
Submitted:
24 February 2023
Posted:
27 February 2023
You are already at the latest version
Abstract
Neurodegeneration is an age-dependent progressive phenomenon with no defined cause. Aging is the main risk factor for neurodegenerative diseases. During aging, activated microglia undergoes phenotypic alterations that can lead to neuroinflammation, which is well accepted event in the pathogenesis of neurodegenerative diseases. Several common mechanisms are shared by genetically or pathologically distinct neurodegenerative diseases, such as excitotoxicity, mitochondrial deficits and oxidative stress, protein misfolding and translational dysfunction, autophagy and microglia activation. Progressive loss of neuronal population due to increased oxidative stress leads to neurodegenerative diseases mostly due to the accumulation of dysfunctional mitochondria. Mitochondrial dysfunction and excessive neuroinflammatory responses are both sufficient to induce pathology in age-dependent neurodegeneration. Therefore, mitochondrial quality control is key determinant for the health and survival of neuronal cells in the brain. Research has been primarily focused to demonstrate the significance of neuronal mitochondrial health, despite the important contributions of non-neuronal cells that constitutes significant portion of the brain volume. Moreover, mitochondrial morphology and function are distinctly diverse in different tissues; however, little is known about their molecular diversity among cell types. Mitochondrial dynamics and quality in different cell types markedly decides the fate of overall brain health, therefore it is not justifiable to overlook non-neuronal cells and their significant and active contribution in facilitating overall neuronal health. In this review article, we aim to discuss the mitochondrial quality control of different cell types in the brain and how important and remarkable is the diversity and highly synchronized connecting property of non-neuronal cells in keeping the neurons healthy to control neurodegeneration.
Keywords:
Astrocytes
; Microglia
; Mitochondria
; Neurons
; Oligodendrocytes
; Oxidative stress
1. Introduction
Mitochondrial dysfunction and oxidative stress are considered as the key central mechanism of progressive cell loss in several neurodegenerative diseases. Accumulation of dysfunctional mitochondria in neurons increases the steady-state levels of reactive oxygen species derived from the leakage of electrons from the electron transport chain. Moreover, neurons are solely dependent on oxidative phosphorylation to keep their energy requirements during healthy conditions. In the nervous system, mitochondria regulate neurite branching and regeneration [1], as well as synaptic strength, stability and signaling [2]. Therefore, mitochondrial quality control and bioenergetic demand in the CNS (central nervous system) are important factors to keep the brain healthy. Although, neurons are predominantly highly specialized cells in the brain due to their unique property of transmitting, processing, and storing information, however they are entirely dependent on supportive glial cells for their bioenergetic demand (Figure 1).
Nonetheless, the effects of mitochondrial dysfunction in non-neuronal cells (astocytes, microglia and oligodenrocytes) and its implication for neuronal homeostasis and brain function has been largely understudied. Research within the last few decades have demonstrated the indispensable role of the glial cells in the CNS function. Unlike neurons and oligodendrocytes, astrocytes and microglia remain actively healthy upon the impairment of their mitochondrial function, due to their higher antioxidant capacity than neurons primarily on account of glycolysis to produce energy. Recent evidence highlights the role of mitochondrial metabolism and signaling in glial cell function. Non-neuronal cells support essential brain functions such as maintenance of neurotransmitter communication, metabolism, trophic support, formation of myelin sheath, wound healing, and immune surveillance [3]. Astrocytes are the major secretory cells characterized by their defined characteristic of releasing factors or transmitters (gliotransmitters) including neurotransmitters (and their precursors), modulators, peptides, hormones, trophic (growth) factors and metabolites. Astrocytic secreted factors are involved in synapse formation, function and plasticity, neuronal growth, differentiation, and survival, as well as in the regulation of the vascular tone and blood flow in the brain. Additionally, astrocytes also mediate synaptic pruning by phagocytosis [4,5]. To prevent any inflammatory impact on neurons, microglia act as the first line of defense. Besides, microglial activation is a dynamic event and plays a central role in neuroinflammation, which can be beneficial or pathogenic for neuronal health. Microglia are tissue resident macrophages (innate immunity) and universal sensors of alterations in CNS physiology. Dynamic healthy microglial mitochondria population is the key player in neuroinflammation. In response to pathogen or damage signals, microglia undergo rapid activation and can acquire different phenotypes exerting neuroprotection or neurotoxicity. In an event of neuroinflammation, activated microglia and astrocytes release soluble mediators such as cytokines, glutamate, and ROS (reactive oxygen species) that negatively affect neuronal function and viability, and thus contribute to neurodegeneration during disease progression. In context to pathogenesis of neurodegenerative diseases, abnormal accumulation of α-syn (alpha-synuclein) aggregates in neurons and glial cells is widely known to be associated with PD (Parkinson’s disease), DLB (dementia with Lewy body) and MSA (multiple system atrophy). Mitochondrial dysfunction in neurons and glia is known as a key feature of α-syn toxicity. Studies aimed at understanding α-syn induced toxicity and its role in neurodegenerative diseases have primarily focused on neurons. However, a growing body of evidence demonstrates that glial cells such as microglia and astrocytes have been implicated in the initial pathogenesis and the progression of synucleinopathy. Furthermore, recent studies highlight the role of mitochondrial metabolism in the normal function of glial cells. Therefore, the complex relationship between glial mitochondria and α-syn mediated neurodegeneration may provide a novel insight into the roles of glial cells in α-syn associated neurodegenerative diseases [6]. Neuronal oxidative stress has been implicated in aging and neurodegenerative disease. Mitochondrial stress related to mtDNA (mitochondrial DNA) dysregulation can produce neuronal dysfunction and death via impaired ETC (electron transport chain) activity, which results in deficient ATP (adenosine triphosphate) production and related increases in mitochondrial ROS production. However, mtDNA dysregulation in post-mitotic neurons may also produce disturbances in mitochondrial homeostasis that are known to impair neuronal function as well. Mitochondria are key contributors to the etiology of diseases associated with neuromuscular defects or neurodegeneration. Well-balanced mitochondrial fission and fusion processes are essential for nervous system development. The mitochondrial anchor protein SNPH (syntaphilin) is a key mitochondrial protein normally expressed in axons to maintain neuronal health by positioning mitochondria along axons for metabolic needs. Mitochondrial dysfunction has been implicated in the degeneration of dopamine neurons in PD. Selective neuronal vulnerability of specific neuroanatomical regions such as frontal cortex and hippocampus is characteristic of age-associated neurodegenerative diseases, however the basis of pathogenesis remains unresolved. In neurons, cofilin1 a key regulator of actin dynamics may contribute to degenerative processes through formation of cofilin-actin rods, and through enhanced mitochondrial fission, mitochondrial membrane permeabilization, and the release of cytochrome C. Overall, mitochondrial impairment induced by dysfunction of actin-regulating proteins such as cofilin1 emerge as important mechanisms of neuronal death with relevance to acute brain injury and neurodegenerative diseases, such as PD or AD (Alzheimer’s disease) [7]. Oligodendrocytes are myelinating cells of the CNS supporting neuronal survival. Oxidants and mitochondrial dysfunction have been suggested as the main causes of oligodendrocyte damage during neuroinflammation as observed in MS (multiple sclerosis). NCX (Na+-Ca2+ exchanger) isoforms constitute the major cellular Ca2+ extruding system in neurons and microglia. Double immunostaining studies showed that in A53T mice (expressing human A53T variant of α-synuclein), NCX1 was exclusively co-expressed in IBA-1-positive microglial cells in the striatum, whereas NCX3 was solely co-expressed in TH-positive neurons in SNpc. in vivo and in vitro findings suggest that the reduction in NCX3 expression and activity in A53T neurons from midbrain may cause mitochondrial dysfunction and neuronal death in this brain area, whereas NCX1 overexpression in microglial cells may promote their proliferation in the striatum [8]. Although some of these cell-type-specific functions are achieved by modulating the well-characterized metabolic roles of mitochondria, less-expected functions emerge, for example, in immune or redox signaling [9], and likely many more remain elusive. Accordingly, mitochondria in different cells or cell types, or even in different parts of the same cell, can behave quite differently.
2. Astrocytic mitochondria and their contribution in neurodegeneration
Astrocytes constitute about 20-40% of the brain cells, depending on the counting methodology and brain region. Due to their dynamic neuroinflammatory activity, astrocytes are often considered as secondary responders to neurodegeneration and are emerging as primary drivers of several brain diseases. Recent evidence suggests that astrocytes, which play a key role in supporting neuronal function and metabolism, might contribute to the development of neurodegenerative diseases [10]. Under activated state, not only microglia but astrocytes also release soluble mediators such as cytokines, glutamate, and ROS that negatively affect neuronal function and viability, and thus contribute to neurodegeneration during disease progression [11]. Mitochondrial dysfunction in astrocytes contributes to neuroinflammatory activation of astrocytes. Likewise, many astrocytic functions, including glutamate metabolism, Ca2+ signaling, fatty acid metabolism, antioxidant production, and inflammation are dependent on healthy mitochondria. Research on astrocytic mitochondrial proteins and their importance in signaling mechanisms have been correlated with neurodegeneration. Astrocytic projections form functionally isolated microdomains, thus facilitating local homeostasis by redistributing ions, removing neurotransmitters, and releasing factors to regulate blood flow and neuronal activity. Microdomains exhibit a spontaneous increase in Ca2+ ensuring local metabolic support to astrocytes for energetically demanding processes [12]. Defective mitochondrial biogenesis and function along with increased ROS generation are important determinants of aging. Also, there is a concomitant decline in circulating levels of IGF-1 (insulin-like growth factor-1) that is closely associated with neuronal aging and neurodegeneration. As regulation of astrocytic mitochondrial function and redox status by IGF-1 is essential to maintain astrocytic function and coordinate hippocampal-dependent spatial learning, downregulation of IGF-1 receptor expression with age is associated with diminished hippocampal-dependent learning and increased gliosis [13]. It has been suggested that impairment of mitochondrial homeostasis in astrocytes contributes to neurodegeneration. The m-AAA (mitochondrial AAA) proteases exerts quality control and regulatory functions vital for mitochondrial homeostasis. AFG3L2, which encodes one of the subunits of the m-AAA protease, is mutated in spinocerebellar ataxia and in infantile syndromes characterized by spastic-ataxia, epilepsy and premature death. Astrocyte dysfunction amplifies both neuroinflammation and glutamate excitotoxicity in patients carrying mutations in AFG3L2, leading to a vicious circle that contributes to neuronal death [14]. FTD3 (Frontotemporal dementia type 3) is amongst the most prevalent early onset dementias which is clinically, pathologically and genetically heterogeneous. Markedly, FTD3 patient derived astrocytes demonstrated the dysregulation of glutamate-glutamine homeostasis, impaired mitochondria function and glutamine hypermetabolism [15]. In another study on FTD3, it was shown that a point mutation in the CHMP2B (charged multivesicular body protein 2B), causes aberrant autophagy that in turn triggers perturbed mitochondrial dynamics with impaired glycolysis, increased ROS and elongated mitochondrial morphology, indicating increased mitochondrial fusion in FTD3 astrocytes. This alteration in astrocyte homeostasis prompts a reactive astrocyte phenotype and elevated release of toxic cytokines, which accumulate in NF-κB (nuclear factor kappa b) pathway activation leading to neurodegeneration [16] (Figure 2).
Reactive astrocytes in ALS (Amyotrophic Lateral Sclerosis) alters their molecular expression pattern and release toxic factors contributing to neurodegeneration and microglial activation. Replenishment of miR-146a in the cortical astrocytes of mSOD1 (symptomatic SOD1-G93A) with pre-miR-146a or by dipeptidyl vinyl sulfone abrogates their phenotypic aberrancies and paracrine deleterious consequences to motor neurons and microglia [17]. MNs (motor neurons) and astrocytes are implicated in the pathogenesis of ALS. In ALS patient-specific VCP (valosin-containing protein)-mutant MNs, a significant increase in cytoplasmic TDP-43 (TAR DNA-binding protein 43) levels and ER (endoplasmic reticulum) stress was observed as primary pathogenic events as well as secondary mitochondrial dysfunction and oxidative stress [18]. In an in vitro model of HD (Huntington’s disease) i.e., 3NP (3-nitropropionic acid, a mitochondrial toxin) treatment induces secretion of soluble neuroprotective factors in response to BDNF (brain-derived neurotrophic factor) in striatal astrocytes that selectively protect neurons expressing mutant huntingtin [19]. Under normal physiological conditions, astrocytes internalize and clears neuronal mitochondria in a specialized process called trans-mitophagy. In AD mouse brains, a significant increase in internalization of neuronal mitochondria in astrocytes and increased degradation of neuronal mitochondria by astrocytes suggest the involvement of S100a4 in impaired mitochondrial transfer between neurons and AD astrocytes along with significant increase in the mitophagy regulator and ROS in aged AD astrocytes [20]. FA (Friedreich’s ataxia) is a recessive, predominantly neurodegenerative disorder caused by mutations in the first intron of the Fxn (frataxin) gene. Fxn is a ubiquitous mitochondrial protein involved in iron-sulfur cluster biogenesis and a decrease in the levels of this protein is responsible for the symptoms observed in the disease. In vitro knockdown of Fxn, leads to a decrease in both mRNA and protein expression, along with mitochondrial superoxide production and present signs of p53-mediated cell cycle arrest and apoptotic cell death in a non-cell autonomous neuron-glia interaction [21]. mtDNA depletion in astrocytes attenuates their primary cilium. Oxidative phosphorylation deficiency in astrocytes stimulates FOXJ1 and RFX transcription factors which are known as master regulators of motile ciliogenesis. During stress, primary cilium elongates and become remarkably distorted due to a chronic activation of the mitochondrial integrated stress response in astrocytes that drives anabolic metabolism and promotes ciliary elongation suggesting metabolic ciliopathy is a novel pathomechanism for mitochondria-related neurodegenerative diseases [22].
3. Microglial mitochondria and their significance in neurodegeneration
Microglia are highly plastic brain cells and perform a distinct range of specialized functions in CNS that require high energy demand regulated by mitochondria. Microglia are extremely metabolically flexible and capable of reprogramming their mitochondrial function upon inflammatory activation to meet their energy demands. Activated microglia promote a neurotoxic, inflammatory environment in the mammalian CNS that drives the pathogenesis of neurodegenerative diseases. Accumulation of microglial mtDNA oxidative damage during aging and increased intracellular ROS production activates the redox-sensitive NF-kB to enhance neuroinflammation (Figure 2). Inflammasome activation of microglia leads to increased ROS generation with a loss of mitochondrial membrane potential and mitochondrial integrity [23]. Mitochondria-derived ROS and cathepsin B, are also necessary for the microglial cell production of interleukin-1β, a key inflammatory cytokine. Supporting the neuroinflammatory role of microglia in neurodegeneration, one of the studies demonstrated an increased expression of cytokines TGFβ1 (transforming growth factor-beta 1) in microglia and an exacerbated inflammatory response, Smad3 signaling to pathological changes in aged mice [24]. In microglia, ROS are generated primarily by NOX2 (NADPH oxidase 2) and activation of NOX2 is associated with mitochondrial DAMPs (damage-associated molecular patterns) signalling, inflammation and amyloid plaque deposition, especially in the cerebrovasculature. Additionally, ROS originating from both NOX and the mitochondria may act as secondary messengers to propagate immune activation; thus intracellular ROS signaling may underlie excessive inflammation and oxidative stress [25]. In a crosstalk study between neuronal and non-neuronal cells, mitochondrial DAMPs have been shown to initiate proinflammatory immune responses from non-neuronal glial cells, including microglia and astrocytes; thereby enlightening their significant contribution to the chronic neuroinflammation and accelerating the neurodegeneration [26]. NLRP3 (NOD-like receptor family pyrin domain-containing 3) inflammasome signaling pathway is a major contributor to the neuroinflammatory process in the CNS. Mitochondrial impairment in microglia can amplify NLRP3 inflammasome signaling to further augment the dopaminergic neurodegenerative process [27]. Release of fragmented and dysfunctional microglial mitochondria has been shown to activate naive astrocytes to A1 state that triggers neuronal death hence, causing neurodegeneration [28]. In reactive astrocytes, mitochondrial DAMPs activate the NLRP3 inflammasome leading to the liberation of IL-1β and IL-18 via the gasdermin D pore, a major pathway to enhance inflammatory responses (Figure 2). Evidence from several preclinical models including the DBA/2J mouse, in addition to patient data, implicates both mitochondrial damage and inflammation in glaucomatous neurodegeneration [29]. Inducible microglial mitochondrial fission has been reported to promote NLRP3-dependent neuroinflammation in hereditary and idiopathic PD [30]. In postnatal organotypic hippocampal slice cultures, microglia transformed into a distinct reactive phenotype by single TLR4 (toll-like receptor 4) stimulation with LPS (lipopolysaccharide), which varied in the pattern of inhibition of the respiratory chain in mitochondria [31]. Mitochondrial UCP2 (uncoupling protein-2) controls mitochondrial functions and is involved in a variety of physiological and pathological processes. LPS treatment of UCP2-silenced microglia activates an enhanced inflammatory response characterized by a greater expression of M1 genes (inducible nitric oxide synthase and tumour necrosis factor-α), while sparing IL-4 (interleukin-4) in inducing M2 genes (mannose receptor 1 and interleukin-10) [32].
4. Mitochondrial quality control in neurons: a promising mechanism of neurodegeneration
Neuronal health is primarily governed by the integrity and functionality of mitochondria. A hierarchical system of cellular surveillance mechanisms protects mitochondria against stress, by precisely monitoring the mitochondrial damage and ensuring the selective removal of dysfunctional mitochondrial proteins or organelles through a highly specialized intracellular event termed “mitophagy”. Neurons critically rely upon healthy mitochondrial function and oxygen supply, as most of the neuronal ATP is produced by oxidative phosphorylation. Moreover, mitochondrial dysfunction plays a central role in glutamate-evoked neuronal excitotoxicity, and mitochondrial fission/fusion dynamics are crucial events for mitochondrial morphology and function. Recent studies highlighted a novel role for actin dynamics in the regulation of mitochondrial morphology and function, for example, through mitochondrial recruitment of Drp1 (dynamin-related protein 1), a key factor in the mitochondrial fission machinery. Neuron-specific, inducible in vivo ablation of the mitochondrial fission protein Drp1 causes ER stress, that activates integrated stress response to inhibit neuronal expression of the cytokine Fgf21 [33]. Another study supporting inevitable role of Drp1 demonstrated that inducible Drp1 ablation in neurons of the adult mouse forebrain results in progressive alterations of mitochondrial morphology in the hippocampal region that are less responsive to antioxidant treatment [34]. Under stress conditions, another key mitochondrial mechanism i.e., Mul1-Mfn2 pathway maintains neuronal mitochondrial integrity, where Mul1 deficiency increases Mfn2 activity that triggers the first phasic mitochondrial hyperfusion and also acts as an ER-Mito tethering antagonist. Overall reduction in ER-Mito coupling triggers an increase in cytoplasmic Ca2+ load that in turn activates calcineurin and induces the second phasic Drp1-dependent mitochondrial fragmentation and mitophagy. Thus, by tightly regulating mitochondrial morphology and ER-Mito contacts, Mul1-Mfn2 pathway plays an early key checkpoint role in maintaining mitochondrial integrity [35]. Mitochondrial protein composition determines the mitochondrial quality. In neurons, nucleoid-associated proteins are highly enriched as a major cargo compared with other mitochondrial components. Axonal Drp1-dependent fission of nucleoid-enriched fragments near the sites of autophagosomes augments their capture to prevent accumulation of mtDNA in the neuron, hence mitigate activation of proinflammatory pathways involved in neurodegeneration [36]. Mitochondrial SNPH protein helps in placing mitochondria along axons to match metabolic needs. In contrast, dendritic SNPH toxicity involves pathologic molecules that trigger toxic misplacement of SNPH into dendrites. Pro-inflammatory cytokines and NMDA (N-methyl-D-aspartate) interact and converge to trigger toxic misplacement of SNPH into dendrites, thus ameliorating diverse inflammatory and excitatory injuries in neurodegenerative diseases [37]. Intracellular migration of mitochondria is tightly regulated to maintain energy homeostasis and prevent oxidative stress inside the cell. Miro (an outer mitochondrial membrane protein) helps in anchoring the mitochondria to microtubule motors and is detached to stop mitochondrial motility as an early step in the clearance of dysfunctional mitochondria. Caspase-cleaved tau may affect mitochondrial migration through the increase of TRAK2 (trafficking kinesin-binding protein 2)-mitochondria binding and reduction of ATP production available for the movement of mitochondria [38]. Metabolic dysfunction and protein aggregation are common characteristics that occur in age-related neurodegenerative disease. Interestingly, α-syn accumulation has been shown to induce upregulation of Miro protein levels due to an interaction via the N-terminus of α-syn [39]. Exposure of agrochemicals selectively triggers a deficit in mitochondrial transport by nitrating the microtubules in neurons harboring the synuclein, SNCA-A53T mutation thus demonstrating a gene environment interaction in PD [40]. Defective mitochondrial biogenesis is another causal event that leads to elevated oxidative stress and neuronal death. Mfn2 ablation and mitochondrial fragmentation in adult neurons cause neurodegeneration through oxidative stress and neuroinflammation in vivo via both apoptosis and aberrant cell-cycle-event-dependent cell death pathways [41]. In motor neurons, glutamate excitotoxicity induces mitochondrial dysfunction by disrupting mitochondrial dynamics via calpain-mediated Mfn2 degradation [42]. Aberrant mitochondrial biogenesis drive adult-onset progressive loss of dopamine neurons and motor deficits in Drosophila models of PINK1 (PTEN induced putative kinase 1) or parkin insufficiency. Such defects result from PARIS (a novel KRAB and zinc finger protein, substrate of Parkin) dependent repression of dopaminergic PGC-1α (Peroxisome proliferator-activated receptor-gamma coactivator) and its downstream transcription factors NRF1 and TFAM that cooperatively promote mitochondrial biogenesis [43]. Selective degeneration of neurons is reasonably due to selective neuronal vulnerable property of the brain. Regional heterogeneity in mitochondrial and other cellular functions contribute to and protect regions such as medulla oblongata, while rendering frontal cortex and hippocampus vulnerable to neurodegenerative diseases [44]. Chronic mitochondrial stress is apparently reported to be associated with major neurodegenerative diseases. Mitochondrial dysfunction and oxidative stress are major contributors to the pathophysiology of other neurodegenerative diseases including AD and PD. Markedly, mitochondrial dysfunction is well documented during the pathogenesis of PD, and increasingly more supportive data in this line of thought suggests abnormal mitochondrial dynamics and quality control as important underlying mechanisms. Alterations in microtubule-dependent transport, mitochondrial dysfunction, and autophagic pathology are involved in neurodegeneration observed in sporadic PD. In addition, animal models of PD utilizing neurotoxins, such as 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, have shown that these toxins disrupt mitochondrial respiration by targeting complex I of the electron transport chain, thereby impairing dopamine neurons in these models. Age-related decline in neuronal health or expression of disease-associated mutations in the pathway may exacerbate the slow kinetics of neuronal mitophagy, leading to neurodegeneration [45]. Mitophagy, the selective removal of damaged mitochondria, is thought to be indispensable to maintain neuronal homeostasis. Mutations of proteins in the pathway cause neurodegenerative diseases, suggesting defective mitochondrial turnover contributes to neurodegeneration. Mutations in PINK1 and parkin cause autosomal recessive PD plausibly due to defective PINK1/Parkin mediated mitophagy [46]. Age-related motor dysfunction and damaged mitochondria pathology in Parkin-deficient mice suggest that impairment of mitochondrial clearance may underlie the pathology of PD [47]. MitoPark mice with defective mitochondria showed greater glutamate release in the dorsal striatum compared to control mice upon KCl stimulation. In addition, glutamate uptake kinetics were slower in MitoPark mice. These findings were coupled with reduced expression of the glutamate re-uptake transporter, GLT-1 (glutamate transporter 1), thus providing an environment suitable for glutamate excitotoxic events, leading to altered physiological function in these mice [48]. Several histone modifications and its mediators have been linked to age dependent neurodegeneration. Sirtuins (SIRT, silent mating type information regulation 2 homolog in yeast) are NAD-dependent histone deacetylases reported to be involved in aging and longevity. Mechanistically, NAD+ metabolism is altered in sporadic PD patient-derived cells, which contributes to Sirtuin-2 activation and subsequent decrease in acetylated-α-tubulin levels [49]. PD-associated VPS35 (Vacuolar protein sorting ortholog 35) mutations show mitochondrial fragmentation and cell death in cultured neurons in vitro, in mouse substantia nigra neurons in vivo and in human fibroblasts from an individual with PD, who has the VPS35(D620N) mutation [50]. Another PD-related protein, LRRK2 (leucine rich repeat kinase 2), promotes Miro removal by forming a complex with Miro. In contrast, pathogenic LRRK2G2019S mutant disrupts this function, thereby delaying the arrest of damaged mitochondria and consequently slowing the initiation of mitophagy. Additionally, Miro degradation and mitochondrial motility are also impaired in sporadic PD patients [51]. PD-linked LRRK2 mutations have been shown to increase dendritic and mitochondrial calcium uptake in cortical neurons and familial PD patient fibroblasts, accompanied by increased expression of the MCU (mitochondrial calcium transporter) [52]. DJ-1 is a multifunctional protein regulating different biological and mitochondrial function. Loss-of-function mutations in DJ-1 were found to cause an autosomal recessive form of PD. Proteins perturbed by the loss of DJ-1 were involved in several mitochondrial functional pathways, including the tricarboxylic acid cycle and electron transport chain. Synaptic mitochondrial respiration showed a significant change due to DJ-1 deficiency [53]. FTD (frontotemporal dementia) is another neurodegenerative disease where mitochondrial quality control is markedly compromised. iPSC-derived neurons from patients with FTDP-17 (frontotemporal dementia and Parkinsonism linked to chromosome 17) present an increased mitochondrial membrane potential, which is partially maintained by the F1Fo ATPase working in reverse mode. Increased mitochondrial membrane potential in FTDP-17 neurons leads to overproduction of the ROS in mitochondria, which in turn causes oxidative stress and cell death [54]. In AD, mitochondria are reportedly involved in Aβ (amyloid-β) deposition. Pathogenic mutation in a Norwegian family in the mitochondrial metallopeptidase PITRM1 (Pitrilysin metallopeptidase 1) is found to underlie a novel mitochondrial neurodegenerative phenotype associated with Aβ accumulation [55]. Mitochondrial localizing protein SIRT3 (Sirtuin 3) protects the cell against oxidative or metabolic stress and it has been observed that SIRT3 mRNA and protein levels were significantly decreased in AD cerebral cortex. SIRT3 downregulation leads to p53-mediated mitochondrial and neuronal damage in AD [56]. By preserving mitochondrial function, SIRT3 protects parvalbumin and calretinin interneurons against Aβ-associated dysfunction and degeneration in AppPs1 AD mice, thus controlling neuronal network hyperactivity [57]. In another pertinent study, SIRT3-knockout mice exhibited poor remote memory, impaired long-term potentiation and decreased neuronal number in the anterior cingulate cortex, which seemed to contribute to their memory deficiencies [58]. Apo (apolipoprotein) E4, the major genetic risk factor for AD, alters mitochondrial function and metabolism early in AD pathogenesis. Global proteomic analysis revealed widespread alterations in mitochondrial functions in apoE4 cells, including reduced levels of numerous respiratory complex subunits and major disruptions to all detected subunits in complex V (ATP synthase). In addition, protein levels linked to mitochondrial endoplasmic reticulum-associated membranes, mitochondrial fusion/fission, mitochondrial protein translocation, proteases, and mitochondrial ribosomal proteins were substantially altered in apoE4 cells [59]. Loss of the mitochondrial m-AAA protease results in accumulation of constitutively active MCU-EMRE (mitochondrial calcium uniporter and essential MCU regulator) channels that leads to mitochondrial Ca2+ overload, mitochondrial permeability transition pore opening, and neuronal death. Mutations in subunits of mitochondrial m-AAA proteases in the inner membrane cause neurodegeneration in SCA28 (spinocerebellar ataxia) and HSP7 (hereditary spastic paraplegia) [60]. Functional studies revealed that SLC25A46 (solute carrier family 25 member 46) may play an important role in mitochondrial dynamics. SLC25A46 deficient mice displayed severe ataxia primarily due to degeneration of Purkinje cells, suggesting the loss of SLC25A46 causes degeneration in neurons by affecting mitochondrial dynamics and energy production [61]. TDP-43 is an RNA-binding protein and a major component of protein aggregates found in ALS and several other neurodegenerative diseases. Both full-length (mitochondrial matrix form) and truncated isoforms (mitochondrial intermembrane space protein) of TDP43 generate toxic aggregates, implying the presence of full-length TDP-43s in the matrix is a primary cause of mitochondrial damage [62]. ALS is an adult-onset neurodegeneration of motor neuron death. Synaptosomes from the spinal cord and motor cortex of SOD1G93A mice (mouse model of ALS) displayed dysfunctional mitochondria exhibiting high activity of hexokinase and phosphofructokinase, key glycolysis enzymes, and of citrate synthase and malate dehydrogenase, key Krebs cycle enzymes [63]. C9orf72 is a mitochondrial inner-membrane-associated protein regulating cellular energy homeostasis via its critical role in the control of oxidative phosphorylation. In C9orf72-ALS model, loss of mitochondrial function is a key mediator of axonal dysfunction [64]. Intriguingly, in C9orf72-linked ALS/FTD patient-derived neurons, impairment of mitochondrial complex I function was reported [65].
5. Significance of oligodendrocytic mitochondria in axonal dysfunction and neurodegeneration
Oligodendrocytes are necessary for the maturation of excitatory domains on the axon including nodes of Ranvier, help buffer potassium, and support neuronal energy metabolism. Disruption of the oligodendrocyte-axon unit in traumatic injuries, AD and demyelinating diseases such as MS results in axonal dysfunction and can ultimately lead in neurodegeneration. MS is a chronic inflammatory demyelinating disease of the CNS and a leading cause of neurological disability worldwide. In general, MS is a heterogeneous autoimmune disease of unknown etiology characterized by inflammation, demyelination, and axonal degeneration that affects both the white and gray matter of CNS. Mitochondria have been increasingly linked to the pathogenesis of MS. Recent studies suggest a role of mitochondrial dysfunction in the neurodegenerative aspects of MS [66]. To date, investigation of mitochondrial dysfunction in MS has focused exclusively on neurons, with no studies exploring whether dysregulation of mitochondrial bioenergetics and/or genetics in oligodendrocytes might be associated with the etiopathogenesis of MS and other demyelinating syndromes. Recent large-scale epidemiological and genomic studies identified several genetic and environmental risk factors for MS. Mitochondria-targeted antioxidants SkQ1 could be promising candidates as components of a combined therapy for MS and related neurological disorders [67]. Oxidative stress in oligodendrocyte’s mitochondria indicates pre-clinical sign of MS-like inflammation and demonstrates that evolving redox and morphological changes in mitochondria accompany oligodendrocyte dysfunction during neuroinflammation [68]. Another classical neurodegenerative disease in context to oligodendrocytes i.e., MSA is neuropathologically characterized by α-syn aggregates in oligodendroglia, and clinically characterized by parkinsonism, ataxia, and autonomic dysfunction. Since abnormal mitochondrial health due to oxidative phosphorylation are reported in MSA, mitochondrial haplogroup background may present the risk of MSA [69]. HSP is characterized by degeneration of CNS axons. PLP (Myelin proteolipid protein) and axon-enriched proteins are mutated HSP proteins that are involved in mitochondrial function, SER (smooth endoplasmic reticulum) structure, and microtubule stability/function. In HSP pathogenesis, Juxtaparanodal mitochondrial degeneration, reduced mitochondria-SER associations and reduced ATP production lead to axonal ovoid formation and axonal degeneration [70]. Oligodendrocyte-specific mtDNA double-strand breaks in PLP:mtPstI mice model cause oligodendrocyte death and demyelination associated with axonal damage and glial activation [71]. VDAC1 (Voltage-dependent anion-selective channel 1) is a mitochondrial porin involved in the cellular metabolism and apoptotic intrinsic pathway in many neuropathological processes. In SCI (spinal cord injury), after the primary cell death, release of pro-inflammatory molecules triggers apoptosis, inflammation, and demyelination leading to the loss of motor functions [72]. In oligodendrocytes, overactivation of AMPARs (AMPA-type ionotropic glutamate receptors) induces intracellular Ca2+ overload and excitotoxic death. It has been shown that AMPARs activation triggers Drp1 mediated mitochondrial fission in oligodendrocytes [73]. Genetically induced mitochondrial or myelin dysfunction experimentation validates that modification in axonal mitochondrial size depends on the thickness of myelin sheath but not vice versa [74]. Under unfavorable bioenergetic scenario, iron deficient oligodendrocytes fail to undergo myelination, thus indicating the regulation of cell metabolism may impact cell fate decisions and maturation [75]. Loss of MnSOD (Mn-Superoxide dismutase) in spinal cord promotes a phenotype of demyelination, inflammation and progressive paralysis that mimics phenotypes associated with progressive multiple sclerosis [76].
Conclusion
Mitochondria are essential for neuronal survival and mitochondrial dysfunction is a hallmark of neurodegeneration. The loss in mitochondrial energy production, oxidative stress, and changes in calcium handling are associated with neurodegenerative diseases; however, different sites and types of mitochondrial dysfunction are linked to distinct neuropathologies. Understanding the causal or correlative relationship between changes in mitochondria and neuropathology will lead to new therapeutic strategies. Mitochondria are promising cellular targets for neuroprotective interventions: They are among the first structures affected in many neuroinflammatory diseases, with mitochondrial impairment ranging from impaired respiratory activity and reduced mitochondrial membrane potential to mitochondrial oxidation and fragmentation. Therefore, the development of neuroprotective strategies might be important in addition to treating inflammation in these diseases.
Acknowledgements
We are grateful to the institutional digital library for permitting the collection of articles cited in this review article. Moreover, we are much thankful to our colleagues for their insightful review and valuable suggestions during the preparation of this manuscript.
References
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenín-von Bernhardi, J.; Flores, B.; Eugenín León, J. Glial Cells and Integrity of the Nervous System. Adv. Exp. Med. Biol. 2016, 949, 1–24. [Google Scholar] [PubMed]
- MacVicar, B.A.; Newman, E.A. Astrocyte Regulation of Blood Flow in the Brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a020388. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Steardo, L.; Peng, L.; Parpura, V. Astroglia, Glutamatergic Transmission and Psychiatric Diseases. Advances in Neurobiology 2016, 307–326. [Google Scholar]
- Jeon, Y.-M.; Kwon, Y.; Jo, M.; Lee, S.; Kim, S.; Kim, H.-J. The Role of Glial Mitochondria in α-Synuclein Toxicity. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, L.; Rust, M.B.; Culmsee, C. Actin(g) on Mitochondria – a Role for cofilin1 in Neuronal Cell Death Pathways. Biol. Chem. 2019, 400, 1089–1097. [Google Scholar] [CrossRef]
- Sirabella, R.; Sisalli, M.J.; Costa, G.; Omura, K.; Ianniello, G.; Pinna, A.; Morelli, M.; Di Renzo, G.M.; Annunziato, L.; Scorziello, A. NCX1 and NCX3 as Potential Factors Contributing to Neurodegeneration and Neuroinflammation in the A53T Transgenic Mouse Model of Parkinson’s Disease. Cell Death Dis. 2018, 9, 725. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Mulica, P.; Grünewald, A.; Pereira, S.L. Astrocyte-Neuron Metabolic Crosstalk in Neurodegeneration: A Mitochondrial Perspective. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Casaril, A.M.; Katsalifis, A.; Schmidt, R.M.; Bas-Orth, C. Activated Glia Cells Cause Bioenergetic Impairment of Neurons That Can Be Rescued by Knock-down of the Mitochondrial Calcium Uniporter. Biochem. Biophys. Res. Commun. 2022, 608, 45–51. [Google Scholar] [CrossRef]
- Agarwal, A.; Wu, P.-H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587–605.e7. [Google Scholar] [CrossRef]
- Logan, S.; Pharaoh, G.A.; Marlin, M.C.; Masser, D.R.; Matsuzaki, S.; Wronowski, B.; Yeganeh, A.; Parks, E.E.; Premkumar, P.; Farley, J.A.; et al. Insulin-like Growth Factor Receptor Signaling Regulates Working Memory, Mitochondrial Metabolism, and Amyloid-β uptake in astrocytes. Mol. Metab. 2018, 9, 141–155. [Google Scholar] [CrossRef]
- Murru, S.; Hess, S.; Barth, E.; Almajan, E.R.; Schatton, D.; Hermans, S.; Brodesser, S.; Langer, T.; Kloppenburg, P.; Rugarli, E.I. Astrocyte-Specific Deletion of the Mitochondrial M-AAA Protease Reveals Glial Contribution to Neurodegeneration. Glia 2019, 67, 1526–1541. [Google Scholar] [CrossRef]
- Aldana, B.I.; Zhang, Y.; Jensen, P.; Chandrasekaran, A.; Christensen, S.K.; Nielsen, T.T.; Nielsen, J.; Hyttel, P.; Larsen, M.R.; Waagepetersen, H.; et al. Glutamate-Glutamine Homeostasis Is Perturbed in Neurons and Astrocytes Derived from Patient iPSC Models of Frontotemporal Dementia.
- Chandrasekaran, A.; Dittlau, K.S.; Corsi, G.I.; Haukedal, H.; Doncheva, N.T.; Ramakrishna, S.; Ambardar, S.; Salcedo, C.; Schmidt, S.I.; Zhang, Y.; et al. Astrocytic Reactivity Triggered by Defective Autophagy and Metabolic Failure Causes Neurotoxicity in Frontotemporal Dementia Type 3. Stem Cell Rep. 2021, 16, 2736–2751. [Google Scholar] [CrossRef]
- Barbosa, M.; Gomes, C.; Sequeira, C.; Gonçalves-Ribeiro, J.; Pina, C.C.; Carvalho, L.A.; Moreira, R.; Vaz, S.H.; Vaz, A.R.; Brites, D. Recovery of Depleted miR-146a in ALS Cortical Astrocytes Reverts Cell Aberrancies and Prevents Paracrine Pathogenicity on Microglia and Motor Neurons. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Hall, C.E.; Yao, Z.; Choi, M.; Tyzack, G.E.; Serio, A.; Luisier, R.; Harley, J.; Preza, E.; Arber, C.; Crisp, S.J.; et al. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. 2017, 19, 1739–1749. [Google Scholar] [CrossRef]
- Saba, J.; Couselo, F.L.; Turati, J.; Carniglia, L.; Durand, D.; de Laurentiis, A.; Lasaga, M.; Caruso, C. Astrocytes from Cortex and Striatum Show Differential Responses to Mitochondrial Toxin and BDNF: Implications for Protection of Striatal Neurons Expressing Mutant Huntingtin. J. Neuroinflammation 2020, 17, 1–15. [Google Scholar] [CrossRef]
- Lampinen, R.; Belaya, I.; Saveleva, L.; Liddell, J.R.; Rait, D.; Huuskonen, M.T.; Giniatullina, R.; Sorvari, A.; Soppela, L.; Mikhailov, N.; et al. Neuron-Astrocyte Transmitophagy Is Altered in Alzheimer's disease. Neurobiol. Dis. 2022, 170, 105753. [Google Scholar] [CrossRef] [PubMed]
- Loría, F.; Díaz-Nido, J. Frataxin Knockdown in Human Astrocytes Triggers Cell Death and the Release of Factors That Cause Neuronal Toxicity. Neurobiol. Dis. 2015, 76, 1–12. [Google Scholar] [CrossRef]
- Ignatenko, O.; Malinen, S.; Rybas, S.; Vihinen, H.; Nikkanen, J.; Kononov, A.; Jokitalo, E.S.; Ince-Dunn, G.; Suomalainen, A. Mitochondrial Dysfunction Compromises Ciliary Homeostasis in Astrocytes. J. Cell Biol. 2022, 222. [Google Scholar] [CrossRef]
- Rawat, P.; Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type-1 Single-stranded RNA Activates the NLRP3 Inflammasome and Impairs Autophagic Clearance of Damaged Mitochondria in Human Microglia. Glia 2018, 67, 802–824. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial Cell Dysregulation in Brain Aging and Neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Mediat. Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial Impairment in Microglia Amplifies NLRP3 Inflammasome Proinflammatory Signaling in Cell Culture and Animal Models of Parkinson’s Disease. npj Park. Dis. 2017, 3, 1–15. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., II; Mochly-Rosen, D. Fragmented Mitochondria Released from Microglia Trigger A1 Astrocytic Response and Propagate Inflammatory Neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Jassim, A.H.; Inman, D.M.; Mitchell, C.H. Crosstalk Between Dysfunctional Mitochondria and Inflammation in Glaucomatous Neurodegeneration. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Lawrence, G.M.; Holley, C.L.; Schroder, K. Parkinson’s Disease: Connecting Mitochondria to Inflammasomes. Trends Immunol. 2022, 43, 877–885. [Google Scholar] [CrossRef]
- Chausse, B.; Lewen, A.; Poschet, G.; Kann, O. Selective Inhibition of Mitochondrial Respiratory Complexes Controls the Transition of Microglia into a Neurotoxic Phenotype in Situ. Brain, Behav. Immun. 2020, 88, 802–814. [Google Scholar] [CrossRef]
- De Simone, R.; Ajmone-Cat, M.A.; Pandolfi, M.; Bernardo, A.; De Nuccio, C.; Minghetti, L.; Visentin, S. The Mitochondrial Uncoupling Protein-2 Is a Master Regulator of Both M1 and M2 Microglial Responses. J. Neurochem. 2015, 135, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Restelli, L.M.; Oettinghaus, B.; Halliday, M.; Agca, C.; Licci, M.; Sironi, L.; Savoia, C.; Hench, J.; Tolnay, M.; Neutzner, A.; et al. Neuronal Mitochondrial Dysfunction Activates the Integrated Stress Response to Induce Fibroblast Growth Factor 21. Cell Rep. 2018, 24, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Oettinghaus, B.; Schulz, J.M.; Restelli, L.M.; Licci, M.; Savoia, C.; Schmidt, A.; Schmitt, K.; Grimm, A.; Morè, L.; Hench, J.; et al. Synaptic Dysfunction, Memory Deficits and Hippocampal Atrophy due to Ablation of Mitochondrial Fission in Adult Forebrain Neurons. Cell Death Differ. 2016, 23, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Cheng, X.-T.; Lin, M.-Y.; Huang, N.; Sheng, Z.-H. Mul1 Restrains Parkin-Mediated Mitophagy in Mature Neurons by Maintaining ER-Mitochondrial Contacts. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.; Ordureau, A.; Harper, J.W.; Holzbaur, E.L. Brain-Derived Autophagosome Profiling Reveals the Engulfment of Nucleoid-Enriched Mitochondrial Fragments by Basal Autophagy in Neurons. Neuron 2022, 110, 967–976.e8. [Google Scholar] [CrossRef] [PubMed]
- Joshi, D.C.; Zhang, C.-L.; Mathur, D.; Li, A.; Kaushik, G.; Sheng, Z.-H.; Chiu, S.-Y. Tripartite Crosstalk between Cytokine IL-1β, NMDA-R and Misplaced Mitochondrial Anchor in Neuronal Dendrites Is a Novel Pathway for Neurodegeneration in Inflammatory Diseases. J. Neurosci. 2022, 42, 7318–7329. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Tapia-Monsalves, C.; Vergara, E.H.; Pérez, M.J.; Aranguiz, A. Truncated Tau Induces Mitochondrial Transport Failure Through the Impairment of TRAK2 Protein and Bioenergetics Decline in Neuronal Cells. Front. Cell. Neurosci. 2020, 14, 175. [Google Scholar] [CrossRef] [PubMed]
- Shaltouki, A.; Hsieh, C.-H.; Kim, M.J.; Wang, X. Alpha-Synuclein Delays Mitophagy and Targeting Miro Rescues Neuron Loss in Parkinson’s Models. Acta Neuropathol. 2018, 136, 607–620. [Google Scholar] [CrossRef]
- Stykel, M.G.; Humphries, K.; Kirby, M.P.; Czaniecki, C.; Wang, T.; Ryan, T.; Bamm, V.; Ryan, S.D. Nitration of Microtubules Blocks Axonal Mitochondrial Transport in a Human Pluripotent Stem Cell Model of Parkinson’s Disease. FASEB J. 2018, 32, 5350–5364. [Google Scholar] [CrossRef]
- Han, S.; Nandy, P.; Austria, Q.; Siedlak, S.L.; Torres, S.; Fujioka, H.; Wang, W.; Zhu, X. Mfn2 Ablation in the Adult Mouse Hippocampus and Cortex Causes Neuronal Death. Cells 2020, 9, 116. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, F.; Li, L.; Tang, F.; Siedlak, S.L.; Fujioka, H.; Liu, Y.; Su, B.; Pi, Y.; Wang, X. MFN2 Couples Glutamate Excitotoxicity and Mitochondrial Dysfunction in Motor Neurons*. J. Biol. Chem. 2015, 290, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, S.K.; Yuan, C.; Khan, M.R.; Karuppagounder, S.S.; Wang, L.; Xiong, Y.; Kang, S.U.; Lee, Y.; Dawson, V.L.; Dawson, T.M. PARIS Induced Defects in Mitochondrial Biogenesis Drive Dopamine Neuron Loss under Conditions of Parkin or PINK1 Deficiency. Mol. Neurodegener. 2020, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Anusha-Kiran, Y.; Mol, P.; Dey, G.; Bhat, F.A.; Chatterjee, O.; Deolankar, S.C.; Philip, M.; Prasad, T.K.; Bharath, M.S.; Mahadevan, A. Regional Heterogeneity in Mitochondrial Function Underlies Region Specific Vulnerability in Human Brain Ageing: Implications for Neurodegeneration. Free. Radic. Biol. Med. 2022, 193, 34–57. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L.F. Degradation of Engulfed Mitochondria Is Rate-Limiting in Optineurin-Mediated Mitophagy in Neurons. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Barodia, S.K.; Creed, R.B.; Goldberg, M.S. Parkin and PINK1 Functions in Oxidative Stress and Neurodegeneration. Brain Res. Bull. 2017, 133, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of Parkin Contributes to Mitochondrial Turnover and Dopaminergic Neuronal Loss in Aged Mice. Neurobiol. Dis. 2019, 136, 104717. [Google Scholar] [CrossRef]
- Farrand, A.Q.; Gregory, R.A.; Bäckman, C.M.; Helke, K.L.; Boger, H.A. Altered Glutamate Release in the Dorsal Striatum of the MitoPark Mouse Model of Parkinson's Disease. Brain Res. 2016, 1651, 88–94. [Google Scholar] [CrossRef]
- Esteves, A.R.; Arduíno, D.M.; Silva, D.F.; Viana, S.D.; Pereira, F.C.; Cardoso, S.M. Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson’s Disease. Mol. Neurobiol. 2017, 55, 1440–1462. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson's Disease-Associated Mutant VPS35 Causes Mitochondrial Dysfunction by Recycling DLP1 Complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; da Cruz, A.B.; Burbulla, L.F.; Lawrence, E.S.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J. Neurosci. 2017, 37, 11151–11165. [Google Scholar] [CrossRef] [PubMed]
- Almikhlafi, M.A.; Stauch, K.L.; Villeneuve, L.M.; Purnell, P.R.; Lamberty, B.G.; Fox, H.S. Deletion of DJ-1 in Rats Affects Protein Abundance and Mitochondrial Function at the Synapse. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial Hyperpolarization in iPSC-Derived Neurons from Patients of FTDP-17 with 10+16 MAPT Mutation Leads to Oxidative Stress and Neurodegeneration. Redox Biol 2017, 12, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; Horvath, R. Amyloid-β in Mitochondrial Disease: Mutation in a Human Metallopeptidase Links Amyloidotic Neurodegeneration with Mitochondrial Processing. EMBO Mol. Med. 2016, 8, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.; Liu, T.; Hwang, Y.J.; Hyeon, S.J.; Im, H.; Lee, K.; Alvarez, V.E.; McKee, A.C.; Um, S.; et al. SIRT3 Deregulation Is Linked to Mitochondrial Dysfunction in Alzheimer's Disease. Aging Cell 2017, 17, e12679. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Wang, J.; Ghena, N.; Zhao, Q.; Perone, I.; King, T.M.; Veech, R.L.; Gorospe, M.; Wan, R.; Mattson, M.P. SIRT3 Haploinsufficiency Aggravates Loss of GABAergic Interneurons and Neuronal Network Hyperexcitability in an Alzheimer's Disease Model. J. Neurosci. 2019, 40, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.; Choi, J.E.; Han, D.; Koh, S.M.; Kim, H.-S.; Kaang, B.-K. Decreased Neuron Number and Synaptic Plasticity in SIRT3-Knockout Mice with Poor Remote Memory. Neurochem. Res. 2017, 44, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Kim, C.; Jimenez-Morales, D.; Newton, B.W.; Johnson, J.R.; Krogan, N.J.; Swaney, D.L.; Mahley, R.W. Neuronal Apolipoprotein E4 Expression Results in Proteome-Wide Alterations and Compromises Bioenergetic Capacity by Disrupting Mitochondrial Function. J. Alzheimer's Dis. 2019, 68, 991–1011. [Google Scholar] [CrossRef] [PubMed]
- König, T.; Tröder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Mühlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The M-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef]
- Li, Z.; Peng, Y.; Hufnagel, R.B.; Hu, Y.-C.; Zhao, C.; Queme, L.F.; Khuchua, Z.; Driver, A.M.; Dong, F.; Lu, Q.R.; et al. Loss of SLC25A46 Causes Neurodegeneration by Affecting Mitochondrial Dynamics and Energy Production in Mice. Hum. Mol. Genet. 2017, 26, 3776–3791. [Google Scholar] [CrossRef]
- Salvatori, I.; Ferri, A.; Scaricamazza, S.; Giovannelli, I.; Serrano, A.; Rossi, S.; D'Ambrosi, N.; Cozzolino, M.; Di Giulio, A.; Moreno, S.; et al. Differential Toxicity of TAR DNA-binding Protein 43 Isoforms Depends on Their Submitochondrial Localization in Neuronal Cells. J. Neurochem. 2018, 146, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Torazza, C.; Bonifacino, T.; Provenzano, F.; Rebosio, C.; Milanese, M.; Usai, C.; Panfoli, I.; Bonanno, G. Altered Glucose Catabolism in the Presynaptic and Perisynaptic Compartments of SOD1G93Amouse Spinal Cord and Motor Cortex Indicates That Mitochondria Are the Site of Bioenergetic Imbalance in ALS. Journal of Neurochemistry 2019, 151, 336–350. [Google Scholar] [CrossRef]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial Bioenergetic Deficits in C9orf72 Amyotrophic Lateral Sclerosis Motor Neurons Cause Dysfunctional Axonal Homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 Regulates Energy Homeostasis by Stabilizing Mitochondrial Complex I Assembly. Cell Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar] [PubMed]
- Fetisova, E.K.; Muntyan, M.S.; Lyamzaev, K.G.; Chernyak, B.V. Therapeutic Effect of the Mitochondria-Targeted Antioxidant SkQ1 on the Culture Model of Multiple Sclerosis. Oxidative Med. Cell. Longev. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Steudler, J.; Ecott, T.; Ivan, D.C.; Bouillet, E.; Walthert, S.; Berve, K.; Dick, T.P.; Engelhardt, B.; Locatelli, G. Autoimmune Neuroinflammation Triggers Mitochondrial Oxidation in Oligodendrocytes. Glia 2022, 70, 2045–2061. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.R.; Heckman, M.G.; Johnson, P.W.; Soto-Beasley, A.I.; Walton, R.L.; Koga, S.; Uitti, R.J.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A. Association of Mitochondrial Genomic Background with Risk of Multiple System Atrophy. Park. Relat. Disord. 2020, 81, 200–204. [Google Scholar] [CrossRef]
- Yin, X.; Kidd, G.J.; Ohno, N.; Perkins, G.A.; Ellisman, M.H.; Bastian, C.; Brunet, S.; Baltan, S.; Trapp, B.D. Proteolipid Protein–deficient Myelin Promotes Axonal Mitochondrial Dysfunction via Altered Metabolic Coupling. J. Cell Biol. 2016, 215, 531–542. [Google Scholar] [CrossRef]
- Madsen, P.M.; Pinto, M.; Patel, S.; McCarthy, S.; Gao, H.; Taherian, M.; Karmally, S.; Pereira, C.V.; Dvoriantchikova, G.; Ivanov, D.; et al. Mitochondrial DNA Double-Strand Breaks in Oligodendrocytes Cause Demyelination, Axonal Injury, and CNS Inflammation. J. Neurosci. 2017, 37, 10185–10199. [Google Scholar] [CrossRef]
- Paschon, V.; Morena, B.C.; Correia, F.F.; Beltrame, G.R.; dos Santos, G.B.; Cristante, A.F.; Kihara, A.H. VDAC1 Is Essential for Neurite Maintenance and the Inhibition of Its Oligomerization Protects Spinal Cord from Demyelination and Facilitates Locomotor Function Recovery after Spinal Cord Injury. Sci. Rep. 2019, 9, 14063. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Quintela-López, T.; Sánchez-Gómez, M.V.; Gaminde-Blasco, A.; Alberdi, E.; Matute, C. Mitochondrial Division Inhibitor 1 Disrupts Oligodendrocyte Ca Homeostasis and Mitochondrial Function. Glia 2020, 68, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Ineichen, B.V.; Zhu, K.; Carlström, K.E. Axonal Mitochondria Adjust in Size Depending on G-Ratio of Surrounding Myelin during Homeostasis and Advanced Remyelination. J. Neurosci. Res. 2020, 99, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Rosato-Siri, M.V.; Adami, P.V.M.; Guitart, M.E.; Verstraeten, S.; Morelli, L.; Correale, J.; Pasquini, J.M. Glial Cell Metabolic Profile Upon Iron Deficiency: Oligodendroglial and Astroglial Casualties of Bioenergetic Adjustments. Mol. Neurobiol. 2023, 60, 1949–1963. [Google Scholar] [CrossRef]
- Bhaskaran, S.; Kumar, G.; Thadathil, N.; Piekarz, K.M.; Mohammed, S.; Lopez, S.D.; Qaisar, R.; Walton, D.; Brown, J.L.; Murphy, A.; et al. Neuronal Deletion of MnSOD in Mice Leads to Demyelination, Inflammation and Progressive Paralysis That Mimics Phenotypes Associated with Progressive Multiple Sclerosis. Redox Biol. 2022, 59, 102550. [Google Scholar] [CrossRef]
Figure 1.
Cross-talk between neurons and non-neuronal cells in the brain. Secretory factors released from activated microglia and reactive astrocytes triggers neuroinflammatory signals in the neurons. Oligodendrocytes support neuronal axons for smooth neurotransmission from dendritic spines. All different types of cells are represented with different colors and specialized networks among these cells depicts the fine tuning to support neuronal health and transmission.
Figure 1.
Cross-talk between neurons and non-neuronal cells in the brain. Secretory factors released from activated microglia and reactive astrocytes triggers neuroinflammatory signals in the neurons. Oligodendrocytes support neuronal axons for smooth neurotransmission from dendritic spines. All different types of cells are represented with different colors and specialized networks among these cells depicts the fine tuning to support neuronal health and transmission.
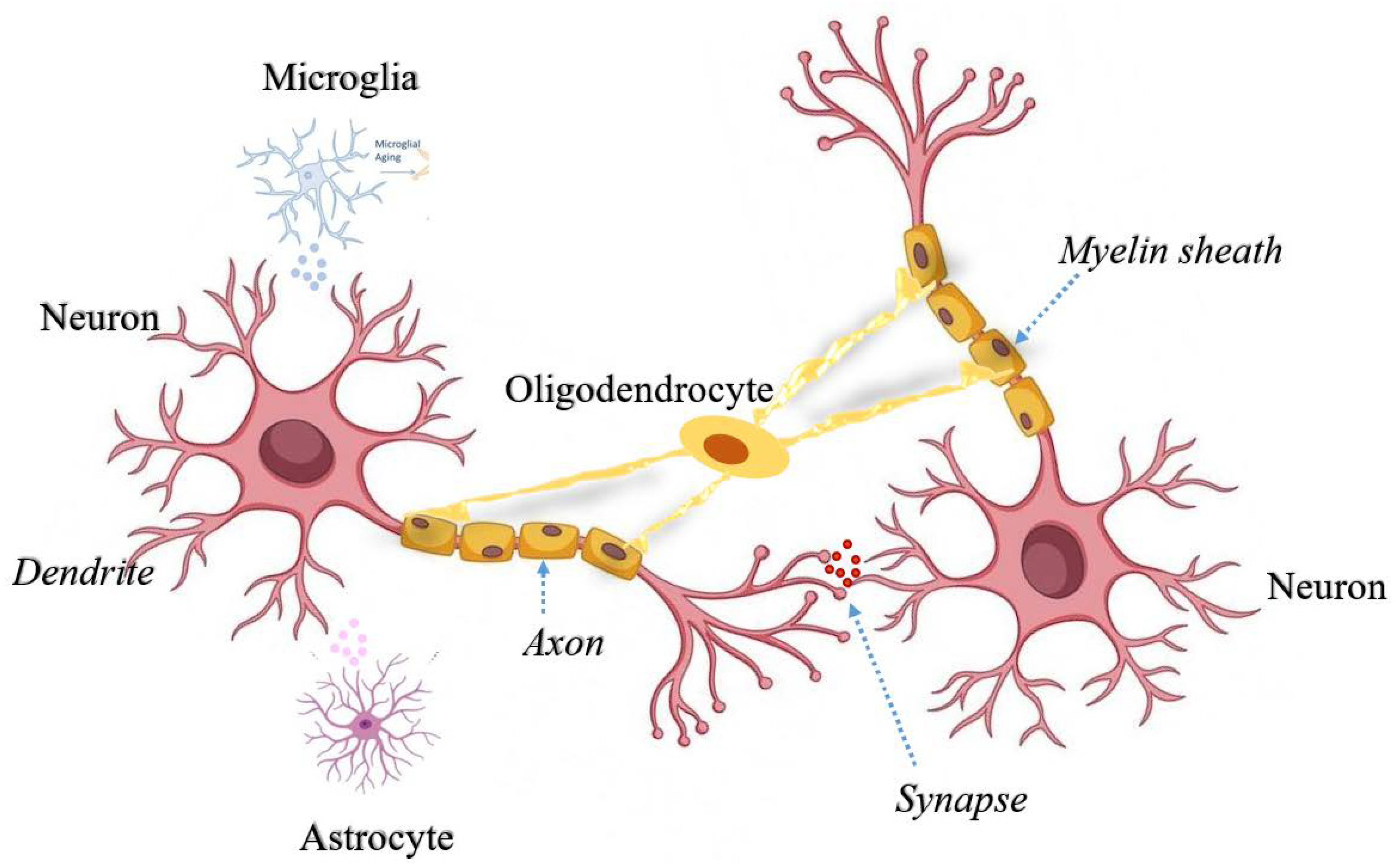
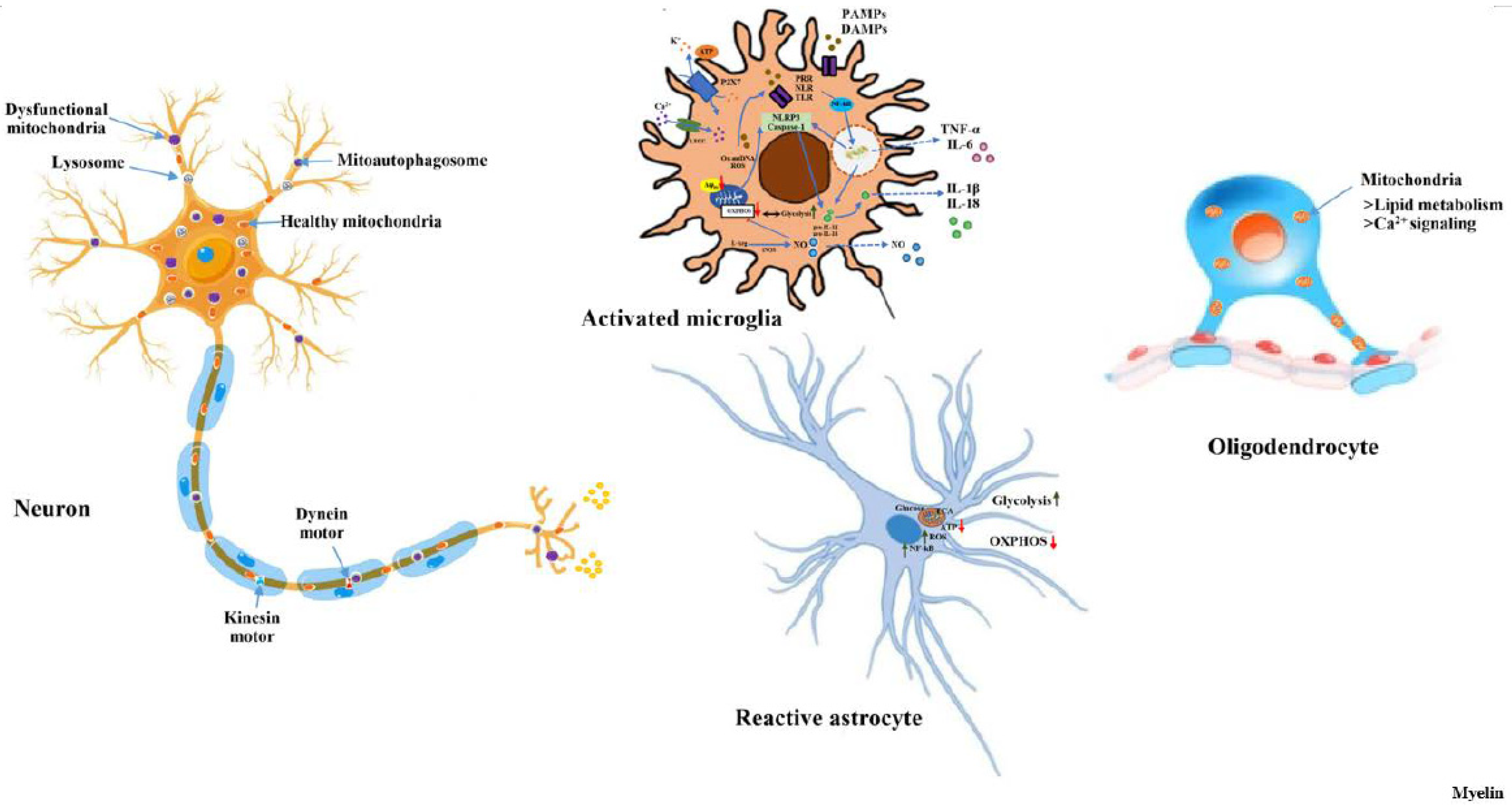
Figure 2.
Mitochondrial dynamics in neurons, microglia, astrocytes and oligodendrocytes. Distinct mitochondrial states, both structural as well as functional in different cell types are represented along with key molecular events of oxidative stress and bioenergetics. Abbreviations used are: PAMPs, pathogen-associated molecular patterns; ATP, adenosine triphosphate; DAMPs, damage-associate molecular patterns; Δψm, mitochondrial membrane potential; IL, interleukin; iNOS, inducible nitric oxide synthase; L-Arg, L-arginine; LTCC, L-type Ca2+ channel; NF-kB, nuclear factor kappa B; NLR, NOD like receptors; NLRP3, NLR family, pyrin domain containing 3; NO, nitric oxide; OXPHOS, oxidative phosphorylation; PRR, pattern-recognition receptors; P2X7, purinergic receptor P2X7; ROS, reactive oxygen species; TCA, tricarboxylic acid; TLR, toll-like receptors; TNF-α, tumur necrosis factors- α.
Figure 2.
Mitochondrial dynamics in neurons, microglia, astrocytes and oligodendrocytes. Distinct mitochondrial states, both structural as well as functional in different cell types are represented along with key molecular events of oxidative stress and bioenergetics. Abbreviations used are: PAMPs, pathogen-associated molecular patterns; ATP, adenosine triphosphate; DAMPs, damage-associate molecular patterns; Δψm, mitochondrial membrane potential; IL, interleukin; iNOS, inducible nitric oxide synthase; L-Arg, L-arginine; LTCC, L-type Ca2+ channel; NF-kB, nuclear factor kappa B; NLR, NOD like receptors; NLRP3, NLR family, pyrin domain containing 3; NO, nitric oxide; OXPHOS, oxidative phosphorylation; PRR, pattern-recognition receptors; P2X7, purinergic receptor P2X7; ROS, reactive oxygen species; TCA, tricarboxylic acid; TLR, toll-like receptors; TNF-α, tumur necrosis factors- α.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



