
Submitted:
12 March 2023
Posted:
13 March 2023
You are already at the latest version
Abstract
The metabolic syndrome, first introduced by Hermann Haller in 1975, was sometimes also known as insulin resistance syndrome, syndrome X, and plurimetabolic syndrome. In 1989 it was rechristened by Kaplan as "Deadly Quartet" based on a consolidation of central obesity, impaired glucose tolerance, dyslipidemia, and systemic hypertension. Metabolic syndrome is positively associated with a pro-inflammatory and pro-thrombotic state, attributed to increased pro-thrombotic and inflammatory markers activity. Moreover, Metabolic syndrome is frequently associated with increased atherosclerotic cardiovascular disease, impaired glucose tolerance, hyperuricemia, obstructive sleep apnea, and chronic kidney disease. Despite concerted endeavors worldwide, the complexity of the pathophysiology of metabolic syndrome is still not clearly understood. Currently, therapeutic possibilities are confined to individual therapy of hyperglycemia, hypertension, hypertriglyceridemia, hyperuricemia, regular physical exercise, and a restricted diet.
Keywords:
metabolic syndrome
; glucose tolerance
; pathophysiology
; hyperglycemia
; diet
1. Introduction
The rapid increase prevalence of Metabolic Syndrome (MS) is emerging as a significant public health concern worldwide. The upward trend of urbanization, high caloric diet uptake, decreased physical activities, and central obesity compounded with a sedentary lifestyle are assigned as the influential underlying factors contributing to the epidemic upsurge of MS. The significant increase in the prevalence of MS and its future challenge to the national and world health scenario is of great importance. Various epidemiological studies have foisted that MS confers a five-fold increased risk of developing Type 2 Diabetes Mellitus (type 2 DM) and a two-fold increase in the risk of developing a cardiovascular disorder (CVD) over the next five to ten years [1]. Moreover, an individual with metabolic syndrome is two to four times more susceptible to developing stroke and at a three to fourfold risk of progressing myocardial infarction (MI) [2]. These events increase two-fold risk of dying compared with those without MS [3]. Earlier, in 2001, the term Metabolic Syndrome became institutionalized with ICD-9 (International Code for Diseases-9) code 277.7 and thought of as a first-order threat for the progression of athero-thrombotic complications.
Visceral obesity and insulin resistance (IR) are recognized as the major intrinsic risk factors for MS. Additionally, decreased physical activities, atherogenic dyslipidemia, calorie-rich dietary intake, as well as hormonal imbalance is the added risk factors for developing MS [4]. The complete pathophysiology of MS is still unclear, however redundant adipose tissues induced persistent low-grade inflammation is considered the crucial underlying cause of developing central obesity-related disorders, like Type 2 Diabetes Mellitus (type 2 DM), cardiovascular diseases (CVD) and Insulin resistance (IR) [5]. The redundant adipose tissue induced low-grade persistent inflammatory condition found to be involved in the progression of diseases related to MS, like atherosclerosis, atherogenic dyslipidemia, hypertension, pro-thrombotic status as well as impaired glucose tolerance [6]
2. Historical background
Though Hermann Haller first introduced the term metabolic syndrome in his scientific literature in 1975 [7], which was sometimes also known as insulin resistance syndrome, and “Syndrome X” by G. M. Reaven in 1980 [8], the documented historical evidence of MS begins with the Italian physician, Morgagni, around four decades earlier [9]. Morgagni noticed a significant association between central obesity, increased arterial blood pressure, atherosclerosis, elevated plasma uric acid, and obstructive sleep apnea. Nicolae Paulescu observed an interconnection between obesity and diabetes and forwarded the statement "most frequently, obese people become glycosuric" in 1920 [10]. In 1927, the Spanish endocrinologist described hypertension and obesity as pre-diabetic conditions [11]. Since its origin, it became able to draw the attention of many researchers worldwide. Later in 1947, J Vague came up with the new concept that central obesity was commonly associated with the metabolic alteration observed in cardiovascular diseases and Type 2 DM [10].
Plurimetabolic syndrome was introduced in the 1960s to the clinical condition, consisting of the frequent and concurrent presence of central obesity, dyslipidemia, Type 2 DM, and systemic increased arterial blood pressure [12]. In 1965 an abstract was presented at the European Association referencing the study in the diabetes annual meeting, which redefined the syndrome characterized by increased arterial blood pressure, hyperglycemia, and central obesity [13]. It is believed that the MS field accelerated forward significantly after the Banting lecture delivered by Reaven, who came up with a cluster of risk factors for diabetes mellitus and cardiovascular diseases. Reaven was the first who introduce the concept of insulin resistance associated with MS [14]. However, he astonishingly passed over the crucial component of visceral obesity, which was later considered a pivotal abnormality. Furthermore, regarding the history of MS, in 1989, it was renamed as "Deadly Quartet" based on the combination of central obesity, increased blood glucose, hypertriglyceridemia and hypertension by Kaplan [14], and further, in 1992, it was retitled as The Insulin Resistance Syndrome [15].
3. Definition and diagnostic criteria
The term syndrome derives from the Greek word “Sundromos” (Sun-syn+dromos= to run), meaning to run together. Metabolic syndrome is characterized by a complicated modification of the metabolism, which involves modification of lipid metabolism (dyslipidemia, obesity), carbohydrate metabolism (glucose intolerance), along with increased arterial blood pressure (hemodynamic disturbance having a metabolic starting point) [16]. Recently, pro-inflammatory, pro-thrombotic state and hormonal factors have also been reported to be involved in MS [17]. These modifications of metabolism are interconnected to each other and are found to be involved in increasing the risk of coronary heart diseases (CHD), cardiovascular atherosclerotic diseases (CVD) and type 2diabetes Mellitus (type 2 DM) and cause mortality [18].
Several documented evidence reveals that many international organizations as well as expert groups have attempted to coin the criteria to diagnose MS. The first attempt was made by World Health Organization (WHO) in 1998 to define the diagnostic criteria for MS, followed by the Europe Group for the Study of Insulin Resistance (EGIR), The National Cholesterol Education Program Adult Treatment Panel (NCEP: ATP III), American Association of Clinical Endocrinologists (AACE) and International Diabetes Federation (IDF). The first criteria developed by the World Health Organization (WHO), in 1998, with the inclusion of the presence of insulin resistance (IR), impaired glucose tolerance (IGT) or type 2 DM as absolutely required factors of MS with at least two of the following factors: waist/hip ratio; male:>0.9 cm; female:>0.85 cm or BMI:>30kg/m2; FBS: ≥110mg/dl or IR or type 2 DM or Rx; TG: ≥150mg/dl; HDL-C: male:<40mg/dl; female:<50mg/dl; BP: Diastolic≥140 and systolic≥90 mmHg; Microalbuminuria [19]. Within a year, the European Group for the study of Insulin Resistance (EGIR) challenged the diagnostic criteria. It modified the WHO definition by excluding microalbuminuria as an essential component of MS, including hyperinsulinemia [20]. EGIR considered insulin resistance as the substantial cause of MS and gives much more importance to obesity than WHO while excluding persons with T2DM. EGIR defines MS as hyperinsulinemia or insulin resistance along with two extra parameters as: FBS≥108.11mg/dl; BP: Diastolic≥140 and systolic≥90 mmHg; TG: ≥150mg/dl; HDL-C:<39mg/dl. Shortly after that, in 2001, the National Cholesterol Education Program Adult Treatment Panel (NCEP: ATP III) released its new criteria for MS, which included waist circumference, blood lipid level, blood pressure and fasting blood glucose level", which differed from both the WHO and EGIR definition. NCEP did not consider IR as a mandatory component of diagnostic criteria and stated that any three of the factors (waist circumference ≥102 cm in males and ≥88 cm in females; TG: ≥150mg/dl and/or on drug treatment; HDL-C: <40 mg/dl in males and <50mg/dl in females; BP: Diastolic≥130 and Systolic≥85mmHg and/or on drug treatment; FBS: ≥100mg/dl and/or on drug treatment) would suffice for a diagnosis of MS [21].

Table 1.
Diagnostic criteria of Metabolic Syndrome, elucidated over the years by different organizations.
Table 1.
Diagnostic criteria of Metabolic Syndrome, elucidated over the years by different organizations.
| Clinical parameters | Criteria | ||||||
|---|---|---|---|---|---|---|---|
| Central obesity | FBS | ↑ TG | ↓ HDL-C | ↑ BP | Other | Diagnosed as MS, if | |
| WHO (1998) [19] |
Waist/hip ratio: Male:>0.9 cm Female:>0.85 or BMI>30kg/m2 |
≥110mg/dl or IR or T2DM or Rx |
≥150mg/dl | Male: <40mg/dl Female: <50mg/dl |
Diastolic≥140 and systolic ≥90 mmHg | Microalbuminuria | Absolutely required IR plus ≥2 criteria |
| EGIR (1999) [20] |
WC Male: ≥94cm Female:≥80cm |
≥108.11 mg/dl | ≥150mg/dl | <39mg/dl | Diastolic≥140 and/or systolic ≥90 mmHg or Rx | Absolutely required IR plus ≥2 criteria | |
| NCEP: ATP III (2005) [40] |
WC Male:≥102cm Female:≥88cm |
≥100 mg/dl or Rx | ≥150 mg/dl | Male: <40mg/dl Female: <50mg/dl |
Diastolic≥130 and/or Systolic≥85 mmHg or Rx |
≥3criteria | |
| IDF (2005) [22] |
WC defined in terms of Ethnicity specific valuesǂ |
≥100 mg/dl or Rx | ≥150 mg/dl or Rx | Male: <40mg/dl Female: <50mg/dl |
Diastolic≥130 and/or Systolic≥85 mmHg or Rx |
Absolutely required central obesity plus ≥ 2 criteria | |
| AHA/NHLBI (2005) [24] |
WC Male: ≥102 cm Female:≥88 cm |
≥100 mg/dl or Rx | ≥150mg/dl or Rx | Male: <40mg/dl Female: <50mg/dl |
Diastolic≥130 and/or Systolic≥85 mmHg or Rx |
≥3criteria | |
| AHA/NHLBI and IDF:2009 [25] |
WC defined in terms of population and country based specific definitionǂǂ | ≥100 mg/dl or Rx | ≥150mg/dl or Rx | Male: <40mg/dl Female: <50mg/dl |
Diastolic≥130 and/or Systolic≥85 mmHg or Rx |
≥3criteria | |
Abbreviations; AHA: American Heart Association; NCEP: ATP III: The National Cholesterol Education Program Adult Treatment Panel III; NHLBI: National Heart, Lung and Blood Institute; BMI: Body Mass Index; IDF: International Diabetes Federation; EGIR: Europe Group for the Study of Insulin Resistance; WHO: World Health Organization; WC: West circumference, IR: Insulin Resistance; FBS: Fasting Blood Sugar; TG: Triglycerides; HDL-C: High Density Lipoprotein-Cholesterol; BP: Blood Pressure; Rx: on medication; T2DM: type 2 Diabetes Mellitus.
Table 2.
Ethnic-specific values for waist circumference (IDF guideline-2005) [22].
Table 2.
Ethnic-specific values for waist circumference (IDF guideline-2005) [22].
| Country/Ethnic group | Waist Circumference | |
|---|---|---|
| Male | Female | |
| Europids | ≥ 94cm | ≥ 80cm |
| South Asians Based on Chinese, Malay and Asian-Indian population |
≥ 90 cm | ≥ 80 |
| Chinese | ≥ 90cm | 80 cm |
| Japanese | ≥ 90cm | ≥ 80cm |
| Ethnic South and Central Americans | Use South Asian recommended until more specific data are available | |
| Sub-Saharan Africans | Use European data until more specific data are available. | |
| Eastern Mediterranean and Middle East (Arab) population | Use European data until more specific data are available. | |
Table 3.
Waist Circumference Thresholds for Abdominal Obesity by different organizations.
| Population | Organization | Recommended waist circumference | |
|---|---|---|---|
| Male | Female | ||
| Europid | “IDF | ≥94 cm | ≥80 cm |
| Caucasian | WHO | ≥94 cm (increased risk) ≥102 cm (still higher risk) |
≥80 cm (increased risk) ≥88 cm (still higher risk) |
| United States | AHA/NHLBI(ATP III”) | ≥102 cm | ≥88 cm |
| Canada | Health Canada | ≥102 cm | ≥88 cm |
| European | European Cardiovascular Societies | ≥102 cm | ≥88 cm |
| Asian (including Japanese) | IDF | ≥90 cm | ≥ 80 cm |
| Asian | WHO | ≥90 cm | ≥ 80 cm |
| Japanese | Japanese Obesity Society |
≥85 cm | ≥90 cm |
| China | CooperativeTask Force | ≥85 cm | ≥80 cm |
| Middle East, Mediterranean | IDF | ≥94 cm | ≥80 cm |
| Sub-Saharan African | IDF | ≥94 cm | ≥80 cm |
| Ethnic Central and South American | IDF | ≥90 cm | ≥80 cm |
More than one diagnostic criterion for MS has created confusion for diagnosis and research. To address the confusion and with the motto to conclude the single definition for MS, the International Diabetes Federation (IDF) proposed criteria that could be included in diagnostic criteria and epidemiological studies as well as in research on MS in April 2005 [22]. Although the pathology of MS, including its every factor, is not entirely understood, central obesity and IR are the major causative factors [23]. According to the new IDF definition, for a person to be characterized as having MS, they essentially have: central obesity (defined as waist circumference with ethnicity-specific values) plus any two of the following four factors, i.e., raised triglycerides, reduced HDL cholesterol, raised blood pressure and raised fasting plasma glucose. In the same year, 2005 American Heart Association (AHA) and the National Heart, Lung and Blood Institute (NHLBI) of the United States introduced the other criteria for MS with the consideration of central obesity adopted by the IDF. They focused on the inflation of metabolic risk factors. According to AHA/NHLBI criteria, a person possessing any three criteria of the following five: waist circumference ≥102 cm in males and ≥88 cm in females; TG: ≥150mg/dl and/or on drug treatment; HDL-C:<40 mg/dl in males and <50mg/dl in females; BP: Diastolic≥130 and Systolic≥85mmHg and/or on drug treatment; FBS: ≥100mg/dl and/or on drug treatment will be characterized as MS [24]. Moreover, in 2009 a joint scientific statement by the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity was published with a common consensus to define MS. According to a joint statement, a person to be characterized as having MS, they must have any three criteria out of following five: central obesity (Population and country based specific definition), TG: ≥150mg/dl and/or on drug treatment; HDL-C:<40 mg/dl in males and <50mg/dl in females; BP: Diastolic≥130 and Systolic≥85mmHg and/or on drug treatment; FBS: ≥100mg/dl and/or on drug treatment [25].
4. Prevalence of Metabolic Syndrome:
More than 3 million hits on Google search engine and nearly 10,000 registered articles in PubMed, resulting from a search for the keyword “Metabolic Syndrome” are evidence of increased concern regarding MS [26]. Such increased concern regarding MS is linked to the increasing prevalence of MS worldwide. The prevalence of MS ranges from <10% to 80%, depending upon the region, urban/rural, composition (ethnicity, sex, age and race) of the study population and the type of accepted definition/criteria for diagnosis of MS [27]. Regardless of any criteria, the prevalence of MS is high in all western societies, which is thought to be the result of the epidemic of central obesity [28]. A study assessed by National Health and Nutrition Examination Survey (NHANES) in 2003-2006 observed that around 34% of the studied population satisfied the criteria for MS, with significant association with age [51]. As reported by IDF, one-fourth of the world population has met the criteria for MS [29]. At the same time, one more study carried out in the United States of America observed nearly 910,000 adolescents qualified for MS diagnosis [30]. In addition, the global prevalence of MS of 8% to 43% in males and 7%-56% in females was reported by NCEP-ATP III in 2001 [31]. Furthermore, despite the relatively lower prevalence of obesity in the Asian population, as per WHO criteria, the rising prevalence of MS is a significant public health problem compared to the western world [32]. China, Hong Kong, Taiwan and Thailand had similar prevalence rates, ranging from 10-15%, whereas higher prevalence was noted in Koreans [33], nevertheless of approximately similar BMI. A significantly high prevalence rate of MS has been reported in India, approximately 31.6% (Male 22.9% and Female 39.9%) compared to other Asian countries [34]. The Prevalence of MS is increasing significantly in developing counties, varying from 9.8% in the male of north urban Indians to 42% in Iranian urban females [35]. A 74% prevalence of MS with no significant differences between male (77.7%, 95% CI:71.0-83.5%) and female (72.2%, 95% CI: 65.2-78.3%) has reported in 2021, in Nepal, using IDF criteria [36]. Another study in India in 2011, applying NCEP: ATP III, reported that nearly 95% of the examined population had at least one abnormal parameter, with the overall prevalence for BMI at 79.1% [37]. A survey carried out in 2007 among Asians in China, Hong Kong, Taiwan, Japan, Philippines and Singapore reported a 10 to 30% prevalence of MetS and most subjects had a history of type 2 DM and CVD [38]. Other Studies reported a 14.0 % ("WHO"), 26.1 % ("ATP III"), 29.8 % ("IDF") and 32.5 % ("modified ATP III") mean prevalence of MS in South Asian regions with a significantly lowered level of HDL cholesterol and hypertension documented as the major risk factor [39].
5. Pathophysiology
Despite concerted efforts worldwide, the intricacy of the pathophysiology of MS is still not clearly understood. Its geographical distribution and increasing prevalence trend in developing countries indicate the association of environmental factors and lifestyle factors such as high caloric diet intake compounded with decreased/lack of physical activities. [40]. There are various hypothetical mechanisms regarding the rudimentary pathophysiology of MS; however, fatty acid flux and insulin resistance, neurohormonal activation, low-grade chronic inflammation and oxidative stress are widely accepted as the crucial underlying factors involved in the initiation, progression and transformation of MS. [41,42,43].
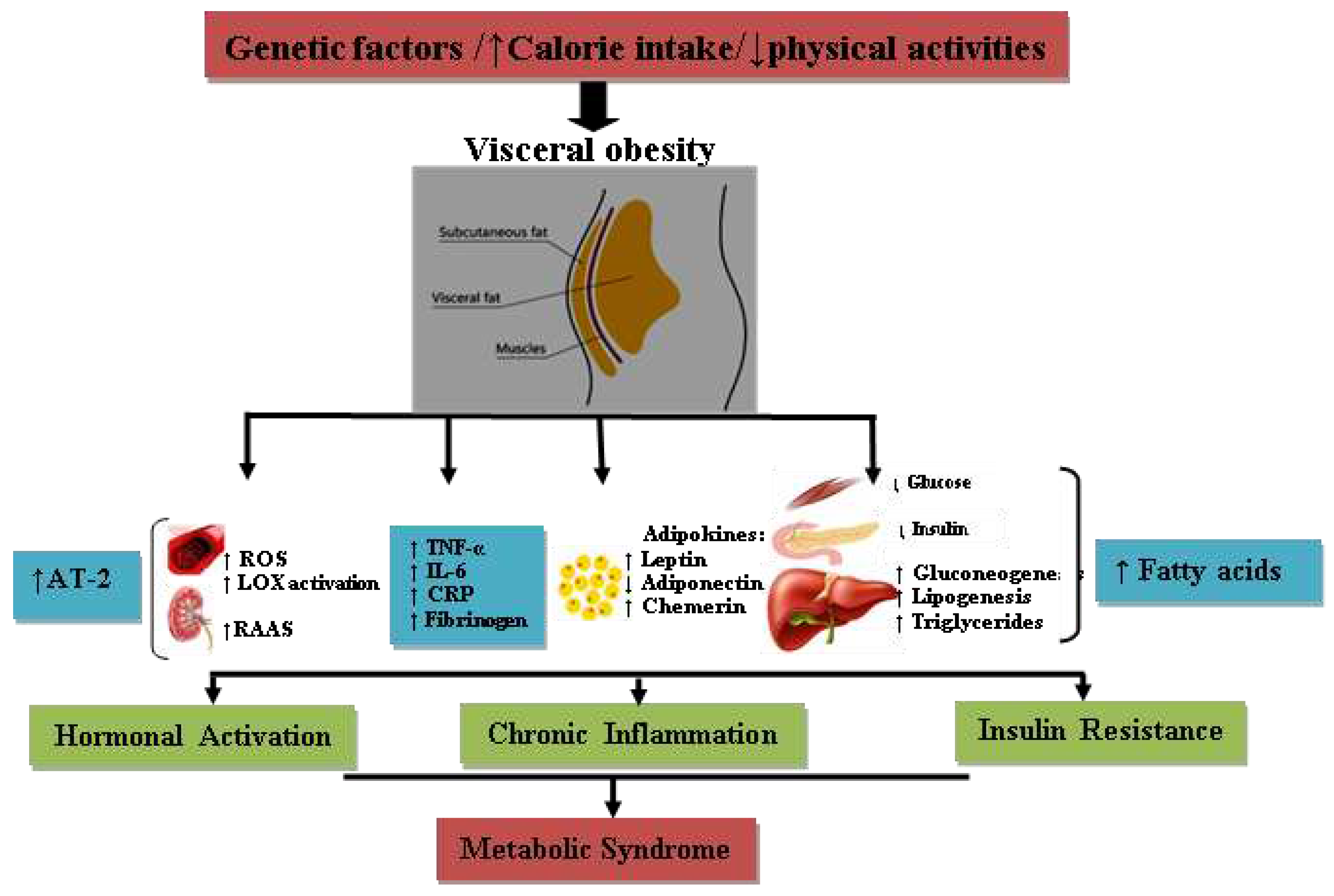
Figure 1.
Schematic illustration of the pathophysiology of metabolic syndrome. ROS; reactive oxygen species, LOX; lipoxygenase, RAAS; Renin-Angiotensin-Aldosterone System, TNF-α; tumor necrosis factor-α, IL-6; interleukin-6, CRP; C-reactive protein; AT-2; angiotensin-2.
Figure 1.
Schematic illustration of the pathophysiology of metabolic syndrome. ROS; reactive oxygen species, LOX; lipoxygenase, RAAS; Renin-Angiotensin-Aldosterone System, TNF-α; tumor necrosis factor-α, IL-6; interleukin-6, CRP; C-reactive protein; AT-2; angiotensin-2.
6. Insulin and Insulin resistance
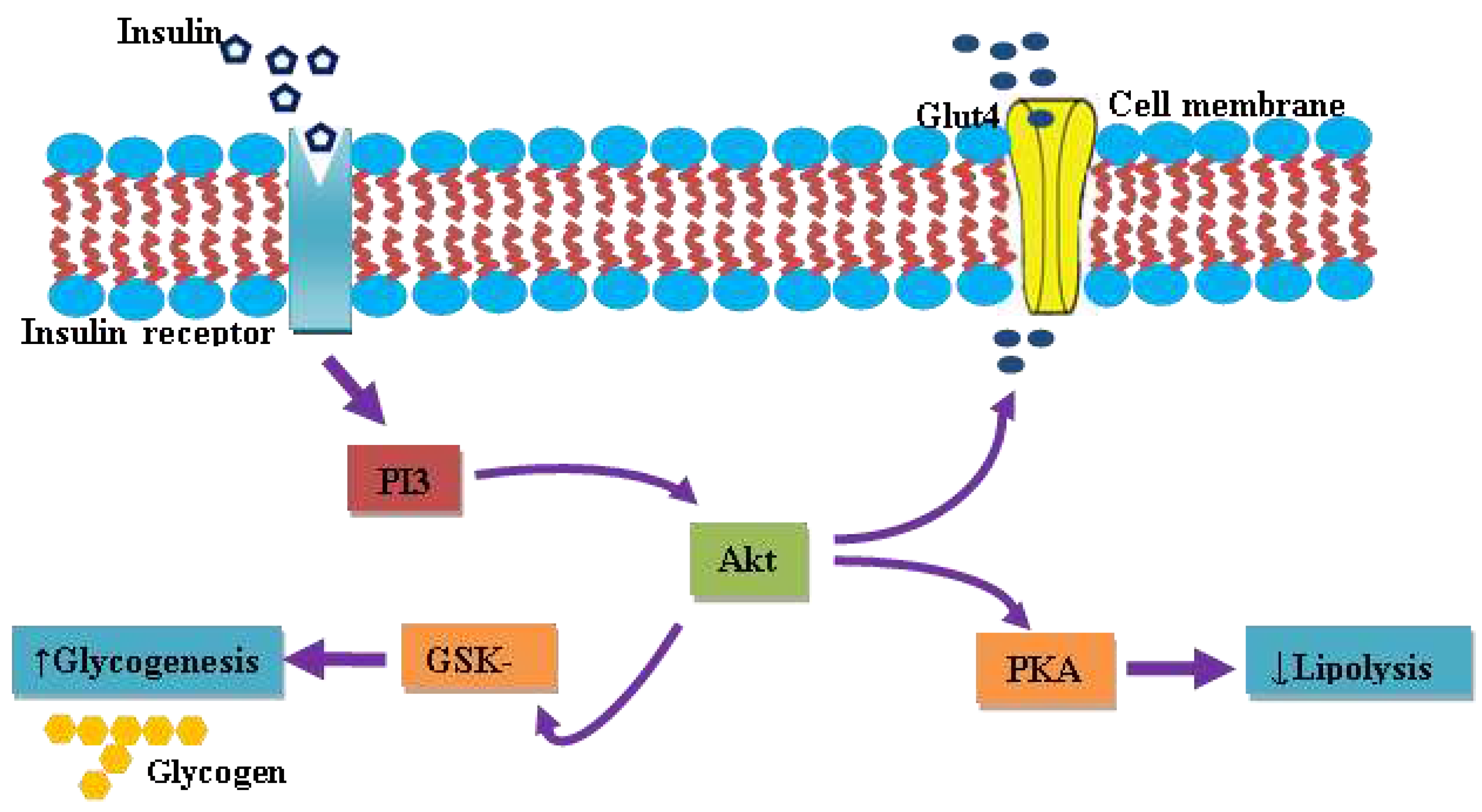
Insulin, the first unearthed peptide hormone secreted by β-cells of the pancreatic islet, is characterized as a major anabolic hormone principally involved in the regulation of carbohydrates, fats, and protein metabolism. Insulin acts via its specific receptor, known as insulin-receptor (IR), abundantly present on the cell membrane of target tissues of the liver, skeletal muscles and adipocytes. The insulin receptor is a homodimer, consisting of two extracellular α-subunits, which facilitate interaction with insulin and two intracellular β-subunit possess tyrosine kinase enzyme activity. The binding of insulin at the extracellular α-unit of IR auto-activates the tyrosine kinase enzyme activity on the intracellular domain of the β-subunit. Subsequently, it phosphorylates the protein kinase, named Insulin Receptor Substrate (IRS). Thus, phosphorylated IRS activates a cascade of signal transduction that leads to the activation of a series of kinases along with transcription factors which mediate the intracellular effects of insulin [44]. The insulin-dependent glucose uptake is mediated by the phosphatidylinositol-3 kinase (PI3) pathway through the Glut4 transporter. A key downstream effecter protein, protein kinase B, gets activated by protein kinase-3-phosphoinositide dependent protein 1, in coordination with another currently unidentified kinase, named 3-phosphoinositide – dependent protein kinase-2, for now. Thus activated Akt eventually phosphorylates and inactivates glycogen synthase kinase-3, promoting glucose storage as glycogen by accelerating glycogenesis [45,46,47]. In addition to promoting glucose storage, insulin inhibits gluconeogenesis and glycogenolysis and subsequently induces the transcription of enzymes of glycolytic as well as a fatty acid synthetic pathway.
Insulin resistance moderated increased circulating free fatty acids (FFAs) are trusted to play a major role in the pathophysiology of MetS [48]. As maintained earlier, insulin normally stimulates muscle and liver glucose uptake while inhibiting lipolysis and liver gluconeogenesis. In adipose tissue, IR impairs insulin-associated lipolysis inhibition, resulting in elevated circulating FFAs, further inactivating the antilipolytic action of insulin. FFAs decrease glucose uptake in muscle by inhibiting protein kinase activation [49]. In contrast, FFAs increase gluconeogenesis and lipolysis by activating liver protein kinase. Studies have also revealed that FFAs alter the redox status and have a pro-inflammatory action that may be linked to IR and pose difficulty for glucose homeostasis and achieving euglycemic states despite hyperinsulinemia. [50].
Figure 2.
Schematics illustration of insulin-dependent glucose uptake and intracellular signaling cascade. PI3k; phosphoinositide 3-kinase, Glut4; glucose transporter 4, PKA; protein kinase A, GSK-3; glycogen synthase kinase 3.
Figure 2.
Schematics illustration of insulin-dependent glucose uptake and intracellular signaling cascade. PI3k; phosphoinositide 3-kinase, Glut4; glucose transporter 4, PKA; protein kinase A, GSK-3; glycogen synthase kinase 3.
7. Adipose tissue and its endocrine activities
In addition to storing excess energy, adipose tissues also function similarly to the endocrine gland and synthesize many biologically active compounds, termed adipocytokines/adipokines, which regulate metabolic homeostasis [51]. A modification in the secretion of pro and anti-inflammatory adipocytokines in subjects with central obesity might be a major factor contributing to the many aspects of MS [52]. The secretion of adipocytokines depended on the adipose tissue's mass and energy state. An abnormal secretion of adipocytokines and various inflammatory adipocytokines has been reported in subjects with central obesity. Such abnormal secretion of adipocytokines is observed to contribute to a chronic inflammatory condition, ultimately involved in the progression of complications related to MS and cardiovascular diseases (CVD). The adipocytokines include proteins that interfere with insulin sensitivity (IL-6, tumor necrosis factor –α [TNF-α], resistin, adiponectin, leptin, chemerin, visfatin) and the proteins affect vascularity (angiotensin and plasminogen inhibitor protein [PAI-1]) [53,54,55].
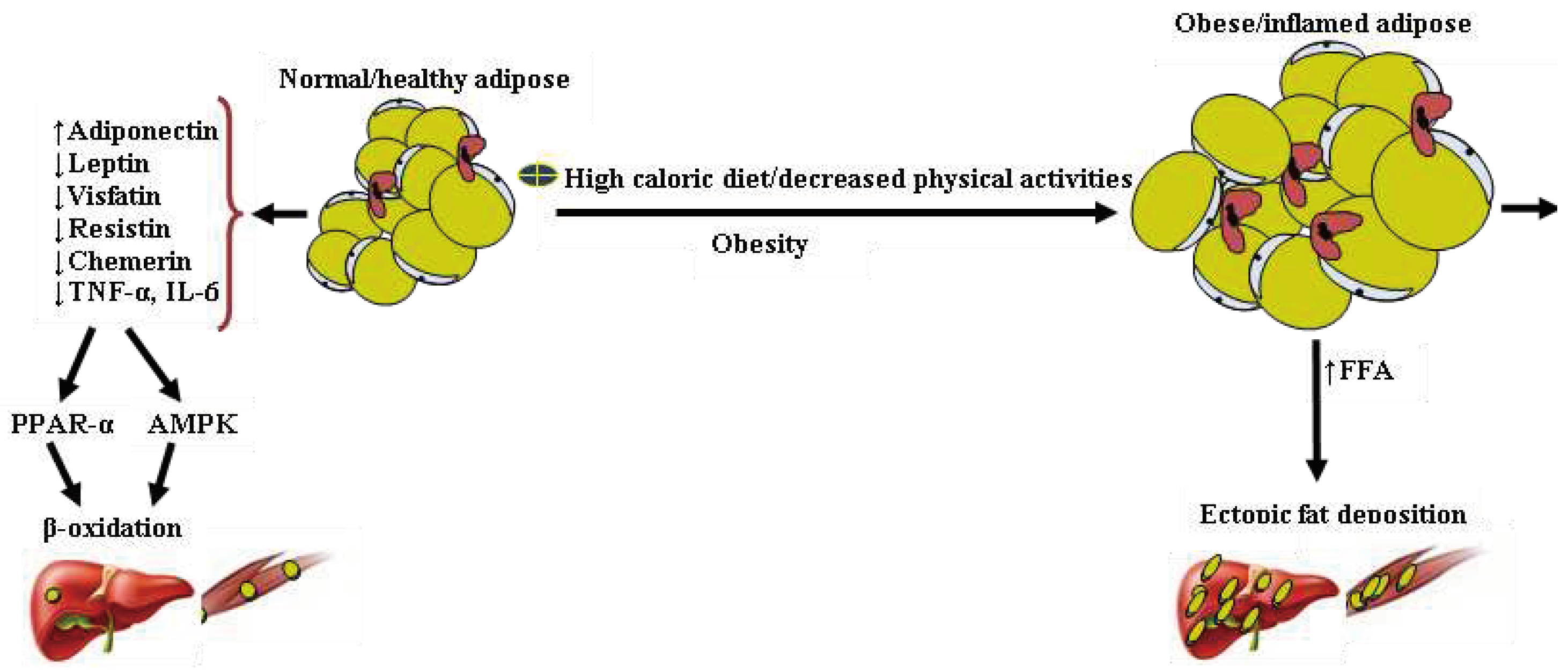
Figure 3.
Alteration in adipocytokines secretion by obese/inflamed adipose results in ectopic fat deposition. Normal adipose tissue secretes relatively high levels of adiponectin and low levels of pro-inflammatory cytokines. Lipids deposition of the liver and muscles are negligible, maintained by an efficient β-oxidation of fatty acid. In contrast, obese/inflamed and insulin-resistant adipose tissue secretes relatively high pro-inflammatory adipocytokines and low levels of adiponectin. These features and the pro-inflammatory condition may affect the β-oxidation of fatty acids and promote ectopic fat deposition in the liver and muscles.
Figure 3.
Alteration in adipocytokines secretion by obese/inflamed adipose results in ectopic fat deposition. Normal adipose tissue secretes relatively high levels of adiponectin and low levels of pro-inflammatory cytokines. Lipids deposition of the liver and muscles are negligible, maintained by an efficient β-oxidation of fatty acid. In contrast, obese/inflamed and insulin-resistant adipose tissue secretes relatively high pro-inflammatory adipocytokines and low levels of adiponectin. These features and the pro-inflammatory condition may affect the β-oxidation of fatty acids and promote ectopic fat deposition in the liver and muscles.
8. Leptin
Leptin, the first adipocytokine, discovered in 1994 by Friedman and Halaas, is a 167-amino acid protein including a 21 amino acid signal peptide [56]. The well-known function of leptin is to regulate long-term food intake, body weight, energy expenditure, and neuroendocrine functions. An overexpressed level of leptin is associated with obesity [57]. A lower leptin level has been reported in lean individuals, and a decreasing level has also been observed associated with fasting. Physiologically, leptin promotes the oxidation of FFAs, reducing ectopic fat accumulation in non-adipose tissue, a favored condition for insulin sensitization [58]. A significant alliance has been observed between plasma leptin concentration, CVD, IR, MS, hypercholesteremia, and inflammatory markers. [59]. Leptin control energy homeostasis regulated in close proximity to the hypothalamus called the stimulator of immune cell activity, the T Helper Cell type 1 pathway [60]. At the molecular level, leptin seals fatty acid entry into mitochondria by malonyl CoA. It inhibits fatty acid oxidation through the activation of adenosine monophosphate-dependent protein kinase in skeletal muscle [61,62].
9. Adiponectin
Adiponectin, also named as adipoQ, ACRp30, apM1, and GBP28, is made up of a collagen tail and a globular head, an adipocyte-secreted adipocytokine [63]. Adiponectin has been reported as a protective adipocytokine, which exhibits anti-inflammatory and anti-atherogenic properties. The central role of this adipocytokine is to improve insulin resistance by accelerating fatty acid oxidation in adipose tissue, resulting in decreased levels of fatty acids in the circulation and of intracellular triglycerides (TG) of the liver and muscle. Furthermore, it has a protective function in the processes of atherosclerosis [64]. At the metabolic level, adiponectin increases insulin sensitivity, mediated through protein kinase A activity, whose activation induces PPARϒ expression, enzymes of fatty acids oxidation, and other enzymes involved in glucose uptake, parallel by inhibiting the enzymes of gluconeogenesis [65]. The circulating level of adiponectin has been observed to be adipose tissue mass-dependent, inversely proportional to the mass of adipose tissue [66]. The plasma level of adiponectin was reduced in subjects with MS, IR, and type 2 DM. Increased plasma adiponectin concentration has been observed to be associated with exercise, weight loss, and thiazolidinedione therapy [67].
10. Resistin
Resistin is a 114-amino acid circulating protein that belongs to the resistin-like family [68]. The precise role of resistin in the human body is still not unfolded. However, it is constrained by growth hormones, glucose and insulin. An increased resistin expression was reported with impaired glucose uptake and glycogen synthesis [69]. A positive interrelation of resistin with the risk factors of MS, such as waist circumference, arterial blood pressure, blood glucose, serum triacylglycerol, serum cholesterol, Serum VLDL, and plasma insulin, has been observed, which strongly supports the underlying synergy between MS and resistin secretion as well as central obesity [70]. Furthermore, resistin has also been positively related to common inflammatory and fibrinolytic biomarkers such as CRP, TNF-α, and IL-6 [71]. Furthermore, several latest studies have implicated the association of resistin not only restricted to central obesity but also associated with CVD, arthritis, atherosclerosis, and various carcinomas [72].
11. Visfatin
Visfatin, a pre-B-cell colony-enhancing factor and nicotinamide phosphoribosyltransferase (Nampt), is an adipocytokine of molecular weight 52KDa. Although it is principally secreted by visceral fat tissue, its secretion by other organs such as bone marrow, activated lymphocytes, liver cells, and skeletal muscle is also found. Recently, several studies have observed increased circulating visfatin in individuals with central obesity, type 2 DM [73,74]. Likely, a positive association of visfatin with obesity and insulin resistance indicates that visfatin might contribute to the progression of MS [88]. Visfatin-induced upregulation of pr-inflammatory cytokines, such as Il-6, TNF-α thought to play a crucial role in the advancement of IR; however, its complete molecular mechanism is still not unfolded [75].
12. Chemerin
Chemerin, initially known as TIG2 (tazarotene-induced gene 2), is encoded by the retinoic acid receptor responder 2 (Rarres2) gene, recently recognized as an adipocytokine [76]. An increased expression of chemerin has been reported in white adipokines, the liver, and the lungs. In humans, chemerin is initially synthesized as a 63 amino acids pre-cursor, which undergoes further extracellular modification to become active chemerin [77]. Active chemerin exhibits the capability to bind with more than one receptor, such as G-protein coupled receptor chemokine-like receptor 1(MKLR 1), G-coupled receptor 1 (GPR 1), and with comparatively low affinity binds to C-C chemokine receptor-like 2 (CCRL 2 [78,79]). It has been reported that chemerin plays a vital role in adipocyte differentiation, triglycerides synthesis, Glut4 expression, and control adiponectin and leptin expression [80]. Furthermore, an increased chemerin expression in white adipose tissue (WAT), which is associated with thermogenesis, suggests that chemerin exerts its effect on weight by regulating adipogenesis rather than thermogenesis [81].
13. Inflammatory status and oxidative stress
The complete mechanism of the onset and progression of MS is not entirely understood. However, central obesity and insulin resistance are reported widely in their etiology, leading to a significant increase in the risk of type 2 DM and CVD. Chronic low-grade inflammation, oxidative stress, and biologically active adipose tissue are central to its pathophysiology [82]. The oxidative stress associated with obesity and insulin resistance leads to the over-activation of the downstream signaling cascade, which is found to be involved in atherogenesis and tissue fibrosis. Secretion of a wide range of inflammatory markers in MS plays a crucial role in the pathogenesis of CVD [83]. In addition, increased infiltration of macrophages in inflamed/obese adipose tissue secretes tumor necrosis factor-alpha [84]. TNF-α induced phosphorylation and inactivation of insulin receptors of adipose tissue and "smooth muscle leads to induction of lipolysis, resulting in increased FFAs with simultaneous inhibition of adiponectin secretion. Interleukin -6 (IL-6), another vital adipocytokine secreted by adipocytes and immune cells, has shown complex regulatory mechanisms [85,86]. Increased secretion of IL-6 has also been reported with central obesity and IR, which acts broadly on the liver, bone marrow, and endothelium [87]. The action of IL-6 on these organs results in increased secretion of acute phase reactants by the liver, including CRP. IL-6 increases fibrinogen levels and lead to a pro-thrombotic state; it promotes the secretion of adhesion molecule by endothelial cells and activates the local RAS pathway. Various studies showed a strong association between CRP levels and the development of MetS, Type 2 DM and CVD [88].
Conclusion
Metabolic syndrome is a common cause of global morbidity and mortality, which has been reported, associated with various factors. Among the possible pathophysiological mechanisms of MS been postulated, insulin resistance, central obesity, low-grade chronic inflammation, pro-inflammatory status, and oxidative stress are considered the most common physiological mechanism. A continued exploration approach is necessary to discover the underlying pathophysiology and factors associated with MS to determine the therapy's absolute target
Author Contributions
Conceptualization, B.K.J., S.K.J., and K.R.P; writing—B.K.J., M.I., and L.A.J.; writing—review and editing, M.Y., and P.M.H.; supervision, S.K.J., and K.R.P.
Funding
This research received no external funding.
Institutional Review Board Statement
Not Applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M. Joint Scientific Statement, Harmonizing the Metabolic Syndrome Circulation. 2009. [Google Scholar]
- Alberti, K.G.M.; Zimmet, P.; Shaw, J. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Olijhoek, J.K.; Van Der Graaf, Y.; Banga, J.-D.; Algra, A.; Rabelink, T.J.; Visseren, F.L.J. The Metabolic Syndrome is associated with advanced vascular damage in patients with coronary heart disease, stroke, peripheral arterial disease or abdominal aortic aneurysm. Eur. Heart J. 2004, 25, 342–348. [Google Scholar] [CrossRef] [PubMed]
- A Comprehensive Review on Metabolic Syndrome. Available online: https://www.researchgate.net/publication/261445761_A_Comprehensive_Review_on_Metabolic_Syndrome (accessed on 21 August 2022).
- Apridonidze, T.; Essah, P.A.; Iuorno, M.J.; Nestler, J.E. Prevalence and characteristics of the metabolic syndrome in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metabolism. 2005, 90, 1929–1935. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiol.-Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar] [CrossRef]
- Haller, H.; Hanefeld, M. Synoptische betrachtung metabolischer risikofaktoren. In Lipidstoffwechselstörungen; Gustav Fischer Verlag: Jena, Germany, 1975; pp. 254–264. [Google Scholar]
- Reaven, G.M. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Crepaldi, G.; Maggi, S. The metabolic syndrome: A historical context. Diabetes Voice 2006, 51, 8–10. [Google Scholar]
- Vague, J. Sexual differentiation; Factor determining forms of obesity. La Presse Médicale 1947, 55, 339. [Google Scholar]
- Paulescu, N. Traité de Physiologie Medicale; Cartea Românească: Bucharest, Romania, 1920. [Google Scholar]
- Avogaro, P.; Crepaldi, G.; Enzi, G.; Tiengo, A. Associazione di iperlipemia, diabete mellito e obesita'di medio grado. Acta Diabetol. Lat. 1967, 4, 572–590. [Google Scholar] [CrossRef]
- Avogaro, P.A.; Crepaldi, G. Essential hyperlipidemia, obesity and diabetes. Diabetologia 1965, 1, 137. [Google Scholar]
- Kaplan, N.M. The deadly quartet and the insulin resistance syndrome: An historical overview. Hypertens. Res. 1996, 19 (Suppl. I), S9–S11. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Valdez, R.A.; Hazuda, H.P.; Mitchell, B.D.; Morales, P.A.; Stern, M.P. Prospective analysis of the insulin-resistance syndrome (syndrome X). Diabetes 1992, 41, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Ionescu-Tîrgovişte, C. Tratat de Diabet Paulescu; Academiei Romane: Bucharest, Romania, 2004; pp. 321–335. [Google Scholar]
- Alessi, M.C.; Juhan-Vague, I. Contribution of PAI-1 in cardiovascular pathology. Arch. Des Mal. Du Coeur Et Des Vaiss. 2004, 97, 673–678. [Google Scholar]
- Hollman, G.; Kristenson, M. The prevalence of the metabolic syndrome and its risk factors in a middle-aged Swedish population—Mainly a function of overweight? Eur. J. Cardiovasc. Nurs. 2008, 7, 21–26. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus. Provisional report of a WHO consultation. Diabet. Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Balkau, B. Comment Comment on the provisional report from the WHO consultation. European Group for the Study of Insulin Resistance (EGIR). Diabet Med. 1999, 16, 442–443. [Google Scholar] [PubMed]
- Expert Panel on Detection, E. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III). JAMA 2001, 285, 2486–2497. [Google Scholar]
- IDF Epidemiology Task Force Consensus Group. International Diabetes Federation: The IDF Consensus Worldwide Definition of the Metabolic Syndrome. 2005. (accessed on). Available online: http://www. idf. org/web data/docs/Metabolic syndrome def. pdf.
- Anderson, P.J.; Critchley, J.A.; Chan, J.C.; Cockram, C.S.; Lee, Z.S.; Thomas, G.N.; Tomlinson, B. Factor analysis of the metabolic syndrome: Obesity vs insulin resistance as the central abnormality. Int. J. Obes. 2001, 25, 1782–1788. [Google Scholar] [CrossRef]
- Yamagishi, K.; Iso, H. The criteria for metabolic syndrome and the national health screening and education system in Japan. Epidemiol. Health 2017, 39, e2017003. [Google Scholar] [CrossRef]
- Alberti, K.G. ; International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; International Association for the Study of Obesity. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; national heart, lung, and blood institute; American heart association; world heart federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar]
- Hansen, B.C.; Bray, G.A. (Eds.) The Metabolic Syndrome: Epidemiology, Clinical Treatment, and Underlying Mechanisms; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Desroches, S.; Lamarche, B. The evolving definitions and increasing prevalence of the metabolic syndrome. Appl. Physiol. Nutr. Metab. 2007, 32, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Hollman, G.; Kristenson, M. The prevalence of the metabolic syndrome and its risk factors in a middle-aged Swedish population—Mainly a function of overweight? Eur. J. Cardiovasc. Nurs. 2008, 7, 21–26. [Google Scholar] [CrossRef]
- IDF Epidemiology Task Force Consensus Group. International Diabetes Federation: The IDF Consensus Worldwide Definition of the Metabolic Syndrome. 2005. (accessed on). Available online: http://www.idf.org/webdata/docs/Metabolic_syndrome_def. pdf.
- Ervin, R.B. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003-2006. Natl. Health Stat. Report 2009, 5, 1–7. [Google Scholar]
- Nestel, P.; Lyu, R.; Low, L.P.; Sheu, W.H.; Nitiyanant, W.; Saito, I.; Tan, C.E. Metabolic syndrome: Recent prevalence in East and Southeast Asian populations. Asia Pac. J. Clin. Nutr. 2007, 16. [Google Scholar]
- Cameron, A.J.; Shaw, J.E.; Zimmet, P.Z. The metabolic syndrome: Prevalence in worldwide populations. Endocrinol. Metab. Clin. 2004, 33, 351–375. [Google Scholar] [CrossRef]
- Yeh, W.T.; Weng, L.C. Epidemiology of metabolic syndrome in Asia. Asia Pac. J. Clin. Nutr. 2008, 17, 37–42. [Google Scholar]
- Nematic, M.; Ahmadpour, F.; Rassouli, Z.B.; Ardabili, H.M.; Azimi-Nezhad, M. A review on underlying differences in the prevalence of metabolic syndrome in the Middle East, Europe and North America. J. Mol. Genet. Med. 2014, 2, 019. [Google Scholar]
- Ko, G.T.; Cockram, C.S.; Chow, C.C.; Yeung, V.; Chan, W.B.; So, W.Y.; Chan, N.N.; Chan, J.C. High prevalence of metabolic syndrome in Hong Kong Chinese—Comparison of three diagnostic criteria. Diabetes Res. Clin. Pract. 2005, 69, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Jha, B.K.; Sherpa, M.L.; Dahal, B.K.; Singh, J.K. Prevalence of Metabolic Syndrome and Its Components in Adults with Central Obesity at Janakpur Zone, Nepal. J. Nepal. Health Res. Counc. 2021, 18, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Deedwania, P.C.; Gupta, A.; Rastogi, S.; Panwar, R.B.; Kothari, K. Prevalence of metabolic syndrome in an Indian urban population. Int. J. Cardiol. 2004, 97, 257–261. [Google Scholar] [CrossRef]
- Misra, A.; Khurana, L. Obesity and the metabolic syndrome in developing countries. J. Clin. Endocrinol. Metab. 2008, 93 (11 Suppl. 1), s9–s30. [Google Scholar] [CrossRef]
- Sawant, A.; Mankeshwar, R.; Shah, S.; Raghavan, R.; Dhongde, G.; Raje, H.; D'souza, S.; Subramanium, A.; Dhairyawan, P.; Tour, S.; et al. Prevalence of metabolic syndrome in urban India. Cholesterol 2011, 2011, 920983. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Wainwright, G.; Mascitelli, L. Is the metabolic syndrome caused by a high fructose, and relatively low fat, low cholesterol diet? Arch. Med. Sci. 2011, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Funahashi, T.; Nakamura, T. The concept of metabolic syndrome: Contribution of visceral fat accumulation and its molecular mechanism. J. Atheroscler. Thromb. 2011, 18, 629–639. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Roberts, C.K.; Hevener, A.L.; Barnard, R.J. Metabolic syndrome and insulin resistance: Underlying causes and modification by exercise training. Compr. Physiol. 2013, 3, 1–58. [Google Scholar]
- Handbook of Diabetes, 4th ed.; Excerpt #4: Normal Physiology of Insulin Secretion and Action; Diabetes In Control; A free weekly diabetes newsletter for Medical Professionals 2014-07-28. Retrieved 2017-06-01; Wiley-Blackwell: Hoboken, NJ, USA, 2010.
- Saltiel, A.R.; Kahn, C.R. Insulin signaling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, J.M.; Alessi, D.R. The insulin signalling pathway. Curr. Biol. 2002, 12, R236–R238. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, P.R. Mechanisms regulating phosphoinositide 3-kinase signaling in insulin-sensitive tissues. Acta Physiol. Scand. 2005, 183, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Tooke, J.E.; Hannemann, M.M. Adverse endothelial function and the insulin resistance syndrome. J. Intern. Med. 2000, 247, 425–431. [Google Scholar] [CrossRef]
- Boden, G.; Shulman, G.I. Free fatty acids in obesity and type 2 diabetes: Defining their role in the development of insulin resistance and β-cell dysfunction. Eur. J. Clin. Investig. 2002, 32, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Mohanty, P.; Dhindsa, S.; Syed, T.; Ghanim, H.; Aljada, A.; Dandona, P. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes 2003, 52, 2882–2887. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Flier, J.S. Adipose tissue as an endocrine organ. Trends Endocrinol. Metab. 2000, 11, 327–332. [Google Scholar] [CrossRef]
- Bernlohr, D.A.; Jenkins, A.E.; Bennaars, A.A. Adipose tissue and lipid metabolism. In New Comprehensive Biochemistry; Elsevier: Amsterdam, The Netherlands, 2002; Volume 36, pp. 263–289. [Google Scholar]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; George, S.R.; O’Dowd, B.F. Unravelling the roles of the apelin system: Prospective therapeutic applications in heart failure and obesity. Trends Pharmacol. Sci. 2006, 27, 190–194. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Muoio, D.M.; Dohn, G.L.; Fiedorek, F.T., Jr.; Tapscott, E.B.; Coleman, R.A. Leptin directly alters lipid partitioning in skeletal muscle. Diabetes 1997, 46, 1360–1363. [Google Scholar] [CrossRef]
- Wallace, A.M. Plasma leptin and the risk of cardiovascular disease in the west of Scotland coronary prevention study (WOSCOPS). Circulation 2004, 44, 1819–1824. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Ohishi, M.; Kihara, S.; Funahashi, T.; Nakamura, T.; Nagaretani, H.; Kumada, M.; Ohashi, K.; Okamoto, Y.; Nishizawa, H.; et al. Association of hypoadiponectinemia with impaired vasoreactivity. Hypertension 2003, 42, 231–234. [Google Scholar] [CrossRef]
- Pischon, T.; Girman, C.J.; Hotamisligil, G.S.; Rifai, N.; Hu, F.B.; Rimm, E.B. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 2004, 291, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Hammer, R.E.; Ikemoto, S.; Brown, M.S.; Goldstein, J.L. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 1999, 401, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, K.; Ogawa, Y.; Masuzaki, H.; Shintani, M.; Miyanaga, F.; Aizawa-Abe, M.; et al. Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes 2001, 50, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Muñoz, F.; García-Macedo, R.; Alarcón-Aguilar, F.; Cruz, M. Adipocinas, tejido adiposo y su relación con células del sistema inmune. Gac. Méd. Méx. 2005, 141, 505–512. [Google Scholar] [PubMed]
- Ukkola, O.; Santaniemi, M. Adiponectin: A link between excess adiposity and associated comorbidities? J. Mol. Med. 2002, 80, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Lee, G.Y.; Chung, J.J.; Ahn, Y.H.; Hong, S.H.; Kim, J.B. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor α. Diabetes 2006, 55, 2562–2570. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.S.; Funahashi, T.; Hanson, R.L.; Matsuzawa, Y.; Tanaka, S.; Tataranni, P.A.; Knowler, W.C.; Krakoff, J. Adiponectin and development of type 2 diabetes in the Pima Indian population. Lancet 2002, 360, 57–58. [Google Scholar] [CrossRef]
- Sánchez, J.C.; López, D.F.; Pinzón, Ó.A.; Sepúlveda, J.C. Adipocinas y síndrome metabólico: Múltiples facetas de un proceso fisiopatológico complejo. Rev. Colomb. De Cardiol. 2010, 17, 167–176. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef]
- Way, J.M.; Görgün, C.Z.; Tong, Q.; Uysal, K.T.; Brown, K.K.; Harrington, W.W.; Oliver, W.R.; Willson, T.M.; Kliewer, S.A.; Hotamisligil, G.S. Adipose tissue resistin expression is severely suppressed in obesity and stimulated by peroxisome proliferator-activated receptor γ agonists. J. Biol. Chem. 2001, 276, 25651–25653. [Google Scholar] [CrossRef]
- Gupta, V.; Singh, A.K.; Gupta, V.; Kumar, S.; Srivastava, N.; Jafar, T.; Pant, A.B. Association of circulating resistin with metabolic risk factors in Indian females having metabolic syndrome. Toxicol. Int. 2011, 18, 168. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.R.; Lazar, M.A. Human resistin: Found in translation from mouse to man. Trends Endocrinol. Metab. 2011, 22, 259–265. [Google Scholar] [CrossRef]
- Tripathi, D.; Kant, S.; Pandey, S.; Ehtesham, N.Z. Resistin in metabolism, inflammation, and disease. FEBS J. 2020, 287, 3141–3149. [Google Scholar] [CrossRef]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef]
- Jurdana, M.; Petelin, A.; Bizjak, M.Č.; Bizjak, M.; Biolo, G.; Jenko-Pražnikar, Z. Increased serum visfatin levels in obesity and its association with anthropometric/biochemical parameters, physical inactivity and nutrition. Clin. Nutr. ESPEN 2013, 8, E59–E67. [Google Scholar] [CrossRef]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with pro-inflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Helfer, G.; Wu, Q.-F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef]
- Mattern, A.; Zellmann, T.; Beck-Sickinger, A.G. Processing, signaling, and physiological function of chemerin. IUBMB Life 2014, 66, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Rourke, J.L.; Muruganandan, S.; Dranse, H.J.; McMullen, N.M.; Signal, C.J. Gpr1 is an active chemerin receptor influencing glucose homeostasis in obese mice. J. Endocrinol. 2014, 222, 201–215. [Google Scholar] [CrossRef]
- De Henau, O.; Degroot, G.N.; Imbault, V.; Robert, V.; De Poorter, C.; Mcheik, S.; Galés, C.; Parmentier, M.; Springael, J.-Y. Signaling properties of chemerin receptors CMKLR1, GPR1 and CCRL2. PLoS ONE 2016, 11, e0164179. [Google Scholar] [CrossRef]
- Chakaroun, R.; Raschpichler, M.; Kloting, N.; Oberbach, A.; Fleming, G.; Kern, M.; Schön, M.R.; Shang, E.; Lohmann, T.; Dreßler, M.; Fasshauer, M. Effects of weight loss and exercise on chemerin serum concentrations and adipose tissue expression in human obesity. Metabolism 2012, 61, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Okla, M.; Kim, J.; Koehler, K.; Chung, S. Dietary factors promoting brown and beige fat development and thermogenesis. Adv. Nutr. 2017, 8, 473–483. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B. Oxidative stress and inflammation: What polyphenols Can Do for Us? Oxid. Med. Cell Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor-alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Deshmukh, A.; GuruMurthy, G.S.; Pothineni, N.V.; Watts, T.E.; Romeo, F.; Mehta, J.L. Inflammation and atherosclerosis—Revisited. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor-alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef]
- Laclaustra, M.; Corella, D.; Ordovas, J.M. Metabolic syndrome pathophysiology: The role of adipose tissue. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 125–139. [Google Scholar] [CrossRef]
- Bernlohr, D.A.; Jenkins, A.E.; Bennaars, A.A. Adipose tissue and lipid metabolism. In New Comprehensive Biochemistry; Elsevier: Amsterdam, The Netherlands, 2002; Volume 36, pp. 263–289. [Google Scholar]
- Wisse, B.E. The inflammatory syndrome: The role of adipose tissue cytokines in metabolic disorders linked to obesity. J. Am. Soc. Nephrol. 2004, 15, 2792–2800. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



