
Submitted:
13 March 2023
Posted:
15 March 2023
You are already at the latest version
Abstract
The intense emergence of mucilage in the Marmara Sea in 2021 has raised concerns due to its potential impact on the global marine system. Considering the potential of mucilage threatening the diverse ecosystems, it is important to define the mucilage microbiome. However, studies dedicated to exploring the biodiversity of mucilage are limited and de novo approaches are needed to understand unexplored biodiversity of mucilage microenvironment. This study aimed to investigate mucilage and water samples from certain stations in Marmara Sea. For this reason, 11 mucilage, 2 pre-mucilage water and 3 clean sea water were collected from 16 stations. DNA isolation, shotgun metagenome sequencing and bioinformatics analysis were performed. It was revealed that the mucilage microbiome and the clean water microbiome were different from each other, and the reads that could not be assigned to any taxon (dark matter) accounted for 58% (p=0.014) of the mucilage metagenome. The clean water had a higher presence of Euryarchaeota (p=0.014), Proteobacteria (p=0.019) and Rhodothermaeota (p=0.034), while Chlamydiae (p=0.014) and Fusobacteria (p=0.034) were found in excess in the mucilage. The study produced the first comparative data on clean sea water and the mucilage microbiome using shotgun metagenomics approaches.
Keywords:
Mucilage microbiome
; Shotgun-metagenome sequencing
; Comparative marine microbiome
1. Introduction
Marine snow (mucilage) formations, a natural structure rich in organic matter, are occasionally observed in the open seas and oceans in different aquatic geolocations of Earth, especially in the Mediterranean [1,2,3]. Anthropogenic causes (liquid and solid wastes, increasing rates of urbanization and industrialization on the coastline, excessive fishing activities, etc.), temperature anomalies due to climate change and the cumulative effect of stratification in the water column, the increase in the abundance of polysaccharides in the aquatic environment and the deterioration of dissolved organic matter balance in the sea. It builds in different forms as accumulation of layers covering its surface, long filamentous/reticulated structures spreading in the upper water column, and thick layers on the seabed settling to the bottom with aging over time [4]. Studies focusing on marine mucilage generally focus on the oceanographic conditions and phytoplanktonic organisms in aquatic environment [1,2,5,6]. Culture-independent genome-based approaches, which have become widespread with the development of next-generation sequencing technologies, have changed the limits of our knowledge about the diversity and ecology of microbial communities in aquatic environments [7,8]. Mapping the microbiome, which is defined as microorganisms in a habitat, their genomes, products and interactions with the environment, at the metagenome scale (whole genomes of all microorganisms) within the scope of combating mucilage through next-generation sequencing technologies seems valuable for the development of complete identification and disposal strategies.
Although the mapping of the total microbiome content of mucilage, which is a rich source of nutrients for microorganisms due to its high organic matter content, seems important for a complete identification and disposal attempts, there are limited studies on the mucilage microbiome in the Sea [9,10,11]. The decomposition of organic substances in the environment, especially nitrogen, of the metabolic pathways operated by bacteria and the excessive reproduction of the degradation products by the bacteria play a role in the first stage of mucilage formation (the formation of a reticulated/filamentous structure). Considering this situation and the role of microbiome elements in polysaccharide accumulation, the scientific dimension of these studies is better understood.
Although the mucilage formation has been seen from time to time in the Marmara Sea since the 1990s, that was reach dimensions that may threaten human health in 2021 [3,12,13,14]. It is thought that revealing the mechanisms is at the beginning of the necessary steps in the elimination of mucilage. When the reports published so far on the definition of the microbial load of mucilage are examined, it is noteworthy that the studies at the metagenome dimension are quite limited. Therefore, what exactly is the microbial content of mucilage, the taxonomic and functional diversity of the mucilage microbiome, the role of ecological association in the network in mucilage development/progression are unanswered research questions. With this study, taxomic mapping of mucilage and Marmara Sea water habitats as well as a metagenome-scale comparative analysis for water and mucilage samples collected from various stations in the Marmara Sea is aimed to be investigated. The study is based on the hypothesis that mucilage dispersal is associated with a significant shift in homeostatic balance in the aquatic microbiome.
2. Materials and Methods
2.1. Fieldwork
Mucilage samples collected from 11 different stations (MS1-MS11) in Marmara Sea between April and September 2021 were included in the current study. 5 g (or 15 ml) of pure mucilage sample was provided in preservatives from ten station and for station 1 the lyophilized mucilage sample was avaliable.
At the beginning of mucilage formations, April 2021, two pre-mucilage water samples (Msu-Y1, Msu-Y2) were taken from areas where mucilage did not reach. In order to represent clean sea water surface sea water samples were taken from three different stations in May 2022, while the mucilage did not exist neither on surface nor through the water column. The stations included in the study were Saros Bay (SAR), Çanakkale Strait (CB) and Special Environmental Protection Area located within the borders of Gökçeada (GEB). All water samples collected from five different locations were taken by means of a sterile nansen water sampler having 5 liter water capacity and were filtered in the field using a vacuum pump through 0.22μM filter paper (Steripak-GP20 filter, Millipore, Bedford, MA) [7]. Then, the filter paper was taken into 15 ml falcon tubes filled with DNA/RNA shiled buffer (Zymo Research). The coordinates for all 16 stations were recorded by GPS. The coordinates of the sampling points are given in Supplement 1.
2.2. Wet laboratory processes
Total DNA isolation was performed using the Qiagen PowerSoil DNA isolation kit in accordance with the manufacturer's procedures. For mucilage samples, approximately 0.2gr sample was collected with sterile tweezers in a BSL2 level biosafety cabinet and DNA isolation was performed in accordance with the kit procedures. For water samples, after the filter papers were cut into small pieces with the help of sterile scalpel in the BSL2 cabinet, they were taken into GarnetBead tubes containing 750ul buffer. After these tubes were treated at 65°C and 95°C for 10 minutes, DNA isolation was performed in accordance with the kit's procedure. The quality of the DNAs obtained after isolation was determined using NanoDrop (Shmadzu BioSpec-nano), and the quantification measurements of DNA samples in terms of double-stranded DNA (dsDNA) amounts were determined in the Qubitfluorometric system (Qubit 2.0 Fluorometer, Life Technologies) using the Qubit dsDNA-BR assay kit. Isolation steps were repeated with modifications for the samples that could not obtain DNA in suitable quality and quantity for sequencing. dsDNA of appropriate quality (260/230nm 2.0-2.2; 260/280nm 1.8-2.0) and density (at least 2ng per microliter).
Shotgun metagenome sequencing was performed using next-generation sequencing technologies for a total of 10 mucilages and 5 water samples (3 clean water and 2 water collected when mucilage was just observed). Metagenome sequencing studies were carried out on Illumina systems in accordance with Nextera-enzymatic digestion technology and Nextera XT DNA Library Preparation Kit protocols (https://genome.med.harvard.edu/documents/libraryPrep/IlluminaNexteraXTProtocol.pdf).
2.3. Dry laboratory processes
2.3.1. Processing of metagenome raw data
Within the aquatic microbiome, of which the highly complex mucilage is a part, it is expected that there will be a significant amount of sequences that have not been previously identified or characterized at the DNA level. For this reason, the DNA data produced in the project was analyzed with a de novo approach. In this direction, firstly, the raw data of the samples were pooled and collectively subjected to metagenome assembly. IDBA-UD assembler [15] was used for this process and contigs were obtained. In order to determine the relative abundances of contigs, each of which is expected to come from the same genome, the total cluster of contigs was indexed with BWA-mem2 [16] tool, and the raw reads were mapped onto the contigs, and the average reads per fragment were determined at the sequence level per million. In order to annotate the genes in the contigs; First, open reading frames were detected by running Prodigal gene detection algorithm [17] in metagenome mode, then each detected frame was annotated at the gene ontology level with eggNOG-mapper [18] and converted to an ontology, as well as functional groups. Unmapped open reading frames were neglected on the assumption that they are not coding genes.
2.3.2. Taxonomic analysis
In order to determine the relative abundance in the contigs obtained in the previous step, Kaiju [19] taxonomic classification program was used. The relative abundance of an assigned taxon was estimated by mapping the sequencing reads onto the associated contigs. Normalization using reads per million base pairs concluded the final relative abundance. The programs are provided with scripts created in the Python programming language.
2.3.3. Comparative Functional analysis
For functional analysis, genes predicted on the contigs by Prodigal were aligned against eggNOG database using Diamond [20]. All eggNOG annotated genes were evaluated for relative abundance for each sample and enriched at the pathway level. As a result of these enrichments, KEGG (Kyoto Encyclopedia of Genes and Genomes) ontologies, CAZy (Carbohydrate-Active enZYmes Database) [21] and protein families classification (PFAM) [22] functional group annotations and their relative abundances are organized by generating specific Python scripts. In order to detect biosynthetic gene clusters, the biosynthetic units in the samples were determined using BiG-MEx (Biosynthetic Gene cluster MEtagenomic eXploration) modules a tool for mining biosynthetic gene clusters’ domains and classes) metabolic cluster mapping tool [23].
The comparisons were primarily conducted with alpha and beta-diversity analyzes on both taxonomic and functional profiles. Bray-Curtis [24] metrics were used as beta-diversity metric and Shannon Index [25] metrics were used as alpha-diversity metric. In order to examine the biodiversity differences between mucilage and clean sea water samples, alpha-diversity distributions were subjected to Mann-Whitney U test and beta-diversity values were subjected to one-way ANOVA test. Mann-Whitney U test and multiple hypothesis test were applied for the relative abundance values of each taxon and functional structure detected in the samples. The differences at taxonomic or functional biodiversity level between the clean sea and mucilage samples were multiply-tested for statistical significance using Mann-Whitney U test. In order to visually observe the genotypic characteristics among the geographic distributions as well as clean water-mucilage difference, the relative anundance vectors were ordinated in 2D using Principal Component Analysis (PCA) performed by Python SciPy library.
3. Results
Because the quality and the amount of isolated dsDNA was not appropriate for shotgun metagenome sequencing, next-generation sequencing could not be achieved for the lyophilized musilage sample (MS1). Thus, shotgun metagenome sequencing were conducted for 15 samples. As a result of sequencing, a total of 58.4Gbp of read data were produced. Sequencing data statistics for all samples are presented in Table 1.
3.1. Taxonomic analyzes
As a result of the taxonomic assignment, it was determined that on avarage 58.2±9.6%of the reads obtained from mucilage samples, 68.9±9.6% from pre-mucilage water samples taken at the very beginning of mucilage formation, and 35.4±2.4% from clean water samples could not be assigned to any taxon. The assignments at species and phylum levels for the assigned reads are presented as Supplement S2 and Supplement S3. According to the taxonomic assingment, environment originated bacteria were dominant in the samples. However, only MS4 was found to be carrying several human pathogens. Table 2 shows the top 5 most common microbial species in each sample, corresponding to more than ten thousand readings.
As a result of the statistical tests carried out for taxonomic assignments of samples, it was determined that the beta diversity of mucilage and clean water was significantly different from each other (Table 3) and the water samples contained a wider range of microbial communities (Figure 1) than mucilages.
It was observed that the clean water samples and mucilage were separated and grouped within themselves, while the two pre-mucilage water samples (Msu-Y1 and MsuY-2) were approached the mucilage group (Figure 2 and Figure 3).
The most common phyla assignments made for clean water samples and mucilage samples are given in Table 4.
Statistically significant differences (p<0.05) were reached for many taxa as a result of the statistical tests performed to reveal whether there is a significant difference at taxonomic level between the samples. However, after error correction (FDR-false discovery rate correction), the phyla found in excess in clean water samples were Euryarchaeota (p=0.014), Proteobacteria (p=0.019) and Rhodothermaeota (p=0.034) while found in the mucilage group as excess level were reported as Chlamydiae (p=0.014) and Fusobacteria (p=0.034). The readings that could not be assigned to any phyla (Unclassified) were found to be significantly higher (p=0.015) in the mucilage group than clean water samples. After error correction, no statistically significant results were found between groups for species level.
3.2. Comparative Functional analysis
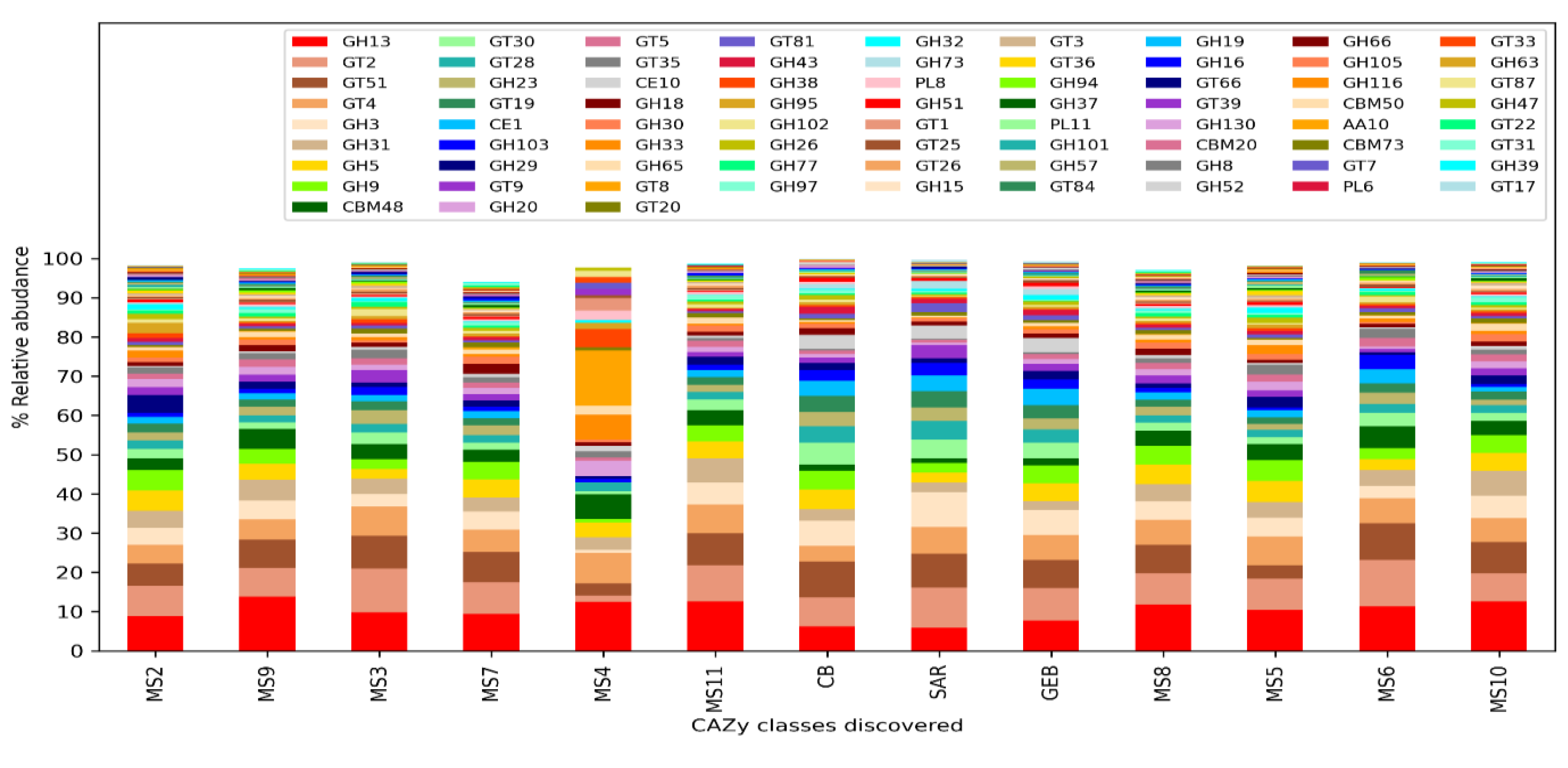
Comparing the clean water and mucilage samples, no statistically significant protein family and KEGG ontologies were detected after the p-value correction. However, the presence and amounts of carbohydrate processing enzyme classes (CAZy) were differing significantly between clean water and mucilage samples. According to the results, it was determined that certain enzyme classes were highly present in the mucilage samples, and that these classes were almost not represented in the mucilage sample MS4, in which human pathogens were dominant.
Two enzyme families (GH3 and GH13, p=0.014) belonging to the glycoside hydrolase (Glycoside Hydrolase, GH) family, three enzyme families (GT8, GT19 and GT28, p=0.015) belonging to the GlycosylTransferase Family (GlycosylTransferase Family, GT) and one family (CE10, p=0.015) from the carbohydrate esterase family (Carbohydrate Esterase Family, CE) were found to be significantly more in clean water then mucilage samples.
One enzyme family (GH31, p=0.022) belonging to the glycoside hydrolase family, four different enzyme families (GT3 (p=0.038), GT30 (p=0.014), GT35 (p=0.016) and GT81 (p=0.014) belonging to the GlycosylTransferase Family one of the enzyme families (CBM 48, p=0.014) with carbohydrate binding activity (carbohydrate-binding modulated) and one family (CE1, p=0.014) from the carbohydrate esterase family were found to be significantly more in mucilage then clean water samples.
Figure 4.
Distribution of carbohydrate processing enzyme (CAZy) classes.
The analysis performed for BiG-Mex are presented in Figure 5 and Figure 6. Among the biosynthetic classes consisting of 25 different gene clusters, terpenes are significantly higher (p=0.03) in clean water, while in domains consisting of 93 different gene pathways, cyanobactin (p=0.006), siderophore (p=0.007), nrps (p=0.007), and bacteriocin (p=0.011) , hserlactone (p=0.017), t3pks (0.017), phosphonate (0.021), ectoine (0.025), t1pks (p=0.025) were detected significantly more in mucilage, whereas sactipeptide more in clean water (p=0.025).
4. Discussion
Mucilage events, the build up of a gelatinous substance on the water's surface and through the water collum, have been observed in different marine environments for more than 200 years [26]. In spite of such a long history of awareness, there have been limited number of studies reporting the microbial content of mucilage [11,27,28]. Thus, the principle aim of the current study was to investigate the microbial content of mucilage and compare it to the marine water. According to the taxonomic assignments achieved on sequencing data within the scope of the study, most of the reads belonging mucilage and pre-mucilage water samples could not be assigned to any taxa. While performing taxonomic analysis, a wide range of sequencing data (obtained from different habitats such as soil, sea, and human) avaliable in public databases as well as in private databases (special permissions had been obatined for this study) and different assignment approaches (nucleotide database search, Kraken, protein family search, Kaiju, sequencing read and contig based recognition approaches etc.) were used. However, despite this effort, more than half of the reads (58% for mucilage, 69% for pre-musilage water) could not be assigned to any taxa and called “dark matter”. This seems to be related to the fact that, many taxa have not yet been defined due to the scarcity of datasets in this field or due to the fact that the mucilage metagenome has hardly been studied, and the inadequacy of studies involving cultureomics approaches in this field. In contrast, the rate of reading that cannot be assigned ant taxon was 34% in clean water. The reads that cannot be assigned to any phylum was found to be significantly higher (p=0.014) in the mucilage group than the clean water samples. In a mucilage sample (MS3) no assigment has been made for a contig over 145k bp and it strengthens the existence of unidentified or new microbial taxa in mucilage microbiome. The presence of unidentified or unknown microbial taxa in mucilage suggests that unknown microorganisms may play an important role in the formation of mucilage and its progress. Therefore, there is a need to large-scale cultureomic data as well as more marine microbiome data to identify taxa in question.
As a result of the alpha diversity analysis, it has been determined that clean water samples contain a wider range of microbial communities than mucilage. However, this most probably related to the lack of identification because more than half of the mucilage metagenome was assigned as dark matter. A study conducted by Rath et al. on the 16S gene region it has been reported that, microbial species detected in mucilage may greatly differ from marine species [29]. Similar result has been obtained in the current study that the beta diversity of the mucilage and clean water was statistically significantly different from each other.
According to the signature of the microbial diversity of the samples, the clean water and mucilage were significantly different from each other. However the water samples taken during the pre-mucilage time appeared to be closer to the mucilage groups than the clean water samples. It can be said that, the network based analysis including unidentified readings (dark matter), provide the information about the fact that the hemostatic balance begins to deteriorate before mucilage dispance all over the area. Therefore, the microbial content of this transition form might be a useful tool to develop early warning strategies in order to alarm early about the formation of mucilage.
It has been reported that the phyla Euryarchaeota, Proteobacteria and Rhodothermaeota are found in high levels in soils [30,31,32]. The highest incidence of these phyla is also encountered in hypersaline environments [33,34]. Of these, while Euryarchaeota is an archae [35] and Rhodothermaeota is a halophilic bacterium that recently defined as a phylum [36]. These phyla were observed at a lower rate in pre-mucilage water than in clean water. Therefore, with the onset of mucilage formation, the rate of archaea and halophiles adapted to extreme conditions began to decline. Another phylum, Proteobacteria, was also significantly decreased in the mucilage. This seems to constitute an important part of the deterioration of marine hemostasis.
Chlamydia is a phylum known to live as intracellular parasites in different living organisims from humans to amoeba causing a variety of serious diseases in humans and animals [37]. In different studies carried out so far, chlamydia has been found in fresh water, sea water, wastewater, soil, rhizosphere, sediments, as well as in the host-associated microbiome (low in general) [38]. In freshwater environments, chlamydiae have been detected at a relative abundance of less than 0.1% and are a rare part of the biosphere, while it was reported to be more common in freshwater than salty water [39,40]. The relative abundance of chlamydia is generally higher in soils and It has also been reported that the rate affected from changing environmental conditions due to deforestation, changing pH and heavy metal pollution [41]. In this study Chlamydia rate was 0.006 in clean water, 0.004 in pre-mucilage water and 0.04 in mucilage samples. The rates obtained within this study seem to be similar to the rates in other studies reporting clean environmental waters. However, In the current study microorganisim belonging Chlamydia was statistically abundant in the mucilage metagenome. Thus, there was a statistically significant difference between clean water samples and mucilage samples. This significant 10-fold difference indicates that the mucilage structure increases the relative abundance of chlamydia and appears to promote positive selection.
Fusobacteria, is a bacterial phylum found in many different habitats. While it has a strong correlation with sand [42], it has been reported to be negatively associated with alluvium [43] in environmental samples. In a study conducted by Gutierrez et al. on the Deepwater Horizon (DWH) oil spill (accepted as the largest oil spill in US waters in 2010), it was reported that the sea surface of oil slicks contains obligate and facultative anaerobic taxa, including members of Fusobacteria [44]. Similar to this study, Fusobacteria in oil-contaminated sediments, deep-sea oil reservoirs, and bacteria found in other oil-contaminated areas thought to have hydrocarbon degrading properties of these bacteria. In the current study, Fusobacteria were found more in mucilage compared to clean water - just as in oil pollution-. Thus a layer that will create an anaerobic environment in the Marmara Sea migth have played a major role for this outcome. However, in the study by Gutierrez et al. the presence of Fusobacteria on the sea surface – this is a highly oxygenated environment- is intriguing. These organisms are found on the seafloor and it has been argued that there is a need for a rising carrier for a vertical transport along the water column from anaerobic/micro-aerophilic zones to the sea surface, and that this carrier is oil. Based on this hypothesis, it was determined that the carrier for Fusobacteria in Marmara Sea could be mucilage.
In the analyzes carried out at the species level in this study, except for one sample, generally marine microbiome members were found. Encountring the human pathogens such as E. faecalis A. baumanni in only one example (MS4) most probably due to sampling the mucilage from the point wehere somehow contaminated with a waste contating high concentration of human pathogens. This sample also contained far fewer Carbohydrate processing enzyme classes than the other mucilage samples. This is most probably due to having fewer environmental microorganism in its microbiome.
As a result of comparative examination, statistically significant results emerged for carbohydrate processing enzyme classes and biosynthetic gene clusters associated with secondary metabolytes between mucilage samples and clean waters. In mucilage formation, especially polysaccharide is an important component of the accumulation. As a result of biosynthetic gene cluster analysis, gene clusters associated with terpene production were found less in mucilage group then clean water. Apart from terpene production, several biosynthetic gene clusters have been identified those can also be at the marker level. In a study conducted by Del Negro et al, it was found that organic matter degradation/decomposition processes are reduced in aged mucilage and new unstable organic pathways originating from phytoplankton of bacterial metabolic pathways is operated for compounds [45]. Therefore, it can be speculated that the biosynthetic pathways such as the siderefor biosynthesis process are exploited for new unstable compounds.
5. Conclusions
As a result of the study, there was a significant difference between the mucilage microbiome and the clean water microbiome, the pre-mucilage water had more similar microbiome to mucilage than clean sea water. Thus, there might be a strong relationship between the deterioration of homeostatic balance in aquatic microbiome and mucilage. The high presence of unidentified or non-assignable reads in each mucilage samples, turned out to be composed of dark matter and mucilage metagenome is in question. With the current knowledge and technology, a full mapping of the mucilage microbiome appears to be difficult at present. However, taking into consideration of pre-mucilage water taxonomic content to follow its formation in stages may be beneficial for early warning approaches. Mucilage disposal strategies such as bacteriophage have been considered to tackle this problem. However, identification and culturing of the target bacteria is essential for bacteriopahge approches. In a largely unidentified habitat such as mucilage, it is hard to determine the keystone elements to be targeted. For this reason, it is important to focus on preventing its formation at this stage.
Although it was detected at very low relative abundance and no species identification was made, the Chlamydiae and Fusobacteria reads in mucilage samples were found to be statistically significantly higher than the clean water samples. In order to reveal the public health risk of mucilage in terms of these potential pathogens, It may be recommended to carry out cultural studies for targeting the pathogen species belonging to these phyla. In addition to that, oil contamination such as petroleum derivative migth have a positive contribution to mucilage formation.
Author Contributions
Conceptualization, AG, OUN and HA; methodology A.G., O.U.N., G.K., I.S., M.N.E, M.H. and H.A; investigation, A.G., G.K., I.S., M.N.E and M.H.; writing—original draft preparation, A.G.; writing-review and editing, A.G. and O.U.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by The Scientific and Technological Research Council of Turkey (TUBITAK) under the project number 121G128.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are stored on Erciyes University, Genome and Stem Cell Center, Metagenomics &Bioinformatics Laboratuary’s data repository and can be provided upon request from the corresponding author.
Acknowledgments
We would like to thank Prof. Dr. Gamze Yıldız from Bursa Uludağ University and Hydrologist Levent Artüz from MAREM (Marmara Environmental Monitoring) for providing two of the mucilage samples included in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- MacKenzie, L.; Sims, I.; Beuzenberg, V.; Gillespie, P. Mass accumulation of mucilage caused by dinoflagellate polysaccharide exudates in Tasmanian Bay, New Zealand. Harmful Algae. 2002, 1, 69–83. [Google Scholar] [CrossRef]
- Nikolaidis, G.; Aligizaki, K.; Koukaras, K.; Moschandreou, K. Mucilage phenomena in North Aegean Sea, Greece: another harmful effect of dinoflagellates? 12th International Conference on Harmful Algae, Copenhagen, Denmark, 2006, 219-222.
- Tüfekçi, V.; Balkis, N.; Beken, C.P.; Ediger, D.; Mantikci, M. Phytoplankton composition and environmental conditions of the mucilage event in the Sea of Marmara. Turk. J. Biol. 2010, 34(2), 199–210. [Google Scholar] [CrossRef]
- Danovaro, R.; Fonda, U.S.; Pusceddu, A. Climate Change and the Potential Spreading of Marine Mucilage and Microbial Pathogens in the Mediterranean Sea”. PLoS One. 2009, 4(9), e7006. [Google Scholar] [CrossRef] [PubMed]
- Liénart, C.; Susperregui, N.; Rouaud, V.; Cavalheiro, J.; David, V.; Del Amo, Y.; Duran, R.; Lauga, B.; Monperrus, M.; Pigot, T.; Bichon, S.; Charlier, K.; Savoye, N. Dynamics of particulate organic matter in a coastal system characterized by the occurrence of marine mucilage–A stable isotope study. J. Sea Res. 2016, 116, 12–22. [Google Scholar] [CrossRef]
- Tas, S.; Kus, D.; Yilmaz, I. N. Temporal variations in phytoplankton composition in the northeastern Sea of Marmara: potentially toxic species and mucilage event. Mediterr. Mar. Sci. 2020, 21(3), 668–683. [Google Scholar] [CrossRef]
- Frias-Lopez, J.; Shi, Y.; Tyson, G.W.; Coleman, M.L.; Schuster, S.C.; Chisholm, S.W.; DeLong, E.F. Microbial community gene expression in ocean surface waters. PNAS. 2008, 105(10), 3805–3810. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Stingl, U. Molecular diversity and ecology of microbial plankton. Nature. 2005, 437, 343–348. [Google Scholar] [CrossRef]
- Ozbayram, E.G.; Akcaalan, R.; Isinibilir, M.; Meric, A. Insights into the bacterial community structure of marine mucilage by metabarcoding. Environ Sci Pollut Res. 2022, 29, 53249–53258. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, S.; Yilmaz, D.K.; Celik, E.S.; Kucuker, M.A. Shotgun Metagenomic Analysis for Mucilage in the Surface Waters of The Çanakkale Strait (Dardanelles): Metabolic Diversity, Microbial Community Structure and Antibiotic Resistance Genes. J. Anatol. Environ Animal Sci. 2021, 6(4), 717–726. [Google Scholar] [CrossRef]
- Rouaud, V.; Susperregui, N.; Fahy, A.; Guyoneaud, R.; Bichon, S.; Liénart, C.; Amo, Y.D.; Savoye, N.; Guadin, P.; Duran, R.; Lauga, B. Dynamics of microbial communities across the three domains of life over an annual cycle with emphasis on marine mucilage in the Southern Bay of Biscay resolved by microbial fingerprinting. Cont. Shelf Res. 2019, 186, 127–137. [Google Scholar] [CrossRef]
- Luglié, A.; Satta, C.; Padedda, B.; Pulina, S.; Sechi, N. What is Chrysophaeum taylorii Lewis & Bryan doing in Sardinia (Tyrrhenian Sea, Mediterranean)? Harmful Algae News. 2008, 36, 4–6. [Google Scholar]
- Karadurmus, U.; Sari, M. Marine mucilage in the Sea of Marmara and its effects on the marine ecosystem: mass deaths. Turk. J. Zool. 2022, 46(1), 93–102. [Google Scholar]
- Kavzoglu, T.; Goral, M. Google Earth Engine for Monitoring Marine Mucilage: Izmit Bay in Spring 2021. Hydrology. 2022, 9(8), 135. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012, 28(11), 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. IEEE International Parallel and Distributed Processing Symposium (IPDPS). 2019, 314–324. [Google Scholar]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11(1), 1–11. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7(1), 1–9. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015, 12(1), 59–60. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res, 2009; 37, (suppl_1), D233-8. [Google Scholar]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.; Studholme, D.J. The Pfam protein families database. Nucleic Acids Res, 2004; 32, (suppl_1), D138-41. [Google Scholar]
- Andreu, V.P.; Augustijn, H.E.; van den Berg, K.; van der Hooft, J.J.; Fischbach, M.A.; Medema, M.H. BiG-MAP: an automated pipeline to profile metabolic gene cluster abundance and expression in microbiomes. bioRxiv. 2020.
- Bray, J.R.; Curtis, J.T. An ordination of upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 4, 325–349. [Google Scholar] [CrossRef]
- Wang, J.; Jiao, Y.; Ren, Y.; Xue, Y.; Ji, Y.; Xu, B. Comparative study on two computing methods for estimating Shannon-Wiener diversity index. J. Fish. China. 2015, 8, 1257–63. [Google Scholar]
- Fonda U. S, Ghirardelli E, Specchi M (1989) Gli episodi di “mare sporco” nell'Adriatico dal 1729 ai giorni nostri. 178 p. Regione Autonoma Friuli-Venezia Giulia. Direzione Regionale Ambiente (ed.).
- Vojvoda, J.; Lamy, D.; Sintes, E.; Garcia, J.A.; Turk, V.; Herndl, G.J. Seasonal variation in marine-snow-associated and ambient-water prokaryotic communities in the northern Adriatic Sea. Aquat. Microb. Ecol. 2014, 3, 221–224. [Google Scholar] [CrossRef]
- Kilias, E.S.; Peeken, I.; Metfies, K. Insight into protist diversity in Arctic sea ice and melt-pond aggregate obtained by pyrosequencing. Polar Res. 2014, 1, 23466. [Google Scholar] [CrossRef]
- Rath, J.; Wu, K.Y.; Herndl, G.J.; De Long, E.F. High phylogenetic diversity in a marine-snow-associated bacterial assemblage. Aquat. Microb. Ecol. 1998, 14, 261–269. [Google Scholar] [CrossRef]
- Canfora, L.; Bacci, G.; Pinzari, F.; Lo Papa, G.; Dazzi, C.; Benedetti, A. Salinity and bacterial diversity: to what extent does the concentration of salt affect the bacterial community in a saline soil? PLoS One. 2014, 9, e106662. [Google Scholar] [CrossRef] [PubMed]
- Hollister, E.B.; Engledow, A.S.; Hammett, A.J.M; Provin, T. L.; Wilkinson, H.H.; Gentry, T.J. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J. 2010, 4, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liu, J.-J.; Banerjee, S.; Zhou, N.; Zhao, Z.-Y.; Zhang, K.; Tian, C.-Y. Soil pH is equally important as salinity in shaping bacterial communities in saline soils under halophytic vegetation. Sci. Rep. 2018, 8, 4550. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Deng, Y.; Zhang, S.; Zhang, W.; Liu, J.; Xie, Y.; Zhang, X.; Huang, H. Prokaryotic community distribution along an ecological gradient of salinity in surface and subsurface saline soils. Sci. Rep. 2017, 7, 13332. [Google Scholar] [CrossRef]
- Fernández, A.B.; Vera-Gargallo, B.; Sánchez-Porro, C.; Ghai, R.; Papke, R.T.; Rodriguez-Valera, F.; Ventosa, A. Comparison of prokaryotic community structure from Mediterranean and Atlantic saltern concentrator ponds by a metagenomic approach. Front. Microbiol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Garrity, G.M.; Holt, J.G. Euryarchaeota phy. nov.". In Whitman WB (ed.). Bergey's Manual of Systematics of Archaea and Bacteria. John Wiley & Sons. 2015. ISBN 9781118960608. [CrossRef]
- Munoz, R.; Rossello-Mora, R.; Amann, R. Revised phylogeny of Bacteroidetes and proposal of sixteen new taxa and two new combinations including Rhodothermaeota phyl. nov"(vol 39, pg 491, 2016). Syst. Appl. Microbiol. 2017, 40, 3–190. [Google Scholar] [CrossRef]
- Benamri, I.; Azzouzi, M.; Sanak, K.; Moussa, A.; Radouani, F. An overview of genes and mutations associated with Chlamydiae species’ resistance to antibiotics. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 59. [Google Scholar] [CrossRef] [PubMed]
- Collingro, A.; Köstlbacher, S.; Horn, M. Chlamydiae in the Environment. Trends Microbiol. 2020, 11, 877–888. [Google Scholar] [CrossRef]
- Pizzetti, I.; Schulz, F.; Tyml, T.; Fuchs, B.M; Amann, R.; Horn, M.; Fazi, S. Chlamydial seasonal dynamics and isolation of 'Candidatus Neptunochlamydia vexilliferae' from a Tyrrhenian coastal lake. Environ. Microbiol. 2016, 18, 2405–2417. [Google Scholar] [CrossRef] [PubMed]
- Héry, M.; Volant, A.; Garing, C.; Luquot, L.; Poulichet, F.E.; Gouze, P. Diversity and geochemical structuring of bacterial communities along a salinity gradient in a carbonate aquifer subject to seawater intrusion. FEMS Microbiol. Ecol. 2014, 90, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Frenne, P.D.; Boon, N.; Brunet, J.; Cousins, S.A.O.; Decocq, G.; Kolb, A.; Lemke, I.; Liira, J.; Naaf, T.; Orczewska, A.; Plue, J.; Wulf, M.; Verheyen, K. Plant species identity and soil characteristics determine rhizosphere soil bacteria community composition in European temperate forests. FEMS Microbiol. Ecol. 2019, 95, fiz063. [Google Scholar] [CrossRef]
- Liu, M.; Li, X.; Zhu, R.; Chen, N.; Ding, L.; Chen, C. Vegetation richness, species identity and soil nutrients drive the shifts in soil bacterial communities during restoration process. Environ. Microbiol. Rep. 2021, 13, 411–424. [Google Scholar] [CrossRef]
- Zhai, W.; Zhang, L.; Liu, H.; Zhang, C.; Liu, D.; Wang, P.; Zhou, Z. Enantioselective degradation of prothioconazole in soil and the impacts on the enzymes and microbial community. Sci. Total Environ. 2022, 824, 153658. [Google Scholar] [CrossRef]
- Gutierrez, T.; Berry, D.; Teske, A.; Aitken, M.D. Enrichment of Fusobacteria in sea surface oil slicks from the deepwater horizon oil spill. Microorganisms. 2016, 3, 24. [Google Scholar] [CrossRef]
- Del Negro, P.; Crevatin, E.; Larato, C.; Ferrari, C.; Totti, C.; Pompei, M.; Giani, M.; Bero, D.; Umani, S.F. Mucilage microcosms. Sci. Total Environ 2005, 353((1-3)), 258–269. [Google Scholar] [CrossRef]
Figure 1.
Plots created over alpha diversity value, a. plot created at the species level, b. plot created at the phylum level.
Figure 1.
Plots created over alpha diversity value, a. plot created at the species level, b. plot created at the phylum level.
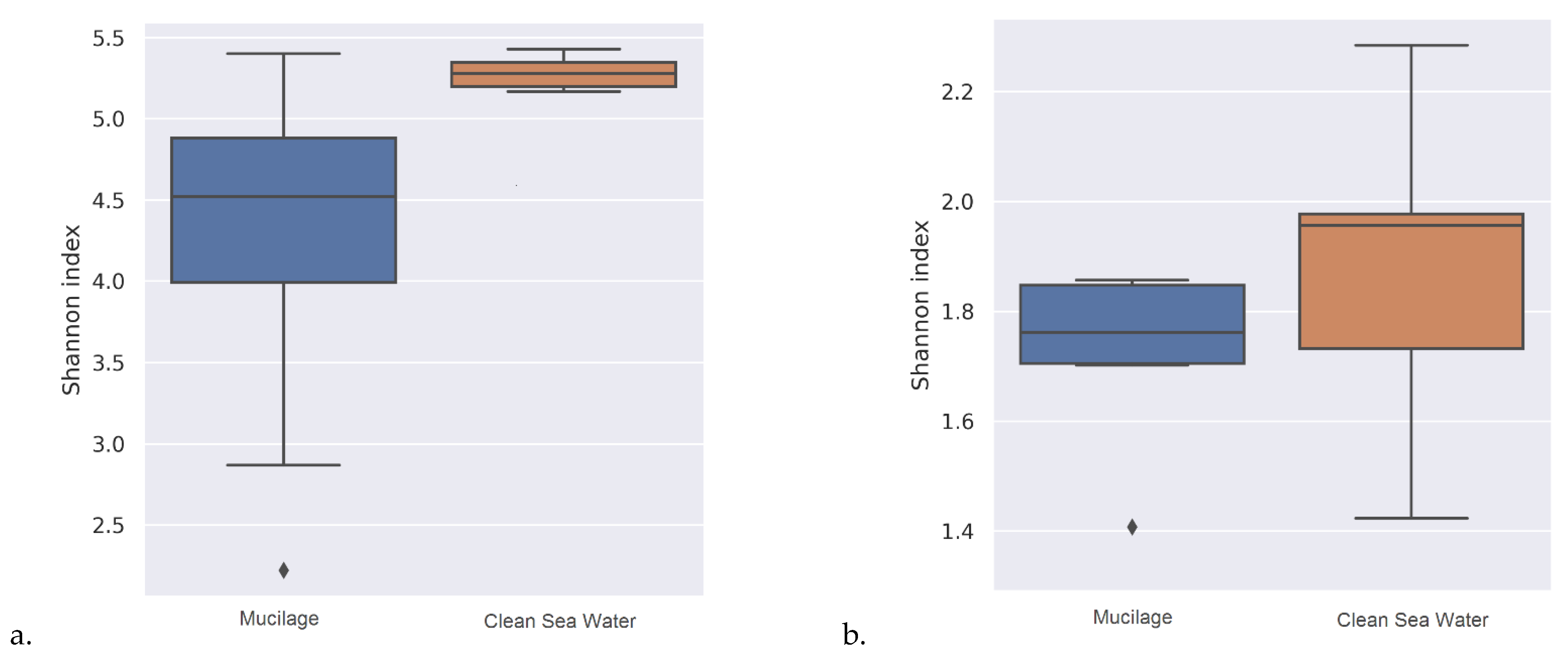
Figure 2.
Plot of principal component analysis (PCA) generated at the phylum level.
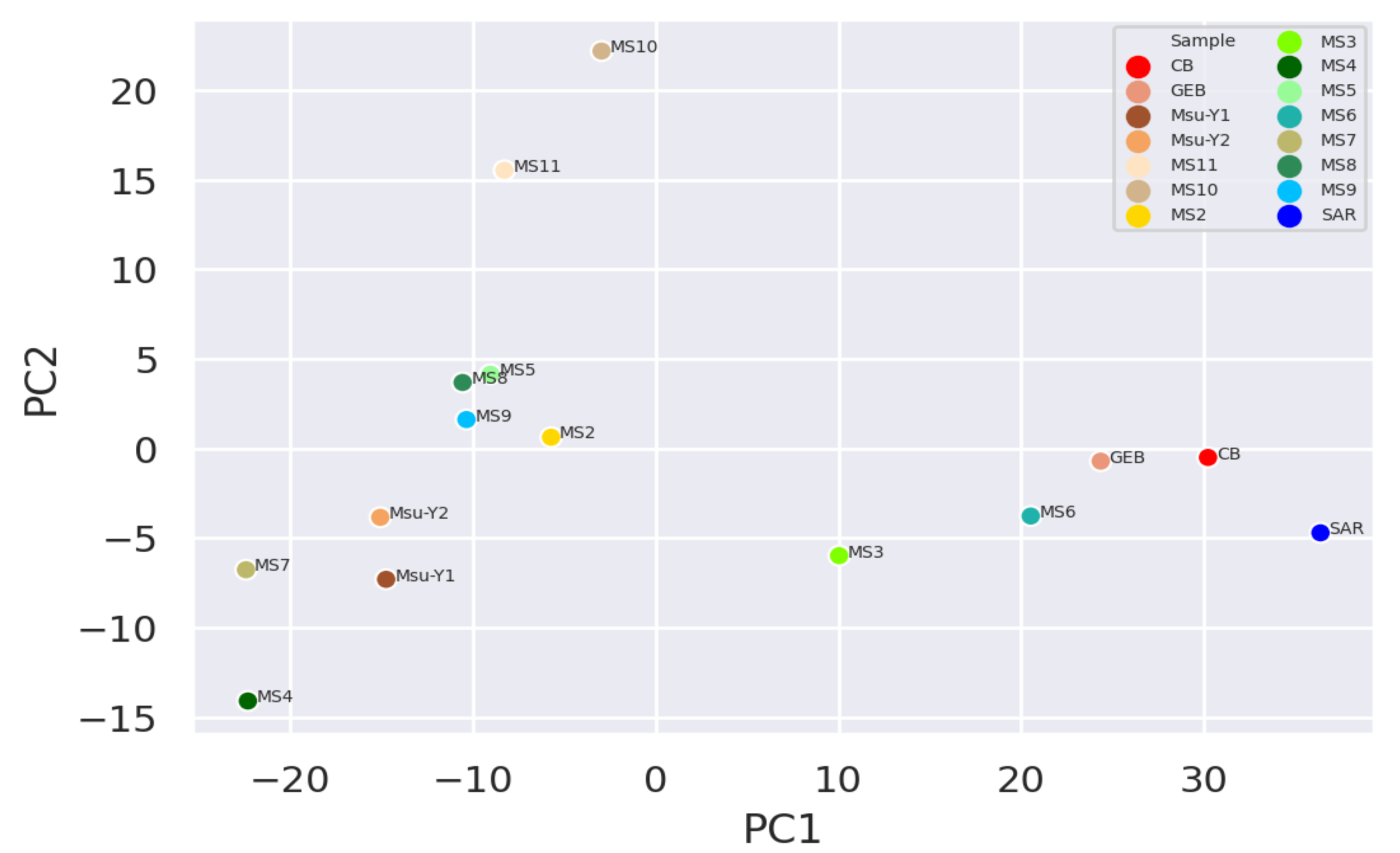
Figure 3.
Plot of principal component analysis (PCA) generated at the species level.
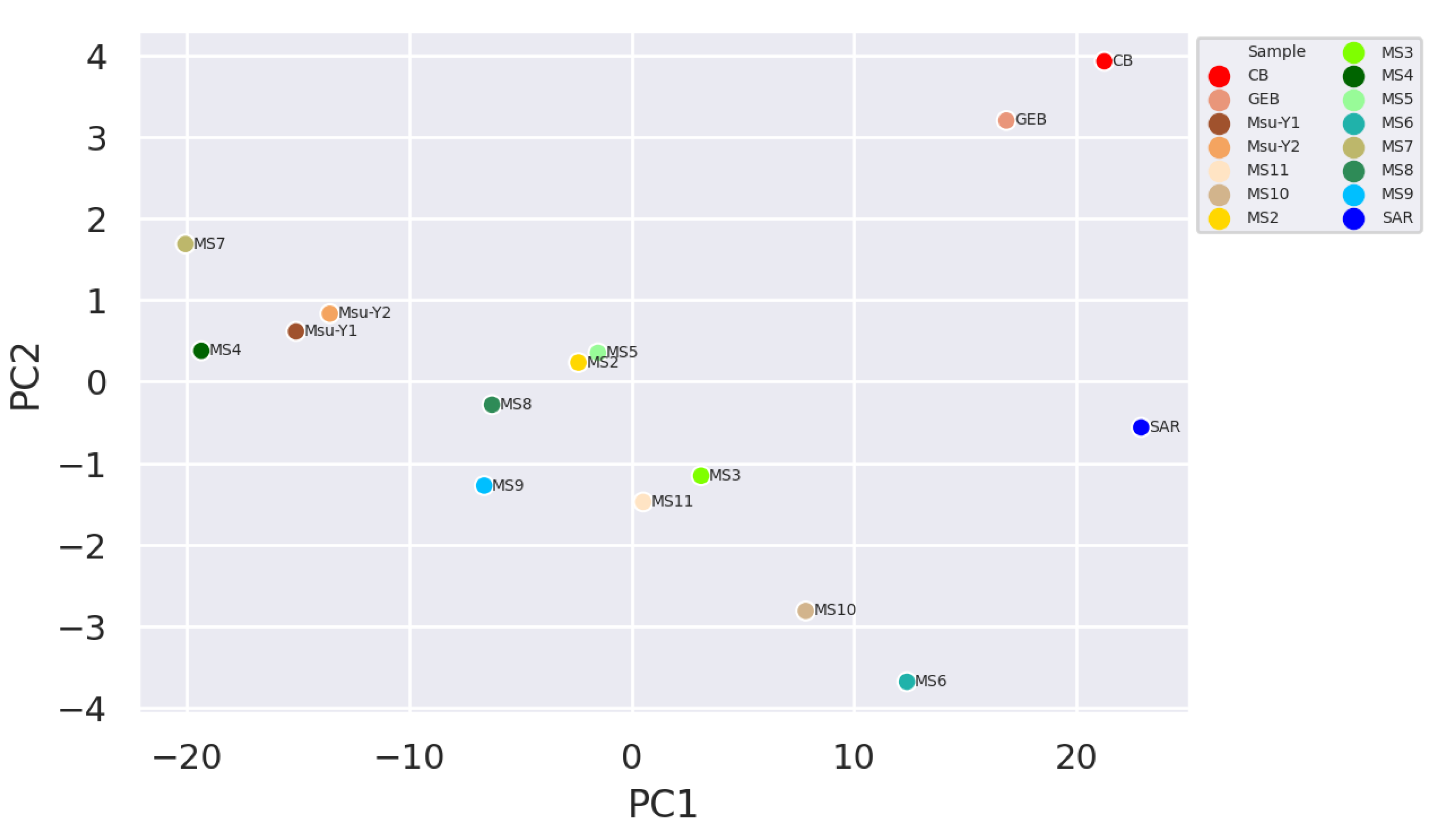
Figure 5.
Class-based distribution of biosynthetic gene clusters (BiG-Mex) per sample.
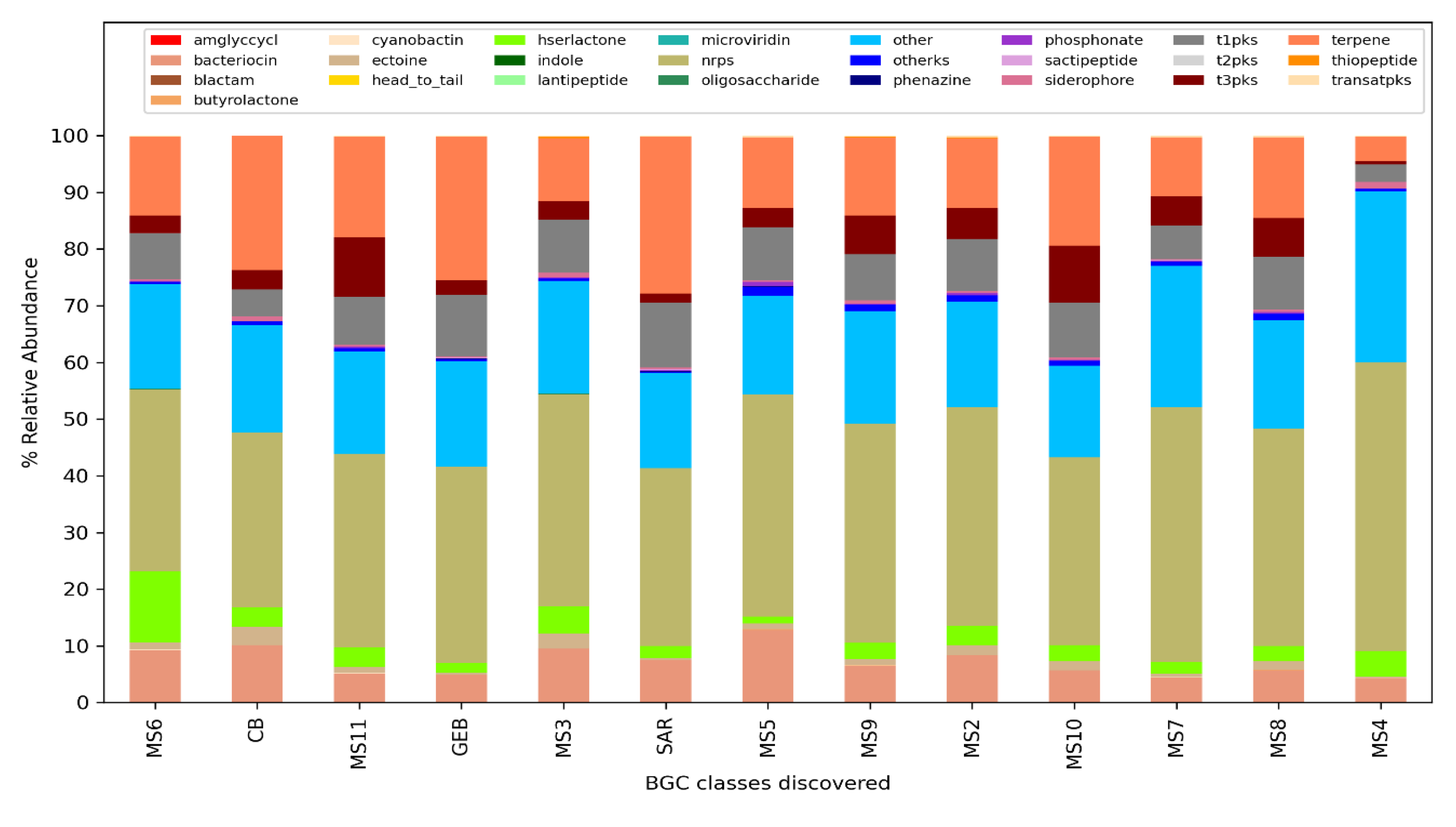
Figure 6.
Domain-based distribution of Biosynthetic gene clusters (BiG-Mex) per sample.
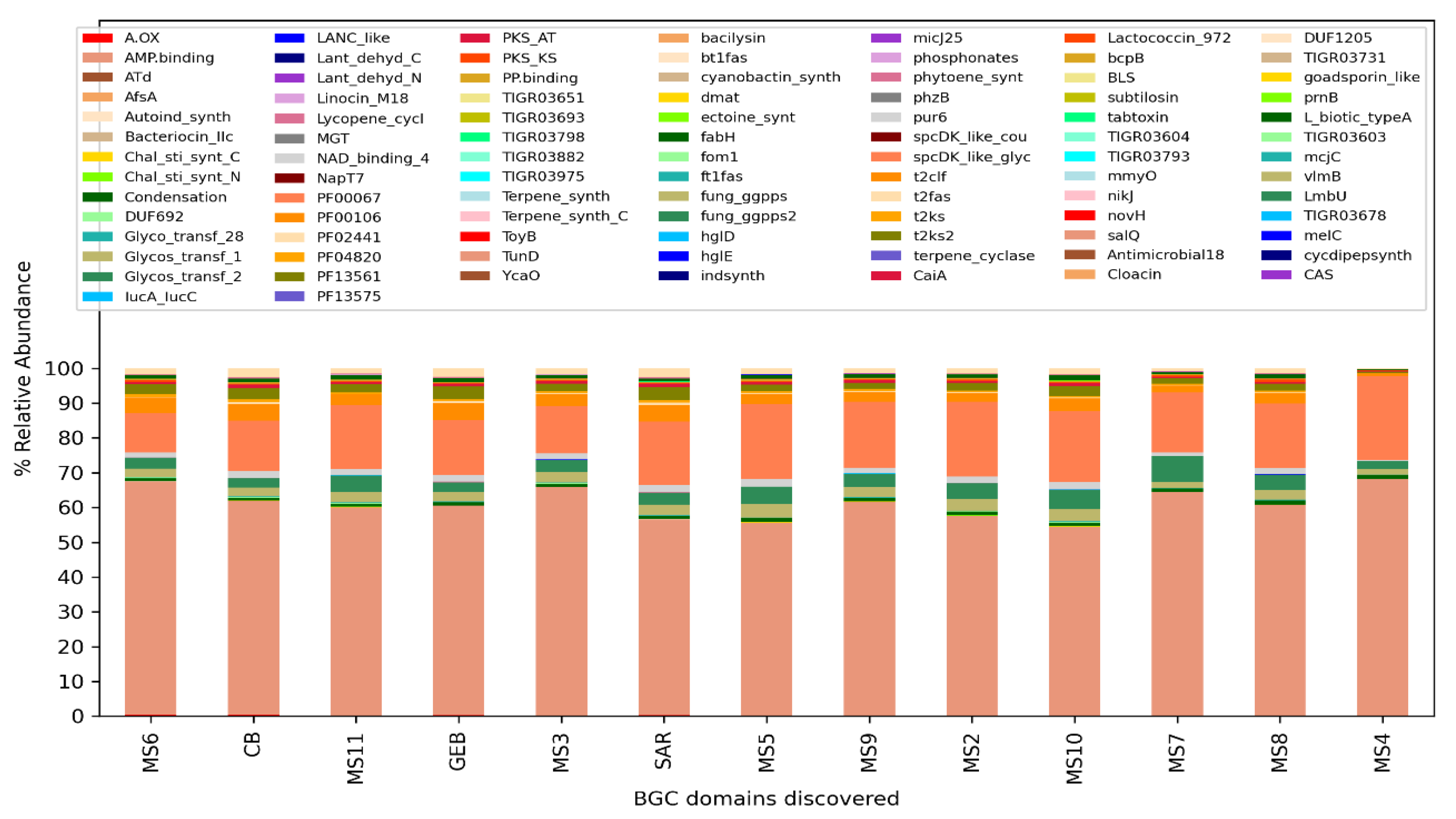

Table 1.
Statistics of sequencing data.
| Sample | Reads | Contigs | N50* | Longest Contig | Average Contig Length | Total Contig Length | N80** |
|---|---|---|---|---|---|---|---|
| MS2 | 21.984.724 | 240.447 | 799 | 88.951 | 697 | 167.812.811 | 426 |
| MS3 | 63.881.640 | 650.720 | 1257 | 442.235 | 940 | 612.212.260 | 537 |
| MS4 | 22.691.910 | 28.712 | 377 | 12.301 | 364 | 10.470.825 | 320 |
| MS5 | 21.714.280 | 249.062 | 823 | 251.373 | 744 | 185.410.840 | 441 |
| MS6 | 23.347.442 | 244.907 | 982 | 107.527 | 823 | 201.558.903 | 479 |
| MS7 | 22.656.394 | 223.788 | 940 | 215.273 | 787 | 176.135.579 | 465 |
| MS8 | 78.719.956 | 795.630 | 1169 | 373.301 | 889 | 707.541.004 | 510 |
| MS9 | 21.926.708 | 203.865 | 1411 | 233.250 | 969 | 197.667.564 | 528 |
| MS10 | 21.991.972 | 302.937 | 670 | 123.372 | 661 | 200.402.569 | 408 |
| MS11 | 24.276.176 | 287.548 | 754 | 73.798 | 685 | 197.096.919 | 422 |
| Msu_Y1 | 27.062.660 | 278.911 | 687 | 6.1121 | 646 | 180.243.434 | 405 |
| Msu_Y2 | 24.864.772 | 314.331 | 583 | 78.131 | 582 | 183.115.265 | 384 |
| GEB | 11.592.104 | 132.904 | 1838 | 219.676 | 1.108 | 147.321.065 | 614 |
| SAR | 8.759.208 | 110.834 | 1005 | 66.910 | 812 | 90.087.134 | 497 |
| CB | 2.040.918 | 37.702 | 953 | 41.650 | 835 | 31.507.945 | 506 |
* When all Contig lengths are listed, the contig length corresponding to the 50th percentile; ** When all Contig lengths are listed, the contig length corresponding to the 80th percentile.
Table 2.
The top five microbial species found in the samples.
| MS2 | Verrucomicrobiaceae bacterium TMED86 | Oceaniferula marina | Lentibacter algarum | Sulfurospirillum arcachonense | Polaribacter porphyrae |
| MS3 | Pseudodesulfovibrio sp. JC047 | Pseudodesulfovibrio sp. zrk46 | Winogradskyella aurantiaca | Hyphomonas sp. Mor2 | Marivita lacus |
| MS4 | Acinetobacter baumannii | Escherichia coli | Bacillus thuringiensis | Pseudomonas viridiflava | Enterococcus faecalis |
| MS5 | Winogradskyella sp. KYW1333 | Candidatus Izimaplasma sp. HR1 | Verrucomicrobiaceae bacterium | Bacteroidetes bacterium | Gammaproteobacteria bacterium |
| MS6 | Sulfitobacter geojensis | Lentilitoribacter sp. Alg239-R112 | Planctomycetes bacterium | Ascidiaceihabitans donghaensis | Sulfitobacter mediterraneus |
| MS7 | Glaciecola sp. KUL10 | Puniceicoccaceae bacterium | Bacteroidetes bacterium | Candidatus Endolissoclinum sp. TMED26 | Marivita lacus |
| MS8 | Bacteroidetes bacterium | Glaciecola sp. KUL10 | Psychroserpens mesophilus | Puniceicoccaceae bacterium | Acidiluteibacter ferrifornacis |
| MS9 | Glaciecola sp. KUL10 | Bacteroidetes bacterium | Roseivirga pacifica | Polaribacter sp. MED152 | Puniceicoccaceae bacterium |
| MS10 | Psychroserpens mesophilus | Bacteroidetes bacterium | Hyphomonas sp. Mor2 | Congregibacter litoralis | gamma proteobacterium NOR5-3 |
| MS11 | Polaribacter vadi | Bacteroidetes bacterium | Nonlabens spongiae | Polaribacter reichenbachii | Polaribacter sp. MED152 |
| Msu_Y1 | Marivita lacus | Bacteroidetes bacterium | Glaciecola sp. KUL10 | Puniceicoccaceae bacterium | Gammaproteobacteria bacterium |
| Msu_Y2 | Bacteroidetes bacterium | Marivita lacus | Glaciecola sp. KUL10 | Puniceicoccaceae bacterium | Gammaproteobacteria bacterium |
| GEB | Planktomarina temperata | Rhodobacteraceae bacterium SB2 | marine gamma proteobacterium HTCC2080 | Halieaceae bacterium | Candidatus Pelagibacter ubique |
| SAR | Alteromonadaceae bacterium TMED101 | Halieaceae bacterium | Litoricolaceae bacterium | Rhodobacteraceae bacterium SB2 | Oceanospirillales bacterium TMED91 |
| CB | Planktomarina temperata | Lentibacter algarum | Rhodobacteraceae bacterium SB2 | Bacteroidetes bacterium | Halieaceae bacterium |
Table 3.
Alpha and beta diversity values.
| Alpha Diversity | P-values |
| Species | 0,0188 |
| Phylum | 0,17 |
| Beta Diversity | |
| Species | 0,000024 |
| Phylum | 0,00164 |
Table 4.
| Phylum | average read ratio found in mucilage samples (%) | average read ratio found in clean water samples (%) |
|---|---|---|
| Proteobacteria | 20.8±9.6 | 47.7±4.1 |
| Bacteroidetes | 13.5±8.8 | 6.9±1.7 |
| Verrucomicrobia | 1.7±1.5 | 2.9±1.01 |
| Planctomycetes | 1.2±1 | 0.33±0.2 |
| Cyanobacteria | 0.5±0.4 | 1.1±0.3 |
| Rhodothermaeota | 0.015±0.012 | 0.4±0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



