
Submitted:
09 February 2023
Posted:
15 February 2023
You are already at the latest version
Abstract
The mechanistic target of rapamycin (mTOR) kinase is one of the top drug targets for promoting health and lifespan extension. Besides rapamycin, only a few other mTOR inhibitors have been developed and shown their ability to slow aging. We used machine learning to predict novel small molecules targeting mTOR. We selected one small molecule, TKA001, based on in-silico predictions of a high on-target probability, low toxicity, favorable physicochemical properties, and preferable ADMET profile. We confirmed TKA001 binding in silico by molecular docking. TKA001 potently inhibits both TOR complex 1 and 2 downstream signaling in vitro. Furthermore, TKA001 inhibits human cancer cell proliferation in vitro and extended the lifespan of C. elegans, suggesting that TKA001 can slow aging in vivo.
Keywords:
AI drug discovery
; mTOR
; rapalog
; C. elegans
; cancer
; longevity
Introduction
One of the most robust interventions to increase healthspan and lifespan in preclinical models is the inhibition of the growth-regulating serine/threonine kinase mechanistic Target of Rapamycin (mTOR). This effect has been demonstrated using genetics in multiple species, including flies, worms, and mice. In Drosophila, suppression of TOR signaling by overexpression of the negative regulators TSC1 or TSC2, or knock-in of dominant negative forms of TOR or S6K significantly extended lifespan [1]. In C. elegans, rapamycin or RNAi-mediated knockdown in adulthood of the conserved TOR pathway components daf-15 (RAPTOR), rheb-1, raga-1, or ragc-1 all robustly extended lifespan [2,3,4,5,6]. In mice, while complete knockout of most mTOR components is embryonically lethal, knock-in of a hypomorphic mTOR allele extends lifespan by ~25% [7].
Pharmacologic inhibition of mTOR has also been proven to be effective in extending lifespan in mice. The mTOR inhibitor rapamycin has repeatedly shown lifespan-extending effects across sex, strain, and dosing regimens. Effective regimens include dosing continuously throughout life, intermittently throughout life, transiently early in life, transiently in midlife, and continuously starting late in life [8,9,10,11,12]. These striking preclinical effects are mediated both directly through suppression of tumorigenesis and indirectly via modulation of lifespan-regulating processes, including insulin and ATF4 signaling pathways [4,13,14].
Rapamycin and structurally related “rapalogs” have also shown promise in human trials against age-associated pathologies. In randomized clinical trials, pharmacological inhibition of mTOR ameliorated the age-related decline of the immune system and reduced skin senescence in elderly humans [15,16].
Despite the clear preclinical promise of rapamycin and related rapalogs, challenges of drug discovery and development have been well-described, with cost estimates of over $100 million over 10 years or more [17]. However, the use of artificial intelligence (AI) assisted methods for early drug discovery can fundamentally transform this process, cutting off years and tens of millions of dollars. We employed an AI method to identify a potent and selective mTOR inhibitor and validated this compound’s anti-cancer and pro-longevity effects in cell culture and C. elegans models.
Results
To identify and generate compounds that inhibit mTOR, we used generative adversarial networks and reinforcement learning methods. We generated more than 1000 small molecules predicted to target mTOR. We performed an independent validation using PASS software [18] to select which of the generated molecules have indeed a high probability of inhibiting mTOR. We narrowed down the candidate list to 132 compounds with a high probability of targeting mTOR (Figure 1A). Since PASS software can be used to predict toxic and adverse effects, we filtered these candidate compounds for their likelihood of low toxicity, of which 29 compounds remained (i.e., 22% of all 132 compounds; Figure 1A). Next, we assessed in-silico these 29 compounds with preferable ADMET profiles (absorption, distribution, metabolism, excretion, toxicity) and found one strong candidate 1-ethyl-3-(4-(4-morpholino-5,7-dihydrofuro[3,4-d]pyrimidin-2-yl)phenyl)urea, which we named TKA001 (Figure 1B-D).
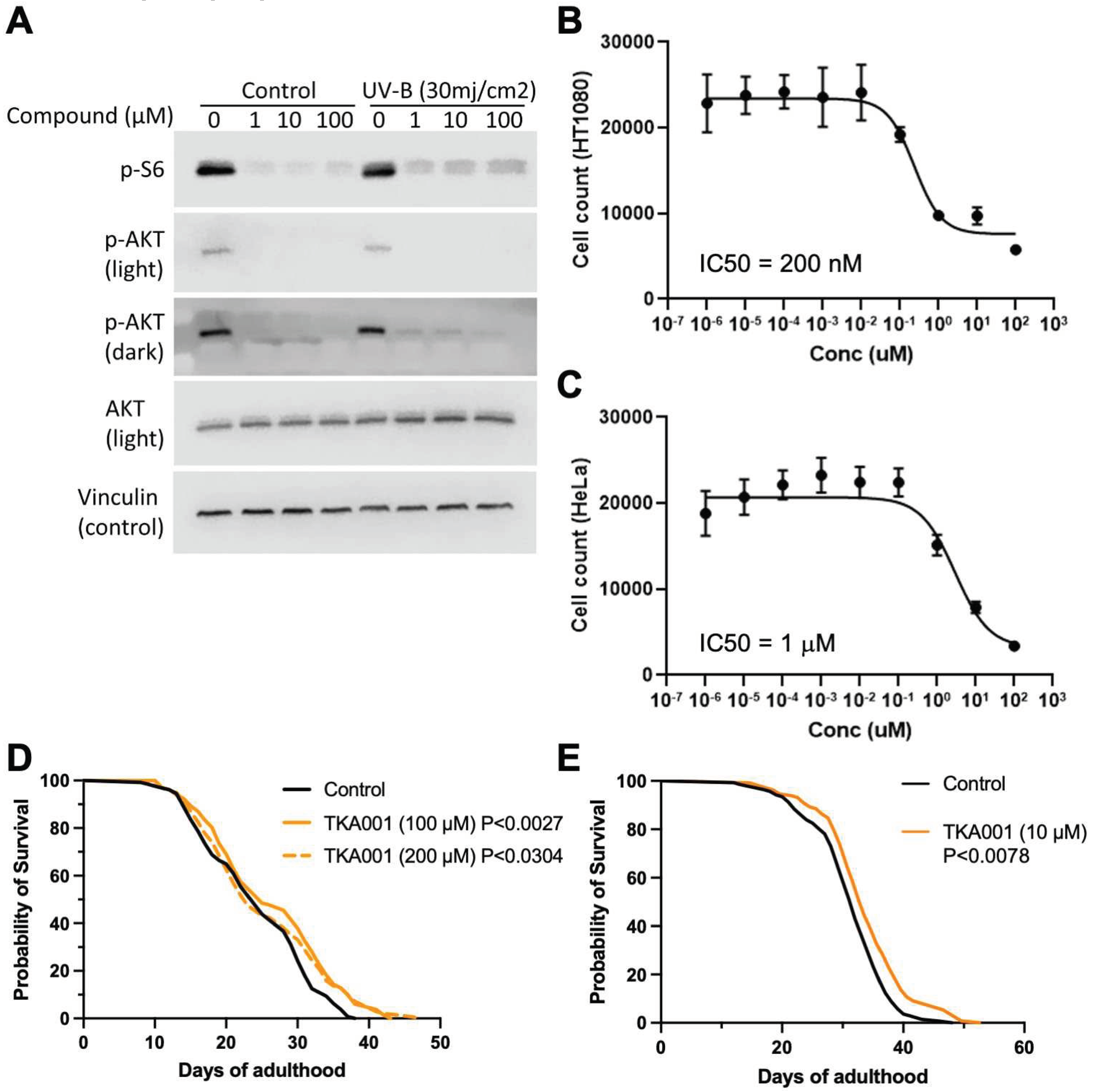
To validate TKA001, we first performed molecular docking of TKA001 to mTOR kinase and found a binding affinity of TKA001 to mTOR of -6.4 kcal/mol (Figure 1E). Next, we assessed mTOR downstream signaling. The mTOR kinase is found in two complexes (mTORC1 and mTORC2; [19]). The mTOR complex 1 (mTORC1) phosphorylates ribosomal protein S6 kinase (S6K; [20]), whereas the mTOR complex 2 (mTORC2) phosphorylates AKT [20]. At 1 µM of TKA001, phosphorylation of S6K and AKT were reduced in HT1080 cells (Figure 2A), suggesting that TKA001 inhibits mTOR kinase in both complexes. To stimulate mTOR signaling, we UV-B irradiated HT1080 cells and still found a strong reduction of S6K and AKT phosphorylation, suggesting that TKA001 efficiently reduces mTOR signaling (Figure 2A).
Our in-silico analysis of TKA001 using CLC-Pred [21] predicted an 85% likelihood as an effective treatment against prostate cancer. We found that TKA001 inhibits cancer cell proliferation of epithelial cancer cells from patients with a fibrosarcoma (HT-1080; half maximal inhibitory concentration (IC50) = 200 nM; Figure 2B) and cervical cancer cells (HeLa; IC50 = 1 µM; Figure 2C). In comparison, IC50 of rapamycin on HT-1080 or HeLa is 1.8 µM and 0.25 µM, respectively [22]. This suggests that TKA001 is a potent inhibitor of cancer cell proliferation in vitro.
Since TKA001 performed well in silico and in vitro, we next wanted to assess the in-vivo efficacy of TKA001 on mTOR inhibition. Genetic inhibition of mTORC1 or mTORC2, knockdown of mTOR/LET-363, or rapamycin treatment increases the lifespan of C. elegans [2,3,4,5,6,23,24,25]. To determine whether TKA001 could also increase the lifespan of C. elegans, we used 100 µM and 200 µM of TKA001 because about 100 µM of rapamycin results in the most robust lifespan extension [2,4,6]. We found that adulthood-specific application of 200 µM of TKA001 only extended the maximal lifespan, whereas 100 µM of TKA001 resulted in both mean and maximum increase of lifespan (Figure 1H). Next, we assessed whether a lower dose would be sufficient to increase lifespan. Indeed, supplementing C. elegans starting from the young adult stage with 10 µM of TKA001 was sufficient to extend the lifespan.
Discussion
The nutrient-sensing mTOR kinase is a master growth regulator essential for development and tissue homeostasis [26]. However, mTOR activity becomes deregulated during aging, showing improper and sustained mTOR signaling in older animals [27,28]. Reducing the function of mTOR increases the lifespans from yeast to mammals [29]. Despite mTOR being the prime target against many age-related and chronic pathologies [30], relatively few mTOR inhibitors have been developed to slow the aging process.
Using deep neuronal artificial learning, we identified many potential mTOR inhibitors. Through in-silico analysis, we selected one mTOR inhibitor with predicted low toxicity and a preferable ADMET profile. We validated on-target by molecular docking to the mTOR kinase, confirming the inhibition of mTOR downstream signaling and cancer cell proliferation in vitro, and increasing lifespan in vivo.
In a series of rational design and classical medicinal chemistry approaches, structurally similar mTOR inhibitors were previously developed based on a quaternary-substituted dihydrofuropyrimidine [31]. The structurally most similar compounds, inhibited mTOR kinase signaling in vitro at 3–4.4 nM [31] and had an IC50 of 31-1700 nM in cancer cell proliferation assays (NCI-PC3, MCF7neo/Her2) [31] compared to our IC50 of 200 nM or 1000 nM in HT-1080 or HeLa, respectively. Given these comparable in-vitro results, reassuringly validates our machine-learning approach to identifying novel small molecules for mTOR inhibition. More importantly, we showed that our TKA001 mTOR inhibitor is able to slow aging in vivo. Further machine-learning approaches hold the potential to speed up drug discovery and facilitate the selection of compounds for future clinical candidates targeting the mTOR-mediated healthy longevity benefits.
Materials and Methods
Reagents
TKA001 is 1-ethyl-3-(4-(4-morpholino-5,7-dihydrofuro[3,4-d]pyrimidin-2-yl)phenyl)urea (C19H23N5O3). Molecular weight: 369.43, Melting point 213°C. TKA001 was synthesized by Otava Chemicals (ZINC71297044 (catalog number 27705871)).
Dimethyl sulphoxide (DMSO) CAS# 67-68-5 BDH Chemicals (VWR) 500 ml, analytical reagent.
In-silico prediction of the mechanism of action and toxicity of candidate compounds
The online web tool PASS [18] was used to predict the pharmacological activities of compounds, as well as their toxicity. PASS indicates the probable activity (Pa) and probable inactivity (Pi) of ‘drug-like’ substances. Using PASS, it is possible to obtain an estimated biological activity profile of a drug-like molecule using only structural formulas. Some of the predicted activities of PASS software are pharmacological effects, mechanism of action, as well as toxic and adverse effects.
In-silico predicted physicochemical properties of TKA001
ADMETlab 2.0 [32] was used to predict the physicochemical properties, drug-likeness, and toxicity of TKA001.
Molecular docking
The SMILES string of TKA001 was converted using the Open Babel tool into the PDBQT format [33]. Docking simulations were performed using PyRx [34]. The selected compound was subjected to docking with mTOR kinase, PDB: 4JSV [35,36,37]. A molecular docking simulation of protein and ligand was performed using PyRx to predict their preferential binding affinity in terms of binding energy (ΔG). Adequate spacing between the grids was ensured so that the ligands could move freely inside. One of the best conformational poses of ligand showing the least ΔG was selected for further analysis using PyMol (Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Human cell lines
Cell lines were maintained at 37°C in a 5% CO2 incubator. Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% Hyclone Cosmic Calf Serum (Cytiva) and 1% penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Cells were routinely passaged at 70% confluence. The following cell lines were used: HT1080: ATCC CCL-121, HeLa: ATCC CRM-CCL-2.
Human cell proliferation
For proliferation assays, cells were seeded in 96-well plates at 2,000 cells per well and treated with vehicle (0.1% DMSO) or TKA001. After 24 hours, cells were stained with Hoechst 33342 (Thermo Fisher Scientific, Waltham, MA, USA) at 10 µg/mL, followed by imaging using a Cytation 5 Cell Imaging Multimode Reader (BioTek, Winooski, VT, USA). Cell count was performed by automated nuclei counting using Gen5 Data Analysis Software (BioTek, Winooski, VT, USA).
Western blots
For western blots, cells were grown to 70% confluence in 6-well plates. Cells were then treated with vehicle (0.1% DMSO) or TKA001 for 2 hours, followed by UV-B irradiation (30 mj/cm2) or control (no irradiation). Thirty minutes after irradiation, cells were placed on ice, washed with cold phosphate-buffered saline (PBS) then harvested in 60 µL of cold RIPA buffer containing protease/phosphatase inhibitors (Thermo Fisher Scientific, Waltham, MA, USA). Cells were incubated on ice for 20 minutes, then centrifuged at 20000× g for 10 minutes, and supernatants were collected. Supernatants were standardized to a concentration of 2 mg/mL and combined with 6x loading buffer containing SDS, glycerol, beta-mercaptoethanol, and bromophenol blue. Samples were boiled at 100°C for 10 minutes, and 15 µL was loaded on a Tris/glycine SDS-polyacrylamide gel. Proteins were separated by SDS-PAGE, transferred to a polyvinylidene diflouride (PVDF) membrane, and immunoblotted with indicated antibodies. Primary antibodies used were pS6 (S240/244): CST 5364, p-AKT (S473): CST 4060, ATK: CST 4691, and Vinculin: CST 13901. Secondary antibody was HRP-linked anti-rabbit IgG (CST 7074). Immunoblots were imaged using an enhanced chemiluminescent detection kit (ECL, Bio-Rad) and visualized on a LiCor Odyssey Fx Imaging System.
C. elegans lifespan
TJ1060 [spe-9(hc88); rrf-3(b26)] C. elegans were grown until the gravid adult stage and synchronized by bleaching [38]. Hatched overnight cultures of L1 C. elegans were seeded onto plates covered with heat-inactivated OP50 bacteria and left at +25°C incubator for 2 days to induce temperature-sensitive spe-9(hc88) mutation to achieve sterility of eggs. Then young adult animals were transferred manually by picking 70-75 individuals onto two 6 cm plates (n ≥ 140 animals per condition) with corresponding TKA001 concentrations in both agar and heat-inactivated OP50. Further lifespan assay was performed at 20°C until death [39]. The TKA001 was dissolved in DMSO, resulting in a 0.2% DMSO final concentration in the NGM plates, and, thus, 0.2% DMSO was used as empty vehicle control. All the plates were normalized by 0.2% DMSO to exclude differences in solvent concentration. Death events were counted every second day and starting from adulthood day 12 (AD12) – on a daily basis. Worms with a vulval protrusion, matricide events, dried on the walls, and abnormally looking were censored. Kaplan-Meier estimator was used for analysis in GraphPad Prism 8 ® software. Log-Rank (Mantel-Cox) test was utilized for statistical analysis (n ≥ 100). Figure 2D: control = 128 death events, TKA001 (100 µM) = 132 death events [ + 8.9% mean lifespan increase], TKA001 (200 µM) = 132 death events [ + 4.7% mean lifespan increase]. Figure 2E: control = 137 death events, TKA001 (10 µM) = 131 death events [ + 6.3% mean lifespan increase].
Author Contributions
All authors participated in analyzing and interpreting the data. T.V. and M.R.M. performed machine learning. T.V. performed molecular dockings. M.R.M. and C.Y.E. designed the experiments. A.D. performed lifespan assays. M.R.M. performed western blots and cell survival assays. C.Y.E. and M.R.M. wrote the manuscript in consultation with the other authors. All authors have read and agreed to the published version of the manuscript.
Funding
Funding from the Swiss National Science Foundation Funding from the SNF P3 Project 190072 to CYE and AD.
Acknowledgments
We thank Ewald and MacArthur lab members for their critical reading and comments. WormBase for curated gene and phenotype information. TJ1060 strain was provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. TV, MRM, and CYE are co-founders of Tinka Therapeutics. CYE is a co-founder and shareholder of Avea Life AG, and is on the Scientific Advisory Board of Maximon AG, Biotein, Longaevus Technologies LTD, and Galyan Bio, INC.
References
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of Lifespan in Drosophila by Modulation of Genes in the TOR Signaling Pathway. Curr Biol 2004, 14, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Robida-Stubbs, S.; Glover-Cutter, K.; Lamming, D.W.; Mizunuma, M.; Narasimhan, S.D.; Neumann-Haefelin, E.; Sabatini, D.M.; Blackwell, T.K. TOR Signaling and Rapamycin Influence Longevity by Regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab 2012, 15, 713–724. [Google Scholar] [CrossRef]
- Zhang, Y.; Lanjuin, A.; Chowdhury, S.R.; Mistry, M.; Silva-García, C.G.; Weir, H.J.; Lee, C.-L.; Escoubas, C.C.; Tabakovic, E.; Mair, W.B. Neuronal TORC1 Modulates Longevity via AMPK and Cell Nonautonomous Regulation of Mitochondrial Dynamics in C. Elegans. Elife 2019, 8, e49158. [Google Scholar] [CrossRef]
- Statzer, C.; Meng, J.; Venz, R.; Bland, M.; Robida-Stubbs, S.; Patel, K.; Petrovic, D.; Emsley, R.; Liu, P.; Morantte, I.; et al. ATF-4 and Hydrogen Sulfide Signalling Mediate Longevity in Response to Inhibition of Translation or MTORC1. Nat Commun 2022, 13, 967. [Google Scholar] [CrossRef]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Müller, F. Genetics: Influence of TOR Kinase on Lifespan in C. Elegans. Nature 2003, 426, 620–620. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Landis, J.N.; Abate, J.P.; Murphy, C.T.; Blackwell, T.K. Dauer-Independent Insulin/IGF-1-Signalling Implicates Collagen Remodelling in Longevity. Nature 2015, 519, 97–101. [Google Scholar] [CrossRef]
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased Mammalian Lifespan and a Segmental and Tissue-Specific Slowing of Aging after Genetic Reduction of MTOR Expression. Cell Reports 2013, 4, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Shindyapina, A.V.; Cho, Y.; Kaya, A.; Tyshkovskiy, A.; Castro, J.P.; Deik, A.; Gordevicius, J.; Poganik, J.R.; Clish, C.B.; Horvath, S.; et al. Rapamycin Treatment during Development Extends Life Span and Health Span of Male Mice and Daphnia Magna. Sci Adv 2022, 8, eabo5482. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin Fed Late in Life Extends Lifespan in Genetically Heterogeneous Mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Strong, R.; Miller, R.A.; Bogue, M.; Fernandez, E.; Javors, M.A.; Libert, S.; Marinez, P.A.; Murphy, M.P.; Musi, N.; Nelson, J.F.; et al. Rapamycin-mediated Mouse Lifespan Extension: Late-life Dosage Regimes with Sex-specific Effects. Aging Cell 2020, 19, e13269. [Google Scholar] [CrossRef]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Baur, J.A.; Boyd, A.R.; de Cabo, R.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nelson, J.F.; et al. Rapamycin, But Not Resveratrol or Simvastatin, Extends Life Span of Genetically Heterogeneous Mice. Journals Gerontology Ser 2011, 66A, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Apelo, S.I.A.; Pumper, C.P.; Baar, E.L.; Cummings, N.E.; Lamming, D.W. Intermittent Administration of Rapamycin Extends the Life Span of Female C57BL/6J Mice. Journals Gerontology Ser 2016, 71, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Chen, D.; Riddle, D.L. The TOR Pathway Interacts with the Insulin Signaling Pathway to Regulate C. Elegans Larval Development, Metabolism and Life Span. Development 2004, 131, 3897–3906. [Google Scholar] [CrossRef] [PubMed]
- Torrence, M.E.; MacArthur, M.R.; Hosios, A.M.; Valvezan, A.J.; Asara, J.M.; Mitchell, J.R.; Manning, B.D. The MTORC1-Mediated Activation of ATF4 Promotes Protein and Glutathione Synthesis Downstream of Growth Signals. Elife 2021, 10, e63326. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Giudice, G.D.; Lattanzi, M.; Valiante, N.M.; Praestgaard, J.; Huang, B.; Lonetto, M.A.; Maecker, H.T.; Kovarik, J.; Carson, S.; et al. MTOR Inhibition Improves Immune Function in the Elderly. Sci Transl Med 2014, 6, 268ra179. [Google Scholar] [CrossRef]
- Chung, C.L.; Lawrence, I.; Hoffman, M.; Elgindi, D.; Nadhan, K.; Potnis, M.; Jin, A.; Sershon, C.; Binnebose, R.; Lorenzini, A.; et al. Topical Rapamycin Reduces Markers of Senescence and Aging in Human Skin: An Exploratory, Prospective, Randomized Trial. Geroscience 2019, 41, 861–869. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the Pharmaceutical Industry: New Estimates of R&D Costs. J Health Econ 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of Activity Spectra for Biologically Active Substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR Complexes, Only One of Which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control. Mol Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat Rev Mol Cell Bio 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Pogodin, P.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A Freely Available Web-Service for in Silico Prediction of Human Cell Line Cytotoxicity for Drug-like Compounds. Plos One 2018, 13, e0191838. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.K.; Sewell, A.K.; Wu, Z.; Han, M. TOR Signaling in Caenorhabditis Elegans Development, Metabolism, and Aging. Genetics 2019, 213, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Taubert, S.; Crawford, D.; Libina, N.; Lee, S.; Kenyon, C. Lifespan Extension by Conditions That Inhibit Translation in Caenorhabditis Elegans. Aging Cell 2007, 6, 95–110. [Google Scholar] [CrossRef]
- Pan, K.Z.; Palter, J.E.; Rogers, A.N.; Olsen, A.; Chen, D.; Lithgow, G.J.; Kapahi, P. Inhibition of MRNA Translation Extends Lifespan in Caenorhabditis Elegans. Aging Cell 2007, 6, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. MTORC1 Controls Fasting-Induced Ketogenesis and Its Modulation by Ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Ham, D.J.; Börsch, A.; Lin, S.; Thürkauf, M.; Weihrauch, M.; Reinhard, J.R.; Delezie, J.; Battilana, F.; Wang, X.; Kaiser, M.S.; et al. The Neuromuscular Junction Is a Focal Point of MTORC1 Signaling in Sarcopenia. Nat Commun 2020, 11, 4510. [Google Scholar] [CrossRef] [PubMed]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.A.; Topisirovic, I.; Hulea, L. MTOR as a Central Regulator of Lifespan and Aging. F1000research 2019, 8, F1000. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. MTOR Is a Key Modulator of Ageing and Age-Related Disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef]
- Cohen, F.; Bergeron, P.; Blackwood, E.; Bowman, K.K.; Chen, H.; DiPasquale, A.G.; Epler, J.A.; Koehler, M.F.T.; Lau, K.; Lewis, C.; et al. Potent, Selective, and Orally Bioavailable Inhibitors of Mammalian Target of Rapamycin (MTOR) Kinase Based on a Quaternary Substituted Dihydrofuropyrimidine. J Med Chem 2011, 54, 3426–3435. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res 2021, 49, gkab255. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Chemical Biology, Methods and Protocols. Methods Mol Biology 2014, 1263, 243–250. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. MTOR Kinase Structure, Mechanism and Regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Costanzo, L.D.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res 2020, 49, D437–D451. [Google Scholar] [CrossRef]
- Teuscher, A.C.; Statzer, C.; Pantasis, S.; Bordoli, M.R.; Ewald, C.Y. Assessing Collagen Deposition During Aging in Mammalian Tissue and in Caenorhabditis Elegans. Methods Mol Biology Clifton N J 2019, 1944, 169–188. [Google Scholar] [CrossRef]
- Statzer, C.; Jongsma, E.; Liu, S.X.; Dakhovnik, A.; Wandrey, F.; Mozharovskyi, P.; Zülli, F.; Ewald, C.Y. Youthful and Age-related Matreotypes Predict Drugs Promoting Longevity. Aging Cell 2021, 20, e13441. [Google Scholar] [CrossRef]
Figure 1.
Characterization of mTOR inhibitor TKA001. (A) In-silico prediction of toxicity of mTOR inhibitor candidate compounds. (B) Lead candidate TKA001 (C19H23N5O3). (C,D) In-silico predicted physiochemical properties of TKA001. (E) Molecular docking of TKA001 and mTOR kinase.
Figure 1.
Characterization of mTOR inhibitor TKA001. (A) In-silico prediction of toxicity of mTOR inhibitor candidate compounds. (B) Lead candidate TKA001 (C19H23N5O3). (C,D) In-silico predicted physiochemical properties of TKA001. (E) Molecular docking of TKA001 and mTOR kinase.
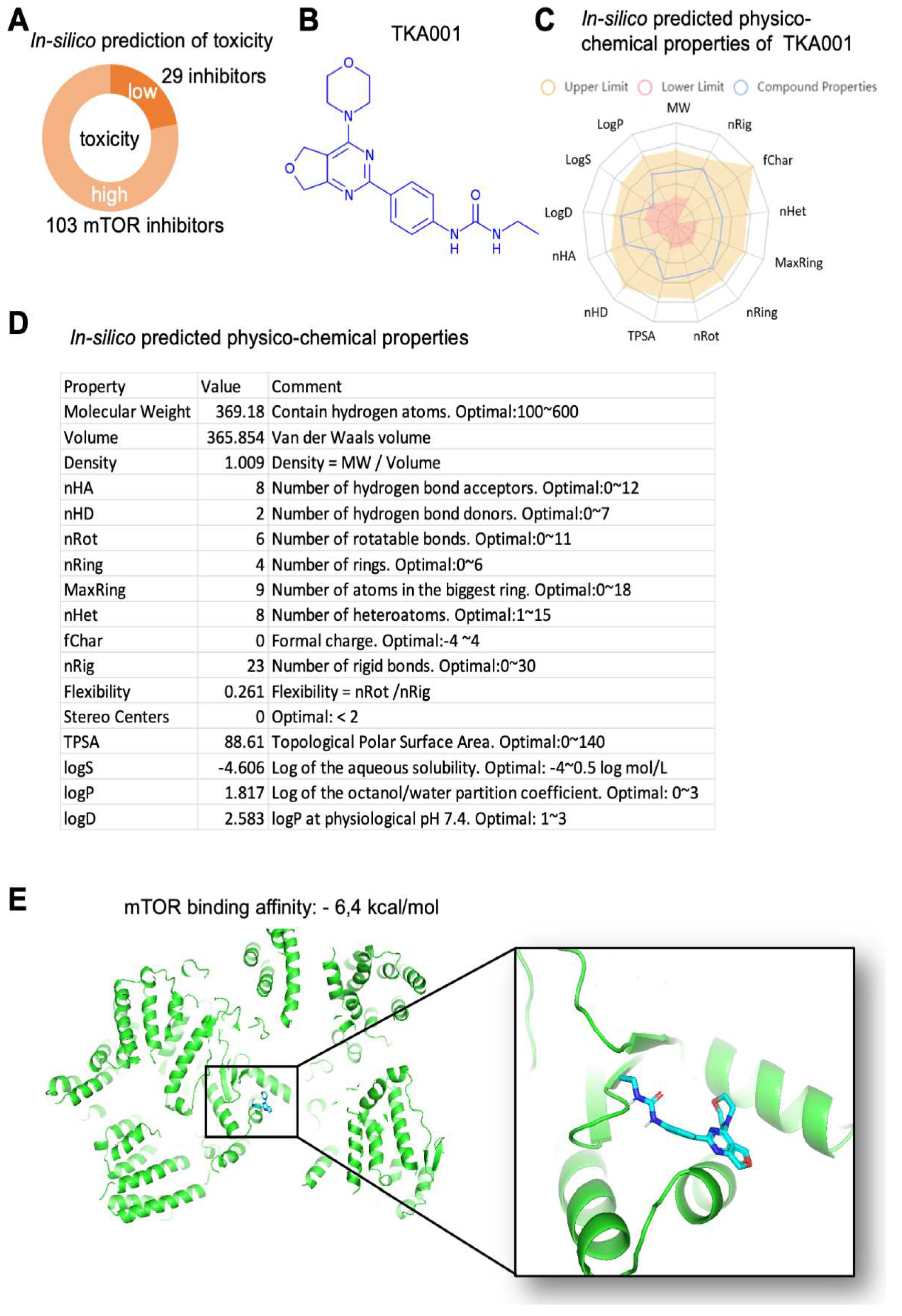
Figure 2.
In-vitro and in-vivo validation of mTOR inhibitor TKA001. (A) Western blot of HT1080 cells pre-treated with increasing concentrations of TKA001 with or without UV-B irradiation. Phospho-S6 (S240/244), phospho-AKT (S473), total AKT, and vinculin loading control. (B) Proliferation assays of TKA001 treatment on HT-1080 cells. Half maximal inhibitory concentration (IC50) = 200 nM. (C) Proliferation assays of TKA001 treatment on HeLa cells. IC50 = 1 µM. (D,E) Feeding 10, 100, or 200 µM TKA001 increased the lifespan of C. elegans. Two independent biological trials. Control = 0.2% DMSO. p-value determined with Log Rank.
Figure 2.
In-vitro and in-vivo validation of mTOR inhibitor TKA001. (A) Western blot of HT1080 cells pre-treated with increasing concentrations of TKA001 with or without UV-B irradiation. Phospho-S6 (S240/244), phospho-AKT (S473), total AKT, and vinculin loading control. (B) Proliferation assays of TKA001 treatment on HT-1080 cells. Half maximal inhibitory concentration (IC50) = 200 nM. (C) Proliferation assays of TKA001 treatment on HeLa cells. IC50 = 1 µM. (D,E) Feeding 10, 100, or 200 µM TKA001 increased the lifespan of C. elegans. Two independent biological trials. Control = 0.2% DMSO. p-value determined with Log Rank.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



