
Submitted:
10 March 2023
Posted:
10 March 2023
You are already at the latest version
Abstract
Microbial biofilms cause several environmental and industrial issues, even on the human health. Although they have represented a threaten due to their resistance to antibiotics, there are currently no approved antibiofilm agents for clinical treatments. The multi-functionality of antimicrobial peptides (AMPs) including the antibiofilm activity and their potentialities to target multiple mi-crobes motivated the synthesis of AMP relatives for developing antibiofilm agents for clinical purposes. Antibiofilm peptides (ABFPs) have been organized in databases that allowed the building of prediction tools which have assisted in the discovery/design of new antibiofilm agents. However, the complex network approach has been explored yet as an assistant tool for this aim. Herein, a kind of similarity networks, called the Half-Space Proximal Network (HSPN) is applied to represent/analyse the chemical space of the ABFPs aimed to identify promising scaf-folds for the development of next generation antimicrobials, able to target both planktonic and biofilm microbial forms. Such analyses also considered the metadata associated to the ABFPs such as origin, other activities, targets, etc. in which the relationships were projected by multilayer networks called metadata networks (METNs). From the complex networks mining, a reduced but informative set of 66 ABFPs representing the original antibiofilm space was extracted. This subset retained from the most central to atypical ABFPs, having some of them, desired properties for developing next generation antimicrobials. So, this subset is advisable for assisting the search/design both new antibiofilm/antimicrobial agents. The provided ABFP motifs list, dis-covered within the HSPN communities, is also useful for the same purpose.

Keywords:
Antibiofilm peptide
; chemical space
; StarPep toolbox
; complex network
; centrality measure
; motif discovery
1. Introduction
Microorganism biofilms have aroused an increasingly interest within the scientific community [1,2]. Biofilms are microbial communities composed by either single or multiple species that can be adhered to different surfaces types allowing their survival to environmental changing conditions caused by the use of biocides and antibiotics in industrial and clinical settings, as well as by other circumstances such as the presence of UV light, heavy metals, anaerobic conditions, salinity, pH gradients, etc [3]. Although, they frequently cause environmental and industrial issues due to their adherence to metals, river rocks, deep-sea vents, plant tissues; they have had a negative impact on human health by settling down in body tissues and medical implant materials [3,4].
According to the National Institute of Health (NIH), biofilms are responsible for more than the 65% of microbial infections in humans and for the 80-90% of the chronic conditions, representing a serious healthcare issue [5,6]. Thus, chronic inflammation, pain and damage in certain tissues or organ systems caused by biofilm-associated infections, may derive in endocarditis, cystic fibrosis, urinary tract infections, and periodontitis [1]. Biofilms are especially hard to remove from implanted medical devices e.g. catheters, stents, prosthetic heart valves, pacemakers, and artificial joints or limbs [7,8]. Furthermore, when planktonic forms (free-living microbes) are detached from biofilms can trigger other complications for patients e.g. bacteraemia, thromboembolism and septic episode [9,10].
Although microbial biofilms have represented a threaten for the human health due to their resistance to antibiotics [11,12], there are currently no approved antibiofilm agents for clinical treatments despite years of research [5]. There are only two antibiofilm candidates in the pipeline of clinical trials, (i) the nitric oxide, a known regulator for biofilms [13] is being proposed for treating chronic rhinosinusitis (Phase 2, NCT04163978) and (ii) the human monoclonal antibody (TRL1068), against the bacterial protein DNABII which stabilizes DNA in the extracellular matrix of biofilms [14] is being evaluated against prosthetic joint infections (Phase 1, NCT04763759) [5]. Currently, the exploration of biofilm formation, mainly by OMICS approaches, and the use of synthetic peptides derived from antimicrobial peptides (AMPs), have allowed to find more promising antibiofilm agent/targets for the clinics [5,15]. The first AMPs that served as templates were the natural human peptide LL-37 and the bovine peptide indolicidin that showed a potent antibiofilm activity in addition to their antibacterial action [16].With the knowledge on molecular signalling in biofilm formation, new entities were targeted by natural and synthetic peptides [5]. For example, the nucleotide second-messenger guanosine tetraphosphate and pentaphosphate ((p)ppGpp) is found in all bacteria as part of the stress response, having also an important role in biofilm formation in many species [17,18]. Therefore, (p)ppGpp is an excellent target for developing antibiofilm agents and surprisingly cationic amphipathic peptides related to the antimicrobial and host defence function can bind directly to (p)ppGpp for its degradation [5,17]. This background points out AMPs and their synthetic relatives as promising alternatives for developing antibiofilm agents, also leveraging the multi-functionality of the AMPs class such as immunomodulatory, anti-inflammatory, wound healing, antifungal and antibacterial activities; especially their potentialities to target multi-drug resistance planktonic strains [19,20]. Thus, the antibiofilm activity has gained attention in the AMPs world, being evaluated in vitro and in vivo models and registered either in general AMP databases such as UniProtKB [21], Antimicrobial Peptide Database (APD) [22], Data Repository of Antimicrobial Peptides (DRAMP) [23], or in others exclusively dedicated to antibiofilm peptides like the Biofilm-active AMPs (BaAMPS) database [24]. This last specific database has been useful for researchers to train machine learning (ML) models to predict the antibiofilm activity [25,26,27,28].
In addition to the efforts for populating AMPs databases and building ML models to assist the discovery/design of peptide drugs, emerging tools and databases based on graph and networks science can also provide new insights to the discovery/design pipeline by analysing the chemical space of bioactive peptides [29,30,31]. Relevant features of the antibiofilm peptides can be approached by applying complex networks, e.g. (i) their structural diversity according to the network topology/modularity (ii) what are the most representative and atypical peptides determined mainly centrality measures, (iii) what other relevant functionalities do they have besides the antibiofilm activity by weighting their associated metadata with centralities measures, (iv) motifs identification in networks communities, (v) the determination of a reduced set of peptides that actually represents the original anti-biofilm chemical space. Here, the Half-Space Proximal Network (HSPN) was applied to represent the structural (sequence-based) space of anti-biofilm peptides registered in the graph-based database (StarPepDB) [32], which probably gathers the most comprehensive subset of antibiofilm peptides collected from both generic AMPs databases and specific ones for antibiofilms (http://mobiosd-hub.com/starpep/). The HSPN is a local proximity graph that extracts a low-degree spanner of the complete graph. Each node is associated with its nearest neighbour, clearing from the complete graph all redundant nodes in the nearest neighbour’s direction by using a half-space hyperplane [33,34]. HSPNs do not consider all the possible pairwise relationships between nodes, instead, these networks apply the half-space proximal test over the set of nodes, obtaining a connected network with a smaller fraction of the maximum number of edges. As they show a lower density; a lesser RAM memory is needed for their construction; so, they were recently applied by our group to study the chemical space of antiparasitic peptides [31].
The estimation of several HSPN parameters e.g. number of communities, centrality measures, metadata associated to central and atypical nodes, neighbourhood analysis along with network visual mining allowed an effective exploration of the chemical space of antibiofilm peptides aimed to assist the discovery/design of next generation antimicrobials targeting both planktonic and biofilm microbial forms. Such new approach was possible by mainly using the network science tools like StarPep toolbox [32] and Gephi [35], complemented with classical bioinformatic analyses.
2. Materials and Methods
2.1. Half-Space Proximal Networks Building
The building of Half-Space Proximal Networks (HSPNs) was described in [31,33,34]. Here, they were applied to represent the chemical/structural space of 174 anti-biofilm peptides (ABFPs) registered in StarPepDB, after removing redundancy at 98% of similarity from the original set of 221 ABFPs by applying the Smith-Waterman algorithm [36]. Thus, the resulting 174 ABFPs were used to generate HSPNs. In doing so, an optimized set of alignment-free (AF) sequence descriptors by feature selection methods were used to represent ABFPs as network nodes [33]. The Euclidean distance metrics with min-max normalization were applied to determine the pairwise similarity relationships among them. ABFPs within the HSPN are clustered by using the modularity optimization algorithm based on the Louvain method [37], thus peptide communities sharing similar features can be displayed in the network. All these steps were performed by using the StarPep toolbox associated to StarPepDB [32].
2.2. Metadata Networks
Metadata networks (METNs) were also explained in [33]. They are another type of network that do not display similarity relationship among the nodes/peptides like the HSPNs. METNs are bi-layer networks represented by the ABFPs and their associated metadata (origin, database, function, target), respectively [32]. Particularly, one layer is composed by the 174 ABFPs and the other by their sourcing database and origin. Therefore, ABFPs belonging from the same database are connected to the same “database node” but additionally can be connected to nodes representing other databases. METNs can be include other layers (metadata) and multi-type links and hierarchical connections may be set.
2.3. Networks similarity cutoff analysis
The optimal similarity threshold for the HSPN was set up by analysing the network parameters behaviour at varying the pairwise similarity cutoff between nodes (peptides). HSPNs representing the ABFPs were built with the Euclidean distance metric but changing similarity threshold from 0 (no cut-off) until 0.95 similarity cutoff. The resulting HSPNs at each similarity cutoff are exported as GraphML files from StarPep toolbox to be imported in Gephi [35]. Gephi allows the calculation of a more comprehensive set of network parameters such as average degree, density, modularity, average clustering coefficient (ACC), number of communities and singletons, network diameter, average path length at each similarity threshold, in order to determine the optimal value. The optimal cutoff should draw the most informative network topology caused by the best trade-off among its parameters. Consequently, it will be considered as the network model representing the ABF chemical space. These exploratory analyses will be addressed in details at the Result and Discussion section.
2.4. Network Visualization
Network visualization is conducted in Gephi by applying layout algorithms to the original HSPNs, allowing a readable network representation where nodes are intended to not overlap [35]. The Fruchterman-Reingold algorithm was applied to layout HSPNs as a circle where communities are represented by different colours while the nodes size were scaled according to centralities measures [38]. Several types of centrality measures were calculated for each node (i) node degree, (ii) harmonic, (iii) betweenness and (iv) hub-bridge. For HSPNs, all these centrality measures were calculated while for the networks with metadata associated to the peptides (e.g. database, origin, target and function) the betweenness is the centrality of choice.
The networks can also be directly shown by using the principal components as the cartesian coordinates of nodes (peptides) represented by a set of AF molecular descriptors. In this case, none layout algorithm is applied.
2.4. Scaffold Extraction by Centrality Measures
The HSPN model selected from the similarity threshold analysis was used to retrieve the most central and atypical ABFPs from its communities and singletons. The ABFPs were ranked down separately according to the harmonic and hub-bridge centrality measures, then a process of redundancy reduction is performed by using the Scaffold extraction plugin from the StarPep toolbox [33]. It consists in applying on peptides bearing similar centrality measures a pairwise identity cutoff value that could range from 0.90 – 0.30. The similarity measure was calculated by the Smith-Waterman local alignment algorithm using default settings [36]. Thus, the resulting extracted sets contain peptides bearing unique values of centrality measures sharing sequence identities under the predefined cutoffs (0.90 – 0.30 of sequence identity). Finally, nodes representing 10% lower centrality than the most central node were also removed in each metric.
2.5. Selection of the Most Representative Extracted Subset
As several subsets were extracted by applying both the harmonic and hub-bridge centralities at different identity cutoffs (0.9 – 0.3), the selection of the subset representing optimally the original HSPN space with the minimum number of ABFPs per metric is need. This selection was performed by visual mining from the HSPN overlapping of the extracted subsets on the HSPN model. The evaluated subsets represented between 30-45% out of the original HSPN space (174 ABFPs). We considered for the selection the spatial distribution (spatial coverage) of the reduced subset on the HSPN model represented by the cartesian coordinates nodes, estimated from the 2 most relevant principal components. Once, the subsets corresponding to the best overlapping having the minimum identity cutoff/number of peptides were found for each metric, the union of the centralities (HC ∪ HB) were also explored at 0.45, 040 and 0.35 identity thresholds. The subsets from the union of 2 centralities should has more coverage than when a single one is applied.
2.6. Motif Discovery
2.6.1. Multiple Sequence Alignments
The motif discovery process was conducted on the ABFPs communities from the HSPN model. A total of 11 communities were analysed, 8 of them showed more than 2 members (clusters 4, 7, 9, 11, 14, 15, 17 and 22) while clusters 6 and 28 only contained 2 peptides. The 20 singletons were gathered in one cluster. The detection of anti-biofilm motifs using alignments were approached as follows:
- -
- Communities with more than 2 ABFPs including the one containing the 20 singletons were aligned independently using multiple sequence alignment (MSA) algorithms. The algorithms of choice were MAFFT (Multiple Alignment using Fast Fourier Transform) v7.487 with the iterative refinement FFT-NS-i option [39] and MUSCLE (Multiple Sequence Comparison by Log- Expectation) v3.8 [40], publicly available at https://www.ebi.ac.uk/Tools/msa/. The conserved motifs were detected by jointly analyzing the consensus sequences and Seq2Logo, implemented in the Jalview v2.11.2.5 program [41] and EMBOSS Cons v6.6.0 available at https://www.ebi.ac.uk/Tools/msa/emboss_cons.
- -
2.6.2. Alignment-Free (AF) Detection
The AF detection of motifs was carried out similarly by communities. They were analysed in STREME (Sensitive, Thorough, Rapid, Enriched Motif Elicitation) to discover ungapped motifs that are enriched with respect to a control set generated by shuffling input peptides [43]. The analyses were performed via MEME webserver (https://meme-suite.org/meme/tools/streme). The motif width was set between 3-5 amino acids length. STREME applies a statistical test at p-value threshold = 0.05 to determine the enrichment of motifs in the input peptides compared to the control set as a stopping criterion.
2.6.3. Motif Enrichment Analysis
Simple Enrichment Analysis (SEA) v5.5.0 from the MEME suite (https://meme-suite.org/meme/tools/sea) was used to validate the motif discovery process by evaluating the enrichment of each listed motif in external benchmark datasets of ABFPs [44]. The relative enrichment ratio of each motif in the query vs. control sequences is defined as:
where, NPOS is the number of positive sequences (query peptides) in the input, and NNEG is the number of control sequences in the input.
3. Results and Discussion
3.1. Half-Space Proximal Network Model
HSPNs of ABFPs were built with the Euclidean distance metric but changing similarity threshold from 0.0 to 0.95. As long the cutoff increases, the network density decreases due to the loss of edges satisfying the similarity cutoff. Therefore, several nodes get disconnected from the giant component of the network and appear as isolated communities or singletons representing atypical sequences. The original HSPNs (with no similarity cutoff) have the particularity that all nodes are fully connected like in the giant components of Half-Space Graphs, however at applying increased similarity cutoffs, increasingly sparser graphs with not connected nodes (singletons) are displayed.
Then, some network parameters were retrieved at different similarity cutoffs in order to determine the optimal one for determining the most informative network topology. In relevance order, network parameters such as density, modularity, average clustering coefficient and number of communities were analysed (Figure 1). The network density is the actual number of edges over the maximum number of possible edges in a network. If the density is too high, the understanding of network topological features gets complicated and by contrary it gets lose useful information; therefore, a compromise between both extremes is need. Generally, network density around 0.1 is acceptable, however as HSPNs show much lower density values (< 0.01) because their node connections are conditioned by a predefined proximal/adjacent space. At increasing the similarity cutoff, networks density tends to drop since only edges weighted with high similarity are retained.
The modularity of the networks was also analysed at each similarity cutoff. This is a network parameter that compares the density within a community with the expected one for the same group of nodes on a random network. We calculated modularity and the number of communities using the modularity optimization clustering algorithm (based on the Louvain method [37]). Unlike the network density, both the modularity and the number of communities/singletons get significantly increased, especially at applying similarity cutoffs from 0.5 (Figure 1).
On the other hand, the average clustering coefficient (ACC) is a global measure of nodes neighbourhood connectivity and can be also used for evaluating network topology changes against similarity thresholds. Although at similarity cutoff of 0.80 all network parameters displayed a dramatical change (Figure 1), the optimal value was selected by jointly analysing all HSPN parameters (File 1SM - Supplementary Materials). The point where the network density dramatically drops but at the same time the modularity up while taking care the trade-off between the number of communities/singletons generated is a good starting criterium for the selection. In this sense, the cutoff value should be between 0.6 – 0.7 where the density and modularity have an inverse behaviour while the ACC did not suffer any dramatical change and the number of communities and singletons are reasonable in order to display informative network topology (Figure 1). Thus, the optimal cutoff of 0.65 was selected by analysing all HSPN parameters displayed in Figure 1 and File 1SM.
In addition, the degree distributions of the HSPN with no similarity cutoff and at 0.65 were plotted to explore the behaviour of these networks as generic models [45]. The node degree distribution of HSPN with not cutoff shows a bell-shaped distribution (Figure 2, File 1SM), revealing the random behaviour of the HSPNs, similarly to the random models. However, when the optimal similarity cutoff is applied the classical normal distribution get loses and several small bell-shape patterns appears along different node degree ranges. Therefore, the HSPN model has not an evident random behaviour which indicates that could be used as a topological network model (Figure 2).
3.2. Network Visual Mining
3.2.1. Visual Mining of HSPNs, the Most Central and Atypical ABFPs
In addition to the numerical characterization of HSPNs by the calculation of network parameters, their visualization also provides new and simple insights to unravel the complex relationships of the objects they represent. In our case, they were used to represent and analyse the chemical space of 174 non-redundant ABFPs by applying AF similarity networks with the application of the half-proximal space graphs [31,33,34]. Network visualization can mirror several network parameters like density, communities, nodes size can be ranked according to different centrality measures e.g. node degree, harmonic, hub-bridge, betweenness, etc. Thus, the most important or central peptides can be highlighted as well as the edges weighed with high or low similarities. This work is aimed to exploit at the top the visual representation of complex networks representing ABFPs for analysing their structural space and their associated metadata that are relevant for the discovery and design of antibiofilm agents [28].
Both the original HSPN (no cutoff) and the HSPN model (cutoff at 0.65) representing the structural space occupied by the 174 ABFPs are depicted in Figure 3, and the File 2SM complement the network projection by numerical characterization. Networks communities are highlighted with different colours and nodes importance was represented by the node degree centrality. The original HSPN shows 5 communities clearly identified by different colour while in the HSPN model bearing 30 communities (20 singletons included), it is more difficult their delineation by colours (see details in File 2SM). However, since the HSPN model is a low-density network with a smaller number of edges (325) than the original HSPN (689) allows a better depiction of the node’s relationships (File 1SM).
Figure 3.
A – HSPNs visualization by using the Fruchterman-Reingold layout without similarity cutoff and B – with a similarity cutoff of 0.65. Peptides communities are represented by different colours while the nodes size was scaled according to node degree. .
Figure 3.
A – HSPNs visualization by using the Fruchterman-Reingold layout without similarity cutoff and B – with a similarity cutoff of 0.65. Peptides communities are represented by different colours while the nodes size was scaled according to node degree. .

The most relevant nodes according to the node degree were clearly identified in the HSPN with no cutoff (e.g. starPep_07526, starPep_02281, starPep_00048, starPep_03668, starPep_08958) whereas at applying the similarity cutoff a significant fraction of graph edges gets lost and the nodes degree decreases. That’s why only two of the most connected nodes were highlighted; the starPep_00048 and starPep_03668. As these two peptides also brought up in the original HSPN in similar locations and clusters, it would be figured it out that changes in the HSPN topology at applying similarity cutoffs do not alter the most popular peptides in both networks. In order to address this question, the top 10 most relevant peptides according to each four centrality measures (node degree, harmonic, betweenness and hub-bridge) were extracted from the HSPNs with and without similarity cutoff (File 2SM). Then, the intersection of the resulting four peptide sets was analysed for each HSPN. Table 1 displays the common peptides identified from the 10-top ranked by four, three and two centrality measures from HSPNs with and without similarity cutoff. The last four rows at each HSPN represent singular peptides identified for each of the four centralities. The cluster/community containing the 10-top ranked ABFPs is also displayed.
Table 1 shows a different composition of common and singular peptides from the 10-top ranked ABFPs by centrality measures at HSPNs with and without similarity cutoff. This observation supports that changes in the HSPN topology by removing edges (similarity relations) not only produce sparser networks with an increased number of clusters, but also lead to a variation of peptides centrality measures and therefore on its topological distribution. However, a small set made up of probably the most relevant peptides were identified for four and three centrality measures in both HSPNs. That are the cases of starPep_00048, starPep_03668, starPep_10922 and starPep_00000. These peptides seem to be very important within the ABF chemical space. The starPep_00048 is a human defensin derivative (HNP1) of 30 amino acid length with several reported interesting bioactivities (antiviral, anti-Gram+/ –, antifungal, anticancer, enzymatic inhibitor) besides its antibiofilm action which probably is exerted by its ability to disrupt membranes and interfere biological process involved in interspecies interaction [46,47]. The starPep_03668 and starPep_10922 are synthetic constructions of 35 and 12 aa length, respectively. The starPep_03668 was designed as pathogen-selective peptide, based on the fusion of a species-specific targeting peptide domain with a wide-spectrum antimicrobial peptide domain. Thus, it showed activity against several communities of Streptococcus species [48]. By contrast, starPep_10922 was designed as a D-enantiomeric peptide aimed to resist proteases degradation and also to prevent the accumulation of (p)ppGpp, which is a key messenger for biofilm formation. StarPep_10922 was able to prevent biofilm formation from P. aeruginosa as well as to disperse and eradicate the bacteria in the resulting mature biofilm [49]. So far, these three central peptides have not been reported as toxic for mammalian cells being lead peptides for developing ABFP drugs. The starPep_00000, a 26 aa length ABFP that was derived from Melittin (bee venom) show up as a promising candidate since several pharmacological activities has been assigned to it besides the antibiofilm one, and has also been extensively evaluated against many targets. However, starPep_00000 has also shown haemolytic activity and toxicity against to eukaryotic cells which may straightforward limit its therapeutic potential, unless its toxicity would be relieved by optimization procedures [50].
The ABFP space is not only represented by the central peptides, there also exist disconnected peptides to the giant component of the network that bear low values of node degree. These peptides are categorized as atypical because they share very low sequence similarities with the central or popular ones, that prevent the estimation of their properties. However; they are remote members of the ABFP family and they also account for the antibiofilm chemical space. In this sense the HSPN with similarity cutoff is very useful to uncover atypical peptides. The cutoff at 0.65 of AF similarity was the optimal to retain a reasonable trade-off between the number of communities and singletons; where the simplest community is considered when 2 nodes (peptides) are connected, and singletons are those that are not connected with any other in the network. Atypical peptides represent singular structures that may represent privilege scaffolds for designing antibiofilm agents.
Since the HSPN with no cutoff is fully connected, no isolated communities and singletons can be identified. Table 2 displays the atypical peptides from the HSPN at similarity cutoff of 0.65, the 20 singletons where all centrality measures reached 0 value and two isolated communities made up of 2 peptides interconnected with node degree 1.
The atypical peptides could have been similarly analysed than the central peptides, but as all singletons displayed 0 values for all centrality measures; then the analysis was carried out by exploring the metadata associated to each peptide by using the StarPep toolbox. Atypical peptides with no reported toxicity to mammalian cells are marked with an asterisk while those that also have a diversity of desired functions contributing to the antibiotic/antibiofilm activity were highlighted in bold. That’s the case of starPep_04044 which is a synthetic peptide of 13 aa length representing a singular structure that can coat successfully titanium surfaces and also can target Gram+ and Gram- bacterial strains in both their planktonic and biofilm forms allowing its utilization for preventing infection-related implant failures in dentistry and orthopaedics. It has been effective on Pseudomonas aeruginosa, Streptococcus gordonii, Porphyromonas gingivalis, Staphylococcus aureus and Escherichia coli [51,52].
3.2.2. Metadata Analysis by Visual Mining
The METNs corresponding to the 174 ABFPs were constructed considering their source database and origin. Similar to the previous visualization, nodes were displayed by colors and size. Red nodes represent source databases and origin, respectively while the ABFPs were in grey (Figure 4A and 4B). Nodes size were scaled according to their betweenness centrality values, which is based on the shortest path between two nodes. Thus, this type of centrality is the number of shortest paths that pass through a target node, particularly on red nodes representing “database” and “origin”.
As shown in Figure 4A, the 174 ABFPs registered in StarPepDB were mainly collected from BaAMP [24], the Structurally Annotated Therapeutic database (SATPdb) [53] and Da-tabase of Antimicrobial Activity and Structure of Peptides (DBAASP) [54] which are represented by larger nodes with the highest connection with the ABFPs. While BaAMP and DBAASP entries were carefully collected from the literature, the SATPdb was built from 22 peptide databases that included BaAMP [24], and others similarly dedicated to specific activities such as AVPdb (antiviral) [55], ParaPep (anti parasitic) [56], Hemolytik (hemolytic) [57], CancerPPD (anticancer) [58], etc; and also from generic AMP databases like DAMPD [59], APD [22], CAMP [60], LAMP [61], DRAMP [62], etc. Such databases that integrated SATPdb were represented as smaller red nodes sharing less edges with the ABFPs.
Most of the ABFPs come from synthetic constructs, represented by the largest node or hub in the network (Figure 4B). However, other natural sources have also provided ABFP scaffolds for further modification/optimization. Among the most contributing taxonomic groups are the Bacteria, Homininae, Similiformes, Pan and Bos (Bos Taurus). Therefore, this information can guide the discovery and design of antibiofilm agents from peptides. Particularly inform on what are the most relevant ABFPs databases and the main sources/origin where to find promising ABFP scaffolds.
3.3. Representing the ABFPs with a reduced subset
3.3.1. The selection of the best representative subset
The original space of 174 ABFPs, illustrated by the HSPN model at the optimal cutoff of 0.65 (Figure 3B), can be further simplified by applying the scaffold extraction algorithm implemented in the StarPep toolbox. This algorithm allows the topological simplification of the network by removing nodes with equal or similar values of centrality measures but retaining those that still share a local similarity below certain cutoff. As the reduction of ABFPs intends to keep the HSPN topology and properties, it is applied to all type of nodes, from the most central to the atypical ones (singletons). The reduction was performed by ranking the harmonic and hub-bridge centrality values of the nodes and by applying similarity cutoffs from relaxed criteria (retain all peptides sharing < 0.9) to more restrictive similarities (< 0.45 of sequence identity). This step produces several subsets for each centrality metrics (Table 3, File 3SM).
As mentioned before, the best subset representing the original space draw by the HSPN model should be composed by the minimum number of ABPFs with a coverage of the original space < 50%. However, the coverage is not the unique criterium to select a representative subset of the original space, its distribution on the bi-dimensional (2D) space occupied by the HSPN model should also be considered. An effective subset should have a topological representativeness over all the network, representing connected, isolated communities and even singletons. In this sense, a subset extracted under the criterium of only one centrality measure might not fulfil the expected 2D coverage. Thus, we decide to fuse the information of the HC and HB centralities since they rank the network nodes according to their position using different definitions. Thus, the union of the subsets 6, 7 and 8 which are highlighted in bold in Table 3 was evaluated. Subsets 6, 7 and 8 display a coverage <50% with a low of number of ABFPs, but promising for their union (HC ∪ HB) at the same cutoff value. It is noteworthy to say the union between two subsets include the common peptides (intersection) and the singular peptides from each subset. The union of subsets 6, 7 and 8 resulted in fasta files (File 3SM) containing 85, 66 and 52 ABFPs representing 49%, 38% and 30% of the HSPN model, respectively. The File 3SM also contain the 221 ABFPs registered in StarPepDB and the 174 used to generate the HSPNs.
Finally, the subsets 6, 7 and 8 from each centrality metric as well as their resulting fusion (HC ∪ HB) were overlapped on the HSPN model to evaluate their 2D coverage/spatial distribution (Figure 1SM). In the three subsets, the union of the centrality measures (HC ∪ HB) at the evaluated similarity cutoffs displayed a better 2D spatial distribution on the HSPN model. Particularly, the HC ∪ HB from subset 7 showed the best trade-off considering the lowest number of peptides with the best 2D coverage (Figure 1SM). Figure 5 summarizes the overlapping of the HC ∪ HB from subset 6, 7 and 8 with the HSPN model. The overlapped subsets are represented by small black nodes over the coloured nodes that represents the communities in the HSPN model.
3.3.2. Visualizing/Analysing the best representative subset with HSPNs
The main goals of extracting a reduced subset representing the complex networks is to allow retrieving useful information from the visual inspection the networks. With reduced number of nodes and edges complex networks turn more legible for the human eyes and the representativeness of the subset result also useful for multi-reference similarity searches against unlabelled peptides. The Figure 6 show the HSPN constructed from the reduced space made up of 66 ABFPs resulted from the HC ∪ HB of the subset 7 (cutoff 0.40). As can be seen in Figure 6A and 6B, most of the main ABFPs identified in the HSPN model were transferred to the reduced space such the cases of starPep_00000, starPep_00042, starPep_00048, starPep_03668, starPep_05561 and starPep_00004, that additionally were among the 10-top relevant ABFPs by the HC and HB centralities (Table 1). Its noteworthy that other ABFPs, not identified among the top-ranked in the HSPN model, brought up significantly when constructing the HSPN with the 66 representatives e.g. starPep_00522 and starPep_00514 (Figure 6A and 6B; File 4SM).
On the other hand, 5 out of the 20 singlentons appeared in the representative subset. They are the following ones starPep_00002, starPep_04044, starPep_05305, starPep_09934, starPep_10637, being all reported as non-toxic except starPep_00002. It was not by chance that the priviliged scaffold of starPep_04044 was also selected, which indicate the scaffold extraction algorithm implemented in StarPep toolbox works.
The subset of 66 representative ABFPs extrated from the HSPN model is described in File 4SM that includes their IDs, sequences, lengths, the cluster where they belonged to in the HSPN model, their centralities measures both in the HSPN model and in the new HSPN constructed (Figure 6). This non-redundat subset of 66 peptides represents the chemical space of the ABFPs, and it is advisable to be used as reference to map new ABFP sequences, also in multi-references similarity searches against unlabel peptide datasets and for design purposes.
3.3.3. Visualizing Mining of the METNs
The reduced subset is also useful for unravelling information from METNs which are even more complex than similarity networks since they include other layers containing additional nodes with associated peptide metadata. Consequently, counting on a representative subset of the original space aids to analyse the huge amount of information associated to the ABFPs. As previosly-mentioned microbial biofilms are responsible for most of chornic and medical device-related infetions as well as for the microbial resistant to several antibiotic classes, thus, the identification of promising ABFP scaffolds is an urgent task.
As an illustrative example of the METNs contribution to the identification of promising ABFPs scaffolds, six relevant ABFPs according to the HC and HB centralities were selected from the representative subset (Figure 6) for visual analysis of their metadata. This time key metadata for the development/design of ABF agents from ABFPs were chosen, e.g. other associated activities of the ABFPs and the targets what they have been evaluated on (Figure 7). The study cases were starPep_00000 (blue), starPep_00193 (yellow), starPep_00004 (green), starPep_00025 (pink), starPep_00514 (black), starPep_00522 (cyan).
The Figure 7A is organized in such way that desired activities were placed outside at the left part of the METN while the undesired ones at the right. The six candidates under study, in addition to the antibiofilm activity, have also the antifungal and the antibacterial, specifically against Gram-positive and Gram-negative strains which are very convinient for developing next-generation antimicrobial agents able to target both planktonic and biofilm microbial forms. However 4 of them were reported as hemolitycs and 5 were “toxic to mammals”. The starPep_00522 (in cyan) is the only one that was neither reported as “haemolytic” nor “toxic to mammals”and therefore its peptidic scaffold can be used for developing antibiofilm agents for clinical purposes. Addtionally, the Figure 7B also shows that starPep_00522 has been evaluated in Escherichia. Coli, Pseudomonas aeruginosa, Candida albicans, Cryptococcus neoformans where the two first targets classified within the ESKAPEE pathogens, considered the most threatening antimicrobial resistant microbes [63]. This visual analysis of the METNs can be extended to all 66 ABFPs of the representative set.
3.4. External Representative ABFPs on the representative Antibiofilm HSPN
In a recent work, Li at al. arrived to 14 representative ABFPs out of a total of 51 peptides with a reported antibiofilm activity either by inhibition of the biofilm formation or by the eradication of pre-formed biofilms. The selection was based on the identification of 14 ABFP classes according to their mechanisms of action. They also evaluated them against the biofilm and planktonic forms of Gram-positive bacterium Streptococcus mutans, the Gram-negative bacterium Pseudomonas aeruginosa, and the fungus Candida albicans. Those ABFPs with MBICs (minimal biofilm inhibitory concentrations) that are lower than their minimal inhibitory concentration (MICs) (minimal inhibitory concentrations) represented promising candidates against biofilm-related infections. Table 4 shows the 14 representative candidates categorized by antibiofilm mechanisms of action (Information taken from Table S2 published in [64].
This set made up of 14 ABFP classes representing different mechanism of action to carry out the antibiofilm activity was mapped on the structural/chemical space of the 66 ABFPs, drawn by the HSPN model (Figure 8A). As the subset of 66 ABFPs showed the best representativeness of the antibiofilm chemical space was used along with the most suitable HSPN projection for the overlapping purpose. The HSPN that plots nodes coordinates from their 2 most relevant principal components, estimated from a non-redundant set of AF descriptors, is the most real approach to display the peptide’s location in the network, allowing a more accurate visual inspection of the similarity and distribution of the 14 representative ABFPs on the reported chemical space (Figure 8A).
Figure 8A shows the node and names corresponding to 14 ABFPs in black colour while the other 66 from StarPepDB were labelled according to the colours assigned to each one of the 5 network communities. As can be observed in Figure 8A, all 14 ABFPs were framed within the antibiofilm HSPN. In fact, the Indolicidin, Protegrin-1 and HBD-3 overlaped perfectly with starPep_00002, starPep_00020 and starPep_00116, respectively. The 14 mechanisms of action classes were distributed among all the 5 HSPN communities, which may indicate a connection between structural patterns (motifs) found within network communities with the antibiofilm mode of action. In order to illustrate this fact, 6 ABFPs that showed antibiofilm activity against both bacteria and fungi (pleurocidin, Pac-525, protegrin-1, TetraF2W-RR, WLBU2, and melittin) overlaped perfeclty or nearly over different commnunities. Pleurocidin, Pac-525, protegrin-1 and melittin are evidently overlaped on the communities coloured in blue, green, pink and light purple. WLBU2 was also placed in the green community as the Pac-525, probably because both act using similar mechanisms of action involving the interaction with the lipopolysaccharides to destroy or penetrate the bacterila membrane. Although, TetraF2W-RR is within the antibifilm space represented by the HSPN, it did not overlap on any specific community. However, it was placed between green and pink communities containing members such as Pac-525, WLBU2, Indolicidin and protegrin-1 which mode of action are closely related to the reported for TetraF2W-RR, bacterial membrane disruption. These 4 ABFPs are ariginine (R)-rich peptides containing repeated units of R allowing the interaction with negatively-charged bacterial membranes, the formation of transmembrane pores and cell penetration [65].
The Figure 8B complements the information extracted from the Figure 8A. It shows those representative ABFPs sharing AF similarities > 0.60 with some of the 66 ABFPs extracted from StarPepDB. Black nodes represent the 9 out of the 14 representative ABFPs that fulfill this condition while the black edges display the similarity relationships from black nodes (origin) to coloured nodes (target). Target nodes labelled as starPep_XXXXX retained the same colour identifying them at the network communities in Figure 8A. The Figure 8B confirms that Indolicidin, Protegrin-1 and HBD-3 share the max. similarity (1.0) with those ABFPs that overlapped with starPep_00002, starPep_00020 and starPep_00116 in Figure 8A. The location of pleurocidin and protegrin-1 was also supported by their highest similarities (0.65 and 1.0) with members of the communities blue (starPep_00496) and pink (starPep_00020), respectively. However, as the pleurocidin shows multiple actions such as membrane disturbance and permeabilization, binding to bacterial DNA and interference with several cellular functions, also display similiarities with members (starPep_00193, starPep_00051) from other 2 communities (Figure 8B).
On the other hand, Figure 8B also served to correct the location of melittin that is actually overlapped over the orange community showing 0.95 of similarity to starPep_00000 (a Melittin derivative), despite in Figure 8A looked over the light purple community. Although the 14 ABFPs could be mapped within the HSPN, the Figure 8B confirmed certain singularity of Pac-525, peptide 1037, TetraF2W-RR, P1 and WLBU2 within the representative ABFP space, at not sharing AF similarities higher than 60%. Such singularity was also confirmed by evaluating the pairwise identity of these last 5 ABFPs against the 66 representative ones (Figue 9B). The 9 ABFPs that were clearly mapped at AF similarities > 0.60 were also compared by pairwise global alignments (Figure 9A)
The inferior part of Figure 9A, framed by the white line, display 5 red dots that corresponds to those ABFPs (Indolicidin, Protegrin-1, HBD-3, Melittin and Nisin) sharing network edges weighted with AF similarities higher than 0.90, the edge weighted with 0.69 is likely represented by the yellow dot and the remaining slightly superior to to 0.60 are depicted in cyan colours. While the Figure 9B confirmed that the similarities shared by the all 5 unmapped ABFPs were actually below 0.60. All dots were moslty coloured in blue and some fews in cyan may represent the values close but below to 0.60.
It is important to say, as the AF and AB similarities are defined under different methodological frameworks, they may characterize the same pairwise relation with different values, despite they are correlated. The file 5SM shows the pariwise identities values of 9 and 5 ABFPs from the 14 mode of action classes against the 66 representing the antibiofilm chemical space.
3.5. Motif Discovery Assisted by Complex Networks
The identification of motifs accounting for the antibiofilm activity can be assisted by the exploration of ABFP similarity networks looking for sequence patterns within network communities. Although, the HSPN representing the ABFP chemical space was built using an AF distance metrics (Euclidean) and the network communities are estimated considering parameters from the nodes and edges properties [37]; such clusters should contain peptides sharing similar features. Thus, the communities from the HSPN model of the 174 ABFPs resulted the source for the motif discovery. The sequence diversity at each community was evaluated by global alignments. The Figure 2SM displays the heatmaps that mirrors the pairwise sequence identities for communities containing more than 2 peptides that correspond to the clusters 4, 7, 9, 11, 14, 15, 17, 22 including singletons clustered (File 3SM). The Figure 2SM evidenced a high sequence diversity within all communities. Consequently, iterative alignment algorithms like MAFFT and MUSCLE were applied to deal with the high sequence diversity. The multiple sequence alignments (MSAs) were visualized with the Jalview which allowed the estimation of their corresponding consensus sequences and Seq2Logos. The consensus sequences from the MSAs were also estimated by the EMBOSS Cons. The full exploration of the MSAs considering their corresponding consensus sequence and Seq2Logos allowed the identification of conserved regions considered as ABF motifs. The strategy carried out for the identification of the motifs in the MSAs performed on cluster 4 of the HSPN is illustrated in Figure 9, while for all communities/clusters is displayed in Figure 3SM.
Figure 9.
Motifs detection by the multiple sequence alignment (MSA) algorithms MAFFT and MUSCLE on the network cluster 4. The MSAs are visualized with the Jalview program which also estimates a Seq2Logo and the consensus from the alignment positions. Another consensus sequence that served as a guide for motif location was estimated by the EMBOSS Con.
Figure 9.
Motifs detection by the multiple sequence alignment (MSA) algorithms MAFFT and MUSCLE on the network cluster 4. The MSAs are visualized with the Jalview program which also estimates a Seq2Logo and the consensus from the alignment positions. Another consensus sequence that served as a guide for motif location was estimated by the EMBOSS Con.
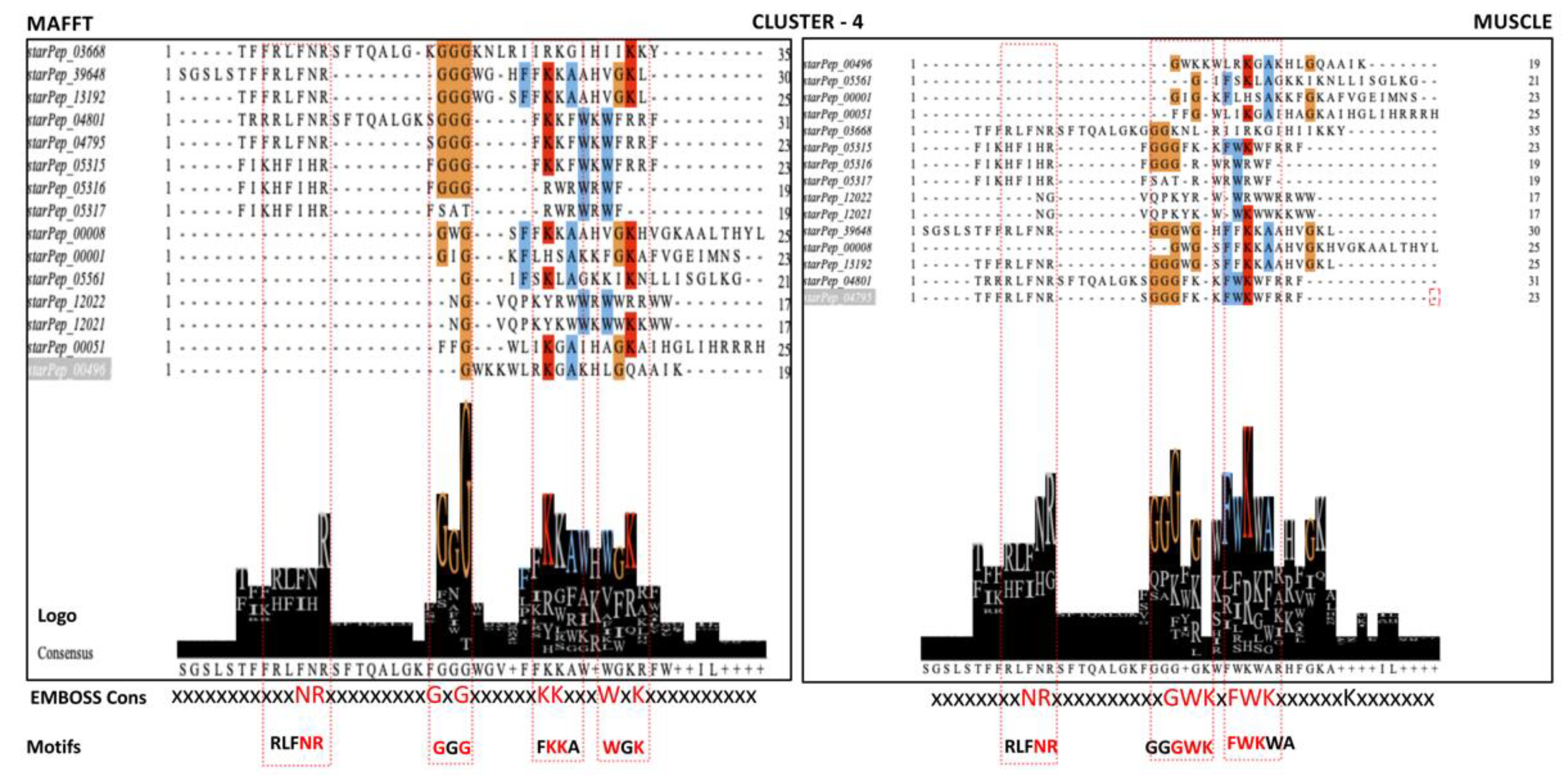
Table 5 listed ABFP motifs identified by each MSA method at each network community or cluster. The consensus estimated by the EMBOSS Cons was the preferred template for motif identification because it gives a more legible output. High scored amino acids/positions are represented by capital letters, less scored but positive residues by lower-case letters while non-consensus positions by x (Table 5).
As part of the motif discovery process, the alignment-based search was complemented by evaluating an AF approach. Based on its high performance and versatility to identify motifs in OMICs data, the STREME algorithm was applied to find unaligned patterns ranging from 3–5 aa length at each network community [43]. STREME computes a score for the detected motifs meeting the statistical significance (p-value threshold < 0.05); set also as a stopping search criterium. Table 6 displays the discriminating motifs against control sequences at each ABFP cluster/community. Motifs appearing in more than 20% of the query peptides are listed according to their statistical significance (score).
Motifs highlighted in bold in Table 5 and Table 6 indicates they are closely related or included into each other. That’s means that both MSA algorithms and STREME showed some degree of agreement in the motif detection discovery. However, both approaches also identify singular motifs, not highlighted. This fact demonstrates that the application of AB and AF approaches was a right choice for a full motif exploration. Given that both methods identified a relative high number of motifs between 33 – 35, enrichment analyses were further performed in order to filter the discovery motifs shown in Table 5 and Table 6.
Motif enrichment analyses are used to determine if a group of sequences contains a statistically significant number of matches to a given motif. In this sense, we used the SEA algorithm [44] to select what motifs from both tables significantly were enriched in two sets of ABFPs. The first set was the reported by Li et al. consisting in 14 representative ABFPs of the antibiofilm modes of action [64], and the second one encompassed 192 non-redundant ABFPs, extracted from the 214 ABFPs registered in BaAMP database [24] (File 3SM). Eight members from the representative subset were included among the 192 ABFPs, but all them have showed antibiofilm activity at different levels. As a screening criterium, ABFPs enriched in both the representative and the extended dataset were selected. Table 7 list the discovered ABFP motifs by the AB and AF approaches at the network communities.
Our motif search approach assisted by the complex networks are not so far from the few findings reported in the literature. Recently, Anastasiu et al. found that the following motifs “RIRV,” “RIVQRIK,” and “IGKEFKR” appeared with more frequency in 242 ABFPs collected from APD and BaAMP databases in respect to a curated negative set [28], when using the “MERCI” software [67]. In this sense, we agree with them in the detection of the “RIRV” which was fully integrated in the RIRVR motif detected in cluster 14 by the MAFFT algorithm, and also enriched in the BaAMP dataset. Although “RIVQRIK” and “IGKEFKR” were not detected as such, we could identify in clusters 15 and among the singletons by the MSA methods, the “RIV” and “FIK” patterns, which are part of them. These two last three-amino acid motifs were also enriched in the extended dataset.
In this previous report, authors also found that the dipeptides “IR/RI”, “WR/RW”, and “KK” were the most common among the selected ABFPs [67]. Certainly, these dipeptides are present in the motifs discovered with the intervention of complex networks, at relatively high frequency. For example, the “IR/RI”, “WR/RW” and “KK” dipeptides appears in 9/7, 7/8 and 12 of the total motifs, respectively. In addition to them, also the “RR” and “KL” dipeptides displayed a similar representation among the motifs.
In a recent report arginine-rich motifs for the antibiofilm activity from peptides designed for sequestering the nucleotide second messenger c-di-GMP, involved in the formation of P. aeruginosa and K. pneumoniae biofilms were revisited [68]. The key role of the DRR and [RK]RxxD motifs from these sequestering peptides (SP) to specifically bind to c-di-GMP was demonstrated by nuclear magnetic resonance (NMR)-based experiments [69]. These motifs associated to SPs are difficult to discover by bioinformatics methods since their sourcing peptides are probably still not registered or underrepresented in databases. However, the peptide R4F4 (RRRRFFFF), with a proven antibiofilm activity on P. aeruginosa through c-di-GMP sequestration [68,70] bears a more frequent motif (RRRR) among the arginine-rich ABFPs. In fact, “RRRR” was detected in our complex network-assisted motif search (Table 5 and 6).
On the other hand, despite the role of WWW motif for disrupting preformed biofilms from methicillin-resistant Staphylococcus aureus was unravelled by NMR and arginine scan experiments in 2017 [71], the WWW motif hardly appears in ABFP databases, being only represented by the designed peptide TetraF2W-RR [72].
Therefore, as the computational motif discovery is highly influenced by the peptide database composition and by the searching algorithm, here we provide new ABFP motifs discovered from combining network science with AB- and AF-based computational tools for motif detection. The motifs listed in 5, 6 and specially in Table 7 are useful for the “in silico” generation of peptide libraries addressed to the antibiofilm activity, as well as for the optimization of antibiofilm candidates. Finally, predicted motifs that actually account/improve the antibiofilm activity could be also used as motif-based descriptors for developing machine-learning models to screen peptide libraries and peptidomes as part of the discovery process.
4. Conclusions
The Half-Space Proximal Networks were successfully introduced to project the pairwise alignment-free similarities from the reported ABFPs. Particularly an HSPN model was obtained at applying an optimal similarity cutoff of 0.65 that allowed an effective delineation of network communities with the respective identification of the most central (relevant) and atypical peptides among the ABFPs. From the topology of the HSPN model, a reduced subset of 66 ABFPs resulting from the union of the harmonic and hub-bridge centralities were extracted by the scaffold extraction algorithm of StarPep toolbox. As these 66 peptides, made up by both the most relevant and atypical ABFPs, were not only selected by the centrality criteria, but also considering their topological distribution and coverage on the original space; they can be considered as representatives of the ABFP chemical space.
Alongside with this previously-mentioned procedure, the metadata associated to both the most central and atypical peptides were analysed by the visual mining of complex networks integrating additional relevant properties for the antibiofilm action and for the discovery/design of next-generation antimicrobial agents, able to combat MDR infections. On the other hand, the proposed network-assisted motif discovery allowed the identification of ABFP motifs by AB and AF approaches within the communities of the HSPN model.
In short, the network-based identification from the most central to the atypical ABFP scaffolds bearing promising antimicrobial activities on MDR targets were mostly transferred to the representative subset of 66 ABFPs. Therefore, this subset together with the discovered ABFP motifs is recommended to be used in the mapping and design of new ABFPs as well as for the development of next-generation antimicrobials.
Supplementary Materials
The Supplementary Information is available free of charge at Zenodo (DOI 10.5281/zenodo.7706555): https://zenodo.org/record/7706555#.ZAef_y8Q1X0.
Author Contributions
G.A.-C. and Y.M.-P. worked mainly on the conceptualization, formal analysis, supervision, validation, writing and reviewing of the manuscript. E.C.-T., F.M.-R., and J.R.V-M. worked on data curation and the design/implementation of the HSPNs and METNs visualization. J.R.-M. and N.-P. were responsible for the motif discovery and peptide diversity analyses. A.A., and C.H.-Z were participated in funding acquisition, writing, and reviewing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Declared none.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The StarPep toolbox software and the respective user manual, are freely available online at http://mobiosd-hub.com/starpep.
Acknowledgments
Y.M.-P. thanks to the USFQ Collaboration Grant 2021-22 (Project ID16897). G.A.-C. and A.A. were supported by national funds through FCT - Foundation for Science and Technology within the scope of UIDB/04423/2020 and UIDP/04423/2020.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vestby, L.K.; Gronseth, T.; Simm, R.; Nesse, L.L. Bacterial Biofilm and its Role in the Pathogenesis of Disease. Antibiotics (Basel) 2020, 9. [Google Scholar] [CrossRef]
- Gloag, E.S.; Fabbri, S.; Wozniak, D.J.; Stoodley, P. Biofilm mechanics: Implications in infection and survival. Biofilm 2020, 2, 100017. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Nunez, C.; Reffuveille, F.; Fernandez, L.; Hancock, R.E. Bacterial biofilm development as a multicellular adaptation: antibiotic resistance and new therapeutic strategies. Curr Opin Microbiol 2013, 16, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Sauer, K.; Stoodley, P.; Goeres, D.M.; Hall-Stoodley, L.; Burmolle, M.; Stewart, P.S.; Bjarnsholt, T. The biofilm life cycle: expanding the conceptual model of biofilm formation. Nat Rev Microbiol 2022, 20, 608–620. [Google Scholar] [CrossRef] [PubMed]
- An, A.Y.; Choi, K.G.; Baghela, A.S.; Hancock, R.E.W. An Overview of Biological and Computational Methods for Designing Mechanism-Informed Anti-biofilm Agents. Front Microbiol 2021, 12, 640787. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J Chin Med Assoc 2018, 81, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Bryers, J.D. Medical biofilms. Biotechnol Bioeng 2008, 100, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Veerachamy, S.; Yarlagadda, T.; Manivasagam, G.; Yarlagadda, P.K. Bacterial adherence and biofilm formation on medical implants: a review. Proc Inst Mech Eng H 2014, 228, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.; Rumbaugh, K. The Consequences of Biofilm Dispersal on the Host. Sci Rep 2018, 8, 10738. [Google Scholar] [CrossRef]
- Rumbaugh, K.P.; Sauer, K. Biofilm dispersion. Nat Rev Microbiol 2020, 18, 571–586. [Google Scholar] [CrossRef]
- Breidenstein, E.B.; de la Fuente-Nunez, C.; Hancock, R.E. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 2011, 19, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Romling, U.; Balsalobre, C. Biofilm infections, their resilience to therapy and innovative treatment strategies. J Intern Med 2012, 272, 541–561. [Google Scholar] [CrossRef]
- Barraud, N.; Hassett, D.J.; Hwang, S.H.; Rice, S.A.; Kjelleberg, S.; Webb, J.S. Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J Bacteriol 2006, 188, 7344–7353. [Google Scholar] [CrossRef]
- Xiong, Y.Q.; Estelles, A.; Li, L.; Abdelhady, W.; Gonzales, R.; Bayer, A.S.; Tenorio, E.; Leighton, A.; Ryser, S.; Kauvar, L.M. A Human Biofilm-Disrupting Monoclonal Antibody Potentiates Antibiotic Efficacy in Rodent Models of both Staphylococcus aureus and Acinetobacter baumannii Infections. Antimicrob Agents Chemother 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Verderosa, A.D.; Totsika, M.; Fairfull-Smith, K.E. Bacterial Biofilm Eradication Agents: A Current Review. Front Chem 2019, 7, 824. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.; Rehm, B.H.; Hancock, R.E. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Nunez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog 2014, 10, e1004152. [Google Scholar] [CrossRef] [PubMed]
- Chavez de Paz, L.E.; Lemos, J.A.; Wickstrom, C.; Sedgley, C.M. Role of (p)ppGpp in biofilm formation by Enterococcus faecalis. Appl Environ Microbiol 2012, 78, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Di Somma, A.; Moretta, A.; Cane, C.; Cirillo, A.; Duilio, A. Antimicrobial and Antibiofilm Peptides. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- Ma, L.; Ye, X.; Sun, P.; Xu, P.; Wang, L.; Liu, Z.; Huang, X.; Bai, Z.; Zhou, C. Antimicrobial and antibiofilm activity of the EeCentrocin 1 derived peptide EC1-17KV via membrane disruption. EBioMedicine 2020, 55, 102775. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 2016, 44, D1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Kang, X.; Dong, F.; Liu, Y.; Zhu, N.; Hu, Y.; Xu, H.; Lao, X.; Zheng, H. DRAMP 3.0: an enhanced comprehensive data repository of antimicrobial peptides. Nucleic Acids Res 2022, 50, D488–D496. [Google Scholar] [CrossRef]
- Di Luca, M.; Maccari, G.; Maisetta, G.; Batoni, G. BaAMPs: the database of biofilm-active antimicrobial peptides. Biofouling 2015, 31, 193–199. [Google Scholar] [CrossRef]
- Sharma, A.; Gupta, P.; Kumar, R.; Bhardwaj, A. dPABBs: A Novel in silico Approach for Predicting and Designing Anti-biofilm Peptides. Sci Rep 2016, 6, 21839. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sharma, A.K.; Jaiswal, S.K.; Sharma, V.K. Prediction of Biofilm Inhibiting Peptides: An In silico Approach. Front Microbiol 2016, 7, 949. [Google Scholar] [CrossRef] [PubMed]
- Fallah Atanaki, F.; Behrouzi, S.; Ariaeenejad, S.; Boroomand, A.; Kavousi, K. BIPEP: Sequence-based Prediction of Biofilm Inhibitory Peptides Using a Combination of NMR and Physicochemical Descriptors. ACS Omega 2020, 5, 7290–7297. [Google Scholar] [CrossRef]
- Bose, B.; Downey, T.; Ramasubramanian, A.K.; Anastasiu, D.C. Identification of Distinct Characteristics of Antibiofilm Peptides and Prospection of Diverse Sources for Efficacious Sequences. Front Microbiol 2021, 12, 783284. [Google Scholar] [CrossRef]
- Aguero-Chapin, G.; Galpert-Canizares, D.; Dominguez-Perez, D.; Marrero-Ponce, Y.; Perez-Machado, G.; Teijeira, M.; Antunes, A. Emerging Computational Approaches for Antimicrobial Peptide Discovery. Antibiotics (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Romero, M.; Marrero-Ponce, Y.; Rodriguez, H.; Aguero-Chapin, G.; Antunes, A.; Aguilera-Mendoza, L.; Martinez-Rios, F. A Novel Network Science and Similarity-Searching-Based Approach for Discovering Potential Tumor-Homing Peptides from Antimicrobials. Antibiotics (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Ayala-Ruano, S.; Marrero-Ponce, Y.; Aguilera-Mendoza, L.; Perez, N.; Aguero-Chapin, G.; Antunes, A.; Aguilar, A.C. Network Science and Group Fusion Similarity-Based Searching to Explore the Chemical Space of Antiparasitic Peptides. ACS Omega 2022, 7, 46012–46036. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Beltran, J.A.; Tellez Ibarra, R.; Guillen-Ramirez, H.A.; Brizuela, C.A. Graph-based data integration from bioactive peptide databases of pharmaceutical interest: toward an organized collection enabling visual network analysis. Bioinformatics 2019, 35, 4739–4747. [Google Scholar] [CrossRef] [PubMed]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Garcia-Jacas, C.R.; Chavez, E.; Beltran, J.A.; Guillen-Ramirez, H.A.; Brizuela, C.A. Automatic construction of molecular similarity networks for visual graph mining in chemical space of bioactive peptides: an unsupervised learning approach. Sci Rep 2020, 10, 18074. [Google Scholar] [CrossRef] [PubMed]
- Chavez, E.; Dobrev, S.; Kranakis, E.; Opatrny, J.; Stacho, L.; Tejeda, H.; Urrutia, J. Half-space proximal: A new local test for extracting a bounded dilation spanner of a unit disk graph. In Proceedings of the Principles of Distributed Systems: 9th International Conference, OPODIS 2005, Revised Selected Papers 9, 2006. Pisa, Italy, December 12-14, 2005; pp. 235–245. [Google Scholar]
- Cherven, K. Network Graph Analysis and Visualization with Gephi; Packt Publishing: 2013.
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J Mol Biol 1981, 147, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. Journal of statistical mechanics: theory and experiment 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Fruchterman, T.M.; Reingold, E.M. Graph drawing by force-directed placement. Software: Practice and experience 1991, 21, 1129–1164. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Bailey, T.L. STREME: Accurate and versatile sequence motif discovery. Bioinformatics 2021, 37, 2834–2840. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Grant, C.E. SEA: Simple enrichment analysis of motifs. BioRxiv, 2008. [Google Scholar]
- Newman, M. Networks; Oxford university press: 2018.
- Martinez, L.R.; Casadevall, A. Cryptococcus neoformans cells in biofilms are less susceptible than planktonic cells to antimicrobial molecules produced by the innate immune system. Infection and immunity 2006, 74, 6118–6123. [Google Scholar] [CrossRef] [PubMed]
- Moazzezy, N.; Asadi Karam, M.R.; Rafati, S.; Bouzari, S.; Oloomi, M. Inhibition and eradication activity of truncated α-defensin analogs against multidrug resistant uropathogenic Escherichia coli biofilm. PLoS One 2020, 15, e0235892. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.; He, J.; Yarbrough, D.K.; Qi, F.; Anderson, M.H.; Shi, W. Targeted killing of Streptococcus mutans by a pheromone-guided "smart" antimicrobial peptide. Antimicrob Agents Chemother 2006, 50, 3651–3657. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Nunez, C.; Reffuveille, F.; Mansour, S.C.; Reckseidler-Zenteno, S.L.; Hernandez, D.; Brackman, G.; Coenye, T.; Hancock, R.E. D-enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem Biol 2015, 22, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Ferrie, R.P.; Ghimire, J.; Ventura, C.R.; Wu, E.; Sun, L.; Kim, S.Y.; Wiedman, G.R.; Hristova, K.; Wimley, W.C. Applications and evolution of melittin, the quintessential membrane active peptide. Biochem Pharmacol 2021, 193, 114769. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hirt, H.; Li, Y.; Gorr, S.U.; Aparicio, C. Antimicrobial GL13K peptide coatings killed and ruptured the wall of Streptococcus gordonii and prevented formation and growth of biofilms. PLoS One 2014, 9, e111579. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, K.V.; Abdolhosseini, M.; Li, Y.; Chen, X.; Gorr, S.U.; Aparicio, C. Bio-inspired stable antimicrobial peptide coatings for dental applications. Acta Biomater 2013, 9, 8224–8231. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Chaudhary, K.; Dhanda, S.K.; Bhalla, S.; Usmani, S.S.; Gautam, A.; Tuknait, A.; Agrawal, P.; Mathur, D.; Raghava, G.P. SATPdb: a database of structurally annotated therapeutic peptides. Nucleic Acids Res 2016, 44, D1119–1126. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: an enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res 2016, 44, 6503. [Google Scholar] [CrossRef]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: a database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res 2014, 42, D1147–1153. [Google Scholar] [CrossRef]
- Mehta, D.; Anand, P.; Kumar, V.; Joshi, A.; Mathur, D.; Singh, S.; Tuknait, A.; Chaudhary, K.; Gautam, S.K.; Gautam, A.; et al. ParaPep: a web resource for experimentally validated antiparasitic peptide sequences and their structures. Database (Oxford) 2014, 2014. [Google Scholar] [CrossRef]
- Gautam, A.; Chaudhary, K.; Singh, S.; Joshi, A.; Anand, P.; Tuknait, A.; Mathur, D.; Varshney, G.C.; Raghava, G.P. Hemolytik: a database of experimentally determined hemolytic and non-hemolytic peptides. Nucleic Acids Res 2014, 42, D444–449. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P. CancerPPD: a database of anticancer peptides and proteins. Nucleic Acids Res 2015, 43, D837–843. [Google Scholar] [CrossRef]
- Seshadri Sundararajan, V.; Gabere, M.N.; Pretorius, A.; Adam, S.; Christoffels, A.; Lehvaslaiho, M.; Archer, J.A.; Bajic, V.B. DAMPD: a manually curated antimicrobial peptide database. Nucleic Acids Res 2012, 40, D1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res 2016, 44, D1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, H.; Lu, H.; Li, G.; Huang, Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS One 2013, 8, e66557. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: a comprehensive data repository of antimicrobial peptides. Sci Rep 2016, 6, 24482. [Google Scholar] [CrossRef] [PubMed]
- Reza, A.; Sutton, J.M.; Rahman, K.M. Effectiveness of Efflux Pump Inhibitors as Biofilm Disruptors and Resistance Breakers in Gram-Negative (ESKAPEE) Bacteria. Antibiotics (Basel) 2019, 8. [Google Scholar] [CrossRef]
- Li, J.; Chen, D.; Lin, H. Antibiofilm peptides as a promising strategy: comparative research. Appl Microbiol Biotechnol 2021, 105, 1647–1656. [Google Scholar] [CrossRef]
- Juretic, D. Designed Multifunctional Peptides for Intracellular Targets. Antibiotics (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Agüero-Chapin, G.; Galpert, D.; Molina-Ruiz, R.; Ancede-Gallardo, E.; Pérez-Machado, G.; De la Riva, G.A.; Antunes, A. Graph Theory-Based Sequence Descriptors as Remote Homology Predictors. Biomolecules 2020, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Vens, C.; Rosso, M.N.; Danchin, E.G. Identifying discriminative classification-based motifs in biological sequences. Bioinformatics 2011, 27, 1231–1238. [Google Scholar] [CrossRef]
- de Souza, C.M.; da Silva, Á.P.; Júnior, N.G.O.; Martínez, O.F.; Franco, O.L. Peptides as a therapeutic strategy against Klebsiella pneumoniae. Trends in Pharmacological Sciences 2022, 43, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Hee, C.S.; Habazettl, J.; Schmutz, C.; Schirmer, T.; Jenal, U.; Grzesiek, S. Intercepting second-messenger signaling by rationally designed peptides sequestering c-di-GMP. Proc Natl Acad Sci U S A 2020, 117, 17211–17220. [Google Scholar] [CrossRef] [PubMed]
- Edwards-Gayle, C.J.C.; Barrett, G.; Roy, S.; Castelletto, V.; Seitsonen, J.; Ruokolainen, J.; Hamley, I.W. Selective Antibacterial Activity and Lipid Membrane Interactions of Arginine-Rich Amphiphilic Peptides. ACS Appl Bio Mater 2020, 3, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Zarena, D.; Mishra, B.; Lushnikova, T.; Wang, F.; Wang, G. The pi Configuration of the WWW Motif of a Short Trp-Rich Peptide Is Critical for Targeting Bacterial Membranes, Disrupting Preformed Biofilms, and Killing Methicillin-Resistant Staphylococcus aureus. Biochemistry 2017, 56, 4039–4043. [Google Scholar] [CrossRef]
- Mishra, B.; Lushnikova, T.; Golla, R.M.; Wang, X.; Wang, G. Design and surface immobilization of short anti-biofilm peptides. Acta Biomater 2017, 49, 316–328. [Google Scholar] [CrossRef]
Figure 1.
Behaviour of HSPNs parameters representing the 174 ABFPs when the similarity cutoff varies from 0 to 0.95.
Figure 1.
Behaviour of HSPNs parameters representing the 174 ABFPs when the similarity cutoff varies from 0 to 0.95.
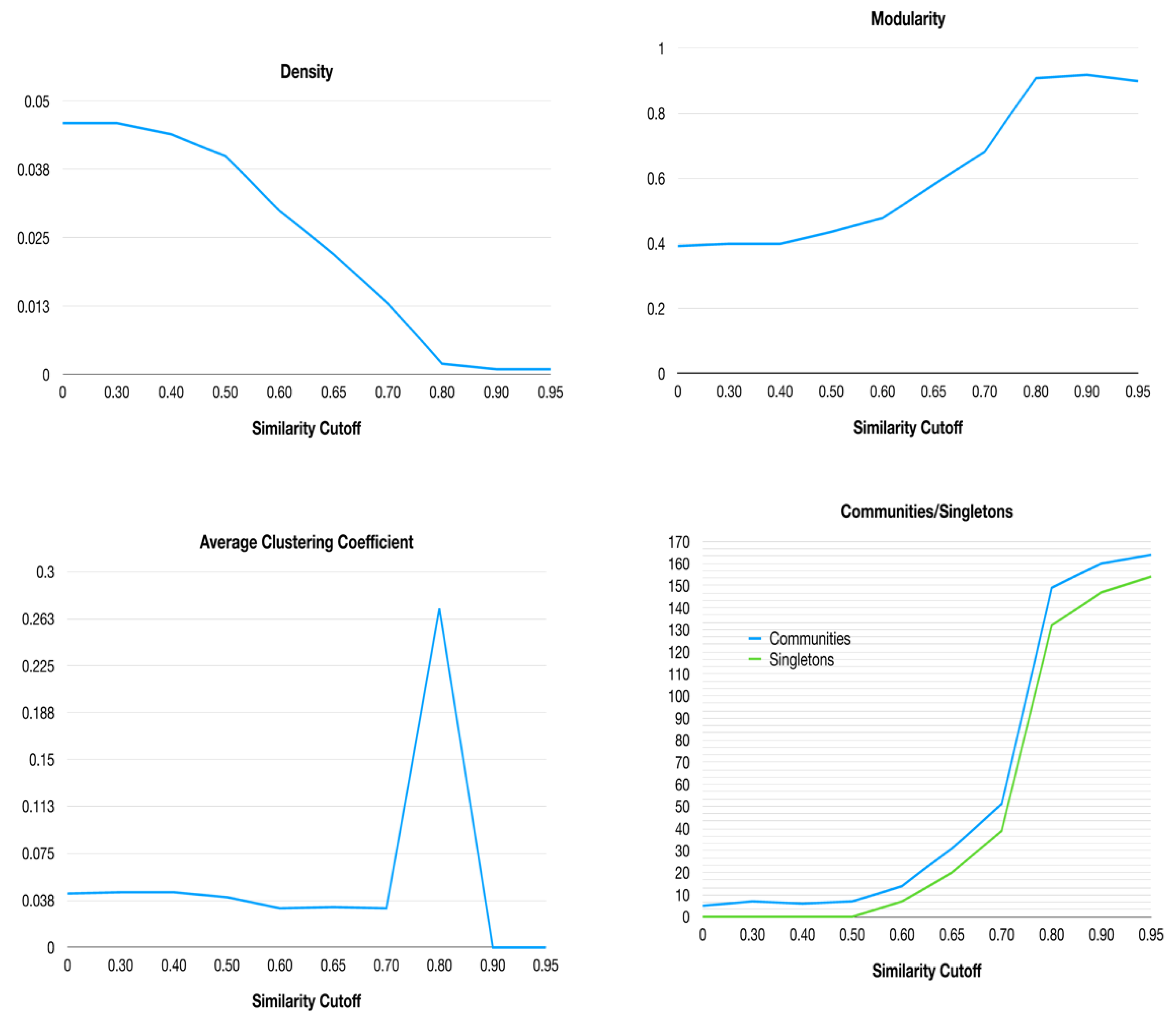
Figure 2.
Degree distribution of the HSPNs corresponding to the 174 ABFPs with no similarity cutoff and after applying a cutoff of 0.65.
Figure 2.
Degree distribution of the HSPNs corresponding to the 174 ABFPs with no similarity cutoff and after applying a cutoff of 0.65.
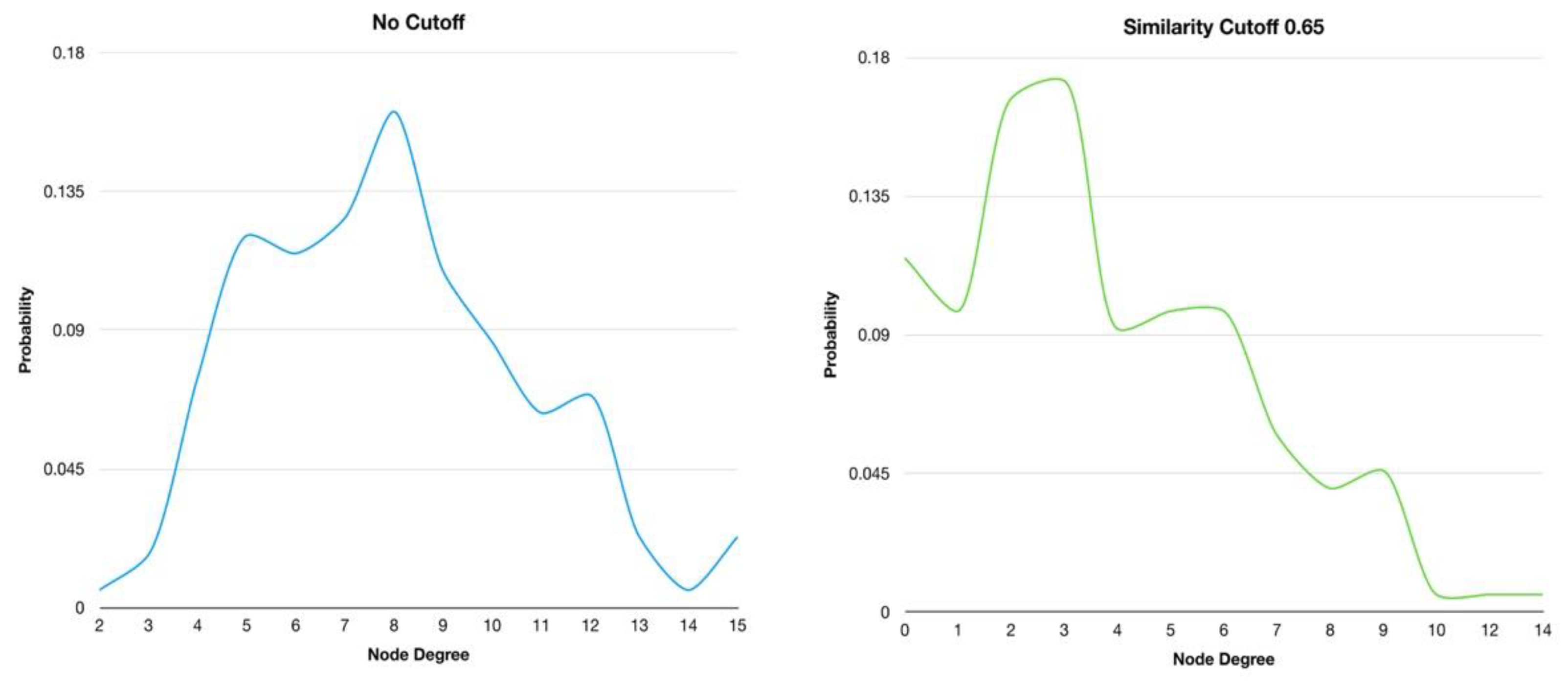
Figure 4.
METNs visualization by using the Fruchterman Reingold layout. Red nodes represent the metadata while the ABFPs are in grey colour. A – METN built with the source database information. B – METN built with the origin information.
Figure 4.
METNs visualization by using the Fruchterman Reingold layout. Red nodes represent the metadata while the ABFPs are in grey colour. A – METN built with the source database information. B – METN built with the origin information.
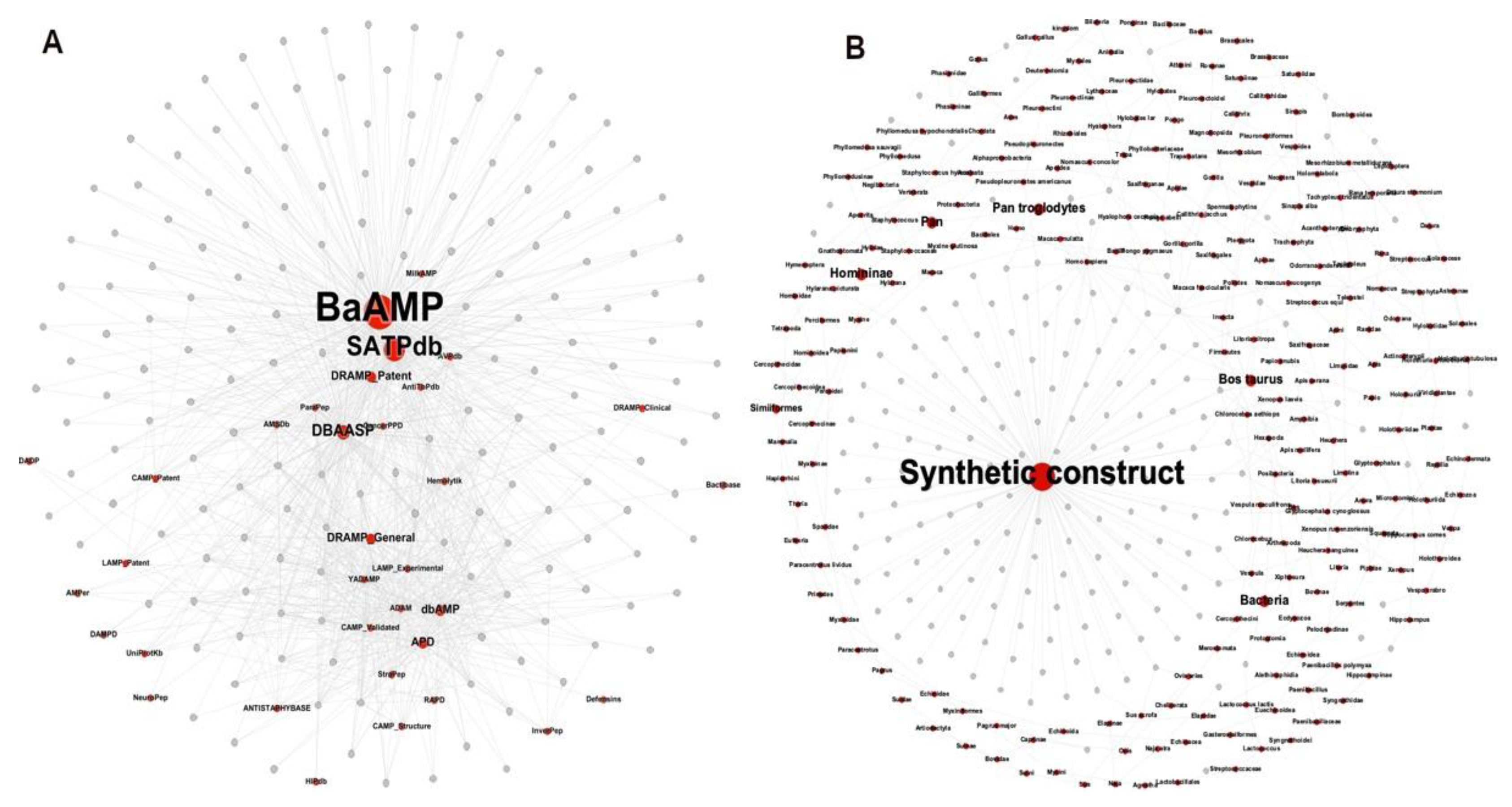
Figure 5.
Overlapping the subsets 6, 7 and 8 resulting from the union of the harmonic and hub-bridge centralities at 0.35, 0.40 and 0.45 over the HSPN model. A – 52 ABFPs from the HC ∪ HB of the subset 8 (cutoff 0.35) over the total 174 ABFPs. B – 66 ABFPs from the HC ∪ HB of the subset 7 (cutoff 0.40) over the total 174 ABFPs and C– 85 ABFPs from the HC ∪ HB of the subset 6 (cutoff 0.45) over the total 174 ABFPs.
Figure 5.
Overlapping the subsets 6, 7 and 8 resulting from the union of the harmonic and hub-bridge centralities at 0.35, 0.40 and 0.45 over the HSPN model. A – 52 ABFPs from the HC ∪ HB of the subset 8 (cutoff 0.35) over the total 174 ABFPs. B – 66 ABFPs from the HC ∪ HB of the subset 7 (cutoff 0.40) over the total 174 ABFPs and C– 85 ABFPs from the HC ∪ HB of the subset 6 (cutoff 0.45) over the total 174 ABFPs.
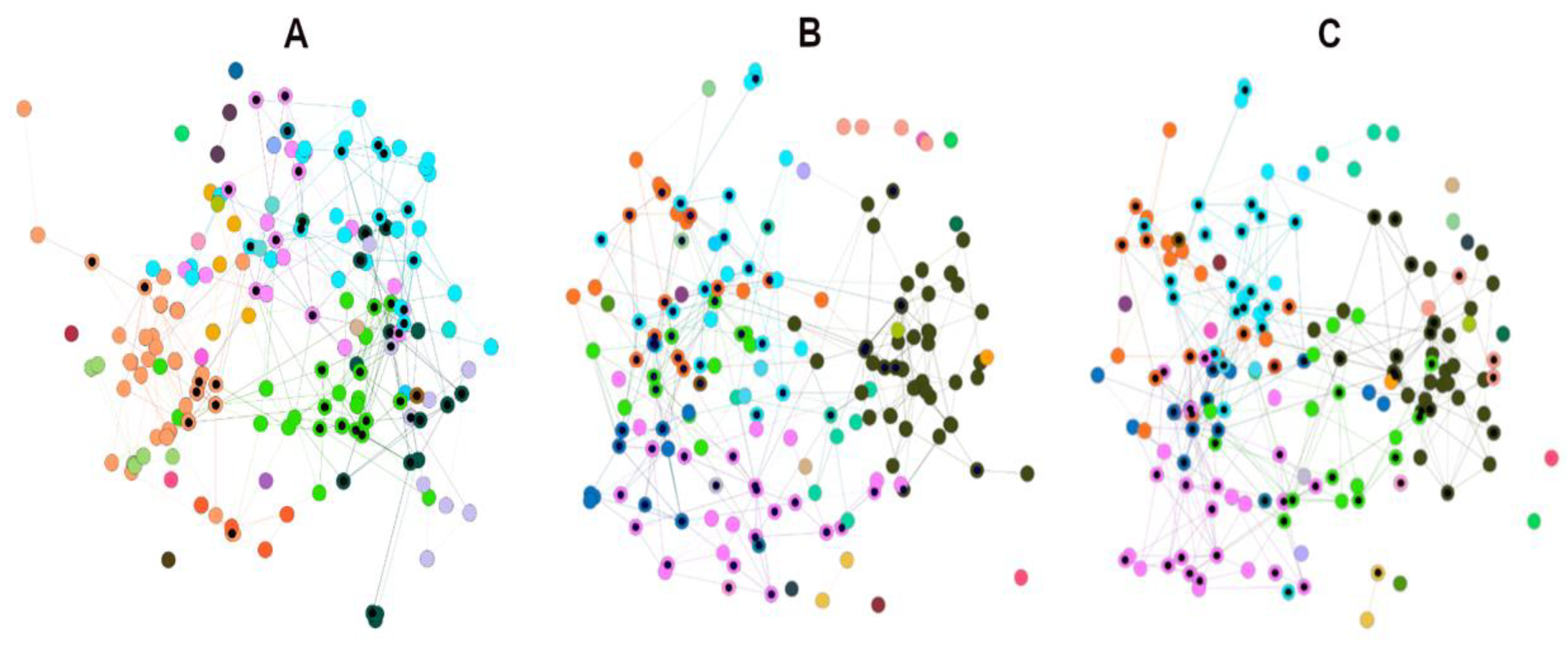
Figure 6.
HSPNs visualization of the representative subset integrated by 66 ABFPs from the HC ∪ HB of the subset 7 (cutoff 0.40) using the Fruchterman-Reingold layout. Peptides communities are represented by different colours while the nodes size was scaled according to harmonic (A) and hub-bridge centrality (B) measures.
Figure 6.
HSPNs visualization of the representative subset integrated by 66 ABFPs from the HC ∪ HB of the subset 7 (cutoff 0.40) using the Fruchterman-Reingold layout. Peptides communities are represented by different colours while the nodes size was scaled according to harmonic (A) and hub-bridge centrality (B) measures.

Figure 7.
METNs visualization of the representative subset integrated by 66 ABFPs from the HC ∪ HB of the subset 7 (cutoff 0.40) using the Fruchterman-Reingold layout. A – Red nodes represent the bioactivities while the ABFPs under study starPep_00000, starPep_00193, starPep_00004, starPep_00025, starPep_00514, starPep_00522 are in blue, yellow, green, pink black and cyan colours, respectively. B – Red nodes represent the targets on what the ABFPs have been evaluated on. ABFPs under study are highlighted with the colour scheme used in A.
Figure 7.
METNs visualization of the representative subset integrated by 66 ABFPs from the HC ∪ HB of the subset 7 (cutoff 0.40) using the Fruchterman-Reingold layout. A – Red nodes represent the bioactivities while the ABFPs under study starPep_00000, starPep_00193, starPep_00004, starPep_00025, starPep_00514, starPep_00522 are in blue, yellow, green, pink black and cyan colours, respectively. B – Red nodes represent the targets on what the ABFPs have been evaluated on. ABFPs under study are highlighted with the colour scheme used in A.
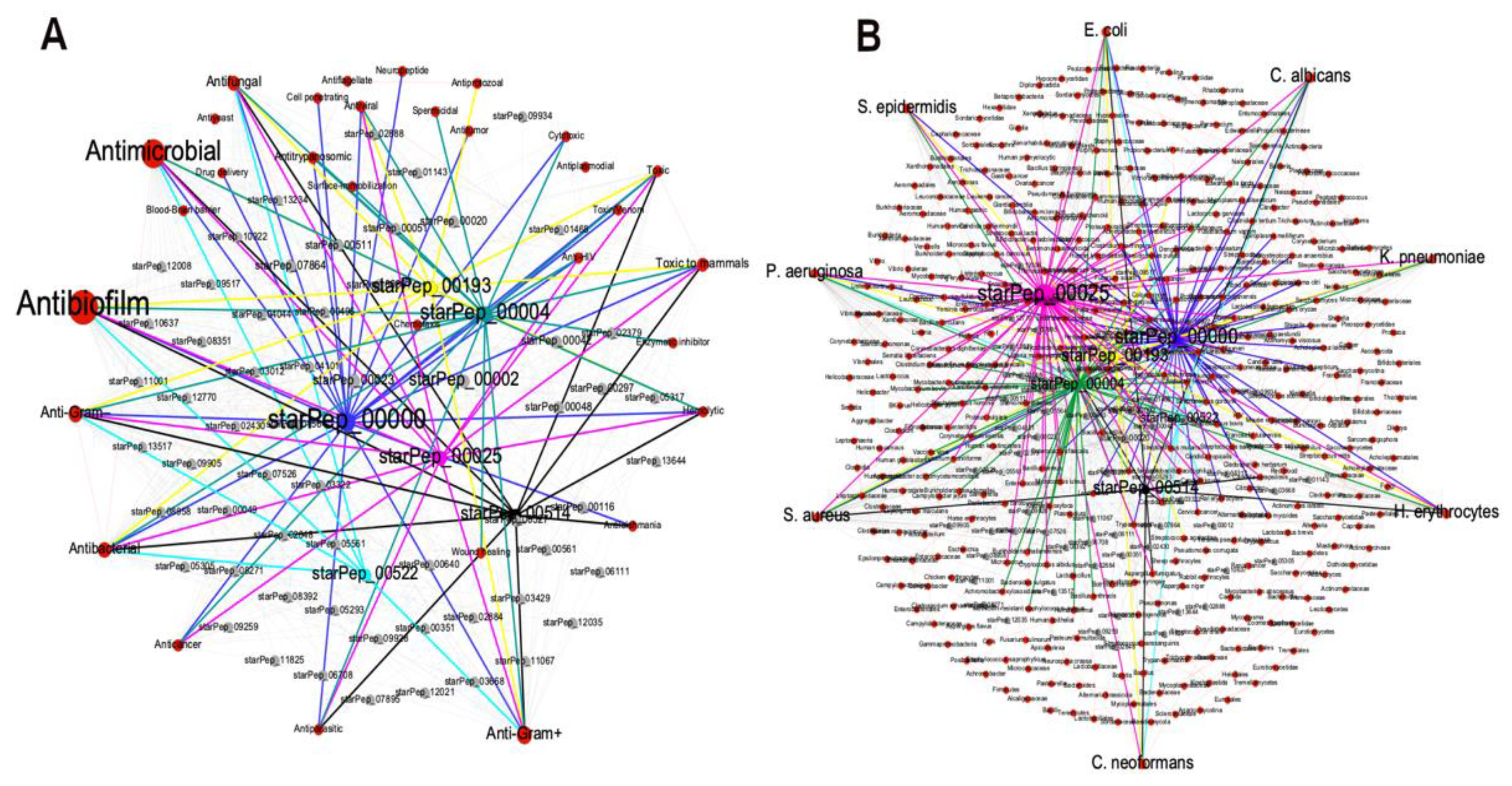
Figure 8.
Visual mining of the 14 ABFPs corresponding to 14 classes of mode of actions on the chemical space of ABFPs. A – Overlapping the 14 representative ABFPs on the set integrated by 66 ABFPs using HSPN. Peptides communities are represented by different colours while the 14 ABFPs nodes/labels were highlighted in black colour. B – Mapping of the representative ABFPs with similarities higher than 0.60 within the antibiofilm HSPN using the Fruchterman-Reingold layout. .
Figure 8.
Visual mining of the 14 ABFPs corresponding to 14 classes of mode of actions on the chemical space of ABFPs. A – Overlapping the 14 representative ABFPs on the set integrated by 66 ABFPs using HSPN. Peptides communities are represented by different colours while the 14 ABFPs nodes/labels were highlighted in black colour. B – Mapping of the representative ABFPs with similarities higher than 0.60 within the antibiofilm HSPN using the Fruchterman-Reingold layout. .
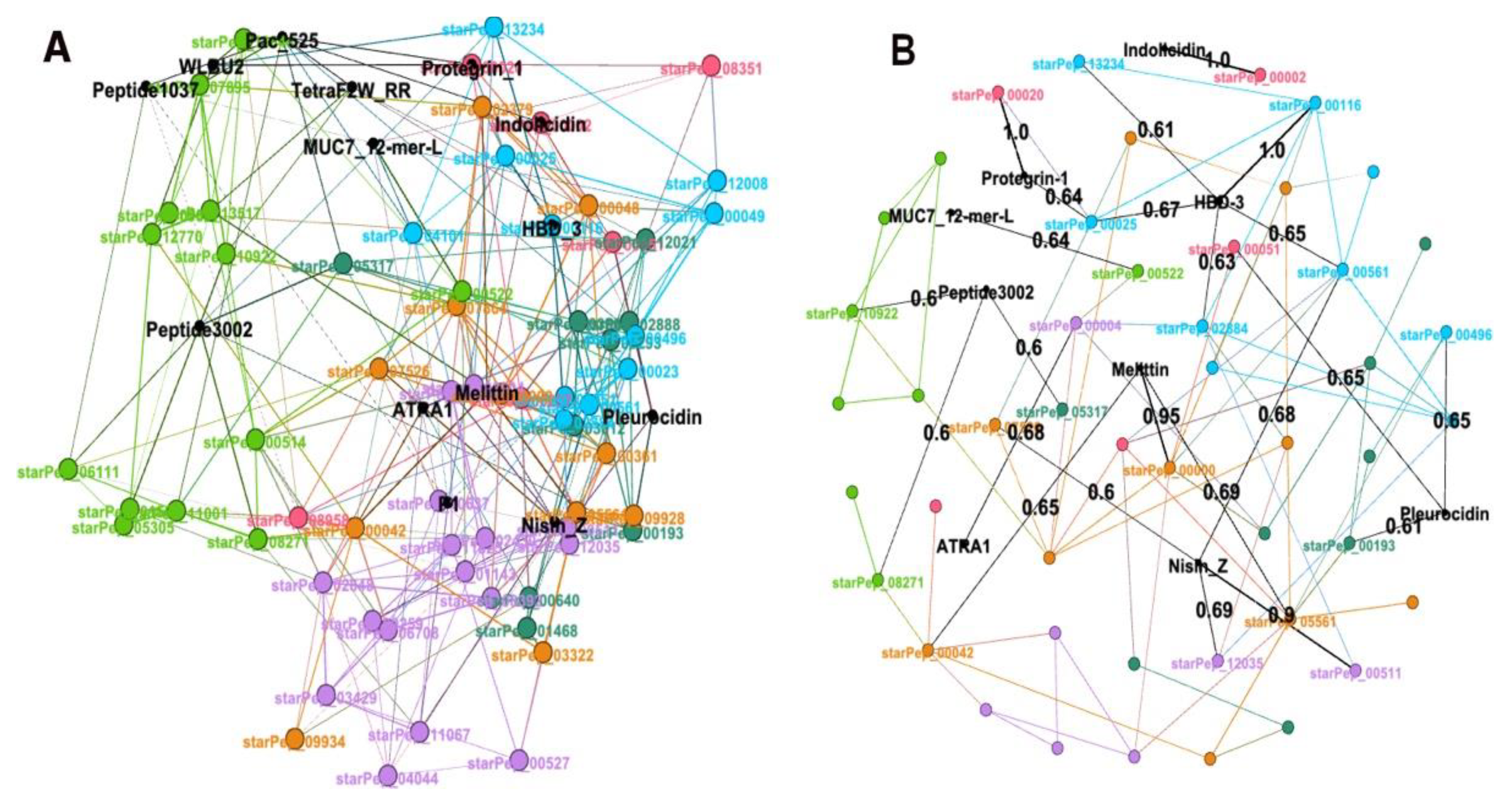
Figure 9.
Heat maps corresponding to the pairwise identities from the comparison of the 14 ABFPs representing the mechanisms of action classes against the 66 representatives of the antibiofilm chemical space. A– the 9 ABFPs that could be mapped on the representative HSPN at AF similarity > 0.60 occupy the from 1 to 9 position in the heatmap. B– the 5 that did not map at AF > 0.60 were placed from 1 to 5 position. The target zone of the heat maps is framed by a white line. All-vs-all global alignments and heat maps visualization were conducted using the SeqDivA software reported in [66].
Figure 9.
Heat maps corresponding to the pairwise identities from the comparison of the 14 ABFPs representing the mechanisms of action classes against the 66 representatives of the antibiofilm chemical space. A– the 9 ABFPs that could be mapped on the representative HSPN at AF similarity > 0.60 occupy the from 1 to 9 position in the heatmap. B– the 5 that did not map at AF > 0.60 were placed from 1 to 5 position. The target zone of the heat maps is framed by a white line. All-vs-all global alignments and heat maps visualization were conducted using the SeqDivA software reported in [66].
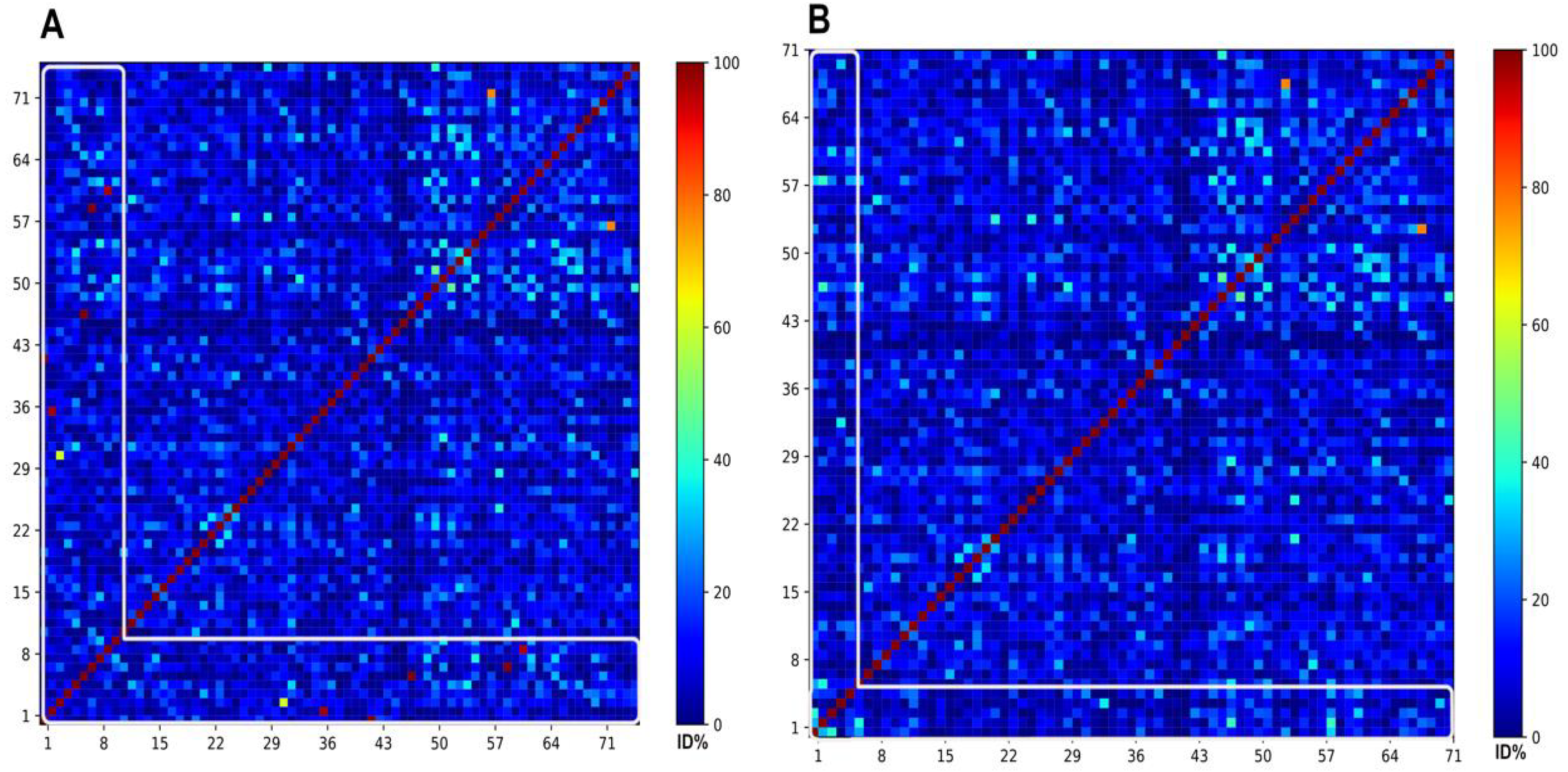

Table 1.
Common and singular peptides from the 10-top ranked ABFPs by four, three, two and one centrality measure (node degree, harmonic, betweenness and hub-bridge) centralities. The cluster number for each peptide at each HSPN is displayed.
Table 1.
Common and singular peptides from the 10-top ranked ABFPs by four, three, two and one centrality measure (node degree, harmonic, betweenness and hub-bridge) centralities. The cluster number for each peptide at each HSPN is displayed.
| HSPN – No Cutoff | |||
| Centrality Measure | Total | Peptide Name | Cluster |
| Node Degree Harmonic Betweenness Hub-Bridge | 1 | starPep_03668 | (1) |
| Node Degree Betweenness Hub-Bridge | 1 | starPep_00048 | (1) |
| Harmonic Hub-Bridge | 2 | starPep_00000 starPep_10922 | (1) (3) |
| Node Degree Betweenness | 7 | starPep_12469 starPep_07526 starPep_00145 starPep_06130 starPep_00042 starPep_08958 starPep_02281 | (3) (2)(2) (3) (0) (2) (2) |
| Node Degree | 1 | starPep_00517 | (0) |
| Harmonic | 7 | starPep_07864 starPep_07895 starPep_02907 starPep_12770 starPep_12531 starPep_13517 starPep_07893 | (0) (3)(3) (3)(3) (3) (2) |
| Betweenness | 1 | starPep_08001 | (3) |
| Hub-Bridge | 6 | starPep_00496 starPep_00561 starPep_00361 starPep_13515 starPep_00193 starPep_05561 | (1) (1) (1) (2) (1) (1) |
| HSPN – Cutoff 0.65 | |||
| Node Degree Harmonic Betweenness Hub-Bridge | 1 | starPep_00048 | (9) |
| Node Degree Betweenness Hub-Bridge | 1 | starPep_00042 | (11) |
| Harmonic Betweenness Hub-Bridge | 1 | starPep_10922 | (15) |
| Node Degree Harmonic Betweenness | 2 | starPep_00000 starPep_03668 | (15) (4) |
| Node Degree Harmonic | 1 | starPep_00361 | (9) |
| Node Degree Betweenness | 1 | starPep_07526 | (17) |
| Node Degree Hub-Bridge | 2 | starPep_00004 starPep_00193 | (11) (9) |
| Harmonic Betweenness | 1 | starPep_02379 | (17) |
| Harmonic Hub-Bridge | 3 | starPep_07895 starPep_02907 starPep_12531 | (15) (15) (15) |
| Node Degree | 2 | starPep_00561 starPep_05561 | (9) (4) |
| Harmonic | 1 | starPep_07893 | (14) |
| Betweenness | 3 | starPep_12530 starPep_04734 starPep_08958 | (15) (17) (15) |
| Hub-Bridge | 2 | starPep_12770 starPep_02908 | (15) (15) |
Table 2.
Atypical peptides identified by the HSPN at similarity cutoff of 0.65. Particularly, the singletons and isolated communities made up of 2 peptides are detailed.
Table 2.
Atypical peptides identified by the HSPN at similarity cutoff of 0.65. Particularly, the singletons and isolated communities made up of 2 peptides are detailed.
| HSPN – Cutoff 0.65 | |||
| Atypical peptides | Total | Peptide Name | Cluster |
| Singletons | 20 | starPep_00002 starPep_00739 starPep_02281 starPep_02383 starPep_02400 starPep_02730 starPep_03693 starPep_04044* starPep_05305* starPep_05447* starPep_05964 starPep_06255 starPep_06358* starPep_08001 starPep_09934* starPep_09989* starPep_10637* starPep_14812* starPep_16445* starPep_18706* | (10) (18) (20) (25) (0) (5) (19) (13) (8) (27) (26) (21) (23) (24) (16) (29) (1) (12) (2) (3) |
| Isolated Community | 2 | starPep_04274 – starPep_04424 ϦstarPep_13860* – starPep_13861* | (6)Ϧ(28) |
*Atypical ABFPs with no toxicity reported. Those highlighted in bold show a variety of activities than the antibiofilm.
Table 3.
ABFP subsets extracted from the HSPN model made up of 174 nodes and 325 edges by using the scaffold extraction algorithm from the StarPep toolbox. The harmonic and hub-bridge centralities were applied for the reduction step at different similarity cutoff values.
Table 3.
ABFP subsets extracted from the HSPN model made up of 174 nodes and 325 edges by using the scaffold extraction algorithm from the StarPep toolbox. The harmonic and hub-bridge centralities were applied for the reduction step at different similarity cutoff values.
| Harmonic (HC) | Hub-Bridge (HB) | ||||||
| Subsets | Cutoff | Edges | Nodes | Coverage1% | Edges | Nodes | Coverage1% |
| 1 | 0.90 | 227 | 154 | 89 | 276 | 154 | 89 |
| 2 | 0.80 | 230 | 138 | 79 | 235 | 137 | 79 |
| 3 | 0.70 | 199 | 122 | 70 | 201 | 125 | 72 |
| 4 | 0.60 | 167 | 103 | 59 | 162 | 104 | 60 |
| 5 | 0.50 | 128 | 80 | 46 | 112 | 80 | 46 |
| 6 | 0.45 | 115 | 74 | 43 | 88 | 68 | 39 |
| 7 | 0.40 | 74 | 54 | 31 | 63 | 51 | 29 |
| 8 | 0.35 | 62 | 45 | 26 | 44 | 40 | 23 |
| 9 | 0.30 | 40 | 32 | 22 | 35 | 34 | 20 |
1 Coverage is the percentage representing each subset of the 174 ABFPs.
Table 4.
Representative ABFPs categorized in 14 classes according to the mechanisms of action elucidated for the antibiofilm activity. The information displayed in this table was gathered from Table S2 published in ref. [64].
| Class-Peptides | Action Mode | Sequence |
| Class1- HBD-3 | Influence on icaAD and icaR genes transcription levels | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK |
| Class2- Nisin Z | Decrease adhesion, kill bacteria, reduce biofilm formation | ITSISLCTPGCKTGALM GCNMKTATCNCSIHVSK |
| Class3- MUC7 12-mer-L | Attracted to bacterial surfaces by the electrostatic bonding | RKSYKCLHKRCR |
| Class4- ATRA1 | Promote biofilm dispersal | KRFKKFFKKLKNSVK KRFKKFFKKLKVIGVT FPF |
| Class5- Pleurocidin | Induces disturbance/permeabilization of the membranes and bind to bacterial DNA, causing interference with cellular functions | GWGSFFKKAAHVGK HVGKAALTHYL |
| Class6- Pac-525 | Ability to enter membranes and to affect the lipopolysaccharides of Gram-negative bacteria | KWRRWVRWI |
| Class7- peptide 1037 | Decreases the attachment of bacterial cells, stimulates twitching motility, and influenced two major quorum sensing systems | KRFRIRVRV |
| Class8- Indolicidin | Induces lipid removal and that mixed indolicidin-lipid patches; membrane permeabilization | ILPWKWPWWPWRR |
| Class9- Protegrin-1* | Forms amyloid fibers to associate with the bacterial membrane and product transmembrane pores | RGGRLCYCRRRFCVCVGR |
| Class10- Peptide 3002 | Blocking (p)ppGpp | ILVRWIRWRIQW |
| Class11-TetraF2W-RR* | Disrupts membranes to kill bacteria rapidly | WWWLRRIW |
| Class12-P1 | Interferes with the proper secretion and/or intermolecular interaction of key extracellular polymers in the biofilm matrix | PARKARAATAATAATAATAATAAT |
| Class13-WLBU2* | LPS-binding property, interfers with bacterial attachment; destroying the bacterial membrane | RRWVRRVRRVWRRVVRVVRRWVRR |
| Class14-Melittin* | Inhibits the expression of biofilm-associated bap genes | GIGAVLKVLTTGLPALISWIKRKRQQ |
* Promising antibiofilm candidates with lower MBICs than MICs.
Table 5.
Discovered motifs by Multiple Sequence Alignment (MSA) at each network cluster/community.
| No | Motif | EMBOSS Cons. | Cluster | Cluster size | MSA Method | Enrichment ratio* |
| 1 | RLFNR | xxxNR | 4 | 15 | MAFFT/MUSCLE | – |
| 2 | GGG | GxG | MAFFT | – | ||
| 3 | GGGWK | xxGWK | MUSCLE | ( –)/(2.25) | ||
| 4 | FKKA | xKKx | MAFFT | (–)/(4.0) | ||
| 5 | FWKWA | FWK | MAFFT | (3.0)/(2.83) | ||
| 6 | WGK | WxK | MAFFT | (–)/(1.41) | ||
| 7 | LLLLLKKK | LLLLLKKK | 6 | 2 | Pair-Aligned | – |
| 8 | LISWIK | lisxik | 7 | 8 | MAFFT | (–)/(2.71) |
| 9 | KNKRK | knkxk | MUSCLE | (3.0)/(2.22) | ||
| 10 | KRKQ | kxkQ | MAFFT | (3.0)/(–) | ||
| 11 | R[GP]RVS | rxRVS | MAFFT | (–)/(3.0) | ||
| 12 | RRPR | RRxR | MUSCLE | – | ||
| 13 | [GR]GG | xGG | MAFFT | (3.0)/(1.90) | ||
| 14 | GGRRRR | GGrrRR | MUSCLE | (–)/(2.0) | ||
| 15 | RRRRR | RRRRR | MAFFT/MUSCLE | – | ||
| 16 | ISGI | Ixxx | 9 | 23 | MAFFT | – |
| 17 | FKKLL | xKKLL | 11 | 27 | MAFFT/MUSCLE | (–)/(2.25) |
| 18 | KKLK | MAFFT | – | |||
| 19 | KKL | MUSCLE | – | |||
| 20 | LKK | LKK | MUSCLE | – | ||
| 21 | RIRVR | RIRVR | 14 | 23 | MAFFT | (–)/(1.58) |
| 22 | RVIR | xRVIR | MAFFT | (–)/(1.32) | ||
| 23 | VRVIR | MUSCLE | (–)/(2.83) | |||
| 24 | R[WL]R | RxR | MUSCLE | (1.57)/(–) | ||
| 25 | RIRRW | RIxRW | 15 | 26 | MAFFT/MUSCLE | (–)/(4.0) |
| 26 | RI[VR]W | (–)/(1.67) | ||||
| 27 | WVV | WVV | MAFFT | (–)/(1.44) | ||
| 28 | I[IR]R | IIxR | MUSCLE | – | ||
| 29 | WLRK | Wxxx | 17 | 23 | MAFFT | (–)/(2.50) |
| 30 | RWK | Rxx | MUSCLE | – | ||
| 31 | KKL | Kxx | MAFFT | – | ||
| 32 | KR[AKL]RK | KRxRK | MUSCLE | (6.0)/(3.0) | ||
| 33 | WR[IV]R | xRWR[IV]R | 22 | 5 | MAFFT/MUSCLE | – |
| 32 | FRWRI | MAFFT | (3.0)/ (–) | |||
| 33 | RWRVR | MUSCLE | (–)/(1.63) | |||
| 34 | YAPWYN | YAPWYN | 28 | 2 | Pair-Aligned | – |
| 35 | [FI][KW]RK | iKrK | Singletons | 20 | MAFFT/MUSCLE | (–)/(1.46) |
*Enrichment ratio was evaluated on the 14 ABFPs categorized by action mode in ref.[64] (first value) and 192 non-redundant ABFPs extracted from 214 reported in BaAMP (second value).
Table 6.
Motifs Identified by STREME at each network cluster.
| No | Motif | Cluster | Cluster size | Matches in ABFPs | Matches in control | Sites (%) | Score | Enrichment ratio* |
| 1 | FKKA | 4 | 15 | 7 | 0 | 46.7 | 3.3e-003 | (–)/(3.33) |
| 2 | GGGR | 7 | 0 | 46.7 | 3.3e-003 | (–)/(2.11) | ||
| 3 | W[KR]WF | 7 | 0 | 46.7 | 3.3e-003 | (–)/(1.38) | ||
| 4 | FIH | 6 | 0 | 40.0 | 8.4e-002 | – | ||
| 5 | RLFNR | 5 | 0 | 33.3 | 2.1e-003 | – | ||
| 6 | KKK | 6 | 2 | 2 | 0 | 100 | 1.7e-001 | – |
| 7 | LLLLL | 2 | 0 | 100 | 1.7e-001 | – | ||
| 8 | RGG | 7 | 8 | 8 | 0 | 100 | 7.8e-005 | (3.0)/(1.56) |
| 9 | ISWIK | 4 | 0 | 50 | 3.8e-002 | (–)/(2.83) | ||
| 10 | NKRKQ | 4 | 0 | 50 | 3.8e-002 | – | ||
| 11 | RPRVS | 3 | 0 | 37.5 | 1.0e-001 | (–)/(3.71) | ||
| 12 | RRRRR | 3 | 0 | 37.5 | 1.0e-001 | – | ||
| 13 | SAC | 9 | 23 | 16 | 1 | 69.6 | 3.3e-006 | – |
| 14 | AKA | 5 | 0 | 21.7 | 2.85e-002 | – | ||
| 15 | CD[VI] | 5 | 0 | 21.7 | 2.85e-002 | – | ||
| 16 | IA[GVK] | 5 | 0 | 21.7 | 2.85e-002 | – | ||
| 17 | LFKKL | 11 | 27 | 9 | 0 | 33.3 | 8.8e-004 | (–)/(2.40) |
| 18 | KVLK | 8 | 0 | 29.6 | 2.1e-003 | (3.0)/(4.0) | ||
| 19 | KRFL | 6 | 0 | 22.2 | 1.1e-002 | (3.0)/(1.8) | ||
| 20 | VRLRI | 14 | 23 | 12 | 0 | 52.2 | 3.5e-005 | – |
| 21 | RVIR | 10 | 0 | 43.5 | 2.8e-004 | (–)/(1.32) | ||
| 22 | VWVI | 15 | 26 | 14 | 3 | 53.8 | 1.3e-003 | (3.0)/(3.0) |
| 23 | VIWRR | 8 | 0 | 30.8 | 2.1e-003 | (–)/(2.50) | ||
| 24 | LRK | 17 | 23 | 9 | 0 | 39.1 | 7.4e-004 | (3.0)/(1.27) |
| 25 | WRRK | 6 | 0 | 26.1 | 1.1e-002 | (–)/(1.67) | ||
| 26 | WRIR | 22 | 5 | 5 | 1 | 100 | 2.4e-002 | (–)/(3.25) |
| 27 | IRR | 2 | 3 | 40.0 | 9.0e-001 | (1.67)/(–) | ||
| 28 | APWTN | 28 | 2 | 2 | 0 | 100 | 1.7e-001 | (–)/(3.0) |
| 29 | KKRK | Singletons | 20 | 2 | 0 | 10.0 | 2.3e-001 | – |
| 30 | KKVVF | 2 | 0 | 10.0 | 2.4e-001 | – | ||
| 31 | LLKLL | 2 | 0 | 10.0 | 2.4e-001 | – | ||
| 32 | VKFK | 2 | 0 | 10.0 | 2.4e-001 | – | ||
| 33 | WRWR | 2 | 0 | 10.0 | 2.4e-001 | (–)/(1.64) |
*Enrichment ratio was evaluated on the 14 ABFPs categorized by action mode in ref.[64]) (first value) and 192 non-redundant ABFPs extracted from 214 reported in BaAMP (second value).
Table 7.
Summary of the discovered ABFP motifs per network community (cluster), enriched in two datasets.
Table 7.
Summary of the discovered ABFP motifs per network community (cluster), enriched in two datasets.
| No | Motif | Cluster | Method | Enrichment ratio* |
| 1 | FWKWA | 4 | MAFFT | (3.0)/(2.83) |
| 2 | KNKRK | 7 | MUSCLE | (3.0)/(2.22) |
| 3 | [GR]GG | 7 | MAFFT/STREME | (3.0)/(1.90) |
| 4 | KVLK | 11 | STREME | (3.0)/(4.0) |
| 5 | KRFL | 11 | STREME | (3.0)/(1.8) |
| 6 | VWVI | 15 | STREME | (3.0)/(3.0) |
| 7 | KR[AKL]RK | 17 | MUSCLE | (6.0)/(3.0) |
| 8 | LRK | 17 | STREME | (3.0)/(1.27) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



