
Submitted:
30 January 2023
Posted:
02 February 2023
You are already at the latest version
Abstract
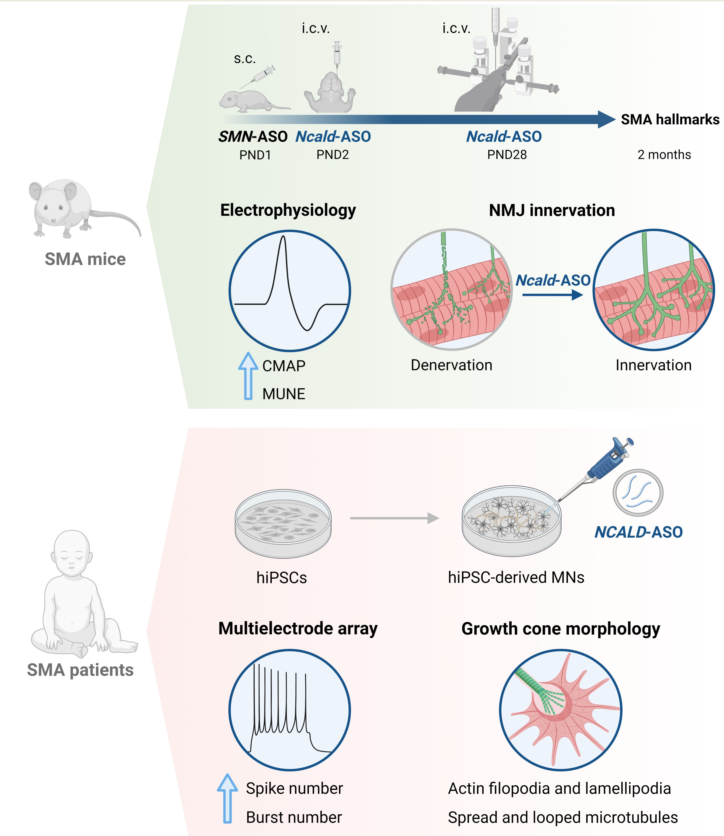
Here we investigated the longer-term effect of Ncald-ASOs by additional i.c.v. bolus injection at PND28. Two weeks after injection of 500 µg Ncald-ASO in wild-type mice, NCALD was significantly reduced in brain and spinal cord and well tolerated. Next, we performed a double-blinded preclinical study combining low-dose SMN-ASO (PND1) with 2x i.c.v. Ncald-ASO or CTRL-ASO (100 µg at PND2, 500 µg at PND28). Ncald-ASO re-injection significantly ameliorated electrophysiological defects and NMJ denervation at 2 months. Moreover, we developed and identified a nontoxic and highly efficient human NCALD-ASO that significantly reduced NCALD in hiPSCs-derived MNs. This improved both neuronal activity and growth cone maturation of SMA MNs, emphasizing the additional protective effect of NCALD-ASO treatment.
Keywords:
Neuromuscular disorder
; NCALD
; SMA
; SMN2
; Antisense oligonucleotide
; Genetic modifier
; Therapy
; hiPSCs
1. Introduction
Spinal muscular atrophy (SMA), a common and devastating neuromuscular disorder, has changed from an early lethal to an efficiently treatable disease for a high proportion of patients in the last years [1,2,3,4,5]. SMA, an autosomal-recessive inherited neurodegenerative disease, has an incidence of 1 in 6,000 – 10,000 with a carrier frequency of 1:51 worldwide and 1:41 in the European population [6]. SMA is caused by biallelic mutations in the survival motor neuron 1 (SMN1) gene, with all patients retaining SMN2, an almost identical copy gene [6]. Approximately 96% of SMA patients show homozygous deletions of exon 7 and 8 or only of exon 7 of SMN1, making genetic testing fast and neonatal screening highly reliable (reviewed by [7,8]). Due to a translationally silent mutation in exon 7 of the SMN2 gene that affects correct splicing, only small amounts of full-length mRNA are produced, leading to a marked decrease in functional SMN protein [6,9,10].
In SMA, the disease severity inversely correlates with the copy number of SMN2; the more, the better the phenotype. Approximately 50% of SMA patients develop SMA type 1; the majority of them carry only two SMN2 copies, are unable to sit or walk and die before 2 years of age. Around 30% of patients have SMA type 2, an intermediate phenotype with patients achieving the ability to sit independently, who usually carry three SMN2 copies. Another ~20% of SMA patients have SMA type 3 with a mild SMA phenotype. These patients are able to walk independently; however, they often become wheelchair-bound with the progression of the disease. SMA type 3 patients normally carry 3-4 SMN2 copies. Rarely, patients, who carry 4-6 SMN2 copies can develop an adult SMA type 4 [11,12].
SMN is a ubiquitously expressed protein with variable expression patterns in different tissues [18], but especially abundant in the lower spinal cord motor neurons (MNs) [13,14]. SMN is a multifunctional protein involved in key cellular processes such as snRNP assembly and splicing, mRNA transport, microRNA biogenesis, local translation, cytoskeleton dynamics, endocytosis, DNA damage repair; mitochondrial biogenesis and others [15,16,17]. Although SMA is considered a MN disorder, the housekeeping functions of SMN explains the multiorgan disfunction in its severe forms, when SMN levels are reduced below a certain threshold [18]. However, why MNs are predominantly vulnerable to reduced SMN protein remains largely unclear. SMN expression levels vary between tissues and stages of development [19], but are particularly required during neuromuscular junction (NMJ) maturation [20]. Moreover, SMN binding to ribosomes and polysomes occurs in a tissue-specific manner in vivo [21] and its deficiency leads to tissue-specific splicing defects that might contribute particularly to MN vulnerability [22]. These findings point towards differential requirements of SMN in each tissue and developmental stage.
Three highly effective SMN-dependent therapies have been developed using gene therapy, antisense oligonucleotides or small molecular (reviewed in [7,8]). If the therapy is applied presymptomatically – requiring neonatal screening – children with 3 and more SMN2 copies meet age-appropriate developmental milestones. Instead, children with only 2 SMN2 copies show delayed or deficient motor milestones [23,24,25,26]. Therefore, additional SMN-independent therapies are more than ever necessary to complement SMN-dependent therapies.
The identification of SMA discordant families with asymptomatic individuals carrying homozygous SMN1 deletions and three to four SMN2 copies led to the discovery of two SMA genetic modifiers, Plastin 3 (PLS3) and NCALD, in humans [27,28]. These findings not only opened the door to a better understanding of crucial molecular pathways disturbed in SMA, but also to the development of combinatorial therapies [28,29,30]. PLS3 is a F-actin binding and bundling protein, and NCALD a neuronal calcium sensor; both act on multiple cellular pathways in MNs including axonal growth, calcium homeostasis, neurotransmission, endocytosis and others [27,28,31,32]. PLS3 overexpression or NCALD reduction ameliorates SMA hallmarks at MN and NMJ level and prolongs survival. The protective effect of both modifiers was corroborated in various genetically modified SMA animal models and by use of gene therapy or antisense oligonucleotides [30,31,33,34,35,36]. A first randomized-blinded preclinical study in SMA mice testing an antisense oligonucleotide-based combinatorial therapy targeting SMN and Ncald, showed a synergistic amelioration of SMA hallmarks, such as electrophysiological and morphological properties of NMJs and muscles at postnatal day (PND) 21 [30]. Interestingly, the effect of Ncald-ASO was rather short and thus comparable to what has been described in a similar therapeutic approach using Chp1-ASO [37], a PLS3 interacting partner and SMA modifier [38], which might explain the failure of a long-term amelioration [30].
In light of these encouraging results, the aim of this study was to determine if Ncald-ASO re-injections could prolong the therapeutic effect observed at PND21 in SMA mice. In addition, we developed human NCALD-ASOs to be tested for efficacy and nontoxicity, and investigated their impact on MN development and neuronal function. Human MNs were differentiated from control and SMA patients’ derived inducible pluripotent stem cells (hiPSC). For both conditions SMA mice and human MNs, we observed a positive effect of NCALD reduction on MN function, further supporting the protective role of NCALD reduction for combinatorial SMA therapies.
2. Results
2.1. Re-injection of Ncald-ASO significantly reduces NCALD in brain and spinal cord
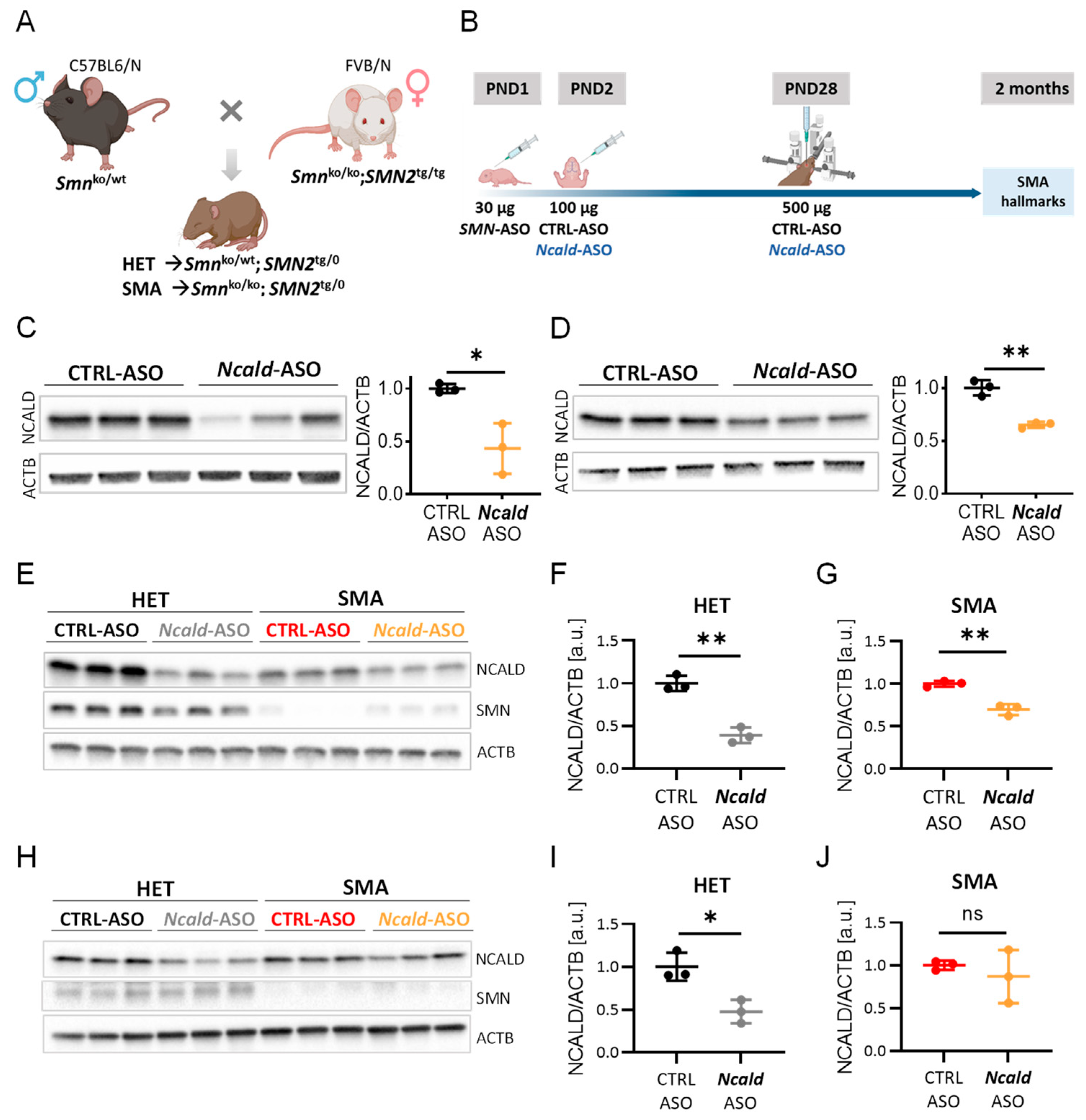
In order to mimic the situation of SMN1-deleted individuals, who remain asymptomatic despite carrying only four SMN2 copies, we injected at PND1 a suboptimal low dose of 30 µg of SMN-ASOs into the severely-affected Taiwanese SMA mice that carry only two human SMN2 copies on null murine background, to produce a mildly affected SMA model that resembles the human situation (Figure 1A) [28,30,37]. As previously demonstrated, this suboptimal SMN-ASO treatment significantly prolongs survival by ameliorating the multiorgan impairment, without fully restoring MN and NMJ function [28,30,33,37], allowing the study of protective modifiers in combinatorial therapies. Since a single Ncald ASOs injection at PND2 had a positive effect on SMA hallmarks but turned out to be rather unstable after 3-4 weeks ([30], here we investigated the longer-term therapeutic effect by reinjecting Ncald-ASOs at one months and studying the mice at two months (Figure 1B).
First, we tested the efficacy and tolerability of i.c.v. bolus injection of 500 µg of CTRL-ASO and Ncald-ASO in adult wild-type mice. For this purpose, animals were injected at PND28 and brain and spinal cord tissues were collected 2-weeks after injection for western blot analysis. In brain lysates NCALD protein levels were reduced to 43% compared to CTRL-ASO injected animals (Figure 1C) and in spinal cord to 63% (Figure 1D). The less pronounced reduction of NCALD in the spinal cord compared to the brain, might be due to the i.c.v. injected ASOs that have to be distributed in the CNS through the cerebrospinal fluid (CSF). Importantly, 500 µg of CTRL- and Ncald-ASO were well tolerated by the animals and no adverse effects were observed.
Because Ncald ASOs applied by i.c.v. bolus injection efficiently knocked down NCALD levels in spinal cord of wildtype animals, we designed a double-blind pre-clinical study in SMA and HET mice as shown in Figure 1B. In detail, at PND1 all animal received 30 µg SMN-ASOs by subcutaneous injection. At PND2, half of the animals received 100 µg CTRL-ASO, the other half 100 µg Ncald-ASO by i.c.v. injection as previously described [30]. At PND28, animals were anesthetized and fixed in a stereotaxic equipment and receive an i.c.v. bolus re-injection with 500 µg CTRL-ASO or Ncald-ASO (Figure 1B). One month after CTRL-ASO or Ncald-ASO re-injection, we determined NCALD downregulation in brain lysates of HET Ncald-ASO (Figure 1E and F) and SMA Ncald-ASO-treated (Figure 1E and G) animals. Both, HET and SMA mice exhibited a significant reduction of NCALD levels when compared to their respective control groups. Moreover, NCALD was significantly reduced in the spinal cord of HET animals 4-weeks after receiving Ncald-ASO re-injection compared to mice treated with CTRL-ASO (Figure 1H and I), however was more variable from one animal to another in SMA mice (Figure 1H and J).
Figure 1.
Experimental scheme and efficacy of 500 µg Ncald-ASO i.c.v bolus re-injection in adult mice. (A) Breeding strategy to obtain intermediate SMA and HET mice on mixed50 background. Figure created with BioRender.com. (B) Scheme showing combinatorial therapy of SMA and HET mice (latter used as controls) with SMN- and either CRTL- or Ncald-ASOs. Figure created with BioRender.com. (C-D) Western blot analysis and quantification of NCALD levels in brain (C) and spinal cord (D) lysates from CRTL- or Ncald-ASO-treated wild-type mice at PND28; samples were collected 2-weeks after injection. 2-weeks post i.c.v bolus injection with 500 µg Ncald-ASO, NCALD levels in the brain and spinal cord are significantly reduced compared to CTRL-ASO-treated animals. Unpaired two-tailed Student‘s t-test. All values reported as mean and error bars represent ± SD. *p ≤ 0.05, **p ≤ 0.01. Each dot in the bar graphs represents an independent animal. (E-G) Western blot analysis and quantification of NCALD levels in brain lysates from ASO-treated experimental animals. Samples were collected 4-weeks after treatment. 4-weeks post i.c.v bolus injection with 500 µg Ncald-ASO, HET and SMA mice showed significantly reduction of NCALD amount compared to CTRL-ASO-treated animals. Unpaired two-tailed Student‘s t-test. All values reported as mean and error bars represent ± SD. **p ≤ 0.01. Each dot in the bar graphs represents an independent animal. (H-J) Western blot analysis and quantification of NCALD levels in spinal cord lysates from ASO-treated experimental animals. Samples were collected 4-weeks after i.c.v bolus injection at PND28 with 500 µg Ncald-ASO, HET but not SMA mice showed significantly reduced NCALD protein amount compared to CTRL-ASO-treated animals. Unpaired two-tailed Student‘s t-test. All values reported as mean and error bars represent ± SD. *p ≤ 0.05. Each dot in the bar graphs represents an independent animal.
Figure 1.
Experimental scheme and efficacy of 500 µg Ncald-ASO i.c.v bolus re-injection in adult mice. (A) Breeding strategy to obtain intermediate SMA and HET mice on mixed50 background. Figure created with BioRender.com. (B) Scheme showing combinatorial therapy of SMA and HET mice (latter used as controls) with SMN- and either CRTL- or Ncald-ASOs. Figure created with BioRender.com. (C-D) Western blot analysis and quantification of NCALD levels in brain (C) and spinal cord (D) lysates from CRTL- or Ncald-ASO-treated wild-type mice at PND28; samples were collected 2-weeks after injection. 2-weeks post i.c.v bolus injection with 500 µg Ncald-ASO, NCALD levels in the brain and spinal cord are significantly reduced compared to CTRL-ASO-treated animals. Unpaired two-tailed Student‘s t-test. All values reported as mean and error bars represent ± SD. *p ≤ 0.05, **p ≤ 0.01. Each dot in the bar graphs represents an independent animal. (E-G) Western blot analysis and quantification of NCALD levels in brain lysates from ASO-treated experimental animals. Samples were collected 4-weeks after treatment. 4-weeks post i.c.v bolus injection with 500 µg Ncald-ASO, HET and SMA mice showed significantly reduction of NCALD amount compared to CTRL-ASO-treated animals. Unpaired two-tailed Student‘s t-test. All values reported as mean and error bars represent ± SD. **p ≤ 0.01. Each dot in the bar graphs represents an independent animal. (H-J) Western blot analysis and quantification of NCALD levels in spinal cord lysates from ASO-treated experimental animals. Samples were collected 4-weeks after i.c.v bolus injection at PND28 with 500 µg Ncald-ASO, HET but not SMA mice showed significantly reduced NCALD protein amount compared to CTRL-ASO-treated animals. Unpaired two-tailed Student‘s t-test. All values reported as mean and error bars represent ± SD. *p ≤ 0.05. Each dot in the bar graphs represents an independent animal.
2.2. Long-term combinatorial treatment with Ncald-ASO and SMN ameliorates electrophysiological defects and NMJ denervation in SMA mice
Electrophysiological defects have been described in SMA patients and animal models, including reduction of compound muscle action potential (CMAP) and motor unit number estimation (MUNE) [30,39,40]]. One single injection of Ncald-ASO at PND2 significantly increased both electrophysiological parameters of the gastrocnemius muscle in SMA mice at PND21, but was not sufficient to prolong the effect for a long-term [30]. Here, SMA mice re-injected with Ncald-ASO showed a significant amelioration of the CMAP response, indicating that prolonged NCALD reduction has a beneficial effect on the functionality of the motor units (Figure 2A and B). Moreover, MUNE analysis confirmed reduced motor unit numbers in SMA compared to HET CTRL-ASO-treated animals, a parameter that was rescued upon Ncald-ASO re-injection (Figure 2C). This data strongly supports that 500 µg Ncald-ASO re-injection in combination with a single low-dose SMN-ASO administration not only prolongs the motor unit function but also prevents the loss of motor unit numbers in SMA mice over time, thus protecting from neurodegeneration.
Since previous work showed that one single injection of Ncald-ASO administered at PND2 improved pathological features of the NMJ in 3-week-old mice but failed to show any amelioration at 3 months [30], we analyzed here NMJ area and innervation of the transversus abdominis muscle upon Ncald-ASO re-injection. To quantify the NMJ area, the size of bungarotoxin (BTX)-positive endplates was measured. To analyze NMJ innervation, NMJs were categorized according to the percentage of nerve terminal that overlaid with the acetylcholine receptors (AChRs) as fully innervated (>80%), partially innervated (>15% - 79%) and non-innervated (< 15%). NMJ area and innervation were affected in SMA mice compared to HET littermates. Strikingly, re-injection with 500 µg Ncald-ASO did not increase NMJ area in SMA animals compared to CTRL-ASO injected SMA mice at 2 months of age (Figure 2D), however denervation of NMJs was ameliorated upon Ncald-ASO re-injection in SMA animals (Figure 2E), emphasizing the key role of NCALD reduction in preventing the progressive retraction of the nerve terminal from its target muscle fiber, hence halting neurodegeneration.
Finally, we analyzed muscle fiber size from gastrocnemius muscles at 2 months of age. Muscle fiber size grouping did not reveal an increase in fiber size upon 500 µg Ncald-ASO reinjection in SMA animals compared to CTRL-ASO SMA mice (Figure 2F).
2.3. Testing of human NCALD-ASOs in MNs derived from SMA and control hiPSCs
Based on our positive results with SMN- and Ncald-ASO long-term combinatorial therapy in SMA mice, we next aimed to develop a similar approach for humans. Therefore, a full battery of human NCALD-ASO were designed and synthesized, which were tested for non-toxicity and efficiency in various cellular systems. Next, the three top hits were investigated for toxicity and tolerability in MNs derived from SMA and control hiPSCs (Figures S1 and S2). The MN differentiation protocol was based on dual SMAD inhibition (Figure S3A) carried out in two SMA type I lines (HGK1 and CS32iSMA) and two control hiPSC (HUVEC and WTC11) lines. At day 15 of the differentiation, MN cultures were composed of about 60% MNs, as verified by staining with the MN marker ISL1 (Figure S3B). Next, we tested the efficacy and tolerability of NCALD-ASO candidates in hiPSC derived MNs. NCALD-ASOs were tested simultaneously in control MNs (HUVEC) derived from the same differentiation, in order to detect possible side effects of the ASOs. MN cultures were treated at day 13 with 60 nM of each NCALD-ASO and proteins were collected at day 20 to assess NCALD reduction by western blot. At day 20 of the differentiation, all NCALD-ASOs significantly downregulated NCALD levels in control MNs as determined by western blot (Figure 3A and B). Treatment with 60 nM of NCALD-ASO55 and NCALD-ASO69 had comparable knockdown efficiencies, reducing NCALD to 51% and 46%, respectively (Figure 3B).
Treatment with 60 nM of NCALD-ASO89 further reduced NCALD to 30% in control MNs (Figure 3B). Surprisingly, the protein amount of the MN marker ISL1 was significantly reduced upon treatment with 60 nM of NCALD-ASO55 and NCALD-ASO89 in comparison to CTRL-ASO and NCALD-ASO69 treated MNs (Figure 3A and 3C). Moreover, SMN was also reduced after NCALD-ASO55 and NCALD-ASO89 treatment when compared to CTRL-ASO or NCALD-ASO69 treated MNs (Figure 3A and D). Strikingly, axonal swelling and some signs of degeneration were observable in the MNs treated with NCALD-ASO55 or NCALD-ASO89 in comparison to CTRL-ASO-treated cells, suggesting some off-target effects that produce toxicity in the neurons (data not shown). This supports the use of 60 nM NCALD-ASO69 to assess the therapeutic effect of NCALD reduction in hiPSC derived MNs.
Next, we tested the efficiency of 60 nM NCALD-ASO69 in the other hiPSC derived MNs, where no significant reduction of ISL1 protein was detected (Figure 3E, F and G). In addition, NCALD reduction was detected in both soma and growth cones of SMA MNs (Figure S4). Thus, we identified NCALD-ASO69 as an efficient and non-toxic ASO that enables NCALD reduction in all four tested hiPSCs derived MNs. Moreover, NCALD downregulation can be detected at the growth cones of the MNs, which are SMA relevant sites. To test the therapeutic potential of NCALD reduction in MNs derived from SMA hiPSCs, we decided to treat the neuronal cultures with 60 nM of NCALD-ASO69 at day 13, and perform the functional and morphological experiments at day 20 of the differentiation.
2.4. Treatment with NCALD-ASO69 Influences Growth Cone Morphology
SMN deficiency leads to axonal outgrowth defects, smaller growth cones and defects in local translation of different actin isoforms, resulting in impaired actin dynamics that affect the presynaptic terminal [41,42]. Interestingly, NCALD is highly abundant in spinal MNs [43] and its reduction promotes neurite outgrowth in Smn-deficient neuronal cells even in the absence of retinoic acid, and axonal growth in smn-depleted zebrafish [28]. Importantly, NCALD directly binds to actin and tubulin, two major components of the cytoskeleton [44]. Here, we investigated if NCALD reduction might have an impact on cytoskeleton dynamics at the growth cone of hiPSC-derived MNs.
First, growth cone structure was classified based on actin morphology in three categories: blunt, filopodia and lamellipodia (Figure 4A) [45]. SMA type I MNs (HGK1 and CS32iSMA) exhibited more growth cones with blunt actin filaments compared to the control lines (Figure 4A). Instead upon NCALD-ASO69 treatment, more growth cones displayed filopodial and lamellipodial actin morphologies and thus were more mature. Surprisingly, NCALD-ASO69 also had an effect in growth cone morphology in one of the control lines (HUVEC), suggesting the NCALD might have a role in actin dynamics independently of the SMA background. Interestingly, no effect was detected in MNs derived from the control WTC11 hiPSC line, suggesting that the effect of NCALD in actin dynamics might differ between individuals.
Second, microtubule morphology was categorized based on whether the microtubules of the central domain displayed a bundled, spread or looped conformation (Figure 4B) [46]. Analysis of the microtubule conformation resulted in marked presence of bundled microtubules in the SMA type I lines compared to the control MNs treated with CTRL-ASO. The number of bundled microtubules was drastically reduced in favor of spread and looped microtubules in the SMA lines upon NCALD-ASO69 treatment. No changes were observed in control lines. These data strongly support the role of NCALD reduction on actin and microtubule cytoskeleton modulation in SMA and therefore, growth cone dynamics, which is crucial for neurite outgrowth and NMJ maturation and function. Moreover, these results go in line with our findings on the improvement of NMJ innervation upon Ncald-ASO treatment in SMA mice, highlighting the potential of NCALD reduction as a promising therapeutic approach that targets one of the key pathological features in SMA, the NMJ.
2.5. Treatment with NCALD-ASO69 Increases Neuronal Activity
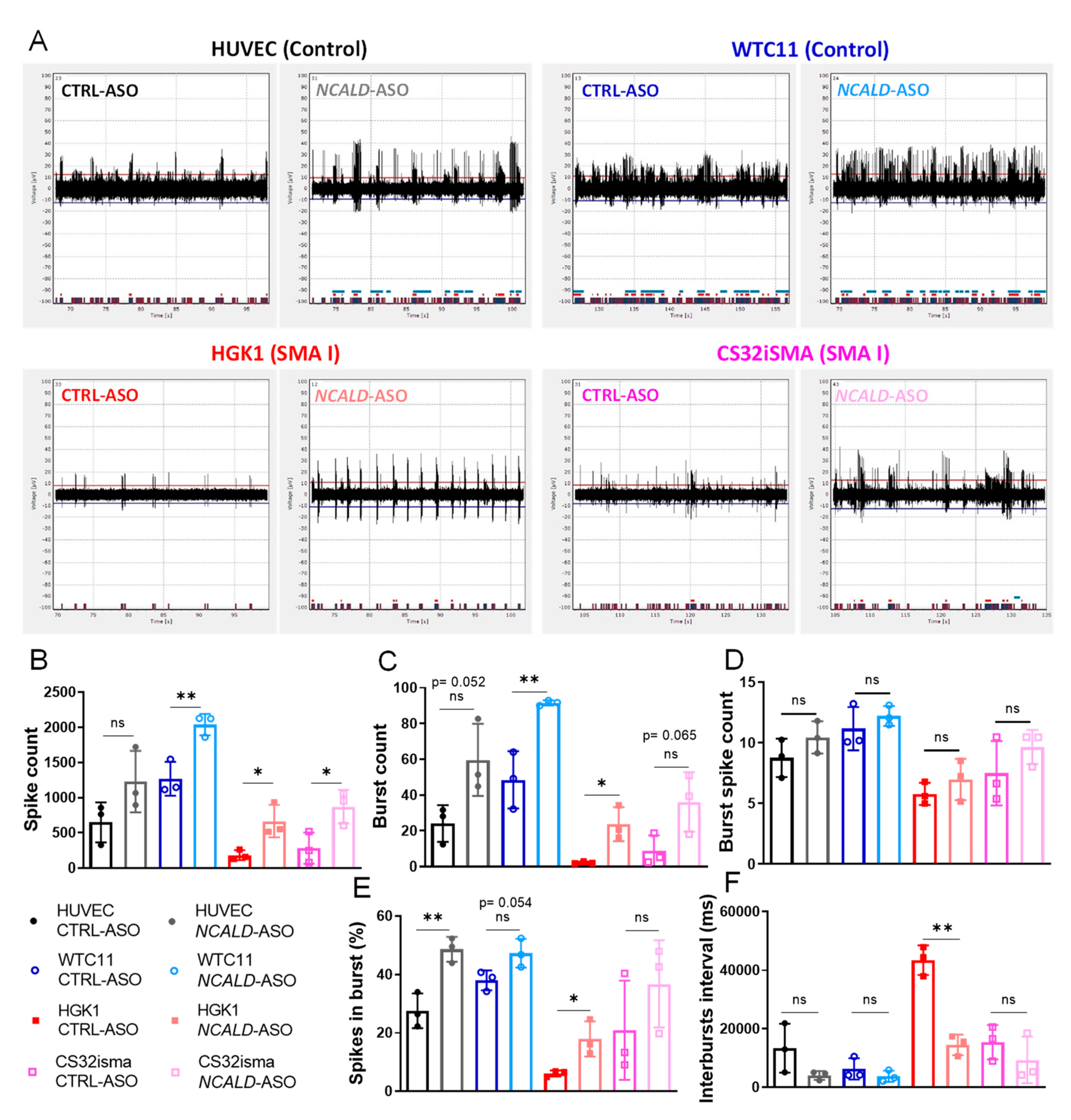
To assess the therapeutic potential of 60 nM NCALD-ASO69 treatment in hiPSC derived MNs from SMA patients, we recorded the spontaneous firing of treated control and SMA type I hiPSC derived MNs at day 20 of differentiation using a multi-electrode array system (MEA). As expected, spontaneous neuronal activity determined by the number of spikes was reduced in SMA type I MNs compared to healthy control MNs treated with CTRL-ASO (Figure 5A, B and Figure S5A). Importantly, both SMA MN lines treated with NCALD-ASO69 exhibited an increase in number of spikes, demonstrating the protective effect of NCALD downregulation on neuronal activity. In addition, SMA MN cultures had fewer active electrodes than the control MN lines, a parameter that was improved after NCALD-ASO69 treatment (Figure S5A). Moreover, the number of bursts were markedly reduced in SMA MNs compared to the healthy control lines (Figure 5B), a parameter that was ameliorated upon NCALD-ASO69 treatment.
Interestingly, an increase in active MNs was observed after NCALD-ASO69 treatment independently of the genotype, as determined by the parameters evaluated: spike count, burst count, burst spike count, percentage of spikes in bursts and interbursts interval (Figure 5B-F; Figure S5A and B). In general, it seems that NCALD reduction increased the spike height in all MN lines (Figure 5A). The control WTC11 MNs and the SMA MNs exhibited a significant increase of spike numbers (Figure 5B) and burst numbers (Figure 5C) but not overall number of burst spike counts (Figure 5D) upon NCALD-ASO69 compared to CTRL-ASO treatment. Furthermore, the control HUVEC line and the SMA type I lines exhibited an increase in the percentage of spikes (Figure 5E) that are present in a burst after treatment with 60 nM NCALD-ASO69. Finally, NCALD-ASO69 had a strong impact in the reduction of the interval between neuronal bursts in the SMA type I line, HGK1 (Figure 5F). These results further support the relevant role of NCALD in neuronal activity and synaptic connectivity, and are consistent with our previous results showing amelioration of the electrophysiological properties of the gastrocnemius muscle in SMA mice upon Ncald-ASO re-injection treatment. Notably, the SMA type I line, HGK1 seems to be more sensitive to NCALD reduction than the SMA type I line, CS32iSMA, and a similar phenomenon is observed between the control MNs, HUVEC and WTC11. These data emphasize the importance of testing compounds in hiPSC derived MNs, which better resemble the human genetic diversity, in order to provide a better understanding of which patients might respond to a treatment and which ones will most probably not.
Figure 5.
Multielectrode array shows increase in neuronal firing upon NCALD-ASO69 treatment. (A) Representative images of the activity detected by one electrode for 30 seconds per condition. (B-F) MEA analysis of various parameter (B) spike count, (C) burst count, (D) burst spike count, (E) spikes in burst and (F) interbursts interval measured at day 20 of CTRL-ASO or NCALD-ASO69-treated MNs. Dots in the graphs represent the average values of the technical replicates of each independent experiment, in total, three independent experiments corresponding to three independent MN differentiations (N=3, n=3-4). Unpaired two-tailed Student’s t test was performed in each line to compare CTRL-ASO versus NCALD-ASO treatment. All values reported as mean and error bars represent ± SD. *p≤0.05, **p ≤0.01.
Figure 5.
Multielectrode array shows increase in neuronal firing upon NCALD-ASO69 treatment. (A) Representative images of the activity detected by one electrode for 30 seconds per condition. (B-F) MEA analysis of various parameter (B) spike count, (C) burst count, (D) burst spike count, (E) spikes in burst and (F) interbursts interval measured at day 20 of CTRL-ASO or NCALD-ASO69-treated MNs. Dots in the graphs represent the average values of the technical replicates of each independent experiment, in total, three independent experiments corresponding to three independent MN differentiations (N=3, n=3-4). Unpaired two-tailed Student’s t test was performed in each line to compare CTRL-ASO versus NCALD-ASO treatment. All values reported as mean and error bars represent ± SD. *p≤0.05, **p ≤0.01.
3. Discussion
Since the discovery of the SMA disease-causing gene in 1995 [6], there have been significant advances in the understanding of the complex pathomechanisms underlying SMA. Most importantly, all the efforts facilitated the development of FDA- and EMA-approved therapies for SMA that changed the natural history of the disease [3,4,47]. Development of drugs that enhance SMN levels are without any doubt the most straightforward therapeutic strategy for SMA, and have shown impressive results in patients. However, even when the therapy is administered at pre-symptomatic stages, SMN-enhancing compounds might be still insufficient to completely counteract disease progression [23,24,25]. Moreover, a recent study has demonstrated that long-term overexpression of AAV9-SMN1 in a SMA mouse model induces a dose-dependent late-onset motor dysfunction characterized by loss of proprioceptive synaptic transmission and neurodegeneration. These observations highlight how crucial it is to understand the temporal requirements of SMN life-long: highest during neonatal life while NMJs undergo maturation and lower during adulthood [20,48]. Combinatorial approaches targeting SMN-dependent and -independent pathways disturbed in SMA might ameliorate functions or symptoms that cannot be improved by the increase of SMN solely. In this regard, SMA protective modifiers represent a unique opportunity to further ameliorate or rescue SMA independently of SMN upregulation. Indeed, pharmacological reduction of the genetic modifier NCALD using ASOs in combination with suboptimal dose of SMN-ASO has shown to ameliorate SMA pathology hallmarks in SMA mice at PND21. However, the therapeutic effect of Ncald-ASOs was rather short due to short-time stability of Ncald-ASOs [30].
In the present study, our main goal was to determine if Ncald-ASO re-injection could prolong the therapeutic effect observed at PND21 in SMA mice. Moreover, we wanted to assess the therapeutic effect of human NCALD-ASOs in hiPSC-derived MNs from control individuals and SMA type I patients. In summary, our main findings demonstrate: (i) Re-injection of 500 µg Ncald-ASO via i.c.v. bolus injection was well tolerated by the animals and significantly reduced NCALD levels in the CNS; (ii) Combinatorial therapy with Ncald-ASO re-injection in SMA mice ameliorates electrophysiological defects and denervation in the long-term compared to a single injection; (iii) Newly developed human NCALD-ASOs significantly downregulated NCALD in hiPSCs-derived MNs, but only NCALD-ASO69 was well tolerated; (iv) NCALD-ASO69 has a positive impact on growth cone cytoskeleton dynamics of SMA type I MNs; (v) NCALD-ASO69 increased neuronal activity in SMA type I MNs.
3.1. Ncald-ASO re-injection prolongs amelioration of electrophysiological defects and NMJ pathology in SMA mice
Electrophysiological measurements (CMAP, MUNE) of the gastrocnemius muscle were significantly reduced in various SMA animal models even upon injection with low-dose SMN-ASOs [30,37,49], implying a decreased motor functionality. In the previous combinatorial therapy approach, using Ncald-ASO and low-dose SMN-ASO, one single injection of Ncald-ASO at PND2 was not sufficient to ameliorate electrophysiological defects long-term [30]. Importantly, here we demonstrated that NCALD reduction achieved by re-injection of Ncald-ASO at PND28, significantly increased CMAP amplitude and motor unit numbers in a long-term fashion. These results indicate that long-term NCALD reduction not only improves the functionality and number of motor units, but also prevents MNs from degeneration. Consequently, preserving neuromuscular function over time most probably halts the progression of the disease, alleviates muscle atrophy and improves muscle function [50]. Interestingly, SMN reduction leads to selective vulnerability of MN pools and muscles [51]. Therefore, it would be essential to investigate the effect of NCALD reduction by analyzing electrophysiological parameters of other affected muscles, for example, the proximal muscle quadratus lumborum.
NMJ denervation and loss of function are key hallmarks of SMA and other neuromuscular disorders such as ALS [52]or Myasthenia gravis [53]that lead to skeletal muscle atrophy. NMJs form during embryonic development and undergo complex maturation steps even following birth [50]. In SMA, MNs and NMJs show early pathological defects, including altered morphology and function that precede MN death. Importantly, MN loss is an irreversible pathogenic event, while NMJs have a strong plasticity and can further improve their functionality by axonal sprouting. In the present work, NMJ area from the TVA muscle was not increased after re-injection with Ncald-ASO compared to NMJ area of animals injected with CTRL-ASO. Instead, long-term treatment of SMA animals with Ncald-ASO showed a significant rescue of the NMJ denervation when compared to animals treated with CTRL-ASO. These results further suggest that the observed amelioration in the electrophysiological parameter CMAP is probably due to an increase of NMJs that are fully innervated and functional. Moreover, the MUNE results indicate that NCALD reduction seems to protect against denervation at the NMJ level (a dying-back phenomenon), which allows the maintenance of motor unit numbers over time. This evidence strongly emphasizes the therapeutic role of NCALD reduction in the NMJ pathology, and the importance of re-injections to prolong the beneficial effect.
3.2. NCALD-ASO69 treatment improves cytoskeleton dynamics and neuronal activity in hiPSC derived MNs
Contrary to SMN-ASOs, which could be developed and then used in both, SMA mice, which carry the human SMN2 gene and in SMA patients [47,54,55] , the orthologous NCALD genes differ in mice and humans and therefore, human specific NCALD-ASOs had to be developed and tested. We designed, developed and tested a full battery of NCALD-ASOs and selected the three best hits showing the least toxicity and highest efficacy to reduce NCALD levels in various cell types. However, when testing these in MNs differentiated from hiPSC lines, only NCALD-ASO69 proved to be non-toxic and well tolerated.
MNs develop very long axons with distal growth cones which are highly plastic and constantly changing structures dependent of cytoskeleton dynamics that during neuronal development respond to intra- and extracellular cues and are the driving force of axonal outgrowth towards the target [56,57]. . Actin cytoskeleton dynamics plays an important role in neurodegeneration through involvement in axonal functions and synapse maintenance [58]. In SMA, SMN reduction leads to axonal outgrowth defects reduced growth cone size and defects in local translation of different actin isoforms, which results in defective actin dynamics [41,42]. Interestingly, NCALD binds to key components of the cytoskeleton, actin and tubulin [44], and its reduction promotes neurite outgrowth [28]. Since MN growth cones represent such a critical component of the future synapse with the skeletal muscle, and MN degeneration implies NMJ pathology [59], we analyzed growth cone morphology upon NCALD-ASO69 treatment in more detail. Analysis of actin morphology revealed a significant reduction of the number of blunt growth cones and an increase in filopodia and lamellipodial terminal ends upon NCALD-ASO69 treatment. These results demonstrate that NCALD reduction has a positive impact on growth cone actin dynamics and decreases the number of neurons with defective axonal outgrowth. Next, microtubule morphology was categorized according to the shape in the central domain of the growth cone: bundled (characterized by thin static microtubules), spread or looped [46]. Excitingly, NCALD-ASO69 treatment resulted in significant increase of spread and looped conformations in both control and SMA hiPSC-derived MNs compared to CTRL-ASO-treated MN cultures, which goes in line with the findings on actin morphology. In addition, this data strongly corroborates the protective role of NCALD reduction at the NMJ level observed in the mouse model.
In addition, we investigated if NCALD-ASO69 treatment has an impact on neuronal activity of MN cultures, since decreased neuronal firing rate is one SMA hallmark, which not only affects MNs but the overall neuronal circuits for motor control [60,61,62,63]. As expected, SMA MNs exhibited reduced spontaneous neuronal activity measured with the multielectrode array at day 20 of the differentiation when compared to control MNs. Moreover, pharmacological NCALD reduction had a significant increase in parameters associated with neuronal activity such as spike count, burst count and percentage of spikes in a burst in SMA and control MNs. Moreover, the time between bursts was significantly reduced in NCALD-ASO69-treated SMA MNs. Interestingly, MNs of the control WTC11 and the SMA HGK1 lines responded better to the treatment than MNs from HUVEC and CS32iSMA. These data emphasize the importance of integrating hiPSC models in the preclinical studies, since hiPSC-derived from patients represent the full array of human genetic variability.
In conclusion, NCALD reduction in hiPSC derived MNs increases neuronal activity, and supports the results obtained in the preclinical study, where the combination of SMN- and Ncald-ASOs ameliorate electrophysiological defects in SMA mice.
4. Materials and Methods
4.1. Mouse model and genotyping
The severely-affected Taiwanese SMA mouse model [FVB.Cg-Tg (SMN2)2Hung Smn1tm1Hung/J, stock number 005058] [64]was purchased from Jackson Laboratory (Bar Harbor, ME, USA). These homozygous SMA Taiwanese mice were originally on congenic FVB/N background, but we backcrossed them for >7 generations with C57BL6/N wild-type mice to obtain a congenic C57BL6/N background [65]. An intermediate mouse model was produced by crossing homozygous females on FVB/N background with male C57BL6/N Smnko/wt to generate the mixed50 SMA mice. This breeding results in a F1 offspring of ~50% severe SMA (Smnko/ko; SMN2tg/0) mice and the corresponding healthy HET (Smnko/wt; SMN2tg/0) mice were generated [12]. Here, we generated a mild SMA mouse model by subcutaneously injecting the F1 offspring with a suboptimal dose of 30µg SMN-ASO in the skin fold of the neck at PND1 using a microliter syringe (Hamilton) [30,33,37] . Approximately equal numbers of male and female mice were used for all the experiments. For genotyping, the primers used were as follows: SmnKOfw: 5’-ATAACACCACCACTCTTACTC-3’; SmnKOrev1: 5’-AGCCTGAAGAACGAGATCAGC-3’; SmnKOrev2: 5’-TAGCCGTGATGCCATTGTCA-3’. Animals were maintained under a 12-hour light/dark cycle with access to food and water ad libitum. Breeding, housing, and experimental use of animals were performed in a pathogen free environment. All mouse experiments were approved by the local animal protection committee LANUV NRW (Landesamt für Natur, Umwelt und Verbraucherschutz) under the reference number 81-02.04.2019.A138.
4.2. Antisense oligonucleotides (ASOs)
ASOs were designed and synthesized by IONIS Pharmaceuticals. Lyophilized stocks of SMN-ASO (ATTCACTTTCATAATGCTGG) were reconstituted with 1X PBS and stored in 10 mg/mL at -20°C for in vivo injections. As a control for the i.c.v. treatment, we used CTRL-ASO (GTTTTCAAATACACCTTCAT). To downregulate Ncald in the CNS, the previously described 5-10-5 2’-O-methoxyethyl (MOE) gapmer with mixed PS/PO backbone Ncald-ASO was used (GTGGTTCTTGTTTTACAGGA) [30]. For in vitro treatment of hiPSCs derived MNs, the following 3-10-3 constrained ethyl (cEt)-modified gapmer NCALD-ASOs were used: NCALD-ASO55 (GACAGATATGACTTCC), NCALD-ASO69 (CACATAGATTAAACCA), NCALD-ASO89 (TCTTTTTGGTCTACCA) and CTRL-ASO (GGCCAATACGCCGTCA).
4.3. ASOs injection in vivo
Neonatal mice (PND2) were i.c.v. injected in the right hemisphere, between the confluence of sinuses with 100 µg of Ncald-ASO or CTRL-ASO using a glass needle [66]. ASOs concentration was determined photometrically (AD260) in order to administer 1.5 µl (1 µl ASO + 0.5 µl of 0.05% w/v trypan blue in PBS). For Ncald-ASO re-injections, i.c.v. bolus injection was performed at PND28. Animals were anesthetized using Ketamine (Ketaset 100 mg/ml, WDT Wirtschaftsgenossenschaft deutscher Tierärzte) / Xylazine (2%, Serumwerk Bernburg AG, Bernburg) and placed in a stereotaxic instrument (Bilaney, Cat# DKI940) with a mouse adaptor (Bilaney, Cat# DKI926) and a micropositioner (Bilaney, Cat#DK5000) using a thermostatic warming plate to maintain the body temperature during the procedure. Next, injection coordinates were determined from the mouse bregma (For HET: X= 1mm, Y= 0.3mm; for SMA X= 0.980mm, Y= 0.250mm; in all the Z= range between -1.6mm and -1.7mm) [67] . To drill a hole in the skull, we used a microdrill (burr size: 0.8 mm, KF Technology). A total of 5 µl of ASO was delivered at a rate of 1 µl/30 seconds.
4.4. Experimental design
All offspring of each litter were blinded-injected and processed by randomized numbering. The experimenter was blinded regarding genotypes of the mice at all steps until final statistical analysis. Experimenter was blinded regarding cell line genotypes during image acquisition, recordings and statistical analysis. We conducted all experiments at least in triplicates. Multielectrode array experiments with hiPSCs correspond to three independent differentiations, each of them with at least three technical replicates. Blinding was performed by randomized numbering by an independent person. To avoid pseudo replication, the mean value per animal and mean value of technical replicates was applied for statistical analysis.
4.5. Western blot
Brain and spinal cord were collected and lysed in RIPA buffer (Sigma) containing protease inhibitors (Complete Mini, Roche). The primary antibodies used: anti-beta-actin HRP-conjugated (Proteintech Cat# HRP-60008, RRID:AB_2819183), anti-ISL1 (mouse, Hybridoma Bank Cat# 39.4D5, RRID:AB_2314683), anti-NCALD (rabbit, Protein tech Cat# 12925-1-AP, RRID:AB_2149410), anti-SMN (mouse, BD Biosciences Cat# 610646, RRID:AB_397973). Signal was detected with rabbit-HRP-conjugated secondary antibody (Cell Signaling Technology Cat# 7074, RRID:AB_2099233) and mouse-HRP-conjugated secondary antibody (ɑ-mouse, Jackson ImmunoResearch Labs Cat# 115-035-003, RRID:AB_10015289) and the chemiluminescence reagent (Thermo Scientific).
4.6. Compound muscle action potential and motor unit number estimation
Compound muscle action potential (CMAP) and motor unit number estimation (MUNE) were recorded as previously described [49]. First, mice were anesthetized with inhaled isofluorane (1 l/min O2 flow rate, 5% isofluorane for induction, 1.5-2% maintenance) and temperature was maintained at 37°C with a thermostatic warming plate. Stimulation of the sciatic nerve at the proximal hind limb was achieved by placing the anode subcutaneously over the sacrum and the stimulation electrode (cathode) positioned subcutaneously at the sciatic notch. The active recording electrode (E1) was located subcutaneously at the proximal gastrocnemius muscle and the reference electrode (E2) was set at the metatarsal region of the right foot. Next, square-wave pulses of 0.1 ms with a <10 mA of intensity were applied to stimulate the sciatic nerve. Five repetitive CMAP responses were recorded per animal and the peak-to-peak amplitude was measured. To estimate MUNE, ten consecutive increments from the initial response were recorded and calculated as previously reported [30,49].
4.7. Analysis NMJ from the transversus abdominis (TVA)
The TVA muscle was fixed in 4% PFA for 20 min and stained according to standard immunohistochemistry protocols. Primary antibodies used: rabbit anti-NF-L (Cell Signaling Technology Cat# 2837, RRID:AB_823575), BTX-594 (Thermo Fisher Scientific Cat# B13423, RRID:AB_2617152). Secondary antibody rabbit AlexaFluor-488 (Thermo Fisher Scientific Cat# A-21206, RRID:AB_2535792). Muscles were mounted on microscope slides with Prolong Gold antifade reagent. NMJ size was quantified using ImageJ (RRID:SCR_003070). NMJ innervation was categorized according to the percentage of NF that innervates each end plate (BTX-positive). Equal or more than 80% was considered fully innervated, between 15% and 79% was counted as partially innervated and less than 15% denervated.
4.8. Muscle fiber analysis
Gastrocnemius muscles were embedded in paraffin and sectioned as described in [65] . Next, haematoxylin (Sigma-Aldrich, #MHS32) and eosin (ScyTek Laboratories, #EYQ999) staining was performed and muscle fiber area was determined with the ZEN software (RRID:SCR_013672 )(Zeiss).
4.9. hiPSCs lines
| Cell line | Phenotype | SMN1/SMN2 copies | Sex | Age sampling | Reprogrammed |
| HUVEC | Healthy control | 2/2 | male | fetal | Retrovirus |
| WTC11 | Healthy control | 2/2 | male | 30 years | Episomal plasmid |
| HGK1 | SMA I | 0/2 | female | 6 months | Retrovirus |
| CS32iSMA | SMA I | 0/2 | male | 7 months | Episomal plasmid |
hiPSCs lines: Healthy control HUVEC hiPSC line was a generous gift from the Kurian Lab [68]. Healthy control WTC11 hiPSC line was generated by the Conklin Lab and purchased from Erasmus MC iPS Core Facility. SMA type 1 HGK1 hiPSC line was generated by iPIERIAN [69]. SMA type 1 CS32iSMA hiPSC line was purchased from Cedars Sinai iPSC Core Facility.
4.10. hiPSC maintenance and differentiation into MNs
Human iPSCs were routinely cultured in feeder-free conditions using Matrigel™-coated plates with mTeSR1 pluripotent media (StemCell Technologies) supplemented with Pen/Strep. On the first day, mTeSR1 media was supplemented with 10 µM Y-27632 (S1049, Selleckchem) to enhance the survival of dissociated cells after passaging or cryopreservation. At 80-90% confluency, hiPSCs were either passaged to a new plate in lower density, cryopreserved or used to start a new MN differentiation. For hiPSC differentiation into MNs, we followed a protocol previously published [70] with some modifications. First, similar size squares (1-2 mm) were generated using a flame-modified Pasteur glass pipette in order to produce the clumps for embryoid body formation and cultures were treated with 3 mg/ml Collagenase type IV (Gibco, Thermofisher) at 37°C. Clumps were gently resuspended in EB formation media containing Essential™6 (Gibco, Thermofisher) medium supplemented with 10 µM Rock inhibitor and transferred to ultra-low attachment flasks (Corning) on an horizontal shaker (40 rpm) in the incubator at 37°C. For the first two days of the differentiation, neuronal basal media (DMEM/F12 Glutamax and Neurobasal supplemented with N2 and B27 without vitamin A) was supplemented with 3 µM CHIR99021 (4423, Tocris Bioscience), 0.2 µM LDN-193189 (S2618, Selleckchem), 40 µg SB431542 (1614, Tocris Bioscience), and 5 µM Y-27632 (S1049, Selleckchem). From day 3 on, neuronal basal media was supplemented with 0.1 µM retinoic acid (Sigma) and 500 nM SAG (Merck Millipore). From day 8 on until the end of the differentiation BDNF (10 ng/ml, Peprotech) and GDNF (10 ng/ml, Peprotech) were added to the media. From day 9 to 11 neuronal media was supplemented with 10 µM DAPT (2634, Tocris Bioscience), and from day 12-16 with 20 µM DAPT. Until day 11 included, media was changed every day. On day 11, clumps with MN progenitors were dissociated into single cells for plating on 20 µg/ml laminin coated wells. Every day, half of the media was changed. From day 17 on, maturation media containing exclusively 10 ng/ml of BDNF, GDNF and CNTF (Peprotech) was added. Every other day media was changed by replacing half of the medium.
4.11. NCALD-ASOs treatment in hiPSCs derived MNs
Cells were plated in a density of 1.2x106 cells in 6-well and 1x105 for multielectrode array experiments. Next, cells were transfected at day 13 with different concentration of NCALD-ASOs or CTRL-ASO using Lipofectamine 3000 transfection reagent, according to the manufacturer‘s protocol.
4.12. hiPSCs and MNs immunohistochemistry
Immunofluorescent stainings of cells were conducted using a standard protocol. For fixation, 4% PFA and 4% PFA supplemented with 4% sucrose was applied to hiPSCs and hiPSC derived MNs, respectively, for 10-15 min at room temperature. The primary antibodies used Anti-OCT 3/4 (mouse, Santa Cruz Cat# sc-5279, RRID:AB_628051), anti-SOX2 (mouse, Santa Cruz Cat# sc-365823, RRID:AB_10842165), anti-SMN (mouse, BD Biosciences Cat# 610646, RRID:AB_397973), anti-ISL1 (mouse, Hybridoma Bank Cat# 39.4D5, RRID:AB_2314683), anti-TUJI (rabbit, Abcam Cat# ab18207, RRID:AB_444319) and Phalloidin conjugated AlexaFluor568 (Thermo Fisher Scientific; A12380). Secondary antibodies used anti-rabbit AlexaFluor488 (Thermo Fisher Scientific Cat# A-21206, RRID:AB_2535792), anti-mouse AlexaFluor568 (Thermo Fisher Scientific Cat# A10042, RRID:AB_2534017).
4.13. Multielectrode array
Electrophysiological recordings at day 20 of the differentiation were performed with a multi-electrode array (Multichannels System) using a 24 well plate containing 12 gold electrodes per well as recently described [71]. Briefly, plates were coated using Poly-L-ornithine and 20 µg/ml laminin. At day 11 of the differentiation, after EBs dissociation, a total of 100.000 cells were seeded in each well, diluted in 60 µl of MN media day 11. Cells were left to settle for 2h before adding 1 ml of the corresponding MN media, in an incubator at 37°C and 5% CO. On day 13 of the differentiation, MNs were treated with 60 nM CTRL-ASO or NCALD-ASO using Lipofectamine 3000. Importantly, measurements at day 20 of the differentiation were taken before media change, since addition of fresh media transiently alters the electrophysiological properties of the MNs. Before each recording, the plate was left for 2 min on the recorder. Experimental recordings took place in a non-humidifier incubator at 37°C for 3 min. Data acquisition and analysis was performed with the Multi-Channel Suite software (Multichannels system) applying the following parameters: threshold for spike detection ± 10 µV, band pass filter with 100 Hz and 3 kHz cut-off frequencies. In order to detect the spikes, an adaptive threshold at 5.5 times the standard deviation of the estimated noise on each electrode was set. Three independent differentiations per line were considered for the statistical analysis.
4.14. Image acquisition and analysis
Fluorescence images of NMJs and cells were acquired with Zeiss microscope (AxioImager.M2) supplied with the Apotome.2 system to mimic a confocal microscope. Images were acquired as Z-stacks with 20X, 40X and 63X objectives. The quantitative analysis was performed with ZEN (RRID:SCR_013672) (Zeiss) and Fiji software. Bright field images for muscle fiber analysis were acquired with Zeiss microscope (Axioskop.2) equipped with AxioCamICc1 and 20X objective.
4.15. Image acquisition and analysis
Fluorescence images of NMJs and proprioceptive inputs were acquired with Zeiss microscope (AxioImager.M2) supplied with the Apotome.2 system to mimic a confocal microscope. Images were acquired as Z-stacks with 20X, 40X objectives. The quantitative analysis was performed with ZEN (RRID:SCR_013672) (Zeiss) and Fiji software. Bright field images for muscle fiber analysis were acquired with Zeiss microscope (Axioskop.2) equipped with AxioCamICc1 and 20X objective. Brain sections were imaged with Leica Slide Scanner (SCN400).
4.16. Statistics
Statistical analysis was performed using the software programs MS Excel 2016 (Microsoft) and GraphPad Prism 9 (GraphPad Software). Unpaired, two tailed Student’s t-tests and one-way ANOVA statistics tests with Tukey posthoc test for multiple comparisons were performed. To compare categories (NMJ innervation and growth cone analysis) χ2 test was performed, and for more than two comparisons, for normalization the p value was multiplied by the total number of comparisons. The figure legends depict the specific statistical test used, sample size and p-values, respectively.
5. Conclusions
Our data here further support the protective effect of NCALD reduction on MN and NMJ function in SMA in mice and in human cellular system. The newly developed human NCALD-ASO, that efficiently downregulates NCALD in human MNs, opens the avenue for combinatorial therapies in SMA patients.
6. Patents
BW holds a US patent 9,988,626 B2 approved June, 5, 2018, Neurocalcin Delta Inhibitors and Therapeutic and Non-Therapeutic Uses Thereof.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1: hiPSCs staining of pluripotency markers OCT3/4 and SOX2; Figure S2: SMN protein in hiPSCs and hiPSC derived MNs. Figure S3: MN differentiation protocol and efficiency. Figure S4: NCALD reduction in MNs upon treatment with NCALD-ASO69. Figure S5: Spikes and burst of healthy and SMA hiPSC derived MNs upon NCALD-ASO69 treatment.
Author Contributions
Conceptualization, B.W and CFB; design of experiments, AMB, RR, KKL and FR; experiments AMB, RR, EZ, KKL and EZ; figures, AMB.; writing, AMB and B.W.; All authors have read, corrected and agreed to the published version of the manuscript.
Funding
BW was supported by the German Research Foundation [Wi 945/17-1 (project 398410809); GRK1960 (project ID 233886668)]; SFB1451 (project 431549029 – A01), and the European Union’s Horizon 2020 Marie Skłodowska-Curie (project 956185; SMABEYOND) and the Center for Molecular Medicine Cologne (project C18).
Institutional Review Board Statement
The animal study protocol was approved by the local animal protection committee LANUV NRW (Landesamt für Natur, Umwelt und Verbraucherschutz) under the reference number 81-02.04.2019.A138.
Informed Consent Statement
Not applicable.
Data Availability Statement
No bioinformatic datasets were generated.
Acknowledgments
The authors thank to all members of the Wirth group for helpful advises, discussion and some reagents.
Conflicts of Interest
AMB, RR and EZ declare no conflict of interest. BW holds a US patent 9,988,626 B2 approved June, 5, 2018, Neurocalcin Delta Inhibitors and Therapeutic and Non-Therapeutic Uses Thereof. KKL, FR and CFB are employee of IONIS Pharmaceuticals.
References
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N Engl J Med 2021, 384, 915–923. [Google Scholar] [CrossRef]
- Darras, B.T.; Masson, R.; Mazurkiewicz-Beldzinska, M.; Rose, K.; Xiong, H.; Zanoteli, E.; Baranello, G.; Bruno, C.; Vlodavets, D.; Wang, Y.; et al. Risdiplam-Treated Infants with Type 1 Spinal Muscular Atrophy versus Historical Controls. N Engl J Med 2021, 385, 427–435. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 2017, 377, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med 2018, 378, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Wirth, B. Spinal Muscular Atrophy: In the Challenge Lies a Solution. Trends Neurosci 2021, 44, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu Rev Genomics Hum Genet 2020, 21, 231–261. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997, 16, 265–269. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord 2018, 28, 197–207. [Google Scholar] [CrossRef]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018, 28, 103–115. [Google Scholar] [CrossRef]
- Battaglia, G.; Princivalle, A.; Forti, F.; Lizier, C.; Zeviani, M. Expression of the SMN gene, the spinal muscular atrophy determining gene, in the mammalian central nervous system. Hum Mol Genet 1997, 6, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Coovert, D.D.; Le, T.T.; McAndrew, P.E.; Strasswimmer, J.; Crawford, T.O.; Mendell, J.R.; Coulson, S.E.; Androphy, E.J.; Prior, T.W.; Burghes, A.H. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 1997, 6, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim Biophys Acta Gene Regul Mech 2017, 1860, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Zilio, E.; Piano, V.; Wirth, B. Mitochondrial Dysfunction in Spinal Muscular Atrophy. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Cuartas, J.; Gangwani, L. R-loop Mediated DNA Damage and Impaired DNA Repair in Spinal Muscular Atrophy. Front Cell Neurosci 2022, 16, 826608. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Gillingwater, T.H. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med 2013, 19, 40–50. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Perenthaler, E.; Courtney, N.L.; Jordan, C.Y.; Shorrock, H.K.; van der Hoorn, D.; Huang, Y.T.; Murray, L.M.; Viero, G.; Gillingwater, T.H. Temporal and tissue-specific variability of SMN protein levels in mouse models of spinal muscular atrophy. Hum Mol Genet 2018, 27, 2851–2862. [Google Scholar] [CrossRef]
- Kariya, S.; Obis, T.; Garone, C.; Akay, T.; Sera, F.; Iwata, S.; Homma, S.; Monani, U.R. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest 2014, 124, 785–800. [Google Scholar] [CrossRef]
- Lauria, F.; Bernabo, P.; Tebaldi, T.; Groen, E.J.N.; Perenthaler, E.; Maniscalco, F.; Rossi, A.; Donzel, D.; Clamer, M.; Marchioretto, M.; et al. SMN-primed ribosomes modulate the translation of transcripts related to spinal muscular atrophy. Nat Cell Biol 2020, 22, 1239–1251. [Google Scholar] [CrossRef]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord 2019, 29, 842–856. [Google Scholar] [CrossRef]
- Vill, K.; Schwartz, O.; Blaschek, A.; Glaser, D.; Nennstiel, U.; Wirth, B.; Burggraf, S.; Roschinger, W.; Becker, M.; Czibere, L.; et al. Newborn screening for spinal muscular atrophy in Germany: clinical results after 2 years. Orphanet J Rare Dis 2021, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Farrar, M.A.; Muntoni, F.; Saito, K.; Mendell, J.R.; Servais, L.; McMillan, H.J.; Finkel, R.S.; Swoboda, K.J.; Kwon, J.M.; et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the Phase III SPR1NT trial. Nat Med 2022, 28, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Farrar, M.A.; Muntoni, F.; Saito, K.; Mendell, J.R.; Servais, L.; McMillan, H.J.; Finkel, R.S.; Swoboda, K.J.; Kwon, J.M.; et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial. Nat Med 2022, 28, 1390–1397. [Google Scholar] [CrossRef]
- Oprea, G.E.; Krober, S.; McWhorter, M.L.; Rossoll, W.; Muller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef]
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Lohr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. American journal of human genetics 2017, 100, 297–315. [Google Scholar] [CrossRef]
- Hosseinibarkooie, S.; Peters, M.; Torres-Benito, L.; Rastetter, R.H.; Hupperich, K.; Hoffmann, A.; Mendoza-Ferreira, N.; Kaczmarek, A.; Janzen, E.; Milbradt, J.; et al. The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype. American journal of human genetics 2016, 99, 647–665. [Google Scholar] [CrossRef]
- Torres-Benito, L.; Schneider, S.; Rombo, R.; Ling, K.K.; Grysko, V.; Upadhyay, A.; Kononenko, N.L.; Rigo, F.; Bennett, C.F.; Wirth, B. NCALD Antisense Oligonucleotide Therapy in Addition to Nusinersen further Ameliorates Spinal Muscular Atrophy in Mice. Am J Hum Genet 2019, 105, 221–230. [Google Scholar] [CrossRef]
- Wolff, L.; Strathmann, E.A.; Muller, I.; Mahlich, D.; Veltman, C.; Niehoff, A.; Wirth, B. Plastin 3 in health and disease: a matter of balance. Cell Mol Life Sci 2021, 78, 5275–5301. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadi, M.; Derdowski, A.; Kalloo, G.; Maginnis, M.S.; O’Hern, P.; Bliska, B.; Sorkac, A.; Nguyen, K.C.; Cook, S.J.; Poulogiannis, G.; et al. Decreased function of survival motor neuron protein impairs endocytic pathways. Proc Natl Acad Sci U S A 2016, 113, E4377–E4386. [Google Scholar] [CrossRef]
- Hosseinibarkooie, S.; Peters, M.; Torres-Benito, L.; Rastetter, R.H.; Hupperich, K.; Hoffmann, A.; Mendoza-Ferreira, N.; Kaczmarek, A.; Janzen, E.; Milbradt, J.; et al. The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype. Am J Hum Genet 2016, 99, 647–665. [Google Scholar] [CrossRef]
- Kaifer, K.A.; Villalon, E.; Osman, E.Y.; Glascock, J.J.; Arnold, L.L.; Cornelison, D.D.W.; Lorson, C.L. Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2017, 2, e89970. [Google Scholar] [CrossRef] [PubMed]
- Alrafiah, A.; Karyka, E.; Coldicott, I.; Iremonger, K.; Lewis, K.E.; Ning, K.; Azzouz, M. Plastin 3 Promotes Motor Neuron Axonal Growth and Extends Survival in a Mouse Model of Spinal Muscular Atrophy. Mol Ther Methods Clin Dev 2018, 9, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.B.; Janzen, E.; Wingrove, E.; Hosseinibarkooie, S.; Muela, N.R.; Davidow, L.; Dimitriadi, M.; Norabuena, E.M.; Rubin, L.L.; Wirth, B.; et al. Genetic modifiers ameliorate endocytic and neuromuscular defects in a model of spinal muscular atrophy. BMC Biol 2020, 18, 127. [Google Scholar] [CrossRef] [PubMed]
- Muinos-Buhl, A.; Rombo, R.; Janzen, E.; Ling, K.K.; Hupperich, K.; Rigo, F.; Bennett, C.F.; Wirth, B. Combinatorial ASO-mediated therapy with low dose SMN and the protective modifier Chp1 is not sufficient to ameliorate SMA pathology hallmarks. Neurobiol Dis 2022, 171, 105795. [Google Scholar] [CrossRef]
- Janzen, E.; Mendoza-Ferreira, N.; Hosseinibarkooie, S.; Schneider, S.; Hupperich, K.; Tschanz, T.; Grysko, V.; Riessland, M.; Hammerschmidt, M.; Rigo, F.; et al. CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis. Brain 2018, 141, 2343–2361. [Google Scholar] [CrossRef]
- Lewelt, A.; Krosschell, K.J.; Scott, C.; Sakonju, A.; Kissel, J.T.; Crawford, T.O.; Acsadi, G.; D’Anjou, G.; Elsheikh, B.; Reyna, S.P.; et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle Nerve 2010, 42, 703–708. [Google Scholar] [CrossRef]
- Arnold, W.D.; Porensky, P.N.; McGovern, V.L.; Iyer, C.C.; Duque, S.; Li, X.; Meyer, K.; Schmelzer, L.; Kaspar, B.K.; Kolb, S.J.; et al. Electrophysiological Biomarkers in Spinal Muscular Atrophy: Preclinical Proof of Concept. Ann Clin Transl Neurol 2014, 1, 34–44. [Google Scholar] [CrossRef]
- Rossoll, W.; Jablonka, S.; Andreassi, C.; Kroning, A.K.; Karle, K.; Monani, U.R.; Sendtner, M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 2003, 163, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Moradi, M.; Sivadasan, R.; Saal, L.; Luningschror, P.; Dombert, B.; Rathod, R.J.; Dieterich, D.C.; Blum, R.; Sendtner, M. Differential roles of alpha-, beta-, and gamma-actin in axon growth and collateral branch formation in motoneurons. J Cell Biol 2017, 216, 793–814. [Google Scholar] [CrossRef]
- Iino, S.; Kobayashi, S.; Hidaka, H. Neurocalcin-immunopositive nerve terminals in the muscle spindle, Golgi tendon organ and motor endplate. Brain Res 1998, 808, 294–299. [Google Scholar] [CrossRef]
- Ivings, L.; Pennington, S.R.; Jenkins, R.; Weiss, J.L.; Burgoyne, R.D. Identification of Ca2+-dependent binding partners for the neuronal calcium sensor protein neurocalcin delta: interaction with actin, clathrin and tubulin. Biochem J 2002, 363, 599–608. [Google Scholar] [CrossRef]
- Khazaei, M.R.; Girouard, M.P.; Alchini, R.; Ong Tone, S.; Shimada, T.; Bechstedt, S.; Cowan, M.; Guillet, D.; Wiseman, P.W.; Brouhard, G.; et al. Collapsin response mediator protein 4 regulates growth cone dynamics through the actin and microtubule cytoskeleton. J Biol Chem 2014, 289, 30133–30143. [Google Scholar] [CrossRef]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urban, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Van Alstyne, M.; Tattoli, I.; Delestree, N.; Recinos, Y.; Workman, E.; Shihabuddin, L.S.; Zhang, C.; Mentis, G.Z.; Pellizzoni, L. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat Neurosci 2021, 24, 930–940. [Google Scholar] [CrossRef]
- Arnold, W.D.; Sheth, K.A.; Wier, C.G.; Kissel, J.T.; Burghes, A.H.; Kolb, S.J. Electrophysiological Motor Unit Number Estimation (MUNE) Measuring Compound Muscle Action Potential (CMAP) in Mouse Hindlimb Muscles. J Vis Exp 2015. [Google Scholar] [CrossRef]
- Boyd, P.J.; Gillingwater, T.H. Axonal and Neuromuscular Junction Pathology in Spinal Muscular Atrophy. In Spinal Musculat Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 133–151. [Google Scholar]
- Woschitz, V.; Mei, I.; Hedlund, E.; Murray, L.M. Mouse models of SMA show divergent patterns of neuronal vulnerability and resilience. Skelet Muscle 2022, 12, 22. [Google Scholar] [CrossRef]
- Gromova, A.; La Spada, A.R. Harmony Lost: Cell-Cell Communication at the Neuromuscular Junction in Motor Neuron Disease. Trends Neurosci 2020, 43, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Tzartos, S.; Evoli, A.; Palace, J.; Burns, T.M.; Verschuuren, J. Myasthenia gravis. Nat Rev Dis Primers 2019, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 2008, 82, 834–848. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef]
- Fan, L.; Simard, L.R. Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet 2002, 11, 1605–1614. [Google Scholar] [CrossRef]
- Nolle, A.; Zeug, A.; van Bergeijk, J.; Tonges, L.; Gerhard, R.; Brinkmann, H.; Al Rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum Mol Genet 2011, 20, 4865–4878. [Google Scholar] [CrossRef]
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neuroscientist 2018, 24, 54–72. [Google Scholar] [CrossRef]
- Monani, U.R.; Coovert, D.D.; Burghes, A.H. Animal models of spinal muscular atrophy. Hum Mol Genet 2000, 9, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.V.; Simon, C.M.; Pagiazitis, J.G.; Chalif, J.I.; Vukojicic, A.; Drobac, E.; Wang, X.; Mentis, G.Z. Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat Neurosci 2017, 20, 905–916. [Google Scholar] [CrossRef]
- Tharaneetharan, A.; Cole, M.; Norman, B.; Romero, N.C.; Wooltorton, J.R.A.; Harrington, M.A.; Sun, J. Functional Abnormalities of Cerebellum and Motor Cortex in Spinal Muscular Atrophy Mice. Neuroscience 2021, 452, 78–97. [Google Scholar] [CrossRef]
- Simon, C.M.; Blanco-Redondo, B.; Buettner, J.M.; Pagiazitis, J.G.; Fletcher, E.V.; Sime Longang, J.K.; Mentis, G.Z. Chronic Pharmacological Increase of Neuronal Activity Improves Sensory-Motor Dysfunction in Spinal Muscular Atrophy Mice. J Neurosci 2021, 41, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, S.; Beck, M.; Lechner, B.D.; Mayer, C.; Sendtner, M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol 2007, 179, 139–149. [Google Scholar] [CrossRef]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A mouse model for spinal muscular atrophy. Nat Genet 2000, 24, 66–70. [Google Scholar] [CrossRef]
- Ackermann, B.; Krober, S.; Torres-Benito, L.; Borgmann, A.; Peters, M.; Hosseini Barkooie, S.M.; Tejero, R.; Jakubik, M.; Schreml, J.; Milbradt, J.; et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet 2013, 22, 1328–1347. [Google Scholar] [CrossRef] [PubMed]
- Glascock, J.J.; Osman, E.Y.; Coady, T.H.; Rose, F.F.; Shababi, M.; Lorson, C.L. Delivery of therapeutic agents through intracerebroventricular (ICV) and intravenous (IV) injection in mice. J Vis Exp 2011. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Miller, T.M. Direct intraventricular delivery of drugs to the rodent central nervous system. J Vis Exp 2013, e50326. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Ruiz, S.; Yi, F.; Herrerias, A.; Batchelder, E.M.; Izpisua Belmonte, J.C. Rapid and highly efficient generation of induced pluripotent stem cells from human umbilical vein endothelial cells. PLoS One 2011, 6, e19743. [Google Scholar] [CrossRef]
- Garbes, L.; Heesen, L.; Holker, I.; Bauer, T.; Schreml, J.; Zimmermann, K.; Thoenes, M.; Walter, M.; Dimos, J.; Peitz, M.; et al. VPA response in SMA is suppressed by the fatty acid translocase CD36. Hum Mol Genet 2013, 22, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovas, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat Commun 2017, 8, 861. [Google Scholar] [CrossRef]
- Delle Vedove, A.; Natarajan, J.; Zanni, G.; Eckenweiler, M.; Muinos-Buhl, A.; Storbeck, M.; Guillen Boixet, J.; Barresi, S.; Pizzi, S.; Holker, I.; et al. CAPRIN1(P512L) causes aberrant protein aggregation and associates with early-onset ataxia. Cell Mol Life Sci 2022, 79, 526. [Google Scholar] [CrossRef]
Figure 2.
Re-injection with 500 µg Ncald-ASO improves electrophysiological defects and NMJ innervation at 2 months. (A) Representative traces of CMAP response in HET and SMA mice in the gastrocnemius muscle at 2 months of age, 4-weeks after re-injection. (B) Quantification of sciatic CMAP response of the gastrocnemius muscle in the four analyzed experimental groups. For CMAP response, peak-to-peak amplitude was quantified. Each dot in the graph represents one animal, N=10-12. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. Error bars represent ± SD. *p≤0.05, **p≤0.01, ***p≤0,001. (C) Quantification of the gastrocnemius muscle MUNE in the four analyzed experimental groups. Each dot in the graph represents one animal, N=10-12. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. Error bars represent ± SD. *p≤0.05, **p≤0.01. (D) Transversus abdominis NMJ area. Representative images and quantification of NMJ area (below) at 2 months of age, showing postsynaptic NMJ region (BTX, magenta) and presynaptic nerve (NF, green) (scale bar: 50 μm). NMJ area was analyzed with ImageJ (N = 6, n = 100 NMJs/mouse). Statistics were performed with mean values of animals per group. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons, **p ≤ 0.01, ***p ≤ 0.001. (E) Transversus abdominis NMJ innervation. Representative images of TVA NMJ innervation in each category, showing the postsynaptic region (BTX, magenta) and presynaptic nerve (NF, green). NMJ innervation was classified in three categories according to the degree of innervation. Quantification (below) of all four experimental groups. HET CTRL-ASO (N=5, n=272), HET Ncald-ASO (N=5, n=270), SMA CTRL-ASO (N=5, n=259), SMA Ncald-ASO (N=6, n=322). Results are presented in percentages. *** denotes statistical significance p ≤ 0.001 (χ2 test). (F) Representative images of H&E staining and quantification (below) of gastrocnemius muscle fibers from HET and SMA animals at 2 months of age. Scale bar 50 µm. Gastrocnemius muscle size categorization according to area intervals of 100 µm2. In total, 5 animals were analyzed per genotype, and 100 muscle fibers per animal.
Figure 2.
Re-injection with 500 µg Ncald-ASO improves electrophysiological defects and NMJ innervation at 2 months. (A) Representative traces of CMAP response in HET and SMA mice in the gastrocnemius muscle at 2 months of age, 4-weeks after re-injection. (B) Quantification of sciatic CMAP response of the gastrocnemius muscle in the four analyzed experimental groups. For CMAP response, peak-to-peak amplitude was quantified. Each dot in the graph represents one animal, N=10-12. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. Error bars represent ± SD. *p≤0.05, **p≤0.01, ***p≤0,001. (C) Quantification of the gastrocnemius muscle MUNE in the four analyzed experimental groups. Each dot in the graph represents one animal, N=10-12. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. Error bars represent ± SD. *p≤0.05, **p≤0.01. (D) Transversus abdominis NMJ area. Representative images and quantification of NMJ area (below) at 2 months of age, showing postsynaptic NMJ region (BTX, magenta) and presynaptic nerve (NF, green) (scale bar: 50 μm). NMJ area was analyzed with ImageJ (N = 6, n = 100 NMJs/mouse). Statistics were performed with mean values of animals per group. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons, **p ≤ 0.01, ***p ≤ 0.001. (E) Transversus abdominis NMJ innervation. Representative images of TVA NMJ innervation in each category, showing the postsynaptic region (BTX, magenta) and presynaptic nerve (NF, green). NMJ innervation was classified in three categories according to the degree of innervation. Quantification (below) of all four experimental groups. HET CTRL-ASO (N=5, n=272), HET Ncald-ASO (N=5, n=270), SMA CTRL-ASO (N=5, n=259), SMA Ncald-ASO (N=6, n=322). Results are presented in percentages. *** denotes statistical significance p ≤ 0.001 (χ2 test). (F) Representative images of H&E staining and quantification (below) of gastrocnemius muscle fibers from HET and SMA animals at 2 months of age. Scale bar 50 µm. Gastrocnemius muscle size categorization according to area intervals of 100 µm2. In total, 5 animals were analyzed per genotype, and 100 muscle fibers per animal.
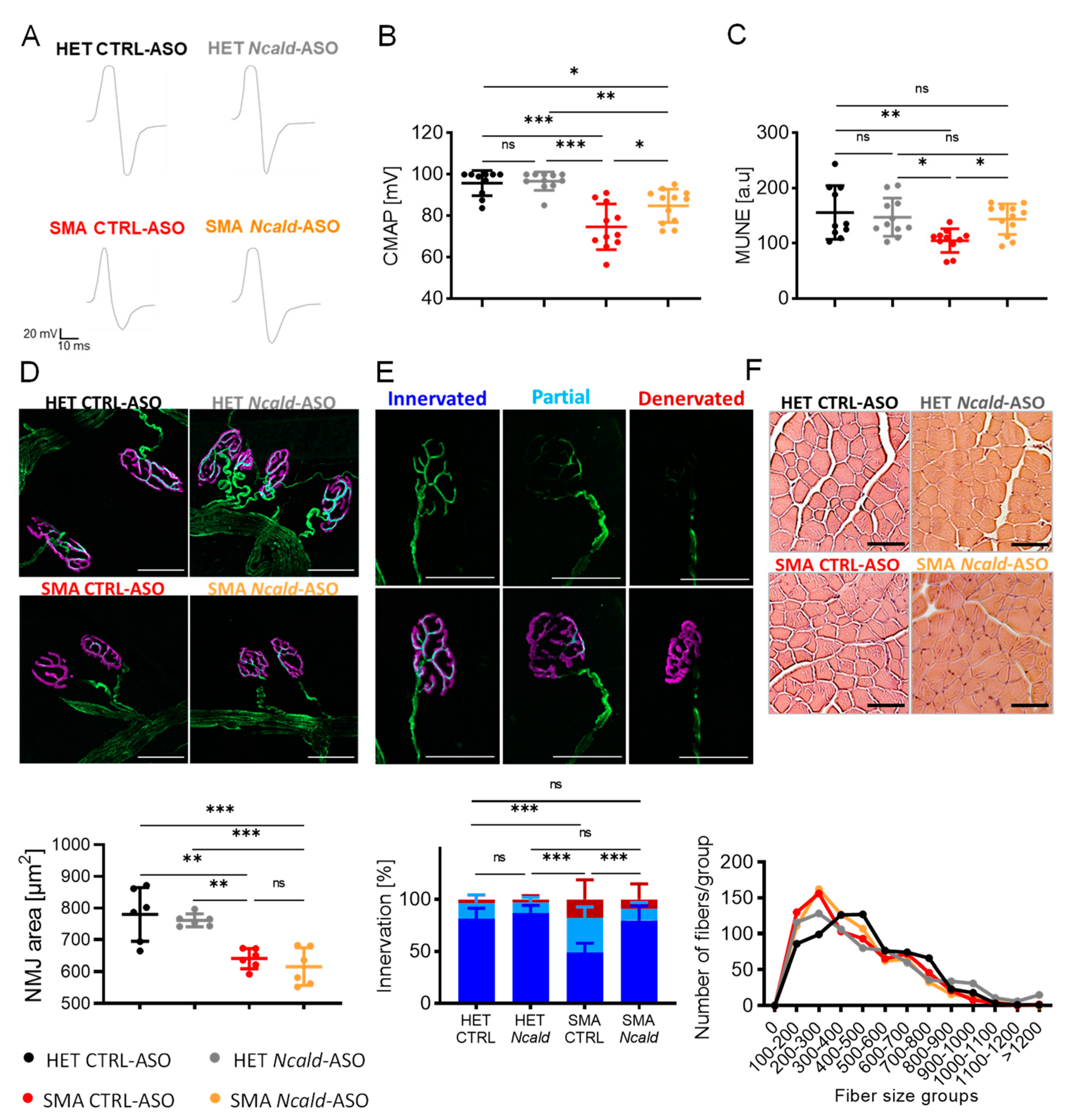
Figure 3.
Efficiency and tolerability of NCALD-ASO candidates 7 days after treatment in hiPSCs derived MNs. (A) Western blot of HUVEC hiPSCs derived MNs treated with 60 nM of CTRL-ASO or the respective NCALD-ASO candidate using Lipofectamine™ at day 13, proteins collected at day 20 of the differentiation. ACTB used as loading control. (B) Quantification of NCALD levels in HUVEC MNs at day 20 after treatment with different NCALD-ASOs. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. (C) Quantification of ISL1 levels in HUVEC MNs at day 20 after treatment with each NCALD-ASOs. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. (D) Quantification of SMN levels in HUVEC MNs at day 20 after treatment with each NCALD-ASOs. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. (E-G) Western blots of control WTC11 (E), SMA type I HGK1 (F) and SMA type I CS32iSMA (G) MNs treated at day 13 of the differentiation with 60 nM of CTRL-ASO or NCALD-ASO69 using Lipofectamine™. Proteins were collected at day 20 of the differentiation. NCALD protein levels are significantly reduced upon NCALD-ASO69 treatment in all lines. No significant reduction of ISL1 protein and thus impact on MNs was detected. Unpaired two-tailed Student’s t test was performed. All values reported as mean and error bars represent ± SD. N=3, each dot in the bar graphs represents an independent well. *p≤0.05, **p≤0.01, ***p≤0,001.
Figure 3.
Efficiency and tolerability of NCALD-ASO candidates 7 days after treatment in hiPSCs derived MNs. (A) Western blot of HUVEC hiPSCs derived MNs treated with 60 nM of CTRL-ASO or the respective NCALD-ASO candidate using Lipofectamine™ at day 13, proteins collected at day 20 of the differentiation. ACTB used as loading control. (B) Quantification of NCALD levels in HUVEC MNs at day 20 after treatment with different NCALD-ASOs. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. (C) Quantification of ISL1 levels in HUVEC MNs at day 20 after treatment with each NCALD-ASOs. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. (D) Quantification of SMN levels in HUVEC MNs at day 20 after treatment with each NCALD-ASOs. Ordinary one-way ANOVA with Tukey posthoc test for multiple comparisons. (E-G) Western blots of control WTC11 (E), SMA type I HGK1 (F) and SMA type I CS32iSMA (G) MNs treated at day 13 of the differentiation with 60 nM of CTRL-ASO or NCALD-ASO69 using Lipofectamine™. Proteins were collected at day 20 of the differentiation. NCALD protein levels are significantly reduced upon NCALD-ASO69 treatment in all lines. No significant reduction of ISL1 protein and thus impact on MNs was detected. Unpaired two-tailed Student’s t test was performed. All values reported as mean and error bars represent ± SD. N=3, each dot in the bar graphs represents an independent well. *p≤0.05, **p≤0.01, ***p≤0,001.
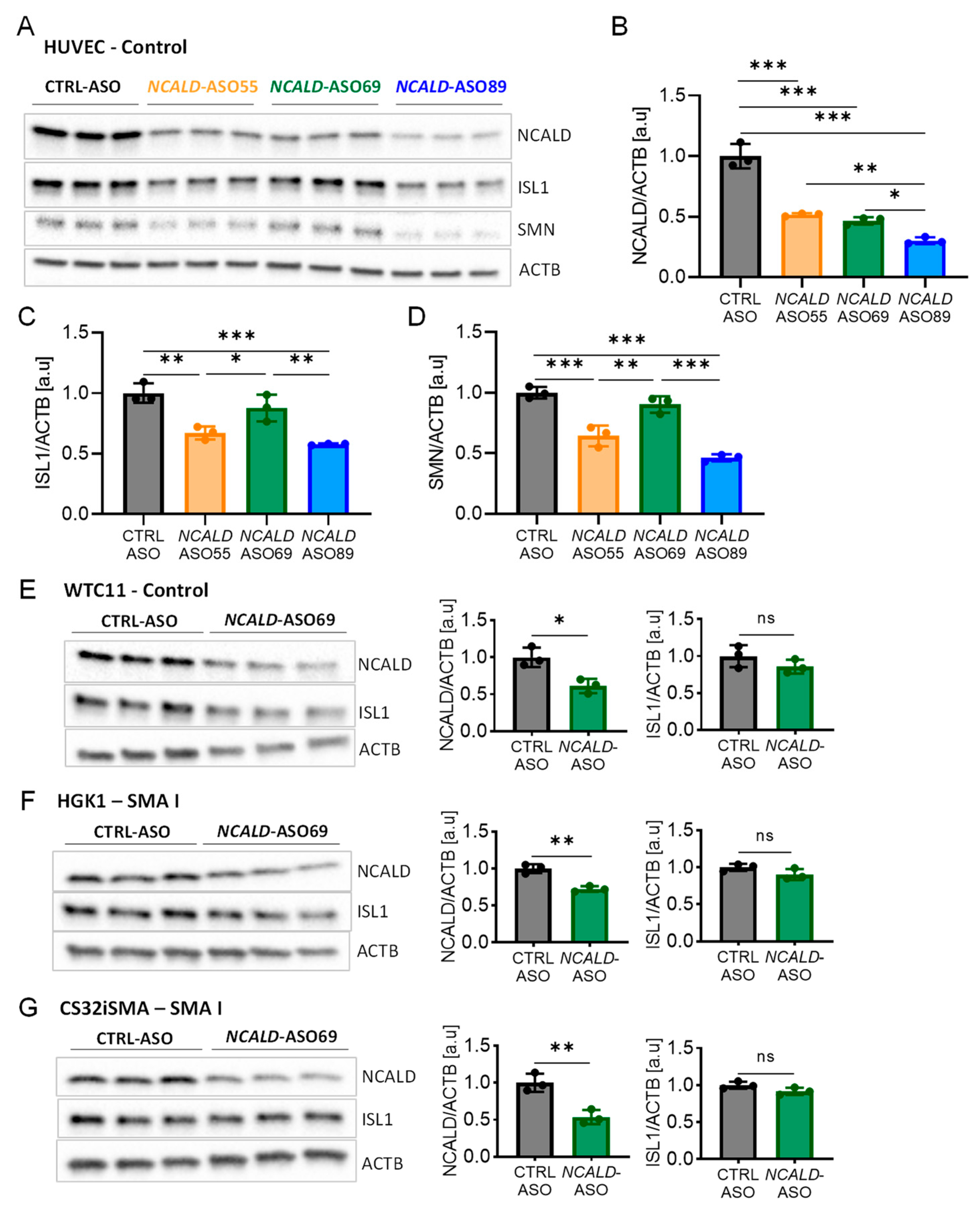
Figure 4.
Analysis of cytoskeleton morphology upon NCALD-ASO69 treatment in hiPSC-derived MNs shows more mature growth cones. (A) For actin morphology analysis, actin was stained using phalloidin (red) and microtubules were stained using β-III-tubulin (TUJ1, white). Actin morphology was categorized as blunt ended, filopodia or lamellipodia based on the morphology of the filaments. Scale bar 10 µm. (B) Microtubules were stained using β-III-tubulin (TUJ1, green), and actin was counterstained using phalloidin (white). Microtubule morphology was categorized as bundled, spread or looped. Scale bar 10 µm. In total, three independent experiments from three independent differentiation were performed per cell line. HUVEC (N=3; CTRL-ASO n=141, NCALD-ASO69 n=103), WTC11 (N=3; CTRL-ASO n=84, NCALD-ASO n=85), HGK1 (N=3; CTRL-ASO n=138, NCALD-ASO69 n=149), CS32isma (N=3; CTRL-ASO n=154, NCALD-ASO69 n=146). Error bars show ± SD. Graphs represent percentages. *p≤0.05, ***p≤0.001 (Chi-square test).
Figure 4.
Analysis of cytoskeleton morphology upon NCALD-ASO69 treatment in hiPSC-derived MNs shows more mature growth cones. (A) For actin morphology analysis, actin was stained using phalloidin (red) and microtubules were stained using β-III-tubulin (TUJ1, white). Actin morphology was categorized as blunt ended, filopodia or lamellipodia based on the morphology of the filaments. Scale bar 10 µm. (B) Microtubules were stained using β-III-tubulin (TUJ1, green), and actin was counterstained using phalloidin (white). Microtubule morphology was categorized as bundled, spread or looped. Scale bar 10 µm. In total, three independent experiments from three independent differentiation were performed per cell line. HUVEC (N=3; CTRL-ASO n=141, NCALD-ASO69 n=103), WTC11 (N=3; CTRL-ASO n=84, NCALD-ASO n=85), HGK1 (N=3; CTRL-ASO n=138, NCALD-ASO69 n=149), CS32isma (N=3; CTRL-ASO n=154, NCALD-ASO69 n=146). Error bars show ± SD. Graphs represent percentages. *p≤0.05, ***p≤0.001 (Chi-square test).
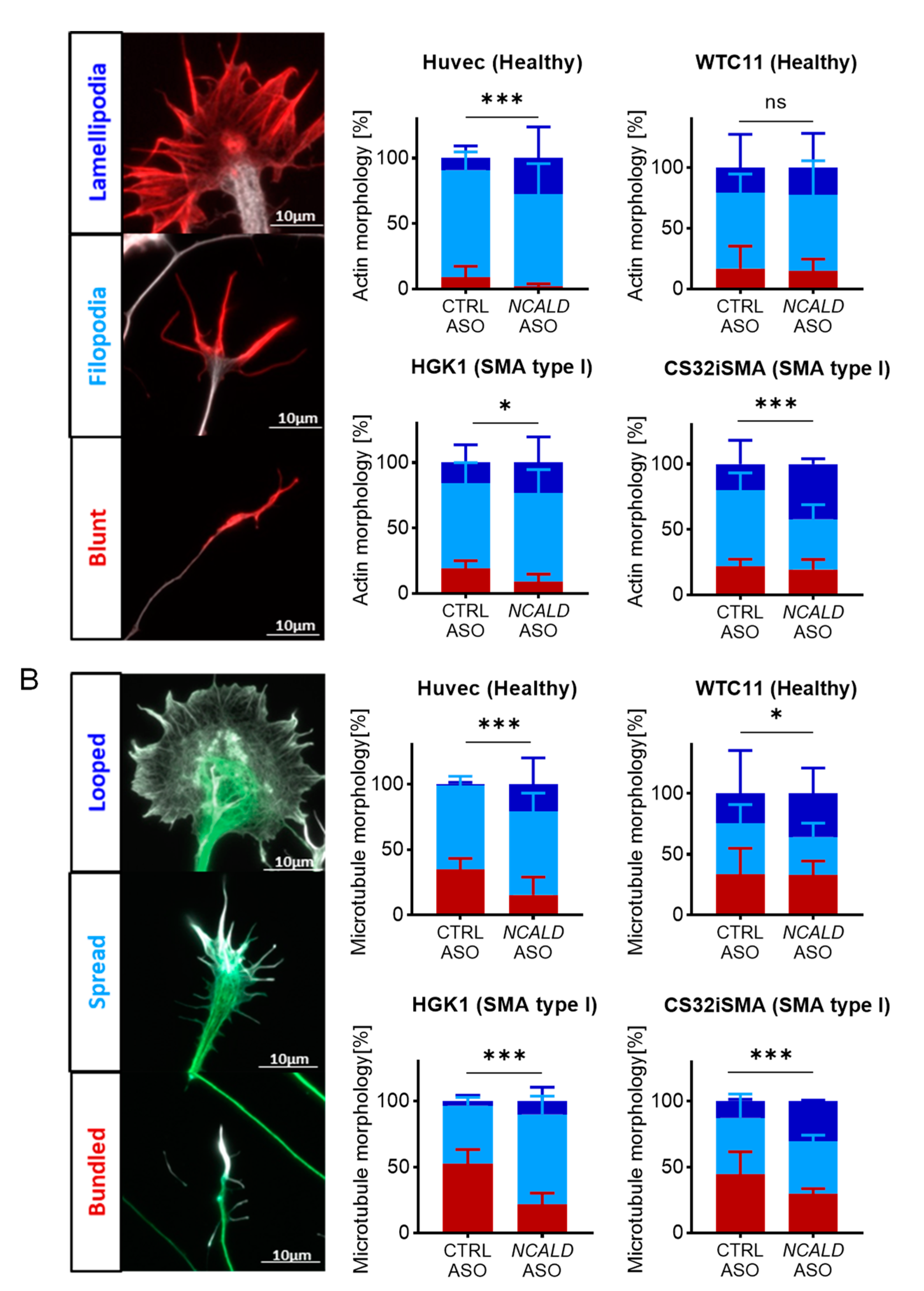
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



