Submitted:
10 February 2023
Posted:
13 February 2023
You are already at the latest version
Abstract
In this study, we de novo assembled whole genomes of two small body-sized West Pacific sciaenids (Larimichthys polyactis and Collichthys lucidus) and compared them with published genome data of two closely-related, large body-sized species (Larimichthys crocea and Miichthys miiuy) and one distantly-related, large body-sized outgroup species (Dicentrarchus labrax). The phylogeny constructed using 7,403 single-copy orthologs shared among the five species indicated that L. crocea and L. polyactis diverged about 42 MYA. The two sibling taxa are more closely-related to C. lucidus than M. miiuy. We further identified four growth-related genes (CDHR2, PGC, PTN and PDGFA) that host five diagnostic amino acid variants on body size traits in the fishes, splitting small-body sized L. polyactis and C. lucidus from large-body sized L. crocea, M. miiuy and D. labrax. The results provide new genomic resources and guidelines to facilitate future endeavors in studying functional genomics and developing selective breeding programs for desirable growth traits in sciaenids.
Keywords:
Larimichthys polyactis
; Collichthys lucidus
; genome
; phylogeny
; ortholog
; growth-related gene
1. Introduction
Sciaenids (Family: Sciaenidae) are a group of fish species in the Order of Perciformes with 50 genera of more than 210 species worldwide, mainly inhabiting in tropical and subtropical coastal sediment and estuary areas [1]. Body size and growth characters vary noticeably in sciaenids. The redlip yellow croaker (Larimichthys polyactis, Bleeker 1877), large yellow croaker (L. crocea, Richardson 1846), spine head croaker (Collichthys lucidus, Richardson 1844) and miiuy croaker (Miichthys miiuy, Basilewsky 1855) are among the most closely related and commercially important marine sciaenid species native to the coastal areas of West Pacific [2,3,4]. Adult L. polyactis and C. lucidus are smaller body-sized, typically reaching up to 20-30cm and 8-16 cm (http://www.fishbase.org), respectively; L. crocea and M. miiuy in contrast have noticeably bigger body sizes and can grow up to 30-40 cm and 45-55 cm in adult size (http://www.fishbase.org), respectively.
Growth is a quantitative (or complex) trait that is mediated by multiple genes distributed across the genome and by environmental factors [5]. It is strongly reflected in body size characters, such as body length, height and weight. In aquaculture, developing growth-related molecular markers is of core commercial interest [6,7].
By taking advantage of latest advances in next-generation genome sequencing and bioinformatics technology, this study was aimed to de novo assemble whole genomes of L. polyactis and C. lucidus and examine genes on body size and growth traits. This research will provide new genomic resources and guidelines to develop molecular-based breeding programs in the sciaenids with desired growth characters and facilitate sustainable marine fish farming and fisheries.
2. Materials and Methods
2.1. Sample and genomic DNA preparation
One L. polyactis sample (adult; 13 cm body length) and one C. lucidus sample (adult; 10 cm body length) were collected from the East China Sea off the coast of Zhejiang, China in 2015 and 2016, respectively, following governmental fishery regulations. The whole fish samples were preserved at the point of capture in 95% ethanol and later transferred and stored at a -40 °C freezer in the laboratory.
Muscle tissue was taken from the fish samples and proceeded with genomic DNA extraction using the standard phenol–chloroform extraction protocol. Genomic DNA was dissolved in TE buffer and stored at -40 °C. The concentrations of DNA in the samples were quantified using spectrophotometry.
We sequenced L. polyactis and C. lucidus genomes using whole genome shotgun approaches. To facilitate the hierarchical assembly, we constructed two short-insert-paired-end (PE) libraries of 200 bp and 450 bp for L. polyactis, and 280 bp and 450 bp for C. lucidus and one mate-pair (MP) library of 5 kb for both species. Followed the library construction, DNA sequencing was carried out with standard procedures (Illumina Inc. California, USA) in Berry Genomics Biotechnology Co., Ltd (Beijing, China).
2.2. Genome assembly
The initial reads were evaluated with FASTQC [8]. Low-quality reads were removed using Trimmomatic 0.36 [9] and subsequently corrected by SOAPec2.01 [10]. We assembled the genomes using the Platanus Assembler v1.2.154 [11]. The short reads (200–500 bp) were used to build contigs with default parameters. Scaffolds were oriented by adding mated-pair sequencing reads (5 kb). Gaps in the initial assembly were subsequently filled with reads from the short-insert libraries using GapCloser [10]. The alignment ratio was calculated with SAMtools v1.659 [12].
2.3. Genome annotation
We used LTR_FINDER [13] and RepeatModeller v1.0.4 (www.repeatmasker.org/ RepeatModeler.html) to detect repetitive sequences, and RepeatMasker v4.0.5 [14] to align them against the de novo Repeat Sequence Library and Repbase TE Library (RepBase16.02). Tandem repeats in the genome were identified using Tandem Repeat Finder v4.04 [15]. RepeatProteinMask [14] was used to identify TE-related protein.
We used homology-based and de novo-based methods to predict protein-coding genes. (i) For the homology prediction, Astatotilapia calliptera (GCF_900246225)[16], Anabas testudineus (GCF_900324465)[17], Boleophthalmus pectinirostris (GCF_000788275)[18], Dicentrarchus labrax (GCA_000689215)[19], Danio rerio (GCF_000002035.6)[20] and Larimichthys crocea (GCF_000972845.2) [21] proteomes were used. We performed TBLASTN v2.2.19 [22] to map genes against L. polyactis and C. lucidus genomes (a cut-off e-value of 1e-5). Genewise v2.2.0 [23] was used to reconstruct gene models on genomic sections that yield hits strong enough to support the presence of a homologous gene (a cut-off e-value of 0.25). (ii) For the de novo prediction, we used SNAP v2006-07-28 [24], GenScan [25], glimmerHMM [26] and Augustus v2.5.5 [27]. The model choice was based on the information of high-quality annotation of L. crocea genes. Finally, we merged the homology and de novo-based models to form a comprehensive and non-redundant set of genes using EvidenceModeler [28].
2.4. Pseudochromosome construction and collinearity analysis
We conducted a synteny analysis to evaluate the quality of the draft whole genome sequences of L. polyactis and C. lucidus. Using the program Ragtag [29], we constructed L. polyactis and C. lucidus pseudochromosome by referring to as chromosome-level genomes of L. crocea (GCA_000972845.2) [21] and C. lucidus (GCA_004119915.2) [30]. We subsequently used “Lastal-p30-m100-e0.05” in LAST v885 [31] to aligned both pseudochromosomes against L. crocea‘s genome and used "maf-swap" to change the sequence order in MAF format to obtain the best pairs of alignment blocks. We depicted syntenic relationships using circle diagrams and other genome basic information, including gene density, repeat element density and GC content by CIRCOS v16 [32].
2.5. Gene family and phylogenetic analysis
Besides the genomes of two small body-sized species (L. polyactis and C. lucidus) obtained in this study, we conducted a comparative analysis using the published genome data of L. crocea (GCF_000972845.2) [21] and M. miiuy (JXSJ00000000.1) [33]. In terms of adult body size, e.g. maximum standard length, L. polyactis (21.8 cm) and C. lucidus (19.8 cm) are noticeably smaller than L. crocea (24.4 cm) and M. miiuy (64.3 cm) [34]. In addition, a species in Perciformes outside of Sciaenidae, D. labrax (GCA_000689215.1) [19] was used as an outgroup species that can grow up to 100 cm at maturity (http://www.fishbase.org). The protein sequences of all the five species were aligned in pairs using the program of BLASTP [35]. The cut-off e-value was set to 1e-5. OrthoMCL 2.0.9 [36] was used in both the gene family and the clustering analysis.
We conducted a phylogenetic analysis using single-copy orthologs shared among the five species. The protein sequences were aligned with MUSCLE [37] and then concatenated using in-house Perl scripts. We used RAxML v 8.2.9 [38] to construct maximum likelihood trees. Finally, four degeneration sites of codons were extracted from the aligned sequences and used for the divergence time calculation for the phylogeny based on the fossil data from http://www.timetree.org/ using baseml and mcmctree in the software package PAML v4.9e [39]
2.6. Growth-related candidate genes
To identify candidate genes contributing to body size differences, we followed the KEGG pathway and GO terms and selected growth-related genes. We then performed all-against-all alignment using BLASTP [35] using an e-value cutoff of 1e-5. We defined reciprocal best hit protein pairs among these taxa as orthologs. We characterized diagnostic amino acid variants between the small body-sized fishes (SMF) composed of L. polyactis and C. lucidus and the large body-sized fishes (LGF) including L. crocea, M. miiuy, and D. labrax. Finally, we simulated 3D structures of the proteins using Phyre2 [40] and then visualized them using UCSF Chimera [41].
3. Results & Discussion
3.1. Genome de novo assembly and assessment
L. polyactis had a sequence depth (coverage of raw sequence data obtained) of 141 x, assembled genome size of 692 Mb, GC content of 41.4%, length of contigs N50 of 7.9 kb, scaffolds N50 of 146 kb, and max sequence length of 1.5 Mb. The genome BUSCO completeness score in L. polyactis is 87.5% with 1.9% duplicated genes; C. lucidus had a sequence depth of 235 x, assembled genome size of 672 Mb, GC content of 41.2%, length of contigs N50 of 12 kb, scaffolds N50 of 664 kb, and max sequence length of 5.9 Mb. The genome BUSCO completeness score in C. lucidus is 90.9% with 2.1% duplicated genes. Table 1 provides detailed information for the genome assembly.
Overall, more than 98% Illumina reads were mapped to the assembled genomes of L. polyactis (98.49%) and C. lucidus (98.59%). The sequencing data were deposited into the NCBI in the Sequence Read Archive (SRA) under the BioProject numbers: L. polyactis, PRJNA587872 (GenBank assembly accession #: GCA_010119295.1) and C. lucidus, PRJNA580353 (GenBank assembly accession #: GCA_009852395.1). The scaffolds were clustered and ordered onto 24 pseudochromosomes of L. polyactis and C. lucidus covering approximately 87.9% and 82.4% of L. crocea‘s genome, respectively (Figure 1).
This study de novo assembled the genomes of L. polyactis and C. lucidus. The genome of L. polyactis was the first built and annotated version. Overall, our results indicated that the two genome assemblies were reasonably complete.
3.2. Repeat sequence and protein-coding gene prediction
On the assembled genomes, we identified 16.8% repetitive sequences (116.4 Mb) in L. polyactis and 18.8% (126.6 Mb) in C. lucidus. These are higher than those in M. miiuy (109.1 Mb) and lower than those in L. crocea (136.1 Mb) and D. labrax (156.5 Mb; Figure 2). These proportions of repeated sequences are within the normal range compared with that of closely related species.
By integrating gene-sets that were predicted based on and homology-based methods, we identified a total of 29,950 protein-coding genes (CDS) with a BUSCO completeness value of 80.1% in L. polyactis genome and 28,601 CDS with a BUSCO completeness value of 85.3% in C. lucidus genome (Table 1). On average, the length of CDS is 1,181.6 bp in L. polyactis and 1,370.7 bp in C. lucidus. The size of exons is 176.3 bp in L. polyactis and 170.2 bp in C. lucidus. The proportions of protein sequences in L. polyactis (75.1%) and in C. lucidus (85.3%) are similar with those in other species in Actinopterygii available in the public databases. All these data manifested that the assembled genomes and annotation results provide useful resources for the further growth trait analysis.
3.3. Phylogenetics and growth-related candidate genes
We compared the gene repertoires to examine gene families in the five species (D. labrax, L. crocea, M. miiuy, C. lucidus and L. polyactis). The clustering analysis identified a total of 19,471 gene families, of which 11,091 were shared among all the five species, while 429 reflected by the overlapping area of circles (Venn diagram in Figure 3) were limited within the sciaenids.
The phylogeny based on sequence data of 7,403 single-copy orthologs that had relatively longer CDS sequences in all the five species indicated that L. crocea and L. polyactis diverged about 42 MYA. The two sibling taxa separated from C. lucidus about 67-43 MYA are more closely related to C. lucidus than M. miiuy (Figure 2). This result is concordant with the hypotheses derived from morphology and molecular data reported previously [2,4].
L. polyactis and C. lucidus differ from L. crocea, M. miiuy and D. labrax noticeably in growth and body size characters. The characters constitute the main target of breeding and reproductive output in fishes and therefore should be related to adaptive variation in natural populations. In this study, we identified four growth-related genes (CDHR2, PGC, PTN and PDGFA) that host diagnostic amino acid variants on body size difference in the fishes, splitting SMF (L. polyactis and C. lucidus) from LGF (L. crocea, M. miiuy and D. labrax; See details in Table 2).
CDHR2 hosts two diagnostic amino acid variants, i.e. Glu1037and Asp1104 in SMF and Asp1037and Glu1104 in LGF including the outgroup (D. labrax). The gene has a cadherin binding activity and a calcium ion binding activity and is involved in several processed, including animal organ development [42]; PGC, PTN and PDGFA each host one diagnostic amino acid variant; PTN has a Met105 in SMF and Leu105 in LGF that can promote cell survival and cell proliferation through MAPK pathway activation in humans [43]; PDGFA has a variant of Arg2Ala3 in SMF and Gly2Thr3 in LGF and is involved in the embryonic cranial skeleton and neurocranium morphogenesis [44,45]; PGC has a Gln295 in SMF and Arg295 in LGF and encodes an aspartic proteinase that is a digestive enzyme produced in the stomach[46]. In addition, our genomic comparisons of SMF against LGF predicted, through 3D structure simulations (See 3D model in Figure 4), the body-size specific variant located in PGC’s functional domain may affect the function by changing the conformation of amino acid residues.
In conclusion, the results provide new genome resources and guidelines to facilitate future endeavors in studying functional genomics and developing selective breeding programs for desirable growth traits in the sciaenids.
Author Contributions
Z. L, J. Y., F. X., Y. W., and Y. C. conceived the study and established the strategy of genome sequencing assemblies and annotations; J. Y., Z. L, F. X., Y. W, and J. M. conducted the bioinformatics analyses; Z. L, J. Y., F. X., Y. C. and W. W. wrote the manuscript; W. W. and W. S. supervised aspects of the work.
Data Accessibility
The whole genome data were deposited at the NCBI in the Sequence Read Archive (SRA) under the BioProject numbers: Collichthys lucidus, PRJNA580353 (GenBank assembly accession #: GCA_009852395.1); Larimichthys polyactis, PRJNA587872 (GenBank assembly accession #: GCA_010119295.1).
Acknowledgments
The research was supported by Zhejiang Provincial Natural Science Foundation of China under Grant No. LY16D060002, the State Key Laboratory of Genetic Resources and Evolution at Kunming Institute of Zoology, Chinese Academy of Sciences under Grant No. GREKF16-03, and the Overseas Scholar Research Foundation of Zhejiang Department of Human Resources & Social Security (2014). We thank Rijin Jiang of Zhejiang Marine Fisheries Research Institute for providing the fish samples.
Conflicts of Interest
The co-authors have no conflict of interest to declare. All the authors approved the final version of the manuscript.
References
- J.S. Nelson, T.C. Grande, M.V. Wilson, Fishes of the World, John Wiley & Sons, 2016. [CrossRef]
- Y. Zhu, Y.-l. Lo, H. Wu, A study on the classification of the Sciaenoid fishes of China, with description of new genera and species, Antiquariaat Junk, 1963.
- R. Liu, J. Liu, Checklist of marine biota of China seas, Science Press, 2008.
- L. Jiang, Y. Su, C. Wu, Y. Chen, A. Zhu, J. Zhang, C. Gu, Phylogenetic estimation of Sciaenidae in the East China Sea inferred from nuclear EPIC DNA sequence variation, Biochemical Systematics and Ecology, 53 (2014) 69-75.
- M. Lynch, B. Walsh, Genetics and analysis of quantitative traits, Sinauer Sunderland, MA, 1998.
- M. Salem, R.L. Vallejo, T.D. Leeds, Y. Palti, S. Liu, A. Sabbagh, C.E. Rexroad, 3rd, J. Yao, RNA-Seq identifies SNP markers for growth traits in rainbow trout, PLoS One, 7 (2012) e36264. [CrossRef]
- W. Song, R. Pang, Y. Niu, F. Gao, Y. Zhao, J. Zhang, J. Sun, C. Shao, X. Liao, L. Wang, Y. Tian, S. Chen, Construction of high-density genetic linkage maps and mapping of growth-related quantitative trail loci in the Japanese flounder (Paralichthys olivaceus), PLoS One, 7 (2012) e50404. [CrossRef]
- R. Schmieder, R. Edwards, Quality control and preprocessing of metagenomic datasets, Bioinformatics, 27 (2011) 863-864.
- A.M. Bolger, M. Lohse, B. Usadel, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, 30 (2014) 2114-2120. [CrossRef]
- R. Luo, B. Liu, Y. Xie, Z. Li, W. Huang, J. Yuan, G. He, Y. Chen, Q. Pan, Y. Liu, J. Tang, G. Wu, H. Zhang, Y. Shi, Y. Liu, C. Yu, B. Wang, Y. Lu, C. Han, D.W. Cheung, S.M. Yiu, S. Peng, Z. Xiaoqian, G. Liu, X. Liao, Y. Li, H. Yang, J. Wang, T.W. Lam, J. Wang, SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler, Gigascience, 1 (2012) 18.
- R. Kajitani, K. Toshimoto, H. Noguchi, A. Toyoda, Y. Ogura, M. Okuno, M. Yabana, M. Harada, E. Nagayasu, H. Maruyama, Y. Kohara, A. Fujiyama, T. Hayashi, T. Itoh, Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads, Genome Res, 24 (2014) 1384-1395.
- H. Li, B. Handsaker, A. Wysoker, T. Fennell, J. Ruan, N. Homer, G. Marth, G. Abecasis, R. Durbin, S. Genome Project Data Processing, The Sequence Alignment/Map format and SAMtools, Bioinformatics, 25 (2009) 2078-2079. [CrossRef]
- Z. Xu, H. Wang, LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons, Nucleic Acids Res, 35 (2007) W265-268.
- N. Chen, Using RepeatMasker to identify repetitive elements in genomic sequences, Curr Protoc Bioinformatics, Chapter 4 (2004) Unit 4 10.
- G. Benson, Tandem repeats finder: a program to analyze DNA sequences, Nucleic Acids Res, 27 (1999) 573-580.
- E.N. Peterson, M.E. Cline, E.C. Moore, N.B. Roberts, R.B. Roberts, Genetic sex determination in Astatotilapia calliptera, a prototype species for the Lake Malawi cichlid radiation, Naturwissenschaften, 104 (2017) 41.
- P. Nath, U. Mukherjee, S. Biswas, S. Pal, S. Das, S. Ghosh, A. Samanta, S. Maitra, Expression of nitric oxide synthase (NOS) in Anabas testudineus ovary and participation of nitric oxide-cyclic GMP cascade in maintenance of meiotic arrest, Mol Cell Endocrinol, 496 (2019) 110544. [CrossRef]
- L.Y. Hong, W.S. Hong, W.B. Zhu, Q. Shi, X.X. You, S.X. Chen, Cloning and expression of melatonin receptors in the mudskipper Boleophthalmus pectinirostris: their role in synchronizing its semilunar spawning rhythm, Gen Comp Endocrinol, 195 (2014) 138-150.
- M. Tine, H. Kuhl, P.A. Gagnaire, B. Louro, E. Desmarais, R.S. Martins, J. Hecht, F. Knaust, K. Belkhir, S. Klages, R. Dieterich, K. Stueber, F. Piferrer, B. Guinand, N. Bierne, F.A. Volckaert, L. Bargelloni, D.M. Power, F. Bonhomme, A.V. Canario, R. Reinhardt, European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation, Nat Commun, 5 (2014) 5770.
- J. Pasquier, C. Cabau, T. Nguyen, E. Jouanno, D. Severac, I. Braasch, L. Journot, P. Pontarotti, C. Klopp, J.H. Postlethwait, Y. Guiguen, J. Bobe, Gene evolution and gene expression after whole genome duplication in fish: the PhyloFish database, BMC Genomics, 17 (2016) 368.
- J. Ao, Y. Mu, L.X. Xiang, D. Fan, M. Feng, S. Zhang, Q. Shi, L.Y. Zhu, T. Li, Y. Ding, L. Nie, Q. Li, W.R. Dong, L. Jiang, B. Sun, X. Zhang, M. Li, H.Q. Zhang, S. Xie, Y. Zhu, X. Jiang, X. Wang, P. Mu, W. Chen, Z. Yue, Z. Wang, J. Wang, J.Z. Shao, X. Chen, Genome sequencing of the perciform fish Larimichthys crocea provides insights into molecular and genetic mechanisms of stress adaptation, PLoS Genet, 11 (2015) e1005118. [CrossRef]
- S.F. Altschul, W. Gish, W. Miller, E.W. Myers, D.J. Lipman, Basic local alignment search tool, Journal of molecular biology, 215 (1990) 403-410.
- E. Birney, M. Clamp, R. Durbin, GeneWise and Genomewise, Genome Res, 14 (2004) 988-995.
- I,Korf, Gene finding in novel genomes, BMC bioinformatics, 5 (2004) 59.
- C. Burge, S. Karlin, Prediction of complete gene structures in human genomic DNA, Journal of molecular biology, 268 (1997) 78-94.
- W.H. Majoros, M. Pertea, S.L. Salzberg, TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders, Bioinformatics, 20 (2004) 2878-2879.
- M. Stanke, O. Keller, I. Gunduz, A. Hayes, S. Waack, B. Morgenstern, AUGUSTUS: ab initio prediction of alternative transcripts, Nucleic Acids Res, 34 (2006) W435-439.
- B.J. Haas, S.L. Salzberg, W. Zhu, M. Pertea, J.E. Allen, J. Orvis, O. White, C.R. Buell, J.R. Wortman, Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments, Genome Biol, 9 (2008) R7.
- M. Alonge, S. Soyk, S. Ramakrishnan, X. Wang, S. Goodwin, F.J. Sedlazeck, Z.B. Lippman, M.C. Schatz, RaGOO: fast and accurate reference-guided scaffolding of draft genomes, Genome Biol, 20 (2019) 224. [CrossRef]
- M. Cai, Y. Zou, S. Xiao, W. Li, Z. Han, F. Han, J. Xiao, F. Liu, Z. Wang, Chromosome assembly of Collichthys lucidus, a fish of Sciaenidae with a multiple sex chromosome system, Sci Data, 6 (2019) 132.
- S.M. Kielbasa, R. Wan, K. Sato, P. Horton, M.C. Frith, Adaptive seeds tame genomic sequence comparison, Genome Res, 21 (2011) 487-493.
- M. Krzywinski, J. Schein, I. Birol, J. Connors, R. Gascoyne, D. Horsman, S.J. Jones, M.A. Marra, Circos: an information aesthetic for comparative genomics, Genome Res, 19 (2009) 1639-1645.
- T. Xu, G. Xu, R. Che, R. Wang, Y. Wang, J. Li, S. Wang, C. Shu, Y. Sun, T. Liu, J. Liu, A. Wang, J. Han, Q. Chu, Q. Yang, The genome of the miiuy croaker reveals well-developed innate immune and sensory systems, Sci Rep, 6 (2016) 21902.
- F. Chen, H. Zhang, Z. Fang, A. Guo, R. Jiang, W. Zhu and Y. Zhou, Length-weight relationships for 15 fish species in the East China Sea mainly captured by the commercial fishery and subelemented by survey samples. J. Appl. Ichthyol. 36 (2020) 536-538.
- C. Camacho, G. Coulouris, V. Avagyan, N. Ma, J. Papadopoulos, K. Bealer, T.L. Madden, BLAST+: architecture and applications, BMC Bioinformatics, 10 (2009) 421.
- L. Li, C.J. Stoeckert, Jr., D.S. Roos, OrthoMCL: identification of ortholog groups for eukaryotic genomes, Genome Res, 13 (2003) 2178-2189.
- R.C. Edgar, MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Res, 32 (2004) 1792-1797.
- A. Stamatakis, RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics, 30 (2014) 1312-1313.
- Z. Yang, PAML 4: phylogenetic analysis by maximum likelihood, Mol Biol Evol, 24 (2007) 1586-1591.
- L.A. Kelley, S. Mezulis, C.M. Yates, M.N. Wass, M.J. Sternberg, The Phyre2 web portal for protein modeling, prediction and analysis, Nat Protoc, 10 (2015) 845-858. [CrossRef]
- E.F. Pettersen, T.D. Goddard, C.C. Huang, G.S. Couch, D.M. Greenblatt, E.C. Meng, T.E. Ferrin, UCSF Chimera--a visualization system for exploratory research and analysis, J Comput Chem, 25 (2004) 1605-1612.
- A. Orts-Del'Immagine, Y. Cantaut-Belarif, O. Thouvenin, J. Roussel, A. Baskaran, D. Langui, F. Koeth, P. Bivas, F.X. Lejeune, P.L. Bardet, C. Wyart, Sensory Neurons Contacting the Cerebrospinal Fluid Require the Reissner Fiber to Detect Spinal Curvature In Vivo, Curr Biol, 30 (2020) 827-839 e824.
- G.E. Stoica, A. Kuo, A. Aigner, I. Sunitha, B. Souttou, C. Malerczyk, D.J. Caughey, D. Wen, A. Karavanov, A.T. Riegel, Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin, Journal of Biological Chemistry, 276 (2001) 16772-16779. [CrossRef]
- N. McCarthy, L. Wetherill, C.B. Lovely, M.E. Swartz, T.M. Foroud, J.K. Eberhart, Pdgfra protects against ethanol-induced craniofacial defects in a zebrafish model of FASD, Development, 140 (2013) 3254-3265.
- N. McCarthy, J.S. Liu, A.M. Richarte, B. Eskiocak, C.B. Lovely, M.D. Tallquist, J.K. Eberhart, Pdgfra and Pdgfrb genetically interact during craniofacial development, Developmental Dynamics, 245 (2016) 641-652.
- M.I. Hassan, A. Toor, F. Ahmad, Progastriscin: structure, function, and its role in tumor progression, J Mol Cell Biol, 2 (2010) 118-127. [CrossRef]
Figure 1.
Syntenic patterns of orthologs on the pseudochromosomes of Larimichthys polyactis (left) and Collichthys lucidus (right) shared with the genome of L_crocea_2.0 (GCA_000972845.2).
Figure 1.
Syntenic patterns of orthologs on the pseudochromosomes of Larimichthys polyactis (left) and Collichthys lucidus (right) shared with the genome of L_crocea_2.0 (GCA_000972845.2).
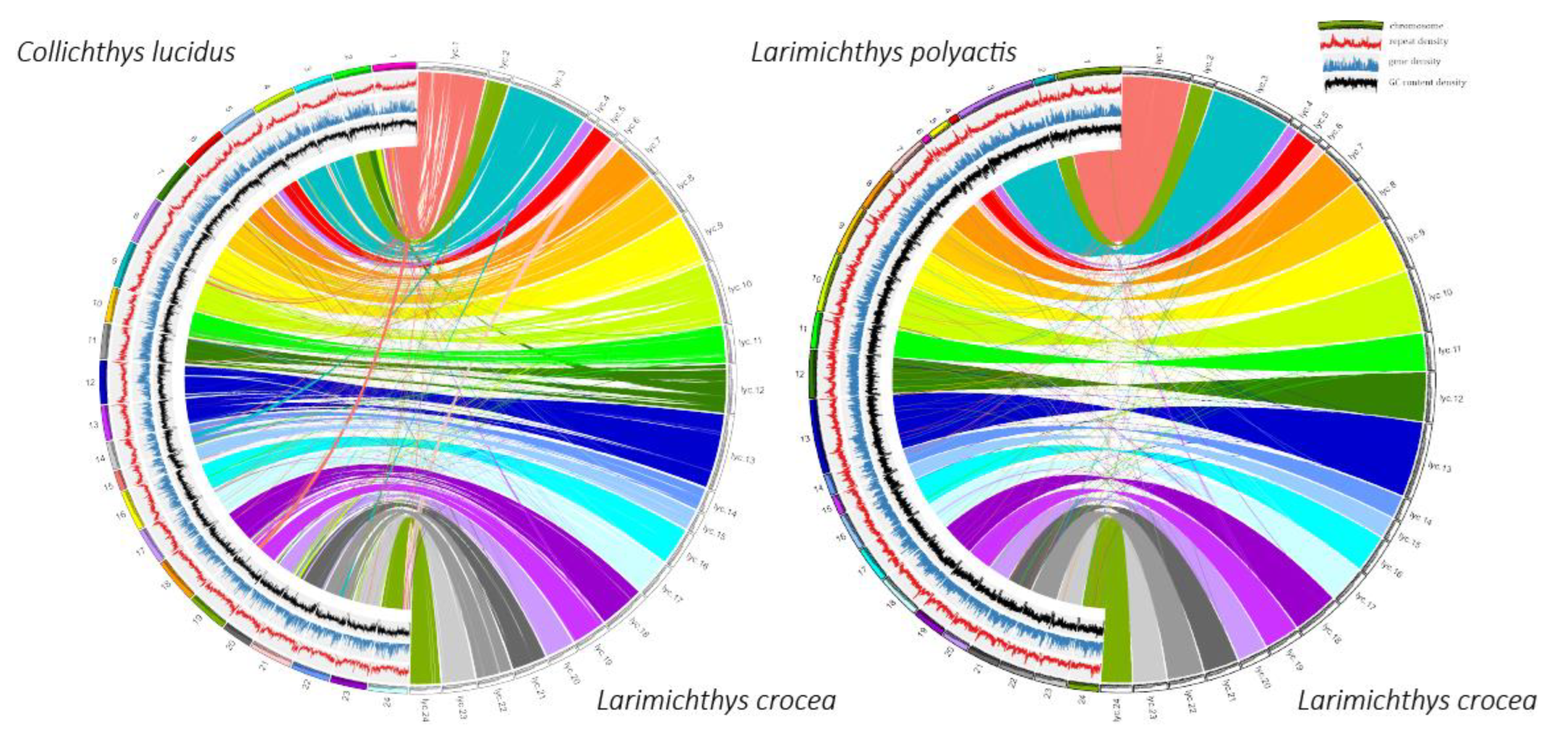
Figure 2.
The number of repetitive sequence elements (repeat sequences) in base pair (right) and maximum likelihood phylogeny based on gene sequences of 7,403 single-copy orthologs (left) of Larimichthys crocea, L. polyactis, Collichthys lucidus, Miichthys miiuy and Dicentrarchus labrax. The number of bootstrap replicates is 2,000.
Figure 2.
The number of repetitive sequence elements (repeat sequences) in base pair (right) and maximum likelihood phylogeny based on gene sequences of 7,403 single-copy orthologs (left) of Larimichthys crocea, L. polyactis, Collichthys lucidus, Miichthys miiuy and Dicentrarchus labrax. The number of bootstrap replicates is 2,000.
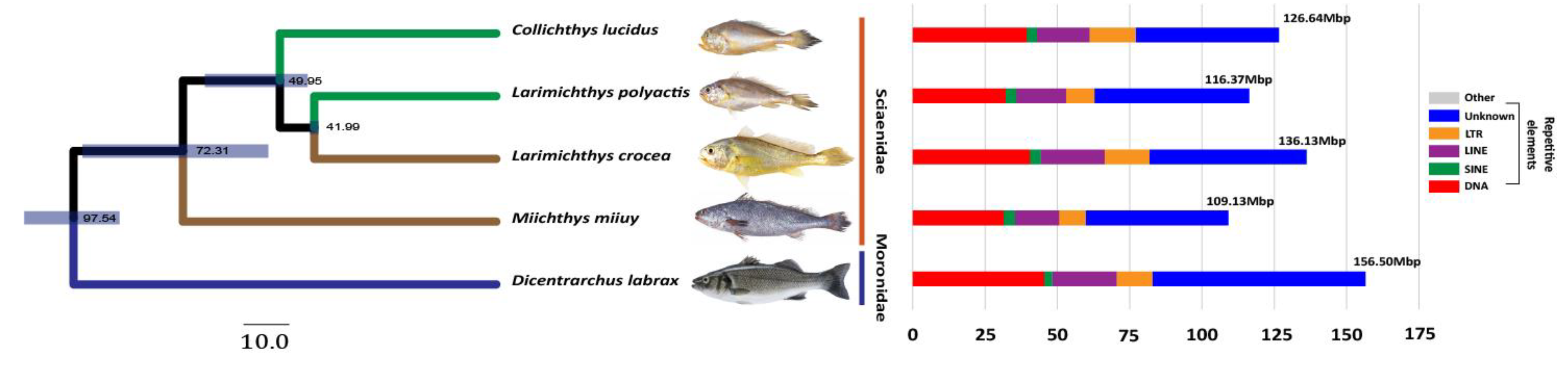
Figure 3.
Venn diagram shows the number of gene families shared among five species in Perciformes, including Larimichthys crocea, L. polyactis, Collichthys lucidus, Miichthys miiuy and Dicentrarchus labrax.
Figure 3.
Venn diagram shows the number of gene families shared among five species in Perciformes, including Larimichthys crocea, L. polyactis, Collichthys lucidus, Miichthys miiuy and Dicentrarchus labrax.
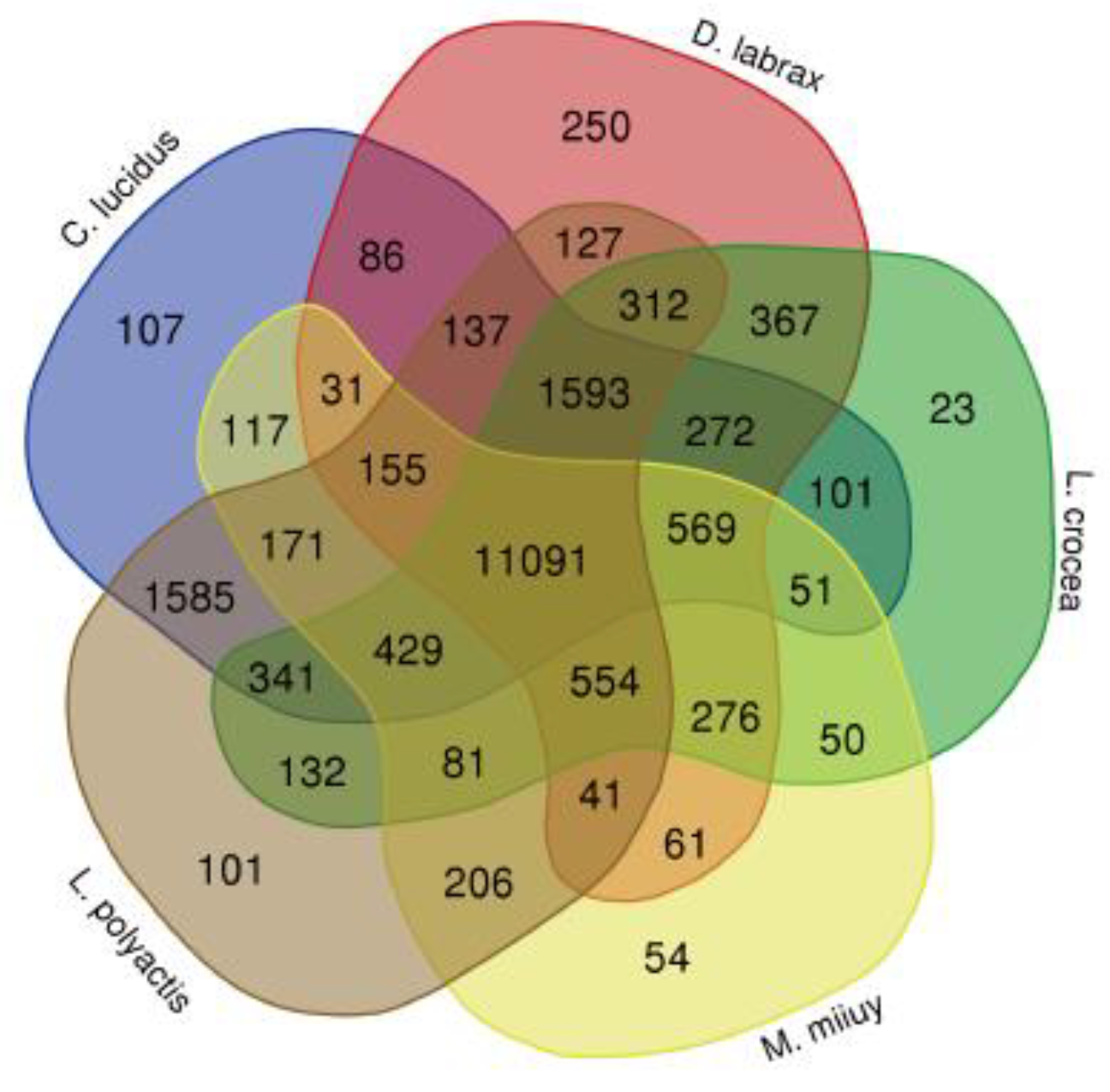
Figure 4.
The simulated 3D structural difference resulted from the diagnostic variant site on the protein of PGC between SMF (Larimichthys polyactis and Collichthys lucidus) and LGF (L. crocea, Miichthys miiuy and Dicentrarchus labrax).
Figure 4.
The simulated 3D structural difference resulted from the diagnostic variant site on the protein of PGC between SMF (Larimichthys polyactis and Collichthys lucidus) and LGF (L. crocea, Miichthys miiuy and Dicentrarchus labrax).
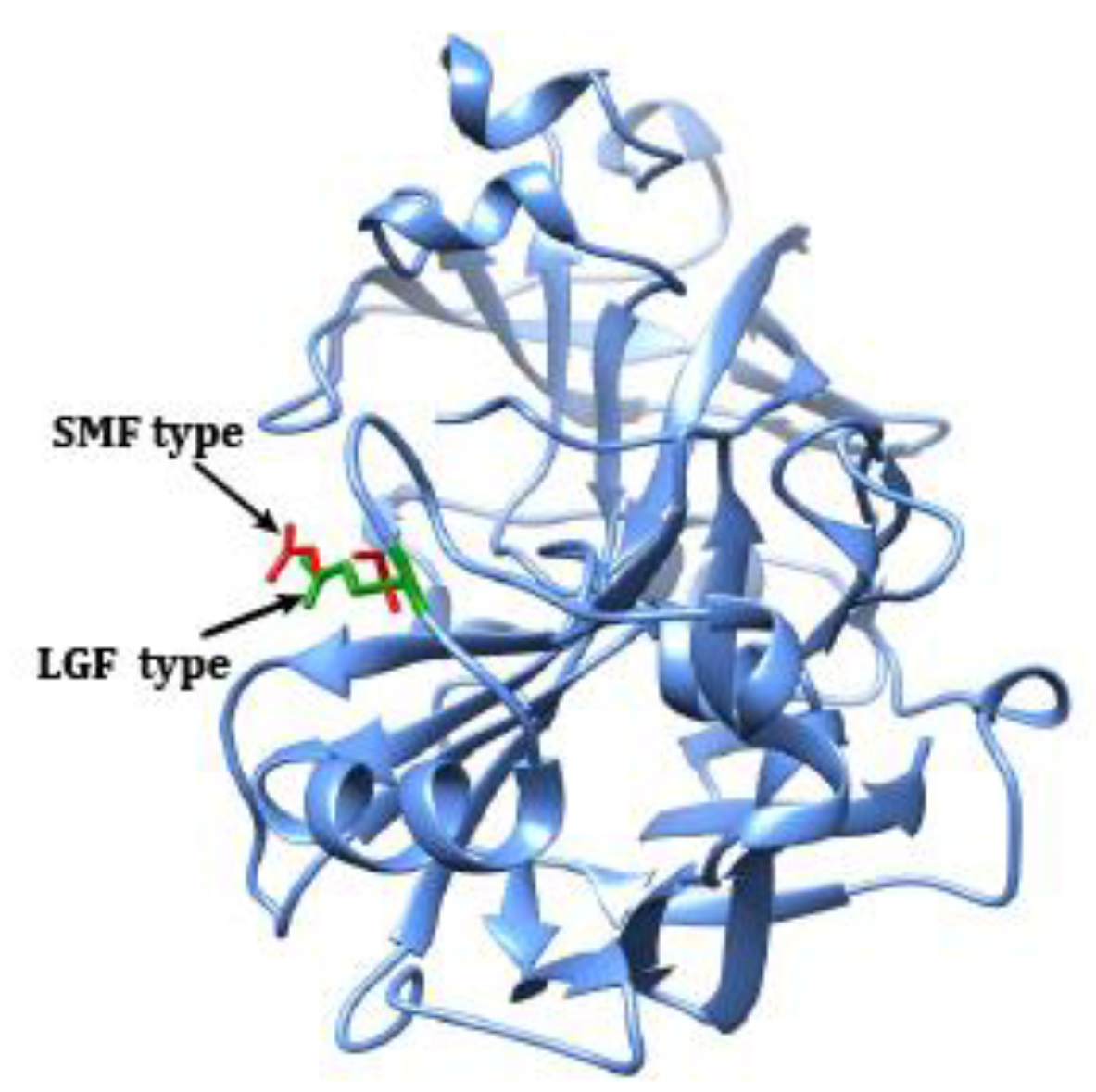

Table 1.
Statistic information of whole genome sequence data obtained in Larimichthys polyactis and Collichthys lucidus.
Table 1.
Statistic information of whole genome sequence data obtained in Larimichthys polyactis and Collichthys lucidus.
| Species | L. polyactis | C. lucidus |
| Sequence depth | 141x | 235x |
| Genome size (BUSCO) | 692 Mb (87.5%) | 672 Mb (90.9%) |
| PE1 (200 bp/280 bp) | 72.0 Gb (200 bp) | 76.8 Gb (280 bp) |
| PE2 (450 bp) | 29.8 Gb | 39.8 Gb |
| MP (5 kb) | 32.8 Gb | 39.5 Gb |
| N50/Contigs (Scaffolds) | 7.9 kb (146 kb) | 12 kb (664 kb) |
| Gene number (BUSCO) | 29,950 (80.1%) | 28,601 (85.3%) |
| Repeat sequence (Ratio) | 116.4 Mb (16.8%) | 126.6 Mb (18.8%) |
Table 2.
The biological functions of four growth-related genes (CDHR2, PGC, PTN and PDGFA) that host diagnostic amino acid variants on body size difference in the fishes, splitting SMF (Larimichthys polyactis and Collichthys lucidus) and LGF (L. crocea, Miichthys miiuy and Dicentrarchus labrax). The genes and their physical base pair positions on specific clusters are defined in the reference genome sequence of L_crocea_2.0 (GCA_000972845.2). The use of amino acids abides by the International Union of Pure and Applied Chemistry (IUPAC) codes.
Table 2.
The biological functions of four growth-related genes (CDHR2, PGC, PTN and PDGFA) that host diagnostic amino acid variants on body size difference in the fishes, splitting SMF (Larimichthys polyactis and Collichthys lucidus) and LGF (L. crocea, Miichthys miiuy and Dicentrarchus labrax). The genes and their physical base pair positions on specific clusters are defined in the reference genome sequence of L_crocea_2.0 (GCA_000972845.2). The use of amino acids abides by the International Union of Pure and Applied Chemistry (IUPAC) codes.
| Cluster | Gene | Position | Diagnostic AAs | Gene annotation | |
| LGF | SMF | ||||
| 8369 | CDHR2 | 1037 | E | D | Cadherin-related family member 2 |
| 1104 | D | E | |||
| 6535 | PGC | 295 | Q | R | Gastricsin |
| 5716 | PTN | 105 | M | L | Growth factor activity mdk-a |
| 11497 | PDGFA | 2-3 | RA | GT | Platelet-derived growth factor subunit A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



