacardio, R.; Cutrin, J.C.; Cavalieri, B.; Mainiero, F.; Strippoli, R.; Villa, P.; Di Girolamo, M.; Martin, F.; Gentile, M.; Santoni, A.; Corda, D.; Poli, G.; Mantovani, A.; Ghezzi, P.; Colotta, F. Noncompetitive Allosteric Inhibitors of the Inflammatory Chemokine Receptors CXCR1 and CXCR2: Prevention of Reperfusion Injury. Proc. Natl. Acad. Sci. USA 2004, 101, 11791–11796. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Choi, J.H.; Kang, Y.J.; Park, S.Y.; Choi, H.C.; Kim, H.S. Reparixin, an Inhibitor of CXCR1 and CXCR2 Receptor Activation, Attenuates Blood Pressure and Hypertension-Related Mediators Expression in Spontaneously Hypertensive Rats. Biol. Pharm. Bull. 2011, 34, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Milanos, L.; Saleh, N.; Kling, R.C.; Kaindl, J.; Tschammer, N.; Clark, T. Identification of Two Distinct Sites for Antagonist and Biased Agonist Binding to the Human Chemokine Receptor CXCR3. Angew. Chem. Int. Ed Engl. 2016, 55, 15277–15281. [Google Scholar] [CrossRef]
- Bernat, V.; Admas, T.H.; Brox, R.; Heinemann, F.W.; Tschammer, N. Boronic Acids as Probes for Investigation of Allosteric Modulation of the Chemokine Receptor CXCR3. ACS Chem. Biol. 2014, 9, 2664–2677. [Google Scholar] [CrossRef]
- Bernat, V.; Brox, R.; Heinrich, M.R.; Auberson, Y.P.; Tschammer, N. Ligand-Biased and Probe-Dependent Modulation of Chemokine Receptor CXCR3 Signaling by Negative Allosteric Modulators. ChemMedChem. 2015, 10, 566–574. [Google Scholar] [CrossRef]
- Brox, R.; Milanos, L.; Saleh, N.; Baumeister, P.; Buschauer, A.; Hofmann, D.; Heinrich, M.R.; Clark, T.; Tschammer, N. Molecular Mechanisms of Biased and Probe-Dependent Signaling at CXC-Motif Chemokine Receptor CXCR3 Induced by Negative Allosteric Modulators. Mol. Pharmacol. 2018, 93, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.H.; Brandolini, L.; Aramini, A.; Bianchini, G.; Silva, R.L.; Zaperlon, A.C.; Verri, W.A.; Alves-Filho, J.C.; Cunha, F.Q.; Teixeira, M.M.; Allegretti, M.; Cunha, T.M. DF2755A, a Novel Non-Competitive Allosteric Inhibitor of CXCR1/2, Reduces Inflammatory and Post-Operative Pain. Pharmacol. Res. 2016, 103, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Brandolini, L.; Aramini, A.; Bianchini, G.; Ruocco, A.; Bertini, R.; Novelli, R.; Angelico, P.; Valsecchi, A.E.; Russo, R.; Castelli, V.; Cimini, A.; Allegretti, M. Inflammation-Independent Antinociceptive Effects of DF2755A, a CXCR1/2 Selective Inhibitor: A New Potential Therapeutic Treatment for Peripheral Neuropathy Associated to Non-Ulcerative Interstitial Cystitis/Bladder Pain Syndrome. Front. Pharmacol. 2022, 13, 854238. [Google Scholar] [CrossRef] [PubMed]
- Sachpatzidis, A.; Benton, B.K.; Manfredi, J.P.; Wang, H.; Hamilton, A.; Dohlman, H.G.; Lolis, E. Identification of Allosteric Peptide Agonists of CXCR4. J. Biol. Chem. 2003, 278, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Sison, E.A.R.; Magoon, D.; Li, L.; Annesley, C.E.; Rau, R.E.; Small, D.; Brown, P. Plerixafor as a Chemosensitizing Agent in Pediatric Acute Lymphoblastic Leukemia: Efficacy and Potential Mechanisms of Resistance to CXCR4 Inhibition. Oncotarget 2014, 5, 8947–8958. [Google Scholar] [CrossRef] [PubMed]
- Hitchinson, B.; Eby, J.M.; Gao, X.; Guite-Vinet, F.; Ziarek, J.J.; Abdelkarim, H.; Lee, Y.; Okamoto, Y.; Shikano, S.; Majetschak, M.; Heveker, N.; Volkman, B.F.; Tarasova, N.I.; Gaponenko, V. Biased Antagonism of CXCR4 Avoids Antagonist Tolerance. Sci. Signal. 2018, 11, eaat2214. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, C.; Benicchi, T.; Otrocka, M.; Mori, E.; Pilli, E.; Ferruzzi, P.; Valensin, S.; Diamanti, D.; Fecke, W.; Varrone, M.; Porcari, V. CXCR4 Antagonists: A Screening Strategy for Identification of Functionally Selective Ligands. J. BioMol. Screen 2014, 19, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, Y.; Meng, X.; Yang, X.; Xiang, Y. The study of targeted blocking SDF-1/CXCR4 signaling pathway with three antagonists on MMPs, type II collagen, and aggrecan levels in articular cartilage of guinea pigs. J. Orthop. Surg. Res. 2020, 15, 195. [Google Scholar] [CrossRef] [PubMed]
- Moriconi, A.; Cunha, T.M.; Souza, G.R.; Lopes, A.H.; Cunha, F.Q.; Carneiro, V.L.; Pinto, L.G.; Brandolini, L.; Aramini, A.; Bizzarri, C.; Bianchini, G.; Beccari, A.R.; Fanton, M.; Bruno, A.; Costantino, G.; Bertini, R.; Galliera, E.; Locati, M.; Ferreira, S.H.; Teixeira, M.M.; Allegretti, M. Targeting the Minor Pocket of C5aR for the Rational Design of an Oral Allosteric Inhibitor for Inflammatory and Neuropathic Pain Relief. Proc. Nat. Acad. Sci. USA 2014, 111, 16937–16942. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.E.; Toy, L.; Schmidt, M.F.; Vogt, H.; Budzinski, J.; Wiefhoff, M.F.J.; Merten, N.; Kostenis, E.; Weikert, D.; Schiedel, M. A Chemical Biology Toolbox Targeting the Intracellular Binding Site of CCR9: Fluorescent Ligands, New Drug Leads and PROTACs. Angew. Chem. Int. Ed. Engl. 2022, 61, e202116782. [Google Scholar] [CrossRef]
- Jaeger, K.; Bruenle, S.; Weinert, T.; Guba, W.; Muehle, J.; Miyazaki, T.; Weber, M.; Furrer, A.; Haenggi, N.; Tetaz, T.; Huang, C.Y.; Mattle, D.; Vonach, J.M.; Gast, A.; Kuglstatter, A.; Rudolph, M.G.; Nogly, P.; Benz, J.; Dawson, R.J.P.; Standfuss, J. Structural Basis for Allosteric Ligand Recognition in the Human CC Chemokine Receptor 7. Cell 2019, 178, 1222–1230.e10. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.P.; Yu, Y.; Chao, J.; Aki, C.; Chao, J.; Biju, P.; Girijavallabhan, V.; Rindgen, D.; Bond, R.; Mayer-Ezel, R.; Jakway, J.; Hipkin, R.W.; Fossetta, J.; Gonsiorek, W.; Bian, H.; Fan, X.; Terminelli, C.; Fine, J.; Lundell, D.; Merritt, J.R.; Rokosz, L.L.; Kaiser, B.; Li, G.; Wang, W.; Stauffer, T.; Ozgur, L.; Baldwin, J.; Taveras, A.G. Discovery of 2-Hydroxy-N,N-Dimethyl-3-{2-[[(R)-1-(5-Methylfuran-2-Yl)Propyl]Amino]-3,4-Dioxocyclobut-1-Enylamino}benzamide (SCH 527123): A Potent, Orally Bioavailable CXCR2/CXCR1 Receptor Antagonist. J. Med. Chem. 2006, 49, 7603–7606. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; Xu Q.; de Waal, P.W.; Ke, J.; Tan, M.H.; Zhang, C.; Moeller, A.; West, G.M.; Pascal, B.D.; Van Eps, N.; Caro, L.N.; Vishnivetskiy, S.A.; Lee, R.J.; Suino-Powell, K.M.; Gu, X.; Pal, K.; Ma, J.; Zhi, X; Boutet, S.; Williams, G.J.; Messerschmidt, M, Gati, C, Zatsepin, NA, Wang, D, James, D, Basu, S, Roy-Chowdhury, S, Conrad, CE, Coe, J, Liu, H.; Lisova, S.; Kupitz, C.; Grotjohann, I.; Fromme, R.; Jiang, Y.; Tan, M.; Yang, H.; Li, J.; Wang, M.; Zheng, Z.; Li, D.; Howe, N.; Zhao, Y.; Standfuss, J.; Diederichs, K.; Dong, Y.; Potter, C.S.; Carragher, B.; Caffrey, M.; Jiang, H.; Chapman, H.N.; Spence, J.C.; Fromme, P.; Weierstall, U.; Ernst, O.P.; Katritch, V.; Gurevich, V.V.; Griffin, P.R.; Hubbell, W.L.; Stevens, R.C.; Cherezov, V.; Melcher, K.; Xu, H.E. Crystal Structure of Rhodopsin Bound to Arrestin by Femtosecond X-Ray Laser. Nature 2015, 523, 561–567. [CrossRef] [PubMed]
- Kang, Y.; Kuybeda, O.; de Waal, P.W.; Mukherjee, S.; van Eps, N.; Dutka, P.; Zhou, X.E.; Bartesaghi, A.; Erramilli, S.; Morizumi, T.; Gu, X.; Yin, Y.; Liu, P.; Jiang, Y.; Meng, X.; Zhao, G.; Melcher, K.; Ernst, O.P.; Kossiakoff, A.A.; Subramaniam, S.; Xu, H.E. Cryo-EM Structure of Human Rhodopsin Bound to an Inhibitory G Protein. Nature 2018, 558, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Tchernychev, B.; Ren, Y.; Sachdev, P.; Janz, J.M.; Haggis, L.; O’Shea, A.; McBride, E.; Looby, R.; Deng, Q.; McMurry, T.; Kazmi, M.A.; Sakmar, T.P.; Hunt, S. 3rd.; Carlson, K.E. Discovery of a CXCR4 Agonist Pepducin That Mobilizes Bone Marrow Hematopoietic Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 22255–22259. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning Receptors on and off with Intracellular Pepducins: New Insights into G-Protein-Coupled Receptor Drug Development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef] [PubMed]
- Ortiz Zacarías, N.V.; Lenselink, E.B.; IJzerman, A.P.; Handel, T.M.; Heitman, L.H. Intracellular Receptor Modulation: Novel Approach to Target GPCRs. Trends Pharmacol. Sci. 2018, 39, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Quoyer, J.; Janz, J.M.; Luo, J.; Ren, Y.; Armando, S.; Lukashova, V.; Benovic, J.L.; Carlson, K.E.; Hunt, S.W.; Bouvier, M. Pepducin Targeting the C-X-C Chemokine Receptor Type 4 Acts as a Biased Agonist Favoring Activation of the Inhibitory G Protein. Proc. Natl. Acad. Sci. USA 2013, 110, E5088–E5097. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Tilley, D.G. Pepducin-Mediated G Protein-Coupled Receptor Signaling in the Cardiovascular System. J. Cardiovasc. Pharmacol. 2022, 80, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.B.; Sansom, O.J. Inhibition of CXCR2 Profoundly Suppresses Inflammation-Driven and Spontaneous Tumorigenesis. J. Clin. Invest. 2012, 122, 3127–3144. [Google Scholar] [CrossRef] [PubMed]
- Kaneider, N.C.; Agarwal, A.; Leger, A.J.; Kuliopulos, A. Reversing Systemic Inflammatory Response Syndrome with Chemokine Receptor Pepducins. Nat. Med. 2005, 11, 661–665. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, K.; Lee, L.; Nguyen, N.; Hsieh, M.-Y.; Kaneider, N.C.; Klein, A.K.; Sprague, K.; van Etten, R.A.; Kuliopulos, A.; Covic, L. Targeting CXCR4 with Cell-Penetrating Pepducins in Lymphoma and Lymphocytic Leukemia. Blood 2012, 119, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Holdfeldt, A.; Winther, M.; Gabl, M.; Dahlgren, C.; Forsman, H. Data on human neutrophil activation induced by pepducins with amino acid sequences derived from β2AR and CXCR4. Data Brief 2016, 8, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.I.; Kim, K.I.; Woo, S.K.; Kim, J.S.; Choi, K.J.; Lee, H.J.; Kim, K.S. Stromal Cell-Derived Factor 1 Protects Brain Vascular Endothelial Cells from Radiation-Induced Brain Damage. Cells 2019, 8, 1230. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, C.J.; Koglin, M.; Olson, W.C.; Marshall, F.H. Opportunities for Therapeutic Antibodies Directed at G-Protein-Coupled Receptors. Nat. Rev. Drug Discov. 2017, 16, 661. [Google Scholar] [CrossRef] [PubMed]
- Riemekasten, G.; Petersen, F.; Heidecke, H. What Makes Antibodies Against G Protein-Coupled Receptors so Special? A Novel Concept to Understand Chronic Diseases. Front. Immunol. 2020, 11, 564526. [Google Scholar] [CrossRef] [PubMed]
- Graßhoff, H.; Fourlakis, K.; Comdühr, S.; Riemekasten, G. Autoantibodies as Biomarker and Therapeutic Target in Systemic Sclerosis. Biomedicines 2022, 10, 2150. [Google Scholar] [CrossRef]
- Recke, A.; Regensburger, A.-K.; Weigold, F.; Müller, A.; Heidecke, H.; Marschner, G.; Hammers, C.M.; Ludwig, R.J.; Riemekasten, G. Autoantibodies in Serum of Systemic Scleroderma Patients: Peptide-Based Epitope Mapping Indicates Increased Binding to Cytoplasmic Domains of CXCR3. Front. Immunol. 2018, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- Weigold, F.; Günther, J.; Pfeiffenberger, M.; Cabral-Marques, O.; Siegert, E.; Dragun, D.; Philippe, A.; Regensburger, A.-K.; Recke, A.; Yu, X.; Petersen, F.; Catar, R.; Biesen, R.; Hiepe, F.; Burmester, G.R.; Heidecke, H.; Riemekasten, G. Antibodies against Chemokine Receptors CXCR3 and CXCR4 Predict Progressive Deterioration of Lung Function in Patients with Systemic Sclerosis. Arthritis Res. Ther. 2018, 20, 52. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-Activated Receptors (PARs) - Focus on Receptor-Receptor-Interactions and Their Physiological and Pathophysiological Impact. Cell Commun. Signal. 2013, 11, 86. [Google Scholar] [CrossRef]
- Nieman, M.T. Protease-Activated Receptors in Hemostasis. Blood 2016, 128, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Covic, L.; Kuliopulos, A. Protease-Activated Receptor 1 as Therapeutic Target in Breast, Lung, and Ovarian Cancer: Pepducin Approach. Int. J. Mol. Sci. 2018, 19, 2237. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Edgington-Mitchell, L.E.; Bunnett, N.W.; Schmidt, B.L. Protease-Activated Receptors in Health and Disease. Physiol. Rev. 2023, 103, 717–785. [Google Scholar] [CrossRef] [PubMed]
- Kyriazis, I.; Ellul, J.; Katsakiori, P.; Panayiotakopoulos, G.; Flordellis, C. The Multiple Layers of Signaling Selectivity at Protease-Activated Receptors. Curr. Pharm. Des. 2012, 18, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Thibeault, P.E.; LeSarge, J.C.; Arends, D.; Fernandes, M.; Chidiac, P.; Stathopulos, P.B.; Luyt, L.G.; Ramachandran, R. Molecular Basis for Activation and Biased Signaling at the Thrombin-Activated GPCR Proteinase Activated Receptor-4 (PAR4). J. Biol. Chem. 2020, 295, 2520–2540. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Covic, L.; Sevigny, L.M.; Kaneider, N.C.; Lazarides, K.; Azabdaftari, G.; Sharifi, S.; Kuliopulos, A. Targeting a Metalloprotease-PAR1 Signaling System with Cell-Penetrating Pepducins Inhibits Angiogenesis, Ascites, and Progression of Ovarian Cancer. Mol. Cancer Ther. 2008, 7, 2746–2757. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.J.; Luo, C.; O’Callaghan, K.; Hinds, P.W.; Covic, L.; Kuliopulos, A. Matrix Metalloprotease-1a Promotes Tumorigenesis and Metastasis. J. Biol. Chem. 2012, 287, 24330–24338. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 Is a Matrix Metalloprotease-1 Receptor That Promotes Invasion and Tumorigenesis of Breast Cancer Cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Gieseler, F.; Kaufmann, R.; Settmacher, U.; Lehnert, H.; Rauch, B.H. Signaling Crosstalk of TGF-β/ALK5 and PAR2/PAR1: A Complex Regulatory Network Controlling Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1568. [Google Scholar] [CrossRef] [PubMed]
- Kaneider, N.C.; Leger, A.J.; Agarwal, A.; Nguyen, N.; Perides, G.; Derian, C.; Covic, L.; Kuliopulos, A. “Role Reversal” for the Receptor PAR1 in Sepsis-Induced Vascular Damage. Nat. Immunol. 2007, 8, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- van der Poll, T.; Levi, M. Crosstalk between Inflammation and Coagulation: The Lessons of Sepsis. Curr. Vasc. Pharmacol. 2012, 10, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Asehnoune, K.; Moine, P. Protease-Activated Receptor-1: Key Player in the Sepsis Coagulation-Inflammation Crosstalk. Crit. Care 2013, 17, 119. [Google Scholar] [CrossRef] [PubMed]
- Lee-Rivera, I.; López, E.; López-Colomé, A.M. Diversification of PAR Signaling through Receptor Crosstalk. Cell Mol. Biol. Lett. 2022, 27, 77. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Trejo, J. Transactivation of the PAR1-PAR2 Heterodimer by Thrombin Elicits β-Arrestin-Mediated Endosomal Signaling. J. Biol. Chem. 2013, 288, 11203–11215. [Google Scholar] [CrossRef] [PubMed]
- Yau, M.-K.; Liu, L.; Suen, J.Y.; Lim, J.; Lohman, R.-J.; Jiang, Y.; Cotterell, A.J.; Barry, G.D.; Mak, J.Y.W.; Vesey, D.A.; Reid, R.C.; Fairlie, D.P. PAR2 Modulators Derived from GB88. ACS Med. Chem. Lett. 2016, 7, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Chandrabalan, A.; Ramachandran, R. Molecular Mechanisms Regulating Proteinase-Activated Receptors (PARs). FEBS J. 2021, 288, 2697–2726. [Google Scholar] [CrossRef] [PubMed]
- Dowal, L.; Sim, D.S.; Dilks, J.R.; Blair, P.; Beaudry, S.; Denker, B.M.; Koukos, G.; Kuliopulos, A.; Flaumenhaft, R. Identification of an Antithrombotic Allosteric Modulator That Acts through Helix 8 of PAR1. Proc. Natl. Acad. Sci. USA 2011, 108, 2951–2956. [Google Scholar] [CrossRef] [PubMed]
- Dockendorff, C.; Aisiku, O.; Verplank, L.; Dilks, J.R.; Smith, D.A.; Gunnink, S.F.; Dowal, L.; Negri, J.; Palmer, M.; Macpherson, L.; Schreiber, S.L.; Flaumenhaft, R. Discovery of 1,3-Diaminobenzenes as Selective Inhibitors of Platelet Activation at the PAR1 Receptor. ACS Med. Chem. Lett. 2012, 3, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Aisiku, O.; Peters, C.G.; de Ceunynck, K.; Ghosh, C.C.; Dilks, J.R.; Fustolo-Gunnink, S.F.; Huang, M.; Dockendorff, C.; Parikh, S.M.; Flaumenhaft, R. Parmodulins Inhibit Thrombus Formation without Inducing Endothelial Injury Caused by Vorapaxar. Blood 2015, 125, 1976–1985. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, D.M.; Majewski, M.W.; Rosas, R.; Kentala, K.; Foster, T.J.; Greve, E.; Dockendorff, C. Characterization of Protease-Activated Receptor (PAR) Ligands: Parmodulins Are Reversible Allosteric Inhibitors of PAR1-Driven Calcium Mobilization in Endothelial Cells. Bioorg Med. Chem. 2018, 26, 2514–2529. [Google Scholar] [CrossRef] [PubMed]
- de Ceunynck, K.; Peters, C.G.; Jain, A.; Higgins, S.J.; Aisiku, O.; Fitch-Tewfik, J.L.; Chaudhry, S.A.; Dockendorff, C.; Parikh, S.M.; Ingber, D.E.; Flaumenhaft, R. PAR1 Agonists Stimulate APC-like Endothelial Cytoprotection and Confer Resistance to Thromboinflammatory Injury. Proc. Natl. Acad. Sci. USA 2018, 115, E982–E991. [Google Scholar] [CrossRef] [PubMed]
- Nazir, S.; Gadi, I.; Al-Dabet, M.M.; Elwakiel, A.; Kohli, S.; Ghosh, S.; Manoharan, J.; Ranjan, S.; Bock, F.; Braun-Dullaeus, R.C.; Esmon, C.T.; Huber, T.B.; Camerer, E.; Dockendorff, C.; Griffin, J.H.; Isermann, B.; Shahzad, K. Cytoprotective Activated Protein C Averts Nlrp3 Inflammasome-Induced Ischemia-Reperfusion Injury via MTORC1 Inhibition. Blood 2017, 130, 2664–2677. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Heo, Y.; Jo, S.; Park, S.-H.; Lee, C.; Chang, J.; Jeon, D.-K.; Kim, T.G.; Han, G.; Namkung, W. Novel Positive Allosteric Modulator of Protease-Activated Receptor 1 Promotes Skin Wound Healing in Hairless Mice. Br. J. Pharmacol. 2021, 178, 3414–3427. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.K.Y.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G.; Brown, G.A.; Cooke, R.M.; Dumelin, C.E.; Doré, A.S.; Geschwindner, S.; Grebner, C.; Hermansson, N.O.; Jazayeri, A.; Johansson, P.; Leong, L.; Prihandoko, R.; Rappas, M.; Soutter, H.; Snijder, A.; Sundström, L.; Tehan, B.; Thornton, P.; Troast, D.; Wiggin, G.; Zhukov, A.; Marshall, F.H.; Dekker, N. Structural Insight into Allosteric Modulation of Protease-Activated Receptor 2. Nature 2017, 545, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ni, B.; Xi, Y.; Chu, X.; Zhang, R.; You, H. Protease-Activated Receptor 2 (PAR-2) Antagonist AZ3451 as a Novel Therapeutic Agent for Osteoarthritis. Aging 2019, 11, 12532–12545. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.J.; Sundström, L.; Geschwindner, S.; Poon, E.K.Y.; Jiang, Y.; Chen, R.; Cooke, R.; Johnstone, S.; Madin, A.; Lim, J.; Liu, Q.; Lohman, R.J.; Nordqvist, A.; Fridén-Saxin, M.; Yang, W.; Brown, D.G.; Fairlie, D.P.; Dekker, N. Protease-Activated Receptor-2 Ligands Reveal Orthosteric and Allosteric Mechanisms of Receptor Inhibition. Commun. Biol. 2020, 3, 782. [Google Scholar] [CrossRef] [PubMed]
- Majewski, M.W.; Gandhi, D.M.; Rosas, R.; Kodali, R.; Arnold, L.A.; Dockendorff, C. Design and Evaluation of Heterobivalent PAR1-PAR2 Ligands as Antagonists of Calcium Mobilization. ACS Med. Chem. Lett. 2019, 10, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yau, M.-K.; Lim, J.; Wu, K.-C.; Xu, W.; Suen, J.Y.; Fairlie, D.P. A Potent Antagonist of Protease-Activated Receptor 2 That Inhibits Multiple Signaling Functions in Human Cancer Cells. J. Pharmacol. Exp. Ther. 2018, 364, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Yau, M.-K.; Lim, J.; Liu, L.; Fairlie, D.P. Protease Activated Receptor 2 (PAR2) Modulators: A Patent Review (2010-2015). Expert. Opin. Ther. Pat 2016, 26, 471–483. [Google Scholar] [CrossRef]
- McIntosh, K.A.; Cunningham, M.R.; Bushell, T.; Plevin, R. The Development of Proteinase-Activated Receptor-2 Modulators and the Challenges Involved. Biochem. Soc. Trans. 2020, 48, 2525–2537. [Google Scholar] [CrossRef]
- Covic, L.; Misra, M.; Badar, J.; Singh, C.; Kuliopulos, A. Pepducin-Based Intervention of Thrombin-Receptor Signaling and Systemic Platelet Activation. Nat. Med. 2002, 8, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Jacques, S.L.; Badar, J.; Kaneider, N.C.; Derian, C.K.; Andrade-Gordon, P.; Covic, L.; Kuliopulos, A. Blocking the Protease-Activated Receptor 1-4 Heterodimer in Platelet-Mediated Thrombosis. Circulation 2006, 113, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Dabek, M.; Ferrier, L.; Annahazi, A.; Bézirard, V.; Polizzi, A.; Cartier, C.; Leveque, M.; Roka, R.; Wittmann, T.; Theodorou, V.; Bueno, L. Intracolonic Infusion of Fecal Supernatants from Ulcerative Colitis Patients Triggers Altered Permeability and Inflammation in Mice: Role of Cathepsin G and Protease-Activated Receptor-4. Inflamm. Bowel Dis. 2011, 17, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Canto, I.; Soh, U.J.K.; Trejo, J. Allosteric Modulation of Protease-Activated Receptor Signaling. Mini Rev. Med. Chem. 2012, 12, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Gruber, A.; Kasuda, S.; Kimmelstiel, C.; O’Callaghan, K.; Cox, D.H.; Bohm, A.; Baleja, J.D.; Covic, L.; Kuliopulos, A. Suppression of Arterial Thrombosis without Affecting Hemostatic Parameters with a Cell-Penetrating PAR1 Pepducin. Circulation 2012, 126, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Habault, J.; Poyet, J.-L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef] [PubMed]
- Michael, E.; Covic, L.; Kuliopulos, A. Lipopeptide Pepducins as Therapeutic Agents. Methods Mol. Biol. 2022, 2383, 307–333. [Google Scholar] [CrossRef] [PubMed]
- Derian, C.K.; Damiano, B.P.; Addo, M.F.; Darrow, A.L.; D’Andrea, M.R.; Nedelman, M.; Zhang, H.-C.; Maryanoff, B.E.; Andrade-Gordon, P. Blockade of the Thrombin Receptor Protease-Activated Receptor-1 with a Small-Molecule Antagonist Prevents Thrombus Formation and Vascular Occlusion in Nonhuman Primates. J. Pharmacol. Exp Ther. 2003, 304, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Covic, L.; Gresser, A.L.; Talavera, J.; Swift, S.; Kuliopulos, A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc. Natl. Acad. Sci. USA 2002, 99, 643–648. [Google Scholar] [CrossRef]
- Abdel-Latif, A.; Smyth, S.S. Preventing Platelet Thrombosis with a PAR1 Pepducin. Circulation 2012, 126, 13–15. [Google Scholar] [CrossRef]
- Kubo, S.; Ishiki, T.; Doe, I.; Sekiguchi, F.; Nishikawa, H.; Kawai, K.; Matsui, H.; Kawabata, A. Distinct Activity of Peptide Mimetic Intracellular Ligands (Pepducins) for Proteinase-Activated Receptor-1 in Multiple Cells/Tissues. Ann. N. Y. Acad. Sci. 2006, 1091, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Bliden, K.P.; Turner, S.E.; Tantry, U.S.; Gesheff, M.G.; Barr, T.P.; Covic, L.; Kuliopulos, A. Cell-Penetrating Pepducin Therapy Targeting PAR1 in Subjects With Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Bergmeier, W.; Mackman, N. Platelet Signaling Pathways and New Inhibitors. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e28–e35. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Boire, A.; Agarwal, A.; Nguyen, N.; O’Callaghan, K.; Tu, P.; Kuliopulos, A.; Covic, L. Blockade of PAR1 Signaling with Cell-Penetrating Pepducins Inhibits Akt Survival Pathways in Breast Cancer Cells and Suppresses Tumor Survival and Metastasis. Cancer Res. 2009, 69, 6223–6231. [Google Scholar] [CrossRef] [PubMed]
- Cisowski, J.; O’Callaghan, K.; Kuliopulos, A.; Yang, J.; Nguyen, N.; Deng, Q.; Yang, E.; Fogel, M.; Tressel, S.; Foley, C.; Agarwal, A.; Hunt, S.W. 3rd.; McMurry, T.; Brinckerhoff, L.; Covic, L. Targeting Protease-Activated Receptor-1 with Cell-Penetrating Pepducins in Lung Cancer. Am. J. Pathol. 2011, 179, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, L.M.; Zhang, P.; Bohm, A.; Lazarides, K.; Perides, G.; Covic, L.; Kuliopulos, A. Interdicting Protease-Activated Receptor-2-Driven Inflammation with Cell-Penetrating Pepducins. Proc. Natl. Acad. Sci. USA 2011, 108, 8491–8496. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, R.; Hascher, A.; Mussbach, F.; Henklein, P.; Katenkamp, K.; Westermann, M.; Settmacher, U. Proteinase-Activated Receptor 2 (PAR(2)) in Cholangiocarcinoma (CCA) Cells: Effects on Signaling and Cellular Level. HistoChem. Cell Biol. 2012, 138, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.M.; Rana, R.; Austin, K.; Baleja, J.D.; Nguyen, N.; Bohm, A.; Covic, L.; Kuliopulos, A. Targeting Liver Fibrosis with a Cell-Penetrating Protease-Activated Receptor-2 (PAR2) Pepducin. J. Biol. Chem. 2016, 291, 23188–23198. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.P.; Garzia, C.; Guha, S.; Fletcher, E.K.; Nguyen, N.; Wieschhaus, A.J.; Ferrer, L.; Covic, L.; Kuliopulos, A. PAR2 Pepducin-Based Suppression of Inflammation and Itch in Atopic Dermatitis Models. J. Invest. Dermatol. 2019, 139, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Holdfeldt, A.; Lind, S.; Hesse, C.; Dahlgren, C.; Forsman, H. The PAR4-Derived Pepducin P4Pal10 Lacks Effect on Neutrophil GPCRs That Couple to Gαq for Signaling but Distinctly Modulates Function of the Gαi-Coupled FPR2 and FFAR2. Biochem. Pharmacol. 2020, 180, 114143. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.; Koziol-White, C.; Zhang, J.; Lam, H.; An, S.S.; Tall, G.G.; Panettieri, R.A.; Benovic, J.L. Interdicting Gq Activation in Airway Disease by Receptor-Dependent and Receptor-Independent Mechanisms. Mol. Pharmacol. 2016, 89, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Kreienbring, K.; Franz, A.; Richter, R.; Dragun, D.; Heidecke, H.; Müller, D.; Mentze, M.; Dechend, R.; Sehouli, J.; Braicu, E.I. The Role of PAR1 Autoantibodies in Patients with Primary Epithelial Ovarian Cancer. Anticancer Res. 2018, 38, 3619–3625. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Lücht, C.; Hosp, I.; Zhao, H.; Wu, D.; Heidecke, H.; Witowski, J.; Budde, K.; Riemekasten, G.; Catar, R. Autoantibodies from Patients with Scleroderma Renal Crisis Promote PAR-1 Receptor Activation and IL-6 Production in Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 11793. [Google Scholar] [CrossRef] [PubMed]
- Tran, F.; Harris, D.M.M.; Scharmacher, A.; Graßhoff, H.; Sterner, K.; Schinke, S.; Käding, N.; Humrich, J.Y.; Cabral-Marques, O.; Bernardes, J.P.; Mishra, N.; Bahmer, T.; Franzenburg, J.; Hoyer, B.F.; Glück, A.; Guggeis, M.; Ossysek, A.; Küller, A.; Frank, D.; Lange, C.; Rupp, J.; Heyckendorf, J.; Gaede, K.I.; Amital, H.; Rosenstiel, P.; Shoenfeld, Y.; Halpert, G.; Rosenberg, A.Z.; Schulze-Forster, K.; Heidecke, H.; Riemekasten, G.; Schreiber, S. Increased Protease-Activated Receptor 1 Autoantibodies Are Associated with Severe COVID-19. ERJ. Open Res. 2022, 8. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Vassart, G.; Pardo, L.; Costagliola, S. A Molecular Dissection of the Glycoprotein Hormone Receptors. Trends Biochem. Sci. 2004, 29, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Bahn, R.S.; Dutton, C.M.; Natt, N.; Joba, W.; Spitzweg, C.; Heufelder, A.E. Thyrotropin Receptor Expression in Graves’ Orbital Adipose/Connective Tissues: Potential Autoantigen in Graves’ Ophthalmopathy. J. Clin. Endocrinol. Metab. 1998, 83, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Latif, R.; Michalek, K.; Davies, T.F. Subunit Interactions Influence TSHR Multimerization. Mol. Endocrinol. 2010, 24, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Neumann, S.; Gershengorn, M.C. Occupancy of Both Sites on the Thyrotropin (TSH) Receptor Dimer Is Necessary for Phosphoinositide Signaling. FASEB J. 2011, 25, 3687–3694. [Google Scholar] [CrossRef]
- Mezei, M.; Latif, R.; Davies, T.F. Modeling TSH Receptor Dimerization at the Transmembrane Domain. Endocrinology 2022, 163, bqac168. [Google Scholar] [CrossRef]
- Kleinau, G.; Worth, C.L.; Kreuchwig, A.; Biebermann, H.; Marcinkowski, P.; Scheerer, P.; Krause, G. Structural-Functional FeatuRes. of the Thyrotropin Receptor: A Class A G-Protein-Coupled Receptor at Work. Front. Endocrinol. (Lausanne) 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Kleinau, G.; Krause, G. Thyrotropin and Homologous Glycoprotein Hormone Receptors: Structural and Functional Aspects of Extracellular Signaling Mechanisms. Endocr. Rev. 2009, 30, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Kleinau, G.; Neumann, S.; Grüters, A.; Krude, H.; Biebermann, H. Novel Insights on Thyroid-Stimulating Hormone Receptor Signal. Transduction. Endocr. Rev. 2013, 34, 691–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.L.; Sugawa, H.; Kosugi, S.; Mori, T. Constitutive Activation of the Thyrotropin Receptor by Deletion of a Portion of the Extracellular Domain. Biochem. Biophys. Res. Commun. 1995, 211, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Vlaeminck-Guillem, V.; Ho, S.-C.; Rodien, P.; Vassart, G.; Costagliola, S. Activation of the CAMP Pathway by the TSH Receptor Involves Switching of the Ectodomain from a Tethered Inverse Agonist to an Agonist. Mol. Endocrinol. 2002, 16, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-R.; Salazar, L.M.; McLachlan, S.M.; Rapoport, B. The Thyrotropin Receptor Hinge Region as a Surrogate Ligand: Identification of Loci Contributing to the Coupling of Thyrotropin Binding and Receptor Activation. Endocrinology 2012, 153, 5058–5067. [Google Scholar] [CrossRef] [PubMed]
- Brüser, A.; Schulz, A.; Rothemund, S.; Ricken, A.; Calebiro, D.; Kleinau, G.; Schöneberg, T. The Activation Mechanism of Glycoprotein Hormone Receptors with Implications in the Cause and Therapy of Endocrine Diseases. J. Biol. Chem. 2016, 291, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Schöneberg, T.; Kleinau, G.; Brüser, A. What Are They Waiting for? Tethered Agonism in G Protein-Coupled Receptors. Pharmacol. Res. 2016, 108, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kleinau, G.; Jäschke, H.; Neumann, S.; Lättig, J.; Paschke, R.; Krause, G. Identification of a Novel Epitope in the Thyroid-Stimulating Hormone Receptor Ectodomain Acting as Intramolecular Signaling Interface. J. Biol. Chem. 2004, 279, 51590–51600. [Google Scholar] [CrossRef]
- Mueller, S.; Kleinau, G.; Jaeschke, H.; Neumann, S.; Krause, G.; Paschke, R. Significance of Ectodomain Cysteine Boxes 2 and 3 for the Activation Mechanism of the Thyroid-Stimulating Hormone Receptor. J. Biol. Chem. 2006, 281, 31638–31646. [Google Scholar] [CrossRef] [PubMed]
- Schihada, H.; Klompstra, T.M.; Humphrys, L.J.; Cervenka, I.; Dadvar, S.; Kolb, P.; Ruas, J.L.; Schulte, G. Isoforms of GPR35 have distinct extracellular N-termini that allosterically modify receptor-transducer coupling and mediate intracellular pathway bias. J. Biol. Chem. 2022, 298, 102328. [Google Scholar] [CrossRef] [PubMed]
- Mezei, M.; Latif, R.; Davies, T.F. Computational Model of the Full-Length TSH Receptor. Elife 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Mezei, M.; Latif, R.; Das, B.; Davies, T.F. Implications of an Improved Model of the TSH Receptor Transmembrane Domain (TSHR-TMD-TRIO). Endocrinology 2021, 162, bqab051. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.-K.; Kleinau, G.; Hoyer, I.; Neumann, S.; Furkert, J.; Rutz, C.; Schülein, R.; Gershengorn, M.C.; Krause, G. Mutations That Silence Constitutive Signaling Activity in the Allosteric Ligand-Binding Site of the Thyrotropin Receptor. Cell Mol. Life Sci. 2011, 68, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Gershengorn, M.C. Small Molecule TSHR Agonists and Antagonists. Ann. Endocrinol. (Paris) 2011, 72, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Kleinau, G.; Haas, A.-K.; Neumann, S.; Worth, C.L.; Hoyer, I.; Furkert, J.; Rutz, C.; Gershengorn, M.C.; Schülein, R.; Krause, G. Signaling-Sensitive Amino Acids Surround the Allosteric Ligand Binding Site of the Thyrotropin Receptor. FASEB J. 2010, 24, 2347–2354. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Nir, E.A.; Eliseeva, E.; Huang, W.; Marugan, J.; Xiao, J.; Dulcey, A.E.; Gershengorn, M.C. A Selective TSH Receptor Antagonist Inhibits Stimulation of Thyroid Function in Female Mice. Endocrinology 2014, 155, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Eliseeva, E.; Boutin, A.; Barnaeva, E.; Ferrer, M.; Southall, N.; Kim, D.; Hu, X.; Morgan, S.J.; Marugan, J.J.; Gershengorn, M.C. Discovery of a Positive Allosteric Modulator of the Thyrotropin Receptor: Potentiation of Thyrotropin-Mediated Preosteoblast Differentiation In Vitro. J. Pharmacol. Exp. Ther. 2018, 364, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Latif, R.; Lau, Z.; Cheung, P.; Felsenfeld, D.P.; Davies, T.F. The “TSH Receptor Glo Assay” - A High-Throughput Detection System for Thyroid Stimulation. Front. Endocrinol. (Lausanne) 2016, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Latif, R.; Realubit, R.B.; Karan, C.; Mezei, M.; Davies, T.F. TSH Receptor Signaling Abrogation by a Novel Small Molecule. Front. Endocrinol. (Lausanne) 2016, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Shpakov, A.O.; Derkach, K.V. The New Pharmacological Approaches for the Regulation of Functional Activity of G Protein-Coupled Receptors. In Evolutionary Physiology and Biochemistry - Advances and Perspectives, InTech: Rijeka, Croatia, 2018. [CrossRef]
- Marcinkowski, P.; Hoyer, I.; Specker, E.; Furkert, J.; Rutz, C.; Neuenschwander, M.; Sobottka, S.; Sun, H.; Nazare, M.; Berchner-Pfannschmidt, U.; von Kries, J.P.; Eckstein, A.; Schülein, R.; Krause, G. A New Highly Thyrotropin Receptor-Selective Small-Molecule Antagonist with Potential for the Treatment of Graves’ Orbitopathy. Thyroid 2019, 29, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Shpakov, A.O. Endogenous and Synthetic Regulators of the Peripheral Components of the Hypothalamo-Hypophyseal-Gonadal and -Thyroid Axes. Neurosci. Behav. Physiol. 2021, 51, 332–345. [Google Scholar] [CrossRef]
- Shpakov, A. Allosteric Modulators of G Protein-Coupled Receptors. Int. J. Mol. Sci. 2022, 23, 2934. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, Y.; Nishihara, E. Thyrotropin Receptor Antagonists and Inverse Agonists, and Their Potential Application to Thyroid Diseases. Endocr. J. 2022, 69, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- van Straten, N.C.R.; Schoonus-Gerritsma, G.G.; van Someren, R.G.; Draaijer, J.; Adang, A.E.P.; Timmers, C.M.; Hanssen, R.G.J.M.; van Boeckel, C.A.A. The First Orally Active Low Molecular Weight Agonists for the LH Receptor: Thienopyr(Im)Idines with Therapeutic Potential for Ovulation Induction. ChemBioChem. 2002, 3, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Jaeschke, H.; Kleinau, G.; Neumann, S.; Costanzi, S.; Jiang, J.; Childress, J.; Raaka, B.M.; Colson, A.; Paschke, R.; Krause, G.; Thomas, C.J.; Gershengorn, M.C. Evaluation of Small-Molecule Modulators of the Luteinizing Hormone/Choriogonadotropin and Thyroid Stimulating Hormone Receptors: Structure-Activity Relationships and Selective Binding Patterns. J. Med. Chem. 2006, 49, 3888–3896. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Neumann, S.; Kleinau, G.; Mueller, S.; Claus, M.; Krause, G.; Paschke, R. An Aromatic Environment in the Vicinity of Serine 281 Is a Structural Requirement for Thyrotropin Receptor Function. Endocrinology 2006, 147, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Neumann, S.; Moore, S.; Thomas, C.J.; Colson, A.-O.; Costanzi, S.; Kleinau, G.; Jiang, J.-K.; Paschke, R.; Raaka, B.M.; Krause, G.; Gershengorn, M.C. A Low Molecular Weight Agonist Signals by Binding to the Transmembrane Domain of Thyroid-Stimulating Hormone Receptor (TSHR) and Luteinizing Hormone/Chorionic Gonadotropin Receptor (LHCGR). J. Biol. Chem. 2006, 281, 9841–9844. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, I.; Haas, A.-K.; Kreuchwig, A.; Schülein, R.; Krause, G. Molecular Sampling of the Allosteric Binding Pocket of the TSH Receptor Provides Discriminative PharmacophoRes. for Antagonist and Agonists. Biochem. Soc. Trans. 2013, 41, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Heitman, L.H.; Ijzerman, A.P. G Protein-Coupled Receptors of the Hypothalamic-Pituitary-Gonadal Axis: A Case for Gnrh, LH, FSH, and GPR54 Receptor Ligands. Med. Res. Rev. 2008, 28, 975–1011. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; IJzerman, A.P. Allosteric Approaches to GPCR Drug Discovery. Drug Discov. Today Technol. 2013, 10, e219–e221. [Google Scholar] [CrossRef] [PubMed]
- Nataraja, S.G.; Yu, H.N.; Palmer, S.S. Discovery and Development of Small Molecule Allosteric Modulators of Glycoprotein Hormone Receptors. Front. Endocrinol. (Lausanne) 2015, 6, 142. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Padia, U.; Cullen, M.J.; Eliseeva, E.; Nir, E.A.; Place, R.F.; Morgan, S.J.; Gershengorn, M.C. An Enantiomer of an Oral Small-Molecule TSH Receptor Agonist Exhibits Improved Pharmacologic Properties. Front. Endocrinol. (Lausanne) 2016, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Huang, W.; Titus, S.; Krause, G.; Kleinau, G.; Alberobello, A.T.; Zheng, W.; Southall, N.T.; Inglese, J.; Austin, C.P.; Celi, F.S.; Gavrilova, O.; Thomas, C.J.; Raaka, B.M.; Gershengorn, M.C. Small-Molecule Agonists for the Thyrotropin Receptor Stimulate Thyroid Function in Human Thyrocytes and Mice. Proc. Natl. Acad. Sci. USA 2009, 106, 12471–12476. [Google Scholar] [CrossRef] [PubMed]
- Bakhtyukov, A.A.; Derkach, K. v; Fokina, E.A.; Sorokoumov, V.N.; Zakharova, I.O.; Bayunova, L. v; Shpakov, A.O. Development of Low-Molecular-Weight Allosteric Agonist of Thyroid-Stimulating Hormone Receptor with Thyroidogenic Activity. Dokl. Biochem. Biophys 2022, 503, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Neumann, S.; Gershengorn, M.C. Small-Molecule Thyrotropin Receptor Agonist Activates Naturally Occurring Thyrotropin-Insensitive Mutants and Reveals Their Distinct Cyclic Adenosine Monophosphate Signal. Persistence. Thyroid 2011, 21, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Latif, R.; Ali, M.R.; Ma, R.; David, M.; Morshed, S.A.; Ohlmeyer, M.; Felsenfeld, D.P.; Lau, Z.; Mezei, M.; Davies, T.F. New Small Molecule Agonists to the Thyrotropin Receptor. Thyroid 2015, 25, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Latif, R.; Morshed, S.A.; Ma, R.; Tokat, B.; Mezei, M.; Davies, T.F. A Gq Biased Small Molecule Active at the TSH Receptor. Front. Endocrinol. (Lausanne) 2020, 11, 372. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Eckstein, A.; Schülein, R. Modulating TSH Receptor Signaling for Therapeutic Benefit. Eur. Thyroid J. 2020, 9, 66–77. [Google Scholar] [CrossRef]
- Kim, S.-M.; Ryu, V.; Miyashita, S.; Korkmaz, F.; Lizneva, D.; Gera, S.; Latif, R.; Davies, T.F.; Iqbal, J.; Yuen, T.; Zaidi, M. Thyrotropin, Hyperthyroidism, and Bone Mass. J. Clin. Endocrinol. Metab. 2021, 106, e4809–e4821. [Google Scholar] [CrossRef]
- Boutin, A.; Neumann, S.; Gershengorn, M.C. Multiple Transduction Pathways Mediate Thyrotropin Receptor Signaling in Preosteoblast-Like Cells. Endocrinology 2016, 157, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Kleinau, G.; Costanzi, S.; Moore, S.; Jiang, J.; Raaka, B.M.; Thomas, C.J.; Krause, G.; Gershengorn, M.C. A Low-Molecular-Weight Antagonist for the Human Thyrotropin Receptor with Therapeutic Potential for Hyperthyroidism. Endocrinology 2008, 149, 5945–5950. [Google Scholar] [CrossRef]
- Turcu, A.F.; Kumar, S.; Neumann, S.; Coenen, M.; Iyer, S.; Chiriboga, P.; Gershengorn, M.C.; Bahn, R.S. A Small Molecule Antagonist Inhibits Thyrotropin Receptor Antibody-Induced Orbital Fibroblast Functions Involved in the Pathogenesis of Graves Ophthalmopathy. J. Clin. Endocrinol. Metab. 2013, 98, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Huang, W.; Eliseeva, E.; Titus, S.; Thomas, C.J.; Gershengorn, M.C. A Small Molecule Inverse Agonist for the Human Thyroid-Stimulating Hormone Receptor. Endocrinology 2010, 151, 3454–3459. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Eliseeva, E.; McCoy, J.G.; Napolitano, G.; Giuliani, C.; Monaco, F.; Huang, W.; Gershengorn, M.C. A New Small-Molecule Antagonist Inhibits Graves’ Disease Antibody Activation of the TSH Receptor. J. Clin. Endocrinol. Metab. 2011, 96, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Pope, A.; Geras-Raaka, E.; Raaka, B.M.; Bahn, R.S.; Gershengorn, M.C. A Drug-like Antagonist Inhibits Thyrotropin Receptor-Mediated Stimulation of CAMP Production in Graves’ Orbital Fibroblasts. Thyroid 2012, 22, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Dik, W.A.; Virakul, S.; van Steensel, L. Current Perspectives on the Role of Orbital Fibroblasts in the Pathogenesis of Graves’ Ophthalmopathy. Exp. Eye Res. 2016, 142, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Derkach, K.V; Bakhtyukov, A.A.; Sorokoumov, V.N.; Shpakov, A.O. New Thieno-[2,3-d]Pyrimidine-Based Functional Antagonist for the Receptor of Thyroid Stimulating Hormone. Dokl. Biochem. Biophys. 2020, 491, 77–80. [Google Scholar] [CrossRef]
- Derkach, K.V; Fokina, E.A.; Bakhtyukov, A.A.; Sorokoumov, V.N.; Stepochkina, A.M.; Zakharova, I.O.; Shpakov, A.O. The Study of Biological Activity of a New Thieno[2,3-D]-Pyrimidine-Based Neutral Antagonist of Thyrotropin Receptor. Bull. Exp. Biol. Med. 2022, 172, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Barbesino, G.; Salvi, M.; Freitag, S.K. Future Projections in Thyroid Eye Disease. J. Clin. Endocrinol. Metab. 2022, 107, S47–S56. [Google Scholar] [CrossRef]
- He, Q.; Dong, H.; Gong, M.; Guo, Y.; Xia, Q.; Gong, J.; Lu, F. New Therapeutic Horizon of Graves’ Hyperthyroidism: Treatment Regimens Based on Immunology and Ingredients From Traditional Chinese Medicine. Front. Pharmacol. 2022, 13, 862831. [Google Scholar] [CrossRef] [PubMed]
- Shpakova, E.A.; Shpakov, A.O.; Chistyakova, O.V.; Moyseyuk, I.V.; Derkach, K.V. Biological Activity in Vitro and in Vivo of Peptides Corresponding to the Third Intracellular Loop of Thyrotropin Receptor. Dokl. Biochem. Biophys. 2012, 443, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Derkach, K.V; Shpakova, E.A.; Titov, A.K.; Shpakov, A.O. Intranasal and Intramuscular Administration of Lysine-Palmitoylated Peptide 612–627 of Thyroid-Stimulating Hormone Receptor Increases the Level of Thyroid Hormones in Rats. Int. J. Pept Res. Ther. 2015, 21, 249–260. [Google Scholar] [CrossRef]
- Tomer, Y.; Huber, A. The Etiology of Autoimmune Thyroid Disease: A Story of Genes and Environment. J. AutoImmun. 2009, 32, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, J.; von Westarp, C. Clinical Applications of Assays for Thyrotropin-Receptor Antibodies in Graves’ Disease. CMAJ. 1986, 134, 1141–1147. [Google Scholar] [PubMed]
- López Ortega, J.M.; Martínez, P.S.; Acevedo-León, D.; Capell, N.E. Anti-TSH Receptor Antibodies (TRAb): Comparison of Two Third Generation Automated Immunoassays Broadly Used in Clinical Laboratories and Results Interpretation. PLoS One 2022, 17, e0270890. [Google Scholar] [CrossRef] [PubMed]
- Wallaschofski, H.; Kuwert, T.; Lohmann, T. TSH-Receptor Autoantibodies - Differentiation of Hyperthyroidism between Graves’ Disease and Toxic Multinodular Goitre. Exp. Clin. Endocrinol. Diab. 2004, 112, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Scappaticcio, L.; Trimboli, P.; Keller, F.; Imperiali, M.; Piccardo, A.; Giovanella, L. Diagnostic Testing for Graves’ or Non-Graves’ Hyperthyroidism: A Comparison of Two Thyrotropin Receptor Antibody Immunoassays with Thyroid Scintigraphy and Ultrasonography. Clin. Endocrinol. (Oxf.) 2020, 92, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.; Miguel, R.N.; Furmaniak, J.; Smith, B.R. TSH Receptor Monoclonal Antibodies with Agonist, Antagonist, and Inverse Agonist Activities. Methods EnzyMol. 2010, 485, 393–420. [Google Scholar] [CrossRef] [PubMed]
- Furmaniak, J.; Sanders, J.; Rees Smith, B. Blocking Type TSH Receptor Antibodies. Auto Immun. Highlights 2013, 4, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Núñez Miguel, R.; Sanders, J.; Sanders, P.; Young, S.; Clark, J.; Kabelis, K.; Wilmot, J.; Evans, M.; Roberts, E.; Hu, X.; Furmaniak, J.; Rees Smith, B. Similarities and Differences in Interactions of Thyroid Stimulating and Blocking Autoantibodies with the TSH Receptor. J. Mol. Endocrinol. 2012, 49, 137–151. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, S.M.; Rapoport, B. Thyrotropin-Blocking Autoantibodies and Thyroid-Stimulating Autoantibodies: Potential Mechanisms Involved in the Pendulum Swinging from Hypothyroidism to Hyperthyroidism or Vice Versa. Thyroid 2013, 23, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.F.; Latif, R. Editorial: TSH Receptor and Autoimmunity. Front. Endocrinol. (Lausanne) 2019, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Morshed, S.A.; Latif, R.; Davies, T.F. Characterization of Thyrotropin Receptor Antibody-Induced Signaling Cascades. Endocrinology 2009, 150, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Morshed, S.A.; Latif, R.; Davies, T.F. Delineating the Autoimmune Mechanisms in Graves’ Disease. Immunol. Res. 2012, 54, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Morshed, S.A.; Ando, T.; Latif, R.; Davies, T.F. Neutral Antibodies to the TSH Receptor Are Present in Graves’ Disease and Regulate Selective Signaling Cascades. Endocrinology 2010, 151, 5537–5549. [Google Scholar] [CrossRef] [PubMed]
- Núñez Miguel, R.; Sanders, P.; Allen, L.; Evans, M.; Holly, M.; Johnson, W.; Sullivan, A.; Sanders, J.; Furmaniak, J.; Rees Smith, B. Structure of Full-Length TSH Receptor in Complex with Antibody K1-70TM. J. Mol. Endocrinol. 2023, 70, e220120. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Latif, R.; Daniel, S.; Eguchi, K.; Davies, T.F. Dissecting Linear and Conformational Epitopes on the Native Thyrotropin Receptor. Endocrinology 2004, 145, 5185–5193. [Google Scholar] [CrossRef] [PubMed]
- Furmaniak, J.; Sanders, J.; Núñez Miguel, R.; Rees Smith, B. Mechanisms of Action of TSHR Autoantibodies. Horm. Metab. Res. 2015, 47, 735–752. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.F.; Andersen, S.; Latif, R.; Nagayama, Y.; Barbesino, G.; Brito, M.; Eckstein, A.K.; Stagnaro-Green, A.; Kahaly, G.J. Graves’ Disease. Nat. Rev. Dis. Primers 2020, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.; Young, S.; Sanders, J.; Kabelis, K.; Baker, S.; Sullivan, A.; Evans, M.; Clark, J.; Wilmot, J.; Hu, X.; Roberts, E.; Powell, M.; Núñez Miguel, R.; Furmaniak, J.; Rees Smith, B. Crystal Structure of the TSH Receptor (TSHR) Bound to a Blocking-Type TSHR Autoantibody. J. Mol. Endocrinol. 2011, 46, 81–99. [Google Scholar] [CrossRef]
- Furmaniak, J.; Sanders, J.; Sanders, P.; Li, Y.; Rees Smith, B. TSH Receptor Specific Monoclonal Autoantibody K1-70TM Targeting of the TSH Receptor in Subjects with Graves’ Disease and Graves’ Orbitopathy-Results from a Phase I Clinical Trial. Clin. Endocrinol. (Oxf.) 2022, 96, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Bandai, S.; Okamura, K.; Fujikawa, M.; Sato, K.; Ikenoue, H.; Kitazono, T. The Long-Term Follow-up of Patients with Thionamide-Treated Graves’ Hyperthyroidism. Endocr. J. 2019, 66, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Puett, D.; Li, Y.; DeMars, G.; Angelova, K.; Fanelli, F. A Functional Transmembrane Complex: The Luteinizing Hormone Receptor with Bound Ligand and G Protein. Mol. Cell Endocrinol. 2007, 260–262, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Menon, K.M.J.; Menon, B. Structure, Function and Regulation of Gonadotropin Receptors - a Perspective. Mol. Cell Endocrinol. 2012, 356, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Arey, B.J. Allosteric Modulators of Glycoprotein Hormone Receptors: Discovery and Therapeutic Potential. Endocrine 2008, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Heitman, L.H.; Kleinau, G.; Brussee, J.; Krause, G.; Ijzerman, A.P. Determination of Different Putative Allosteric Binding Pockets at the Lutropin Receptor by Using Diverse Drug-like Low Molecular Weight Ligands. Mol. Cell Endocrinol. 2012, 351, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Gentry, P.R.; Sexton, P.M.; Christopoulos, A. Novel Allosteric Modulators of G Protein-Coupled Receptors. J. Biol. Chem. 2015, 290, 19478–19488. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kobilka, B.K.; Steyaert, J. Nanobodies to Study G Protein-Coupled Receptor Structure and Function. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 19–37. [Google Scholar] [CrossRef]
- Anderson, R.C.; Newton, C.L.; Millar, R.P. Small Molecule Follicle-Stimulating Hormone Receptor Agonists and Antagonists. Front. Endocrinol. (Lausanne) 2018, 9, 757. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Zariñán, T.; Dias, J.A.; Conn, P.M. Mutations in G Protein-Coupled Receptors That Impact Receptor Trafficking and Reproductive Function. Mol. Cell Endocrinol. 2014, 382, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Ulloa-Aguirre, A.; Zariñán, T.; Jardón-Valadez, E. Misfolded G Protein-Coupled Receptors and Endocrine Disease. Molecular Mechanisms and Therapeutic Prospects. Int. J. Mol. Sci. 2021, 22, 12329. [Google Scholar] [CrossRef] [PubMed]
- Ulloa-Aguirre, A.; Zariñán, T.; Gutiérrez-Sagal, R.; Tao, Y.-X. Targeting Trafficking as a Therapeutic Avenue for Misfolded GPCRs Leading to Endocrine Diseases. Front. Endocrinol. (Lausanne) 2022, 13, 934685. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.L.; Whay, A.M.; McArdle, C.A.; Zhang, M.; van Koppen, C.J.; van de Lagemaat, R.; Segaloff, D.L.; Millar, R.P. Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. USA 2011, 108, 7172–77176. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.L.; Anderson, R.C.; Kreuchwig, A.; Krause, G.; Katz, A.A.; Millar, R.P. Rescue of Function of Mutant Luteinising Hormone Receptors with Deficiencies in Cell Surface Expression, Hormone Binding, and Hormone Signalling. Neuroendocrinology 2021, 111, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.L.; Anderson, R.C. Pharmacoperones for Misfolded Gonadotropin Receptors. Handb. Exp. Pharmacol. 2018, 245, 111–134. [Google Scholar] [CrossRef] [PubMed]
- Heitman, L.H.; Oosterom, J.; Bonger, K.M.; Timmers, C.M.; Wiegerinck, P.H.G.; Ijzerman, A.P. [3H]-Org 43553, the First Low-Molecular-Weight Agonistic and Allosteric Radioligand for the Human Luteinizing Hormone Receptor. Mol. Pharmacol. 2008, 73, 518–524. [Google Scholar] [CrossRef] [PubMed]
- van Koppen, C.J.; Zaman, G.J.R.; Timmers, C.M.; Kelder, J.; Mosselman, S.; van de Lagemaat, R.; Smit, M.J.; Hanssen, R.G.J.M. A Signaling-Selective, Nanomolar Potent Allosteric Low Molecular Weight Agonist for the Human Luteinizing Hormone Receptor. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 378, 503–514. [Google Scholar] [CrossRef] [PubMed]
- van de Lagemaat, R.; Timmers, C.M.; Kelder, J.; van Koppen, C.; Mosselman, S.; Hanssen, R.G.J.M. Induction of Ovulation by a Potent, Orally Active, Low Molecular Weight Agonist (Org 43553) of the Luteinizing Hormone Receptor. Hum. Reprod. 2009, 24, 640–648. [Google Scholar] [CrossRef] [PubMed]
- van de Lagemaat, R.; van Koppen, C.J.; Krajnc-Franken, M.A.M.; Folmer, B.J.B.; van Diepen, H.A.; Mulders, S.M.; Timmers, C.M. Contraception by Induction of Luteinized Unruptured Follicles with Short-Acting Low Molecular Weight FSH Receptor Agonists in Female Animal Models. Reproduction 2011, 142, 893–905. [Google Scholar] [CrossRef] [PubMed]
- van de Lagemaat, R.; Raafs, B.C.; van Koppen, C.; Timmers, C.M.; Mulders, S.M.; Hanssen, R.G.J.M. Prevention of the Onset of Ovarian Hyperstimulation Syndrome (OHSS) in the Rat after Ovulation Induction with a Low Molecular Weight Agonist of the LH Receptor Compared with HCG and Rec-LH. Endocrinology 2011, 152, 4350–4357. [Google Scholar] [CrossRef] [PubMed]
- Gerrits, M.; Mannaerts, B.; Kramer, H.; Addo, S.; Hanssen, R. First Evidence of Ovulation Induced by Oral LH Agonists in Healthy Female Volunteers of Reproductive Age. J. Clin. Endocrinol. Metab. 2013, 98, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Derkach, K.V; Dar’in, D.V; Lobanov, P.S.; Shpakov, A.O. Intratesticular, Intraperitoneal, and Oral Administration of Thienopyrimidine Derivatives Increases the Testosterone Level in Male Rats. Dokl. Biol. Sci. 2014, 459, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Derkach, K.V; Dar’in, D.V.; Bakhtyukov, A.A.; Lobanov, P.S.; Shpakov, A.O. In Vitro and in Vivo Studies of Functional Activity of New Low Molecular Weight Agonists of the Luteinizing Hormone Receptor. Biochem. (Mosc.) Suppl. Ser. A Membr. Cell Biol. 2016, 10, 294–300. [Google Scholar] [CrossRef]
- Derkach, K.V; Legkodukh, A.S.; Dar’in, D.V; Shpakov, A.O. The Stimulating Effect of Thienopyrimidines Structurally Similar to Org 43553 on Adenylate Cyclase Activity in the Testes and on Testosterone Production in Male Rats. Cell Tissue Biol. 2017, 11, 73–80. [Google Scholar] [CrossRef]
- Derkach, K.V.; Bakhtyukov, A.A.; Dar’in, D.V.; Golovanova, N.E.; Shpakov, A.O. Novel Thienopyrimidine Derivatives with an Activity of Full and Inverse Agonists of the Luteinizing Hormone Receptor. J. Evol. Biochem. Physiol. 2019, 55, 414–418. [Google Scholar] [CrossRef]
- Derkach, K.V; Dar’in, D.V; Shpakov, A.O. Low-Molecular-Weight Ligands of Luteinizing Hormone Receptor with the Activity of Antagonists. Biochem. (Mosc.) Suppl. Ser. A Membr. Cell Biol. 2020, 14, 223–231. [Google Scholar] [CrossRef]
- Shpakov, A.O.; Derkach, K.V.; Dar’in, D.V.; Lobanov, P.S. [Activation of Adenylyl Cyclase in Rats Testes and Ovaries Using Thienopyrimidine Derivatives]. Tsitologiia 2014, 56, 346–352. [Google Scholar] [PubMed]
- Bakhtyukov, A.A.; Derkach, K.V.; Gureev, M.A.; Dar’in, D.V.; Sorokoumov, V.N.; Romanova, I.V.; Morina, I.Y.; Stepochkina, A.M.; Shpakov, A.O. Comparative Study of the Steroidogenic Effects of Human Chorionic Gonadotropin and Thieno[2,3-D]Pyrimidine-Based Allosteric Agonist of Luteinizing Hormone Receptor in Young Adult, Aging and Diabetic Male Rats. Int. J. Mol. Sci. 2020, 21, 7493. [Google Scholar] [CrossRef]
- Bakhtyukov, A.A.; Derkach, K.V.; Romanova, I.V.; Sorokoumov, V.N.; Sokolova, T.V..; Govdi, A.I.; Morina, I.Yu.; Perminova, A.A.; Shpakov, A.O. Effect of Low-Molecular-Weight Allosteric Agonists of the Luteinizing Hormone Receptor on Its Expression and Distribution in Rat Testes. J. Evol. Biochem. Physiol. 2021, 57, 208–220. [Google Scholar] [CrossRef]
- Bakhtyukov, A.A.; Derkach, K.V.; Sorokoumov, V.N.; Stepochkina, A.M.; Romanova, I.V.; Morina, I.Y.; Zakharova, I.O.; Bayunova, L.V.; Shpakov, A.O. The Effects of Separate and Combined Treatment of Male Rats with Type 2 Diabetes with Metformin and Orthosteric and Allosteric Agonists of Luteinizing Hormone Receptor on Steroidogenesis and Spermatogenesis. Int. J. Mol. Sci. 2021, 23, 198. [Google Scholar] [CrossRef] [PubMed]
- Derkach, K.V; Romanova, I. V.; Bakhtyukov, A.A.; Morina, I.Y.; Dar’in, D.V.; Sorokoumov, V.N.; Shpakov, A.O. The Effect of Low-Molecular-Weight Allosteric Agonist of Luteinizing Hormone Receptor on Functional State of the Testes in Aging and Diabetic Rats. Bull. Exp Biol. Med. 2021, 171, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Stepochkina, A.M.; Bakhtyukov, A.A.; Derkach, K. v.; Sorokoumov, V.N.; Shpakov, A.O. A Comparative Study of the Steroidogenic Effect of 5-Amino-N-Tert-Butyl-2-(Methylthio)-4-(3-(Nicotinamido)Phenyl)Thieno[2,3-d]-Pyrimidine-6-Carboxamide and Chorionic Gonadotropin with Different Methods of Administration to Male Rats. J. Evol. Biochem. Physiol. 2022, 58, 54–63. [Google Scholar] [CrossRef]
- Bakhtyukov, A.A.; Morina, I.Yu.; Derkach, K.V.; Romanova, I.V.; Sorokoumov, V.N.; Shpakov, A.O. Development of Approaches to Reducing the Effective Gonadotropin Dose in Treating Androgen Insufficiency in Male Rats with Type 1 Diabetes Mellitus. J. Evol. Biochem. Physiol. 2022, 58, 1503–1513. [Google Scholar] [CrossRef]
- Derkach, K.V; Bakhtyukov, A.A.; Bayunova, L.V; Zorina, I.I.; Shpakov, A.O. Normalization of Testicular Steroidogenesis and Spermatogenesis in Male Rats with Type 2 Diabetes Mellitus under the Conditions of Metformin Therapy. Dokl. Biol. Sci. 2020, 493, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Derkach, K.V; Bakhtyukov, A.A.; Morina, I.Y.; Romanova, I.V; Bayunova, L.V; Shpakov, A.O. Comparative Study of the Restoring Effect of Metformin, Gonadotropin, and Allosteric Agonist of Luteinizing Hormone Receptor on Spermatogenesis in Male Rats with Streptozotocin-Induced Type 2 Diabetes Mellitus. Bull. Exp. Biol. Med. 2022, 172, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Bakhtyukov, A.A.; Derkach, K.V.; Fokina, E.A.; Lebedev, I.A.; Sorokoumov, V.N.; Bayunova, L.V.; Shpakov, A.O. Effect of various luteinizing hormone receptor agonists on steroidogenesis in the ovaries of sexually mature female rats. J. Evol. Biochem. Physiol. 2023, 59, 55–65. [Google Scholar] [CrossRef]
- Jorand-Lebrun, C.; Brondyk, B.; Lin, J.; Magar, S.; Murray, R.; Reddy, A.; Shroff, H.; Wands, G.; Weiser, W.; Xu, Q.; McKenna, S.; Brugger, N. Identification, Synthesis, and Biological Evaluation of Novel Pyrazoles as Low Molecular Weight Luteinizing Hormone Receptor Agonists. Bioorg. Med. Chem. Lett. 2007, 17, 2080–2085. [Google Scholar] [CrossRef] [PubMed]
- Heitman, L.H.; Narlawar, R.; de Vries, H.; Willemsen, M.N.; Wolfram, D.; Brussee, J.; IJzerman, A.P. Substituted Terphenyl Compounds as the First Class of Low Molecular Weight Allosteric Inhibitors of the Luteinizing Hormone Receptor. J. Med. Chem. 2009, 52, 2036–2042. [Google Scholar] [CrossRef]
- Shpakova, E.A.; Derkach, K.V.; Shpakov, A.O. Biological Activity of Lipophilic Derivatives of Peptide 562-572 of Rat Luteinizing Hormone Receptor. Dokl. Biochem. Biophys. 2013, 452, 248–250. [Google Scholar] [CrossRef]
- Derkach, K.V; Shpakova, E.A.; Shpakov, A.O. Palmitoylated Peptide 562-572 of Luteinizing Hormone Receptor Increases Testosterone Level in Male Rats. Bull. Exp. Biol. Med. 2014, 158, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Shpakova, E.A.; Sorokoumov, V.N.; Akent’ev, A.V.; Derkach, K.V.; Tennikova, T.B.; Shpakov, A.O. The Relationship between Micelle Formation and Biological Activity of Peptide 562–572 of Luteinizing Hormone Receptor Modified with Decanoyl Radicals. Cell tissue Biol. 2017, 11, 227–233. [Google Scholar] [CrossRef]
- Schniewind, H.A.; Sattler, L.-M.; Haudum, C.W.; Münzker, J.; Minich, W.B.; Obermayer-Pietsch, B.; Schomburg, L. Autoimmunity to the Follicle-Stimulating Hormone Receptor (FSHR) and Luteinizing Hormone Receptor (LHR) in Polycystic Ovarian Syndrome. Int. J. Mol. Sci. 2021, 22, 13667. [Google Scholar] [CrossRef] [PubMed]
- Chiauzzi, V.A.; Bussmann, L.; Calvo, J.C.; Sundblad, V.; Charreau, E.H. Circulating Immunoglobulins That Inhibit the Binding of Follicle-Stimulating Hormone to Its Receptor: A Putative Diagnostic Role in Resistant Ovary Syndrome? Clin. Endocrinol. (Oxf.) 2004, 61, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.D.; Lind, C.; De Pascali, F.; Penn, R.B.; MacKerell, A.D. Jr; Deshpande, D.A. In silico identification of a β2-adrenoceptor allosteric site that selectively augments canonical β2AR-Gs signaling and function. Proc. Natl. Acad. Sci. USA 2022, 119, e2214024119. [Google Scholar] [CrossRef] [PubMed]
- Bekker, G.J.; Araki, M.; Oshima, K.; Okuno, Y.; Kamiya, N. Accurate Binding Configuration Prediction of a G-Protein-Coupled Receptor to Its Antagonist Using Multicanonical Molecular Dynamics-Based Dynamic Docking. J. Chem. Inf. Model. 2021, 61, 5161–5171. [Google Scholar] [CrossRef] [PubMed]
- Abiko, L.A.; Dias Teixeira, R.; Engilberge, S.; Grahl, A.; Mühlethaler, T.; Sharpe, T.; Grzesiek, S. Filling of a water-free void explains the allosteric regulation of the β1-adrenergic receptor by cholesterol. Nat. Chem. 2022, 14, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, J.; Yuan, Y.; Chen, X.; Zhang, F.; Pu, X. Molecular Mechanisms of Diverse Activation Stimulated by Different Biased Agonists for the β2-Adrenergic Receptor. J. Chem. Inf. Model. 2022, 62, 5175–5192. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Yin, J.; Yao, Q.; Wang, T.; Chen, J.; Liang, Q.; Li, Q.; Zhao, X. Development of an Allostery Responsive Chromatographic Method for Screening Potential Allosteric Modulator of Beta2-adrenoceptor from a Natural Product-Derived DNA-Encoded Chemical Library. Anal. Chem. 2022, 94, 9048–9057. [Google Scholar] [CrossRef]
- Yen, H.Y.; Liko, I.; Song, W.; Kapoor, P.; Almeida, F.; Toporowska, J.; Gherbi, K.; Hopper, J.T.S.; Charlton, S.J.; Politis, A.; Sansom, M.S.P.; Jazayeri, A.; Robinson, C.V. Mass spectrometry captures biased signalling and allosteric modulation of a G-protein-coupled receptor. Nat. Chem. 2022, 14, 1375–1382. [Google Scholar] [CrossRef]
- Ahn, S.; Pani, B.; Kahsai, A.W.; Olsen, E.K.; Husemoen, G.; Vestergaard, M.; Jin, L.; Zhao, S.; Wingler, L.M.; Rambarat, P.K.; Simhal, R.K.; Xu, T.T.; Sun, L.D.; Shim, P.J.; Status, D.P.; Huang, L.Y.; Franch, T.; Chen, X.; Lefkowitz, R.J. Small-Molecule Positive Allosteric Modulators of the β2-Adrenoceptor Isolated from DNA-Encoded Libraries. Mol. Pharmacol. 2018, 94, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Meng, K.; Shim, P.; Wang, Q.; Zhao, S.; Gu, T.; Kahsai, A.W.; Ahn, S.; Chen, X. Design, Synthesis, and Functional Assessment of Cmpd-15 Derivatives as Negative Allosteric Modulators for the β2-Adrenergic Receptor. Bioorg. Med. Chem. 2018, 26, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Masoudi, A.; Kahsai, A.W.; Huang, L.Y.; Pani, B.; Staus, D.P.; Shim, P.J.; Hirata, K.; Simhal, R.K.; Schwalb, A.M.; Rambarat, P.K.; Ahn, S.; Lefkowitz, R.J.; Kobilka, B. Mechanism of β2AR regulation by an intracellular positive allosteric modulator. Science 2019, 364, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Pani, B.; Ahn, S.; Rambarat, P.K.; Vege, S.; Kahsai, A.W.; Liu, A.; Valan, B.N.; Staus, D.P.; Costa, T.; Lefkowitz, R.J. Unique Positive Cooperativity Between the β-Arrestin-Biased β-Blocker Carvedilol and a Small Molecule Positive Allosteric Modulator of the β2-Adrenergic Receptor. Mol. Pharmacol. 2021, 100, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hua, T.; Liu, Z.-J. Structural Features of Activated GPCR Signaling Complexes. Curr. Opin. Struct. Biol. 2020, 63, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.; Du, Y.; Quoyer, J.; Panettieri, R.A.; Janz, J.M.; Bouvier, M.; Kobilka, B.K.; Benovic, J.L. Development and Characterization of Pepducins as Gs-Biased Allosteric Agonists. J. Biol. Chem. 2014, 289, 35668–35684. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.; Schilling, J.; Song, J.; Carter, R.L.; Du, Y.; Yoo, S.M.; Traynham, C.J.; Koch, W.J.; Cheung, J.Y.; Tilley, D.G.; Benovic, J.L. β-Arrestin-Biased Signaling through the β2-Adrenergic Receptor Promotes Cardiomyocyte Contraction. Proc. Natl. Acad. Sci. USA 2016, 113, E4107–16. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; Thomas, T.P.; Carter, R.L.; de Lucia, C.; Gao, E.; Koch, W.J.; Benovic, J.L.; Tilley, D.G. Pepducin-Mediated Cardioprotection via β-Arrestin-Biased β2-Adrenergic Receptor-Specific Signaling. Theranostics 2018, 8, 4664–4678. [Google Scholar] [CrossRef] [PubMed]
- Okyere, A.D.; Song, J.; Patwa, V.; Carter, R.L.; Enjamuri, N.; Lucchese, A.M.; Ibetti, J.; de Lucia, C.; Schumacher, S.M.; Koch, W.J.; Cheung, J.Y.; Benovic, J.L.; Tilley, D.G. Pepducin ICL1-9-Mediated β2-Adrenergic Receptor-Dependent Cardiomyocyte Contractility Occurs in a Gi Protein/ROCK/PKD-Sensitive Manner. Cardiovasc. Drugs Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Boivin-Jahns, V.; Jahns, R. GPCR-Autoantibodies in Chronic Heart Failure. Front. BioSci. (Landmark Ed.) 2018, 23, 2065–2081. [Google Scholar] [CrossRef] [PubMed]
- Wölfel, A.; Sättele, M.; Zechmeister, C.; Nikolaev, V.O.; Lohse, M.J.; Boege, F.; Jahns, R.; Boivin-Jahns, V. Unmasking featuRes. of the auto-epitope essential for β1-adrenoceptor activation by autoantibodies in chronic heart failure. ESC Heart Fail, 2020; 7, 1830–1841. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Naga Prasad, S. v Autoantibodies and Cardiomyopathy: Focus on Beta-1 Adrenergic Receptor Autoantibodies. J. Cardiovasc. Pharmacol. 2022, 80, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Jahns, R.; Boivin, V.; Siegmund, C.; Inselmann, G.; Lohse, M.J.; Boege, F. Autoantibodies Activating Human Beta1-Adrenergic Receptors Are Associated with Reduced Cardiac Function in Chronic Heart Failure. Circulation 1999, 99, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Jane-wit, D.; Altuntas, C.Z.; Johnson, J.M.; Yong, S.; Wickley, P.J.; Clark, P.; Wang, Q.; Popović, Z.B.; Penn, M.S.; Damron, D.S.; Perez, D.M.; Tuohy, V.K. Beta1-Adrenergic Receptor Autoantibodies Mediate Dilated Cardiomyopathy by Agonistically Inducing Cardiomyocyte Apoptosis. Circulation 2007, 116, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Bornholz, B.; Roggenbuck, D.; Jahns, R.; Boege, F. Diagnostic and Therapeutic Aspects of β1-Adrenergic Receptor Autoantibodies in Human Heart Disease. AutoImmun. Rev. 2014, 13, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Soave, M.; Cseke, G.; Hutchings, C.J.; Brown, A.J.H.; Woolard, J.; Hill, S.J. A monoclonal antibody raised against a thermo-stabilised β1-adrenoceptor interacts with extracellular loop 2 and acts as a negative allosteric modulator of a sub-set of β1-adrenoceptors expressed in stable cell lines. Biochem. Pharmacol. 2018, 147, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Bornholz, B.; Weidtkamp-Peters, S.; Schmitmeier, S.; Seidel, C.A.M.; Herda, L.R.; Felix, S.B.; Lemoine, H.; Hescheler, J.; Nguemo, F.; Schäfer, C.; Christensen, M.O.; Mielke, C.; Boege, F. Impact of Human Autoantibodies on β1-Adrenergic Receptor Conformation, Activity, and Internalization. Cardiovasc. Res. 2013, 97, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Wallukat, G.; Schimke, I. Agonistic Autoantibodies Directed against G-Protein-Coupled Receptors and Their Relationship to Cardiovascular Diseases. Semin. ImmunoPathol. 2014, 36, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, Y.; Marullo, S.; Hoyer, S.; Waagstein, F.; Andersson, B.; Vahlne, A.; Guillet, J.G.; Strosberg, A.D.; Hjalmarson, A.; Hoebeke, J. Mapping of a Functional Autoimmune Epitope on the Beta 1-Adrenergic Receptor in Patients with Idiopathic Dilated Cardiomyopathy. J. Clin. Invest. 1990, 86, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Wallukat, G.; Wollenberger, A.; Morwinski, R.; Pitschner, H.F. Anti-Beta1-Adrenoceptor Autoantibodies with Chronotropic Activity from the Serum of Patients with Dilated Cardiomyopathy: Mapping of Epitopes in the First and Second Extracellular Loops. J. Mol. Cell Cardiol. 1995, 27, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Behr, B.; Hoffmann, C.; Ottolina, G.; Klotz, K.-N. Novel Mutants of the Human Beta1-Adrenergic Receptor Reveal Amino Acids Relevant for Receptor Activation. J. Biol. Chem. 2006, 281, 18120–18125. [Google Scholar] [CrossRef] [PubMed]
- Boivin, V.; Beyersdorf, N.; Palm, D.; Nikolaev, V.O.; Schlipp, A.; Müller, J.; Schmidt, D.; Kocoski, V.; Kerkau, T.; Hünig, T.; Ertl, G.; Lohse, M.J.; Jahns, R. Novel Receptor-Derived Cyclopeptides to Treat Heart Failure Caused by Anti-β1-Adrenoceptor Antibodies in a Human-Analogous Rat Model. PLoS One 2015, 10, e0117589. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Bai, Y.; Zhang, S.; Xu, W.; Xu, J.; Zhou, Y.; Zhang, S.; Wu, Y.; Yu, H.; Cao, N.; Liu, H.; Wang, W. Cyclic Peptide RD808 Reduces Myocardial Injury Induced by β1-Adrenoreceptor Autoantibodies. Heart Vessels 2019, 34, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Mijares, A.; Lebesgue, D.; Wallukat, G.; Hoebeke, J. From Agonist to Antagonist: Fab Fragments of an Agonist-like Monoclonal Anti-Beta(2)-Adrenoceptor Antibody Behave as Antagonists. Mol. Pharmacol. 2000, 58, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zuccolo, J.; Kem, D.C.; Zillner, C.; Lee, J.; Smith, K.; James, J.A.; Cunningham, M.W.; Yu, X. Implications of a Vasodilatory Human Monoclonal Autoantibody in Postural Hypotension. J. Biol. Chem. 2013, 288, 30734–30741. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Allosteric regulation of chemokine receptors.
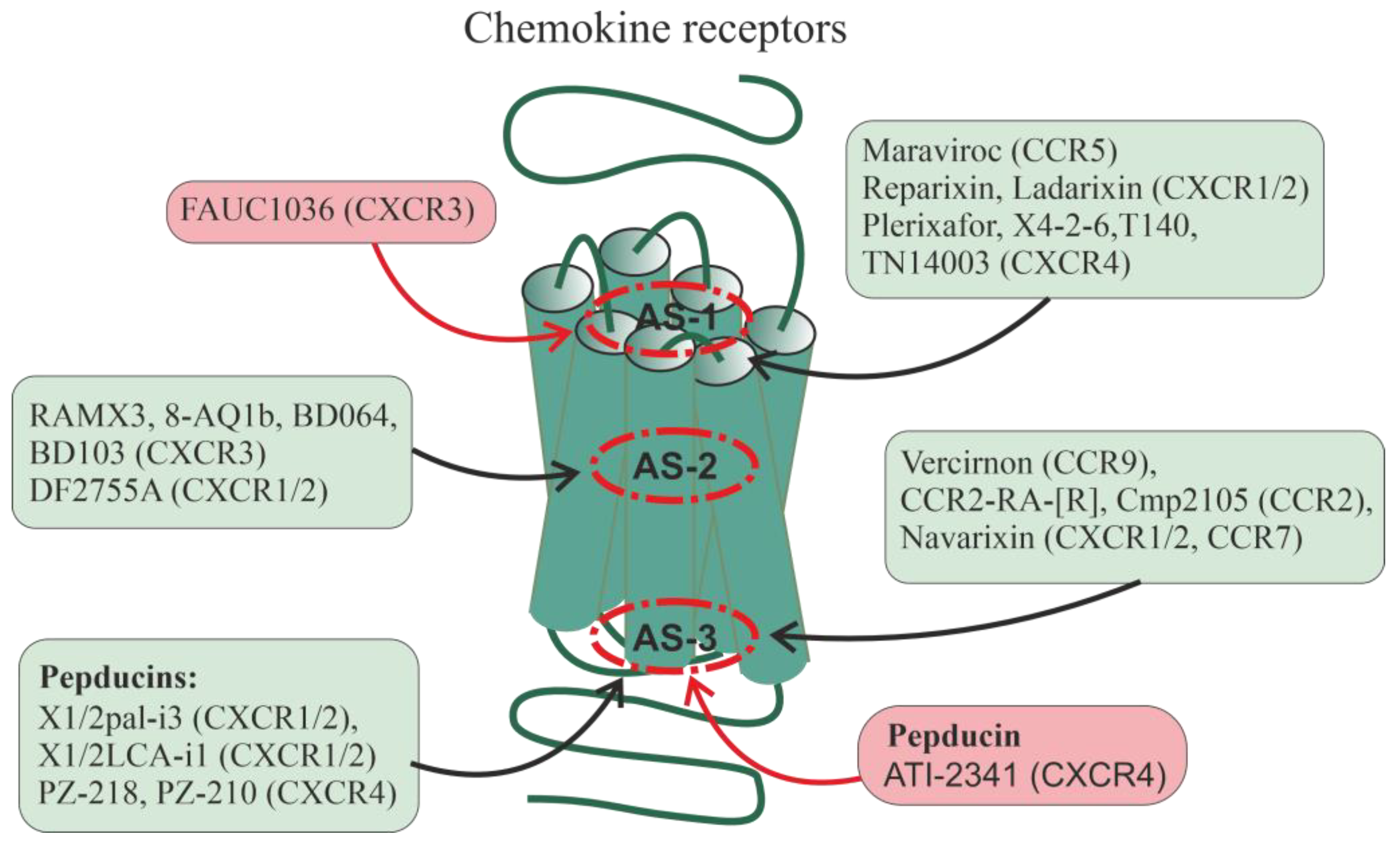
Figure 2.
Allosteric regulation of proteinase-activated receptors.
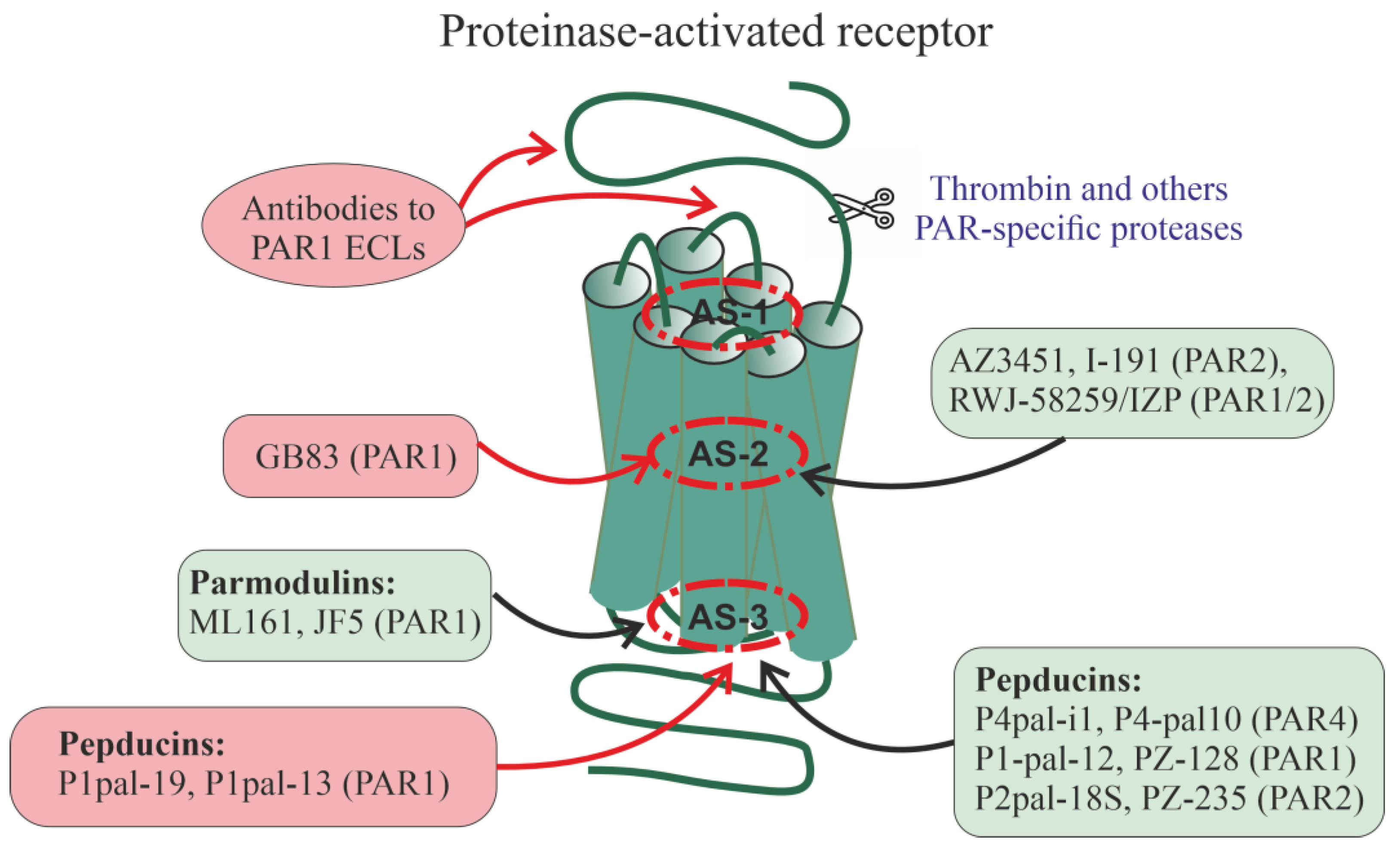
Figure 3.
Allosteric regulators of thyroid-stimulating hormone receptor.
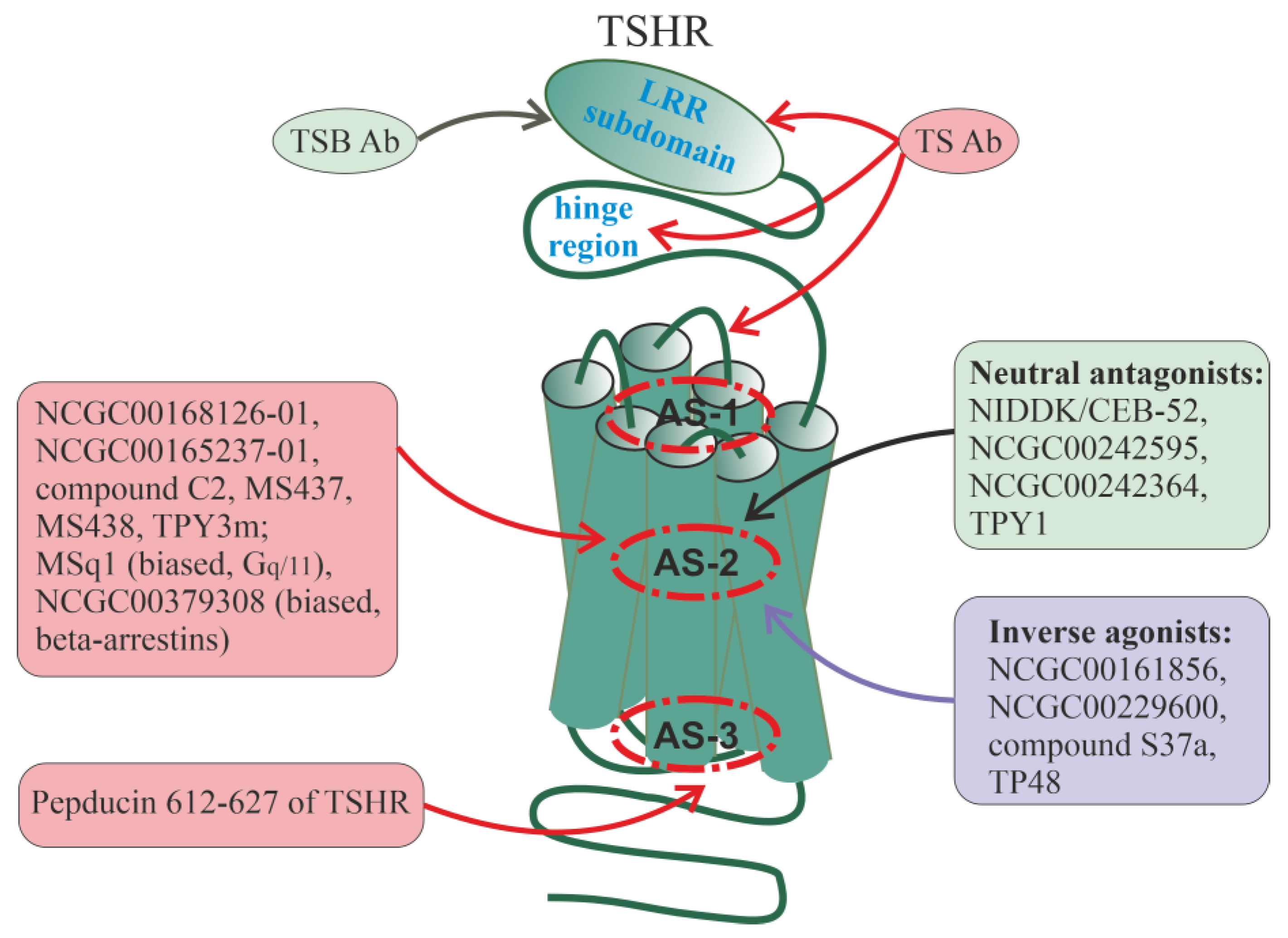
Figure 4.
Allosteric regulators of luteinizing hormone receptor.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



