
Submitted:
21 February 2023
Posted:
23 February 2023
You are already at the latest version
Abstract
The ever-growing repertoire of genomic techniques continues to expand our understanding of true diversity and richness of prokaryotic genomes. Riboproteogenomics laid the foundation for dynamic studies of previously overlooked genomic elements. Most strikingly, bacterial genomes were revealed to harbour robust repertoires of small open reading frames (sORFs) encoding a diverse and broadly expressed range of small proteins, or sORF-encoded polypeptides (SEPs). In recent years, continuous efforts led to great improvements in annotation and characterization of such proteins, yet many challenges remain to fully understand the pervasive nature of small proteins and their impact on bacterial biology. In this work we review recent developments in the dynamic field of bacterial genome reannotation, catalogue important biological roles carried out by small proteins and identify challenges obstructing the way to full understanding of these elusive proteins.
Keywords:
small ORF encoded polypeptides (SEPs)
; riboproteogenomics
; bacterial pathogens
; protein tagging
; proteomics
Glossary
Initiating ribosome: a ribosome which integrates the interaction of initiation factors, mRNA and initiator tRNA (Formyl-Met-tRNA) for optimal start codon selection, and concomitantly reading frame determination for translation of an mRNA sequence into a protein product [1].
N-terminal proteomics (N-terminomics): the discipline of mapping protein N-termini of a given proteome sample, also known as the N-terminome. Next to representing the start of proteolytically processed proteins, N-termini may also serve as proxies of translation initiation, thereby greatly contributing to the process of genome annotation.
Proteoforms: multiple molecular forms of proteins that describe the biological protein variability at the level of protein primary structure and thus protein isoforms that are expressed from a single gene. Therefore, proteoforms generally contribute to the increased complexity of proteomes [2]. While post-translational modifications can give rise to expressed proteoforms, N-terminal proteoforms specifically point to proteoforms generated co-translationally by alternative translation initiation and/or differential co-translational initiator methionine (iMet) excision.
Retapamulin (RET): a member of the pleuromutilins, a class of antibiotics known for their ability to act as bacterial protein synthesis inhibitors by their specific interaction with the 50S subunit of bacterial initiating ribosomes. Retapamulin is known to obstruct the first steps of translation elongation thereby arresting initiating ribosomes at start codons. By complementing ribosome profiling (Ribo-seq) with retapamulin treatment (Ribo-RET), the signal-to-noise ratio can be improved specifically for calling of (alternative) translation start sites [3]. However, Gram-negative bacteria, including the important model organisms Escherichia coli (E. coli) and Salmonella Typhimurium (S. Typhimurium), are partially protected against retapamulin action thanks to their TolC multidrug efflux pump, requiring tolC deletion strains for optimal Ribo-RET performance [4]. Lefamulin, however, is a newer pleuromutilin which might eventually outcompete retapamulin for Ribo-RET purposes as for this drug higher activity has been reported in Gram-negative bacteria [5].
Riboproteogenomics: a term used to refer to the combination of systematic complementary ribosome profiling, proteomics (e.g. N-terminal proteomics) and genomics for studying translation (initiation) landscapes [6].
Ribosome profiling (by sequencing) (Ribo-seq): the identification of open reading frames (ORFs) by deep-sequencing of ribosome-protected mRNA fragments [7]. By relying on ribosome-mRNA binding, Ribo-seq offers a genome-wide view on the translational landscape of an organism (translatome). The standard ribosome profiling procedure in prokaryotes employs chloramphenicol treatment to specifically stall elongating ribosomes [8], but this step can be omitted by performing flash-freezing of the samples [5].
Shotgun proteomics: composition analysis of complex peptide samples (obtained either through digestion or small polypeptide-enrichment) through the complementary use of high-performance liquid chromatography (HPLC) and mass spectrometry (MS), for peptide separation and peptide/protein identification, respectively.
sORFeome: the small ORF collection of the ORFeome, which refers to the totality of ORFs harboured by a species’ genome. In this review, the sORFeome considers all ORFs shorter in length than 300 base pairs (bp).
sORFs and SEPs: small in size but not in importance
In the continuous effort to improve bacterial genome annotations, the development of ribosome profiling by next-generation sequencing [7], Ribo-seq (see Glossary) in short, allowed the recent discovery of a plethora of small open reading frames (sORFs). Classified as open reading frames built of no more than 300 nucleotides (nt.), these newly discovered genes potentially give rise to their encoded small proteins; referred to as sORF encoded polypeptides (SEPs). By providing direct evidence of many sORFs harbouring ribosomal activity [7,9,10], Ribo-seq freed these specific ORFs from their status as ‘noise’ during the process of gene prediction and genome annotation. Currently, multiple Ribo-seq datasets have been published for model bacterial species like the Gram-negative Escherichia coli (E. coli) [3,11,12,13] and the Gram-positive Bacillus subtilis (B. subtilis) [14]. Similar efforts were also reported for specific bacterial human pathogens including the model species Salmonella enterica subspecies enterica serovar Typhimurium (S. Typhimurium) [15,16,17] and more recently for Streptococcus pneumoniae (S. pneumoniae) [18], Mycobacterium tuberculosis (M. tuberculosis) [19], Staphylococcus aureus (S. aureus) [20] and Campylobacter jejuni (C. jejuni) [21]. A comprehensive overview of available prokaryotic ribosome profiling studies has been compiled by Vazquez-Laslop et al [5] and ribosome profiling traces corresponding to (some of) these and other studies can be consulted via the online genome browser GWIPS-viz [22].
Since the aforementioned studies report on the discovery of novel, putative sORFs, these recent efforts all contributed to a now exhaustive list of hypothetical bacterial SEPs. In aid of gene annotation, Ribo-seq studies provide translational evidence for only a small subset of in silico predicted sORFs, making their consideration in genome (re-)annotation efforts more straightforward. However, functional characterization has only been reported for a small portion of putative sORFs and their encoding SEPs, leaving an enormous world of the sORFeome (see Glossary) uncharted. With documented bacterial SEP functions falling within diverse categories of basic and essential bacterial physiology [23,24,25,26,27,28] as well as infection biology [29,30,31,32], the need for more large-scale validation and functional characterization efforts is high. In this context, it is noteworthy that difficulties in biochemical detection and therefore validation of SEPs are known and have been extensively documented [33,34,35], but that also recent bacterial SEP validation studies fail to fully address many of the challenges in small protein detection [35], such as their proposed low expression or low stability [36]. Nonetheless, as expression detection is a prerequisite for functional investigations, further improvement in SEP detection might turn out to be of great value to expand our current understanding of bacterial (infection) biology. Fundamentally, because of the lack of standard work-flows to go from computationally predicted sORFs to functionally annotated SEPs, a whole piece might be missing out of the puzzle the bacterial life is known to be.
Studying bacterial biology in the genomics era
Sequencing revolution demands annotation evolution
The initial genome sequencing efforts were mainly driven by the desire to broaden knowledge on bacterial pathogens, with the very first genome sequence, which is 25 years old by now, originating of the free-living bacterium Haemophilus influenzae (H. influenzae) [37,38]. This milestone for the microbiology field was established through whole genome shotgun sequencing (WGS), a technique that heralded the first-generation sequencing revolution [37,38,39]. In the meantime, advances in sequencing techniques under the form of high-throughput, next-generation sequencing (NGS) and supporting bioinformatics have resulted in the exponential increase in the number of bacterial genomes available to date by significantly lowering both sequencing time and costs [37,38,39]. This progress for genomics was rightly marked as the second or NGS revolution, but some of the remaining pitfalls were recently surpassed with the advent of high-resolution, single-molecule, long-read sequencing, permitting genome assemblies of unparalleled quality initiating the third revolution [40].
Genome sequences serve as a starting point for a better understanding of bacterial functioning, evolution and interaction with the environment. These studies are of special importance for (human) (opportunistic) pathogenic bacteria including both culturable and non-culturable bacteria (e.g. Eubacterium saphenum (Eubacteriaceae), a species of the periodontitis oral microbiome [41]). Moreover, the ever-augmenting number of available gene sequences evoked a switch from forward to reverse genetics approaches [42]. However, the massive accumulation of genome sequences entailed a new challenge. Data on microbial genomes are now being generated faster than they can be manually processed to extract valuable information from, making automatization of annotation an urging challenge for the current revolution in sequencing technology. The need for more accurate annotations of bacterial genomes is reflected by the numerous recent tools for annotation purposes reported like DFAST [43] and Bakta [44] focussing on annotation speed, and Pseudofinder for an improved discrimination between genes and pseudogenes [45], besides the continuous evolution of commonly employed toolkits like PGAP [46]. Further, (ribo)proteogenomics (see Glossary) has become a fixed value for genome annotation as it can already link gene prediction to expression [6,35,47,48].
Automatic genome annotation requires manual curation: a frustrating paradox
The applied principles in popular, prokaryotic annotation tools like RAST, Prokka and PGAP [49] illustrate the central idea of automatic genome annotation, being the search for homology with annotated genes, proteins or domains in databases. Hence, annotation of new genomes is strongly influenced by the information inherently present in available databases and consequently, is steered by the annotation principles that have been used thus far. Once introduced, annotation errors propagate in these databases that are assumed to assist annotation [50]. This situation illustrates why the ‘automatic’ character of used annotation pipelines is not that absolute, as human intervention or metadata is still heavily required to manually curate their performance [51]. Lobb et al demonstrated the incompleteness of bacterial genome annotations by determining the percentage of the average bacterial proteome that can be functionally annotated based on homology with database annotated proteins and domains, indicating a range between 52% and 79%, depending on the annotation tool used [52]. Self-evidently, because of research bias, the contrasting status of model and understudied bacterial species plays a pivotal role in the value of this percentage, leaving no less than 90% of the proteome unannotated for some of the latter class of intriguing bacterial species [52].
sORFs: the weak spots of automated bacterial genome annotation
The annotation bias is especially challenging when it comes to the detection of sORFs. These coding sequences (CDS) of arbitrarily no more than 150-300 nt. encode small proteins or SEPs, with the interpretation for ‘small’ ranging from smaller than (or equal to) 50 [53,54] to 100 amino acids (AA) [55]. SEPs distinguish themselves from canonical small peptides in the fact that their origin is translational and no proteolytic processing step is required to make them this small. In this way, the typical length cut-offs for gene prediction and annotation, chosen based on the strong belief that genes should be of sufficient length to be functional, turned out to be too short-sighted thereby obstructing sORF identification. Within existing genome annotations, a grant majority of sORFs encode ribosomal SEPs, which are significantly more conserved than newly discovered sORFs [33]. One of the explanations for their generally lower conservation can be found in their origin, being constant genomic evolution events from larger genes [33].
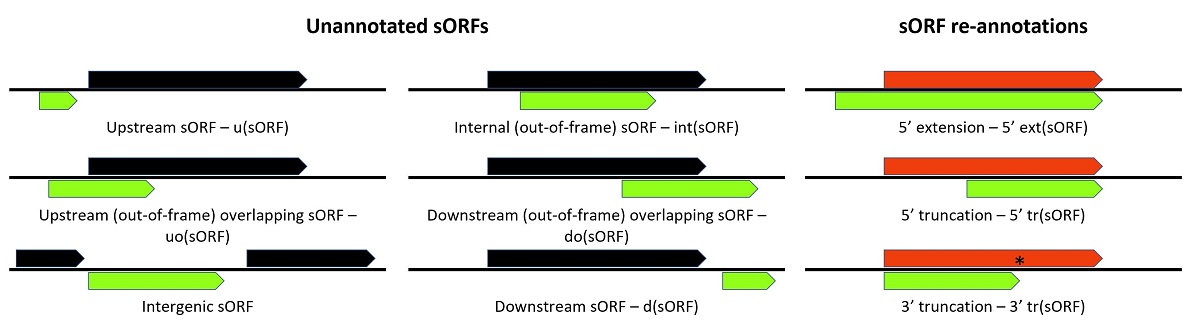
In line with previous ORF annotations, newly discovered sORFs can be classified as intergenic sORFs, upstream (overlapping) (regulatory) sORFs (u(o)sORFs), internal (out-of-frame) sORFs (intsORFs) or downstream (out-of-frame) (overlapping) (regulatory) sORFs (d(o)sORFs); a classification based on the relation between the genome-orientation of the newly discovered sORF and existing gene annotations (Figure 1A) [33,36,56]. For bacterial genomes, the genomic positioning of annotations is especially informative in case of polycistronic mRNAs, frequently encoding gene product(s) with a strong interplay.
Figure 1.
Small ORF (sORF) re-annotations and categorization of unannotated sORFs discovered by riboproteogenomics. A. The annotation of newly discovered sORFs is based on the relation between the genomic location of the novel sORF and existing (s)ORF annotations. Especially for the typical bacterial polycistronic gene organization, positional ORF annotations in the context of transcripts are meaningful as the interaction of the resulting gene products can be regulatory in nature. B. The implementation of riboproteogenomics for genome annotation can also result in the (conditional) re-annotation of previously annotated genes. Under given circumstances, ORFs can appear as 3’ (3’tr(sORF)) or 5’ (5’tr(sORF)) truncated variants, meaning that ORFs can turn into sORF annotations, or sORFs into shorter sORF annotations (resulting in N-terminally truncated SEP translation products). For sORFs, 5’ extensions (5’ext(sORF)) can exist still resulting in N-terminally extended SEP translation products.
Figure 1.
Small ORF (sORF) re-annotations and categorization of unannotated sORFs discovered by riboproteogenomics. A. The annotation of newly discovered sORFs is based on the relation between the genomic location of the novel sORF and existing (s)ORF annotations. Especially for the typical bacterial polycistronic gene organization, positional ORF annotations in the context of transcripts are meaningful as the interaction of the resulting gene products can be regulatory in nature. B. The implementation of riboproteogenomics for genome annotation can also result in the (conditional) re-annotation of previously annotated genes. Under given circumstances, ORFs can appear as 3’ (3’tr(sORF)) or 5’ (5’tr(sORF)) truncated variants, meaning that ORFs can turn into sORF annotations, or sORFs into shorter sORF annotations (resulting in N-terminally truncated SEP translation products). For sORFs, 5’ extensions (5’ext(sORF)) can exist still resulting in N-terminally extended SEP translation products.

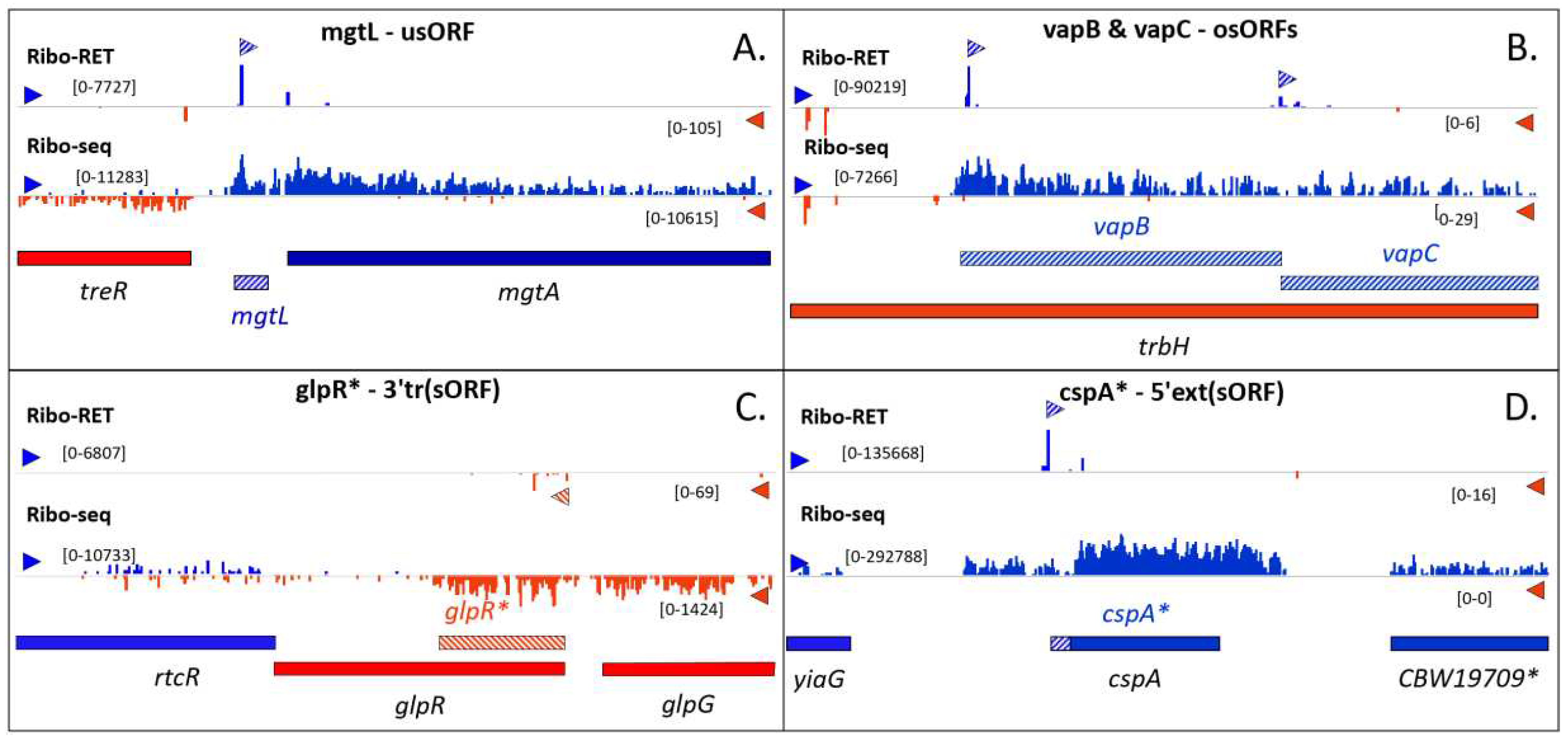
Figure 2.
Riboproteogenomics-supported novel and re-annotated Salmonella sORFs. Ribo-seq/Ribo-RET profiles of the Salmonella Typhimurium strain SL1344 are shown. A. MgtL was delineated as a new sORF in the SL1344 genome, but was found to have matching annotations in related genomes [15]. Moreover, peptide evidence is available for this MgtA regulatory leader peptide [15]. B. VapB and VapC, an upstream and downstream osORF respectively, take part in the plasmid-encoded Vap toxin-antitoxin system and were also only recently annotated in the SL1344 genome [57]. C. For the pseudogene glpR, a 3’ truncated version of GlpR (GlpR*) was predicted with the same start site [57]. D. CspA was found to have an in-frame upstream alternative start encoding a 5’ extended proteoform (CspA*), supported by peptide evidence [15]. * is indicative of an ORF re-annotation.
Figure 2.
Riboproteogenomics-supported novel and re-annotated Salmonella sORFs. Ribo-seq/Ribo-RET profiles of the Salmonella Typhimurium strain SL1344 are shown. A. MgtL was delineated as a new sORF in the SL1344 genome, but was found to have matching annotations in related genomes [15]. Moreover, peptide evidence is available for this MgtA regulatory leader peptide [15]. B. VapB and VapC, an upstream and downstream osORF respectively, take part in the plasmid-encoded Vap toxin-antitoxin system and were also only recently annotated in the SL1344 genome [57]. C. For the pseudogene glpR, a 3’ truncated version of GlpR (GlpR*) was predicted with the same start site [57]. D. CspA was found to have an in-frame upstream alternative start encoding a 5’ extended proteoform (CspA*), supported by peptide evidence [15]. * is indicative of an ORF re-annotation.
sORFs were generally overlooked until increasingly more SEPs were identified – rather by chance – across all domains of life as well as viruses [58]. Moreover, sORFs and their encoding SEPs turned out to be of considerable biological importance for the respective organisms, further strengthened by Lluch-Senar et al who identified the genomic class of sORFs as being the most frequently essential one in case of the genome-reduced bacterium Mycoplasma pneumoniae [59]. Bacterial SEPs are, among other functions, known to be involved in basic (essential) processes underlying bacterial functioning, including cell division (e.g. MciZ [23]), transport of molecules (e.g. KdpF [25]) and signal transduction (e.g. SafA [27]) and to act as chaperones (e.g. MntS [60]). The discovery of the unexpected coding potential of bacterial sRNAs – not surprisingly – took place through mining the E. coli genome [53,61,62]. In this regard, the bacterial operon gene structure surely deserves some credit for the initial, unintended discovery of the functional potential of small proteins. In 1999, Gaβel et al discovered, at that time, the smallest E. coli protein KdpF (29 AA) through extensive examination of the K+-transporter complex encoding KdpABC operon, and co-purified KdpF with the complex emphasizing the possibility for small proteins to take active roles in protein complexation and bacterial functioning [63].
SEPs as a novel research hotspot for the study of bacterial biology
SEPs as accomplices in bacterial (infection) biology
With existing examples of SEPs acting as virulence factors and toxins [64,65,66], functional investigations of SEPs belonging to the proteome of pathogenic bacterial species paved the way for research endeavours focussing on SEPs as potential novel therapeutic targets. For Listeria monocytogenes, N-terminal proteomics (N-terminomics) (see Glossary) linked Prli42 (31 AA) to survival in macrophages by acting as a stressosome player [31,67]. Another human pathogen, S. aureus, expresses the membrane peptide toxin PepA1 (31 AA) that has been hypothesized to be implicated in regulation of survival after internalization into immune cells [32]. The 59 AA SEP Yp1 encoded by the bubonic plague pathogen Yersinia pestis (Y. pestis), was found to regulate virulence through modulation of the expression of type III secretion system (T3SS) components and in the same study, a high conservation of SEPs was noted between different pathogenic Yersinia strains, so SEPs can be expected to confer pathogenesis-related benefits [48]. Intriguingly, numerous novel SEPs are predicted transmembrane proteins [13,18,68]. The hydrophobic nature of this specific SEP class [69,70] has already been postulated to be implicated in host-pathogen interactions exerted via cell-cell interactions [48].
The SEP arsenal of Salmonella
S. Typhimurium has served as a model species for the study of SEP expression in general and within the framework of bacteria-host interactions [15,16,17,35,71]. The divergence of the genus Salmonella from E. coli, estimated to have taken place 160-120 million years ago [72], was established through multiple horizontal gene transfer events, which left their marks in the genome in the shape of Salmonella pathogenicity islands (SPI) and gave the genus the capacity to develop into the successful pathogen as we know it today. In addition to local gastroenteritis, S. Typhimurium leads to a systemic disease in mice reminiscent of typhoid fever caused exclusively in humans by Salmonella enterica subspecies enterica serovar Typhi (S. Typhi). As such, S. Typhimurium infection in mice has been extensively used as a model system mimicking human typhoid fever [73].
For Salmonella, some of the few functionally characterized SEPs represent convincing links with virulence. As a first Salmonella SEP study case, MgtR (30 AA) was shown to be indirectly involved in intramacrophage survival through regulation of the virulence protein MgtC [29]. Further, two cold shock proteins, CspC (69 AA) and CspE (70 AA), were proven to be indispensable for Salmonella pathogenicity [74]. For MgrB (47 AA), it has been shown that it binds and thereby inhibits the PhoQ kinase [30] which takes part in the PhoPQ two-component system involved in regulation of virulence. Recently, a direct link between MgrB and virulence has been demonstrated through creation of an ∆mgrB mutant, which failed in infecting macrophages and epithelial cells [17]. What’s more, Venturini et al showed more than half of the SEPs identified in their study to be differentially expressed upon infection, which is evident in case of the T3SS apparatus or injectisome protein members SsaS (88 AA) and SsaI (82 AA) [17]. Accordingly, a great deal of studies found the expression of a significant part of previously unannotated sORFs of bacterial pathogens to follow infection-relevant expression patterns [15,16,35].
Ribo-seq: a game changer for genome (re)annotation
The intriguing bacterial SEP functions reported highlight the need for new advances to ‘enrich’ bacterial genome annotations for sORFs. Ribo-seq revolutionized the study of translation by deep sequencing of ribosome-protected mRNA fragments [75]. Ribosomes cover approximately 30 nt. when bound onto mRNA, causing these ‘ribosome protected parts’ to be resistant towards nuclease degradation (Figure B). Sequencing of these footprints gives clues on the whereabouts of ribosomes along translated mRNAs while additionally enabling to demarcate boundaries of translation, and thus delineation of translated ORFs. Recently, Ribo-seq was tailored towards identification of prokaryotic translation initiation sites (TISs) by stalling initiating ribosomes (see Glossary) [3] through the action of retapamulin (RET) (see Glossary) (Ribo-RET) [7] or alternatively, the newer pleuromutilin lefamulin, as especially in Gram-negative bacteria lefamulin exceeds retapamulin activity [5].
The more precise TIS delineation further enabled the discovery of overlapping (Figure 2B) (s)ORFs besides the discovery of ORFs translated as distinct protein isoforms or N-terminal proteoforms (see Glossary) (Figure 2C-D) [3,47], features that challenge standard annotation algorithms [76,77] and that are widespread among sORFs. Ribo-RET together with Ribo-seq data are at the basis of (conditional) gene reannotations [15,35,57]. Besides revealing differential expression, conditional Ribo-seq and -RET profiles (e.g. when comparing diverse bacterial growth conditions) can further disclose the existence of (conditional) gene extensions and truncations by showing differential Ribo-seq coverage patterns (3’ truncations and 5’ extensions) or alternative translations starts (5’ truncations and 5’ extensions) across the tested conditions (Figure 2D). More recently, Ribo-seq protocols were developed to search the genome for ribosomal activity at stop codons (Figure 3C) using the terminating ribosome bound release factor sequestrator apidaecin (API) (Ribo-API) in combination with puromycin, the latter to remedy the obstacles of stop codon read through and ribosome queuing inherent to the use of API [5,21,36].
Since ribosomal protection does not necessarily point to translation, combining Ribo-RET with Ribo-API data (Figure 3) may prove valuable for the finding of truly translated ORFs [36], while additionally enabling the discovery of translational particularities such as ribosomal frameshifting events (e.g. intsORF in E. coli sfsA [4]). Ribo-seq data in turn can fuel de novo machine learning algorithms, like ribosome profiling assisted (re)annotation (REPARATION) [15] and the modular algorithm smORFer [20] for the delineation of translated prokaryotic ORFs. In particular for sORFs, that are so difficult to find in genomes through standard annotation tools, Ribo-seq has been proven instrumental to uncover their translation potential [5,15,16,18,35]. SmProt offers a dedicated platform for the structured storage of SEPs from diverse model organisms, including E. coli SEPs, which have been experimentally or computationally identified (by Ribo-seq) [78]. As befit every technical application, Ribo-seq has its difficulties and shortcomings [79] and SEP detection by means of (immuno)blotting, most commonly following epitope tagging, often serves as a sole means of experimental, biochemical validation of Ribo-seq-derived newly discovered SEPs and can therefore be used to filter out likely false positive SEP candidates [80].
SEPs: the thorns in the eye of standard protein detection methods
Empirical SEP discovery is hindered by biochemical peculiarities
From a biochemical perspective, SEPs are inherently more difficult to study than average sized proteins, a statement also applying to proteins significantly larger than average. Traditional two-dimensional gel electrophoresis (2DE), a technique profiting from the charge- and molecular weight (MW)-based separation of proteins respectively by isoelectric focusing (IEF) and sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), fails in detecting proteins of extreme sizes, either at the extreme large or short end of the spectrum [81,82], while the category of larger proteins in regular gel-free shotgun proteomics (see Glossary) is rather overrepresented by the theoretical increased peptide coverage. The relative higher hydrophobicity indices in the class of recently discovered SEPs [35,69,70] on the other hand, offers yet another explanation for their absence on 2DE gels [81,82,83] and proteomics datasets in general. The fact that nine out of the ten most hydrophobic Salmonella proteins fall within the category of SEPs definitely supports the previous observation [35].
With respect to mass spectrometry (MS), the short size of SEPs heavily constraints the number of peptides produced after trypsin digestion [35], as this scarcity of peptides is outnumbered by the numerous peptides originating from larger non-SEPs in the complete pool of tryptic peptides. While SEPs with lengths shorter than 100 AA are accountable for 10% of the annotated S. Typhimurium proteome, the theoretically identifiable tryptic peptides originating from SEPs merely compose 2,5% of the totality of identified tryptic peptides in Salmonella [35]. The significant technical limitations that SEPs bring along for empirical protein discovery methods like 2DE and MS, are further accountable for the long-time ignorance towards SEPs, again explaining the underrepresentation of sORFs in genome annotations.
Experimental SEP validation suffers the same flaws
Independent from their empirical discovery, the aforementioned SEP-specific peculiarities have also hindered experimental validation, either through MS- or blotting-based detection, and even Ribo-seq-based sORF predictions. Computational analysis of riboproteogenomics data on putative translated sORFs and identified SEPs provided insights into the intrinsic MS-detectability of SEPs with specific attention for correlations with SEP size, abundance, stability and hydrophobicity [35]. Based on an S. Typhimurium dataset of complementary translatomics (Ribo-seq and -RET) and shotgun proteomics data [6], AP3 - an algorithm designed for the prediction of MS-detectability of theoretical peptides [84] - was implemented in an attempt to explain the obvious discrepancy between these two experimental omics datasets, i.e. the hits of the proteomic pipeline only covered 65% of the by Ribo-seq identified translated proteome. The trend observed showed a clear correlation between the number of theoretical detectable peptides and the length of the protein from which the theoretical peptide descends, a conclusion logically linking SEP detection difficulties to size.
Protocols aiming at high-molecular-weight protein depletion [85] or low-molecular-weight protein enrichment [34] have been proposed to increase peptide identification rate and coverage of the limited theoretically identifiable peptide arsenal originating from SEPs [86], but these technologies forego the quantitative aspect of proteomics data and are therefore not generally applicable. Also the use of a more diverse set of MS-sequencing proteases for proteome digestion might benefit SEP identification through increased sequence coverage as was done during a recent proteogenomic study of the Y. pestis genome [48]. For example, for S. Typhimurium, choosing chymotrypsin over the standardly used trypsin, 30% more SEPs could in theory be picked up through shotgun proteomics [35]. Do’s and don’ts for MS-based small protein discovery were comprehensively reviewed by Ahrens et al [87].
Unlike the proteome-wide characteristic of MS, immunoblotting is a common go-to for detection and quantification of epitope-tagged proteins, but here, the initial fractionation of the proteins by means of 1D SDS-PAGE, is already troublesome when dealing with SEPs [83] as mentioned for 2DE [81,82]. On top, the ensuing blotting step is also problematic as the small size of the SEPs permits them to more easily move through the blotting membrane, a phenomenon known as membrane blow through [88]. Moreover, the associated incubation steps for the purpose of immunodetection, which often take place under shaking conditions in voluminous incubation and washing solutions, make the conventional immunoblotting procedure far less favourable for SEP detection. Low MW proteins distinguish themselves under these conditions by the ability to easily detach from the membrane and to get lost in the discarded incubation fluids [89].
All considering, SEPs can generally blame their small sizes for giving gene prediction, expression and validation analyses a hard time. Some voices, however, state that sORFs specifically come with low expression levels translating into the low abundance of SEPs [16,68,90,91] and on top of that, SEPs are postulated to be highly unstable because of rapid SEP degradation [16,19,36], features that are all believed (in)direct consequences of the small sizes of SEPs. Experiments exploring the positive effect of the ClpP protease inhibitor bortezomib on blotting-based SEP expression validation attempts seemed to corroborate the SEP instability assumption as inhibitor usage allowed the blotting-based validation of 3 additional SEPs under study [36]. A more critical view on the presented data however should acknowledge a more general instead of a SEP-specific protein stabilization of the compound [35]. Nonetheless, conformational studies of bacterial and archaeal SEPs by means of NMR spectroscopy may be in support of this decreased stability assumption as the results suggested the majority of studied SEPs to go through life without a well-defined structure [92] and intrinsically disordered proteins have been experimentally linked to higher proteolytic degradation susceptibility [93].
Contrastingly, based on bioinformatics predictions, Kubatova et al reported that folding of the SEPs might require complexation and so that not all these apparently unstructured SEPs are intrinsically disordered under physiological relevant conditions [92]. The fact that many SEPs have been found to engage in larger cytosolic and membrane protein complexes is further supportive of this [17,54]. New light on this discussion was moreover shed by the use of a multivariate logistic regression model for the prediction of SEP MS-detection probability including the number and detectability (AP3 score) of SEP tryptic peptides and translational abundance (Ribo-seq expression values in RPKM), stability (instability index) and hydrophobicity of the SEPs. Here, 75% of variation in the model was explained by the scarcity of (unique) tryptic peptides and poor peptide detectability as the major factors limiting MS-detectability of SEPs [35], at least pointing towards stability not being the main driver when considering detection of the SEPs under study.
Current trends in dealing with sORFs and their encoded SEPs
State-of-the-art in the genomic discovery of sORFs
Currently, Ribo-seq is considered the most comprehensive method to scan genomes for expressed sORFs (Table 1). Ribo-seq offers a plethora of advantages over all other techniques that have been exploited to improve genome annotations, with its main strengths being the genome-wide and high-throughput character complemented with its independence from existing annotations [17]. Moreover, the wealth of knowledge obtained on sORFs on behalf of Ribo-seq approaches created opportunities for the development of dedicated (s)ORF prediction algorithms [94], like REPARATION [15]. The wealth of data on putative translated sORFs, however, brings the strong need for experimental validation for Ribo-seq (and computationally) predicted sORFs [80] as mRNA protection per se cannot be considered as a proxy for SEP identification. Bluntly put, Ribo-seq provides direct evidence of the interaction between a transcript and a ribosome, whereas MS and alternative protein-based validation can truly relate transcripts with their ultimate products – translated proteins.
Viewing the clear and circumventable irreconcilabilities between SEPs and MS- or (immuno)blotting-based expression validation (Table 1) [35,83,88,89] and because of the computational complications inherent to Ribo-seq data analysis [95], it can be stated that comprehensive and robust sORF and SEP detection technologies do not exist for the time being. This is especially so in the case of bacterial Ribo-seq data, because of the general poorer resolution and resulting inadequacy of translation-specific features like its inherent triplet periodicity [79]. It is well-established that ribosomal footprints generated in bacterial samples display less consistent lengths, a phenomenon attributed to both intrinsic properties of bacterial ribosomes and sequence specificity of employed nucleases. Clearly both experimental and computational improvements to the technology are needed to fully address these challenges.
State-of-the-art in experimental SEP validation and functional SEP studies
Some of the molecular size-related detection obstacles for SEPs are conveniently circumvented by using protein tags or translational reporters (e.g. superfolder green fluorescent protein (sfGFP) [18]) with corresponding molecular weights making the translational fusions exceeding the class of SEPs. Based on literature of bacterial SEP validation and characterization, the sequential peptide affinity (SPA) tag has been the go-to epitope tag for immunodetection of putative (bacterial) SEPs [3,13,16,17,21,53]. Studies resorting to this tag reported the epitope to permit the visualization of the expression of many reported SEPs. The tag combines the calmodulin binding peptide (CBP) and three consecutive FLAG-tags [96], separated by a tobacco etch virus (TEV) protease cleavage site, together accounting for a MW increase of about 6.3 kDa [97]. When dealing with SEPs, a tag of 6.3 kDa will often be as voluminous as – if not more voluminous than – the protein under study. However, while aiding detection, such a tag may alertly interfere with the physiological function/localization of the SEP [98]. Nevertheless, epitope interference has also been reported for smaller peptide tags like the highly positively charged His-tag [99,100], while contrarily, large (globular) tags were frequently shown to be innocuous [98].
The ultimate aim: functional characterization of validated bacterial SEPs
As it are typically only few SEP validations that corroborate genome-wide bacterial SEP discoveries, there is definitely a need for more general, unbiased sORFeome-wide validation efforts. For example, an extensive study of the translational landscape of S. pneumoniae connected the SEP of only one of their newly discovered sORFs, rio3, to bacterial host colonization through targeted endogenous mutagenesis (Table 1) [18], as also done in the pathogenic bacterium Y. pestis and in the extremophilic bacterium Deinococcus radiodurans (D. radiodurans) for the functional characterization of SEP-yp1 and SEP-yp2 [48] and SEP068184 [101], respectively implicating these SEPs in regulation of antiphagocytic capability and regulation of oxidative resistance.
Prior to functional studies, motif or domain prediction might provide a first hint towards the biological implication of the newly discovered SEPs (Table 1). Bioinformatics prediction of hydrophobic transmembrane motifs is relatively straightforward and widely exploited for the exploratory study of novel SEPs [21,35,48,101]. The short primary SEP structures are however no ideal subjects for functional domain searches [3,36,62], which is explained by the average size of protein domains coinciding with the upper length threshold of SEPs (100 AA) [102]. When contrasting bacterial domain annotations of SEPs versus non-SEPs for the SL1344 and LT2 S. Typhimurium strains as well as the K12 E. coli strain (Figure 4), the annotation ranged from 7% to 18% for SEPs, while for non-SEPs these percentages varied from 28% to 62%. While remarkably big discrepancies in the percentages of domain annotations between these related species/strains could be observed, SEP versus non-SEP domain annotations were in each case shown to be 3- to 4-fold lower. Large-scale SEP studies reporting Pfam domain predictions for high-confidence, novel SEPs are in line with these lower percentages of domain annotation [48,101]. Conservation analysis [12,36] of (the genomic surrounding of) predicted sORFs (e.g. gene co-occurrence in case of polycistrons) might also help to prioritize functional SEPs (Table 1) [33]. For the genomic context of the putative sORF start codons, higher RNA secondary structure predictions (as compared to the start codon region itself) may further also serve as indicators of translation initiation [36].
Specifically for sORF discovery in pathogenic bacteria, exploring putative SEP sequences for transmembrane domains and signal peptides is commonly exerted [18,48] as cell contact and secreted molecules are the major interaction routes between bacterial pathogens and their host cells [48]. These predictions are also used for the discovery of novel quorum sensing systems and players in Gram-negatives, for which peptides are known to fulfil the role of quorum sensing pheromones [18]. Cao et al performed differential expression analysis of a set of putative sORFs interrogating different host environment-mimicking conditions [48]. The same principle can be applied for all kinds of alternative stress conditions to find SEPs involved in different bacterial defence mechanisms and responses [33,101], but also expression differences over standard growth conditions may suggest a biological impact [12]. A recent study on SEP profiling specifically focussed on identification of stress response SEPs through the choice for the extremophilic bacterium D. radiodurans. Being a model organism for studying bacterial extreme stress responses, bacteria were subjected to ionizing radiation and oxidative stress resulting in the identification of 19 and 11 out of 109 newly identified SEPs as being upregulated under the respective stress condition [101].
Concluding Remarks and Future Perspectives
In recent years, well-recognized bacterial SEP examples have been drawing the attention of microbiologists to sORFs as new study subjects in the context of general microbiology as well as host-pathogen interactions. As sORFs are generally under annotated in genomes, Ribo-seq efforts should be appreciated for the contributions they made to the wealth of (putative) SEPs uncovered over the past years. However, experimental SEP validation is deemed required prior to including them in existing bacterial genome annotations, with epitope protein tagging and MS as the most commonly used methods. Besides the inherent small size and often hydrophobic nature of SEPs as confounding features hindering their detection, the – sometimes highly specific – expression conditions of SEPs might further complicate the validation process [16]. Conditional gene expression programmes of Salmonella are clearly illustrated in the continuous cataloguing effort – the SalCom repository [103]. Whether the common assumption of lower stability and abundance of SEPs serves a general additional hindering factor however, remains to be firmly established. Of note for SEP validation is that the rapid development of more sensitive high-throughput MS developments (e.g. data independent acquisition (DIA) and ion mobility MS) is expected to further aid bottom-up as well as top-down SEP detection [104].
While nonetheless both go hand-in-hand, overcoming the obstacles of SEP validation is one thing, but framing these small proteins within the host’s biology is another. Studies hunting for SEPs in diverse bacterial proteomes often focus on functional investigation of few individual SEPs through targeted endogenous mutagenesis and no efforts to collectively address the functionality of the small proteome have been undertaken [18,48,101]. There is, consequently, no doubt that the largest part of the sORFeome remains to be functionally explored. Although when discussing biologically meaningful SEPs, not too much emphasis must be placed on the word ‘function’, a term used for proteins that contribute to cell fitness and that are under purifying selection [105], as even merely the act of translating a sORF can influence the expression of its genomic context, much like operon leader peptides (e.g. the threonine operon leader peptide thrL) [3,62]. Lately, peptide evidence for newly annotated leader peptides, like for the MgtA leader peptide MgtL, has been changing the perception on this type of genomic elements as being exclusively regulatory (Figure 1A) [15].
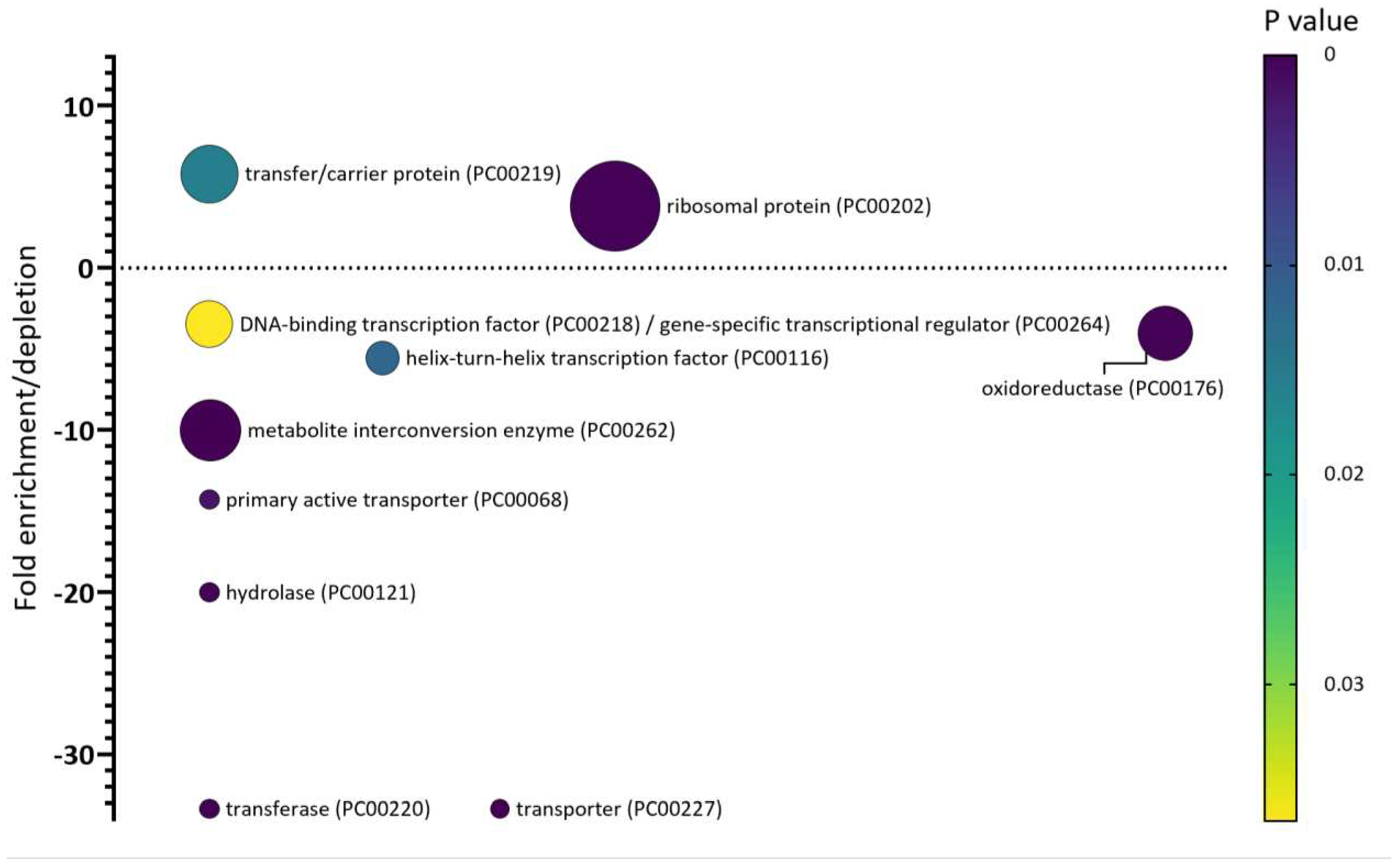
With the functional knowledge at hand, and irrespective of ribosomal SEPs, bacterial SEPs can be concluded to be extensively engaged in protein complexation with an important fraction represented by (trans)membrane complexes [62]. Many of the known small proteins function through binding and resulting regulation of standard-sized proteins [3]. From this viewpoint, the current missing factor in bacterial SEP characterization is an interactomics-oriented approach. As protein-protein interactions (PPIs) might in particular play an important role for SEPs containing transmembrane domains, the underrepresentation of members of the e.g. (transmembrane) transporter protein class (Figure ) and domain annotations in general for UniProt SEP annotations (Figure 44) might again indicate that the biological occupations of (transmembrane) SEPs rather goes through binding and regulation of other proteins or protein complexes, as also evident from the overrepresentation of transfer/carrier proteins among SEPs (Figure 5), while the same analysis also revealed that unclassified proteins were about 2-fold overrepresented in the category of SEPs, providing an interesting niche for future functional SEP discoveries. Also, established E. coli SEP interactomes show the SEP players to locate on the periphery of complexes, suggesting SEPs to take part in transient and differing interactions in multiple complexes, again providing some evidence for SEPs as regulators [62].
The interactome-associated SEP aspects ‘hydrophobicity’ and ‘transientness’ eventually bring the concept of in vivo proximity-dependent biotinylation (PDB) to the forefront. Unlike affinity purification (coupled to MS, AP-MS), PDB approaches have the power to catch weak and transient protein-protein interactions and equally important for the SEP protein class shown to be enriched for transmembrane proteins, are capable of handling less soluble proteins viewing its compatibility with the solubilization of membrane-(associated) proteins [106,107]. APEX, a PDB method exploiting the enzymatic activity of peroxidases for the biotinylation of proteins [107], was already successfully applied in bacteria for the elucidation of the type VI secretion biogenesis process in E. coli [108]. BioID, standing for proximity-dependent biotin identification, is another implementation of the PDB principle and requires the translational fusion of the protein of interest to a promiscuous biotin ligase (PBL) for the biotinylation of proximal and interacting proteins. As the overlap between APEX and BioID interactomes has been claimed to be limited, BioID could thus potentially offer interesting complementary perspectives into bacterial SEP biology [107].
High-throughput phenotypic screening is also emerging as an initiative to characterize gene products through the use of phenotypic microarrays interrogating the metabolization of compounds deterministic for unique molecular pathways [109]. The construction of knock-out libraries by means of CRISPR/Cas9 or alternative recombineering strategies could offer a valuable approach to enable sORF/SEP phenotyping at larger scales [42,110]. Combining data gathered through a diverse range of omics techniques like PBL and phenotyping should enable small protein research to finally piece together the functionalities of bacterial SEPs encoded by newly discovered sORFs.

Table 1.
Available toolkit for sORF prediction, sORF annotation and SEP expression validation and functional characterization. MW; molecular weight, MS; mass spectrometry.
Table 1.
Available toolkit for sORF prediction, sORF annotation and SEP expression validation and functional characterization. MW; molecular weight, MS; mass spectrometry.
| State-of-the-art | Pros | Cons | Suggested improvements | |
|---|---|---|---|---|
| sORF prediction and annotation | Ribo-seq | - Genome-wide - Independent from existing annotations - Indicative of ribosomal activity - Broadly applicable - Improved resolution for detection of start (Ribo-RET) and stop codons (Ribo-API) |
- Requirement for experimental SEP validation - Computationally intensive and complex - Poor data resolution inherent to bacterial Ribo-seq |
Refinement of bacterial Ribo-seq protocols and data-analysis |
| SEP expression validation | MS | - SEP abundance data - Proteome-wide technique suited for empirical SEP discovery |
- Limited number of tryptic SEP peptides - Hydrophobic character of peptides - Sensitivity of detection |
- Use of alternative proteases (e.g. chymotrypsin) - High-MW protein depletion or low-MW protein enrichment |
| (Immuno) blotting | - Information on MW and thus SEP integrity - Quantification of SEP expression |
- Tag interference on SEP function/localization - Small SEP size - Sensitivity not adequate for low SEP abundances |
- Use of smaller, charge-neutral tags (e.g. HiBiT) - SEP specific customization (e.g., blotting membrane (type, pore size), blotting buffer and method) - In solution detection of SEPs |
|
| SEP functional characterization | Conservation analysis | - first impression of SEP functioning - High-throughput screening |
Lower conservation of SEPs | - Interrogation of gene co-occurrence - RNA secondary structure analysis |
| Domain prediction | First impression of SEP functioning and localization | Too short primary SEP sequences for domain prediction | Motif prediction (e.g. transmembrane motifs) | |
| Mutation analysis | Targeted and multiplex approach | Laboursome | ||
| Expression analysis | Conditional impact of expression unknown | Conditional expression maps |
Acknowledgments
This research was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (PROPHECY grant agreement no. 803972) and the Research Foundation Flanders (FWO-Vlaanderen) project numbers G051120N and G045921N to P.V.D.
References
- Gualerzi, C.O. and Pon, C.L. (2015) Initiation of mRNA translation in bacteria: Structural and dynamic aspectsCellular and Molecular Life Sciences, 72Birkhauser Verlag AG, 4341–4367.
- Gawron, D. et al. (2014) The proteome under translational controlProteomics, 14Wiley-VCH Verlag, 2647–2662.
- Weaver, J. et al. (2019) Identifying small proteins by ribosome profiling with stalled initiation complexes. mBio 10.
- Meydan, S. et al. (2019) Retapamulin-Assisted Ribosome Profiling Reveals the Alternative Bacterial Proteome. Mol Cell 74, 481-493.e6. [CrossRef]
- Vazquez-Laslop, N. et al. (2022) Identifying Small Open Reading Frames in Prokaryotes with Ribosome Profiling. in Journal of Bacteriology, 204. [CrossRef]
- Willems, P. et al. (2022) To New Beginnings: Riboproteogenomics Discovery of N-Terminal Proteoforms in Arabidopsis Thaliana. Front Plant Sci 12, 1–18. [CrossRef]
- Ingolia, N.T. et al. (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science (1979) 324, 218–223. [CrossRef]
- Buskirk, A.R. and Green, R. (2017) Ribosome pausing, arrest and rescue in bacteria and eukaryotesPhilosophical Transactions of the Royal Society B: Biological Sciences, 372Royal Society.
- Guo, H. et al. (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840. [CrossRef]
- Ingolia, N.T. et al. (2011) Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802. [CrossRef]
- Li, G.W. et al. (2014) Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635. [CrossRef]
- Hucker, S.M. et al. (2017) Discovery of numerous novel small genes in the intergenic regions of the Escherichia coli O157:H7 Sakai genome. PLoS One 12. [CrossRef]
- VanOrsdel, C.E. et al. (2018) Identifying New Small Proteins in Escherichia coli. Proteomics 18, 1–8.
- Li, G.W. et al. (2012) The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 484, 538–541. [CrossRef]
- Ndah, E. et al. (2017) REPARATION: Ribosome profiling assisted (re-)annotation of bacterial genomes. Nucleic Acids Res 45. [CrossRef]
- Baek, J. et al. (2017) Identification of unannotated small genes in Salmonella. G3: Genes, Genomes, Genetics 7, 983–989.
- Venturini, E. et al. (2020) A global data-driven census of Salmonella small proteins and their potential functions in bacterial virulence . microLife 1, 1–20. [CrossRef]
- Laczkovich, I. et al. (2022) Discovery of Unannotated Small Open Reading Frames in Streptococcus pneumoniae D39 Involved in Quorum Sensing and Virulence Using Ribosome Profiling. mBio 13. [CrossRef]
- Smith, C. et al. (2022) Pervasive translation in Mycobacterium tuberculosis. 11, 73980. [CrossRef]
- Bartholomaus, A. et al. (2021) SmORFer: A modular algorithm to detect small ORFs in prokaryotes. Nucleic Acids Res 49. [CrossRef]
- Froschauer, K. et al. Complementary Ribo-seq approaches map the translatome and provide a small protein census in the foodborne pathogen Campylobacter jejuni. [CrossRef]
- Michel, A.M. et al. (2014) GWIPS-viz: Development of a ribo-seq genome browser. Nucleic Acids Res 42. [CrossRef]
- Araújo-Bazán, L. et al. (2019) Synthetic developmental regulator MciZ targets FtsZ across Bacillus species and inhibits bacterial division. Mol Microbiol 111, 965–980. [CrossRef]
- Burby, P.E. and Simmons, L.A. (2020) Regulation of cell division in bacteria by monitoring genome integrity and DNA replication statusJournal of Bacteriology, 202American Society for Microbiology.
- Sweet, M.E. et al. (2021) Structural basis for potassium transport in prokaryotes by KdpFABC. Proc Natl Acad Sci U S A 118, 1–9. [CrossRef]
- Wang, H. et al. (2017) Increasing intracellular magnesium levels with the 31-amino acid MgtS protein. Proc Natl Acad Sci U S A 114, 5689–5694. [CrossRef]
- Yoshitani, K. et al. (2019) Identification of an internal cavity in the PhoQ sensor domain for PhoQ activity and SafA-mediated control. Biosci Biotechnol Biochem 83, 684–694.
- Xu, J. et al. (2019) MgrB affects the acid stress response of Escherichia coli by modulating the expression of iraM. FEMS Microbiol Lett 366.
- Olvera, M.R. et al. (2019) Synthetic hydrophobic peptides derived from MgtR weaken Salmonella pathogenicity and work with a different mode of action than endogenously produced peptides. Sci Rep 9, 1–13. [CrossRef]
- Yadavalli, S.S. et al. (2020) Functional determinants of a small protein controlling a broadly conserved bacterial sensor kinase. J Bacteriol 202. [CrossRef]
- Williams, A.H. et al. (2019) The cryo-electron microscopy supramolecular structure of the bacterial stressosome unveils its mechanism of activation. Nat Commun 10. [CrossRef]
- Sur, V.P. et al. (2022) Dynamic study of small toxic hydrophobic proteins PepA1 and PepG1 of Staphylococcus aureus. Int J Biol Macromol. [CrossRef]
- Gray, T. et al. (2021) Small Proteins; Big Questions. J Bacteriol. [CrossRef]
- Fijalkowski, I. et al. (2021) Small Protein Enrichment Improves Proteomics Detection of sORF Encoded Polypeptides. Front Genet 12, 2042. [CrossRef]
- Fijalkowski, I. et al. (2022) Hidden in plain sight : challenges in proteomics detection of small ORF-encoded polypeptides. [CrossRef]
- Stringer, A. et al. (2022) Identification of Novel Translated Small Open Reading Frames in Escherichia coli Using Complementary Ribosome Profiling Approaches. [CrossRef]
- Loman, N.J. and Pallen, M.J. (2015) Twenty years of bacterial genome sequencing. Nat Rev Microbiol 13, 787–794. [CrossRef]
- Land, M. et al. (2015) Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics 15, 141–161. [CrossRef]
- Dorado, G. et al. (2021) Analyzing modern biomolecules: The revolution of nucleic-acid sequencing-review. Biomolecules 11, 1–18. [CrossRef]
- van Dijk, E.L. et al. (2018) The Third Revolution in Sequencing Technology. Trends in Genetics 34, 666–681.
- Ye, C. et al. (2020) Unculturable and culturable periodontal-related bacteria are associated with periodontal inflammation during pregnancy and with preterm low birth weight delivery. Sci Rep 10. [CrossRef]
- Fels, U. et al. (2020) Bacterial Genetic Engineering by Means of Recombineering for Reverse Genetics. Front Microbiol 11, 1–19. [CrossRef]
- Tanizawa, Y. et al. (2018) DFAST: A flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34, 1037–1039. [CrossRef]
- Schwengers, O. et al. (2021) Bakta: Rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom 7. [CrossRef]
- Syberg-Olsen, M.J. et al. (2021) Pseudofinder : detection of pseudogenes in prokaryotic genomes. bioRxiv 39, 1–7. [CrossRef]
- Zhao, Y. et al. (2018) PGAP-X: Extension on pan-genome analysis pipeline. BMC Genomics 19. [CrossRef]
- Fijalkowska, D. et al. (2020) Bacterial riboproteogenomics: The era of N-terminal proteoform existence revealed. FEMS Microbiol Rev 44, 418–431. [CrossRef]
- Cao, S. et al. (2021) Proteogenomic discovery of sORF-encoded peptides associated with bacterial virulence in Yersinia pestis. Commun Biol 4.
- Dong, Y. et al. (2021) Genome annotation of disease-causing microorganisms. Brief Bioinform 22, 845–854. [CrossRef]
- Danchin, A. et al. (2018) No wisdom in the crowd: genome annotation in the era of big data – current status and future prospects. Microb Biotechnol 11, 588–605. [CrossRef]
- Dziurzynski, M. et al. (2021) Simple, Reliable, and Time-Efficient Manual Annotation of Bacterial Genomes with MAISEN. Methods in Molecular Biology 2242, 221–229.
- Lobb, B. et al. (2020) An assessment of genome annotation coverage across the bacterial tree of life. Microb Genom 6. [CrossRef]
- Hemm, M.R. et al. (2010) Small stress response proteins in Escherichia coli: Proteins missed by classical proteomic studies. J Bacteriol 192, 46–58. [CrossRef]
- Storz, G. et al. (2014) Small proteins can no longer be ignored. Annu Rev Biochem 83, 753–777. [CrossRef]
- Andrews, S.J. and Rothnagel, J.A. (2014) Emerging evidence for functional peptides encoded by short open reading framesNature Reviews Genetics, 15Nature Publishing Group, 193–204.
- Mudge, J.M. et al. (2022) Standardized annotation of translated open reading framesNature Biotechnology, 40Nature Research, 994–999.
- Willems, P. et al. (2020) Lost and Found: Re-searching and Re-scoring Proteomics Data Aids Genome Annotation and Improves Proteome Coverage. [CrossRef]
- Finkel, Y. et al. (2018) Viral Short ORFs and Their Possible Functions. Proteomics 18, 1–8. [CrossRef]
- Lluch-Senar, M. et al. (2015) Defining a minimal cell: essentiality of small ORF s and nc RNA s in a genome-reduced bacterium . Mol Syst Biol 11, 780. [CrossRef]
- Martin, J.E. et al. (2015) The Escherichia coli Small Protein MntS and Exporter MntP Optimize the Intracellular Concentration of Manganese. PLoS Genet 11, 1–31. [CrossRef]
- Wassarman, K.M. et al. (2001) Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 15, 1637–1651. [CrossRef]
- Hemm, M.R. et al. (2020) Escherichia coli Small Proteome . EcoSal Plus 9.
- Gaßel, M. et al. (1999) The KdpF Subunit Is Part of the K-translocating Kdp Complex of Escherichia coli and Is Responsible for Stabilization of the Complex in Vitro*. [CrossRef]
- Andresen, L. et al. (2020) The small toxic salmonella protein timp targets the cytoplasmic membrane and is repressed by the small rna timr. mBio 11, 1–16. [CrossRef]
- Wang, N. et al. (2021) sORF-encoded polypeptide SEP1 Is a novel virulence factor of phytophthora pathogens. Molecular Plant-Microbe Interactions 34, 157–167.
- Fozo, E.M. et al. (2008) Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol Microbiol 70, 1076–1093. [CrossRef]
- Impens, F. et al. (2017) N-terminomics identifies Prli42 as a membrane miniprotein conserved in Firmicutes and critical for stressosome activation in Listeria monocytogenes. Nat Microbiol 2.
- Miravet-Verde, S. et al. (2019) Unraveling the hidden universe of small proteins in bacterial genomes. Mol Syst Biol 15, 1–17. [CrossRef]
- Garai, P. and Blanc-Potard, A. (2020) Uncovering small membrane proteins in pathogenic bacteria: Regulatory functions and therapeutic potential. Mol Microbiol 114, 710–720. [CrossRef]
- Yadavalli, S.S. and Yuan, J. (2022) Bacterial Small Membrane Proteins: The Swiss Army Knife of Regulators at the Lipid Bilayer. in Journal of Bacteriology, 204. [CrossRef]
- Giess, A. et al. (2017) Ribosome signatures aid bacterial translation initiation site identification. BMC Biol 15, 1–14. [CrossRef]
- Ochman, H. et al. (1999) Calibrating bacterial evolution. Proc Natl Acad Sci U S A 96, 12638–12643.
- Chaudhuri, D. et al. (2018) Salmonella Typhimurium infection leads to colonization of the mouse brain and is not completely cured with antibiotics. Front Microbiol 9, 1–12. [CrossRef]
- Michaux, C. et al. (2017) RNA target profiles direct the discovery of virulence functions for the cold-shock proteins CspC and CspE. Proc Natl Acad Sci U S A 114, 6824–6829. [CrossRef]
- Wang, Y. et al. (2020) Recent advances in ribosome profiling for deciphering translational regulation. Methods 176, 46–54. [CrossRef]
- Pavesi, A. et al. (2018) Overlapping genes and the proteins they encode differ significantly in their sequence composition from non-overlapping genes. PLoS One 13, 1–24. [CrossRef]
- Wright, B.W. et al. (2022) Overlapping genes in natural and engineered genomes. Nat Rev Genet 23, 154–168. [CrossRef]
- Olexiouk, V. et al. (2018) An update on sORFs.org: A repository of small ORFs identified by ribosome profiling. Nucleic Acids Res 46, D497–D502. [CrossRef]
- Mohammad, F. et al. A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. [CrossRef]
- Fremin, B.J. and Bhatt, A.S. (2020) Structured RNA Contaminants in Bacterial Ribo-Seq. mSphere 5. [CrossRef]
- Meleady, P. (2018) Two-dimensional gel electrophoresis and 2D-DIGE. Methods in Molecular Biology 1664, 3–14.
- Lee, P.Y. et al. (2020) The evolution of two-dimensional gel electrophoresis - from proteomics to emerging alternative applications. J Chromatogr A 1615, 460763. [CrossRef]
- Kielkopf, C.L. et al. (2021) Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of proteins. Cold Spring Harb Protoc 2021, 494–504. [CrossRef]
- Gao, Z. et al. (2019) AP3: An Advanced Proteotypic Peptide Predictor for Targeted Proteomics by Incorporating Peptide Digestibility. Anal Chem 91, 8705–8711. [CrossRef]
- Cassidy, L. et al. (2019) Depletion of High-Molecular-Mass Proteins for the Identification of Small Proteins and Short Open Reading Frame Encoded Peptides in Cellular Proteomes. J Proteome Res 18, 1725–1734. [CrossRef]
- Becher, D. et al. (2020) Optimized proteomics workflow for the detection of small proteins. J Proteome Res 19, 4004–4018. [CrossRef]
- Ahrens, C.H. et al. (2022) A Practical Guide to Small Protein Discovery and Characterization Using Mass Spectrometry. [CrossRef]
- Kurien, B.T. and Hal Scofield, R. (2015) Western Blotting: An Introduction. Methods Mol Biol 1312, 17.
- Tomisawa, S. et al. (2013) A new approach to detect small peptides clearly and sensitively by Western blotting using a vacuum-assisted detection method. Biophysics (Japan) 9, 79–83. [CrossRef]
- Olexiouk, V. et al. (2016) SORFs.org: A repository of small ORFs identified by ribosome profiling. Nucleic Acids Res 44, D324–D329. [CrossRef]
- Peeters, M.K.R. and Menschaert, G. (2020) The hunt for sORFs: A multidisciplinary strategy. Exp Cell Res 391. [CrossRef]
- Kubatova, N. et al. (2020) Rapid Biophysical Characterization and NMR Spectroscopy Structural Analysis of Small Proteins from Bacteria and Archaea. ChemBioChem 21, 1178–1187. [CrossRef]
- Uversky, V.N. (2017) Paradoxes and wonders of intrinsic disorder: Stability of instability. Intrinsically Disord Proteins 5, e1327757. [CrossRef]
- Yu, J. et al. (2021) Comprehensive evaluation of protein-coding sORFs prediction based on a random sequence strategy. Frontiers in Bioscience - Landmark 26, 272–278. [CrossRef]
- Gelhausen, R. et al. (2022) RiboReport - Benchmarking tools for ribosome profiling-based identification of open reading frames in bacteria. Brief Bioinform 23, 1–22. [CrossRef]
- Hopp, T.P. et al. (1988) A short polypeptide marker sequence useful for recombinant protein identification and purification. Bio/Technology 6, 1204–1210. [CrossRef]
- Zeghouf, M. et al. (2004) Sequential Peptide Affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J Proteome Res 3, 463–468. [CrossRef]
- Vandemoortele, G. et al. (2019) Pick a Tag and Explore the Functions of Your Pet Protein. Trends Biotechnol 37, 1078–1090. [CrossRef]
- Booth, W.T. et al. (2018) Impact of an N-terminal polyhistidine tag on protein thermal stability. ACS Omega 3, 760–768. [CrossRef]
- Munadziroh, E. et al. (2020) Effect of poly-histidine tag position toward inhibition activity of secretory leukocyte protease inhibitor as candidate for material wound healing. Avicenna J Med Biotechnol 12, 32–36.
- Zhou, C. et al. (2022) Probing the sORF-Encoded Peptides of Deinococcus radiodurans in Response to Extreme Stress. Molecular & Cellular Proteomics 21, 100423.
- Xiong, J. (2006) Protein Motifs and Domain Prediction. Essential Bioinformatics. [CrossRef]
- Srikumar, S. et al. (2015) RNA-seq Brings New Insights to the Intra-Macrophage Transcriptome of Salmonella Typhimurium. PLoS Pathog 11. [CrossRef]
- Kitata, R.B. et al. (2022) Advances in data-independent acquisition mass spectrometry towards comprehensive digital proteome landscapeMass Spectrometry ReviewsJohn Wiley and Sons Inc.
- Keeling, D.M. et al. The meanings of “function” in biology and the problematic case of de novo gene emergence. [CrossRef]
- Liu, X. et al. (2020) Combined proximity labeling and affinity purification−mass spectrometry workflow for mapping and visualizing protein interaction networks, 15. [CrossRef]
- Samavarchi-Tehrani, P. et al. (2020) Proximity dependent biotinylation: Key enzymes and adaptation to proteomics approaches. Molecular and Cellular Proteomics 19, 757–773. [CrossRef]
- Santin, Y.G. et al. (2018) In vivo TssA proximity labelling during type VI secretion biogenesis reveals TagA as a protein that stops and holds the sheath. Nat Microbiol 3, 1304–1313. [CrossRef]
- Guard, J. (2022) Through the Looking Glass: Genome, Phenome, and Interactome of Salmonella entericaPathogens, 11MDPI.
- Todor, H. et al. (2021) Bacterial CRISPR screens for gene functionCurrent Opinion in Microbiology, 59Elsevier Ltd, 102–109.
Figure 3.
Ribo-seq toolset for the discovery of translated sORFs in bacterial genomes. In general, ribosome profiling by sequencing (Ribo-seq) (B) relies on binding of ribosomes onto mRNA molecules as evidence for their translation into proteins [7]. When performing bacterial Ribo-seq, micrococcal nuclease (MNase) is added to (ribosome-bound) mRNA. Nuclease digestion by MNase only proceeds when no ribosomes are bound onto the mRNA molecule. Deep sequencing of isolated, ribosome-protected intact mRNA fragments enables delineation of translated genomic regions. Retapamulin-assisted Ribo-seq (Ribo-RET) (A) [3] and apidaecin-assisted Ribo-seq (Ribo-API) (C) [21] are variants of the standard Ribo-seq protocol which make use of the antibiotic retapamulin and the antimicrobial peptide apidaecin (Api137) for specific halting of initiating and terminating ribosomes, allowing for the more accurate assignment of translation initiation and termination sites, respectively. Recently, the pleuromutilin lefamulin has been introduced as an alternative for retapamulin with higher activity in Gram-negative bacteria and therefore general and wider applicability [5]. Combining Ribo-seq deep sequencing patterns with Ribo-RET and Ribo-API-derived profiles can be used to more precisely delineate start and stop codons of newly discovered (small) ORFs.
Figure 3.
Ribo-seq toolset for the discovery of translated sORFs in bacterial genomes. In general, ribosome profiling by sequencing (Ribo-seq) (B) relies on binding of ribosomes onto mRNA molecules as evidence for their translation into proteins [7]. When performing bacterial Ribo-seq, micrococcal nuclease (MNase) is added to (ribosome-bound) mRNA. Nuclease digestion by MNase only proceeds when no ribosomes are bound onto the mRNA molecule. Deep sequencing of isolated, ribosome-protected intact mRNA fragments enables delineation of translated genomic regions. Retapamulin-assisted Ribo-seq (Ribo-RET) (A) [3] and apidaecin-assisted Ribo-seq (Ribo-API) (C) [21] are variants of the standard Ribo-seq protocol which make use of the antibiotic retapamulin and the antimicrobial peptide apidaecin (Api137) for specific halting of initiating and terminating ribosomes, allowing for the more accurate assignment of translation initiation and termination sites, respectively. Recently, the pleuromutilin lefamulin has been introduced as an alternative for retapamulin with higher activity in Gram-negative bacteria and therefore general and wider applicability [5]. Combining Ribo-seq deep sequencing patterns with Ribo-RET and Ribo-API-derived profiles can be used to more precisely delineate start and stop codons of newly discovered (small) ORFs.
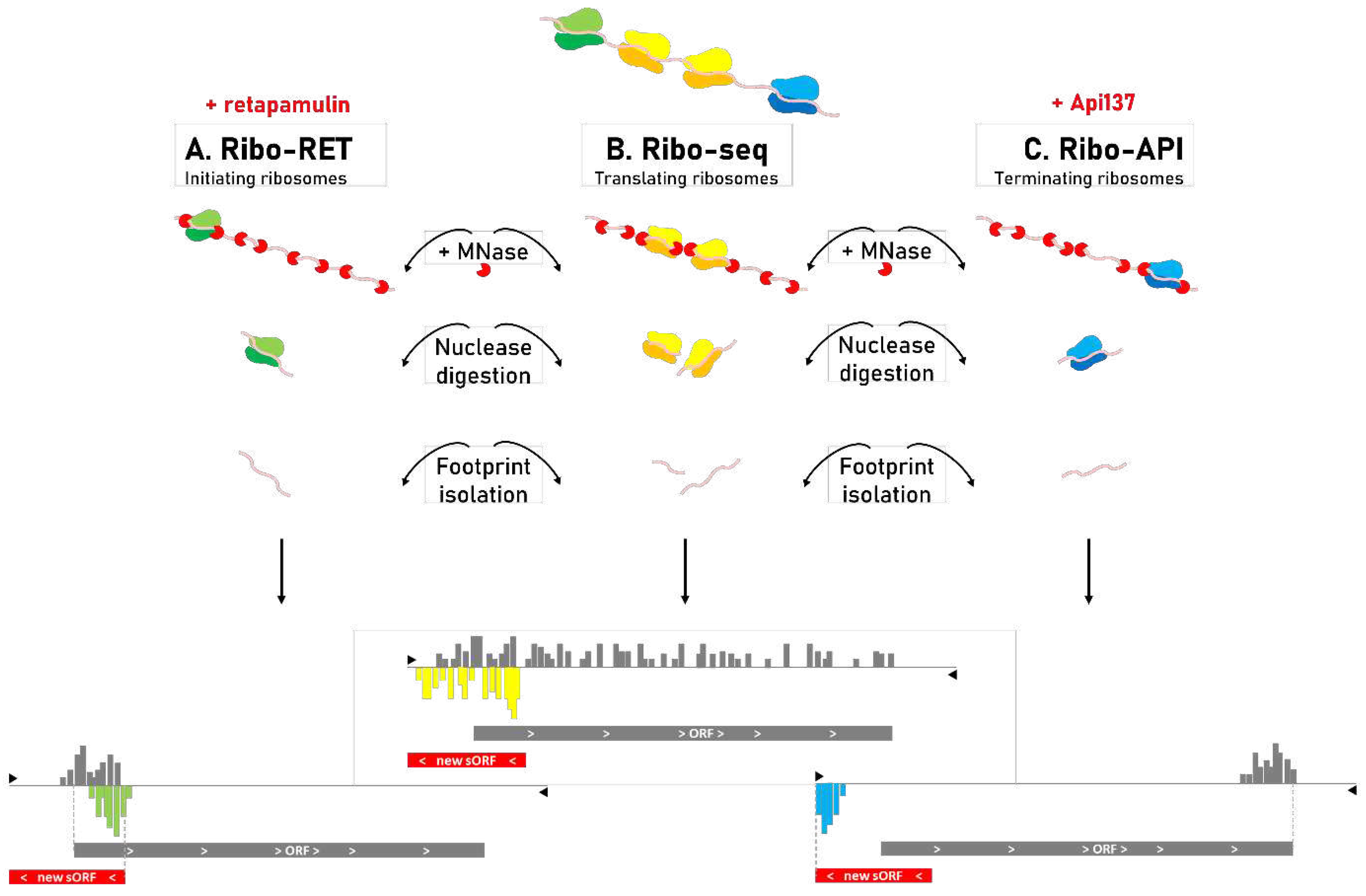
Figure 4.
Domain annotations for bacterial SEPs compared to non-SEPs. Database protein entries of S. Typhimurium strains SL1344 (#4659, UP000008962) and LT2 (#4533, UP000001014) and E. coli K12 (#4448, UP000000625) were interrogated for domain annotations in the category of SEPs and non-SEPs with the % domain annotation colour coded (blue scale). Categorical percentages of proteome occurrence for SEPs and non-SEPs are indicated in the pie charts.
Figure 4.
Domain annotations for bacterial SEPs compared to non-SEPs. Database protein entries of S. Typhimurium strains SL1344 (#4659, UP000008962) and LT2 (#4533, UP000001014) and E. coli K12 (#4448, UP000000625) were interrogated for domain annotations in the category of SEPs and non-SEPs with the % domain annotation colour coded (blue scale). Categorical percentages of proteome occurrence for SEPs and non-SEPs are indicated in the pie charts.
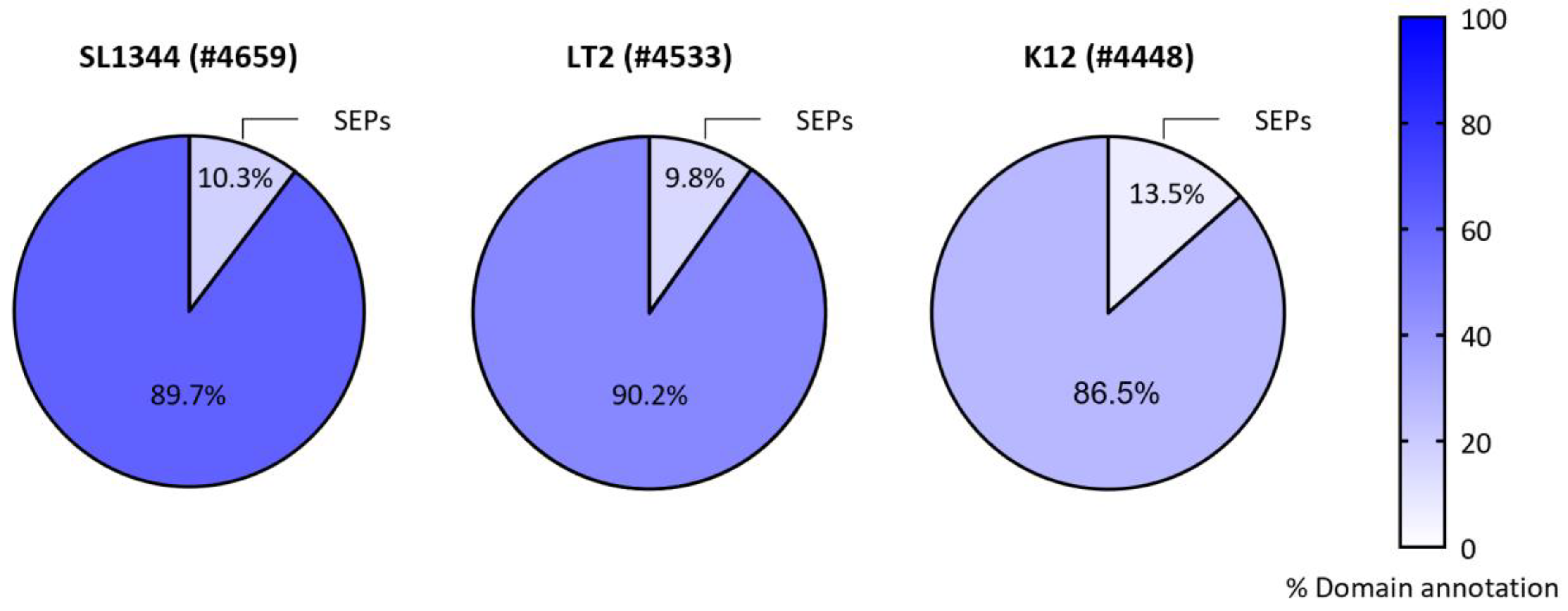
Figure 5.
Multiple variable bubble plot representations of significantly over- or under-represented protein classes in the category of annotated S. Typhimurium SEPs. Overrepresentation test of SEP annotations – represented as fold enrichment/depletion - of S. Typhimurium strain LT2 (#4533, UP000001014) was determined using PANTHER (version 17.0, released 2022-02-22) and p values were corrected for multiple hypotheses testing using Bonferroni correction for multiple testing. Only corrected p values < 0.05 were considered. The bubble size corresponds to the number of proteins corresponding to the indicated PANTHER protein class (smallest bubble size corresponds to 7 members) and the colour code represents the p value scale.
Figure 5.
Multiple variable bubble plot representations of significantly over- or under-represented protein classes in the category of annotated S. Typhimurium SEPs. Overrepresentation test of SEP annotations – represented as fold enrichment/depletion - of S. Typhimurium strain LT2 (#4533, UP000001014) was determined using PANTHER (version 17.0, released 2022-02-22) and p values were corrected for multiple hypotheses testing using Bonferroni correction for multiple testing. Only corrected p values < 0.05 were considered. The bubble size corresponds to the number of proteins corresponding to the indicated PANTHER protein class (smallest bubble size corresponds to 7 members) and the colour code represents the p value scale.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



