
Submitted:
08 February 2023
Posted:
13 February 2023
You are already at the latest version
Abstract
Background: Traumatic brain injury (TBI) remains a significant risk factor for post-traumatic epilepsy (PTE). The pathophysiological mechanisms underlying the injury-induced epileptogenesis are under under investigation. The dentate gyrus, a structure highly susceptible to injury, and has been implicated in the evolution of seizure development. Methods: Utilizing the murine unilateral focal control cortical impact (CCI) injury, we evaluated seizure onset using 24/7 EEG video analysis at 2-4 months post-injury. Cellular changes in the dentate gyrus and hilus of the hippocampus were quantified by non-biased stereology and Imaris image analysis to evaluate Prox1-positive cell migration, astrocyte branching and morphology, as well as neuronal loss at four months post-injury. Isolation of region-specific astrocytes and RNA-seq was performed to determine differential gene expression in PTE+ vs. PTE- that may comport with the epileptogenic process. Results: CCI injury resulted in 37% PTE+-incidence, which increased with injury severity and hippocampal damage. Histological assessments uncovered a significant loss of hilar interneurons that coincided with aberrant migration of Prox1-positive granule cells and reduced astroglial branching in PTE+ compared to PTE- mice. We uniquely identified Cst3 as a PTE+-specific gene signature in astrocytes across all brain regions. Conclusions: These findings suggest that epileptogenesis may emerge following TBI due to distinct aberrant cellular remodeling events and key molecular changes in the dentate gyrus of the hippocampus.
Keywords:
Traumatic brain injury
; hippocampus
1. Introduction
Acquired brain injury caused by sudden trauma to the brain disrupts normal function [1,2]. Traumatic brain injury (TBI) remains a significant and growing source of mortality and permanent disability among roughly 2.87 million people yearly in the USA, resulting in a profound health and economic burden [3]. TBI substantially increases the risk of developing comorbidities such as epilepsy, depression, post-traumatic stress disorder, and dementia [4]. Post-traumatic epilepsy (PTE) is a type of acquired epilepsy that represents one of the most common TBI sequelae, affecting up to 50% of individuals after severe TBI and representing up to one-fifth of all acquired epilepsies [5,6,7]. PTE manifests clinically as spontaneous unprovoked, recurrent seizures with devastating consequences for patient care, recovery, and overall health [7,8,9].
TBI produces direct and secondary damage to the brain that includes neuronal loss, cerebral blood flow disruption, neuroinflammation, reactive oxidative damage, and blood-brain barrier disruption [10,11,12,13]. These pathophysiological changes may contribute to circuit imbalances that favor excitatory over inhibitory synaptic function [14,15], formation of spontaneous epileptic foci, and PTE [15,16]. However, the details about the epileptiform process are not well understood from its origin and progression. PTE remains a leading cause of death years after TBI [17,18]. Most patients with PTE have focal with secondary generalized seizures [19,20,21], but other seizure types are related to hippocampal sclerosis and primary generalized seizures [22,23]. A variety of antiepileptic drugs (AED), including phenytoin [24], levetiracetam [25], lamotrigine, gabapentin, and valproic acid, have been developed to reduce the onset of seizures post-injury with relative success [26]. While AEDs can be effective in controlling acute or symptomatic seizures (<1 week post-TBI), they do not prevent post-traumatic epileptogenesis and ~30% of individuals with PTE will develop drug resistance [27,28]. Importantly, these therapies do not target the epileptogenesis process thus, do not prevent the late development or progression of PTE [29] and a higher percentage will show AED-related adverse effects that compromise the quality of life, making AED treatment options ineffective [16,30,31].
Despite rigorous clinical and pre-clinical research in TBI and PTE, there are no current therapies to prevent the development of PTE and there is a strong need for better biomarkers and clinical tools that can reliably predict which individuals will develop PTE. Finally, only 7-8% of patients are potential candidates for surgical resection of the epileptogenic foci [32], but surgery complications and difficulty identifying the seizure foci compromise the surgery outcome [16]. The lack of effective treatment highlights the need for robust and reproducible animal models of PTE with spontaneous seizures that are not induced by chemoconvulsants to understand the cellular and molecular changes underlying seizure development after TBI [33].
In the current study, we establish that murine focal controlled cortical impact (CCI) injury, an established model of PTE [34,35,36,37,38], results in PTE onset whose incidence is correlated with injury severity. Importantly, we observed significant cellular changes in the hippocampus, namely aberrant Prox1-positive neuroblast migration, excessive hilar interneuron loss, and altered astrogliosis with divergent transcriptomic signatures that are region specific. These findings add to the body of work describing hippocampal alterations associated with the development of PTE.
2. Materials and Methods
Animals. CD1 male mice at P60-90 from Charles River were housed in an AALAC-accredited facility with a 12 h light-dark cycle, food, and water ad libitum. For EEG/video recording, animals were housed individually in a 12.5” x 12.5” x 15.5” polycarbonate cage (AAA Plastic Products, Birmingham, AL) with corncob bedding and nesting material. All experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and with the approval of the Virginia Tech Institutional Animal Care and Use Committee (IACUC; #17-138).
Controlled Cortical Impact (CCI). Mice were injured as previously described [39,40,41,42,43]. Briefly, ketamine 100mg/kg, xylazine 10mg/kg, and Buprenorphine SR (0.5 mg/kg) were administered subcutaneously for anesthetic and analgesia before surgery. Hair on the scalp was removed. Mice were then positioned in a stereotaxic frame at 37˚C using a homeothermic blanket system (Harvard apparatus, Lewes, DE). A Φ=4mm craniectomy was drilled over the right parietal bone, and injury induced using a Φ=3-mm flat tip, connected to an eCCI-6.3 device (Custom Design & Fabrication, LLC) at a velocity of 5.0 m/s, 250 ms impact duration, and depth of 2.0 mm (n=20) or 2.5 mm (n=23; 15 of which were used for astrocyte isolation). Two severe injured mice (2.5mm depth) died unexpectedly during the course of the study and were threfore excluded. Kwik-sil (WPI, Sarasota, FL, USA) was applied to cover the craniectomy, and the incision was closed with 4.0 PDO sutures (AD surgical, Sunnyvale, CA, USA).
EEG Implantation. Electrode placement was performed 60 days after CCI injury as previously described [44]. Using a stereotaxic micromotor drill (Stoelting) equipped with a 0.7 mm carbon steel burr drill bit (FST), two holes were drilled through the skull for reference electrodes at coordinates (1.00 ML, 1.00 AP) and (-1.00 ML, -1.00 AP), and a ground electrode (-1.00 ML, -5.00 AP). Two more holes were drilled partially through the skull, and screws were inserted as anchors. A 0.125mm diameter platinum-iridium electrode coated in Teflon (Plastics One, Roanoke VA) was implanted intracranially within 0.5 mm from the surface of the dura. Dental cement (Stoelting Wood dale IL) was applied to secure the electrode. Animals were excluded from the experiment if they had profuse hemorrhage during EEG implementation or lost more than 20% of their body weight throughout the study. Sham animals were subject to craniectomy surgery and electrode implantation. Electrodes were connected to a commutator (Plastics One) using EEG cables (Plastics One) and then to an amplifier (EEG100C, BioPac) with a gain of 5000, a 100 Hz lowpass filter, 0.5 Hz high pass filter, and 500 Hz sampling rate and recorded continuously for two months using BioPac’s AcqKnowledge software.
EEG and video analysis. EEG data were analyzed using a Matlab [45] automated algorithm and manually in BioPac’s AcqKnowledge software (Goleta, CA, USA). We used five criteria to identify seizures. The event duration had to be over 5 seconds and consisted of spikes (20-70 ms), sharp waves (70-200 ms), poly spikes, or slow-wave complexes. We further looked for waveform asymmetry across the x-axis, the evolution of the amplitude and frequency of the spikes over the time course of the event, and post-ictal suppression of the waveform. When an electrographic seizure was observed, the corresponding video was referenced to determine if there were any behavioral correlates, and scratching, grooming, or feeding behavior was observed as sources of artifacts. Mice were considered PTE+ if they had 2 or more epileptic episodes during the study.
Brain tissue preparation, serial sectioning, and staining. Tissue handling was performed as previously described [46,47]. Briefly, Mice were euthanized with isoflurane followed by transcardial perfusion with 1X PBS then 4% paraformaldehyde. Brains were placed in 4% PFA at 4oC overnight, followed by cryopreservation, embedded in optimal cutting temperature compound (OCT; Fisher Scientific), and stored at -80oC. Five serial coronal sections of 30μm, spaced 450μm apart, were mounted on pre-coated, charged slides using CryoStar Cryostat NX70 (Thermo Fisher Scientific).
Brain tissue preparation for astrocyte isolation and RNA extraction. Isolation of cortical and hippocampal astrocytes was performed as described previously [48,49,50]. Briefly, the ipsilateral and contralateral cortices and hippocampi from 15 CCI-injured mice (2.5mm depth) and 5 shams were microdissected and separated in ice-cold ACSF (120 mM NaCl, 3.0 mM KCl, 2 mM MgCl, 0.2 mM CaCl, 26.2 mM NaHCO3, 11.1 mM glucose, 5.0 mM HEPES, 3 mM AP5, 3 mM CNQX) bubbled with 95% oxygen. Tissue was minced, dissociated for 15–30 min using Worthington Papain Dissociation Kit, and subsequently triturated and filtered through a 70 µM filter until a single-cell suspension was then used to isolate astrocytes utilizing Miltenyi Biotec’s ACSA-2+ MicroBead kit then placed on RNAlater (ThermoFisher).
RNA extraction and sequencing analysis. RNA isolation was performed on isolated brain-derived astrocytes [48,49]. RNA sequencing was performed by MedGenome. Libraries were prepared using the TakaraSMARTSeq V4 ultra-low input RNA kit. Sequencing was performed on a NovSeq instrument. Paired-end reads 2 x 100 bp sequencing runs were performed with an average of 55 million reads per sample. Bases with quality scores less than 30 and adapters were trimmed from raw sequencing reads by Trim Galore (v 0.6.4). After trimming, only reads with lengths greater than 30bp were mapped to mm10 by RSEM (v1.2.28) with bowtie2 (v 2.4.1), with an average 81.5% mapping efficiency. The raw counts were used to identify differentially expressed genes by DESeq2 (v 1.36.0). Only genes with an average TPM greater than 5 in at least one group, p-value less than 0.05, and at least 1.5 fold change were considered differentially expressed genes. All the differentially expressed genes were used for GO enrichment with R package clusterProfiler (v4.4.4) and org.Mm.eg.db (v3.15.0). The top 10 most significant biological processes (BP) terms were used to generate GO circle plot with R package GOplot (v1.0.2).
Immunohistochemistry, StereoInvestigator analysis and lesion volume. Coronal serial sections were fixed with 10% buffered formalin, washed 3 times in 1XPBS and blocked in 2% cold water fish gelatin (Sigma, Inc) with 0.2% triton, then incubated overnight with primary antibodies, goat IgG anti-doublecortin (DCX) 1/200 dilution (Santa Cruz sc-8066) and rabbit IgG anti-Prox-1 at 1/200 dilution (ECM Biosciences, #CM4961) in block buffer at 4oC, washed with PBS 3 times, incubated with secondary antibodies and mounted in media with DAPI (SouthernBiotech). Images were acquired using a Nikon ECLIPSE Ti2 Inverted confocal microscope with a motorized stage and a Nikon C2 laser system. Five coronal sections were analyzed by a blinded investigator using the Optical Fractionator probe from MBF StereoInvestigator software (MicroBrightField, Williston, VT, USA) and an upright Olympus BX51TRF motorized microscope (Olympus America, Center Valley, PA, USA) as previously described [39,40,42,43]. Contours for the DG and hilus were created, and the optical fractionator's grid size was set at 150x150mm with a 75x75mm counting frame. The estimated number of cells was then divided by the contoured volume in planimetry and represented as the estimated number of cells per mm3. Lesion volume was assessed on five serial coronal sections stained with Cresyl violet acetate (Electron Microscopy Sciences, Hatfield, PA) by a blinded investigator using the Cavalieri Estimator from StereoInvestigator (MicroBrightField, Williston, VT, USA) on an upright Olympus BX51TRF motorized microscope (Olympus America, Center Valley, PA, USA) as previously described [41,51,52].
Astrocyte morphology analysis. Sofware-based analysis was used to estimate the coverage, morphology, and branching of the astrocytes in the hilus. Five serial sections were stained for Glial fibrillary acidic protein (GFAP) as previously described [43]. 20X z-stack images were used to determine astrocyte Hilar coverage using volumetric analysis whereas 40X z-stack images were used for morphology and branching analysis. 3D surface reconstructions of GFAP+ astrocytes within the hilus were created in Imaris software (Oxford Instruments) based on absolute threshold intensity with a smoothing detail of .250µm for morphology analysis or no smoothing detail for volumetric analysis. For morphology analysis, GFAP-positive astrocytes were separated by using seed points that were approximately equal in size to a typical cell astrocyte soma, calculated in slicer mode of Imaris. Any surfaces with a voxel size of 10 were excluded from the data set. Cells that extended outside the region of interest were excluded from analysis.
Sholl analysis of astrocytes in Imaris. Following surface morphology analysis, astrocytes were selected at random to be skeletonized using Imaris’ filament tracer. Filaments were created using the auto path algorithm. Using the surface reconstruction as a guide, a region of interest was created around the selected astrocyte. The starting point of the astrocyte was selected by isolating one point at the center of the astrocyte, which could be determined using DAPI, and subsequent seed points were selected by the Imaris quality index with a thresholding of +/-100. Dendrite diameter was selected by Imaris local contrast +/-.5 and the shortest distance from distance map algorithm. Following filament creation, 3D Sholl data was extracted from Imaris with a 1µm increasing shell size.
Statistical analysis. Student’s two-tailed t-test was used for comparison of the two experimental groups. For three or more groups, multiple comparisons were done using one-way and two-way ANOVA, with post hoc test for multiple pairwise examinations. For Sholl analysis, significance was tested using a Mixed-Effects model with a Dunnett's Post-hoc analysis for multiple comparisons. Data were graphed using GraphPad Prism, version 9 (GraphPad Software, Inc., San Diego, CA). Mean values were reported together with the standard error of the mean (SEM).
3. Results
Murine CCI-induced PTE is associated with injury severity and hippocampal damage
We utilized the controlled cortical impact (CCI) injury model of TBI to monitor and quantify the incidence of PTE in adult male CD1 mice after moderate or severe injury, 2mm or 2.5mm impact depth, respectively. Two months after the injury, subdural electrodes were implanted. EEG and video recording were performed continuously from 2 to 4 months after injury (Figure 1A) utilizing a bipolar montage with two recording electrodes and one ground electrode (Figure 1B). Electrographic seizures were defined as high-amplitude rhythmic discharges with an amplitude and frequency evolution, and a duration minimum 5s, epileptiform (interictal) spiking sometimes were present before the onset of the seizure to finalize with postictal depression. Generalized seizures were observed, showing typical interictal spikes, seizure activity, and post-seizure depression (Figure 1C). Using an automated MatLab algorithm [45], each EEG event was manually confirmed together with a video review. We demonstrate that 37% of all mice tested were PTE+, with a higher incidence seen in mice whose histopathology showed evidence of hippocampal damage or displacement and categorized as severe (Figure 1D). No seizures were demonstrated in sham-injured mice. Typical epileptic behavior included rapid tremors and muscular spasms from anterior to posterior, lateral recumbency, and rapid tail movements, other mild behaviors were also noted such as uncoordinated movements, or a sudden stop of locomotion, grooming, or eating that correlated to the duration of EEG activity.
Using a 3mm flat tip impactor at 2.5mm depth, we found that severe-injured mice showed extensive cortical and hippocampal loss with limited posterior intact hippocampal tissue remaining (A/P -2.85) (Figure 2A,E). We also categorized the 2mm impact depth as resulting in 2 histological subtypes, a moderate/severe phenotype with cortical and partial hippocampal loss (A/P -1.95) (Figure 2B) and a moderate phenotype with cortical tissue loss with the intact hippocampus (Figure 2C). The use of a round tip impactor at 2mm depth did not result in any observable hippocampal loss and seizure occurrence was rare (data not shown). The quantified lesion volume shows moderate injury (5.4mm2 +/- 0.52) with limited PTE incidence compared to moderate/severe (6.8mm2 +/- 0.64) or severe injury (10.8mm2 +/- 0.52) (Figure 2D). We noted that increased injury severity that included hippocampal damage showed a positive correlation with PTE incidence, i.e., moderate/severe or severe injury (Figure 2D). Only 5% of CCI-injured mice developed PTE and whose pathology was consistent with moderate injury compared to 13% that displayed moderate/severe injury and 17% with severe injury (Figure 1D). Thus, CCI injury that impacts the hippocampus to some degree correlated with 86% of the PTE+ mice in this study. We conclude that hippocampal function may play a significant role in the development of PTE after focal trauma to the brain.
PTE+ mice show aberrant migration of Prox1-neuroblast in the dentate gyrus and hilus
PTE incidence correlates with hippocampal damage, a region partly responsible for ongoing adult neurogenesis in the subgranular zone (SGZ). The lengthy process of generating new excitatory neurons coincides with the onset of PTE and, therefore, is a likely candidate for aberrant changes in cellular remodeling. We quantified the number of doublecortin (DCX)-positive neuroblasts in the upper 2/3 of the dentate gyrus (DG) and those expressing Prox1, an excitatory granule cell marker [53], in the hilar region in the contralateral and ipsilateral hemispheres of PTE- and PTE+ mice (Figure 3A–D). Under physiological conditions, DCX+ neuroblasts are present in the bottom third of the DG, here using non-biased stereological analysis, we found a significant increase of aberrant DCX+ cells at four months post-CCI injury in the ipsilateral >2/3 DG (p=0.001) which is further increased in PTE+ compared to PTE- mice (p=0.009) (Figure 3H).
No significant difference, albeit trending, was found in the estimated number of DCX+ or double-labeled DCX+/Prox1+ neuroblasts in the contralateral (data not shown) or ipsilateral hilus (Figure 3I). However, we detected a significant increase in total Prox1+ granule cells in the ipsilateral hilus of injured mice compared to sham (p<0.0001). The PTE+ mice had a slightly higher presence of Prox1+ cells, although not significantly different than PTE- (p=0.23). These findings are the first to show the misguidance of Prox1+ granule cells in the hilus, which likely differentiated from immature neuroblasts at four months post-CCI injury. The occurrence of PTE coincides with disruption to the neurogenic process.
PTE+ mice display increased hilar expression of cFos alongside an excessive loss of inhibitory neurons
To evaluate whether neurogenic-mediated changes observed in PTE+ mice coincide with altered neuronal activity, we quantified c-fos expression, a trans-synaptic marker of neuronal activity whose expression is increased after seizures [54,55,56,57] in the DG and hilus. We found a trend towards increased numbers of c-fos+ cells in the contralateral DG in PTE- (p=0.36) and PTE+ (p=0.18) compared to sham that did not reach statistical significance. We further observed a statistical increase in the ipsilateral (PTE-, p=0.029; PTE+, p=0.002) c-fos levels following CCI injury compared to sham levels (Figure 4A). Interestingly, we also found a significant increase in c-fos+ cells in the ipsilateral hilus of PTE- (p=0.028) and PTE+ (p<0.0001) mice compared to sham (Figure 4B,C–E). However, the presence of c-fos+ cells was statistically more significant in PTE+ (Figure 4B,E) compared to PTE- (Figure 4B,D) (P=0.03).
To characterize neuronal changes in the hilus that may further delineate c-fos expression in PTE+ and PTE- mice, we quantified the number of NeuN-positive, a pan-neuronal marker, Prox1+/NeuN+, excitatory granule neurons [58] and Prox1-/NeuN+, hilar inhibitory neurons [59]. Injury reduced the number of NeuN+ cells in the PTE+ ipsilateral hilus (p=0.06) and was statistically reduced in the PTE+ contralateral hilus (p=0.004) compared to sham (Figure 5A,C–E). While the number of Prox1/NeuN co-labeled excitatory neurons was increased in both PTE- (p=0.01) and PTE+ (p=0.0005) ipsilateral hilus compared to sham, no difference was observed between injury groups (p=0.58). In contrast, both PTE- (p=0.0008) and PTE+ (p<0.0001) mice show reduced Prox1-/NeuN+ inhibitory neurons in the ipsilateral and contralateral (p=0.01 PTE-; p<0.0001 PTE+) hilus compared to sham. Interestingly, the loss of inhibitory neurons was more significant in PTE+ (Figure 5A,I–K) compared to PTE- mice (Figure 5A,F–H) in the ipsilateral hilus (p=0.04). There was also a trend towards reduced numbers in the contralateral PTE+ hilus (Figure 5B) (p=0.16).
Finally, the proportions of hilar NeuN+ cells that are Prox1+ granule cells (GCs) represent only < 7% of all neurons in sham conditions. However, CCI injury dramatically elevates this to 40% GCs (Figure 5M, purple), while reducing Prox1-/NeuN+ inhibitory neurons to 60% (Figure 5M, blue) of the population in the ipsilateral PTE- hilus. Most notably, PTE+ mice show the opposite trend with 70% GCs vs. 30% inhibitory neurons. The alterations in GC proportions were also observed in the contralateral hilus between PTE- and PTE+ mice, but to a lesser extent. These changes correlate with increased c-fos positive neurons in the hilus of PTE+ mice. This data suggests that a more significant loss of hilar inhibitory interneurons, complemented by the replacement of excitatory granule cells originating from the neurogenic compartment, may result from or elicit the onset of seizures after TBI.
Post-traumatic epilepsy alters hilar astrogliosis and morphometric properties
Astrocytes show structural plasticity in response to synaptic activity and behavior, which contributes to remodeling the surrounding synapses and influences seizure development [60,61,62,63]. Understanding astrocyte morphology is essential to clarify the molecular basis of PTE. We evaluated the % of hilar astrocyte coverage in the hilus of PTE+, PTE–, and sham mice by calculating the ratio between the GFAP volume and the volume per tissue section. Our results suggest that CCI induces an increase in astrocytic coverage in the ipsilateral hilus of PTE- (p=0.03) and PTE+ (P=0.002) mice compared to the corresponding contralateral hemisphere (Figure 6A). Additionally, PTE+ mice fail to increase the astrocyte coverage, displaying a lower percentage (%) of astrocytic coverage in the ipsilateral hilus compared to PTE- (p=0.04), suggesting that PTE may affect astrocytic activation/proliferation.
Analysis of astrocyte morphology using Imaris showed altered astrocytic shape in CCI-injured cells in hilus that was not significantly different between PTE+ and PTE- mice. Analysis of sphericity, prolation, and oblation was performed to compare the morphological complexity of the astrocytes. We observed a trend in reduced sphericity in hilar ipsilateral CCI-injured astrocytes. However, significance was not reached, and no differences in prolate or oblate morphologies were found (Figure 6B–D). To further characterize the changes in the morphology of astrocytes, we performed a Sholl analysis. Interestingly, we found that PTE+ hilar astrocytes showed a reduction in branch complexity at 1 and 6 µm from cell soma compared to PTE- (p<0.05) and at 4 to 15 µm compared to sham (p>0.05). Additionally, hilar astrocytes from PTE+ mice displayed reduced branching compared to sham at 2 to 12 µm from the soma (p<0.05). (Figure 6G). These findings demonstrate that as assessed by GFAP immunostaining, astrocytes alter their coverage in the hilus post CCI injury, with a reduction in branching complexity observed in PTE+ mice.
Transcriptomic signature of forebrain PTE+ astrocytes following CCI injury
Astrocyte dysfunction has been implicated in the pathophysiology of epilepsy [64,65,66,67], post-traumatic aberrant neurogenesis [68,69], and interneuron degeneration [70]. To gain insight into the molecular pathways associated with astrocyte dysfunction in PTE, we performed RNA-seq on astrocytes isolated from the ipsilateral and contralateral hippocampus of sham, PTE+ and PTE- mice at 4 months post-CCI injury. We assessed transcriptomic changes with gene ontology pathway enrichment to detect the top most significant DEGs and biological processes (BP), respectively. Overall, comparing PTE- to PTE+ we detected 273 total dysregulated protein genes in the contralateral hippocampus, 77 upregulated (top five Log2FC: Cd209d, Sema3f, Slo4a1, and Apol9b) and 196 downregulated (top five Log2FC: Cd3e, Ccnf, Exosc6, Ccdc69 and 2810408A11Rik) (Figure 7E,F, respectively). We identified 160 genes in the ipsilateral hippocampus (Figure 7B), 25 upregulated (top five Log2FC: Spdl1, Slc16a4, Slitrk4, Kctd9 and Gbp2b) and 135 downregulated (top five Log2FC: Tlr13, Trim6l, Lilrb4a, Alox5ap and Slc47a1) (Figure 7B,F and Supplemental Table S1). Cystatin 3, (Cst3) was the only gene altered in PTE+ astrocytes in both the contralateral and ipsilateral hippocampus compared to PTE- (-0.63, -0.72 Log2FC; respectively <0.05 p-value). Circo plots of the top 10 region-specific GO pathways included those associated with interferon-gamma, response to hypoxia, glucose metabolic processes, and mRNA regulation among others (Figure 7C,D). No common biological processes emerged across the contralateral and ipsilateral hippocampus. Of note, we identified several ‘pan reactive’ markers [71] of astrocytes including Lcn2, Timp1, Cxcl10, and Vim that were differentially expressed when comparing Sham to PTE- in the ipsilateral hippocampus, these differences were not observed in the contralateral hippocampus. These data identify key transcriptomic differences in forebrain astrocytes in mice that develop seizures post-CCI injury.
4. Discussion
Post-traumatic epilepsy (PTE) accounts for 20% of all symptomatic epilepsy and is represented by reoccurring generalized or focal, with secondary generalization, seizures [19,20,21]. Brain contusions and subdural hematomas are the strongest risk factors for seizures, and this increased risk persists for years [5,6,7]. The mechanism by which contusion injury to the brain leads to the subsequent onset of seizures remains under investigation. Using a CCI injury to model cortical contusions, we find that CCI injury that includes all severity types, results in an overall 37% incidence of PTE. In mice that display histopathogical evidence of hippocampal damage or displacement, we observed greater occurance of seizures. This data comports with clinical indications that describe the severity of the injury as a significant risk factor for PTE [4,72] and pre-clinical findings showing that CCI injury results in roughly 40% of male mice developing spontaneous electrographic seizures [37,73]. We further detail the histopathology of injury severity, including moderate, moderate/severe, and severe that was associated with the onset of PTE. We demonstrate that hippocampal changes, well-known as playing a pivotal role in epilepsy [74,75,76], may be central to the development of PTE. Indeed, previous studies show structural and functional network changes in the dentate gyrus coincide with PTE following CCI injury [37,77,78,79,80,81].
Our findings show selective loss of Prox1-negative hilar interneurons after CCI injury and that PTE+ mice demonstrate a more significant reduction in this cell population than PTE- mice. Prior studies also report a bi-lateral selective vulnerability of dentate hilar somatostatin-positive (SST+) interneurons following TBI alongside dentate granule cell hyperexcitability [75] and GAD-67 interneurons are shown to be reduced in the hilus with increasing CCI injury severity [82]. Here, we show that excessive hilar interneuron loss is associated with mice that develop spontaneous seizures compared to those that do not, suggesting a threshold of interneuron cell death must be reached and that targeting this cell population may aid in the prevention of PTE. This data is supported by the fact that hippocampal transplantation of GABA-ergic progenitors can restore TBI-induced synaptic inhibition and seizure onset [83]. Further studies are needed to discriminate the subtypes of interneurons, like mossy or SST+ cells, that are selectively lost in PTE, the mechanisms driving their demise, and conditions associated with TBI.
We reveal that PTE+ mice show aberrant migration of Prox1+/DCX+ immature excitatory granule neuroblasts in the upper 2/3 of the DG of PTE+ compared to PTE- mice. Importantly, CCI-injury results in a significant increase in the presence of hilar Prox1+ excitatory granule neurons, a fraction of which are Prox1+/DCX+, indicating that these cells may have matured from SGZ-derived progenitors that were aberrantly migrated [84,85,86] or were granule neurons that dispersed from the DG [87]. It is plausible to assume that this shift from a predominate Prox1-/NeuN+ interneuron populated region (92-97%) to one where Prox1+/NeuN+ granule neurons reside (72%) may disrupt the excitatory vs. inhibitory balance of the DG circuitry [88,89]. Indeed, selective ablation of SGZ neurogenesis can reduce chronic seizure frequency [89], suggesting that mis-migration of immature excitatory granule neurons may contribute to aberrant network reorganization. Importantly, this proportional alteration may represent a reliable histological biomarker that could indicate the existence of seizure susceptible mice.
Interestingly, epileptogenic vulnerable inhibitory interneuron subsets express reelin, whose loss contributes to disrupted neuroblast chain migration in the DG [90]. The interneuron loss may precede and even supports aberrant neurogenesis in PTE. Overall, these findings suggest that excessive hilar interneuron loss coincides with excitatory granule cell replacement, partly from the neighboring neurogenic SGZ, which may contribute to the genesis of post-traumatic epilepsy.
Finally, our interrogation of astroglial remodeling in the DG revealed that CCI injury results in increased ipsilateral GFAP coverage, indicative of astrogliosis. This increase was not due to the CCI-induced expansion of the GFAP-expressing stem cell population as demonstrated by the lack of overlap between GFAP and nestin (Supp. Figure S1). While no difference was observed between PTE- and PTE+ hilar area coverage, we did observe significant changes in the branching complexity in PTE+ hilar astrocytes. This data was consistent with their divergent transcriptomic signature that included changes in Cystatin 3 (Cst3), a cysteine protease inhibitor, one of the most highly expressed genes in astrocytes [91,92], across all brain regions. Glial expression of Cst3 is also associated with abnormal neuroblast migration and neurodegeneration in the epileptic dentate gyrus [69,93], suggesting alterations in astroglial-specific Cst3 may play a role in network reorganization in the DG of PTE mice. In addition, we identified that a nubmer of the genes altered in PTE+ astrocytes are known to be involved in differentiation, migration, and cell morphology (Supplemental Table S2), of which a number of then are asociated with epilepsy Eml1[94], Map2, Map1b [95], Sema3f [96] and Ptn [97]. At present, little is known about the role of astrocytes in the health and function of interneurons. Future studies evaluating astrocytic-specific mechanisms in hilar interneuron survival and immature dentate granule cell migration will expand our understanding of epileptogenesis in traumatic brain injury.
Author Contributions
Conceptualization, MT, MO, PV, HS, SR; experimentation, EKGB, AS, TV, DP, AK, FG.; EEG analysis, EKGB, AS, TV, DP, JD; Data analysis, EKGB, CK, JB, XW, FG; writing, EKGB, MT,MO, review and editing, MT, MO SR, HS, PV.
Funding
This research was funded by CURE EPILEPSY foundation-XWH-15-2-0069 (MO, SR, HS, MT, PV).
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We would like to thank the following graduate students for assistance in semi-automated EEG analysis, Beatriz Torres-Ceja, Raymundo Hernandez, and Jiangtao Li.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Golub, V.M. and D.S. Reddy, Post-Traumatic Epilepsy and Comorbidities: Advanced Models, Molecular Mechanisms, Biomarkers, and Novel Therapeutic Interventions. Pharmacol Rev, 2022. 74(2): p. 387-438. [CrossRef]
- Timofeev, I. , et al., Decompressive craniectomy—operative technique and perioperative care, in Advances and technical standards in neurosurgery. 2012, Springer. p. 115-136.
- Taylor, C.A. , et al., Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths - United States, 2007 and 2013. MMWR Surveill Summ, 2017. 66(9): p. 1-16.
- DeGrauw, X. , et al., Epidemiology of traumatic brain injury-associated epilepsy and early use of anti-epilepsy drugs: An analysis of insurance claims data, 2004-2014. Epilepsy Res, 2018. 146: p. 41-49. [CrossRef]
- Asikainen, I., M. Kaste, and S. Sarna, Early and late posttraumatic seizures in traumatic brain injury rehabilitation patients: brain injury factors causing late seizures and influence of seizures on long-term outcome. Epilepsia, 1999. 40(5): p. 584-9. [CrossRef]
- Frey, L.C. , Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia, 2003. 44(s10): p. 11-7. [CrossRef]
- Hauser, W.A., J. F. Annegers, and L.T. Kurland, Prevalence of epilepsy in Rochester, Minnesota: 1940–1980. Epilepsia, 1991. 32(4): p. 429-445. [CrossRef]
- Chen, J.W. , et al., Posttraumatic epilepsy and treatment. J Rehabil Res Dev, 2009. 46(6): p. 685-96. [CrossRef]
- Small, C. , et al., Examining the role of astrogliosis and JNK signaling in post-traumatic epilepsy. Egypt J Neurosurg, 2022. 37. [CrossRef]
- Mukherjee, S. , et al., Neuroinflammatory mechanisms of post-traumatic epilepsy. J Neuroinflammation, 2020. 17(1): p. 193. [CrossRef]
- Ping, X. and X. Jin, Transition from Initial Hypoactivity to Hyperactivity in Cortical Layer V Pyramidal Neurons after Traumatic Brain Injury In Vivo. J Neurotrauma, 2016. 33(4): p. 354-61. [CrossRef]
- Pitkänen, A. , et al., Epilepsy biomarkers–toward etiology and pathology specificity. Neurobiology of disease, 2019. 123: p. 42-58. [CrossRef]
- Greer, K. , et al., Abrogation of atypical neurogenesis and vascular-derived EphA4 prevents repeated mild TBI-induced learning and memory impairments. Scientific Reports, 2020. 10(1): p. 15374. [CrossRef]
- Hunt, R.F., J. A. Boychuk, and B.N. Smith, Neural circuit mechanisms of post-traumatic epilepsy. Front Cell Neurosci, 2013. 7: p. 89. [CrossRef]
- Dudek, F.E. and T.P. Sutula, Epileptogenesis in the dentate gyrus: a critical perspective. Prog Brain Res, 2007. 163: p. 755-73. [CrossRef]
- Larkin, M. , et al., Post-Traumatic, Drug-Resistant Epilepsy and Review of Seizure Control Outcomes from Blinded, Randomized Controlled Trials of Brain Stimulation Treatments for Drug-Resistant Epilepsy. Cureus, 2016. 8(8): p. e744. [CrossRef]
- Rao, V.R. and K.L. Parko. Clinical approach to posttraumatic epilepsy. in Seminars in neurology. 2015. Thieme Medical Publishers. [CrossRef]
- Lucke-Wold, B.P. , et al., Traumatic brain injury and epilepsy: Underlying mechanisms leading to seizure. Seizure, 2015. 33: p. 13-23. [CrossRef]
- Agrawal, A. , et al., Post-traumatic epilepsy: An overview. Clinical Neurology and Neurosurgery, 2006. 108(5): p. 433-439. [CrossRef]
- Englander, J. , et al., Analyzing risk factors for late posttraumatic seizures: a prospective, multicenter investigation. Arch Phys Med Rehabil, 2003. 84(3): p. 365-73. [CrossRef]
- Haltiner, A.M., N. R. Temkin, and S.S. Dikmen, Risk of seizure recurrence after the first late posttraumatic seizure. Arch Phys Med Rehabil, 1997. 78(8): p. 835-40. [CrossRef]
- Diaz-Arrastia, R. , et al., Neurophysiologic and neuroradiologic features of intractable epilepsy after traumatic brain injury in adults. Arch Neurol, 2000. 57(11): p. 1611-6. [CrossRef]
- Gupta, P.K. , et al., Subtypes of post-traumatic epilepsy: clinical, electrophysiological, and imaging features. J Neurotrauma, 2014. 31(16): p. 1439-43. [CrossRef]
- Temkin, N.R. , et al., A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med, 1990. 323(8): p. 497-502. [CrossRef]
- Szaflarski, J.P., Y. Nazzal, and L.E. Dreer, Post-traumatic epilepsy: current and emerging treatment options. Neuropsychiatr Dis Treat, 2014. 10: p. 1469-77. [CrossRef]
- Zaccara, G., S. Lattanzi, and F. Brigo, Which treatment strategy in patients with epilepsy with focal seizures uncontrolled by the first anti-seizure medication? Epilepsy Behav, 2021. 121(Pt A): p. 108031.
- Marion, D. , Management of traumatic brain injury: past, present, and future. Clinical neurosurgery, 1999. 45: p. 184-191.
- Brodie, M.J. , Road to refractory epilepsy: the Glasgow story. Epilepsia, 2013. 54 Suppl 2: p. 5-8. [CrossRef]
- Bratton, S.L. , et al., XIII. antiseizure prophylaxis. Journal of neurotrauma, 2007. 24(Supplement 1): p. S-83-S-86. [CrossRef]
- Kwan, P., S. C. Schachter, and M.J. Brodie, Drug-resistant epilepsy. New England Journal of Medicine, 2011. 365(10): p. 919-926. [CrossRef]
- Semah, F. , et al., Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology, 1998. 51(5): p. 1256-1262. [CrossRef]
- Irimia, A. and J.D. Van Horn, Epileptogenic focus localization in treatment-resistant post-traumatic epilepsy. Journal of clinical neuroscience, 2015. 22(4): p. 627-631. [CrossRef]
- Pitkanen, A. , et al., From traumatic brain injury to posttraumatic epilepsy: what animal models tell us about the process and treatment options. Epilepsia, 2009. 50 Suppl 2: p. 21-9. [CrossRef]
- Bolkvadze, T. and A. Pitkanen, Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. J Neurotrauma, 2012. 29(5): p. 789-812. [CrossRef]
- Butler, C.R., J. A. Boychuk, and B.N. Smith, Effects of Rapamycin Treatment on Neurogenesis and Synaptic Reorganization in the Dentate Gyrus after Controlled Cortical Impact Injury in Mice. Front Syst Neurosci, 2015. 9: p. 163. [CrossRef]
- Guo, D. , et al., Rapamycin attenuates the development of posttraumatic epilepsy in a mouse model of traumatic brain injury. PLoS One, 2013. 8(5): p. e64078. [CrossRef]
- Hunt, R.F., S. W. Scheff, and B.N. Smith, Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol, 2009. 215(2): p. 243-52. [CrossRef]
- Reddy, D.S. , et al., A Comprehensive and Advanced Mouse Model of Post-Traumatic Epilepsy with Robust Spontaneous Recurrent Seizures. Curr Protoc, 2022. 2(6): p. e447. [CrossRef]
- Kowalski, E.A. , et al., Monocyte proinflammatory phenotypic control by ephrin type A receptor 4 mediates neural tissue damage. JCI Insight, 2022. 7(15).
- Soliman, E. , et al., Conditional Deletion of EphA4 on Cx3cr1-Expressing Microglia Fails to Influence Histopathological Outcome and Blood Brain Barrier Disruption Following Brain Injury. Front Mol Neurosci, 2021. 14: p. 747770. [CrossRef]
- Kowalski, E.A. , et al., Peripheral loss of EphA4 ameliorates TBI-induced neuroinflammation and tissue damage. J Neuroinflammation, 2019. 16(1): p. 210. [CrossRef]
- Theus, M.H. , et al., Loss of NLRX1 Exacerbates Neural Tissue Damage and NF-kappaB Signaling following Brain Injury. J Immunol, 2017. 199(10): p. 3547-3558. [CrossRef]
- Brickler, T.R. , et al., Angiopoietin/Tie2 Axis Regulates the Age-at-Injury Cerebrovascular Response to Traumatic Brain Injury. J Neurosci, 2018. 38(45): p. 9618-9634. [CrossRef]
- Patel, D.C., E. G. Thompson, and H. Sontheimer, Brain-Derived Neurotrophic Factor Inhibits the Function of Cation-Chloride Cotransporter in a Mouse Model of Viral Infection-Induced Epilepsy. Front Cell Dev Biol, 2022. 10: p. 961292. [CrossRef]
- Shandra, O. , et al., Repetitive Diffuse Mild Traumatic Brain Injury Causes an Atypical Astrocyte Response and Spontaneous Recurrent Seizures. J Neurosci, 2019. 39(10): p. 1944-1963. [CrossRef]
- Okyere, B. , et al., EphA4/Tie2 crosstalk regulates leptomeningeal collateral remodeling following ischemic stroke. J Clin Invest, 2020. 130(2): p. 1024-1035. [CrossRef]
- Okyere, B. , et al., Temporal remodeling of pial collaterals and functional deficits in a murine model of ischemic stroke. J Neurosci Methods, 2018. 293: p. 86-96. [CrossRef]
- Holt, L.M. and M.L. Olsen, Novel Applications of Magnetic Cell Sorting to Analyze Cell-Type Specific Gene and Protein Expression in the Central Nervous System. PLoS One, 2016. 11(2): p. e0150290. [CrossRef]
- Holt, L.M., S. T. Stoyanof, and M.L. Olsen, Magnetic Cell Sorting for In Vivo and In Vitro Astrocyte, Neuron, and Microglia Analysis. Curr Protoc Neurosci, 2019. 88(1): p. e71. [CrossRef]
- Holt, L.M. , et al., Astrocyte morphogenesis is dependent on BDNF signaling via astrocytic TrkB.T1. Elife, 2019. 8.
- Theus, M.H. , et al., EphrinB3 blocks EphB3 dependence receptor functions to prevent cell death following traumatic brain injury. Cell Death Dis, 2014. 5: p. e1207. [CrossRef]
- Brickler, T. , et al., Nonessential Role for the NLRP1 Inflammasome Complex in a Murine Model of Traumatic Brain Injury. Mediators Inflamm, 2016. 2016: p. 6373506. [CrossRef]
- Iwano, T. , et al., Prox1 postmitotically defines dentate gyrus cells by specifying granule cell identity over CA3 pyramidal cell fate in the hippocampus. Development, 2012. 139(16): p. 3051-3062. [CrossRef]
- Barros, V.N. , et al., The pattern of c-Fos expression and its refractory period in the brain of rats and monkeys. Frontiers in Cellular Neuroscience, 2015. 9. [CrossRef]
- Szyndler, J. , et al., Mapping of c-Fos expression in the rat brain during the evolution of pentylenetetrazol-kindled seizures. Epilepsy & Behavior, 2009. 16(2): p. 216-224. [CrossRef]
- Dragunow, M. and H. Robertson, Generalized seizures induce c-fos protein (s) in mammalian neurons. Neuroscience letters, 1987. 82(2): p. 157-161. [CrossRef]
- Simler, S. , et al., C-fos expression after single and kindled audiogenic seizures in Wistar rats. Neuroscience letters, 1994. 175(1-2): p. 58-62. [CrossRef]
- Kempermann, G., H. Song, and F.H. Gage, Neurogenesis in the Adult Hippocampus. Cold Spring Harb Perspect Biol, 2015. 7(9): p. a018812. [CrossRef]
- Lake, B.B. , et al., Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science, 2016. 352(6293): p. 1586-1590. [CrossRef]
- Hayatdavoudi, P. , et al., The role of astrocytes in epileptic disorders. Physiol Rep, 2022. 10(6): p. e15239. [CrossRef]
- Aoki, Y. , et al., Altered expression of astrocyte-related receptors and channels correlates with epileptogenesis in hippocampal sclerosis. Pediatric and Developmental Pathology, 2019. 22(6): p. 532-539. [CrossRef]
- Leiter, I. , et al., Attenuation of epileptogenesis by 2-deoxy-d-glucose is accompanied by increased cerebral glucose supply, microglial activation and reduced astrocytosis. Neurobiology of disease, 2019. 130: p. 104510. [CrossRef]
- Torres-Ceja, B. and M.L. Olsen, A closer look at astrocyte morphology: Development, heterogeneity, and plasticity at astrocyte leaflets. Current Opinion in Neurobiology, 2022. 74: p. 102550. [CrossRef]
- Zhang, H. , et al., Dynamic Transitions of Epilepsy Waveforms Induced by Astrocyte Dysfunction and Electrical Stimulation. Neural Plast, 2020. 2020: p. 8867509. [CrossRef]
- Steinhauser, C., G. Seifert, and P. Bedner, Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia, 2012. 60(8): p. 1192-202. [CrossRef]
- Seifert, G., G. Carmignoto, and C. Steinhauser, Astrocyte dysfunction in epilepsy. Brain Res Rev, 2010. 63(1-2): p. 212-21. [CrossRef]
- Heinemann, U., D. Kaufer, and A. Friedman, Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia, 2012. 60(8): p. 1251-7. [CrossRef]
- Robinson, C., C. Apgar, and L.A. Shapiro, Astrocyte Hypertrophy Contributes to Aberrant Neurogenesis after Traumatic Brain Injury. Neural Plast, 2016. 2016: p. 1347987. [CrossRef]
- Pirttila, T.J. , et al., Cystatin C expression is associated with granule cell dispersion in epilepsy. Ann Neurol, 2005. 58(2): p. 211-23. [CrossRef]
- Liu, X. , et al., Manganese-induced neurotoxicity: the role of astroglial-derived nitric oxide in striatal interneuron degeneration. Toxicol Sci, 2006. 91(2): p. 521-31. [CrossRef]
- Liddelow, S.A. , et al., Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 2017. 541(7638): p. 481-487. [CrossRef]
- Kharatishvili, I. and A. Pitkanen, Association of the severity of cortical damage with the occurrence of spontaneous seizures and hyperexcitability in an animal model of posttraumatic epilepsy. Epilepsy Res, 2010. 90(1-2): p. 47-59. [CrossRef]
- Hunt, R.F., S. W. Scheff, and B.N. Smith, Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J Neurophysiol, 2010. 103(3): p. 1490-500. [CrossRef]
- Lothman, E.W., E. H. Bertram, 3rd, and J.L. Stringer, Functional anatomy of hippocampal seizures. Prog Neurobiol, 1991. 37(1): p. 1-82. [CrossRef]
- Lowenstein, D.H. , et al., Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci, 1992. 12(12): p. 4846-53. [CrossRef]
- Golarai, G. , et al., Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci, 2001. 21(21): p. 8523-37. [CrossRef]
- Shapiro, L.A., L. Wang, and C.E. Ribak, Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia, 2008. 49 Suppl 2: p. 33-41. [CrossRef]
- Golub, V.M. and D.S. Reddy, Contusion brain damage in mice for modelling of post-traumatic epilepsy with contralateral hippocampus sclerosis: Comprehensive and longitudinal characterization of spontaneous seizures, neuropathology, and neuropsychiatric comorbidities. Exp Neurol, 2022. 348: p. 113946. [CrossRef]
- Di Sapia, R. , et al., In-depth characterization of a mouse model of post-traumatic epilepsy for biomarker and drug discovery. Acta Neuropathol Commun, 2021. 9(1): p. 76. [CrossRef]
- Butler, C.R., J. A. Boychuk, and B.N. Smith, Differential effects of rapamycin treatment on tonic and phasic GABAergic inhibition in dentate granule cells after focal brain injury in mice. Exp Neurol, 2016. 280: p. 30-40. [CrossRef]
- Bugay, V. , et al., A Mouse Model of Repetitive Blast Traumatic Brain Injury Reveals Post-Trauma Seizures and Increased Neuronal Excitability. J Neurotrauma, 2020. 37(2): p. 248-261. [CrossRef]
- Frankowski, J.C., Y. J. Kim, and R.F. Hunt, Selective vulnerability of hippocampal interneurons to graded traumatic brain injury. Neurobiol Dis, 2019. 129: p. 208-216. [CrossRef]
- Zhu, B., J. Eom, and R.F. Hunt, Transplanted interneurons improve memory precision after traumatic brain injury. Nat Commun, 2019. 10(1): p. 5156. [CrossRef]
- Kron, M.M., H. Zhang, and J.M. Parent, The developmental stage of dentate granule cells dictates their contribution to seizure-induced plasticity. J Neurosci, 2010. 30(6): p. 2051-9. [CrossRef]
- Parent, J.M. , et al., Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci, 1997. 17(10): p. 3727-38. [CrossRef]
- Covolan, L. , et al., Cell damage and neurogenesis in the dentate granule cell layer of adult rats after pilocarpine- or kainate-induced status epilepticus. Hippocampus, 2000. 10(2): p. 169-80.
- Houser, C.R. , Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res, 1990. 535(2): p. 195-204. [CrossRef]
- Jinde, S., V. Zsiros, and K. Nakazawa, Hilar mossy cell circuitry controlling dentate granule cell excitability. Front Neural Circuits, 2013. 7: p. 14. [CrossRef]
- Cho, K.O. , et al., Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat Commun, 2015. 6: p. 6606. [CrossRef]
- Gong, C. , et al., Reelin regulates neuronal progenitor migration in intact and epileptic hippocampus. J Neurosci, 2007. 27(8): p. 1803-11. [CrossRef]
- Farhy-Tselnicker, I. , et al., Activity-dependent modulation of synapse-regulating genes in astrocytes. Elife, 2021. 10. [CrossRef]
- Hasegawa, A. , et al., Regulation of glial development by cystatin C. J Neurochem, 2007. 100(1): p. 12-22. [CrossRef]
- Pirttila, T.J. , et al., Cystatin C modulates neurodegeneration and neurogenesis following status epilepticus in mouse. Neurobiol Dis, 2005. 20(2): p. 241-53. [CrossRef]
- Gavrilovici, C. , et al., Postnatal Role of the Cytoskeleton in Adult Epileptogenesis. Cereb Cortex Commun, 2020. 1(1): p. tgaa024. [CrossRef]
- Perveen, N. , et al., Temporal Lobe Epilepsy: What do we understand about protein alterations? Chemical Biology & Drug Design, 2021. 98(3): p. 377-394. [CrossRef]
- do Canto, A.M. , et al., Neuroproteomics in Epilepsy: What Do We Know so Far? Frontiers in Molecular Neuroscience, 2021. 13.
- Zhang, S. , et al., Elevated expression of pleiotrophin in pilocarpine-induced seizures of immature rats and in pentylenetetrazole-induced hippocampal astrocytes in vitro. Acta Histochem, 2014. 116(2): p. 415-20. [CrossRef]
Figure 1.
Evaluation of seizure onset following CCI injury. (A) Control cortical impact (CCI) injury was induced using either 2.0mm or 2.5mm impactor depth over the right parietal cortex of male adult CD1 mice at day 0. Continuous 24/7 EEG and video recording was performed at two to four months post-injury. (B) EEG electrode implant was performed utilizing a bipolar montage composed of two recording electrodes (ipsilateral and contralateral to injury), one ground electrode (green), and two anchor screws to improve retention. (C) EEG readings display generalized seizures with typical interictal spikes, seizure activity, and post-seizure depression. (D) CCI injury was performed on 41 mice (21 at 2.5mm depth and 20 at 2.0mm depth) and 12 shams. We observed electrographic and behavioral seizures, in 15 CCI-injured and 0 in sham-injured mice. The histological assessment of tissue damage and injury depth for each animal was used to stratify the number of PTE+ mice based on their severity of injury.
Figure 1.
Evaluation of seizure onset following CCI injury. (A) Control cortical impact (CCI) injury was induced using either 2.0mm or 2.5mm impactor depth over the right parietal cortex of male adult CD1 mice at day 0. Continuous 24/7 EEG and video recording was performed at two to four months post-injury. (B) EEG electrode implant was performed utilizing a bipolar montage composed of two recording electrodes (ipsilateral and contralateral to injury), one ground electrode (green), and two anchor screws to improve retention. (C) EEG readings display generalized seizures with typical interictal spikes, seizure activity, and post-seizure depression. (D) CCI injury was performed on 41 mice (21 at 2.5mm depth and 20 at 2.0mm depth) and 12 shams. We observed electrographic and behavioral seizures, in 15 CCI-injured and 0 in sham-injured mice. The histological assessment of tissue damage and injury depth for each animal was used to stratify the number of PTE+ mice based on their severity of injury.
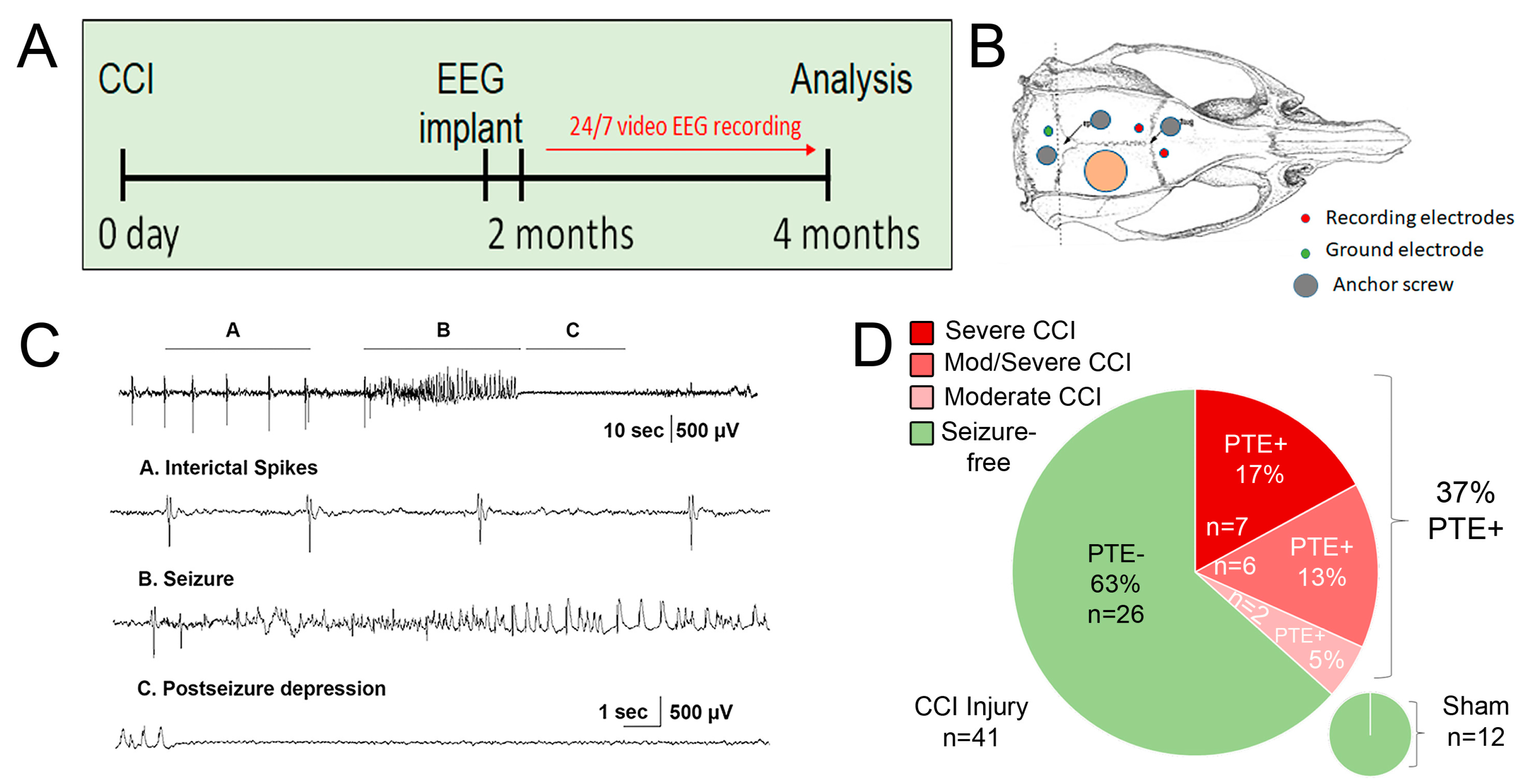
Figure 2.
Histological comparison of injury severity in CCI-injured mice. (A-C) Cresyl violet acetate staining of serial coronal section of mouse brain from anterior to posterior encompassing the brain lesion. Based on the quantification of the cavity volume, and hippocampal displacement, there were 3 categories, moderate (C), moderate/severe (B), and severe (A) lesions in the cortex with or without hippocampal pathology. (D ) Quantified data displaying lesion volume (mm3) based on the three categories of histopathology. Each dot represents a single animal, and PTE+ animals marked by a red square. (E) Gross pictures showing gross pathology of cavitation induced by unilateral CCI injury between 2.5mm depth (top panel) and 2.0mm depth (bottom panel). Total n=15 receiving 2.0mm depth and n=13 receiving 2.5mm depth were processed for Nissl staining and stratified accordingly.
Figure 2.
Histological comparison of injury severity in CCI-injured mice. (A-C) Cresyl violet acetate staining of serial coronal section of mouse brain from anterior to posterior encompassing the brain lesion. Based on the quantification of the cavity volume, and hippocampal displacement, there were 3 categories, moderate (C), moderate/severe (B), and severe (A) lesions in the cortex with or without hippocampal pathology. (D ) Quantified data displaying lesion volume (mm3) based on the three categories of histopathology. Each dot represents a single animal, and PTE+ animals marked by a red square. (E) Gross pictures showing gross pathology of cavitation induced by unilateral CCI injury between 2.5mm depth (top panel) and 2.0mm depth (bottom panel). Total n=15 receiving 2.0mm depth and n=13 receiving 2.5mm depth were processed for Nissl staining and stratified accordingly.
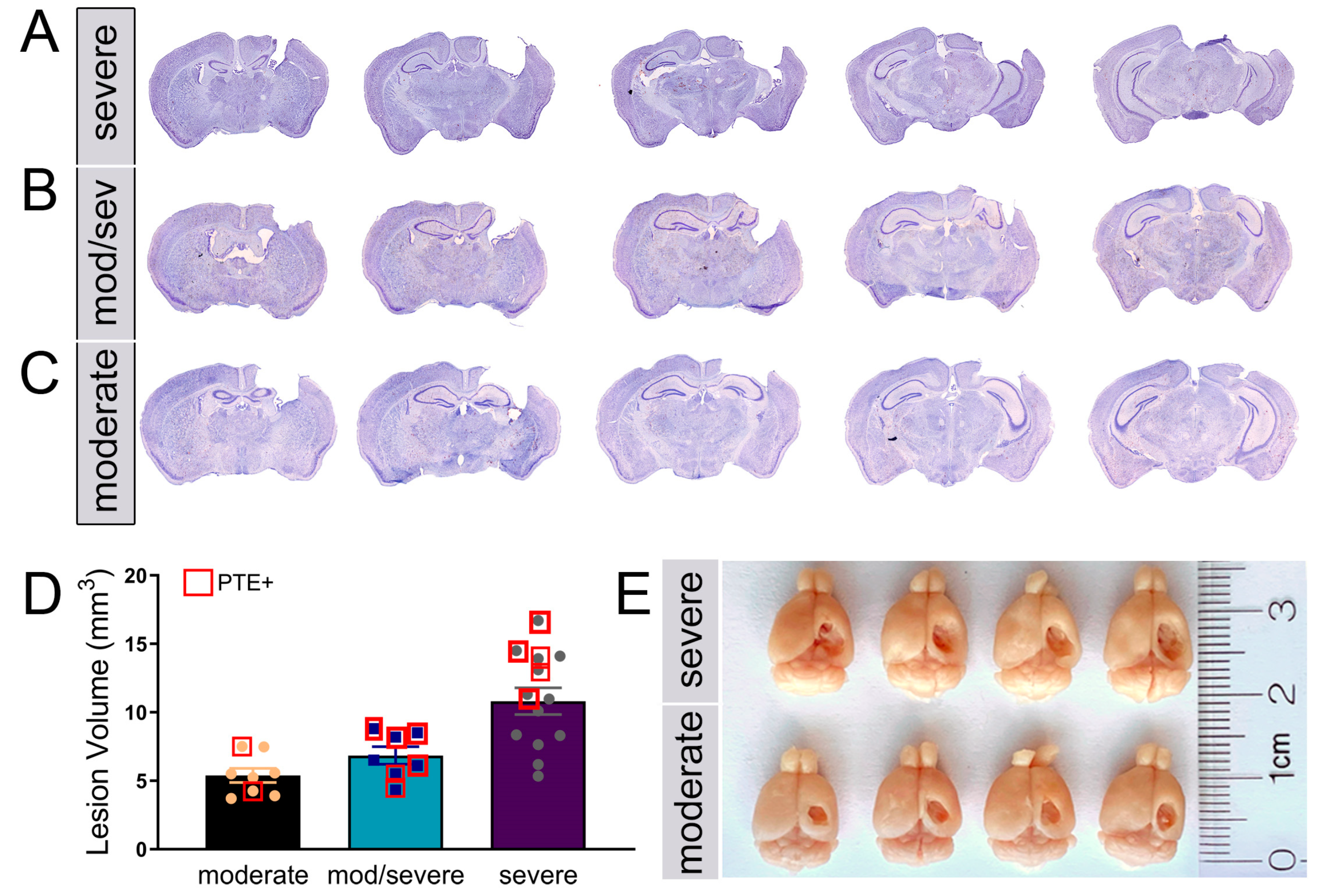
Figure 3.
PTE+ mice show increased aberrant migration of DCX+/Prox1+ cells in the dentate gyrus. (A) Representative max z-projected confocal image of the contralateral PTE- dentate gyrus (DG) showing Prox1 and DCX immunofluorescence. (B-G) High magnificantion max z-projected confocal image at four months in PTE- (B-D) and PTE+ (E-G) ispsilateral DG. This shows the presence of Prox1 and Prox1/DCX double-labeled cells in the upper 2/3 of the DG (blue arrows)and in the hilus (white arrows). (H) Quantified graph showing PTE+ mice had a greater increase in Prox1/DCX+ cells compared to PTE- (P=0.008). (I) Quantified graph showing a significant increase in Prox1+ cells in the ipsilateral hilus of CCI-injured mice compared to sham (P<0.0001) but not compared to PTE- mice(P=0.23). **P<0.01; ***P<0.001; ****P<0.00001. One-way ANOVA with Bonferroni post-hoc). n=5-9/group.
Figure 3.
PTE+ mice show increased aberrant migration of DCX+/Prox1+ cells in the dentate gyrus. (A) Representative max z-projected confocal image of the contralateral PTE- dentate gyrus (DG) showing Prox1 and DCX immunofluorescence. (B-G) High magnificantion max z-projected confocal image at four months in PTE- (B-D) and PTE+ (E-G) ispsilateral DG. This shows the presence of Prox1 and Prox1/DCX double-labeled cells in the upper 2/3 of the DG (blue arrows)and in the hilus (white arrows). (H) Quantified graph showing PTE+ mice had a greater increase in Prox1/DCX+ cells compared to PTE- (P=0.008). (I) Quantified graph showing a significant increase in Prox1+ cells in the ipsilateral hilus of CCI-injured mice compared to sham (P<0.0001) but not compared to PTE- mice(P=0.23). **P<0.01; ***P<0.001; ****P<0.00001. One-way ANOVA with Bonferroni post-hoc). n=5-9/group.
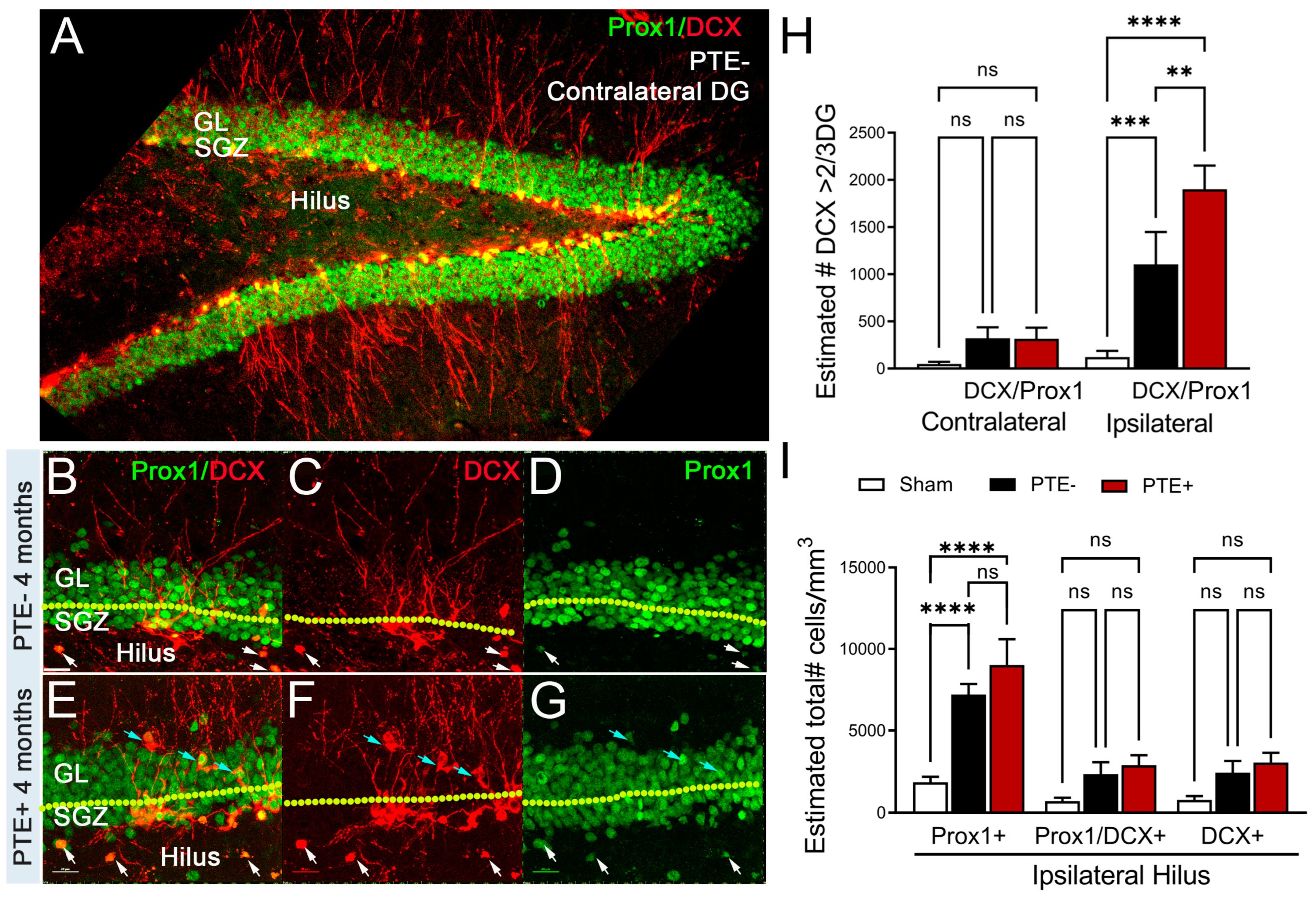
Figure 4.
PTE+ mice display altered cFos in the dentate gyrus. (A) Stereological quantification of cFos-positive, a neuronal activation marker, cells in the contralateral and ipsilateral DG. Injury induced an increase in the number of cFos expressing neurons regardless of PTE status relative to sham (PTE- p=0.02; PTE+ p=0.002). (B) Quantified graph showing no change in contralateral hilare cFos, however, PTE+ (p=0.0001) and PTE- (p=0.02) are significant compared to sham in the ipsilateral hilus. PTE+ display a greater increase in cFos compared to PTE- (p=0.03). (C-E) Representative max z-projection of the dentate gyrus from sham (C), PTE- (D), and PTE+ (E) stained with DAPI in blue and cFos in green. *P<0.05; **P<0.01; ****P<0.00001. One-way ANOVA with Bonferroni post-hoc. Scale bar=100µm. n=5-9/group.
Figure 4.
PTE+ mice display altered cFos in the dentate gyrus. (A) Stereological quantification of cFos-positive, a neuronal activation marker, cells in the contralateral and ipsilateral DG. Injury induced an increase in the number of cFos expressing neurons regardless of PTE status relative to sham (PTE- p=0.02; PTE+ p=0.002). (B) Quantified graph showing no change in contralateral hilare cFos, however, PTE+ (p=0.0001) and PTE- (p=0.02) are significant compared to sham in the ipsilateral hilus. PTE+ display a greater increase in cFos compared to PTE- (p=0.03). (C-E) Representative max z-projection of the dentate gyrus from sham (C), PTE- (D), and PTE+ (E) stained with DAPI in blue and cFos in green. *P<0.05; **P<0.01; ****P<0.00001. One-way ANOVA with Bonferroni post-hoc. Scale bar=100µm. n=5-9/group.
Figure 5.
PTE+ mice show altered neuronal composition in the hilus. (A) Quantified data showing PTE+ mice display a trend towards reduced overall NeuN-positive cells in the ipsilateral hilus, increased Prox1/NeuN double-labled granule cells (p=0.005)and reduced Prox1-negative, NeuN-postive inhibitory neurons (p=0.0001) compared to sham. A significant reduction in Prox1-/NeuN+ cells in PTE+ mice is obsereved compared to PTE- (p=0.04). (B) Quantified data showing a reduction in overall numbers of NeuN+ cells in the contralateral hilus of PTE+ mice compared to sham (p=0.004) and a reduction in Prox1-/NeuN+ inhibitory neurons in CCI-injured PTE+ (p=0.0001) and PTE- (p=0.01) mice compared to sham. (C-E) Representative max z-projected confocal image of Prox1 (green) and NeuN (red) in the PTE- contralateral, (F-H) PTE- ipsilateral , and (I-K) PTE+ ipsilateral hippocampus. (L) Proportion of Prox1+/NeuN+ granule cells/total NeuN vs. Prox1-/NeuN+ inhibitory neurons/total NeuN in the ipsilateral hilus of sham, PTE-, and PTE+ mice. (M) Similar proportions displayed for the contralateral hilus. *P<0.05; **P<0.01; ***P<0.001; ****P<0.00001. One-way ANOVA with Bonferroni post-hoc. Scale bar=100µm. n=5-9/group.
Figure 5.
PTE+ mice show altered neuronal composition in the hilus. (A) Quantified data showing PTE+ mice display a trend towards reduced overall NeuN-positive cells in the ipsilateral hilus, increased Prox1/NeuN double-labled granule cells (p=0.005)and reduced Prox1-negative, NeuN-postive inhibitory neurons (p=0.0001) compared to sham. A significant reduction in Prox1-/NeuN+ cells in PTE+ mice is obsereved compared to PTE- (p=0.04). (B) Quantified data showing a reduction in overall numbers of NeuN+ cells in the contralateral hilus of PTE+ mice compared to sham (p=0.004) and a reduction in Prox1-/NeuN+ inhibitory neurons in CCI-injured PTE+ (p=0.0001) and PTE- (p=0.01) mice compared to sham. (C-E) Representative max z-projected confocal image of Prox1 (green) and NeuN (red) in the PTE- contralateral, (F-H) PTE- ipsilateral , and (I-K) PTE+ ipsilateral hippocampus. (L) Proportion of Prox1+/NeuN+ granule cells/total NeuN vs. Prox1-/NeuN+ inhibitory neurons/total NeuN in the ipsilateral hilus of sham, PTE-, and PTE+ mice. (M) Similar proportions displayed for the contralateral hilus. *P<0.05; **P<0.01; ***P<0.001; ****P<0.00001. One-way ANOVA with Bonferroni post-hoc. Scale bar=100µm. n=5-9/group.
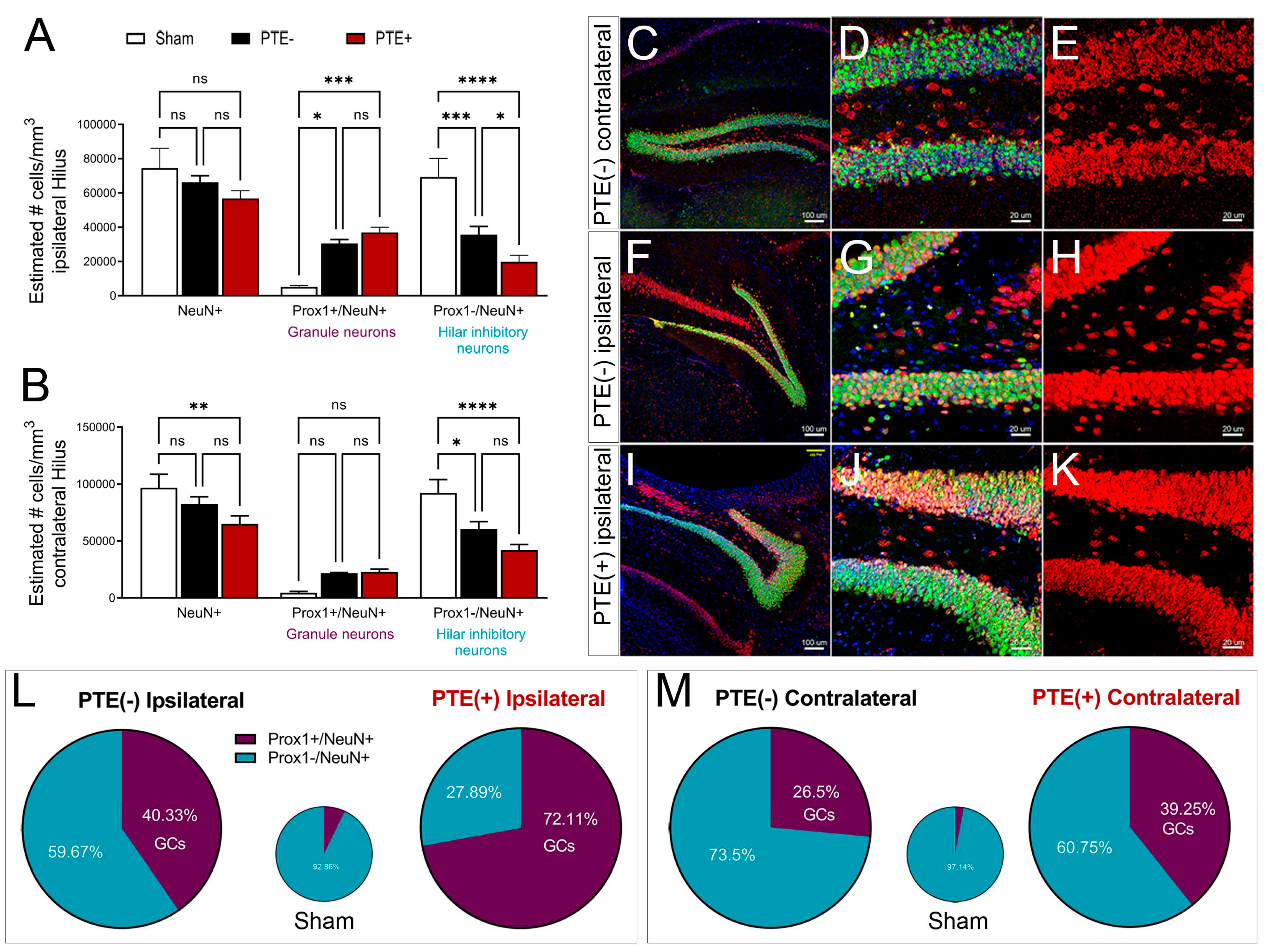
Figure 6.
Alterations in hilar astrocyte morphology shows distinct change after CCI injury and redcued branching in PTE+ mice. (A) Quantified data displaying the ratio of hilar astrocyte coverage. CCI induced an increase in astrocytic coverage in the ipsilateral hilus of PTE – ( P=0.03) and PTE+ mice (P=0.02) compared to the corresponding contralateral hemisphere. (B-D). Morphological changes using Imaris software analysis reveals no significant differences in sphericyty (B), and prolate (C). Although hilar astrocytes from ipsilateral injury on PTE+ and PTE- have a trend towards reduced oblate values ((D). (E-F) Representative 3D confocal max z-projected images from PTE- and PTE+ hippocampus, respectively. (G) Sholl analysis comparing the ipsilateral hemispheres of sham, PTE+, and PTE- animals. PTE+ astrocytes show a reduction in branching, within 1 and 6 um from cell soma compared to PTE- (p<0.05), and within 4 to15um compared to sham (p<0.05). Astrocytes from PTE+ had reduced branching compared to sham within 2 to 12 um from the soma (p<0.05). (H) 3D graphical representation of hilar astrocytes in Imaris, displaying an oblong shape and reduced cell processes in injured mice compared to sham. *P<0.05; **P<0.01; One-way ANOVA with Bonferroni post-hoc. n=4-8/group.
Figure 6.
Alterations in hilar astrocyte morphology shows distinct change after CCI injury and redcued branching in PTE+ mice. (A) Quantified data displaying the ratio of hilar astrocyte coverage. CCI induced an increase in astrocytic coverage in the ipsilateral hilus of PTE – ( P=0.03) and PTE+ mice (P=0.02) compared to the corresponding contralateral hemisphere. (B-D). Morphological changes using Imaris software analysis reveals no significant differences in sphericyty (B), and prolate (C). Although hilar astrocytes from ipsilateral injury on PTE+ and PTE- have a trend towards reduced oblate values ((D). (E-F) Representative 3D confocal max z-projected images from PTE- and PTE+ hippocampus, respectively. (G) Sholl analysis comparing the ipsilateral hemispheres of sham, PTE+, and PTE- animals. PTE+ astrocytes show a reduction in branching, within 1 and 6 um from cell soma compared to PTE- (p<0.05), and within 4 to15um compared to sham (p<0.05). Astrocytes from PTE+ had reduced branching compared to sham within 2 to 12 um from the soma (p<0.05). (H) 3D graphical representation of hilar astrocytes in Imaris, displaying an oblong shape and reduced cell processes in injured mice compared to sham. *P<0.05; **P<0.01; One-way ANOVA with Bonferroni post-hoc. n=4-8/group.
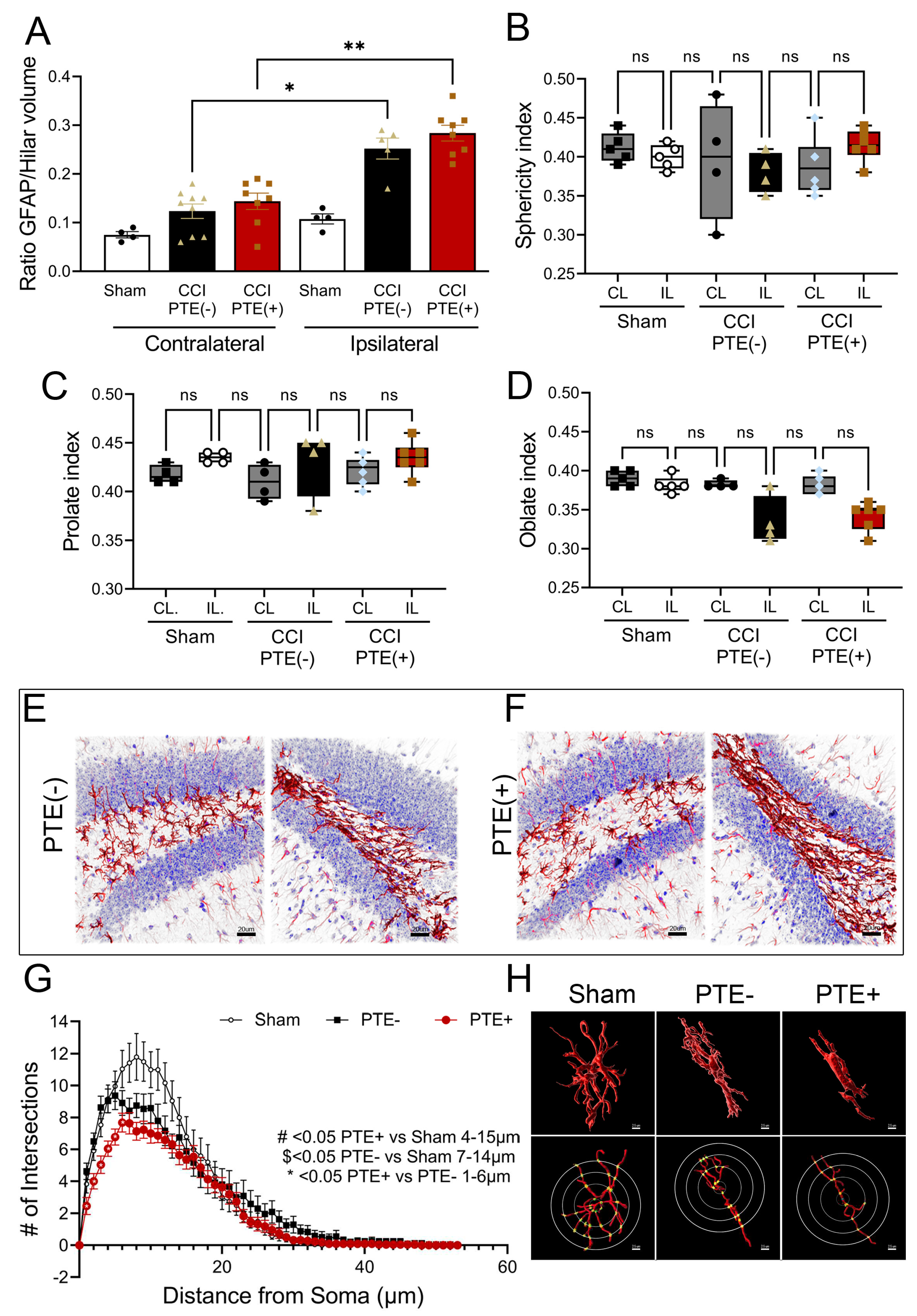
Figure 7.
Transcriptomic analysis of hippocampal astrocytes demsontrate key differences in PTE+ mice. RNA sequencing was performed on purified astrocytes from the hippocampus. (A) Venn diagram showing 156 unique DEGs between sham and PTE- mice, and 244 between PTE- and PTE+ with only 29 common between these comparisons in the contralateral hippocampus,. (B) Venn diagram showing 190 unique DEGs identified between sham and PTE- vs. 125 between PTE- and PTE+; 35 of which were common between comparisons in the ipsilateral hippocampus. (C,D) Circo plots show the top 10 gene ontology enrichment terms in PTE- vs PTE+ astrocytes from the hippocampus. These include GO terms associated with cellular response to hypoxia, cognition, and response to interferon-gamma. (E) Tope 5 up- and down-regulated genes in the contralateral and (F) ipsilateral PTE+ astrocytes compared to PTE-. n=5 sham and PTE+; n=10 PTE-.
Figure 7.
Transcriptomic analysis of hippocampal astrocytes demsontrate key differences in PTE+ mice. RNA sequencing was performed on purified astrocytes from the hippocampus. (A) Venn diagram showing 156 unique DEGs between sham and PTE- mice, and 244 between PTE- and PTE+ with only 29 common between these comparisons in the contralateral hippocampus,. (B) Venn diagram showing 190 unique DEGs identified between sham and PTE- vs. 125 between PTE- and PTE+; 35 of which were common between comparisons in the ipsilateral hippocampus. (C,D) Circo plots show the top 10 gene ontology enrichment terms in PTE- vs PTE+ astrocytes from the hippocampus. These include GO terms associated with cellular response to hypoxia, cognition, and response to interferon-gamma. (E) Tope 5 up- and down-regulated genes in the contralateral and (F) ipsilateral PTE+ astrocytes compared to PTE-. n=5 sham and PTE+; n=10 PTE-.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



