
Submitted:
14 February 2023
Posted:
14 February 2023
You are already at the latest version
Abstract
Phthalate esters (PAEs) are a ubiquitous kind of environmental endocrine-disrupting chemicals, causing environmental and health issues. EstJ6 is an effective phthalate-degrading hydrolase, and its mutant with a combination of three non-conservative distal mutations has an improved activity against PAEs with unknown molecular mechanisms. Herein, we attempt to fill the significant gap between distal mutations and the activity of this enzyme using computational approaches. We find mutations result in a redistribution of enzyme’s preexisting conformational states and dynamics changes of key functional regions, especially the lid over the active site. The outward motion of the lid upon mutations should make it easier for substrates or products to enter or exit. Additionally, the stronger substrate binding affinity and conformational rearrangements of catalytic reaction-associated residues in mutant, accompanied by the strengthened communication within the protein, might contribute to the elevated catalytic efficiency. Finally, an attempt has been done to improve the thermostability of EstJ6 upon introducing a distal disulfide bond between residues A23 and A29, and the simulation results are as expected. Together, our work explored the allosteric effects caused by distal mutations, which could provide insights into the rational design of esterases for industrial applications in the future.
Keywords:
esterases
; phthalate esters
; rational design of proteins
; distal mutations
; molecular dynamics simulation
; toxicity
; allostery
1. Introduction
Phthalate esters (PAEs) are extensively used in the plastic manufactures to increase flexibility and durability of materials [1,2,3]. Usually, PAEs are combined with materials physically, so they are easily released into air, water, and soil during the processes of application, transportation, and stockpile [4,5], and have become a major environmental hazard. They can stably exist and accumulate in the environment because of their structural stability [6]. PAEs in the environment derive mostly from artificial synthesis, and a very small part are from plant synthesis, for example, freshwater algae and cyanobacteria are capable of producing dibutyl phthalate (DBP) [7,8]. Six kinds of PAEs (Figure 1A), including dibutyl phthalate (DBP), diethyl phthalate (DEP), diethyl hexyl phthalate (DEHP), butyl benzyl phthalate (BBP), dioctyl phthalate phthalate (DnOP), and dimethyl phthalate (DMP), are listed as priority pollutants by the US Environmental Protection Agency [9]. DEHP with large sidechains and DBP with short sidechains are two of the most widely used PAEs, and are more contaminated than other types [10]. Generally, the exposure routes of human to PAEs include intruding digestive system via the food chain, inhaling into the lungs by breathing, and direct skin contact [9,11]. Then, they will quickly participate in metabolism in the body and pose a serious threat to human health [10]. At very low concentrations, they interfere with the endocrine systems of humans and animals, even causing male reproductive abnormalities and precocious puberty in children. Each year, the global use of PAEs is about eight million tons, and this number still continues to grow [11]. Therefore, it is forwardly needed to develop an efficient, economical, and environmentally friendly approach to decontaminating PAEs-polluted environment [12].
Under no-interference condition, the degradation process of PAEs, via hydrolysis or photolysis, is time-consuming. Biodegradation is the principal approach to removing PAEs in the environment, with the advantages of high efficiency, low consumption and environmental safety. Especially, the leading role of bacterial degradation in the removal of phthalates has been widely explored [13,14]. 99% natural bacteria are unable to be cultured under experimental conditions, however, meta-genome technology resolves this obstacle, to a large extent, in excavating the esterase resource [15,16]. In terms of the general metabolic pathway of phthalates, it can be divided into two main steps, namely, the hydrolysis of the two ester bonds [17]. Some strains have the ability to hydrolyze phthalate esters to phthalic acid monoester but cannot further utilize the latter. Based on the above problems, researchers chose the sludge of lotus pond as the object to extract total DNA in order to establish a meta-genome library and unearth some new kinds of esterases [18]. Interestingly, one novel esterase named EstJ6 was discovered, which possesses of the ability to hydrolyze both ester bonds of PAEs, sequentially.
Generally, the native esterases have limited hydrolysis ability and thermostability, which can’t satisfy the industry development demands. For the purpose of acquiring the more practical esterases, directed evolution emerges as time requires. Error-prone PCR was performed to construct a library of EstJ6 mutants, following by substrate screening to obtain the optimal mutants. Finally, ET2.2 with mutations (A67V, T91M, and V249I) was obtained with strongest stability and best function [19]. Due to the absence of EstJ6 crystal structure, homology modeling assists us to construct one credible substitute (Figure 1B). Based on the structure of EstJ6, the entrance of the active site could be roughly divided into the lid and cap regions. The difference is that the former one is relatively static while the latter is very flexible. According to the modelled structure of EstJ6, the mutated residues in ET2.2 are away from the active pocket. However, it is not clear how these distal mutations increase enzyme activity.
At present, the research on biodegradation of PAEs mainly stays on the screening of degrading strains, but few efforts focus on the implied molecular mechanisms of related PAEs degrading enzymes. Classical biological methods have many limitations in the study of protein functions and the most nonnegligible point is that they cannot accurately describe the kinetics behavior of proteins. Now the development of computational biology technology provides us with a new perspective to understand protein function from both conformational and dynamics changes upon mutation [20].
In many cases, remote mutations could change the non-covalent interaction network within proteins, which results in a redistribution of the enzyme’s preexisting conformational states or changes of the conformational dynamics of key functional structural elements, such as loops and covers around active sites [21], and further affects the function of enzymes. In this study, the allosteric regulation of EstJ6 upon three combined distal mutations was uncovered by using computational methods, such as homology modeling, molecular docking and molecular dynamics simulations. DBP, the best substrate of EstJ6 and ET2.2, is applied here as a probe to analyze the relationship between structure and function of the phthalate-degrading hydrolase EstJ6. Our results not only provide insights into the rational design of PAEs-degrading hydrolases based on EstJ6 but might also contribute to the expansion the existing enzymes upon distal mutations in the future.
2. Materials and Methods
2.1. Sequence Alignment and Co-Evolution Analysis
The sequence of EstJ6 was collected from the online site of National Center for Biotechnology Information (NCBI). Based on the sequence similarity, the first nine sequences (sequence similarity to EstJ6 ≥ 57%) were collected from uniport [22] (https://www.uniprot.org/) by using the Blast module. The Clustalw server (https://www.genome.jp/tools-bin/clustalw) was then applied for further sequence alignment, and then ESPript [23] was used for figure prettification. By means of evolutionary sequence covariation, the EV-couplings website (https://evcouplings.org/) was employed to predict co-evolution pairs, conservative residues, and mutational effects of EstJ6 with default parameters. The sequence alignment result obtained from Ev-coupling website was uploaded to WebLogo3 (https://weblogo.threeplusone.com/) to generate a sequence logo figure.
2.2. Homology Modeling and Molecular Docking
The initial three-dimensional structure of EstJ6 (residues 2-297) was constructed using the automated Swiss-Model web service. The template (PDB ID: 6Y9K [24] ) with the highest sequence identity (86.53%) was used. The rationality of the structure was verified by the Ramachandran plot. In order to obtain the mutant ET2.2, we performed in silico mutations using PyMOL [25]. Molecular docking of EstJ6 and ET2.2 with DBP was then carried out by AutoDock Vina [26]. Semi-flexible docking method was used, which means the protein is rigid and the ligand is flexible [27]. The optimal binding modes were searched within a box of 20×20×20 Å3 with the triads and oxyanion-hole residues as the box center. The best binding poses for EstJ6 and ET2.2 with DBP were preserved and presented.
2.3. Conventional Molecular Dynamics Simulation
All-atom classic molecular dynamics simulations (CMD) for EstJ6, ET2.2, EstJ6-DBP, and ET2.2-DBP (hereafter referred to as WT-apo, Mut-apo, WT-com and Mut-com), were performed using the AMBER 18 suite [28] for three 500-ns replicates in explicit water. Tleap [28] was used to generate topology and coordinate files using the ff14SB force field [29]. Na+ or Cl– ions were added to maintain the neutrality of the systems. The models were solvated in a rectangular box filled with TIP3P water molecules [30]. The distances between the solute and the box borders were at least 10 Å. The files for molecular dynamics simulation had been ready. Subsequently, each system underwent energetically minimized using a multistep process. Then, these systems were gradually heated from 0 to 300 K by 10,000 steps, followed by a 200-ps equilibration was performed at 300 K and 1.0 atm for all systems. Afterward, MD production simulations were performed. The time step for all production simulations was set to 2-fs, and a nonbonded cutoff of 8 Å was used with periodic boundary conditions. The Particle Mesh Ewald (PME) method [31] was used to handle the long range electrostatic interactions. All bonds involving hydrogen atoms were constrained using the SHAKE algorithm [32]. The coordinates were saved every 4-ps.
Considering the large pocket size of EstJ6, it is difficult to capture a stabilized transition state of DBP and residues involved in the catalytic reaction at the active site. Hence, DBP was confined to certain space by conducting simulations with distance restraints. In addition, the states of triads were also considered. In detail, three distance restraints were applied with a 5.5-Å upper bound during our restrained simulations in order to simulate the catalytic state: i) DBP and S146; ii) S146 and H270; iii) H270 and E240. The specific atoms in restrictions include a certain carbonyl carbon of DBP and atoms in triads involved in the hydrogen-bond network. The harmonic biased potential with a force constant of 32 kcal/mol/Å2 was used. The WT-com and Mut-com systems were simulated for three 600-ns replicates.
2.4. Trajectory Analyses
The root-mean-square deviations (RMSDs) of all Cα atoms were calculated along the simulation time referring to the initial structure to evaluate the structural stability and the equilibrium state of MD simulations. Residual flexibilities of the proteins were detected by calculating Cα root-mean-square fluctuations (RMSFs) during. The last 100-ns simulations using the average structures as references. Statistical analyses, such as residue-residue correlation map, Rg, cluster analysis, and principal component analysis (PCA), were performed over the last 100-ns trajectories using the CPPTRAJ module [33] of the AMBER 18 software. The residues within 5 Å of DBP were considered as pocket residues. The binding free energy between protein and ligand was calculated using the MM-GBSA approach [34]. Dynamical network analysis [35] was carried out using VMD [36] with the default settings.
2.5. Disulfide-Bond Design
Disulfide by Design2 (DbD2) [37] (http://cptweb.cpt.wayne.edu/DbD2/index.php) is an online version of the original DbD website for assessing disulfide bonds. The homology-modelling structure of Estj6 was uploaded to DbD2 as the template. The criterion for the χ3 torsion angles was specified to the smallest range of -87°/+97° ± 30° and for the Cα-Cβ-Cϒ bond angles of 114.6 ± 10°. Among all candidates, we chose the more promising residue pairs for disulfide bond design based on the sequence and structure analysis.
3. Results
3.1. Sequential and Structural Analyses of EstJ6
Both sequence alignment and co-evolution analysis were performed to recognize highly conservative residues and residue-residue coupling information of EstJ6. Alignment result involving high similarity sequences shown in Figure S1 unveils some crucial conservative residues in EstJ6, such as the oxyanion hole residues 75-HGGG-80, conservative helical region 143-GXSXG-149 [38] and catalytic triads (S146, E240, and H270) [18], which play essential roles in ligand binding and catalytic process for family IV of hydrolyses. We then employed WebLogo 3 [39] to perform a sequence logo generation based on multiple sequence comparison of EstJ6 obtained from Ev-coupling to investigate the residue conservativeness and the results are shown in Figure 2 and Figure S2. Not surprisingly, residues of the mentioned two crucial motifs and catalytic triads have high frequency (Figure 2, Figure S2 and Table S1). For example, the first X in the GXSXG conserved motif customarily is composed of acid residues, such as glutamate (11.25%) and aspartate (62.27%). As for the second X, its frequency of alanine is 82.75%. The occurrence probability of residue 240 for acidic residues in the catalytic triads including glutamate and aspartate is 23.88% and 74.13%, respectively. During the hydrolysis process, the position determination of H270 is assisted by E240 via hydrogen-bond interaction [18,40]. Noteworthy, E240 and H270 belong to coupling evolutionary residues pairs according to the Ev-coupling analysis. As for the three mutational sites, all of them have low occurrence frequency during evolution process (Figure 2 and Table S1).
Further to investigate the structural characteristic of EstJ6, its three-dimensional structure was modelled using homology modeling method [41] with the structure (PDB ID: 6Y9K [24]; sequence identity to EstJ6: 86.53%) as a template (Figure 1B). Ramachandran plot (Figure S3) displays that 90.9% residues reside in the most favored regions, which indicates the conformational rationality of the modeling product. EstJ6 presents a typical α/β hydrolase folding structure, and possesses of typical cap and lid regions, which are vital for substrate selectivity and binding [42]. The binding pocket is mainly formed by hydrophobic residues (Figure S4), which is beneficial for the binding of PAEs, which are hydrophobic molecules.
Referring to the structure of EstJ6, three mutations of ET2.2 (A67V, T91M, and V249I) are all distal from the active site. Mutational analysis from an online server named Ev-coupling predicted the effect strength of these three mutations: T91M (3.942), A67V (-2.782) and V249I (-1.384) (< 0, damaging substitution; = 0, neutral substitution; > 0, beneficial substitution). Although, two of them might be negative to the function of EstJ6 in the condition of a single mutation, wet experiments suggest that their combination is beneficial to the esterase. For sake of exploring local structure influence caused by mutations, we compared the property of these mutation sites. After mutation, all of the three sites reinforce their hydrophobicity, and hence we speculate that these changes might affect overall conformational dynamics of EstJ6 and further its function. To confirm our speculation, molecular dynamics simulations were performed to gain atomic-level insights into the influence of the combination of three distal mutations on the structure and dynamics of EstJ6.
3.2. Global and Local Conformational Variations of EstJ6 Induced by Mutations and Substrate Binding
To explore the allosteric regulation mechanism of distal mutations on EstJ6, total four systems were considered for molecular dynamics (MD) simulations, including wild type and mutated EstJ6 with and without DBP (name as WT-apo, WT-com, Mut-apo, and Mut-com). Each system was conducted for three 500-ns simulation replications. MD simulation allows the protein to be completely relaxed in solvent, collecting dynamics information unattainable from static situation [43]. First, the whole MD trajectories were monitored by the root-mean-square deviations of Cα atoms (Cα RMSDs) referring to the starting structure, to evaluate whether the systems are stable and converged (Figure S5). Almost all simulations are equilibrated after 300-ns, undulating around 1.5 Å~2 Å, indicating that the structures don’t deviate much from the original structure and are stable during the simulations.
In order to further investigate the flexibility of individual residue, the analysis of root-mean-square fluctuations of Cα atoms (Cα RMSFs) for the last 100-ns simulations was performed (Figure 3A). The lid (residues 182-199) over the active site exhibits highest RMSF values in all systems. Our results suggest that ligand binding makes the lid more rigid, while mutations result in a more flexible lid region. The subsequent peak in WT-apo involving residues G211-S216 is reduced in other systems, which might be advantageous to maintain the stability of the hydrophobic pocket. In order to intuitively observe how the changes caused by mutations in RMSF are distributed spatially, we mapped the RMSF differences between the two apo systems onto the protein structure (Figure 3B). A significant increase of flexibility in lid region is easily observed. In addition, the first helix of the cap region and a loop close to the entrance of the ligand binding pocket also become slightly more flexible in Mut-apo, while regions deeper in the pocket are found with moderately enhanced rigidification.
Proteins are dynamic, and the relationships among residues play important roles in protein functions [44], as well as allosteric regulation of enzymes [45,46]. Through constructing dynamics cross-correlation matrix (DCCM) [47], the correlations of each pair of Cα atoms are intuitively vivid. The results of DCCMs for EstJ6-apo and ET2.2-apo calculated based on the last 100-ns trajectories are presented in Figure 4A-B. The correlation map of WT-apo is quite disordered, while color deepens in most areas after mutations, revealing the increased residue-residue communication within the protein upon mutations. In details, residues become more positively correlated with neighboring ones, indicating the enhanced self-correlation of some regions, especially residues 220-297 containing triad residues E240 and H270. Moreover, an obvious negative correlation between the areas containing residues 175-200 and most of the rest is observed, which might be related to the enhanced stretching movements of lid region. Additionally, the internal correlations among lid residues are enhanced positively, which might help to control the pocket in a more coordinated way.
To further delve into the patency for substrate entry into the binding pocket, we analyzed the contact information between lid region and a motif involving residues from 265 to 272. As presented in Figure 4C-D, for WT-apo system, there are very high contact intensities between H270 with residues 195-197 of the lid region. These contacts can generate a certain spatial barrier for DBP entering into pocket, whereas most of these contacts disappeared or were weakened upon mutations, replaced by several low-frequency contacts. The additional contacts, such as 191-267, 189-267, and 189-270, might be formed when the lid region stretching up away from the pocket.
3.3. Mutations Increase the Flexibility of the Lid Region Resulting in a More Open Pocket
Next, to depict the conformational discrepancies of EstJ6 upon DBP binding and mutations, principal component analysis (PCA) was performed for the last 100-ns MD trajectories of all systems considering three replicates together. The results are displayed in the free-energy surface using the first two principal components (called as PC1 and PC2). As illustrated in Figure 5A, all systems exhibit decentralized or broad conformational sampling. The structure of WT-apo mainly distributes along PC1, while the others are along PC2. To intuitively recognize the global motions captured by PC1 and PC2, the residue displacements were mapped onto protein structure (Figure 5B-C), and the conformational motions along PC1 and PC2 are also shown in Supplementary Videos S1-S2.
The most mobile region captured by both PC1 and PC2 is the lid region (Figure 5B-C), which is consistent with the RMSF analysis (Figure 3A). In detail, according to Supplementary Video S1, PC1 mainly captures the open-to-closed motion of the lid. The inward or closure motion of the lid is accompanied by negligible changes of the rest of the protein. The lid region, controlling the opening and closing of the pocket, plays an important role in substrate recognition and binding. The open state should be easier for the entrance of substrates and the release of product, while a relatively stable and closed active site maybe benefit substrate binding and further catalytic reaction. Additionally, PC2 illustrates the stretch and shrink motion of the lid region, as well as an overall conformational rearrangement of EstJ6, as displayed in the Supplementary Video S2.Finally, a more confined pocket can be noticed along PC2. As a result, WT-apo prefers a more smaller binding pocket, which might be not conducive to ligand binding or positioning.
Based on the PCA results, the global motions of EstJ6 include the opening of the entrance and the expansion of the inside space of the pocket. For apo EstJ6, mutations result in the opening of the pocket with an intermediate size space. Upon substrate binding, the lid swings between open and closed states, and the closed one prefers a large inside space of the active site, especially for the Mut-com one. Therefore, the opening state upon mutations is beneficial for the entering of ligands, meanwhile ligand binding needs a spacious interior space.
As discussed above, the high flexibility of the lid region has been unveiled. To further investigate the opening and closing of active-site entrance, the displacements of the lid were monitored according to a self-defined coordinate system (Figure 6A). The open-closed motion is mainly captured by the z-axis, while the x/y-axis captures the twist motion. During the simulations, distinct distribution of lid conformations in the four systems are observed. As indicated in Figure 6, the lid region in the Mut-apo system present more decentralized conformational sampling compared to those in other systems. The entrance in WT-apo is closed most, then mutations result in an outward movement along the z-axis, while ligand binding induces the lid having a larger motion along the positive direction of the y-axis, especially the Mut-com system. It seems that lid region of Mut-com tends to cover above the pocket. We can conclude that mutations and ligand binding do influence the lid motion to regulate the pocket opening and closing, which is consistent with PCA result.
3.4. Effects of Distal Mutations on the Active Site
As discussed above, the overall and partial structural stabilities and motions of the EstJ6 system behave differently upon ligand binding and/or mutations, especially the open-closed motion of the lid over the active site. To further explore the properties of the active pocket, the stability of hydrophobic residues within 5 Å of DBP was analyzed (Figure 7A-B and Table S2). As can be seen, mutations result in a more compact and stable substrate binding pocket for apo EstJ6. Due to the low polarity of DBP and a relatively large pocket of EstJ6, the residues of pocket are disturbed in the presence of substrate based on the results of pocket RMSDs (Figure 7A and Table S2). Moreover, ligand binding results in a more spacious pocket compared to the corresponding apo one, consistent with the PCA results suggesting by PC2.
The catalytic reaction of esterases belongs to the classic acid-base catalysis mechanism, mostly dominated by the catalytic triads. As for EstJ6, residues (S146, E240 and H270) have been confirmed as its catalytic triads [18]. In the catalytic process, an electron is transferred from E240 to S146 via H270 through the hydrogen-bond network among them. Then, H270 accepts H+ from Ser146, generating an alkyl oxide ion, whose nucleophilicity is much stronger than that of the original hydroxyl group, to attack the carbonyl carbon of DBP. Therefore, the orientation of triads is very important for the catalytic reaction. Herein, the side-chain distances for S146-H270 and E240-H270 pairs were calculated during the last 100-ns trajectories of complex systems (Figure 7C-D). Interestingly, it is obtained that the average S146-H270 distances are 5.34 ± 0.39 Å for the WT-com system, 4.96 ± 0.38 Å for Mut-com, and the average E240-H270 distances are 6.87 ± 1.05 Å for the WT-com, 6.3 ± 1.01 Å for the Mut-com, respectively. Combining the distance means and distribution plots, it delineates that compared with the wild-type system, distal mutations result in conformational shift of the triads, which become closer to each other. Although this shift is minor, it might be enough to induce significant effects on the catalytic efficiency [48].
3.5. The Discrepancy of Residue-Residue Communications among All Systems
In order to identify the conformational dynamics influence of distal mutations on EstJ6, community network analysis was performed using the Network-View [49] plugin in VMD [36]. This method estimates the motional relevance among residues and classifies them into different sub-groups or communities. The inter-community message flow is determined as the time-average communication between communities during simulations, which unveils the strength of inter-community coupling represented by the thickness of edges between the two communities.
As illustrated in Figure 8, an interesting and highly reproducible community among all systems is group 2, mainly contributed by the lid region. Comparing with the WT-apo system, it is found that the dynamic communications between group 2 with other regions are decreased upon mutations. That is to say, the communication between the lid and the rest is weakened in Mut-apo, which might render more freedom for lid motion. This is consistent with our previous observations.
Generally, the number of communities can reflect intra-/inter-molecular coupling degree during the dynamics process. the four systems vary in the number of communities. It is noted that an overall enhancement of intra-protein communication is observed for both mutant systems compared to the related WT one, indicated by both the reduced number of communities and the reinforced betweenness among them. As for the two mutant systems, their protein dynamics networks share a very similar frame, but ligand binding highly strengthens the residue-residue coupling among communities. The enhanced communications around ligand binding pocket might be favorable for substrate binding. Especially, stronger communication between substrate and the active center in mutant system should increase the collision probability of reactants, and further contributes to the improvement of enzyme activity.
Based on the community analysis, it is observed that mutation allosterically affects the inter-communication of EstJ6. Combined differences discussed above, it explains the increased stability and hydrolysis capacity of mutant in terms of dynamic coupling.
3.6. Distal Mutations Enhance the Binding of Substrate to EstJ6
After investigating the pocket stability in each system and triads peculiarity in complex systems, we find that the distal mutations stabilize the active pocket of EstJ6. In view of the ligand features and the large pocket size of EstJ6, it is not easy to capture the transition state for catalytic reaction. Herein, we attempted to apply distance restraints to limit the sampling space of substrate and triads.
First, the convergence of MD trajectories was monitored by the Cα RMSDs referring to their initial coordinates (Figure S6). Second, to explore the effects of mutations on the flexibility of protein in detail, we calculated the RMSFs based on the last 100-ns MD trajectories (Figure 9and Figure S7). It can be noticed that the lid region in mutant still exhibits higher flexibility than WT one, which is consistent with no-restraint simulations. The high flexibility of the lid improves the exchange efficiency of product and reactant. Most residues around binding pocket are rendered slightly in blue, which means the pocket a little more rigid in Mut-com, especially the cap region protecting the hydrophobic pocket. In order to harvest dominant conformations, cluster analysis was performed over the last 100-ns trajectories. Both systems were divided into two major clusters, accounting for 66% and 33%, respectively. Based on the representative structure of the most dominant cluster, the carbonyl carbon of DBP in mutant system is more vulnerable to be attacked by S146 (Figure S8).
To further explore the allosteric effects of distal mutations on the binding of DBP to the receptor, the MM-GBSA method [34] was used to measure the binding free energy for the DBP-bound systems. The average binding free energies, as well as contributions of various energy terms, are shown in Table 1. As can be seen, compared with wild-type system, the DBP binding is enhanced upon mutations. In detail, the critical and powerful contributions to the binding free energy mainly come from the van der Waals and electrostatic interactions, and the nonpolar solvation interaction also plays a slightly beneficial role. Then, the representative structures of the complex systems were used for molecular docking by the Autodock vina [26]. The mutant system presents lower docking score than the WT one, which is consistent with the MM-GBSA results (Table 1). Therefore, it is certified that distal mutations make significant allosteric consequences on the binding of DBP.
As we all know, the per-residue contribution has been widely applied to investigate the details of protein-ligand interaction at the atomic level, and the computational results demonstrate good relevance with the experimental binding free energy differences upon alanine mutation. Here, by decomposing the total binding free energy to individual residues, we try to further recognize hotspot residues (per residue contribution > 0.5 kcal/mol) responsible for ligand binding (Figure 10 and Table S3). For Mut-com, six residues (G78, G79, Y80, S146, A147, and W176) have larger contributions than those in the WT-com system. The increased contributions of these residues mainly come from hydrophobic interactions. L201 close to the lid region plays a larger role for DBP binding in WT-com system. However, because of the high flexibility of the lid, the binding with L201 is not conducive to the stability of the substrate.
3.7. Rational Designed Disulfide Bonds Increase the Thermostability of EstJ6
Based on the above analysis, we have learnt how the distal mutations upgrade the enzymatic properties of EstJ6, while thermostability is another essential factor for the application of industrial enzymes. Disulfide bonds is vital for the folding, stability and function of many proteins, and the introduction of disulfide bonds is a common strategy in rational design of proteins or enzymes to improve the thermostability [50]. Hence, here we intend to adopt disulfide bond engineering to elevate EstJ6 thermostability.
Potential residue pairs that may form disulfide bonds were predicted using a web-accessible method DbD2 [37] (http://cptweb.cpt.wayne.edu/DbD2/index.php). There are 35 residue pairs satisfying the default criteria of DbD2 (Table S4). Predicted substitutions may be located in essential functional areas that possibly harm catalytic processes. Hence, residues within 10 Å of the catalytic triad and 5 Å of the other important regions (such as the lid region and binding pocket) were excluded. After filtering, 16 potential residue pairs were prepared for reasonability assessment. The P217 and A224 pair is located at the downstream of lid region, and hence might influence lid motion. The alteration of residues A250 and L262 may be not conducive to the correct positioning of H270. High conservative residues including A53, A58, P167 and P231 are abandoned to minimize detrimental effects on enzyme activity.
Among the remaining 11 potential residue pairs, based on sequence and structure information, the A23C-A29C disulfide bond was chosen as promising candidate for site-directed mutagenesis to improve the thermostability of EstJ6. Notably, this mutated pair is located at the cap region which is a cover over the binding pocket (Figure 11A). Cap region accounts for pocket hydrophobicity and substrate selectivity [51]. Sequence alignment result suggests that the sites said above often are hydrophilic residues. These residue substitutions may cause negligible impact on substrate identification.
Finally, a A23C/A29C mutant was modeled with the introduction of a disulfide bond between them, and molecular dynamics simulations were applied to investigate protein stability. First, we analyzed the Rg of the overall structure. As Figure 11B shows, more compact structure emerges after the introduction of disulfide bond. Then, in order to further investigate the stability of cap region, the RMSDs of this region were calculated. As depicted in Figure 11C, the new disulfide bond improves the stability of cap region. These performances are similar as ET2.2 in stability aspect. In the future, these mutations need to be further validated by biochemical and biophysical experimental characterization.
4. Discussion
Mutations, including distal ones, are sophisticated for the conformational and functional regulation of proteins. Understanding the implied molecular mechanism will be helpful for the further improvement of enzyme resources. In this work, we attempt to clarify how distal mutations improve the activity of an esterase via computational approaches. Based on the above observations, two following aspects deserve to be discussed further.
4.1. The Conformational Changes of the Lid Closely Related to the Activity of Esterases
The lid region, shared by most lipases and some esterases, controls the opening and closing of the substrate binding pocket. The outward motion of the lid makes the pocket accessible, allowing substrates to enter the active site and be hydrolyzed [52]. Researchers found that the movement tendency of lid region would influence substrate binding channel in lipase [53]. Hence, many former researches focused on modifying the lid to improve the properties of lipase. For example, Karkhane [54] and Panizza [55] applied mutations on the lid region of lipase and harvested mutants with enhanced activity. Li et al. developed an activity-enhanced esterase mutant with an instable lid-like structure [56].
In this study, we also observed that the lid region in the EstJ6 mutant showed higher dynamics and a stronger tendency to move away from the active site, further proving that the lid region is a key factor affecting esterase activity. In the future, the rational design of esterases is suggested to focus on lid reconstruction for activity improvement and substrate specificity.
4.2. The Potential and Challenge of Computation Biology in Rational Design of Proteins
Natural enzymes often need to be improved or optimized for large-scale industrial applications. In order to overcome the problems of existing enzymes with poor features and difficulties in mining new ones, researchers generally modify and optimize the existing ones via protein evolution. Right now, rational design employing computational approaches provides an incisive modification scheme with time- and labor-saving screening features. With the development of protein engineering methodology and in silico tools, the discovery of new enzymes has resulted in increased industrial interests.
In the past, active site was the major focus for enzyme design. The application of local modification strategies, focusing on the chemical steps through transition state stabilization in active site of existing enzymes, plays a pleasing role in the improvement of stability, activity and substrate selectivity [57,58]. In addition, the platform Rosetta [59], invented by David baker group, started a new road for protein rational design. Based on this platform, many de novo designed enzymes came into existence [60,61].
Mutations, located all around the enzyme structure, are also very common in many laboratory-evolved enzymes. Increasing evidence indicates that distal mutations are also significant for protein function, such as increased activity and drug resistance [62,63,64]. As indicated by many recent studies, conformational dynamics of enzymes is crucial for substrate binding, product release, as well as allosteric regulation. Hence, unlike active-site mutations directly influencing ligand binding or activity, distal mutations usually produce marked effects on the redistribution of protein conformational states or inner-protein residue interaction network, and then alter enzyme function. Dramatic changes on protein conformational stabilities after mutation, including distal ones, favor conformational states vital for the preferring functionality.
Of note is that mutations located all around the enzyme structure contrasts with most of the computational design strategies that reduce the problem into active-site alterations [21]. Hence, it is promising to comprehend of the effect caused by distal mutations on protein engineering, as reported by Shaikh et al [65]. Now considering the rapid growth of computer performance and the gradual recognition of molecular mechanism involved in enzyme design, it is likely that rational design, including distal-site modifications, will eventually make a very significant contribution to protein engineering.
5. Conclusions
In the present study, we investigated the possible allosteric regulation mechanism of distal mutations on EstJ6. Our results demonstrate that distal mutations play a critical role in the dynamical behavior of EstJ6, including the opening motion of the lid region, as well as the strengthened residue coupling around the pocket. The opening of the pocket entrance should benefit the entering of substrate and the exit of product. Additionally, the stronger ligand binding, accompanied by the enhancement of local hydrogen-bond network involved in catalytic reaction upon mutations, might greatly increase the occurrence of catalytic reaction. Hence, our present study filled the gap between distal mutations and the improved enzyme activity. As rational design has become the most rapid evolution method for the development of industrial enzymes, next we in silico designed a new EstJ6 mutant with the introduction of a disulfide bond based on sequence and structure information to improve protein stability. Fortunately, further simulations confirm our prediction. These findings are of instructive significance for the exploration of new highly efficient industry esterases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1-2 and Table S1: Sequence conservative analysis for EstJ6; Figure S3: The Ramachandran map of EstJ6 structure predicted by Swiss-model server; Figure S4: The hydropathicity scores of EstJ6 residues; Figure S5: Root-mean-square deviations (RMSDs) of protein Cα atoms in the four systems; Figure S6: Cα Root-mean-square deviations (RMSDs) in simulations with distance restraints; Figure S7: Changes in RMSF upon mutations for complex systems during distance restricted simulations; Figure S8: The first representative binding state of complex systems; Table S2: The means and deviations of RMSD and Rg values of pocket residues; Table S3: The binding free energy contribution of essential residues in the pocket; Table S4: Predictions of potential disulfide bonds in EstJ6; Supplementary Videos S1.mp4: Detailed motions along PC1; Supplementary Videos S2.mp4: Detailed motions along PC2.
Author Contributions
J.G. and L.X. conceived and designed the study. R.X. simulated the systems. J.G., L.X., R.X., Y.Z., M.L. and Y.B. analyzed the data and wrote the manuscript.
Funding
This work was financially supported by Macao Polytechnic University (RP/CAI-01/2022), National Natural Science Foundation of China (82160676), and the Science and Technology Planning Project of Guizhou Province [Qian Ke He Foundation ZK (2021) General 539].
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- A, X.L.; A, Z.S.; A, G.C.; A, W.Z.; A, Y.C.; A, R.K.; B, X.W.; B, Y.S.; C, J.Y.A.B. Determination of phthalate esters in environmental water by magnetic Zeolitic Imidazolate Framework-8 solid-phase extraction coupled with high-performance liquid chromatography. Journal of Chromatography A 2015, 1409, 46–52. [Google Scholar] [CrossRef]
- Zeng; Ping; Song; Yonghui; Tan; Ruijie; Liu; Ruixia; Bin; Gao. Spatial distribution and ecological risk assessment of phthalic acid esters and phenols in surface sediment from urban rivers in Northeast China. Environmental Pollution 2016. [Google Scholar] [CrossRef]
- Fischer, A.; Theuerkorn, K.; Stelzer, N.; Gehre, M.; Thullner, M.; Richnow, H.H. Applicability of stable isotope fractionation analysis for the characterization of benzene biodegradation in a BTEX-contaminated aquifer. Environ Sci Technol 2007, 41, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Sayyad, G.; Price, G.W.; Sharifi, M.; Khosravi, K. Fate and transport modeling of phthalate esters from biosolid amended soil under corn cultivation. Journal of hazardous materials 2017, 323, 264–273. [Google Scholar] [CrossRef]
- Net, S.; Sempéré, R.; Delmont, A.; Paluselli, A.; Ouddane, B. Occurrence, fate, behavior and ecotoxicological state of phthalates in different environmental matrices. Environmental science & technology 2015, 49, 4019–4035. [Google Scholar] [CrossRef]
- Xu, G.; Li, F.; Wang, Q. Occurrence and degradation characteristics of dibutyl phthalate (DBP) and di-(2-ethylhexyl) phthalate (DEHP) in typical agricultural soils of China. Sci Total Environ 2008, 393, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Babu, B.; Wu, J.T. Production of phthalate esters by nuisance freshwater algae and cyanobacteria. Sci Total Environ 2010, 408, 4969–4975. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y. Biosynthesis of di-(2-ethylhexyl) phthalate (DEHP) and di-n-butyl phthalate (DBP) from red alga--Bangia atropurpurea. Water research 2004, 38, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Meeker, J.D.; Sathyanarayana, S.; Swan, S.H. Phthalates and other additives in plastics: human exposure and associated health outcomes. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 2009, 364, 2097–2113. [Google Scholar] [CrossRef]
- Hauser, R.; Calafat, A.M. Phthalates and human health. Occup Environ Med 2005, 62, 806–818. [Google Scholar] [CrossRef]
- Julinová, M.; Slavík, R. Removal of phthalates from aqueous solution by different adsorbents: a short review. J Environ Manage 2012, 94, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Hu, R.W.; Zhao, H.M.; Huang, H.B.; Xiang, L.; Liu, B.L.; Feng, N.X.; Li, H.; Li, Y.W.; Cai, Q.Y. , et al. Mechanistic insight into esterase-catalyzed hydrolysis of phthalate esters (PAEs) based on integrated multi-spectroscopic analyses and docking simulation. J Hazard Mater 2021, 408, 124901. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.K.; Kundapur, R.; Shouche, Y.S.; Karegoudar, T.B. Degradation of plasticizer di-n-butylphthalate by Delftia sp. TBKNP-05. Curr Microbiol 2006, 52, 369–374. [Google Scholar] [CrossRef]
- Cheng, X.; Dong, S.; Chen, D.; Rui, Q.; Guo, J.; Dayong, W.; Jiang, J. Potential of esterase DmtH in transforming plastic additive dimethyl terephthalate to less toxic mono-methyl terephthalate. Ecotoxicol Environ Saf 2020, 187, 109848. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.H. Antibacterial drugs: Discovering antibiotics through soil metagenomics. Nat Rev Drug Discov 2018, 17, 240–241. [Google Scholar] [CrossRef]
- Jansson, J.K.; Hofmockel, K.S. The soil microbiome-from metagenomics to metaphenomics. Curr Opin Microbiol 2018, 43, 162–168. [Google Scholar] [CrossRef]
- Nahurira, R.; Ren, L.; Song, J.; Jia, Y.; Wang, J.; Fan, S.; Wang, H.; Yan, Y. Degradation of Di(2-Ethylhexyl) Phthalate by a Novel Gordonia alkanivorans Strain YC-RL2. Current microbiology 2017, 74, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhang, Y.; Shi, Y.; Jiang, J.; Wu, S.; Li, L.; Shao, Y.; Xin, Z. Identification and characterization of a novel phthalate-degrading hydrolase from a soil metagenomic library. Ecotoxicol Environ Saf 2020, 190, 110148. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Yang, H.; Shao, Y.; Li, L.; Sun, S.; Wang, L.; Tan, Y.; Xin, Z. Enhancing the activity and thermal stability of a phthalate-degrading hydrolase by random mutagenesis. Ecotoxicology and environmental safety 2021, 209, 111795. [Google Scholar] [CrossRef]
- Maria-Solano, M.A.; Serrano-Hervás, E.; Romero-Rivera, A.; Iglesias-Fernández, J.; Osuna, S. Role of conformational dynamics in the evolution of novel enzyme function. Chem Commun (Camb) 2018, 54, 6622–6634. [Google Scholar] [CrossRef]
- Osuna, S. The challenge of predicting distal active site mutations in computational enzyme design. WIREs Computational Molecular Science 2020, 11. [Google Scholar] [CrossRef]
- UniProt Consortium, T. UniProt: the universal protein knowledgebase. Nucleic Acids Res 2018, 46, 2699. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 2014, 42, W320–324. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.; Becker, A.-K.; Palm, G.J.; Berndt, L.; Badenhorst, C.P.S.; Godehard, S.P.; Reisky, L.; Lammers, M.; Bornscheuer, U.T. Sequence-Based Prediction of Promiscuous Acyltransferase Activity in Hydrolases. Angewandte Chemie International Edition 2020, 59, 11607–11612. [Google Scholar] [CrossRef]
- Pan, L.; Aller, S.G. Tools and procedures for visualization of proteins and other biomolecules. Curr Protoc Mol Biol 2015, 110, 19–12. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Li, X.; Ye, L.; Wang, X.; Wang, X.; Liu, H.; Qian, X.; Zhu, Y.; Yu, H. Molecular docking, molecular dynamics simulation, and structure-based 3D-QSAR studies on estrogenic activity of hydroxylated polychlorinated biphenyls. Sci Total Environ 2012, 441, 230–238. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J Comput Chem 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Harvey, M.J.; De Fabritiis, G. An Implementation of the Smooth Particle Mesh Ewald Method on GPU Hardware. J Chem Theory Comput 2009, 5, 2371–2377. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Journal of Computational Physics 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. Journal of Chemical Theory and Computation 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc Natl Acad Sci U S A 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J Mol Graph 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics 2013, 14, 346. [Google Scholar] [CrossRef]
- Nardini, M.; Dijkstra, B.W. α/β Hydrolase fold enzymes: the family keeps growing. Current Opinion in Structural Biology 1999, 9, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: a sequence logo generator. Genome Res 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- de Carvalho Lima Torres, A.; de Lima, L.N.; Tardioli, P.W.; de Sousa Júnior, R. Mathematical modeling of enzymatic syntheses of biosurfactants catalyzed by immobilized lipases. Reaction Kinetics, Mechanisms and Catalysis 2020, 130, 699–712. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T. A P.; Rempfer, C.; Bordoli, L., et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.A.; Salleh, A.B.; Leow, T.C.; Yahaya, N.M.; Abdul Rahman, M.B. Site-directed mutagenesis: role of lid region for T1 lipase specificity. Protein Eng Des Sel 2018, 31, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Dehury, B.; Patra, M.C.; Maharana, J.; Sahu, J.; Sen, P.; Modi, M.K.; Choudhury, M.D.; Barooah, M. Structure-based computational study of two disease resistance gene homologues (Hm1 and Hm2) in maize (Zea mays L.) with implications in plant-pathogen interactions. PLoS One 2014, 9, e97852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, J.; Li, L.; Liu, X.; Yao, X.; Liu, H. The solvent at antigen-binding site regulated C3d-CR2 interactions through the C-terminal tail of C3d at different ion strengths: insights from molecular dynamics simulation. Biochim Biophys Acta 2016, 1860, 2220–2231. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, H.X. Allosteric activation of SENP1 by SUMO1 β-grasp domain involves a dock-and-coalesce mechanism. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xu, Y.; Guo, J. Insights into the negative regulation of EGFR upon the binding of an allosteric inhibitor. Chem Biol Drug Des 2022, 99, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Hünenberger, P.H.; Mark, A.E.; Gunsteren, W.F.v. Fluctuation and Cross-correlation Analysis of Protein Motions Observed in Nanosecond Molecular Dynamics Simulations. Journal of Molecular Biology 1995, 252. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, X.; Xu, Y. [Improvement of catalytic activity of Aspergillus terreus lipase by site-directed mutagenesis]. Sheng wu gong cheng xue bao = Chinese journal of biotechnology 2018, 34, 1091–1105. [Google Scholar] [CrossRef]
- Eargle, J.; Luthey-Schulten, Z. NetworkView: 3D display and analysis of protein.RNA interaction networks. Bioinformatics 2012, 28, 3000–3001. [Google Scholar] [CrossRef]
- Ning, L.; Guo, J.; Jin, N.; Liu, H.; Yao, X. The role of Cys179-Cys214 disulfide bond in the stability and folding of prion protein: insights from molecular dynamics simulations. J Mol Model 2014, 20, 2106. [Google Scholar] [CrossRef]
- Secundo, F.; Carrea, G.; Tarabiono, C.; Gatti-Lafranconi, P.; Brocca, S.; Lotti, M.; Jaeger, K.-E.; Puls, M.; Eggert, T. The lid is a structural and functional determinant of lipase activity and selectivity. Journal of Molecular Catalysis B: Enzymatic 2006, 39, 166–170. [Google Scholar] [CrossRef]
- Barbe, S.; Lafaquière, V.; Guieysse, D.; Monsan, P.; Remaud-Siméon, M.; André, I. Insights into lid movements of Burkholderia cepacia lipase inferred from molecular dynamics simulations. Proteins 2009, 77, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Pleiss, J.; Fischer, M.; Schmid, R.D. Anatomy of lipase binding sites: the scissile fatty acid binding site. Chemistry and Physics of Lipids 1998, 93, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Karkhane, A.A.; Yakhchali, B.; Jazii, F.R.; Bambai, B. The effect of substitution of Phe181 and Phe182 with Ala on activity, substrate specificity and stabilization of substrate at the active site of Bacillus thermocatenulatus lipase. Journal of Molecular Catalysis B: Enzymatic 2009, 61, 162–167. [Google Scholar] [CrossRef]
- Paola; Panizza; Silvia; Cesarini; Pilar; Diaz; Sonia; Rodríguez; Giordano. Saturation mutagenesis in selected amino acids to shift Pseudomonas sp. acidic lipase Lip I.3 substrate specificity and activity. Chemical Communications 2015. [Google Scholar] [CrossRef]
- Ren, L.-Q.; Chang, T.-T.; Ren, D.-P.; Zhou, Y.; Ye, B.-C. Rational design to improve activity of the Est3563 esterase from Acinetobacter sp. LMB-5. Enzyme and Microbial Technology 2019, 131, 109331. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.M.; Bolduc, J.M.; Gallaher, J.L.; Stoddard, B.L.; Baker, D. Alteration of enzyme specificity by computational loop remodeling and design. Proc Natl Acad Sci U S A 2009, 106, 9215–9220. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wijma, H.J.; Song, L.; Cui, Y.; Otzen, M.; Tian, Y.; Du, J.; Li, T.; Niu, D.; Chen, Y. , et al. Computational redesign of enzymes for regio- and enantioselective hydroamination. Nat Chem Biol 2018, 14, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef]
- Jiang, L.; Althoff, E.A.; Clemente, F.R.; Doyle, L.; Röthlisberger, D.; Zanghellini, A.; Gallaher, J.L.; Betker, J.L.; Tanaka, F.; Barbas, C.F. , 3rd, et al. De novo computational design of retro-aldol enzymes. Science 2008, 319, 1387–1391. [Google Scholar] [CrossRef]
- Röthlisberger, D.; Khersonsky, O.; Wollacott, A.M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J.L.; Althoff, E.A.; Zanghellini, A.; Dym, O. , et al. Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Tyukhtenko, S.; Rajarshi, G.; Karageorgos, I.; Zvonok, N.; Gallagher, E.S.; Huang, H.; Vemuri, K.; Hudgens, J.W.; Ma, X.; Nasr, M.L. , et al. Effects of Distal Mutations on the Structure, Dynamics and Catalysis of Human Monoacylglycerol Lipase. Scientific Reports 2018, 8, 1719. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Schnell, J.R.; McElheny, D.; Bae, S.-H.; Duggan, B.M.; Benkovic, S.J.; Dyson, H.J.; Wright, P.E. A Distal Mutation Perturbs Dynamic Amino Acid Networks in Dihydrofolate Reductase. Biochemistry 2013, 52, 4605–4619. [Google Scholar] [CrossRef] [PubMed]
- Ragland, D.A.; Nalivaika, E.A.; Nalam, M.N.L.; Prachanronarong, K.L.; Cao, H.; Bandaranayake, R.M.; Cai, Y.; Kurt-Yilmaz, N.; Schiffer, C.A. Drug Resistance Conferred by Mutations Outside the Active Site through Alterations in the Dynamic and Structural Ensemble of HIV-1 Protease. Journal of the American Chemical Society 2014, 136, 11956–11963. [Google Scholar] [CrossRef]
- Shaikh, F.A.; Withers, S.G. Teaching old enzymes new tricks: engineering and evolution of glycosidases and glycosyl transferases for improved glycoside synthesis. Biochemistry and Cell Biology 2008, 86, p–169. [Google Scholar] [CrossRef]
Figure 1.
The 2D structures of six common PAEs and the 3D structure of EstJ6. (A) The 2D structures of six common PAEs including BBP, DEHP, DBP, DMP, DnOP and DEP. (B) Overview of EstJ6 stereo structure and mutation sites for ET2.2. Magenta sticks represent triads S146, E240, and H270. Essential motifs including lid and cap regions are colored by purple and yellow, respectively. The substrate binding pocket is highlighted by an oval.
Figure 1.
The 2D structures of six common PAEs and the 3D structure of EstJ6. (A) The 2D structures of six common PAEs including BBP, DEHP, DBP, DMP, DnOP and DEP. (B) Overview of EstJ6 stereo structure and mutation sites for ET2.2. Magenta sticks represent triads S146, E240, and H270. Essential motifs including lid and cap regions are colored by purple and yellow, respectively. The substrate binding pocket is highlighted by an oval.
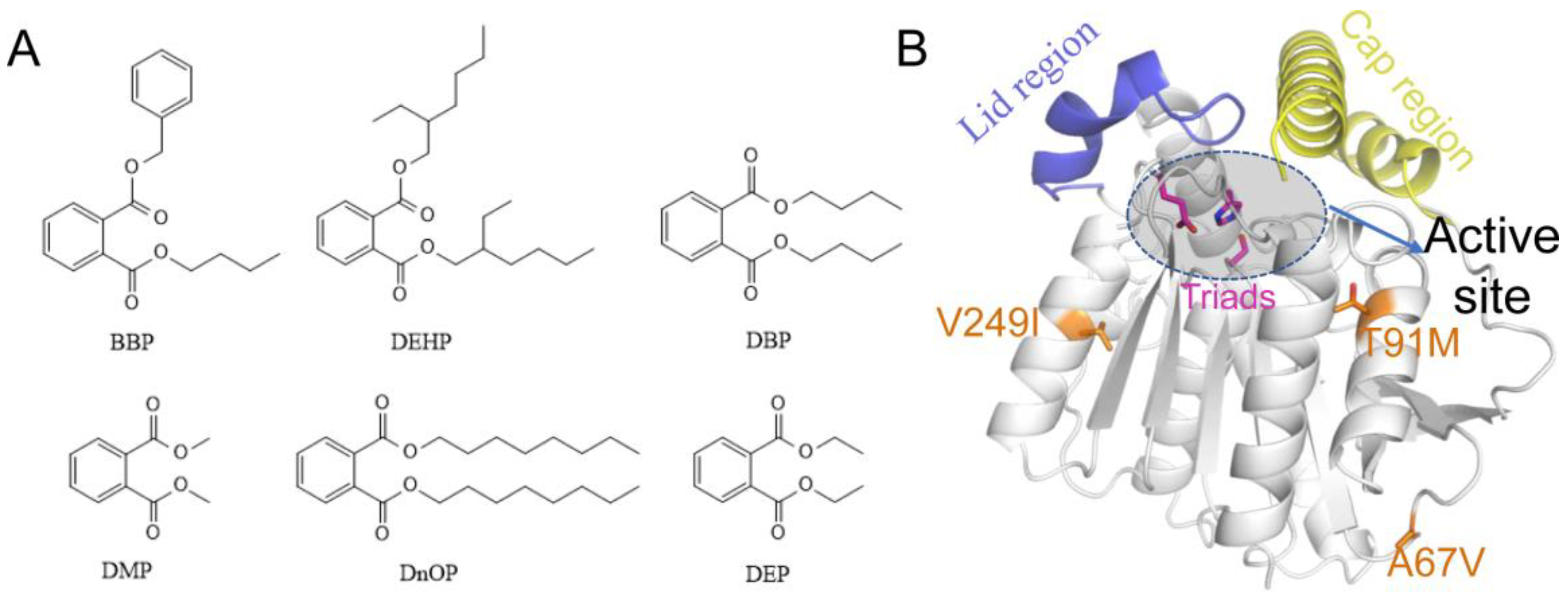
Figure 2.
Graphical representation of sequence conservation of partial EstJ6. The sequence logo was generated by the WebLogo3 server after sequence alignment. The mutated sites, triads, and three conservative motifs are highlighted in orange, cyan and yellow, respectively. The size of logos represents the relative frequency of each residue.
Figure 2.
Graphical representation of sequence conservation of partial EstJ6. The sequence logo was generated by the WebLogo3 server after sequence alignment. The mutated sites, triads, and three conservative motifs are highlighted in orange, cyan and yellow, respectively. The size of logos represents the relative frequency of each residue.

Figure 3.
System stability of all systems. (A) Cα RMSFs based on the last 100-ns MD trajectories for each system using their average conformations as references. The results of all systems are averaged over the three replicate simulations. The cap and lid regions are highlighted in yellow and purple background, respectively. (B) Changes in RMSF upon mutations displayed on the apo structure according to a color scale (lower and higher flexibilities depicted in blue and red, respectively).
Figure 3.
System stability of all systems. (A) Cα RMSFs based on the last 100-ns MD trajectories for each system using their average conformations as references. The results of all systems are averaged over the three replicate simulations. The cap and lid regions are highlighted in yellow and purple background, respectively. (B) Changes in RMSF upon mutations displayed on the apo structure according to a color scale (lower and higher flexibilities depicted in blue and red, respectively).
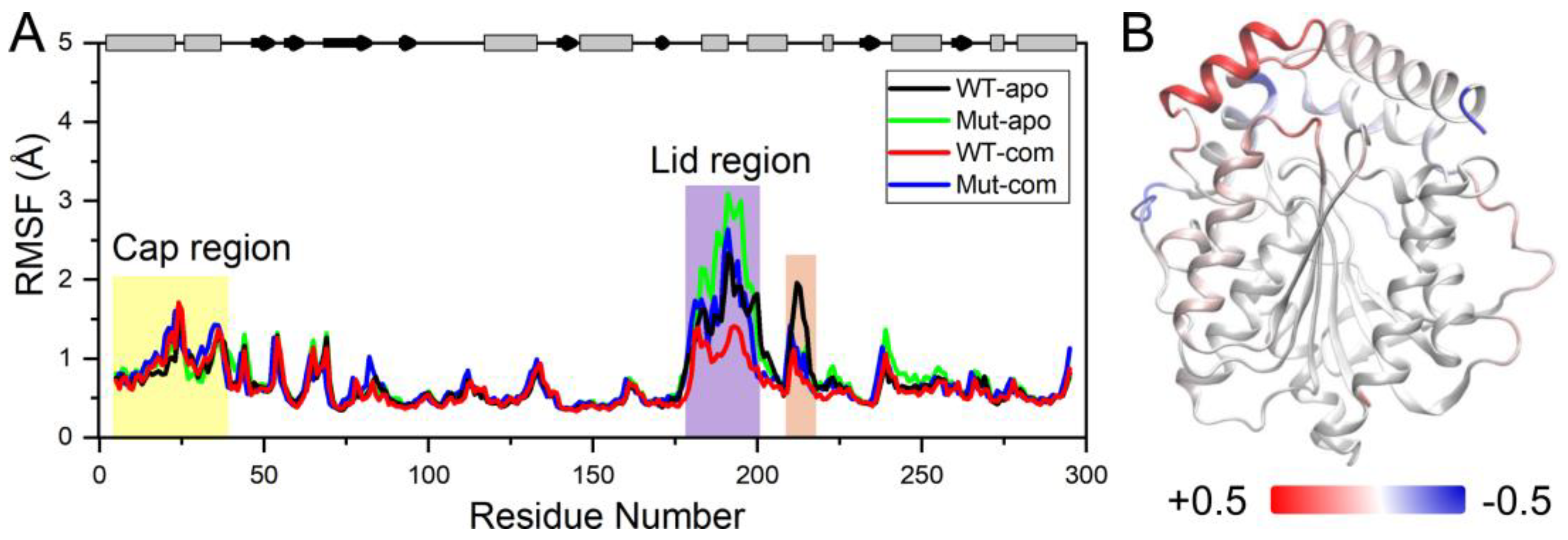
Figure 4.
The residue-residue correlation information of the entire protein and the contact information of the pocket entrance regions in the two apo systems. (A-B) Dynamical cross-correlation maps for the Cα atom pairs according to the last 100-ns MD trajectories: WT-apo (A) and Mut-apo (B). Areas with strong variations between apo systems are emphasized with black boxes. (C-D) Contact map between the lid region and residues 265-272: WT-apo (C) and Mut-apo (D). (E) The overall structure of EstJ6. The pocket entrance residues are indicated by blue and red spheres, and residues 200-297 are depicted in red.
Figure 4.
The residue-residue correlation information of the entire protein and the contact information of the pocket entrance regions in the two apo systems. (A-B) Dynamical cross-correlation maps for the Cα atom pairs according to the last 100-ns MD trajectories: WT-apo (A) and Mut-apo (B). Areas with strong variations between apo systems are emphasized with black boxes. (C-D) Contact map between the lid region and residues 265-272: WT-apo (C) and Mut-apo (D). (E) The overall structure of EstJ6. The pocket entrance residues are indicated by blue and red spheres, and residues 200-297 are depicted in red.
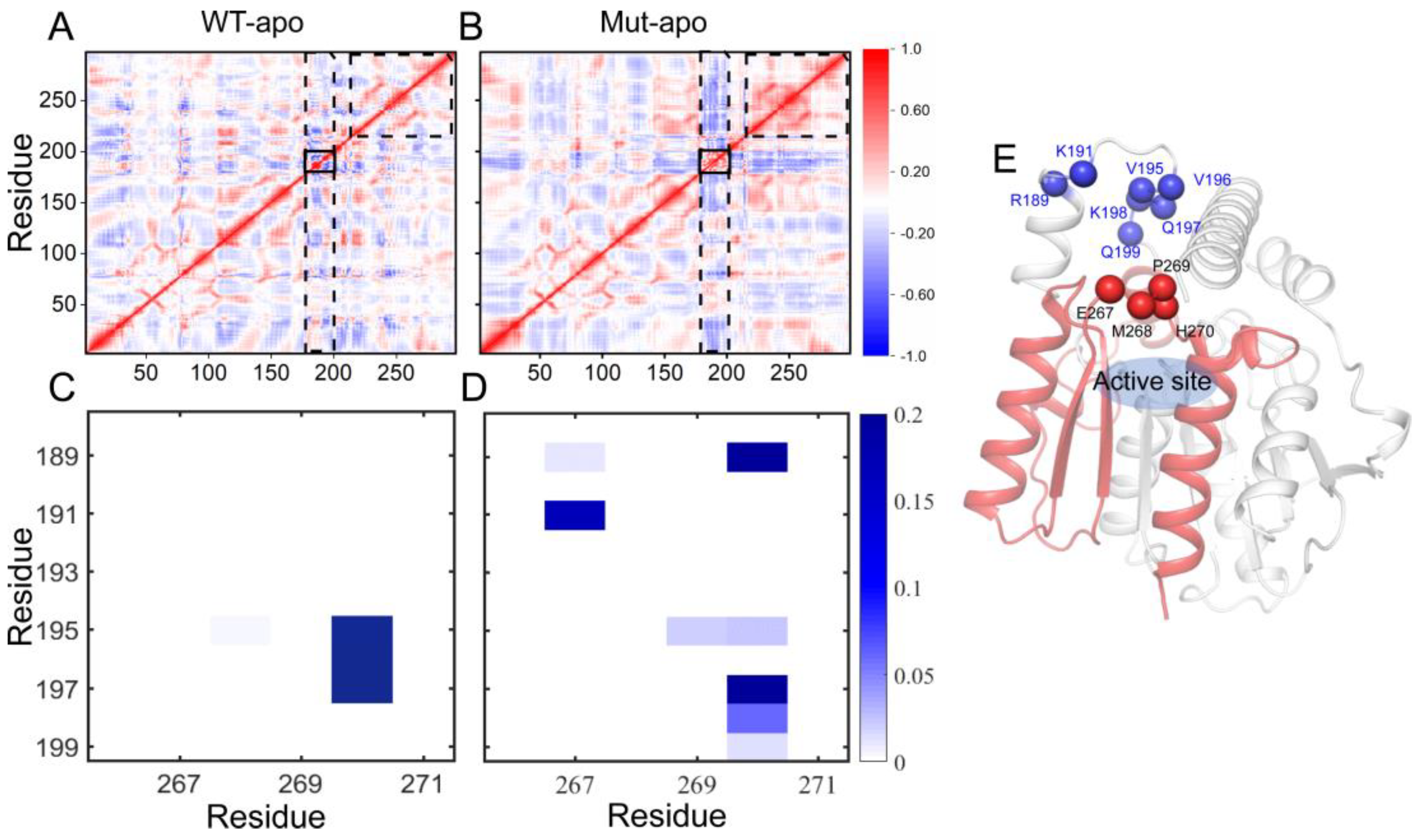
Figure 5.
Free energy profiles over the first two principal components (PC1 and PC2) in all systems: WT-apo, Mut-apo, WT-com, and Mut-com (A). The displacements of protein along PC1 (B) and PC2 (C) are mapped onto the structure. The detailed motions along the two PCs are displayed in videos in the Supplementary Videos 1 (PC1) and 2 (PC2). In the videos, the lid and cap regions are colored in purple and yellow, respectively. And the Cα atoms of essential pocket residues are depicted in green spheres, including oxygen hole residues and triads.
Figure 5.
Free energy profiles over the first two principal components (PC1 and PC2) in all systems: WT-apo, Mut-apo, WT-com, and Mut-com (A). The displacements of protein along PC1 (B) and PC2 (C) are mapped onto the structure. The detailed motions along the two PCs are displayed in videos in the Supplementary Videos 1 (PC1) and 2 (PC2). In the videos, the lid and cap regions are colored in purple and yellow, respectively. And the Cα atoms of essential pocket residues are depicted in green spheres, including oxygen hole residues and triads.
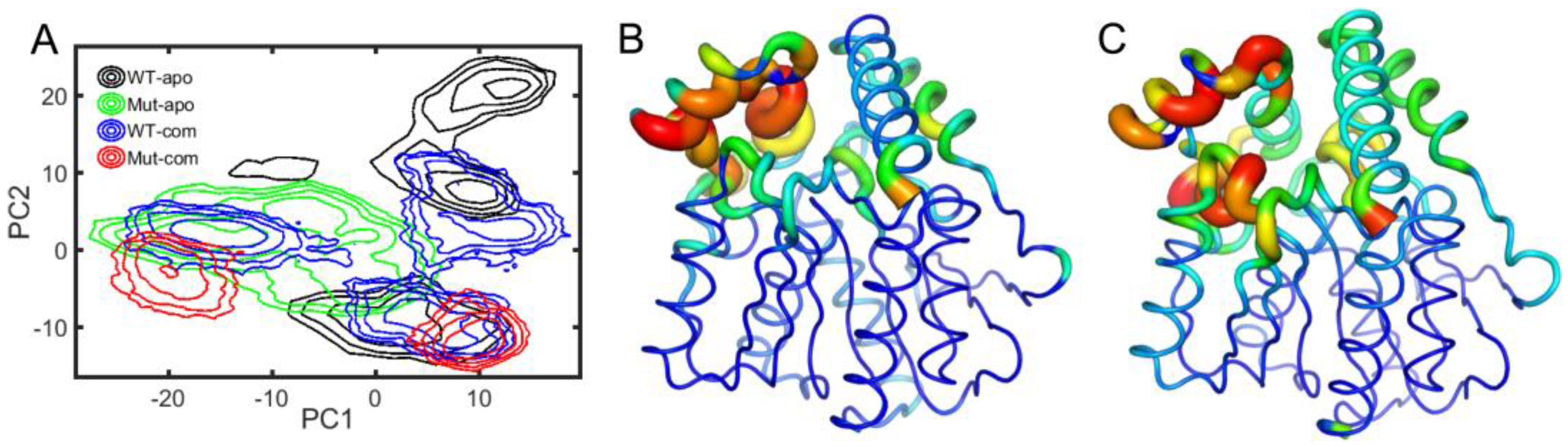
Figure 6.
Displacements of the lid region (Cα atoms of residues 192-196, colored in purple) according to a self-defined coordinate system. The Cα atom of S146 is set as the origin, and the x- and y-axes go through the Cα atoms of Q244 and D284, respectively. The three Cα atoms are shown as yellow spheres.
Figure 6.
Displacements of the lid region (Cα atoms of residues 192-196, colored in purple) according to a self-defined coordinate system. The Cα atom of S146 is set as the origin, and the x- and y-axes go through the Cα atoms of Q244 and D284, respectively. The three Cα atoms are shown as yellow spheres.
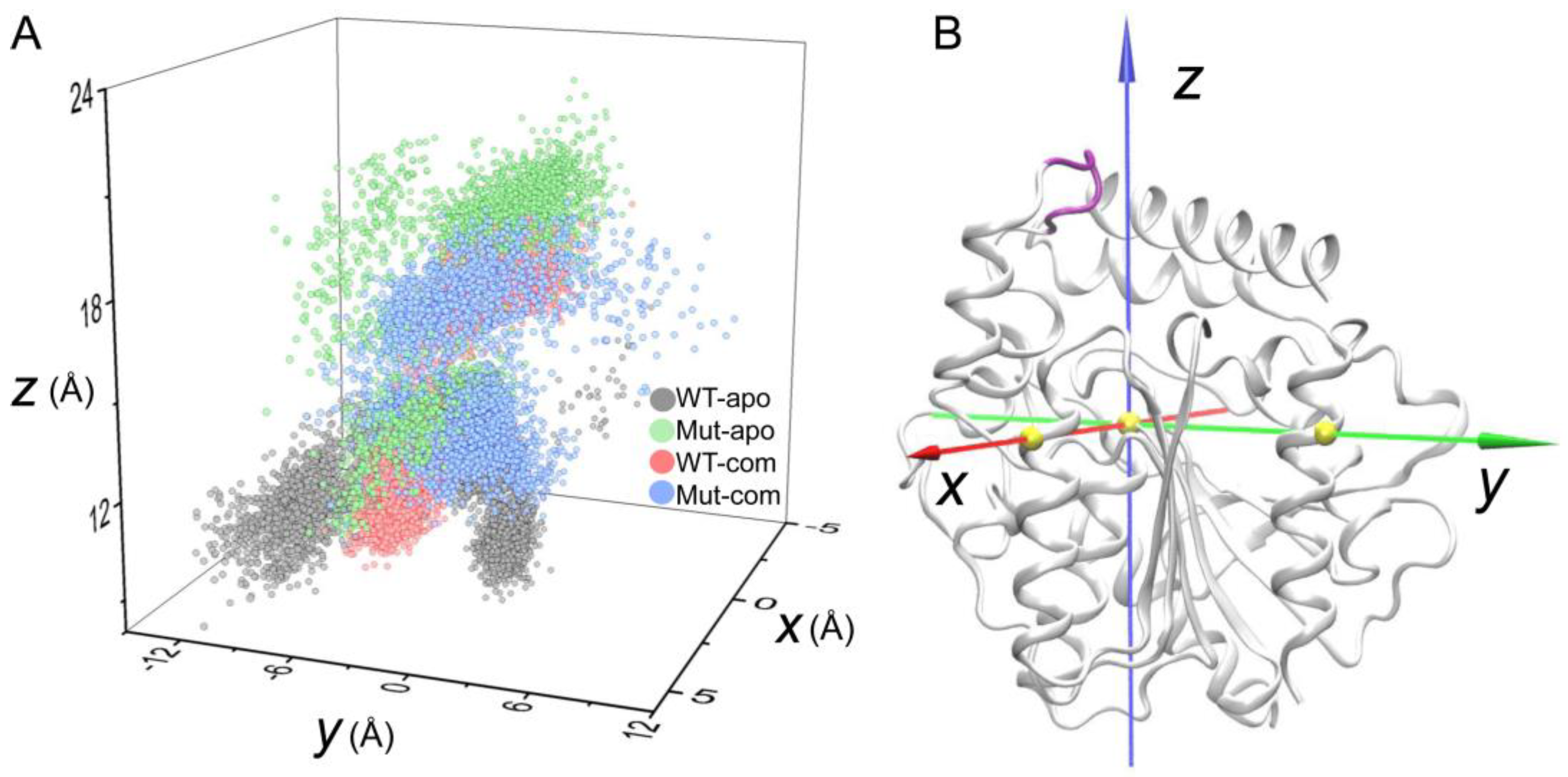
Figure 7.
Substrate binding pocket analyses. The distribution of RMSD (A) and Rg (B) of hydrophobic residues in the binding site. The free energy landscape distribution of the side-chain distances of the catalytic triad including S146-H270 (d1) and E240-H270 (d2) with lower energy minima basins colored in deep blue for WT-com (C) and Mut-com (D). (E) The local structure and hydrogen-bond network of the active site involving the catalytic triads.
Figure 7.
Substrate binding pocket analyses. The distribution of RMSD (A) and Rg (B) of hydrophobic residues in the binding site. The free energy landscape distribution of the side-chain distances of the catalytic triad including S146-H270 (d1) and E240-H270 (d2) with lower energy minima basins colored in deep blue for WT-com (C) and Mut-com (D). (E) The local structure and hydrogen-bond network of the active site involving the catalytic triads.
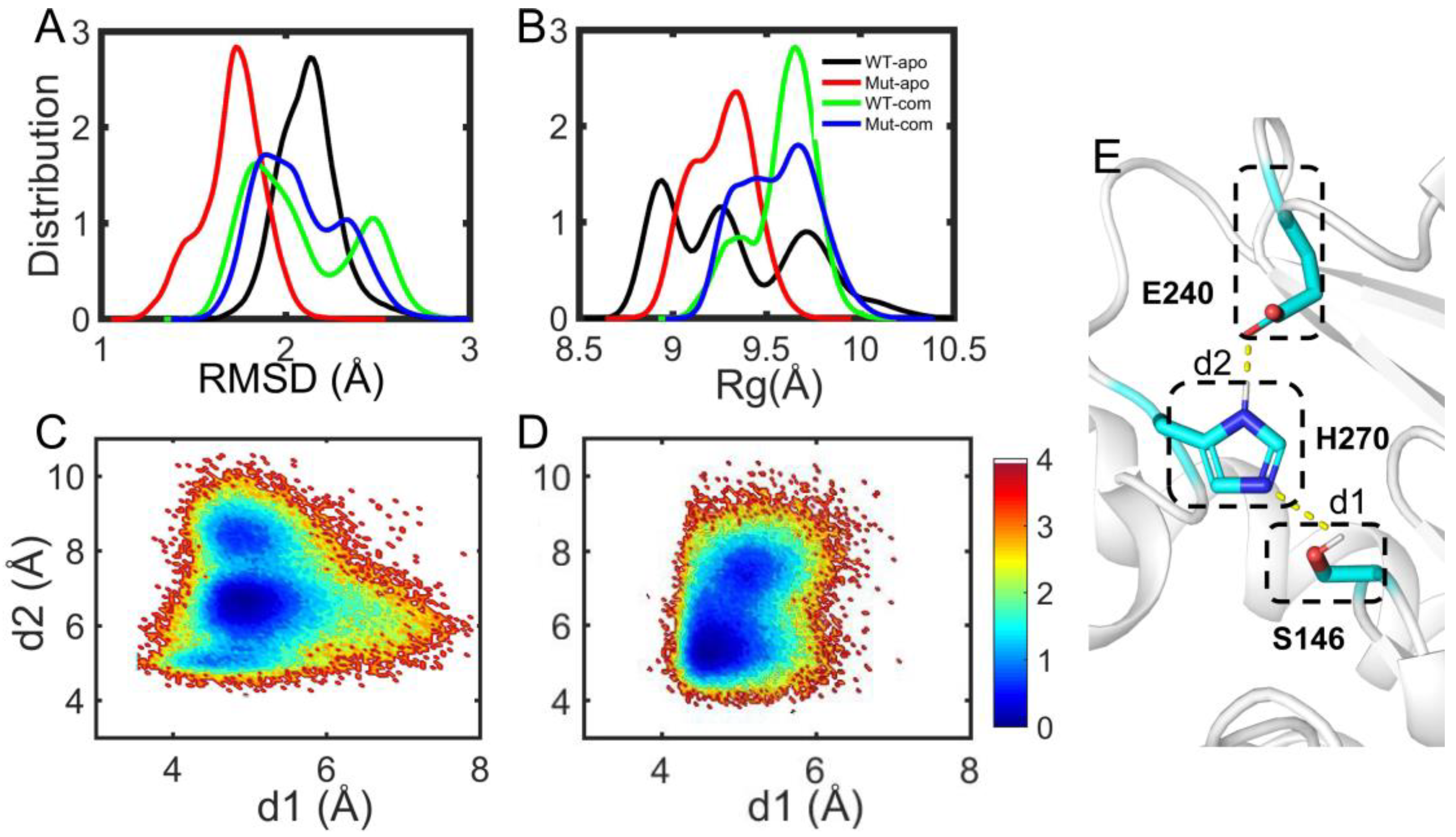
Figure 8.
Community networks of all systems are displayed in the tertiary structure (top panels) or in schematic two-dimensional representation (bottom panels) with corresponding colors: (A) WT-apo; (B) Mut-apo; (C) WT-com; (D) Mut-com. The size of a node is proportional to the number of residues within it, and the thickness of an edge is indicative of the communication strength of the two connected nodes.
Figure 8.
Community networks of all systems are displayed in the tertiary structure (top panels) or in schematic two-dimensional representation (bottom panels) with corresponding colors: (A) WT-apo; (B) Mut-apo; (C) WT-com; (D) Mut-com. The size of a node is proportional to the number of residues within it, and the thickness of an edge is indicative of the communication strength of the two connected nodes.
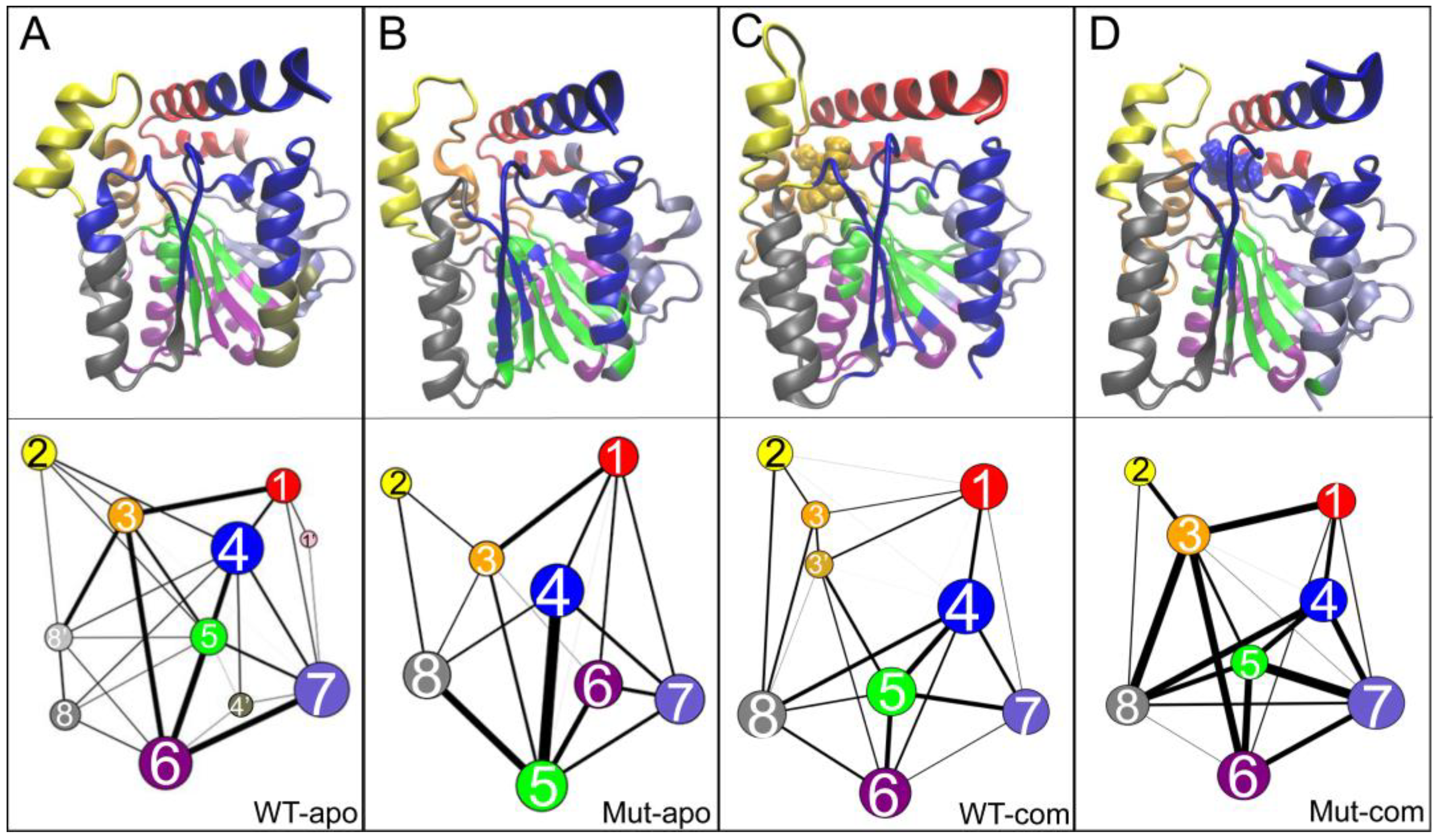
Figure 9.
Cα RMSFs based on the last 100-ns distance-limited MD trajectories.
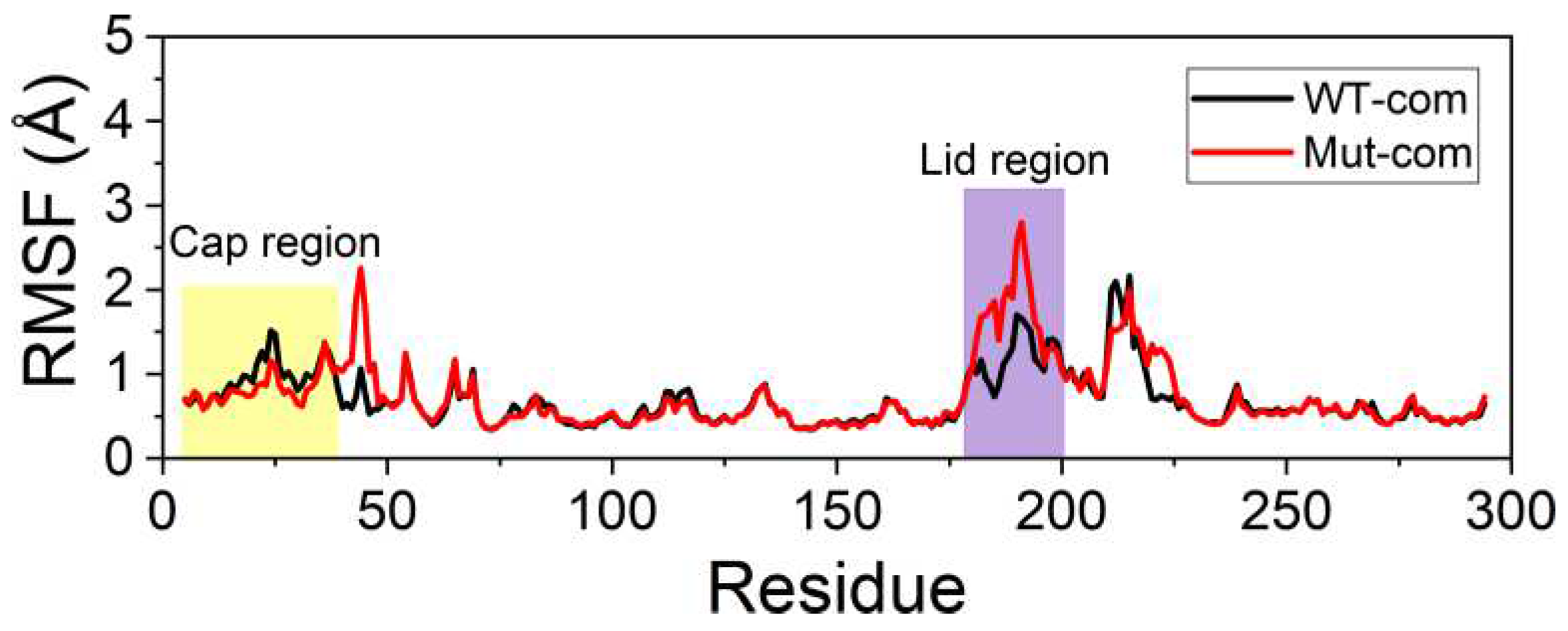
Figure 10.
Residue contribution to the binding of DBP: WT-com and Mut-com (A), and the key residues are marked. (B) The key residues contributing to DBP binding are displayed in sticks.
Figure 10.
Residue contribution to the binding of DBP: WT-com and Mut-com (A), and the key residues are marked. (B) The key residues contributing to DBP binding are displayed in sticks.
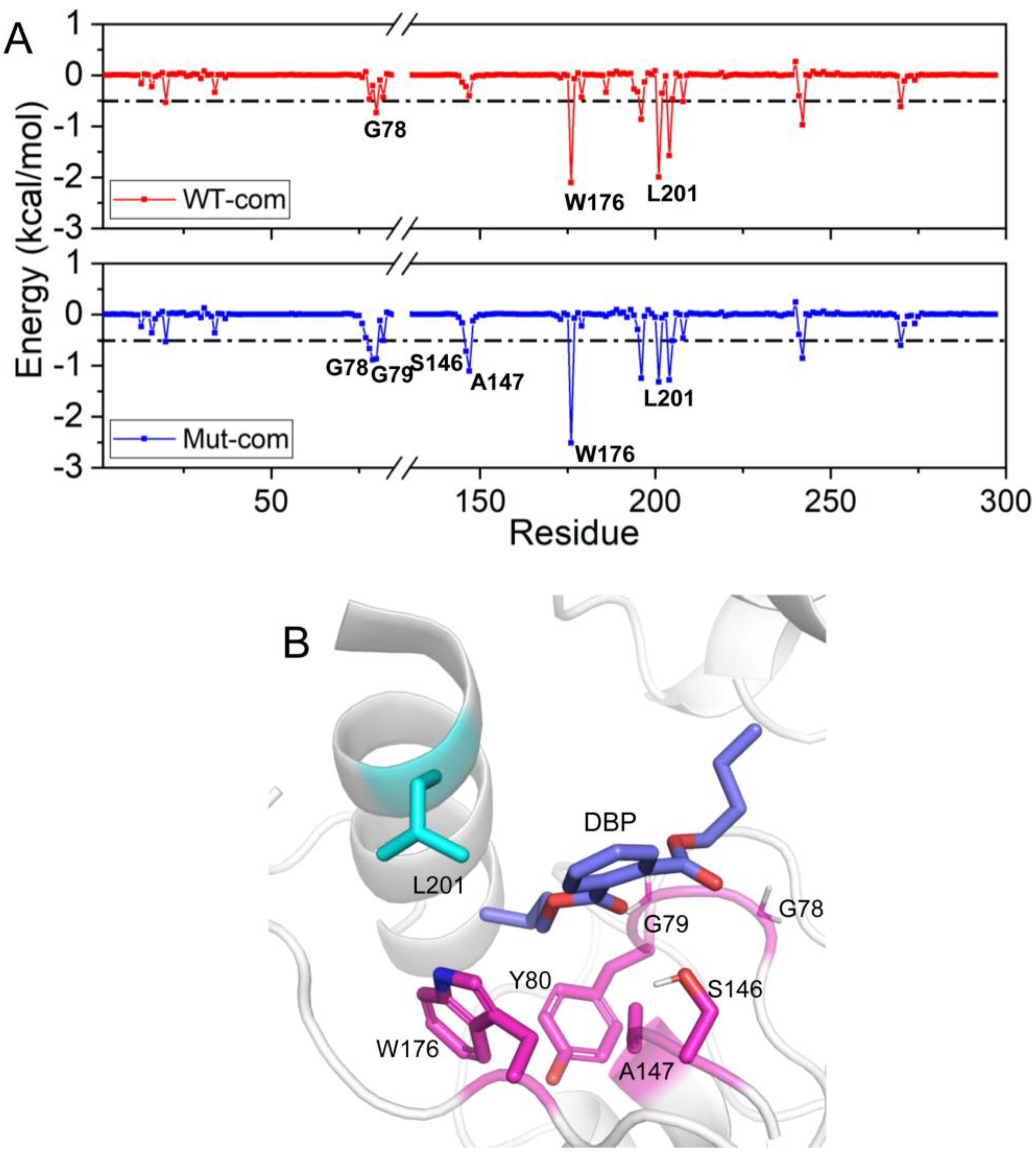
Figure 11.
Structure and dynamics information of mutant. (A) The mutation sites A23C and A29C are depicted in sticks. (B) The distribution plot of Rg about overall structure including wild type and mutantA23C-A29C. (C) The RMSD distribution plot of cap region for wild type and mutantA23C-A29C.
Figure 11.
Structure and dynamics information of mutant. (A) The mutation sites A23C and A29C are depicted in sticks. (B) The distribution plot of Rg about overall structure including wild type and mutantA23C-A29C. (C) The RMSD distribution plot of cap region for wild type and mutantA23C-A29C.
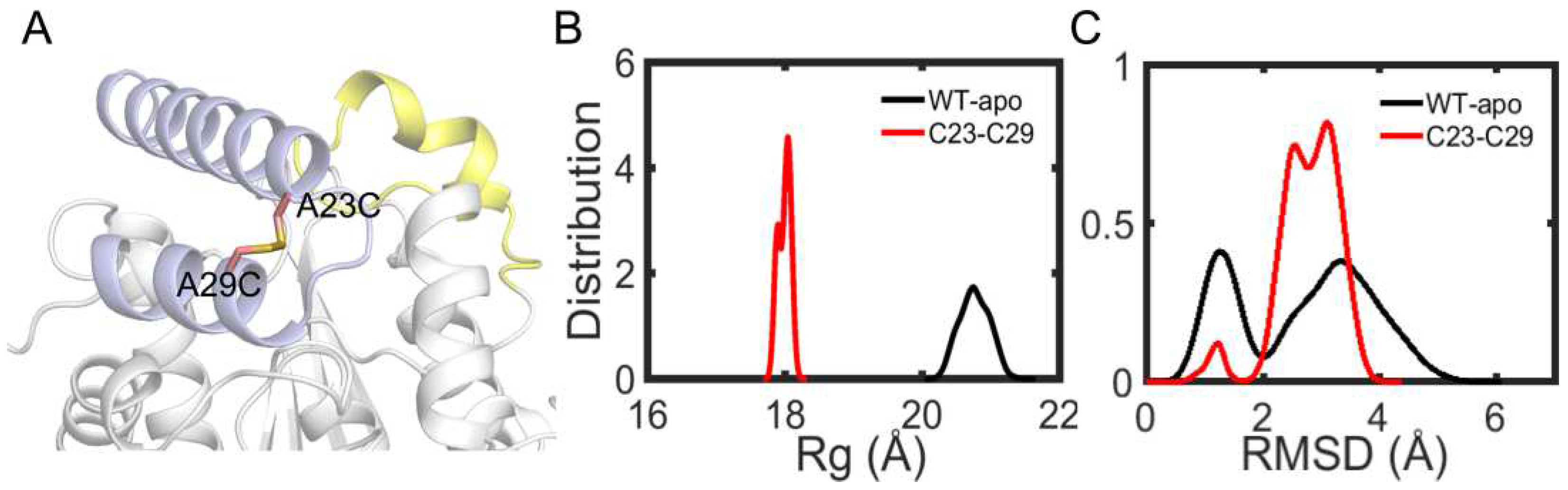

Table 1.
Terms of the binding free energy between WT-com and Mut-com averaged over the last 20 ns of the replicate runs (kcal/mol).
Table 1.
Terms of the binding free energy between WT-com and Mut-com averaged over the last 20 ns of the replicate runs (kcal/mol).
| Contributions | WT-com (kcal/mol) | Mut-com (kcal/mol) |
| ΔEvdw | -41.34 ± 4.24 | -44.50 ± 0.55 |
| ΔEele | -7.37 ± 6.98 | -12.07 ± 10.13 |
| ΔGpol,sol | 19.85 ± 6.35 | 23.02 ± 5.83 |
| ΔGnpol,sol | -5.83 ± 0.43 | -6.24 ± 0.09 |
| ΔEMM | -48.71 ± 9.14 | -56.57 ± 10.19 |
| ΔGsol | 14.02 ± 6.26 | 16.78 ± 5.92 |
| ΔGtotal | -34.68 ± 4.47 | -39.79 ± 4.70 |
| Docking score | -5.5 ± 2.12 | -6.45 ± 0.92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



