Submitted:
23 February 2023
Posted:
24 February 2023
You are already at the latest version
Abstract
Autoimmune disorders and some types of blood cancer originate when B lymphocytes malfunction. In particular, when B cells produce antibodies recognizing the body’s proteins, it leads to various autoimmune disorders. And when B cells of various developmental stages transform into cancer cells, it results in blood cancers, including multiple myeloma, lymphoma, and leukemia. Thus, new methods of targeting B cells are required for various patient groups. Here, we used protein kinase inhibitors alectinib, brigatinib, ceritinib, crizotinib, entrectinib, and lorlatinib previously approved as drugs treating anaplastic lymphoma kinase (ALK)-positive lung cancer cells. We hypothesized that the same inhibitors will efficiently target leukocyte tyrosine kinase (LTK)-positive, actively protein-secreting mature B lymphocytes, including plasma cells. We isolated CD19-positive human B cells from the blood of healthy donors and used two alternative methods to stimulate cell maturation toward plasma cells. Using cell proliferation and flow cytometry assays, we found that ceritinib and entrectinib eliminate plasma cells from B cell populations. Alectinib, brigatinib, and crizotinib also inhibited B cell proliferation, while lorlatinib had no or limited effect on B cells. More generally, we concluded that several drugs previously developed to treat ALK-positive malignant cells can be also used to treat LTK-positive B cells.
Keywords:
B-cell
; autoimmune disease
; drug repurposing
1. Introduction
B lymphocytes initiate their development in the bone marrow and complete maturation in peripheral lymphoid organs [1]. Cell development starts with pre-progenitor(pro)-B cells and undergoes through pro-B and pre-B cells to immature and mature B lymphocytes expressing B cell receptors (BCR). Later on, B cells express antibodies (immunoglobulins) and then develop into larger plasma cells, which are specialized in antibody secretion [2]. While antibodies are crucial during the immune response to many diseases, some antibodies recognize polypeptides naturally expressed by the normal cells in the body resulting in various autoimmune diseases [3].
Early stage B cell maturation is associated with the V(D)J recombination process, when Variable (V), Diversity (D), and Joining (J) gene segments assemble through DNA recombination resulting in immunoglobulin heavy (IgH) and light (IgL) chains of antibodies [1,4,5]. The V(D)J recombination process depends on the DNA double-strand breaks (DSBs) initiated by recombination-activating gene (RAG)1/2 and then repaired in an error-prone manner by the non-homologous DNA end-joining (NHEJ) molecular pathway. The NHEJ is initiated by Ku70/Ku80(Ku86) factors recruited to the DSB and then facilitating recruitment of downstream proteins, such as DNA-dependent protein kinase, catalytic subunit (DNA-PKcs), DNA ligase 4 (LIGIV), X-ray repair cross-complementing protein 4 (XRCC4), XRCC4-like factor (XLF), Paralogue of XRCC4 and XLF (PAXX), and Modulator of Retrovirus Infection (MRI) [5,6]. Mature B cells can change the constant region of immunoglobulins in another recombination process called class switch recombination (CSR). The CSR is initiated by DNA lesions introduced by activation-induced cytidine deaminase (AID), and DNA breaks are repaired by NHEJ [1,4,5,6].
There are several strategies used to treat autoimmune diseases, including targeting the antibody-expressing B cells and surgically removing peripheral lymphoid organs [3], although these methods are not ideal and patient management needs further improvement. One strategy is to target antibody-producing B cells (plasma cells) via enzymes specifically expressed in large amounts. One such protein is leukocyte tyrosine kinase (LTK) [7,8]. It was recently suggested that LTK-positive cancer cells as well as plasma cell-mediated diseases can be treated using tyrosine kinase inhibitors [9].
LTK was identified as an endoplasmic reticulum (ER)-bound protein required for efficient secretion (including antibodies), and it was proposed to be a potential target for the development of new medicines [7]. LTK localizes to the ER and regulates ER export [7]. The LTK is a tyrosine kinase, which is very similar in structure to the anaplastic lymphoma kinase (ALK) [7]. The ALK kinase is a known target in cancer therapy [10], and the available medicines targeting ALK might also target the LTK.
Drug repurposing is an important direction in using developing medicines to treat new diseases by targeting the same or different pathways [11,12,13]. While a lot of drug repurposing research was made focusing on anti-viral drug treatments and combinations [14,15,16,17,18,19,20,21,22], here, we focused on drugs approved to treat ALK-positive cancers (alectinib, ceritinib, crizotinib, brigatinib, entrectinib, lorlatinib [10]) to target ALK-negative but LTK-positive mature B cells.
Gene fusions are an important driver of oncogenesis, and their detection has improved patient outcomes. However, oncogenic drivers remain unknown in a substantial proportion of lung cancers [23]. Examples of genes participating in oncogenic fusion include ALK, ROS1, and RET [24]. CLIP1-LTK fusion is a recently-reported translocation associated with lung cancer [25]. Because of similarity between ALK and LTK structure, it was possible to use medicines previously approved to treat ALK-positive lung cancers to target LTK-positive ones [25]. In particular, five FDA-approved drugs (alectinib, brigatinib, ceritinib, crizotinib, and lorlatinib) as well as multikinase inhibitors entrectinib and repotrectinib, were efficient in targeting LTK-positive lung cancer cells [25]. As B lymphocytes are known to express LTK (but not ALK), we proposed that the drugs listed above could target B cells that would allow drug repurposing to treat B-cell-related disorders [9].
Here, we exposed human B cells to tyrosine kinase inhibitors and found that alectinib, brigatinib, ceritinib, crizotinib, and entrectinib inhibit the growth of B lymphocytes. Furthermore, we validated the findings using two alternative B-cell stimulation systems. Pre-clinical studies involving in vivo models will be needed to find out more optimal medicines and concentrations for patient treatment.
2. Materials and Methods
2.1. Purification of CD19-positive B lymphocytes
A cluster of differentiation 19 (CD19)-positive B lymphocytes (total B cells) were purified from buffy coats derived from healthy donors using magnetic microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany #130-050-301), according to the manufacturer’s instructions. Samples from twenty-two donors are described in this study. Samples from additional donors were used to optimize the protocols and are not included in the manuscript. The purity of resulting B cells was validated using flow cytometry staining and analysis for CD19 and IgM, as previously described [26,27,28,29,30,31].
2.2. Stimulation of B cells
Purified CD19-positive B lymphocytes were kept in cell culture at 1.0-2.5×106 cells/mL in 200 µL of Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% heat-inactivated fetal bovine serum and penicillin/streptomycin. Recombinant human interleukins IL-4 (20 ng/mL; Cat# 200-04), IL-21 (50 ng/mL; Cat# 200-21; both Peprotech, Cranbury, NJ, USA), and soluble recombinant human CD40L (100ng/mL; MegaCD40L, Enzo LifeSciences, Farmingdale, NY, USA, ALX-522-120-C010) were used to keep the lymphocyte alive and to increase the purity of mature B cells. For differentiation toward antibody-secreting cells, B lymphocytes were stimulated (0.25×106 cells/well) in the presence of CD40L and IL-4/IL-21 for 5 days [26]. Then, the tyrosine kinase inhibitors alectinib (CH5424802, AF-802, RG-7853), brigatinib (AP26113), ceritinib (LDK378; S7083), crizotinib (PF-02341066), entrectinib (RXDX-101), and lorlatinib (PF-6463922) (all from Selleckchem, Houston, TX, USA) were added (0.1-30 µM, as indicated), and the cells were incubated for two additional days. Equal volumes of vehicle (DMSO/EtOH, EtOH, DMSO) was used in the control wells. Fixable Viability Stain 510 (BD Biosciences; Franklin Lakes, NJ, USA; Cat# 564406) was used to include life cells in the analyses. Samples from different donors were used to generate every data point.
The inhibitor concentration range was chosen both based on the pilot experiments and the capacity of the chemicals to dissolve (solubility). Moreover, the concentrations reported in the literature are between 1nM and 10µM [25]. In our experimental settings, the concentrations lower than 0.1-1.0µM had minor or no effect on the cells in our model system (Figure 1), while concentrations of 10.0-30.0µM resulted in nearly complete inhibition of cell growth. Thus, we focused on the concentration range of 0.1-10.0µM. For this, original 20µM inhibitor solutions were made as a pre-mix, and 3-fold dilutions followed (10.0, 3.3, 1.1, 0.37, and 0.12µM). The pre-mixed inhibitor-containing solutions were then combined with an equal volume of cell suspension.
2.3. Stimulation of cells using Immunocult kit
Alternatively, the purified CD19-positive B lymphocytes were stimulated using an Immunocult human B cell expansion kit (StemCell, Cambridge, UK; Cat# 100-0645), according to the manufacturer’s instructions. Briefly, frozen PBMCs from healthy donors were reconstituted in phosphate-buffered saline (PBS) buffer and washed twice in the centrifuge at 4°C, 700g for 5 minutes. CD19-positive B cells were isolated as described above (Section 2.1.). The cells were expanded and differentiated using a human B cell expansion medium from the ImmunoCult kit (StemCell, Cambridge, UK; Cat# 100-0645).
A human B cell expansion medium was prepared as described below. At room temperature (15-25°C), 2mL of ImmunoCult-ACF Human B cell expansion supplement was added to 98 mL of ImmunoCult-XF B cell base medium. The solution was mixed by inverting the bottle several times.
B lymphocytes were diluted to 105 cells per mL. The cells were incubated at standard conditions, 37°C, and 5% CO2 in a humidified incubator. The cell concentrations were adjusted to 100 000 per mL using the same solution every three days by adding fresh Human B cell expansion medium. On day ten, the tyrosine kinase inhibitors ceritinib (LDK378; S7083) and entrectinib (RXDX-101) (both from Selleckchem, Houston, TX, USA) were added at 1-10 µM, as indicated. Equal volumes of vehicle (DMSO/EtOH, EtOH, DMSO) was added to the control wells. Fixable Viability Stain 510 (BD Biosciences; Franklin Lakes, NJ, USA; Cat# 564406) was used to include life cells in the analyses. Finally, live cells were analyzed using a flow cytometry assay to identify mature B lymphocytes (section 2.4). Samples from different donors were used to generate every data point.
2.4. Flow cytometry
The cells were washed in 98% PBS/2% fetal calf serum (FCS) buffer. Next, the cells were stained with anti-human monoclonal antibodies on ice, then washed twice in the same buffer (98%PBS/2%FCS) and once in PBS. For detection of intracellular B-lymphocyte induced maturation protein 1 (BLIMP1) and interferon regulatory factor 4 (IRF4) expression, the BD Pharmingen Transcription Buffer Set (BD Biosciences; Franklin Lakes, NJ, USA; Cat# 562574) was used. BLIMP1 was detected using PE Rat monoclonal anti-human/mouse BLIMP1 (clone 6D3; BD Pharmingen; Franklin Lakes, NJ, USA; Cat# 564702). IRF4 was detected using Alexa Fluor 488 Rat monoclonal anti-human/mouse IRF4 (clone IRF4.3E4; Biolegends, San Diego, CA, USA; Cat# 646405). Finally, CD19-positive cells were gated using CD19 Monoclonal Antibody (SJ25C1), PerCP-eFluor™ 710, eBioscience™ (Invitrogen, ThermoFisher Scientific, Waltham, MA, USA; Cat# 46-0198-42). Flow cytometry analyses were performed using Attune NxT (ThermoFisher Scientific; Waltham, MA, USA) flow cytometer, and FlowJo software was used to analyze the cells.
2.5. Mature B lymphocyte inhibition assay
Incorporation of 3H-Thymidine (Montebello Diagnostics, Oslo, Norway, MT6032) was used to test functions of tyrosine kinase inhibitors (alectinib, brigatinib, ceritinib, crizotinib, entrectinib, and lorlatinib), as previously described in [32]. A proportion of inhibition was expressed as 100% (“1.0”) when there was no 3H-signal detected, while inhibition was 0% (“0.0”) in the mock-treated cells.
Briefly, the cells were incubated in the RPMI medium complemented with IL-4, IL-21, and CD40L, as described in Section 2.2. Following 5 days of incubation, 3H Thymidine (1:200) and tyrosine kinase inhibitors (when indicated) were added to the solution, and the cells were incubated for another 2 days (48 hours), similar to the scheme in Figure 2A, except the last stage. For radioactivity detection, B cells were lysed in water by osmotic pressure and were transferred from 96 well plates to glass fiber filtermat (8x12 Filtermat A, GF/C, 100/pk, #1450-421, Perkin Elmer). The filters were rinsed with water and sealed in plastic bags with a scintillation cocktail (OptiScint® LLT, #6013461, PerkinElmer). Radioactivity was detected and analyzed using TopCount NXT microplate counter (SKU: IC 10005, PerkinElmer).
3. Results
3.1. ALK inhibitors prevent accumulation of thymidine in CD19-positive B cell populations
Peripheral blood mononuclear cells (PBMC) from healthy donors were used to isolate CD19-positive B lymphocytes and further stimulate these cells for maturation, as described in the section “Materials and Methods (2.1 and 2.2.)”. We then exposed the mature B cells to tyrosine kinase (ALK) inhibitors, i.e. alectinib, brigatinib, ceritinib, crizotinib, entrectinib, and lorlatinib, when indicated (Figure 1). The experiments were performed at least six times in triplicates, and the average effective concentrations expected to inhibit half of the cells (EC50) were the following: 2.5 µM for alectinib, 6.0 µM for brigatinib, 1.8 µM for ceritinib, 7.1 µM for crizotinib, and 3.4 µM for entrectinib. Opposingly, even higher concentrations of lorlatinib, i.e. 10 µM in 7 experiments and 30 µM in 5 experiments, did not result in the inhibition of at least 50% of cells (e.g., Figure 1).
Figure 1.
Inhibition of CD19-positive B lymphocyte populations proliferation expressed as a percentage normalized to mock-treated controls. The efficiency of tyrosine kinase inhibitors alectinib (A), brigatinib (B), ceritinib (C), crizotinib (D), entrectinib (E), and lorlatinib (F) was measured using incorporation of H3-Thymidine by B lymphocytes as counts per minute (CPM). Three representative samples from three healthy donors (1,2, and 3) are shown. The values were first normalized to the mock-treated controls that are taken as “1”. Inhibition was then calculated as: “Inhibition=1-normalized CPM”. Thus, lack of inhibition in mock-treated cells is expressed as 1-1=0 (0% inhibition), while lack of radioactivity was taken as a total inhibition and resulted in 1-0=1 (100% inhibition). The experiment was performed at least 6 times with selected inhibitors, i.e. with alectinib (n=7), brigatinib (n=6), ceritinib (n=10), crizotinib (n=6), entrectinib (n=10), and lorlatinib (n=7). (G) Summary of six to ten experiments, as indicated, showing distribution of values. Average±SEM is as follows: alectinib (2.5±0.2), brigatinib (6.0±1.5), ceritinib (1.8±0.1), crizotinib (7.1±0.3), entrectinib (3.4±0.5). The table containing experimental data for each experiment are available in Supplementary information (Figure S3). Each data point represents an individual donor sample.
Figure 1.
Inhibition of CD19-positive B lymphocyte populations proliferation expressed as a percentage normalized to mock-treated controls. The efficiency of tyrosine kinase inhibitors alectinib (A), brigatinib (B), ceritinib (C), crizotinib (D), entrectinib (E), and lorlatinib (F) was measured using incorporation of H3-Thymidine by B lymphocytes as counts per minute (CPM). Three representative samples from three healthy donors (1,2, and 3) are shown. The values were first normalized to the mock-treated controls that are taken as “1”. Inhibition was then calculated as: “Inhibition=1-normalized CPM”. Thus, lack of inhibition in mock-treated cells is expressed as 1-1=0 (0% inhibition), while lack of radioactivity was taken as a total inhibition and resulted in 1-0=1 (100% inhibition). The experiment was performed at least 6 times with selected inhibitors, i.e. with alectinib (n=7), brigatinib (n=6), ceritinib (n=10), crizotinib (n=6), entrectinib (n=10), and lorlatinib (n=7). (G) Summary of six to ten experiments, as indicated, showing distribution of values. Average±SEM is as follows: alectinib (2.5±0.2), brigatinib (6.0±1.5), ceritinib (1.8±0.1), crizotinib (7.1±0.3), entrectinib (3.4±0.5). The table containing experimental data for each experiment are available in Supplementary information (Figure S3). Each data point represents an individual donor sample.
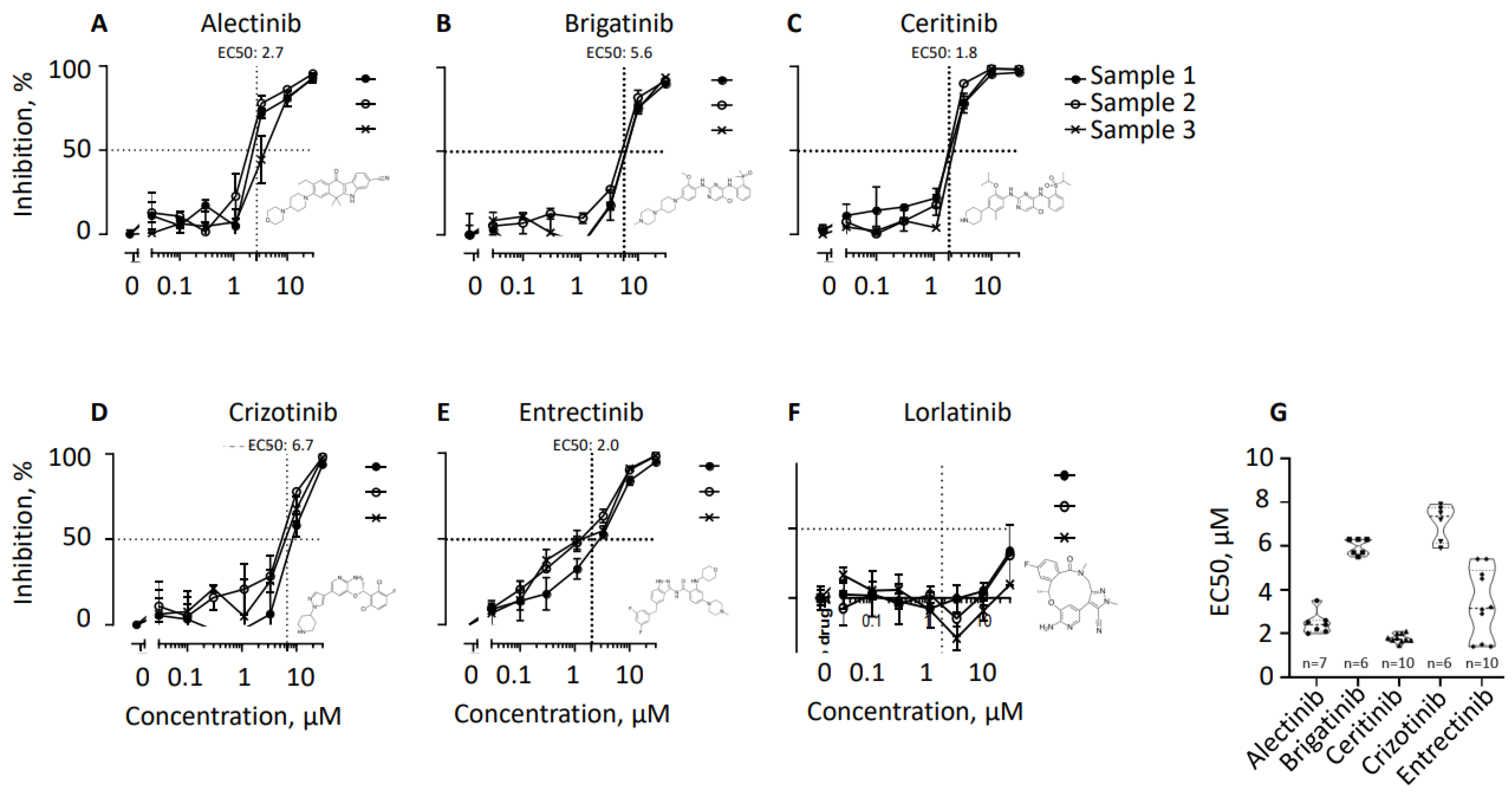
The summary of tyrosine kinase inhibitors EC50s is represented in Figure 1G and Figure S3, as a graph visualizing distribution (Figure 1G) and numbers with average and SEM (Figure S3). Altogether, alectinib, ceritinib, and entrectinib demonstrated the lowest EC50 and thus are potential drugs for further pre-clinical studies, when depletion of mature B cells is required, for example, during autoimmune disorders and multiple myeloma cancer.
3.2. Ceritinib and entrectinib eliminate BLIMP1/IRF4 double-positive B cell populations
We then focused on the effects of two tyrosine kinase inhibitors on mature B cell populations, i.e. ceritinib and entrectinib. These drugs demonstrated prominent inhibitory features (Figure 1), left high proportions of life cells at the end of the experiment, and are of commercial interest. Both ceritinib and entrectinib inhibited mature B cell populations in multiple experiments (summarized in Figure 1), with ceritinib showing lower EC50 concentrations and more consistent numbers (Figure 1). We then focused on live cell populations of these CD19-positive B cells and analyzed the presence of mature BLIMP1/IRF4 double-positive lymphocytes, i.e. plasma cells [26,33] (Figure 2). We found that majority of live CD19-positive lymphocytes in our experiments with tyrosine kinase inhibitors were BLIMP1/IRF4 double-positive (62±7.7%, Figure 2). When the cells were incubated for 48 hours in the presence of ceritinib, the proportion of BLIMP1/IRF4 double-positive cells reduced, e.g. to 36±8.2% when exposed to 3 µM of ceritinib, to 7±3.3% when exposed to 10 µM of ceritinib, and 30±7.1% and 14±3% when exposed to 3 µM and 10 µM of entrectinib, respectively. Thus, we concluded that ceritinib and entrectinib eliminate BLIMP1/IRF4 double-positive mature B cell populations, and have lower effects on live CD19-positive but BLIMP1- and IRF4-negative cells.
Figure 2.
Ceritinib and entrectinib inhibit the proliferation of B lymphocytes in cell culture. (A) Schematic overview of the experiment. (B) Examples of flow cytometry detecting live, CD19-positive B lymphocytes expressing BLIMP1 and IRF4 (plasma cells). Stimulated mature B lymphocyte populations included 64% of BLIMP1/IRF4 double-positive cells. Following 48 hours of treatment with 3 µM and 10 µM of ceritinib, the proportion of BLIMP1/IRF4 double-positive cells reduced to 36 % and 4 %, respectively. Similarly, following 48 hours of treatment with 3 µM and 10 µM of entrectinib, the proportion of BLIMP1/IRF4 double-positive cells reduced to 27% and 11%, respectively. (C) Summary of five experiments similar to one shown in (B) with standard deviation. Samples from different donors were used to generate every data point.
Figure 2.
Ceritinib and entrectinib inhibit the proliferation of B lymphocytes in cell culture. (A) Schematic overview of the experiment. (B) Examples of flow cytometry detecting live, CD19-positive B lymphocytes expressing BLIMP1 and IRF4 (plasma cells). Stimulated mature B lymphocyte populations included 64% of BLIMP1/IRF4 double-positive cells. Following 48 hours of treatment with 3 µM and 10 µM of ceritinib, the proportion of BLIMP1/IRF4 double-positive cells reduced to 36 % and 4 %, respectively. Similarly, following 48 hours of treatment with 3 µM and 10 µM of entrectinib, the proportion of BLIMP1/IRF4 double-positive cells reduced to 27% and 11%, respectively. (C) Summary of five experiments similar to one shown in (B) with standard deviation. Samples from different donors were used to generate every data point.
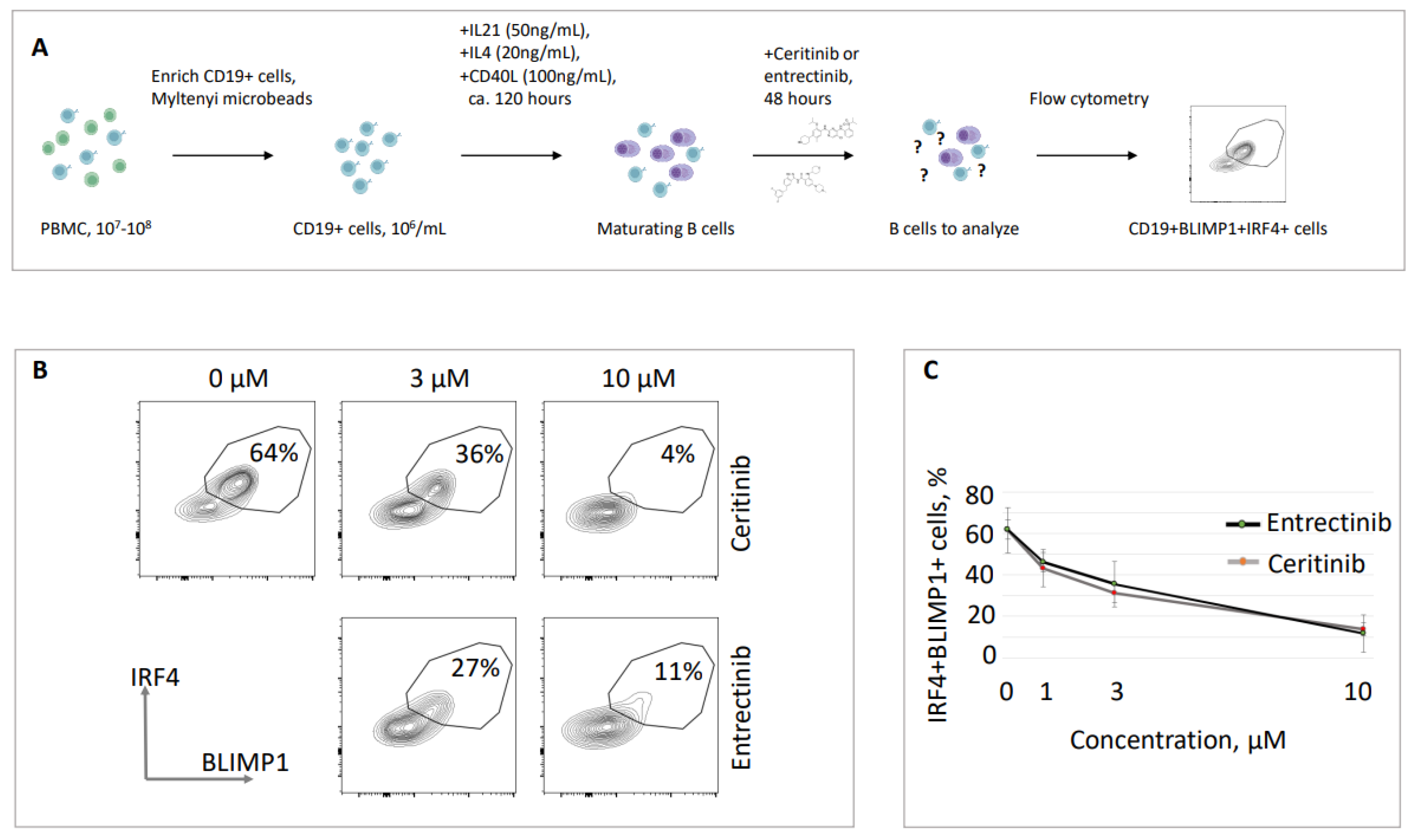
3.3. Validation of ceritinib and entrectinib inhibitory effects on plasma cells
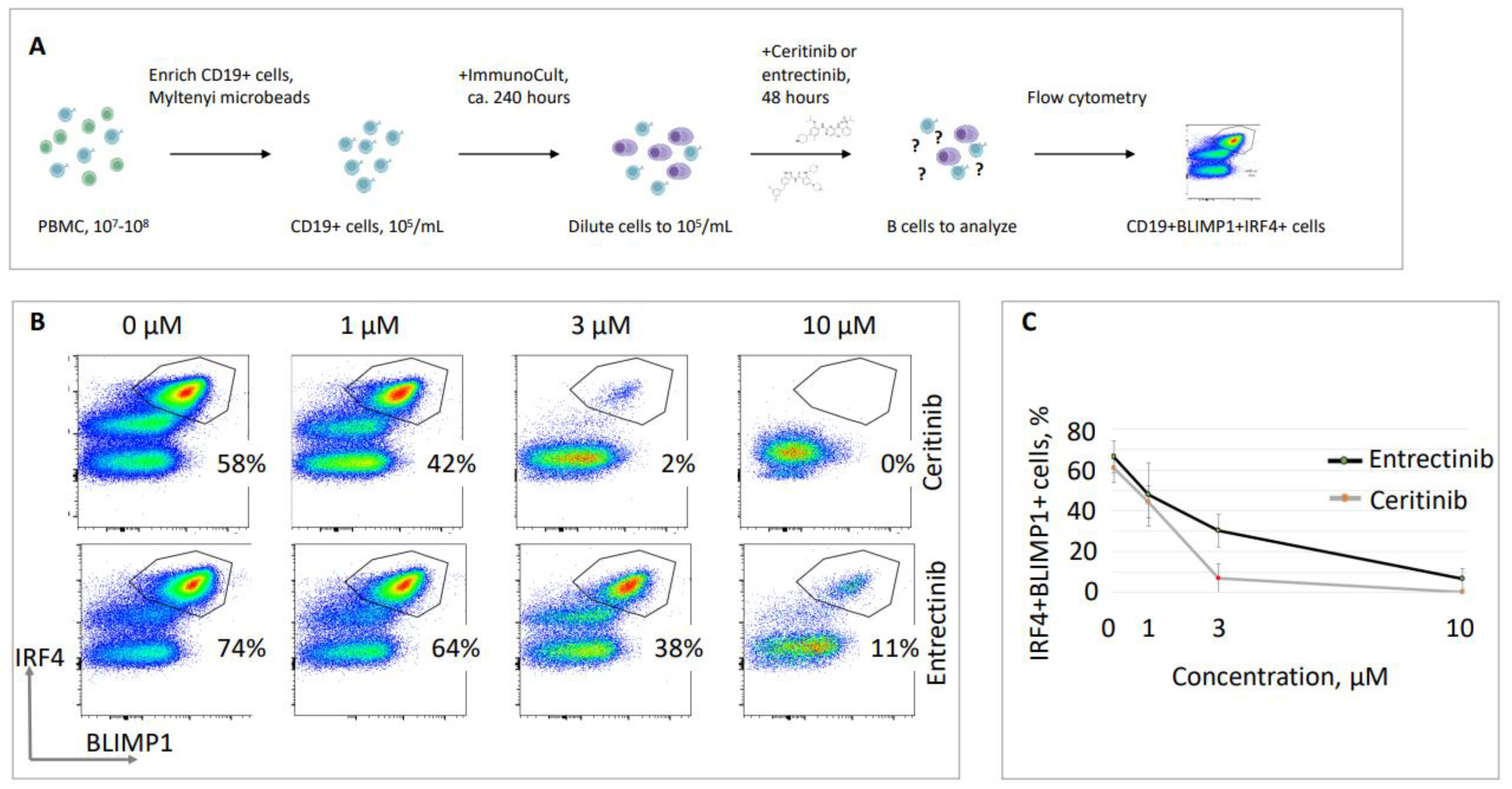
We then validated the previously obtained data on the inhibitory effects of ceritinib and entrectinib using alternative B cell stimulation methods described in Materials and Methods section 2.3, with a commercially available kit. We isolated CD19-positive B cells, stimulated the cells over ten days in cell culture, and then analyzed BLIMP1/IRF4 double-positive populations (plasma cells) inside the live CD19 populations (Figure 3). We detected relatively high proportions of BLIMP1/IRF4 populations (58% and 74%, as in the example in Figure 3). However, BLIMP1/IRF4 double-positive populations reduced after 48 hours of cell exposure to ceritinib, i.e. only 44±4.5% of cells were detected at a concentration of 1 µM, 7±4.0% at 3 µM, and 0±0.0% at 10 µM. Similarly, exposure to entrectinib resulted in only 33±4.6% of BLIMP1/IRF4 double-positive cells at 3 µM, and 7±4.0% at 10 µM (Figure 3). The experiments were performed at least three times using samples from different donors, and the representative populations are shown (Figure 3).
Altogether, we concluded that entrectinib and ceritinib inhibit the overall proliferation of CD19-positive B lymphocytes as measured by thymidine incorporation (Figure 1), as well as result in reduced populations of mature BLIMP1/IRF4 B cell populations (antibody-expressing plasma cells), stimulated using two alternative protocols (Figure 2 and Figure 3).
4. Discussion
Antibody-expressing B lymphocytes, including a more mature form, plasma cells, are therapeutic targets during severe autoimmune disorders, e.g. immune thrombocytopenia [3]. The treatment options include corticosteroids, rituximab targeting the majority of B cell populations expressing CD20, daratumumab targeting mature B cells expressing CD38, and sometimes splenectomy [3]. New and more effective methods of antibody-expressing B cells targeting are required.
Here, we tested several known tyrosine kinase inhibitors developed and approved to inhibit ALK-expressing cells. We used these inhibitors, however, to target ALK-negative and LTK-positive plasma cells that secrete a lot of proteins (antibodies) using ER [7]. The populations of BLIMP1/IRF4 double-positive plasma cells were reduced correlating with inhibitor concentrations (Figure 2 and Figure 3). Nevertheless, BLIMP1/IRF4-negative B cells, previously selected as CD19-positive, were alive (Figure 2 and Figure 3), suggesting some level of specificity against antibody-secreting cells.
We used the tyrosine kinase inhibitor concentrations available in literature [25] to optimize the protocols and to find suitable conditions for our experiments (Figure 1). The radioactivity assay based on the accumulation of 3H Thymidine was used for decades in different variations. However, it was criticised by several authors due to several limitations, i.e. irradiation of the cell components by the radioactivity itself [34]. Nevertheless, this assay is still widely used (e.g. [32,35,36,37]), in combination with other methods. Thus, we also used two flow cytometry-based methods to analyze the effects of tyrosine kinase inhibitors on stimulated B cells (Figure 2 and Figure 3).
ALK and LTK are structurally similar protein tyrosine kinases, involved in various processes, including oncogenic transformations and autoimmune disorders [38]. Recently, CLIP1-LTK fusion was identified as a driver of non-small cell lung cancer (NSCLC) [25]. While ALK is a well-established translocation partner in lung cancer, only initial knowledge is available regarding LTK-dependent lung cancers [23,25]. Similar to ALK-positive patients, some of the LTK-positive lung cancer patients had metastases to the central nervous system, and the smoking history in LTK-positive patients was variable [23,25]. Identification of LTK as a marker and target for lung cancer patients has several implications, including development of new drugs specifically targeting LTK, which in turn can boost the therapy of other LTK-positive pathologies. All the drugs used in our current study, i.e. alectinib, brigatinib, ceritinib, crizotinib, entrectinib, and lorlatinib, were originally developed to target ALK, and appeared to reduce LTK phosphorylation in the context of CLIP1-LTK [25]. However, lorlatinib was selected as the best drug against CLIP1-LTK-induced cancer. Contrary, in our studies, lorlatinib had the lowest effect on B cell proliferation (Figure 1).
Previously, we demonstrated that multiple drugs can be reused as potential antivirals [11,12,13,14,15,16,17,18,19,39,40,41,42,43]. Our current study is the first one to demonstrate that ALK inhibitors are acting on ALK-negative but LTK-positive [8] plasma cells. It opens options for further drug repurposing, potentially considering all available inhibitors against ALK to be used against LTK-positive cells, and, in particular, in cases of B-cell dependent disorders, such as autoimmune diseases (e.g., immune thrombocytopenia) or cancer (e.g., multiple myeloma).
Moreover, during the B lymphocyte development, in the processes known as the V(D)J recombination and class switch recombination, there are many cases of known genetic interaction [4,5,6]. Previously, synthetic lethality between the DNA repair proteins Poly(ADP-Ribose) Polymerase 1 (PARP1) and Breast cancer type 1 (BRCA1) resulted in the development of an anti-cancer drug, olaparib [44]. Today, genetic interaction is known in B lymphocytes between NHEJ factors and DNA damage response proteins (DDRs), for example, between XLF and Ataxia telangiectasia mutated (ATM), histone H2AX [45], a mediator of DNA damage checkpoint protein 1 (MDC1) [29], p53-binding factor 1 (53BP1) [46], PAXX [31], and MRI [47]. Thus, it is also possible to look for the B lymphocyte targeting options using known and to be discovered genetic interactions, including synthetic lethality, in the pathways required for B cell maturation, including the V(D)J recombination and class switching.
During the first screening (Figure 1), we identified several tyrosine kinase inhibitors resulting in reduced rate of proliferation of B cell populations, including alectinib, brigatinib, ceritinib, crizotinib, and entrectinib. To validate the findings using two other methods, we focused on ceritinib and entrectinib. However, alectinib, brigatinib, and crizotinib are also good candidates to be tested in the following studies, in addition to multiple similar tyrosine kinase inhibitors available and reported in the literature (e.g., [25]). The cells respond to ceritinib (3 µM) differently in different experimental settings (Figure 2 and Figure 3). However, it is hard to compare these data points because the experiments were performed using samples from different donors and the timing of the experiments differs (seven days for Figure 2 and twelve days for Figure 3). In both cases, ceritinib had a clear effect on B cell populations, validating the data in Figure 1.
The populations of the cells presented in Figure 2 and Figure 3 are not identical. We explain the different shapes of the cell populations for the following reasons. First, the stimulation cocktail used was different, i.e. as described in the Materials and Methods for Figure 2, and a commercially available kit (ImmunoCult) for Figure 3. Moreover, the cells described in Figure 2 were incubated for five days before the inhibitors were added for another two days. Differently, the cells in Figure 3 were incubated ten days before the inhibitors were added. The cell fixation and population gating procedures, nevertheless, were the same for both types of experiments (Materials and Methods, and Supplementary Figures S1 and S2). In particular, dead cells and debris were always excluded from the analyses, and each data point represents an individual donor.
In this study, we focused on tyrosine kinase inhibitors targeting human B lymphocytes. However, it is possible that the same inhibitors will be targeting other cell types, e.g. T lymphocytes, fibroblasts, etc. It cannot be excluded, because LTK can be expressed by various cell types, and because the inhibitors are promiscuous and can potentially target different enzymes, including in B cells. What is clear, however, is that these inhibitors do not inhibit proliferation of all the cells (Figure 2 and Figure 3), but rather some cell populations are more sensitive (e.g., BLIMP1+IRF+) than others (e.g., BLIMP1-IRF-).
Further studies will include broader use of ALK inhibitors against LTK-positive cells, and potentially vice versa. Following the cell-based experiments, pre-clinical studies will be done on animal models. However, the drugs are already approved to be used in human patients, although to treat different diseases, which is why future approval of the treatment options would be simpler.
5. Conclusions
Tyrosine kinase inhibitors alectinib, brigatinib, ceritinib, crizotinib, and entrectinib lead to reduced proliferation levels of mature human B cells in cell culture. Ceritinib and entrectinib eliminate BLIMP1/IRF4 double-positive B cell populations (antibody-secreting plasma cells) at concentrations that allow BLIMP1/IRF4-negative B cells to survive. We validated that ceritinib and entrectinib target plasma cells using two independent B-cell stimulation methods.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Institutional Review Board Statement
Buffy coat samples were obtained from healthy adult blood donors. Ethical approval was obtained from the Regional Ethics Committee in South-Eastern Norway (2014/840/REK and 2016/905/REK). Informed consent was obtained from all subjects prior to donation.
Conflicts of Interest
The authors declares no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Alt, F.W.; Zhang, Y.; Meng, F.L.; Guo, C.; Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013, 152, 417-429. [CrossRef]
- Rajewsky, K. Clonal selection and learning in the antibody system. Nature 1996, 381, 751-758. [CrossRef]
- Terrell, D.R.; Neunert, C.E.; Cooper, N.; Heitink-Polle, K.M.; Kruse, C.; Imbach, P.; Kuhne, T.; Ghanima, W. Immune Thrombocytopenia (ITP): Current Limitations in Patient Management. Medicina (Kaunas) 2020, 56. [CrossRef]
- Kumar, V.; Alt, F.W.; Oksenych, V. Functional overlaps between XLF and the ATM-dependent DNA double strand break response. DNA Repair (Amst) 2014, 16, 11-22. [CrossRef]
- Castaneda-Zegarra, S.; Fernandez-Berrocal, M.; Tkachov, M.; Yao, R.; Upfold, N.L.E.; Oksenych, V. Genetic interaction between the non-homologous end-joining factors during B and T lymphocyte development: In vivo mouse models. Scand J Immunol 2020, 92, e12936. [CrossRef]
- Wang, X.S.; Lee, B.J.; Zha, S. The recent advances in non-homologous end-joining through the lens of lymphocyte development. DNA Repair (Amst) 2020, 94, 102874. [CrossRef]
- Centonze, F.G.; Reiterer, V.; Nalbach, K.; Saito, K.; Pawlowski, K.; Behrends, C.; Farhan, H. LTK is an ER-resident receptor tyrosine kinase that regulates secretion. J Cell Biol 2019, 218, 2470-2480. [CrossRef]
- Roll, J.D.; Reuther, G.W. ALK-activating homologous mutations in LTK induce cellular transformation. PLoS One 2012, 7, e31733. [CrossRef]
- Farhan, H.O., NO), Munthe, Ludvig (Fjellhamar, NO), Tasken, Kjetil (Rykkinn, NO), Skanland, Sigrid (Oslo, NO), Giliberto, Mariaserena (Oslo, NO), Hellem Schjesvold, Fredrik (Jar, NO), Driessen, Christoph (St. Gallen, CH), Besse, Lenka (St. Gallen, CH), Besse, Andrej (St. Gallen, CH). ALK INHIBITORS FOR TREATMENT OF ALK-NEGATIVE CANCER AND PLASMA CELL-MEDIATED DISEASES. 2022.
- Tao, J.; Zheng, C.; Zhang, C.; Zhou, L.; Liu, Z.; Zhou, Y.; Huang, X.; Lin, L.; Zhai, L. First-line treatments for patients with advanced ALK gene rearrangements in NSCLC: a systematic review and network meta-analysis. J Int Med Res 2022, 50, 3000605221132703. [CrossRef]
- Ianevski, A.; Simonsen, R.M.; Myhre, V.; Tenson, T.; Oksenych, V.; Bjoras, M.; Kainov, D.E. DrugVirus.info 2.0: an integrative data portal for broad-spectrum antivirals (BSA) and BSA-containing drug combinations (BCCs). Nucleic Acids Res 2022. [CrossRef]
- Ianevski, A.; Yao, R.; Simonsen, R.M.; Myhre, V.; Ravlo, E.; Kaynova, G.D.; Zusinaite, E.; White, J.M.; Polyak, S.J.; Oksenych, V.; et al. Mono- and combinational drug therapies for global viral pandemic preparedness. iScience 2022, 25, 104112. [CrossRef]
- Ianevski, A.; Zusinaite, E.; Tenson, T.; Oksenych, V.; Wang, W.; Afset, J.E.; Bjoras, M.; Kainov, D.E. Novel Synergistic Anti-Enteroviral Drug Combinations. Viruses 2022, 14. [CrossRef]
- Ianevski, A.; Yao, R.; Lysvand, H.; Grodeland, G.; Legrand, N.; Oksenych, V.; Zusinaite, E.; Tenson, T.; Bjoras, M.; Kainov, D.E. Nafamostat-Interferon-alpha Combination Suppresses SARS-CoV-2 Infection In Vitro and In Vivo by Cooperatively Targeting Host TMPRSS2. Viruses 2021, 13. [CrossRef]
- Ianevski, A.; Yao, R.; Zusinaite, E.; Lello, L.S.; Wang, S.; Jo, E.; Yang, J.; Ravlo, E.; Wang, W.; Lysvand, H.; et al. Synergistic Interferon-Alpha-Based Combinations for Treatment of SARS-CoV-2 and Other Viral Infections. Viruses 2021, 13. [CrossRef]
- Ianevski, A.; Yao, R.; Zusinaite, E.; Lysvand, H.; Oksenych, V.; Tenson, T.; Bjoras, M.; Kainov, D. Active Components of Commonly Prescribed Medicines Affect Influenza A Virus-Host Cell Interaction: A Pilot Study. Viruses 2021, 13. [CrossRef]
- Oksenych, V.; Kainov, D.E. Broad-Spectrum Antivirals and Antiviral Drug Combinations. Viruses 2022, 14. [CrossRef]
- Kainov, D.; Oksenych, V. Broad-Spectrum Antivirals and Antiviral Combinations: An Editorial Update. Viruses 2022, 14. [CrossRef]
- Ianevski, A.; Kulesskiy, E.; Krpina, K.; Lou, G.; Aman, Y.; Bugai, A.; Aasumets, K.; Akimov, Y.; Bulanova, D.; Gildemann, K.; et al. Chemical, Physical and Biological Triggers of Evolutionary Conserved Bcl-xL-Mediated Apoptosis. Cancers (Basel) 2020, 12. [CrossRef]
- Ianevski, A.; Yao, R.; Biza, S.; Zusinaite, E.; Mannik, A.; Kivi, G.; Planken, A.; Kurg, K.; Tombak, E.M.; Ustav, M., Jr.; et al. Identification and Tracking of Antiviral Drug Combinations. Viruses 2020, 12. [CrossRef]
- Ianevski, A.; Yao, R.; Fenstad, M.H.; Biza, S.; Zusinaite, E.; Reisberg, T.; Lysvand, H.; Loseth, K.; Landsem, V.M.; Malmring, J.F.; et al. Potential Antiviral Options against SARS-CoV-2 Infection. Viruses 2020, 12. [CrossRef]
- Ianevski, A.; Ahmad, S.; Anunnitipat, K.; Oksenych, V.; Zusinaite, E.; Tenson, T.; Bjoras, M.; Kainov, D.E. Seven classes of antiviral agents. Cell Mol Life Sci 2022, 79, 605. [CrossRef]
- Cooper, A.J.; Sequist, L.V.; Johnson, T.W.; Lin, J.J. LTK fusions: A new target emerges in non-small cell lung cancer. Cancer Cell 2022, 40, 23-25. [CrossRef]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet 2021, 398, 535-554. [CrossRef]
- Izumi, H.; Matsumoto, S.; Liu, J.; Tanaka, K.; Mori, S.; Hayashi, K.; Kumagai, S.; Shibata, Y.; Hayashida, T.; Watanabe, K.; et al. The CLIP1-LTK fusion is an oncogenic driver in non-small-cell lung cancer. Nature 2021, 600, 319-323. [CrossRef]
- Szodoray, P.; Andersen, T.K.; Heinzelbecker, J.; Imbery, J.F.; Huszthy, P.C.; Stanford, S.M.; Bogen, B.; Landsverk, O.B.; Bottini, N.; Tveita, A.; et al. Integration of T helper and BCR signals governs enhanced plasma cell differentiation of memory B cells by regulation of CD45 phosphatase activity. Cell Rep 2021, 36, 109525. [CrossRef]
- Dewan, A.; Xing, M.; Lundbaek, M.B.; Gago-Fuentes, R.; Beck, C.; Aas, P.A.; Liabakk, N.B.; Saeterstad, S.; Chau, K.T.P.; Kavli, B.M.; et al. Robust DNA repair in PAXX-deficient mammalian cells. FEBS Open Bio 2018, 8, 442-448. [CrossRef]
- Gago-Fuentes, R.; Xing, M.; Saeterstad, S.; Sarno, A.; Dewan, A.; Beck, C.; Bradamante, S.; Bjoras, M.; Oksenych, V. Normal development of mice lacking PAXX, the paralogue of XRCC4 and XLF. FEBS Open Bio 2018, 8, 426-434. [CrossRef]
- Beck, C.; Castaneda-Zegarra, S.; Huse, C.; Xing, M.; Oksenych, V. Mediator of DNA Damage Checkpoint Protein 1 Facilitates V(D)J Recombination in Cells Lacking DNA Repair Factor XLF. Biomolecules 2019, 10. [CrossRef]
- Castaneda-Zegarra, S.; Huse, C.; Rosand, O.; Sarno, A.; Xing, M.; Gago-Fuentes, R.; Zhang, Q.; Alirezaylavasani, A.; Werner, J.; Ji, P.; et al. Generation of a Mouse Model Lacking the Non-Homologous End-Joining Factor Mri/Cyren. Biomolecules 2019, 9. [CrossRef]
- Castaneda-Zegarra, S.; Xing, M.; Gago-Fuentes, R.; Saeterstad, S.; Oksenych, V. Synthetic lethality between DNA repair factors Xlf and Paxx is rescued by inactivation of Trp53. DNA Repair (Amst) 2019, 73, 164-169. [CrossRef]
- Fauskanger, M.; Haabeth, O.A.W.; Skjeldal, F.M.; Bogen, B.; Tveita, A.A. Tumor Killing by CD4(+) T Cells Is Mediated via Induction of Inducible Nitric Oxide Synthase-Dependent Macrophage Cytotoxicity. Front Immunol 2018, 9, 1684. [CrossRef]
- Khoenkhoen, S.; Adori, M.; Pedersen, G.K.; Karlsson Hedestam, G.B. Flow Cytometry-Based Protocols for the Analysis of Human Plasma Cell Differentiation. Front Immunol 2020, 11, 571321. [CrossRef]
- Hu, V.W.; Black, G.E.; Torres-Duarte, A.; Abramson, F.P. 3H-thymidine is a defective tool with which to measure rates of DNA synthesis. FASEB J 2002, 16, 1456-1457. [CrossRef]
- Jacobsen, J.; Haabeth, O.A.; Tveita, A.A.; Schjetne, K.W.; Munthe, L.A.; Bogen, B. Naive idiotope-specific B and T cells collaborate efficiently in the absence of dendritic cells. J Immunol 2014, 192, 4174-4183. [CrossRef]
- Tveita, A.A.; Schjesvold, F.; Haabeth, O.A.; Fauskanger, M.; Bogen, B. Tumors Escape CD4+ T-cell-Mediated Immunosurveillance by Impairing the Ability of Infiltrating Macrophages to Indirectly Present Tumor Antigens. Cancer Res 2015, 75, 3268-3278. [CrossRef]
- Tveita, A.A.; Schjesvold, F.H.; Sundnes, O.; Haabeth, O.A.; Haraldsen, G.; Bogen, B. Indirect CD4+ T-cell-mediated elimination of MHC II(NEG) tumor cells is spatially restricted and fails to prevent escape of antigen-negative cells. Eur J Immunol 2014, 44, 2625-2637. [CrossRef]
- De Munck, S.; Provost, M.; Kurikawa, M.; Omori, I.; Mukohyama, J.; Felix, J.; Bloch, Y.; Abdel-Wahab, O.; Bazan, J.F.; Yoshimi, A.; et al. Structural basis of cytokine-mediated activation of ALK family receptors. Nature 2021, 600, 143-147. [CrossRef]
- Andersen, P.I.; Ianevski, A.; Lysvand, H.; Vitkauskiene, A.; Oksenych, V.; Bjoras, M.; Telling, K.; Lutsar, I.; Dumpis, U.; Irie, Y.; et al. Discovery and development of safe-in-man broad-spectrum antiviral agents. Int J Infect Dis 2020, 93, 268-276. [CrossRef]
- Ianevski, A.; Zusinaite, E.; Shtaida, N.; Kallio-Kokko, H.; Valkonen, M.; Kantele, A.; Telling, K.; Lutsar, I.; Letjuka, P.; Metelitsa, N.; et al. Low Temperature and Low UV Indexes Correlated with Peaks of Influenza Virus Activity in Northern Europe during 2010(-)2018. Viruses 2019, 11. [CrossRef]
- Bosl, K.; Ianevski, A.; Than, T.T.; Andersen, P.I.; Kuivanen, S.; Teppor, M.; Zusinaite, E.; Dumpis, U.; Vitkauskiene, A.; Cox, R.J.; et al. Common Nodes of Virus-Host Interaction Revealed Through an Integrated Network Analysis. Front Immunol 2019, 10, 2186. [CrossRef]
- Ianevski, A.; Zusinaite, E.; Kuivanen, S.; Strand, M.; Lysvand, H.; Teppor, M.; Kakkola, L.; Paavilainen, H.; Laajala, M.; Kallio-Kokko, H.; et al. Novel activities of safe-in-human broad-spectrum antiviral agents. Antiviral Res 2018, 154, 174-182. [CrossRef]
- Bulanova, D.; Ianevski, A.; Bugai, A.; Akimov, Y.; Kuivanen, S.; Paavilainen, H.; Kakkola, L.; Nandania, J.; Turunen, L.; Ohman, T.; et al. Antiviral Properties of Chemical Inhibitors of Cellular Anti-Apoptotic Bcl-2 Proteins. Viruses 2017, 9. [CrossRef]
- Helleday, T. PARP inhibitor receives FDA breakthrough therapy designation in castration resistant prostate cancer: beyond germline BRCA mutations. Ann Oncol 2016, 27, 755-757. [CrossRef]
- Zha, S.; Guo, C.; Boboila, C.; Oksenych, V.; Cheng, H.L.; Zhang, Y.; Wesemann, D.R.; Yuen, G.; Patel, H.; Goff, P.H.; et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2011, 469, 250-254. [CrossRef]
- Oksenych, V.; Alt, F.W.; Kumar, V.; Schwer, B.; Wesemann, D.R.; Hansen, E.; Patel, H.; Su, A.; Guo, C. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci U S A 2012, 109, 2455-2460. [CrossRef]
- Castaneda-Zegarra, S.; Zhang, Q.; Alirezaylavasani, A.; Fernandez-Berrocal, M.; Yao, R.; Oksenych, V. Leaky severe combined immunodeficiency in mice lacking non-homologous end joining factors XLF and MRI. Aging (Albany NY) 2020, 12, 23578-23597. [CrossRef]
Figure 3.
Reduced proportions of IRF4+BLIMP1+ cells in the presence of tyrosine kinase inhibitors ceritinib and entrectinib. (A) Schematic overview of the experiment. (B) Representative live CD19-positive B cell populations labeled with BLIMP1 and IRF4 to select mature antibody-secreting B cells (plasma cells). Exposure to increasing concentrations of ceritinib and entrectinib (1 µM, 3 µM, and 10 µM) resulted in reduced populations of BLIMP1/IRF4 double-positive mature B lymphocytes. (C) Summary of three experiments similar to one shown in (B) with standard deviation. Samples from different donors were used to generate every data point.
Figure 3.
Reduced proportions of IRF4+BLIMP1+ cells in the presence of tyrosine kinase inhibitors ceritinib and entrectinib. (A) Schematic overview of the experiment. (B) Representative live CD19-positive B cell populations labeled with BLIMP1 and IRF4 to select mature antibody-secreting B cells (plasma cells). Exposure to increasing concentrations of ceritinib and entrectinib (1 µM, 3 µM, and 10 µM) resulted in reduced populations of BLIMP1/IRF4 double-positive mature B lymphocytes. (C) Summary of three experiments similar to one shown in (B) with standard deviation. Samples from different donors were used to generate every data point.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



