Submitted:
31 January 2023
Posted:
02 February 2023
You are already at the latest version
Abstract
In cystic fibrosis (CF), pulmonary infection with Pseudomonas aeruginosa is a cause of increased morbidity and mortality, especially in patients for whom infection becomes chronic and there is reliance on long-term suppressive therapies. Current antimicrobials, though varied mechanistically and by mode of delivery, are inadequate not only due to eradication failure in many cases, but also because they do not halt the progression of lung function decline over time. One of the reasons for this failure is thought to be the biofilm mode of growth of P. aeruginosa, wherein self-secreted exopolysaccharides (EPSs) provide physical protection against antibiotics and an array of niches with resulting metabolic and phenotypic heterogeneity. Targeting the three EPSs secreted by P. aeruginosa (alginate, Psl and Pel) is currently under investigation as a way of disrupting the biofilm extracellular matrix to potentiate the action of antibiotics. In this review, we look at each EPS as a potential therapeutic target for combatting pulmonary infection with P. aeruginosa in CF, with a particular focus on the current evidence for these emerging therapies and barriers to bringing these therapies into clinic.
Keywords:
Cystic fibrosis
; Pseudomonas aeruginosa
; antibiotic adjunct
; anti-biofilm
; biofilm-degrading enzymes
; matrix exopolysaccharides
; alginate oligosaccharide
1. Introduction
Pseudomonas aeruginosa is a ubiquitous, Gram-negative bacteria that occupies environmental niches and is an opportunistic pathogen that poses risks to certain vulnerable groups of patients. Examples include people who are immunocompromised (including those with human immunodeficiency virus or neutropenia stemming from cancer chemotherapy), patients who are post-surgery, burn victims, and individuals with respiratory illnesses, including chronic obstructive pulmonary distress, primary ciliary dyskinesia, and most notably, cystic fibrosis (CF) [1,2,3,4]. CF is a genetic, multisystem disease that results in improper epithelial chloride ion transport. This leads to a dehydrated lung environment, impaired mucociliary escalator function, and the production of a sticky mucus that creates suitable grounds for colonization by a number of pathogens, including P. aeruginosa [5].
Within the lung, P. aeruginosa forms sputum-suspended aggregates (or biofilms) composed of exopolysaccharides (EPSs), proteins, and extracellular DNA (eDNA). This EPS matrix provides P. aeruginosa biofilm with a scaffolding framework which generates chemical and nutrient gradients that differentially affect cells, giving rise to the incredible metabolic and phenotypic diversity observed through the biofilm [6]. The EPS matrix also provides physical protection against antimicrobials and host immune cells. In vitro work with antibiotics has consistently demonstrated that biofilm-grown P. aeruginosa possess minimum inhibitory concentrations (MICs) that are many orders of magnitude higher than their planktonic counterparts [7,8]. Neutrophils face tremendous difficult in eradicating biofilm infections, which results in the accumulation of immune-mediated debris and an aberrant inflammatory response that progressively damages the lung tissue [9]. Managing biofilm infections is an arms race, and as P. aeruginosa gradually accumulates adaptations that strengthens its defenses and survivability, our antibiotic arsenal dwindles. In cases where attempts at eradication fail, the infection becomes chronic, leading to further inflammation and scarring. This damage clinically manifests as progressive lung function decline, decreased quality of life, and increased mortality in patients infected with this organism [10,11].
Current strategies for eradicating and suppressing P. aeruginosa biofilms rely on combination high dose antibiotic therapy to overcome its naturally high tolerance. However, this approach has several drawbacks including the risk of accumulated side-effects (such as oto- or nephrotoxicity with aminoglycoside use) and the emergence of multidrug resistance (MDR) [12,13,14,15]. In 2021, the U.S. CF Foundation reported that amongst individuals who cultured P. aeruginosa, 12.3% were classified as having an MDR strain [16]. This percentage increases with patient age and is thought to represent the cumulative effect of mutagenic adaptation and antibiotic use in the form of chronic suppressive therapy with nebulized antibiotics and courses of antibiotics (nebulized, oral, and intravenous) prescribed in the setting of chronic suppressive therapy as well as acute pulmonary exacerbations. The emergence of MDR not only limits therapeutic options, but is associated with worse clinical outcomes in CF [17]. Given the scarcity of new antibiotics, there has been a surge of interest in compounds that aim to increase the efficacy of treatments currently in clinical use (antibiotic adjuncts), some of which target components specific to P. aeruginosa.
The use of biofilm degrading compounds to increase the efficacy of antibiotics is an approach with a well-established evidence base in CF. For instance, the use of the mucolytic human recombinant dornase alfa (rhDNase), an EPS-disruptor that degrades eDNA within biofilms, has been shown to be effective at reducing the rate of lung function decline and the number of pulmonary exacerbations in meta-analyses of clinical trials [18]. Targeting matrix EPSs specific to the biofilms of P. aeruginosa is a promising adjuvant strategy, as these polysaccharides plays a prominent role in substrate adhesion, cell aggregation, and community architecture, factors which confer survival benefits and lead to increased P. aeruginosa tolerance to antimicrobials and host defenses [19,20]. In this review, we focus on the disruption of the three P. aeruginosa-specific matrix EPSs as novel therapeutic targets for increasing the efficacy of antibiotics currently in clinical use. We summarize the current evidence base for EPS degrading enzymes and other strategies with a focus on the eventual clinical use alongside antibiotics for CF lung infection.
2. Matrix Exopolysaccharides of P. aeruginosa biofilms
To fully appreciate the potential EPS targets and strategies that will be discussed in this review, we begin with a summary of P. aeruginosa biofilm development. The life cycle of the P. aeruginosa biofilm is characterized by four stages, depicted in Figure 1: attachment, microcolony formation, biofilm maturation, and dispersal of biofilm bacteria to recolonize and propagate new biofilm development.
The life cycle described in Figure 1 begins when P. aeruginosa attaches to a substratum (which may include other cells) and shifts from a highly virulent, motile phenotype to a more sessile phenotype. This transition is characterized by the slowing of twitching motility and secretion of EPSs that anchor bacterial cells [21]. As P. aeruginosa proliferates, structured communities called microcolonies form and grow, eventually resulting in the development of mature biofilms. Maturing biofilms are identified by profound changes in the expression of genes that control twitching motility, EPS secretion, and quorum sensing systems, resulting in an intricately structured, highly responsive, heterogeneous bacterial community [22]. Moreover, the biofilm structure is also influenced by the complex interactions between the bacteria and the host environment. Genomic studies have revealed that differing rates of up and down-regulation of genes encoding EPSs depend on the particular set of conditions in the bacterial microenvironment [23,24]. When appropriate, the final stage of the biofilm life cycle is characterized by the dispersal of the mature biofilm, releasing virulent cells and non-surface attached aggregates to colonize new areas in the CF lung [25].
The three EPSs of P. aeruginosa biofilms, Psl, Pel, and alginate, are all distinct in structure and function within the biofilm, from enabling surface attachment or bacterial aggregation, to maintaining architecture and nourishment in the biofilm, to offering physical protection from antibiotics and host defenses. Their specific properties and physiological significance in the CF lung will be elaborated upon below.
2.1. Psl and Pel
Psl is a branched neutral pentasaccharide composed of repeating d-mannose, d-glucose and l-rhamnose subunits and is involved in all stages of P. aeruginosa biofilm development [26]. Psl synthesis requires proteins encoded in the psl operon, which consists of 15 co-transcribed genes (pslA – O) [27,28]. Upon encountering a surface or substrate, Psl anchors P. aeruginosa and connects neighboring cells [26,29]. As P. aeruginosa swims, it deposits Psl trails that enhance the surface motility of subsequent cells, aiding in the formation of microcolonies [30,31]. Sensing Psl further stimulates biofilm development by increasing levels of an important intracellular secondary messenger, cyclic di-guanosine monophosphate (cGMP) [32,33,34]. As the biofilm matures, Psl provides structural integrity by cross-linking CdrA, an extracellular adhesin, and eDNA [21,35,36,37]. Psl limits the penetration of antibiotics by sequestration, and weakens host immunity by preventing efficient complement deposition, thereby inhibiting the production of neutrophil reactive oxygen species (ROS) and efficient phagocytosis of P. aeruginosa [38]. Clinically, Psl is associated with increased P. aeruginosa tolerance to tobramycin and is associated with aggregation in the sputum of children with CF, and this has been put forth as a reason for failure of eradication therapy with this antibiotic [39,40,41]. Although Psl is primarily produced by non-mucoid bacteria, studies have shown it remains utilized in mucoid strains, contributing to biofilm development, maintaining architecture and stability, and evading host immune effectors [42,43,44].
Pel is a cationic polymer important for biofilm formation, specifically pellicle development at the air-liquid interface, and like Psl, it is primarily associated with non-mucoid P. aeruginosa biofilms [45,46]. Pel is synthesized by proteins expressed by the pel operon (pelA – G), and structurally, it is composed of galactosamine and N-acetyl galactosamine residues linked by partially de-N-acetylated alpha 1-4 glycosidic linkages [47]. In strains that rely on Pel for biofilm formation, such as PA14, the EPS acts as a primary scaffold, maintaining cell-to-cell interactions between bacterial cells in a similar fashion as Psl in PAO1 [36,48,49,50].The positively charged Pel has been observed to cross-link the negatively charged eDNA through ionic interactions, conferring tolerance against positively charged antibiotics like tobramycin, but not neutrally charged antibiotics, such as ciprofloxacin [39,51]. Moreover, Pel-eDNA interactions protect eDNA from digestion by DNase I, and thus, (in theory) rendering nuclease-based therapies less effective [39,51].
Although P. aeruginosa can produce both Psl and Pel, their relative contribution to biofilm formation and structural integrity is strain dependent. In certain strains understood to be more reliant on Pel such as PA14, knockout Pel mutants are unable to form viable biofilms [52]. However, in strains that rely more on Psl, such as PAO1, knockout Psl mutants inhibit biofilm formation [52]. The localization of Psl and Pel within the biofilm is also strain dependent. In PAO1, Psl is localized to the periphery of mature microcolonies and bacterial aggregates while Pel is localized to the base of mature microcolonies [39]. However, in PA14, it is Pel that localizes to the periphery of mature microcolonies [21,39]. The ability to synthesize both Psl and Pel allows for redundancy in which EPS is incorporated into the matrix, highlighting the adaptive capacity of P. aeruginosa. For example, it has been observed that Psl-deficient PAO1 mutants upregulate Pel expression in the peripheral region after several repeat cultures [39,52]. Based on these findings, four classes of P. aeruginosa strains have been proposed by Colvin et al.: I) strains that rely on Pel as the dominant matrix polysaccharide (e.g., PA14), II) strains that rely on Psl as the dominant matrix polysaccharide (e.g., PAO1), III) strains that produce an EPS redundant matrix where single psl or pel mutations lead to impaired biofilm formation, and IV) strains that produce significantly elevated matrix levels such that a single psl or pel mutation has minimal impact on biofilm biomass [52,53,54].
2.2. Alginate
Alginate is the most studied and extensively characterized EPS secreted by P. aeruginosa. In contrast to Psl and Pel, both of which are specific to P. aeruginosa, it must be noted that alginate genes can be found in many different bacterial species [55,56,57]. Alginate is an anionic linear polymer with a high molecular weight (MW: 120 – 480 kDa), and is composed of β-1,4-linked D-mannuronic and α-L-guluronic acids [20,58,59]. The overproduction of alginate results in a mucoid phenotype and is a significant virulence factor in P. aeruginosa CF lung infections. [20,60,61]. Clinically, there has been a noted correlation between infection with mucoid P. aeruginosa and faster rates of lung function decline than with non-mucoid strains [62,63,64]. Alginate overproduction is often attributed to an inactivating mutation in the mucA gene, which encodes an anti-sigma factor that sequesters AlgT, an activator of alginate biosynthesis. Thus, mucA mutations result in constitutive alginate synthesis and the appearance of the mucoid phenotype [61,65,66,67].
Although wild-type P. aeruginosa has the genetic ability to over-produce alginate, it is rarely seen outside the CF lung and arises due to selection pressures in this specific niche. The mucoid phenotype has drastic effects on biofilm structure and the perseverance of infection within the lung, a fact that is corroborated by the observation that clinical isolates from CF patients often possess alginate-overproducing mucoid phenotypes [68,69,70,71]. Using whole genome sequencing, Marvig et al. followed the evolution of 474 P. aeruginosa isolates collected longitudinally from 34 patients with CF, beginning at initial infection and continuing for a mean timespan of 4.8 years. They found significant convergent, within-host evolution of 52 pathoadaptive genes, especially those responsible for alginate overproduction [72].
This observed survival advantage in mucoid P. aeruginosa has been attributed to several factors, specifically the thick and viscous structure of alginate-overproducing biofilms opposed to the flat and uniform colonies observed in their non-mucoid counterparts [61,73]. In the dehydrated lung mucus of patients with CF, alginate helps maintain the hydration of the bacterial cells, a vital function for cell survival and maintenance [68,74]. Furthermore, alginate serves as a protective barrier against antimicrobial therapy and host-mediated immune responses like macrophage phagocytosis [75,76]. This evidence supports the hypothesis that alginate overproduction reduces the clinical efficacy of antibiotic therapy in patients with CF, and for this reason alginate-specific therapeutics will be explored further in this review [77,78,79,80].
3. Therapies
3.1. Matrix degrading enzymes
Proposed anti-biofilm therapies employing matrix degrading enzymes include PslG glycoside hydrolase (PslGh), PelA glycoside hydrolase (PelAh), and alginate lyase (AgLase). While PslGh and PelAh are both synthesized by and specific to P. aeruginosa matrix EPSs, AgLase can be isolated from a variety of sources including algae, mollusks, bacteria, viruses, and fungi [81]. The known properties, effects, and toxicities of these compounds (including OligoG discussed later in this review) are summarized in Figure 2.
PslG is a 48.11 kDa molecular weight (MW) periplasmic protein involved in Psl synthesis, and is co-transcribed with 15 other genes in the psl operon [28,82]. PslG contains an electronegative glycoside hydrolase domain (PslGh) that cleaves Psl chains by targeting adjacent mannose residues [28,82]. PelA is a 101.3 kDa multidomain, periplasmic protein encoded by the first gene of the pel operon and contains a deacetylase domain and a glycoside hydrolase domain with distinct functions [52,83]. Its deacetylase domain is essential to Pel synthesis as it gives Pel a positive charge, allowing it to associate with other charged elements in the biofilm matrix such as eDNA [39,83,84]. Its glycoside hydrolase domain (MW: 30.58 kDa), herein PelAh, cleaves cationic, partially deacetylated residues within Pel residues and is involved in biofilm dispersal [85,86,87]. Given the highly selective action of PslGh and PelAh against their substrates Psl and Pel, which have critical roles in biofilm development and functioning, PslGh and PelAh have been studied as adjuvant therapies for eradicating P. aeruginosa biofilms.
Several lines of evidence from in vitro studies support the potential of PslGh and PelAh for the treatment of P. aeruginosa biofilms. When supplied exogenously, PslGh and PelAh can disrupt P. aeruginosa biofilms at nanomolar concentrations with extremely high specificity [82,86]. In developing biofilms, PslGh impairs the ability of P. aeruginosa to associate with surfaces and other bacteria, leading to more rapid, scattered movement of P. aeruginosa, delaying microcolony formation and inhibiting biofilm development [88]. The half maximal effective concentration (EC50) values of PslGh and PelAh to disrupt mature PAO1 biofilms after 1 hour were 12.9 ± 1.1 and 35.7 ± 1.1 nM, respectively [86]. When combined, PslGh - PelAh improved the penetration and efficacy of ciprofloxacin, tobramycin, colistin, neomycin, and polymyxin, allowing for smaller antibiotic dosages [89,90]. PslGh also works remarkably quickly, completely disrupting all Psl fibers within 5 minutes of administration and triggering sudden biofilm dispersal [82]. Moreover, PslGh -dispersed bacteria has a lower MIC for tobramycin and ciprofloxacin compared to planktonic bacteria [82]. PslGh further sensitizes the host immune system towards P. aeruginosa biofilms, increasing the deposition of C3 complement proteins which stimulates macrophage and neutrophil phagocytosis and neutrophil ROS production [28,82,90]. When tested in vitro, both PslGh and PelAh were observed to be non-toxic to neutrophils, lung fibroblasts, and red blood cells, and PslGh was additionally non-toxic to colonic epithelial cells and macrophages. [28,82,90]. Experiments using animal models of P. aeruginosa have validated in vitro findings of the efficacy and safety of PslGh and PelAh. When injected locally, PslGh potentiated tobramycin killing of mature P. aeruginosa biofilms on mouse peritoneum implants [82]. In a wound infection model, prophylactic treatment with PslGh showed an additive killing effect when tobramycin was added 24 hours later and was not toxic to the wound [90]. PelAh embedded in a bacterial cellulose membrane effectively destabilized P. aeruginosa biofilms when topically applied to infected murine chronic wounds [91]. Although a majority of studies have utilized wound and implant models, the results demonstrate the proof of concept for topical (i.e. nebulized) application in pulmonary infection in CF.
In the most comprehensive pre-clinical evaluation of PslGh and PelAh to date, Ostapska et al. prophylactically administered PslGh and PelAh intratracheally to mice with P. aeruginosa lung infections with or without antibiotics [89]. They found that the antimicrobial effects of ciprofloxacin, but not ceftazidime, were potentiated when administered every 8 hours following initial treatment with PslGh - PelAh. In healthy mice, a single intratracheal dose of up to 250/250 ug of PslGh - PelAh was well tolerated, with no changes in weight, temperature, mortality, makers of pulmonary injury, or numbers of macrophage, eosinophils, and neutrophils cells compared to buffer-treated mice. However, there was an increase of pulmonary lymphocytes following PslGh - PelAh treatment, highlighting the need to further investigate the adaptive immune response to this therapy. When administered alone, PslGh has a half-life of 18 hours and PelAh has a half-life of 3 hours. However, PelAh stability increased to 5 hours when co-administered with PslGh and catalytic activity was retained >24 hours. However, PslGh - PelAh treatment of P. aeruginosa infected mice in the absence of antibiotics triggered pulmonary inflammation and lethal septicemia, underscoring the necessity of antibiotic co-treatment [89].
It is worth mentioning several technological developments that may enhance the feasibility of PslGh and PelAh in clinical applications. Lipid liquid crystal nanoparticles encapsulating PslGh and tobramycin offer protection against proteolytic degradation and are releasde upon encountering P. aeruginosa. This product was seen to be 10-100 fold more effective at eradicating P. aeruginosa infections in vivo using a Caenorhabditis elegans infection model [92]. PslGh constructs with significantly enhanced trypsin resistance have also been developed, which may extend the half-life of PslGh, allowing for smaller and less frequent doses [93]. PslGh can also be immobilized on the lumen surface of medical-grade polyethylene, polyurethane, and polydimethylsiloxane (silicone) catheter tubing, reducing initial attachment of P. aeruginosa. The bacterial burden of PslGh -modified catheters decreased by 3 logs for up to 11 days under dynamic flow culture conditions and 1.5 logs when used in in vivo rat infection models [94].
Published research has reported that mucoid, alginate-rich P. aeruginosa isolates produce heterogenous biofilms with a high tolerance to antibiotics [61]. Many anti-biofilm strategies have been designed and examined for increasing the susceptibility of antibiotics through disrupting the alginate EPS. One such promising therapeutic technique is using the enzyme AgLase, which can be derived from various sources, and weigh anywhere between 25-60 kDa [81]. Depending on its source, AgLase can degrade alginate at a variety of cleavage points, disrupting established mucoid P. aeruginosa biofilms, and enhancing antibiotic penetration and host immune functions [81]. AgLase has been reported in vitro and in vivo to disrupt biofilms, enhancing the efficacy of amikacin, tobramycin, ciprofloxacin, and gentamicin [75,95,96,97]. AgLase co-administration with DNase has been found to potentiate antimicrobial biofilm eradication to a greater effect than seen with either agent on its own [97,98]. AgLase enhances neutrophil killing, macrophage phagocytosis, and alveolar macrophage efferocytosis [76,99]. It is worth noting, however, the conflicting evidence for AgLase as an antibiotic adjunct. One study reported that AgLase did not have any effect on pure or mixed cultures of P. aeruginosa biofilms, a fact which authors attributed in certain instances to alginate not being the main contributing component of the biofilm and in others to protection from enzymatic degradation due to the presence of other molecules [100]. Another study compared two AgLases to proteins similar in structure, but without the capacity to enzymatically degrade alginate, and observed equal rates of bacterial biofilm disruption and antibiotic synergy between these compounds, suggesting that the potentiation effects of AgLases are uncoupled to their catalytic activity [101]. AgLase was also unable to degrade mucoid P. aeruginosa biofilms embedded in sputum [102].
Though the evidence is unclear if and how AgLases combat bacterial biofilms, AgLase products with therapeutic implications are under development. AgLase-polyethylene glycol conjugates have been developed that significantly reduce its immunoreactivity [103]. AgLase functionalized chitosan nanoparticles of ciprofloxacin are novel delivery carriers, which has demonstrated enhanced biofilm degradation in in vitro experiments with a mucoid clinical P. aeruginosa strain, without toxicity to human lung epithelial cells or rat lung tissues [104]. In another distinct approach, it has long been known that bacteriophage (viruses that infect and lyse bacterial cells) employ polysaccharide depolymerases to migrate through polysaccharide-rich biofilms and infect embedded bacterial cells [105,106]. One study using a mucoid P. aeruginosa isolate revealed that P. aeruginosa-specific bacteriophage could reduce the viscosity of purified CF alginate by up to 40% and penetrate through the mucoid alginate matrix to lyse biofilm-associated bacteria [107]. In a subsequent study, Glonti et al. isolated an alginate lyase responsible for the alginate-degrading properties of a P. aeruginosa bacteriophage, and reported activity against alginate purified from many sources, including from clinical CF P. aeruginosa isolates [108]. Finally, a P. aeruginosa phage isolated from hospital sewage was able to disrupt mature P. aeruginosa biofilms and potentiate serum bactericidal activity [109]. A number of clinical trials involving the use of bacteriophage for P. aeruginosa lung infection in CF are currently underway, and although the contribution of specific depolymerases to the antibiofilm properties of P. aeruginosa bacteriophages are to be determined, it will be interesting to see whether such enzymes will play a role in the success or failure of these therapies (NCT04596319; NCT04684641; NCT05010577; NCT05453578) [110,111,112,113].
3.2. Other novel strategies
Alongside the development of matrix degrading enzymes as adjunctive therapies for patients CF, other novel therapeutics that may enhance airway clearance and lung function are being investigated. One such example is the 3.2 kDa alginate oligosaccharide, OligoG CF-5/20 (OligoG), which is sourced from the brown seaweed, Laminaria hyperborean and can reduce the viscoelasticity of CF sputum [59,114,115]. Being relatively small, OligoG can also readily diffuse through mucoid biofilms and disrupt EPS within biofilm matrices [59,116]. OligoG disrupts biofilms in a time and dose dependent manner and can acts as an antibiotic potentiator, increasing the penetration and efficacy of tobramycin, erythromycin, colistin, and ciprofloxacin [59,116,117,118]. Mice intratracheally infected with a mucoid clinical P. aeruginosa isolate showed a 2.5 log reduction in bacterial colony-forming units (CFUs) following treatment with 5% OligoG [117]. Like the aforementioned matrix disruptors, as OligoG is not bactericidal, it is unlikely to create an adaptive pressure, a theory supported by studies showing prolonged treatment with OligoG does not induce resistance [114]. Furthermore, OligoG-colistin conjugates have been developed that can produce a sustained biofilm inhibitory effect, while limiting the toxicity of colistin [119]. There are also novel compounds under development that combine the mucoactive properties of alginate oligomers with nitric oxide release as an added antibiofilm agent to improve the efficacy of antibiotics [120].
Importantly, OligoG is the first inhaled polymer therapy that has been investigated in humans as a potential novel therapeutic approach for airway clearance in CF patients. AlgiPharma, the lead developer of OligoG, has been spearheading efforts to develop OligoG as a novel CF therapeutic and have completed several clinical trials, although the results of only one Phase 2b trial has been published so far. In this randomized, double-blind, placebo-controlled, multi-centered, crossover study, 90 adult CF patients were screened, and 65 patients were randomly allocated to receive either OligoG via dry powder inhalation (1050 mg per day) or a placebo. The primary endpoint was forced expiratory volume in 1 second (FEV1), measured at the end of the 28-day treatment regimen [121]. This study revealed that OligoG administered thrice daily via dry powder inhalation was well tolerated, with no significant differences in serious adverse events between treatment and placebo groups. Furthermore, concentration in plasma was in the range of 0.5-8.98 µgmL-1, with no detectable OligoG in plasma after 28 days of washout (day 56 of the study). Despite this reassuring safety data, however, no significant improvement in FEV1 was observed in the treatment group. Interestingly, post hoc exploratory analyses indicated that patients on inhaled tobramycin and patients <25 years of age showed positive trends in lung function, highlighting that further studies of OligoG are essential [121]. A prospective clinical study is currently being planned under the framework of HORIZON2020 by the European Clinical Trial Network [122]. Whatever the outcome, these trials will shed new light on the therapeutic potential of disrupting alginate biofilms in P. aeruginosa pulmonary infection.
4. Discussion
Successful treatment of P. aeruginosa CF pulmonary infections is difficult to achieve, in part due to the heterogeneity of bacterial populations within the biofilm (cells with differing metabolism, mutation rate, phenotypic expression of virulence factors, etc.) that limits the effectiveness of broad-spectrum antibiotics at our disposal. The agents described in this review all have the potential to disrupt the biofilm structure. Although not bactericidal by themselves, by disrupting the extracellular matrix, the hope is that an adjuvant effect will result from exposing bacterial cells to both effectors of the immune system (like alveolar macrophages and neutrophils), as well as to antibiotics. Additionally, their selectivity for targeting P. aeruginosa infections make them appealing adjuncts in an era of personalized medicine, while limiting off-target effects. These agents are summarized in Table 1, which focuses on the challenges and considerations that exist as these compounds transition from laboratory bench to CF patient, or from clinical trial to effective therapy.
One should note that some of the compounds mentioned in Table 1 are further along the development pipeline than others, with OligoG being the only compound administered to humans in clinical trial. Although this trial failed to meet its primary endpoint (improvement in FEV1), there were several trends in post-hoc analyses that should be taken into consideration for clinical trials in development, including the use of a placebo inhaler unexpectedly reduced bacterial burden in sputum, and some promising trends in certain patient groups.
The matrix degrading enzymes PslGh, PelAh, and AgLase all have shown efficacy in vitro and in vivo as potent biofilm-disruptors and antibiotic potentiators in their own right, although it is clear from Table 1 that if these compounds are to be developed into viable antibiotic adjuncts in CF patients, more research is needed. Specifically, investigations across many different clinical strains are required to demonstrate whether there is the potential for widespread applicability or whether the promising effects that have been observed are specific to particular bacterial strains. To overcome this high level of specificity, studies bringing these therapies to animal models have focused on co-administration of certain compounds, whether it is a DNase or another glycoside hydrolase [97,124]. Combining therapy of different matrix-degrading enzymes may have many benefits, from targeting multiple EPSs at once and achieving broader therapeutic coverage, to extending the half-life of the compounds, as observed when combining PelAh with PslGh [124]. Moreover, there is no reason that antibiotics with different mechanisms of action should all act in additive or synergistic ways with these compounds, and this is illustrated by some of the preliminary in vitro work showing additional benefit when PslGh was administered with the fluoroquinolone ciprofloxacin, but not with the third generation cephalosporin ceftazidime – both commonly used agents in P. aeruginosa infection [124].
Additional information is also needed for optimal dose concentrations for safety and efficacy, as well as whether specific antibiotic combinations have more synergistic potential than others. Although PslGh and PelAh have been demonstrated to be non-cytotoxic to mammalian cells in all the studies included in this review, Ostapska et al. showed an increase in the number of pulmonary lymphocytes following glycoside hydrolase administration, warranting further study, as these compounds may trigger specific adaptive immune responses over-time. Of critical importance is the hematological dissemination and lethal septicemia that has been seen in animal models if glycoside hydrolases are used in the absence of antibiotics [124]. Additionally, most of the in vivo studies use wound or implant infection models, leaving a gap in knowledge concerning the efficacy of these compounds in pulmonary infection models, which better represent CF lung infection.
Concerning the mode of administration for these compounds, with the exception of OligoG, it is unknown whether (i) they are stable when nebulized or inhaled and (ii) can be administered in sufficient quantity to exert clinically significant antibiotic potentiation. Given that the glycoside hydrolases PslGh and PelAh disperse biofilms almost immediately, it seems reasonable to develop these compounds either for administration prior to or for co-administration with antibiotics. Furthermore, there is lacking information on the stability of these compounds in CF sputum, where it is possible that the presence of proteases and other specifics of the environmental niche (e.g. temperature, pH, etc.) could denature these enzymes before they have the ability to degrade the biofilm. Some of these concerns may be addressed by technological modifications briefly introduced in this review, such as novel lipid liquid crystal and chitosan nanoparticles to enhance delivery, protease-resistant hydrolase/lyases that may increase enzymatic longevity, and polyethylene glycol-conjugation that may decrease immunoreactivity and improve safety profiles.
Lastly, we must address the chasm that exists between clinical outcomes in patients with CF lung infection and in vitro laboratory methods for testing antibiotic sensitivity [125]. This is likely due to the simplicity of in vitro assays and their inadequacy of simulating a biofilm mode of growth and the myriad of conditions encountered in the human lung. However, even when clinical trials have assessed choice of antibiotic based on assays that have used bacterial biofilms, there has been no link with patient outcome [126,127]. There are a variety of assays that have been developed/are in development that aim to rectify this disconnect, including the use of artificial or CF sputum, animal models of pulmonary infection, as well as the polymicrobial nature of the lung environment.
Concerning the latter, P. aeruginosa matrix EPSs have also been noted to play important roles in mixed-species communities, conferring both communal benefits and competitive advantages. For instance, Psl is required to integrate P. aeruginosa into mixed-species biofilms with Pseudomonas protegens and Klebsiella pneumoniae and also confers communal stress resistance [128]. Psl and Pel promote the development of mixed-species biofilms with Staphylococcus aureus and can further grant antibiotic-tolerance to Psl non-producers such as Escherichia coli and to Staphylococcus aureus by binding secreted staphylococcal protein A [129,130,131]. The alginate overproduction of mucoid P. aeruginosa isolates from the CF lung not only render them more permissive to the growth of S. aureus [132], but have been seen to protect Burkholderia cenocepacia from inflammatory responses and host immune effectors [133]. Thus, disruption of P. aeruginosa matrix polysaccharides may yield a greater therapeutic effect beyond the scope of the primary infection [134].
5. Conclusion
Despite tremendous progress in our understanding of bacterial biofilms in recent years, P. aeruginosa lung infection remains a significant cause of morbidity and mortality in the CF population [16]. The recent introduction of highly effective modulator therapy (HEMT) in CF has shown great promise in restoring some CFTR protein function and thus impacting mucus and the CF lung environment in clinically meaningful ways, improving pulmonary function, BMI, patient-reported quality of life, and decreasing rates of pulmonary exacerbations [135]. Additionally, there have been reports that the modulator ivacaftor (used in most of the currently available HEMT) might possess some antimicrobial activity both directly [136], and when combined with common antibiotics [137,138]. Though it is not clear yet what impact these therapies will have on lung infection generally, by “normalizing” the lung environment, the hope is that younger patients starting on these therapies will not be affected by common CF pathogens. However, current evidence suggests that even with HEMT, CF lung disease still progresses, albeit slower, and organisms like P. aeruginosa will continue to be problematic for the majority of individuals with well-established lung disease [139,140,141]. Additionally, the high cost of HEMT and lack of uniform provision across different health systems means that many people in developing countries with CF will not be able to benefit from these compounds in the short term [135]. There also remains a population of CF patients that possess CFTR mutations not amenable to treatment by current HEMT, and patients have withdrawn from HEMT due to adverse hepatic, somatic, or psychiatric symptoms [139]. It is thus important to continue searching for novel antimicrobial strategies and compounds to improve outcomes in patients.
The three EPSs produced by P. aeruginosa (Psl, Pel and alginate) each represent novel targets for antibiotic adjuvant therapy. As we enter an era of more personalized medicine, tailored therapies may hold the key to antibiotic potentiation that we have been searching for, whether the target is an alginate over-producing mucus isolate or a Psl/Pel-rich one. Despite this promise, the only compound discussed in this review that has been given in clinical trials (OligoG) failed to meet the primary endpoint of increased lung function at the end of the trial period. Many of the other compounds, including matrix-degrading enzymes, have only been administered in animals and data are lacking on applicability across clinical strains found in the CF lung. Hence, there are several steps before such compounds come to human clinical trials. Further in vitro work will need to establish efficacy of these compounds across a variety of clinical strains, in addition to considering other clinically relevant questions, including optimizing concentrations, and assessing mode and timing of delivery in relation to other therapies.
Author Contributions
Conceptualization, J.C. and I.M..; investigation, J.C, S.E., S.P., and A.M.; data curation, J.C..; writing—original draft preparation, J.C, S.E., S.P. and A.M.; writing—review and editing, J.C. and I.M.; visualization, J.C.; supervision, I.M. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to acknowledge Cystic Fibrosis Canada for funding a clinical fellowship for IM, as well as the Canadian Glycomics Network (GlycoNet), which has provided research funding for JC and AM.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Maisetta, G.; Grassi, L.; Esin, S.; Kaya, E.; Morelli, A.; Puppi, D.; Piras, M.; Chiellini, F.; Pifferi, M.; Batoni, G. Targeting Pseudomonas aeruginosa in the Sputum of Primary Ciliary Dyskinesia Patients with a Combinatorial Strategy Having Antibacterial and Anti-Virulence Potential. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Bellettato, C.M.; Braccioni, F.; Romagnoli, M.; Casolari, P.; Caramori, G.; Fabbri, L.M.; Johnston, S.L. Infections and Airway Inflammation in Chronic Obstructive Pulmonary Disease Severe Exacerbations. Am. J. Respir. Crit. Care Med. 2006, 173, 1114–1121. [Google Scholar] [CrossRef]
- Mulcahy, L.R.; Isabella, V.M.; Lewis, K. Pseudomonas Aeruginosa Biofilms in Disease. Microb. Ecol. 2014, 68, 1. [Google Scholar] [CrossRef] [PubMed]
- Peleg, A.Y.; Hooper, D.C. Hospital-Acquired Infections Due to Gram-Negative Bacteria. N. Engl. J. Med. 2010, 362, 1804. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of Cystic Fibrosis Lung Disease. N. Engl. J. Med. 2015, 372, 351. [Google Scholar] [CrossRef] [PubMed]
- Limoli, D.H.; Jones, C.J.; Wozniak, D.J. Bacterial Extracellular Polysaccharides in Biofilm Formation and Function. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.C.; Ruseska, I.; Wright, J.B.; Costerton, J.W. Tobramycin Resistance of Pseudomonas aeruginosa Cells Growing as a Biofilm on Urinary Catheter Material. Antimicrob. Agents Chemother. 1985, 27, 619. [Google Scholar] [CrossRef] [PubMed]
- Thöming, J.G.; Häussler, S. Pseudomonas Aeruginosa Is More Tolerant Under Biofilm Than Under Planktonic Growth Conditions: A Multi-Isolate Survey. Front. Cell. Infect. Microbiol. 2022, 12, 113. [Google Scholar] [CrossRef] [PubMed]
- Rada, B. Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis. Pathogens 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and Management of Pulmonary Infections in Cystic Fibrosis. 2012, 168, 918–951. [CrossRef]
- Lund-Palau, H.; Turnbull, A.R.; Bush, A.; Bardin, E.; Cameron, L.; Soren, O.; Wierre-Gore, N.; Alton, E.W.F.W.; Bundy, J.G.; Connett, G.; et al. Pseudomonas aeruginosa Infection in Cystic Fibrosis: Pathophysiological Mechanisms and Therapeutic Approaches. Expert Rev. Respir. Med. 2016, 10, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Barat, L.; Ciofu, O.; Kragh, K.N.; Pressler, T.; Johansen, U.; Motos, A.; Torres, A.; Hoiby, N. Phenotypic Shift in Pseudomonas aeruginosa Populations from Cystic Fibrosis Lungs after 2-Week Antipseudomonal Treatment. J. Cyst. Fibros. 2017, 16, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Llor, C.; Bjerrum, L. Antimicrobial Resistance: Risk Associated with Antibiotic Overuse and Initiatives to Reduce the Problem. Ther. Adv. Drug Saf. 2014, 5, 229. [Google Scholar] [CrossRef] [PubMed]
- Mowat, E.; Paterson, S.; Fothergill, J.L.; Wright, E.A.; Ledson, M.J.; Walshaw, M.J.; Brockhurst, M.A.; Winstanley, C. Pseudomonas aeruginosa Population Diversity and Turnover in Cystic Fibrosis Chronic Infections. Am. J. Respir. Crit. Care Med. 2011, 183, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Mingeot-Leclercq, M.P.; Tulkens, P.M. Aminoglycosides: Nephrotoxicity. Antimicrob. Agents Chemother. 1999, 43, 1003. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry: 2021 Annual Data Report. Bethesda, MD. 2022.
- Fainardi, V.; Neglia, C.; Muscarà, M.; Spaggiari, C.; Tornesello, M.; Grandinetti, R.; Argentiero, A.; Calderaro, A.; Esposito, S.; Pisi, G. Multidrug-Resistant Bacteria in Children and Adolescents with Cystic Fibrosis. Children 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Montgomery, M. Dornase Alfa for Cystic Fibrosis. Cochrane Database Syst. Rev. 2021, 2021. [Google Scholar] [CrossRef]
- Maunders, E.; Welch, M. Matrix Exopolysaccharides; the Sticky Side of Biofilm Formation. FEMS Microbiol. Lett. 2017, 364, 120. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Hayes, D.; Wozniak, D.J. Cystic Fibrosis and Pseudomonas aeruginosa: The Host-Microbe Interface. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Conover, M.; Lu, H.; Parsek, M.R.; Bayles, K.; Wozniak, D.J. Assembly and Development of the Pseudomonas aeruginosa Biofilm Matrix. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef]
- Sauer, K.; Camper, A.K.; Ehrlich, G.D.; Costerton, J.W.; Davies, D.G. Pseudomonas aeruginosa Displays Multiple Phenotypes during Development as a Biofilm. J. Bacteriol. 2002, 184, 1140. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Falcone, M.; Molin, S.; Johansen, H.K. High-Resolution in Situ Transcriptomics of Pseudomonas aeruginosa Unveils Genotype Independent Patho-Phenotypes in Cystic Fibrosis Lungs. Nat. Commun. 2018 91 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kordes, A.; Grahl, N.; Koska, M.; Preusse, M.; Arce-Rodriguez, A.; Abraham, W.R.; Kaever, V.; Häussler, S. Establishment of an Induced Memory Response in Pseudomonas aeruginosa during Infection of a Eukaryotic Host. ISME J. 2019, 13, 2018–2030. [Google Scholar] [CrossRef] [PubMed]
- Sauer, K.; Stoodley, P.; Goeres, D.M.; Hall-Stoodley, L.; Burmølle, M.; Stewart, P.S.; Bjarnsholt, T. The Biofilm Life Cycle: Expanding the Conceptual Model of Biofilm Formation. Nat. Rev. Microbiol. 2022 2010 2022, 20, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lu, H.; Sprinkle, A.; Parsek, M.R.; Wozniak, D.J. Pseudomonas aeruginosa Psl Is a Galactose- and Mannose-Rich Exopolysaccharide. J. Bacteriol. 2007, 189, 8353. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.Z.; Wang, D.; Liu, Y.; Zhang, Z.; Wozniak, D.J. Regulation of Biofilm Exopolysaccharide Biosynthesis and Degradation in Pseudomonas aeruginosa. Annu. Rev. Microbiol. 2022, 76, 413–433. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.; Whitfield, G.B.; Hill, P.J.; Little, D.J.; Pestrak, M.J.; Robinson, H.; Wozniak, D.J.; Howell, P.L. Characterization of the Pseudomonas aeruginosa Glycoside Hydrolase PslG Reveals That Its Levels Are Critical for Psl Polysaccharide Biosynthesis and Biofilm Formation. J. Biol. Chem. 2015, 290, 28374–28387. [Google Scholar] [CrossRef] [PubMed]
- Byrd, M.S.; Sadovskaya, I.; Vinogradov, E.; Lu, H.; Sprinkle, A.B.; Richardson, S.H.; Ma, L.; Ralston, B.; Parsek, M.R.; Anderson, E.M.; et al. Genetic and Biochemical Analyses of the Pseudomonas aeruginosa Psl Exopolysaccharide Reveal Overlapping Roles for Polysaccharide Synthesis Enzymes in Psl and LPS Production. Mol. Microbiol. 2009, 73, 622–638. [Google Scholar] [CrossRef]
- Zhao, K.; Tseng, B.S.; Beckerman, B.; Jin, F.; Gibiansky, M.L.; Harrison, J.J.; Luijten, E.; Parsek, M.R.; Wong, G.C.L. Psl Trails Guide Exploration and Microcolony Formation in Pseudomonas aeruginosa Biofilms. Nature 2013, 497, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Parsek, M.R.; Wozniak, D.J.; Ma, L.Z. A Spider Web Strategy of Type IV Pili-Mediated Migration to Build a Fibre-like Psl Polysaccharide Matrix in Pseudomonas aeruginosa Biofilms. Environ. Microbiol. 2013, 15, 2238–2253. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.-G.; O’Toole, G.A. C-Di-GMP and Its Effects on Biofilm Formation and Dispersion: A Pseudomonas aeruginosa Review. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Irie, Y.; Borlee, B.R.; O’Connor, J.R.; Hill, P.J.; Harwood, C.S.; Wozniak, D.J.; Parsek, M.R. Self-Produced Exopolysaccharide Is a Signal That Stimulates Biofilm Formation in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 2012, 109, 20632–20636. [Google Scholar] [CrossRef] [PubMed]
- Borlee, B.R.; Goldman, A.D.; Murakami, K.; Samudrala, R.; Wozniak, D.J.; Parsek, M.R. Pseudomonas aeruginosa Uses a Cyclic-Di-GMP-Regulated Adhesin to Reinforce the Biofilm Extracellular Matrix. Mol. Microbiol. 2010, 75, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Le Mauff, F.; Razvi, E.; Reichhardt, C.; Sivarajah, P.; Parsek, M.R.; Howell, P.L.; Sheppard, D.C. The Pel Polysaccharide Is Predominantly Composed of a Dimeric Repeat of α-1,4 Linked Galactosamine and N-Acetylgalactosamine. Commun. Biol. 2022, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Reichhardt, C.; Jacobs, H.M.; Matwichuk, M.; Wong, C.; Wozniak, D.J.; Parsek, M.R. The Versatile Pseudomonas aeruginosa Biofilm Matrix Protein CdrA Promotes Aggregation through Different Extracellular Exopolysaccharide Interactions. J. Bacteriol. 2020, 202. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, X.; Liu, H.; Zhang, L.; Guo, Y.; Yu, S.; Wozniak, D.J.; Ma, L.Z. The Exopolysaccharide Psl–EDNA Interaction Enables the Formation of a Biofilm Skeleton in Pseudomonas aeruginosa. Environ. Microbiol. Rep. 2015, 7, 330. [Google Scholar] [CrossRef]
- Mishra, M.; Byrd, M.S.; Sergeant, S.; Azad, A.K.; Parsek, M.R.; McPhail, L.; Schlesinger, L.S.; Wozniak, D.J. Pseudomonas aeruginosa Psl Polysaccharide Reduces Neutrophil Phagocytosis and the Oxidative Response by Limiting Complement-Mediated Opsonization. Cell. Microbiol. 2012, 14, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.K.; Storek, K.M.; Ledvina, H.E.; Coulon, C.; Marmont, L.S.; Sadovskaya, I.; Secor, P.R.; Tseng, B.S.; Scian, M.; Filloux, A.; et al. Pel Is a Cationic Exopolysaccharide That Cross-Links Extracellular DNA in the Pseudomonas aeruginosa Biofilm Matrix. Proc. Natl. Acad. Sci. USA. 2015, 112, 11353–11358. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.J.; Jackson, L.; CW Yau, Y.; Reichhardt, C.; Beaudoin, T.; Uwumarenogie, S.; Guttman, K.M.; Lynne Howell, P.; Parsek, M.R.; Hoffman, L.R.; et al. The Role of Psl in the Failure to Eradicate Pseudomonas aeruginosa Biofilms in Children with Cystic Fibrosis. NPJ Biofilms Microbiomes 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.J.; Yau, Y.C.W.; Park, S.; Eisha, S.; McDonald, N.; Parsek, M.R.; Howell, P.L.; Hoffman, L.R.; Nguyen, D.; DiGiandomenico, A.; et al. Pseudomonas aeruginosa Aggregation and Psl Expression in Sputum Is Associated with Antibiotic Eradication Failure in Children with Cystic Fibrosis. Sci. Reports 2022, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.J.; Wozniaka, D.J. Psl Produced by Mucoid Pseudomonas aeruginosa Contributes to the Establishment of Biofilms and Immune Evasion. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Huse, H.K.; Kwon, T.; Zlosnik, J.E.A.; Speert, D.P.; Marcotte, E.M.; Whiteley, M. Pseudomonas aeruginosa Enhances Production of a Non-Alginate Exopolysaccharide during Long-Term Colonization of the Cystic Fibrosis Lung. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Ma, L.; Wang, S.; Wang, D.; Parsek, M.R.; Wozniak, D.J. The Roles of Biofilm Matrix Polysaccharide Psl in Mucoid Pseudomonas aeruginosa Biofilms. FEMS Immunol. Med. Microbiol. 2012, 65, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.; Kolter, R. Genes Involved in Matrix Formation in Pseudomonas aeruginosa PA14 Biofilms. Mol. Microbiol. 2004, 51, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, D.J.; Wyckoff, T.J.O.; Starkey, M.; Keyser, R.; Azadi, P.; O’Toole, G.A.; Parsek, M.R. Alginate Is Not a Significant Component of the Extracellular Polysaccharide Matrix of PA14 and PAO1 Pseudomonas aeruginosa Biofilms. Proc. Natl. Acad. Sci. USA. 2003, 100, 7907. [Google Scholar] [CrossRef] [PubMed]
- Le Mauff, F.; Razvi, E.; Reichhardt, C.; Sivarajah, P.; Parsek, M.R.; Howell, P.L.; Sheppard, D.C. The Pel Polysaccharide Is Predominantly Composed of a Dimeric Repeat of α-1,4 Linked Galactosamine and N-Acetylgalactosamine. Commun. Biol. 2022 51 2022, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jackson, K.D.; Landry, R.M.; Parsek, M.R.; Wozniak, D.J. Analysis of Pseudomonas aeruginosa Conditional Psl Variants Reveals Roles for the Psl Polysaccharide in Adhesion and Maintaining Biofilm Structure Postattachment. J. Bacteriol. 2006, 188, 8213. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hu, Y.; Liu, Y.; Zhang, J.; Ulstrup, J.; Molin, S. Distinct Roles of Extracellular Polymeric Substances in Pseudomonas aeruginosa Biofilm Development. Environ. Microbiol. 2011, 13, 1705–1717. [Google Scholar] [CrossRef]
- Colvin, K.M.; Gordon, V.D.; Murakami, K.; Borlee, B.R.; Wozniak, D.J. The Pel Polysaccharide Can Serve a Structural and Protective Role in the Biofilm Matrix of Pseudomonas aeruginosa. PLoS Pathog 2011, 7, 1001264. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.K.; Dreifus, J.E.; Reichhardt, C.; Storek, K.M.; Secor, P.R.; Wozniak, D.J.; Hisert, K.B.; Parsek, M.R. Pseudomonas aeruginosa Aggregates in Cystic Fibrosis Sputum Produce Exopolysaccharides That Likely Impede Current Therapies. Cell Rep. 2021, 34, 108782. [Google Scholar] [CrossRef]
- Colvin, K.M.; Irie, Y.; Tart, C.S.; Urbano, R.; Whitney, J.C.; Ryder, C.; Howell, P.L.; Wozniak, D.J.; Parsek, M.R. The Pel and Psl Polysaccharides Provide Pseudomonas aeruginosa Structural Redundancy within the Biofilm Matrix. Environ. Microbiol. 2012, 14, 1913–1928. [Google Scholar] [CrossRef] [PubMed]
- Kirisits, M.J.; Prost, L.; Starkey, M.; Parsek, M.R. Characterization of Colony Morphology Variants Isolated from Pseudomonas aeruginosa Biofilms. Appl. Environ. Microbiol. 2005, 71, 4809. [Google Scholar] [CrossRef] [PubMed]
- Häußler, S.; Ziegler, I.; Löttel, A.; Götz, F.V.; Rohde, M.; Wehmhöhner, D.; Saravanamuthu, S.; Tümmler, B.; Steinmetz, I. Highly Adherent Small-Colony Variants of Pseudomonas aeruginosa in Cystic Fibrosis Lung Infection. J. Med. Microbiol. 2003, 52, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Cote, G.L.; Krull, L.H. Characterization of the Exocellular Polysaccharides from Azotobacter Chroococcum. Carbohydr. Res. 1988, 181, 143–152. [Google Scholar] [CrossRef]
- Fett, W.F.; Wijey, C.; Lifson, E.R. Occurrence of Alginate Gene Sequences among Members of the Pseudomonad RRNA Homology Groups I-IV. FEMS Microbiol. Lett. 1992, 78, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Gacesa, P. Bacterial Alginate Biosynthesis - Recent Progress and Future Prospects. Microbiology 1998, 144, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.R.; Linker, A. Production and Characterization of the Slime Polysaccharide of Pseudomonas aeruginosa. J. Bacteriol. 1973, 116, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.C.; Pritchard, M.F.; Ferguson, E.L.; Powell, K.A.; Patel, S.U.; Rye, P.D.; Sakellakou, S.M.; Buurma, N.J.; Brilliant, C.D.; Copping, J.M.; et al. Targeted Disruption of the Extracellular Polymeric Network of Pseudomonas aeruginosa Biofilms by Alginate Oligosaccharides. NPJ Biofilms Microbiomes 2018, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Govan, J.R.; Deretic, V. Microbial Pathogenesis in Cystic Fibrosis: Mucoid Pseudomonas aeruginosa and Burkholderia Cepacia. Microbiol. Rev. 1996, 60, 539–574. [Google Scholar] [CrossRef] [PubMed]
- Hentzer, M.; Teitzel, G.M.; Balzer, G.J.; Heydorn, A.; Molin, S.; Givskov, M.; Parsek, M.R. Alginate Overproduction Affects Pseudomonas aeruginosa Biofilm Structure and Function. J. Bacteriol. 2001, 183, 5395–5401. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, M.; Rabsch, P.; Von Der Hardt, H. Long-Term Follow up of Changes in FEV1 and Treatment Intensity during Pseudomonas aeruginosa Colonisation in Patients with Cystic Fibrosis. Thorax 1998, 53, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Parad, R.B.; Gerard, C.J.; Zurakowski, D.; Nichols, D.P.; Pier, G.B. Pulmonary Outcome in Cystic Fibrosis Is Influenced Primarily by Mucoid Pseudomonas aeruginosa Infection and Immune Status and Only Modestly by Genotype. Infect. Immun. 1999, 67, 4744. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.L.; Mellis, C.M.; Petrovic, L. Mucoid Pseudomonas aeruginosa Is a Marker of Poor Survival in Cystic Fibrosis. Pediatr. Pulmonol. 1992, 12, 158–161. [Google Scholar] [CrossRef] [PubMed]
- DeVries, C.A.; Ohman, D.E. Mucoid-to-Nonmucoid Conversion in Alginate-Producing Pseudomonas aeruginosa Often Results from Spontaneous Mutations in AlgT, Encoding a Putative Alternate Sigma Factor, and Shows Evidence for Autoregulation. J. Bacteriol. 1994, 176, 6677. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, C.D.; Ye, R.W.; Parsek, M.R.; Xie, Z.D.; Chakrabarty, A.M. The AlgT (AlgU) Gene of Pseudomonas aeruginosa, a Key Regulator Involved in Alginate Biosynthesis, Encodes an Alternative Sigma Factor (Sigma E). Proc. Natl. Acad. Sci. USA. 1995, 92, 7941–7945. [Google Scholar] [CrossRef] [PubMed]
- Whitney, J.C.; Howell, P.L. Synthase-Dependent Exopolysaccharide Secretion in Gram-Negative Bacteria. Trends Microbiol. 2013, 21, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Hay, I.D.; Wang, Y.; Moradali, M.F.; Rehman, Z.U.; Rehm, B.H.A. Genetics and Regulation of Bacterial Alginate Production. Environ. Microbiol. 2014, 16, 2997–3011. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C. Airway Surface Dehydration in Cystic Fibrosis: Pathogenesis and Therapy. Annu. Rev. Med. 2007, 58, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.W.; Schurr, M.J.; Mudd, M.H.; Govan, J.R.W.; Holloway, B.W.; Deretic, V. Mechanism of Conversion to Mucoidy in Pseudomonas aeruginosa Infecting Cystic Fibrosis Patients. Proc. Natl. Acad. Sci. USA. 1993, 90, 8377. [Google Scholar] [CrossRef] [PubMed]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung Infections Associated with Cystic Fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Marvig, R.L.; Sommer, L.M.; Molin, S.; Johansen, H.K. Convergent Evolution and Adaptation of Pseudomonas aeruginosa within Patients with Cystic Fibrosis. Nat. Genet. 2014 471 2014, 47, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Troxler, R.B.; Hoover, W.C.; Britton, L.J.; Gerwin, A.M.; Rowe, S.M. CLEARANCE OF INITIAL MUCOID PSEUDOMONAS AERUGINOSA IN PATIENTS WITH CYSTIC FIBROSIS. Pediatr. Pulmonol. 2012, 47, 1113. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.S.; Van De Mortel, M.; Nielsen, L.; De Guzman, G.N.; Li, X.; Halverson, L.J. Alginate Production by Pseudomonas Putida Creates a Hydrated Microenvironment and Contributes to Biofilm Architecture and Stress Tolerance under Water-Limiting Conditions. J. Bacteriol. 2007, 189, 8290–8299. [Google Scholar] [CrossRef] [PubMed]
- Alkawash, M.A.; Soothill, J.S.; Schiller, N.L. Alginate Lyase Enhances Antibiotic Killing of Mucoid Pseudomonas aeruginosa in Biofilms. APMIS 2006, 114, 131–138. [Google Scholar] [CrossRef] [PubMed]
- McCaslin, C.A.; Petrusca, D.N.; Poirier, C.; Serban, K.A.; Anderson, G.G.; Petrache, I. Impact of Alginate-Producing Pseudomonas aeruginosa on Alveolar Macrophage Apoptotic Cell Clearance. J. Cyst. Fibros. 2015, 14, 70–77. [Google Scholar] [CrossRef]
- Høiby, N. Prospects for the Prevention and Control of Pseudomonal Infection in Children with Cystic Fibrosis. Paediatr. Drugs 2000, 2, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Koch, C. Early Infection and Progression of Cystic Fibrosis Lung Disease. Pediatr. Pulmonol. 2002, 34, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.L.; Emerson, J.; Mayer-Hamblett, N.; Burns, J.L.; McNamara, S.; Accurso, F.J.; Konstan, M.W.; Chatfield, B.A.; Retsch-Bogart, G.; Waltz, D.A.; et al. Duration of Treatment Effect after Tobramycin Solution for Inhalation in Young Children with Cystic Fibrosis. Pediatr. Pulmonol. 2007, 42, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Sang, S.Y.; Coakley, R.; Lau, G.W.; Lymar, S.V.; Gaston, B.; Karabulut, A.C.; Hennigan, R.F.; Hwang, S.H.; Buettner, G.; Schurr, M.J.; et al. Anaerobic Killing of Mucoid Pseudomonas aeruginosa by Acidified Nitrite Derivatives under Cystic Fibrosis Airway Conditions. J. Clin. Invest. 2006, 116, 436–446. [Google Scholar] [CrossRef]
- Zhu, B.; Yin, H. Alginate Lyase: Review of Major Sources and Classification, Properties, Structure-Function Analysis and Applications. Bioengineered 2015, 6, 125. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Su, T.; Wu, H.; Liu, S.; Wang, D.; Zhao, T.; Jin, Z.; Du, W.; Zhu, M.J.; Chua, S.L.; et al. PslG, a Self-Produced Glycosyl Hydrolase, Triggers Biofilm Disassembly by Disrupting Exopolysaccharide Matrix. Cell Res. 2015 2512 2015, 25, 1352–1367. [Google Scholar] [CrossRef]
- Marmont, L.S.; Whitfield, G.B.; Rich, J.D.; Yip, P.; Giesbrecht, L.B.; Stremick, C.A.; Whitney, J.C.; Parsek, M.R.; Harrison, J.J.; Lynne Howell, P. PelA and PelB Proteins Form a Modification and Secretion Complex Essential for Pel Polysaccharide-Dependent Biofilm Formation in Pseudomonas aeruginosa. J. Biol. Chem. 2017, 292, 19411. [Google Scholar] [CrossRef] [PubMed]
- Colvin, K.M.; Alnabelseya, N.; Baker, P.; Whitney, J.C.; Lynne Howell, P.; Parsek, M.R. PelA Deacetylase Activity Is Required for Pel Polysaccharide Synthesis in Pseudomonas aeruginosa. J. Bacteriol. 2013, 195, 2329. [Google Scholar] [CrossRef] [PubMed]
- François Le Mauff, X.; Natalie Bamford, X.C.; Alnabelseya, N.; Zhang, Y.; Baker, P.; Robinson, H.; Codée, J.D.; Lynne Howell, P.; Sheppard, D.C.; Whitfield, C. Molecular Mechanism of Aspergillus Fumigatus Biofilm Disruption by Fungal and Bacterial Glycoside Hydrolases. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Baker, P.; Hill, P.J.; Snarr, B.D.; Alnabelseya, N.; Pestrak, M.J.; Lee, M.J.; Jennings, L.K.; Tam, J.; Melnyk, R.A.; Parsek, M.R.; et al. Exopolysaccharide Biosynthetic Glycoside Hydrolases Can Be Utilized to Disrupt and Prevent Pseudomonas aeruginosa Biofilms. Sci. Adv. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Snarr, B.D.; Baker, P.; Bamford, N.C.; Sato, Y.; Liu, H.; Lehoux, M.; Gravelat, F.N.; Ostapska, H.; Baistrocchi, S.R.; Cerone, R.P.; et al. Microbial Glycoside Hydrolases as Antibiofilm Agents with Cross-Kingdom Activity. [CrossRef]
- Zhang, J.; He, J.; Zhai, C.; Ma, L.Z.; Gu, L.; Zhao, K. Effects of PslG on the Surface Movement of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- Ostapska, H.; Raju, D.; Corsini, R.; Lehoux, M.; Lacdao, I.; Gilbert, S.; Sivarajah, P.; Bamford, N.C.; Baker, P.; Gravelat, F.N.; et al. Preclinical Evaluation of Recombinant Microbial Glycoside Hydrolases as Antibiofilm Agents in Acute Pulmonary Pseudomonas aeruginosa Infection. Antimicrob. Agents Chemother. 2022, 66. [Google Scholar] [CrossRef] [PubMed]
- Pestrak, M.J.; Baker, P.; Dellos-Nolan, S.; Hill, P.J.; Passos da Silva, D.; Silver, H.; Lacdao, I.; Raju, D.; Parsek, M.R.; Wozniak, D.J.; et al. Treatment with the Pseudomonas aeruginosa Glycoside Hydrolase PslG Combats Wound Infection by Improving Antibiotic Efficacy and Host Innate Immune Activity. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, M.; Karakulska, J.; Sobolewski, P.; Kowalska, U.; Grygorcewicz, B.; Böttcher, D.; Bornscheuer, U.T.; Drozd, R. Glycoside Hydrolase (PelAh) Immobilization Prevents Pseudomonas aeruginosa Biofilm Formation on Cellulose-Based Wound Dressing. Carbohydr. Polym. 2020, 246. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.R.; Raju, D.; Lacdao, I.; Gilbert, S.; Sivarajah, P.; Howell, P.L.; Prestidge, C.A.; Thomas, N. Protective Liquid Crystal Nanoparticles for Targeted Delivery of PslG: A Biofilm Dispersing Enzyme. ACS Infect. Dis. 2021, 7, 2102–2115. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; He, J.; Li, N.; Liu, S.; Xu, S.; Gu, L. A Rational Designed PslG With Normal Biofilm Hydrolysis and Enhanced Resistance to Trypsin-Like Protease Digestion. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Asker, D.; Awad, T.S.; Raju, D.; Sanchez, H.; Lacdao, I.; Gilbert, S.; Sivarajah, P.; Andes, D.R.; Sheppard, D.C.; Howell, P.L.; et al. Preventing Pseudomonas aeruginosa Biofilms on Indwelling Catheters by Surface-Bound Enzymes. ACS Appl. bio Mater. 2021, 4, 8248. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.S.; Park, S.; Ramos, M.C.; Nast, C.C.; Eftekhar, F.; Schiller, N.L. Effects of Alginase on the Natural History and Antibiotic Therapy of Experimental Endocarditis Caused by Mucoid Pseudomonas aeruginosa. Infect. Immun. 1992, 60, 3979–3985. [Google Scholar] [CrossRef] [PubMed]
- Daboor, S.M.; Rohde, J.R.; Cheng, Z. Disruption of the Extracellular Polymeric Network of Pseudomonas aeruginosa Biofilms by Alginate Lyase Enhances Pathogen Eradication by Antibiotics. J. Cyst. Fibros. 2021, 20, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Suntres, Z.E.; Omri, A. Importance of DNase and Alginate Lyase for Enhancing Free and Liposome Encapsulated Aminoglycoside Activity against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2009, 64, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Aspe, M.; Jensen, L.; Melegrito, J.; Sun, M. The Role of Alginate and Extracellular DNA in Biofilm-Meditated Pseudomonas aeruginosa Gentamicin Resistance. J. Exp. Microbiol. Immunol. 2012, 16, 42–48. [Google Scholar]
- Eftekhar, F.; Speert, D.P. Alginase Treatment of Mucoid Pseudomonas aeruginosa Enhances Phagocytosis by Human Monocyte-Derived Macrophages. Infect. Immun. 1988, 56, 2788–2793. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.E.; Ertesvåg, H.; Beyenal, H.; Lewandowski, Z. Resistance of Biofilms Containing Alginate-producing Bacteria to Disintegration by an Alginate Degrading Enzyme (Algl). Biofouling 2009, 17, 203–210. [Google Scholar] [CrossRef]
- Lamppa, J.W.; Griswold, K.E. Alginate Lyase Exhibits Catalysis-Independent Biofilm Dispersion and Antibiotic Synergy. 2013. [CrossRef]
- Mahajan, S.; Sunsunwal, S.; Gautam, V.; Singh, M.; Ramya, T.N.C. Biofilm Inhibitory Effect of Alginate Lyases on Mucoid P. Aeruginosa from a Cystic Fibrosis Patient. Biochem. Biophys. reports 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Lamppa, J.W.; Ackerman, M.E.; Lai, J.I.; Scanlon, T.C.; Griswold, K.E. Genetically Engineered Alginate Lyase-PEG Conjugates Exhibit Enhanced Function and Reduced Immunoreactivity. PLoS ONE 2011, 6, 17042. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Tripathi, M.; Pandey, N.; Agrawal, A.K.; Gade, S.; Anjum, M.M.; Tilak, R.; Singh, S. Alginate Lyase Immobilized Chitosan Nanoparticles of Ciprofloxacin for the Improved Antimicrobial Activity against the Biofilm Associated Mucoid P. Aeruginosa Infection in Cystic Fibrosis. Int. J. Pharm. 2019, 563, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-Encoded Virion-Associated Enzymes to Overcome the Carbohydrate Barriers during the Infection Process. Appl. Microbiol. Biotechnol. 2017, 101, 3103. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.W. Polysaccharide Lyases. FEMS Microbiol. Rev. 1995, 16, 323–347. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, G.W.; Denyer, S.P.; Olliff, C.J.; Ibrahim, L.J. Reduction in Exopolysaccharide Viscosity as an Aid to Bacteriophage Penetration through Pseudomonas aeruginosa Biofilms. Appl. Environ. Microbiol. 2001, 67, 2746. [Google Scholar] [CrossRef] [PubMed]
- Glonti, T.; Chanishvili, N.; Taylor, P.W. Bacteriophage-Derived Enzyme That Depolymerizes the Alginic Acid Capsule Associated with Cystic Fibrosis Isolates of Pseudomonas aeruginosa. J. Appl. Microbiol. 2010, 108, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Liu, Y.; Wang, C.; He, T.; Gao, S.; Xing, S.; Huang, Y.; Fan, H.; Zhang, X.; Yu, W.; et al. Identification of a Lytic Pseudomonas aeruginosa Phage Depolymerase and Its Anti-Biofilm Effect and Bactericidal Contribution to Serum. Virus Genes 2019, 55, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Ph 1/2 Study Evaluating Safety and Tolerability of Inhaled AP-PA02 in Subjects With Chronic Pseudomonas aeruginosa Lung Infections and Cystic Fibrosis. Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT04596319. (accessed on 28 January 2023).
- CYstic Fibrosis BacterioPHage Study at Yale (CYPHY). Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT04684641. (accessed on 28 January 2023).
- Nebulized Bacteriophage Therapy in Cystic Fibrosis Patients With Chronic Pseudomonas aeruginosa Pulmonary Infection. Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT05010577. (accessed on 28 January 2023).
- A Phase 1b/2 Trial of the Safety and Microbiological Activity of Bacteriophage Therapy in Cystic Fibrosis Subjects Colonized With Pseudomonas aeruginosa. Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT05453578. (accessed on 28 January 2023).
- Khan, S.; Tøndervik, A.; Sletta, H.; Klinkenberg, G.; Emanuel, C.; Onsøyen, E.; Myrvold, R.; Howe, R.A.; Walsh, T.R.; Hill, K.E.; et al. Overcoming Drug Resistance with Alginate Oligosaccharides Able To Potentiate the Action of Selected Antibiotics. Antimicrob. Agents Chemother. 2012, 56, 5134. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, M.F.; Powell, L.C.; Menzies, G.E.; Lewis, P.D.; Hawkins, K.; Wright, C.; Doull, I.; Walsh, T.R.; Onsøyen, E.; Dessen, A.; et al. A New Class of Safe Oligosaccharide Polymer Therapy To Modify the Mucus Barrier of Chronic Respiratory Disease. Mol. Pharm. 2016, 13, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Stokniene, J.; Varache, M.; Rye, P.D.; Hill, K.E.; Thomas, D.W.; Ferguson, E.L. Alginate Oligosaccharides Enhance Diffusion and Activity of Colistin in a Mucin-Rich Environment. Sci. Reports 2022 121 2022, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hengzhuang, W.; Song, Z.; Ciofu, O.; Onsøyen, E.; Rye, P.D.; Høiby, N. OligoG CF-5/20 Disruption of Mucoid Pseudomonas aeruginosa Biofilm in a Murine Lung Infection Model. Antimicrob. Agents Chemother. 2016, 60, 2620–2626. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, M.F.; Powell, L.C.; Jack, A.A.; Powell, K.; Beck, K.; Florance, H.; Forton, J.; Rye, P.D.; Dessen, A.; Hill, K.E.; et al. A Low-Molecular-Weight Alginate Oligosaccharide Disrupts Pseudomonal Microcolony Formation and Enhances Antibiotic Effectiveness. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Stokniene, J.; Powell, L.C.; Aarstad, O.A.; Aachmann, F.L.; Rye, P.D.; Hill, K.E.; Thomas, D.W.; Ferguson, E.L. Bi-Functional Alginate Oligosaccharide–Polymyxin Conjugates for Improved Treatment of Multidrug-Resistant Gram-Negative Bacterial Infections. Pharm. 2020, Vol. 12, Page 1080 2020, 12, 1080. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, M.J.R.; Dorrier, J.M.; Schoenfisch, M.H. Antibiofilm Efficacy of Nitric Oxide-Releasing Alginates against Cystic Fibrosis Bacterial Pathogens. ACS Infect. Dis. 2019, 5, 1327. [Google Scholar] [CrossRef] [PubMed]
- Koningsbruggen-Rietschel, S. van; Davies, J.C.; Pressler, T.; Fischer, R.; MacGregor, G.; Donaldson, S.H.; Smerud, K.; Meland, N.; Mortensen, J.; Fosbøl, M.Ø.; et al. Inhaled Dry Powder Alginate Oligosaccharide in Cystic Fibrosis: A Randomised, Double-Blind, Placebo-Controlled, Crossover Phase 2b Study. ERJ Open Res. 2020, 6, 00132–02020. [Google Scholar] [CrossRef] [PubMed]
- A Pivotal Phase IIb Clinical Trial of Inhaled Alginate Oligosaccharide (OligoG) for Cystic Fibrosis. Available online: Https://Cordis.Europa.Eu/Project/Id/755234. (accessed on 28 January 2023).
- Christensen, B.E.; Ertesvag, H.; Haluk, B.; Lewandowski, Z. Biofouling Resistance of Biofilms Containing Alginate-Producing Bacteria to Disintegration by an Alginate Degrading Enzyme (Algl). 2001. [CrossRef]
- Ostapska, H.; Raju, D.; Corsini, R.; Lehoux, M.; Lacdao, I.; Gilbert, S.; Sivarajah, P.; Bamford, N.C.; Baker, P.; Gravelat, F.N.; et al. Preclinical Evaluation of Recombinant Microbial Glycoside Hydrolases as Antibiofilm Agents in Acute Pulmonary Pseudomonas Aeruginosa Infection. Antimicrob. Agents Chemother. 2022, 66. [Google Scholar] [CrossRef] [PubMed]
- LiPuma, J.J. The Sense and Nonsense of Antimicrobial Susceptibility Testing in Cystic Fibrosis. J. Pediatric Infect. Dis. Soc. 2022, 11 (Suppl. S2), S46–S52. [Google Scholar] [CrossRef] [PubMed]
- Waters, V.; Ratjen, F. Standard versus Biofilm Antimicrobial Susceptibility Testing to Guide Antibiotic Therapy in Cystic Fibrosis. Cochrane Database Syst. Rev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Waters, V.; Jahnke, N.; Ratjen, F. Standard versus Biofilm Antimicrobial Susceptibility Testing to Guide Antibiotic Therapy in Cystic Fibrosis. Cochrane Database Syst. Rev. 2020, 2020. [Google Scholar] [CrossRef]
- Periasamy, S.; Nair, H.A.S.; Lee, K.W.K.; Ong, J.; Goh, J.Q.J.; Kjelleberg, S.; Rice, S.A. Pseudomonas aeruginosa PAO1 Exopolysaccharides Are Important for Mixed Species Biofilm Community Development and Stress Tolerance. Front. Microbiol. 2015, 6, 851. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, T.; Yau, Y.C.W.; Stapleton, P.J.; Gong, Y.; Wang, P.W.; Guttman, D.S.; Waters, V. Staphylococcus Aureus Interaction with Pseudomonas Aeruginosa Biofilm Enhances Tobramycin Resistance. NPJ Biofilms Microbiomes 2017, 3, 25. [Google Scholar] [CrossRef]
- Billings, N.; Ramirez Millan, M.; Caldara, M.; Rusconi, R.; Tarasova, Y.; Stocker, R.; Ribbeck, K. The Extracellular Matrix Component Psl Provides Fast-Acting Antibiotic Defense in Pseudomonas aeruginosa Biofilms. PLOS Pathog. 2013, 9, e1003526. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.C.; Kundukad, B.; Seviour, T.; Van der Maarel, J.R.C.; Yang, L.; Rice, S.A.; Doyle, P.; Kjelleberg, S. Dynamic Remodeling of Microbial Biofilms by Functionally Distinct Exopolysaccharides. MBio 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Limoli, D.H.; Whitfield, G.B.; Kitao, T.; Ivey, M.L.; Davis, M.R.; Grahl, N.; Hogan, D.A.; Rahme, L.G.; Howell, P.L.; O’Toole, G.A.; et al. Pseudomonas aeruginosa Alginate Overproduction Promotes Coexistence with Staphylococcus Aureus in a Model of Cystic Fibrosis Respiratory Infection. MBio 2017, 8. [Google Scholar] [CrossRef]
- Chattoraj, S.S.; Murthy, R.; Ganesan, S.; Goldberg, J.B.; Zhao, Y.; Hershenson, M.B.; Sajjan, U.S. Pseudomonas aeruginosa Alginate Promotes Burkholderia Cenocepacia Persistence in Cystic Fibrosis Transmembrane Conductance Regulator Knockout Mice. Infect. Immun. 2010, 78, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Salsgiver, E.L.; Fink, A.K.; Knapp, E.A.; LiPuma, J.J.; Olivier, K.N.; Marshall, B.C.; Saiman, L. Changing Epidemiology of the Respiratory Bacteriology of Patients With Cystic Fibrosis. Chest 2016, 149, 390. [Google Scholar] [CrossRef] [PubMed]
- McBennett, K.A.; Davis, P.B.; Konstan, M.W. Increasing Life Expectancy in Cystic Fibrosis: Advances and Challenges. Pediatr. Pulmonol. 2022, 57 (Suppl. S1), S5. [Google Scholar] [CrossRef] [PubMed]
- Reznikov, L.R.; Abou Alaiwa, M.H.; Dohrn, C.L.; Gansemer, N.D.; Diekema, D.J.; Stoltz, D.A.; Welsh, M.J. Antibacterial Properties of The Cftr Potentiator Ivacaftor. J. Cyst. Fibros. 2014, 13, 515. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.E.; Dubois, A.V.; Ingram, R.J.; Weldon, S.; Taggart, C.C.; Elborn, J.S.; Tunney, M.M. Activity of Innate Antimicrobial Peptides and Ivacaftor against Clinical Cystic Fibrosis Respiratory Pathogens. Int. J. Antimicrob. Agents 2017, 50, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.Y.; Lim, D.J.; Mackey, C.; Skinner, D.; Zhang, S.; McCormick, J.; Woodworth, B.A. Ivacaftor, a Cystic Fibrosis Transmembrane Conductance Regulator Potentiator, Enhances Ciprofloxacin Activity Against Pseudomonas Aeruginosa. Am. J. Rhinol. Allergy 2019, 33, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, R.V.E.; Su, V.C.H.; Quon, B.S. Real-World Safety of CFTR Modulators in the Treatment of Cystic Fibrosis: A Systematic Review. J. Clin. Med. 2021, 10, 1–56. [Google Scholar] [CrossRef] [PubMed]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Durfey, S.L.; Pipavath, S.; Li, A.; Vo, A.T.; Ratjen, A.; Carter, S.; Morgan, S.J.; Radey, M.C.; Grogan, B.; Salipante, S.J.; et al. Combining Ivacaftor and Intensive Antibiotics Achieves Limited Clearance of Cystic Fibrosis Infections. MBio 2021, 12. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic overview of the P. aeruginosa life cycle and the role of matrix exopolysaccharides Psl, Pel, and alginate. The biofilm life cycle begins with Psl-mediated surface/substrate attachment of P. aeruginosa. The association of P. aeruginosa cells results in microcolony formation as motile cells transition to a sessile phenotype. As the biofilm develops, Psl, Pel, or both EPSs are incorporated in the biofilm architecture to form large 3D macrostructures. The mature biofilm is characterized by the differential expression of these EPSs, the activation of quorum sensing, increasing population heterogeneity, and eventual dispersal to new areas of the lung. Created with Biorender.com.
Figure 1.
Schematic overview of the P. aeruginosa life cycle and the role of matrix exopolysaccharides Psl, Pel, and alginate. The biofilm life cycle begins with Psl-mediated surface/substrate attachment of P. aeruginosa. The association of P. aeruginosa cells results in microcolony formation as motile cells transition to a sessile phenotype. As the biofilm develops, Psl, Pel, or both EPSs are incorporated in the biofilm architecture to form large 3D macrostructures. The mature biofilm is characterized by the differential expression of these EPSs, the activation of quorum sensing, increasing population heterogeneity, and eventual dispersal to new areas of the lung. Created with Biorender.com.
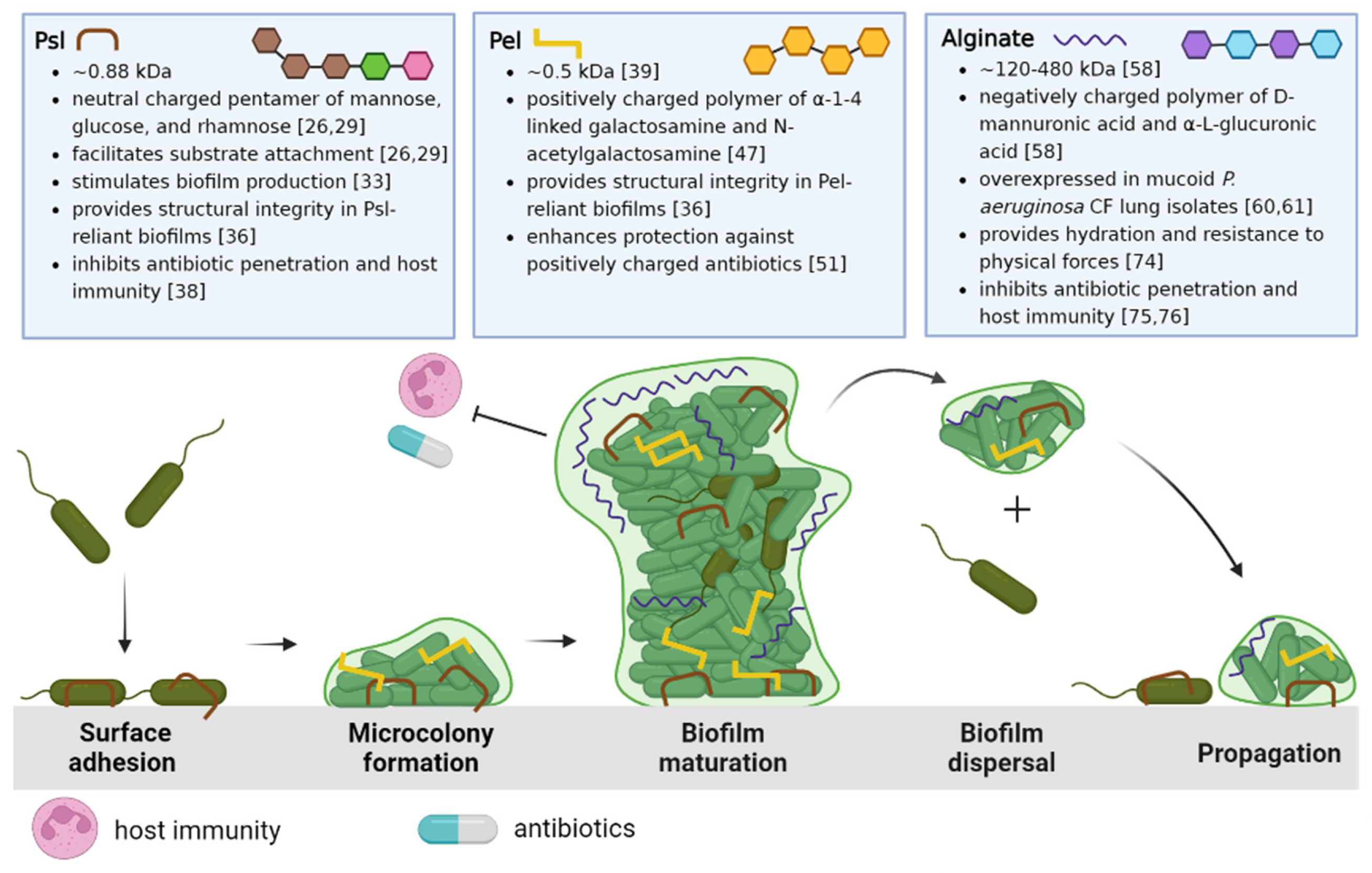
Figure 2.
Overview of therapeutics targeting matrix EPSs of P. aeruginosa biofilms suspended in CF airway sputum. Outlined are potential therapeutic applications, drug mechanisms, half-life, synergistic antibiotics, immune effects, safety, and methods of delivery for PslGh (top left), PelAh (top right), AgLase (bottom left), and OligoG (bottom right).
Figure 2.
Overview of therapeutics targeting matrix EPSs of P. aeruginosa biofilms suspended in CF airway sputum. Outlined are potential therapeutic applications, drug mechanisms, half-life, synergistic antibiotics, immune effects, safety, and methods of delivery for PslGh (top left), PelAh (top right), AgLase (bottom left), and OligoG (bottom right).
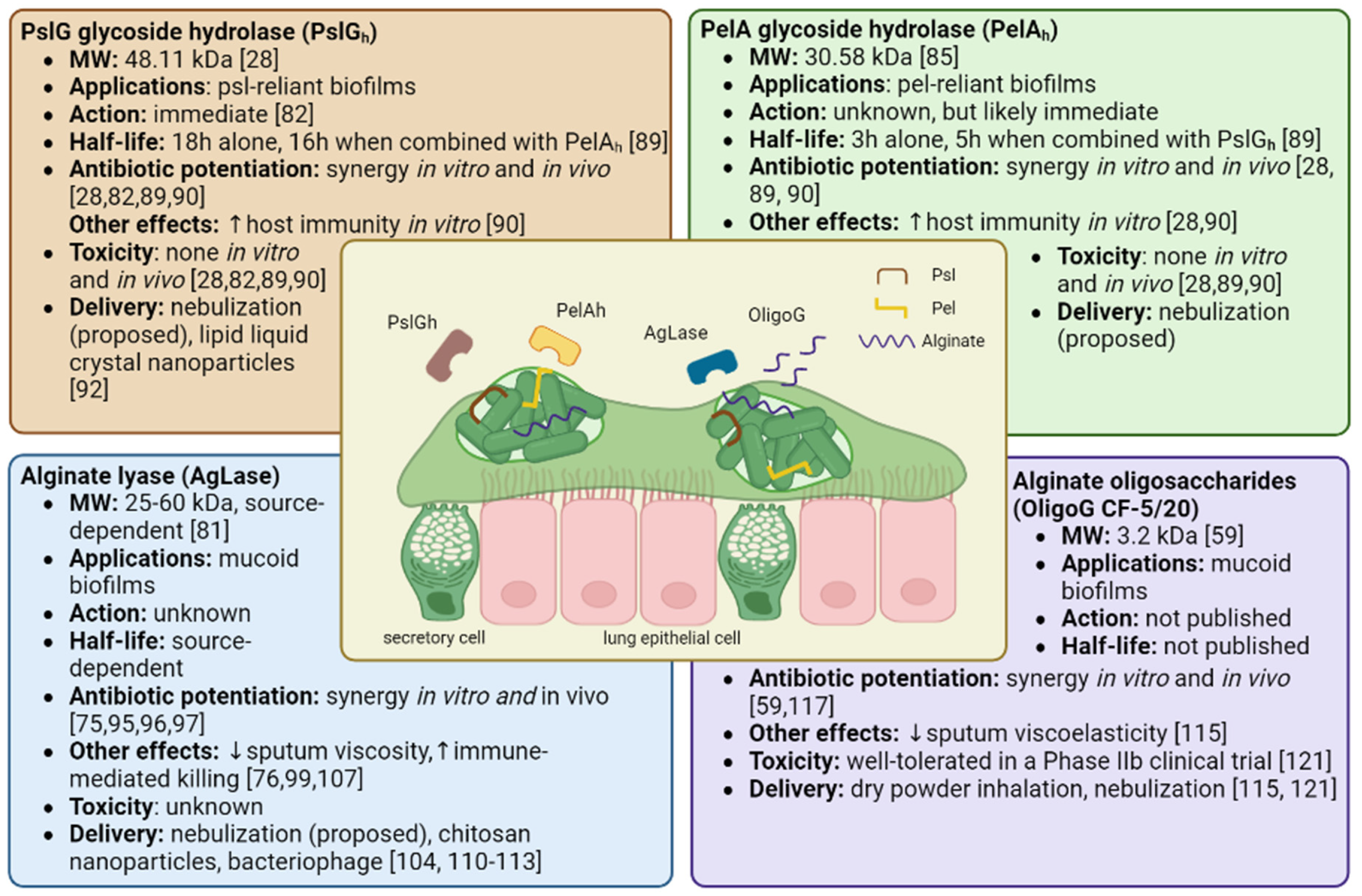

Table 1.
EPS-Targeted Therapies of P. aeruginosa Biofilms in CF.
| Product | Therapeutic Challenges and Considerations |
|---|---|
| PslG glycoside hydrolase (PslGh) |
|
| PelA glycoside hydrolase (PelAh) |
|
| Alginate Lyase (AgLase) |
|
|
Alginate Oligosaccharide (OligoG, CF-5/20) |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



