
Submitted:
30 March 2023
Posted:
31 March 2023
Read the latest preprint version here
Abstract
Sesquiterpene lactones (STLs) are a large group of terpenoids most commonly found in plants of the Asteraceae family, e. g. in chicory plants, displaying a wide range of interesting biological activities. However, further studies on the biological potential of chicory derived STLs and analogues are challenging as only four of these molecules are commercially available (as analytical standards), and to date there are no published or patented simple extraction-purification processes capable of large scale STLs isolation. In this work we describe a novel three-step large scale extraction and purification method for the simultaneous purification of 11,13-dihydrolactucin (DHLc) and lactucin (Lc) starting from a chicory genotype rich in these STLs and in the corresponding glycosyl and oxalyl conjugated forms. After a small-scale screening on 100 mg of freeze-dried chicory root powder, the best results were achieved with a 17 h water maceration at 30°C. With these conditions we managed to increase the content of DHLc and Lc, at the same time favouring the hydrolysis of their conjugated forms. On a larger scale, the extraction of 750 g of freeze-dried chicory root powder, followed by a liquid-liquid extraction step and a reversed-phase chromatography, allowed the recovery of 642.3 ± 76.3 mg of DHLc and 175.3 ± 32.9 mg of Lc. The two pure STLs were subsequently used in the context of semi-synthesis to generate analogues for biological evaluation as antibacterial agents. In addition, other described chicory STLs that are not commercially available were also synthesized or extracted to serve as analytical standards for the study. In particular, lactucin-oxalate and 11,13-dihydrolactucin-oxalate were synthesized in two steps starting from Lc and DHLc, respectively. On the other hand, 11β,13-dihydrolactucin-glycoside was obtained after a MeOH/H2O (70/30) extraction, followed by a liquid-liquid extraction step and a reversed-phase chromatography. Together, this work will help facilitate the evaluation of the biological potential of chicory derived STLs and their semisynthetic analogues.
Keywords:
chicory
; sesquiterpene lactones
; 11
; 13-dihydrolactucin
; lactucin
; plant extraction
; semi-synthesis of analytical standards
1. Introduction
Sesquiterpene lactones (STLs) are a large group of naturally occurring terpenoids characterized by a fifteen-carbon (C15) backbone. Commonly found in plants like chicory and other members of the Asteraceae family, these molecules are accumulated in their leaves and roots as specialized metabolites[1]. Among the identified compounds, lactucin (Lc), lactucopicrin (Lp), 8-deoxylactucin and the corresponding 11β,13-dihydro (DH) derivatives were the principal ones found[2]. Depending on chicory species, previous studies also reported the presence of 15-glycosyl, as well as 15-oxalates conjugates[3] (Table 1).
These specialized metabolites can protect the plant acting as anti-herbivory and antimicrobials or inhibiting growth of competing plants[4]. In addition, these molecules are also well known to be responsible for the bitter taste of chicory[2].
The wide range of biological activities exhibited by STLs is mainly attributed to the 𝛼-methylene-𝛾-lactone group present in their structure[5]. In particular, if present, this moiety can act as a Michael acceptor and react with nucleophiles (sulfhydryl or amino groups) in enzymes, transcription factors, and other proteins, alkylating them irreversibly[6].
Of particular interest is that several biological activities have been reported for some chicory derived STLs. For instance, antimicrobial and antifungal activities were described for lactucopicrin, which showed an interesting activity against both S. aureus and P. aeruginosa, and for 11β,13-dihydrolactucin, which showed a promising activity against P. aeruginosa and five different strains of Candida[7]. Furthermore, an anti-adipogenesis effect both in vivo and in vitro and an anticancer activity were reported for lactucin[8,9]. Bischoff et al. found lactucin and lactucopicrin able to prevent the growth of the HB3 clone of strain Honduras-1 of Plasmodium falciparum[10]. Moreover, lactucin, 11β,13-dihydrolactucin and lactucopicrin were evaluated for analgesic and sedative properties in mice by Wesołowska et al. and the three compounds showed analgesic effects in both the two models of nociception used[11]. In addition, Rojas-Silva P et al. reported the first findings of leishmanicidal activity of bitter STLs isolated from chicory roots[12]. Recently, lactucin and lactucopicrin were also evaluated as acetylcholinesterase inhibitors[13].
Nevertheless, further studies on the biological potential of chicory derived STLs and semi-synthetic analogues are challenging as only four of these molecules (Lc, DHLc, Lp, DHLp) are commercially available (as analytical standards), and to date, there are no published or patented simple extraction-purification processes capable of large scale STLs isolation.
On this basis, we focused our attention on 11β,13-dihydrolactucin and lactucin large-scale extraction. Indeed, the presence of two hydroxyl groups, an exocyclic double bond and a ketone in their structure make these two STLs an interesting starting point for the semi-synthesis of other chicory derived STLs (expensive or not commercially available) and original potentially bioactive analogues.
Our presented work describes a novel three-step large scale extraction and purification method for the simultaneous purification of both 11β,13-dihydrolactucin and lactucin starting from a chicory genotype rich in the corresponding conjugated forms, i.e., 11β,13-dihydrolactucin-glycoside (DHLc-gly), 11β,13-dihydrolactucin-oxalate (DHLC-ox) and lactucin-oxalate (Lc-ox). The process is simple, cheap and characterized by a limited number of steps and solvents involved (ultra-pure water, ethyl acetate, acetonitrile) (Figure 1). Eventually, the two extracted STLs were subsequently used in the context of semi-synthesis to obtain analogues for biological evaluation as antibacterial agents. In addition, other chicory STLs that are not commercially available were also synthesized to serve as analytical standards, including DHLc-ox and Lc-ox.
2. Results
2.1. Identification of the Main STLs
At the beginning of our study, before the screening of different parameters to find the best conditions for DHLc and Lc isolation, we wanted to confirm the identity of the two targeted STLs and their conjugated forms by LC-QTOF-HRMS analysis. Back then, a different chicory genotype richer in DHLc and Lc was available for the experiment. A 70 minute-gradient allowed a nice separation of the main STLs present in chicory root and showed the rich and complex profile of a water chicory extract (Figure 2).
The diagnostic fragment ions obtained for the five molecules in positive ionization mode were in accordance with the ones reported in the literature[3,14] (Table 2). DHLc and Lc showed [M+H]+ ions at m/z 279 and 277, respectively and the [M+Na]+ adduct was the most abundant. DHLc-gly showed [M+H]+ ion at m/z 441 and the [M+Na]+ adduct was the most abundant. DHLc-ox and Lc-ox showed [M+H]+ ions at m/z 351 and 349, respectively. For DHLc-ox, the molecular ion [M+H]+ was the most abundant. For Lc-ox, the fragment corresponding to the loss of oxaloyl and a molecule of water was the most abundant.
2.2. Method Development
2.2.1. Extraction Conditions Determination
Considered the high quantity of conjugated STLs, i.e., DHLc-glycoside, DHLc-oxalate and Lc-oxalate, present in our chicory genotype, the aim of our study was to find suitable extraction conditions leading to high concentrations of DHLc and Lc, the two targeted molecules, at the same time favouring the hydrolysis of their conjugated forms (Figure 3). Indeed, oxalyl and glycosyl functionalities can undergo chemical hydrolysis following the addition of a water molecule under different pH conditions. Moreover, we assumed that an enzymatic hydrolysis could also take place during the extraction following the action of specific endogenous chicory root enzymes. Interestingly, in support of our theory, few studies in the literature suggested the instability of these conjugated forms after treatment with enzymes or acidified solvents, making difficult their identification in chicory roots extracts[3,14].
With this goal in mind, we planned to study the impact of different extraction solvents, times and temperatures on the level of the five STLs. At the beginning of our study, only commercially available DHLc and Lc were accessible as analytical standards. Therefore, to choose between different extraction conditions, the peak areas of the five STLs measured by UPLC-DAD analysis were compared. Later on, after DHLc-gly extraction-purification (paragraph 2.5) and the synthesis of DHLc-ox and Lc-ox (paragraph 2.6), five calibration curves were performed and STLs concentrations after different extraction conditions were measured and are reported in Table 3 (means of three samples ± standards deviation).
We started our study performing a maceration with the conventional solvents found in the literature. In general, given their hydrophilic features, higher concentrations of the two free STLs were measured using 100% of water (0.050 mM ± 0.003 for DHLc and 0.024 mM ± 0.002 for Lc). With pure methanol and ethanol, the quantity of STLs significantly decreased (0.005 mM ± 0 for DHLc and 0.009 mM ± 0 for Lc) (Figure 4 A and B). Methanol/water mixtures efficiently extracted conjugated STLs (Figure 4 B), but led to a decrease in DHLc and Lc content (Figure 4 A). For instance, the use of water led to an increase of the extraction yield by a factor of 2 for free STLs and no increase for conjugated STLs compared to the use of MeOH/H2O 50/50 mixtures. Moreover, the water extraction avoided the presence of polyphenols such as 3-caffeoylquinic acid (3-CQA) and 3,5-dicaffeoylquinic acid (3,5-DiCQA) whose raw material is rich, normally extracted by alcoholic solvents[15,16,17,18]. These molecules can coelute with the targeted STLs, making the purification step more challenging (Figure S1, supplementary information).
After the selection of water as the best extraction solvent, the effect of the extraction temperature on the levels of the two free molecules and their conjugated forms was studied. Overall, the concentrations of the two free STLs were found higher when lower temperatures were chosen. In particular, from 30°C to 50°C, the levels of DHLc increased by a factor of 1.5 (from 0.051 mM ± 0.001 to 0.076 mM ± 0.001) (Figure 4 C). This phenomenon can be partially explained by the concomitant hydrolysis of DHLc-gly, which decreased by a factor of 1.15 (from 0.550 mM ± 0.006 to 0.478 mM ± 0.010) (Figure 4 D). On the contrary, in this temperature range, Lc, Lc-ox and DHLc-ox concentrations did not increase. Surprisingly, from 60°C to 80°C, DHLc-gly level increased by a factor of 1.08 (from 0.565 mM ± 0.013 to 0.612 mM ± 0.005) and DHLc level decreased by a factor of 1.64 (from 0.041 mM ± 0 to 0.025 mM ± 0). From 90°C to 100°C, a decrease in DHLc-gly and Lc-ox content was found. In contrast, Lc and DHLc concentrations, probably because of the hydrolysis of the corresponding conjugated STLs, started to increase. Despite the interesting levels of Lc observed at 100°C (increased by a factor of 2.12 compared to 30°C), these conditions would be too energy-consuming for the scale up of the process. Therefore, 30°C or 50°C were considered to be a good compromise for DHLc and Lc extraction and selected for further studies.
At last, the effect of the extraction time was studied at 50°C (Figure 4 E and F) and 30°C (Figure 4 G and H). The collected data showed that the concentrations of the two free STLs increased with the extraction time, with a maximum at 17h (increased by a factor of 10 and 5 at 30°C and 50°C, respectively, for the sum of DHLc and Lc). On the contrary, the levels of conjugated molecules dramatically decreased. In particular, compared to 50°C, 30°C allowed a greater hydrolysis of the conjugated STLs (decrease by a factor of 12 and a factor of 2 at 30°C and 50°C, respectively). In addition to favouring the hydrolysis of the conjugated forms, the increasing extraction time could also play a role on the extraction efficiency, allowing the solvent to longer penetrate the plant cell walls.
In conclusion, a 17 h maceration by water at 30°C represented the best condition for DHLc and Lc extraction and for the concomitant hydrolysis of their conjugated forms and was selected for further studies and for the scale-up of the extraction method.
2.3. Scaling-up
1) Water Extraction. The extraction conditions determined on a small scale (water, 17 h, 30°C, 1/10 ratio, w/v) were selected for the scaling up phase. After the extraction of 750 g of chicory root powder and the centrifugation step, the supernatant was evaporated to directly obtain a crude extract to be purified.
2) Ethyl-acetate Extraction. Because of the large amount of sugars present in chicory roots (e.g., inulin, glucose, fructose, sucrose), the concentrated sample was caramel-like, very difficult to handle and to use in the further purification step. Therefore, we planned to introduce a liquid-liquid extraction step with a suitable organic solvent. Ethyl acetate, together with chloroform, butanol and hexanol were tested. As the collected data show, after three ethyl acetate extractions almost all 11β,13-dihydrolactucin and lactucin passed in the organic phase (recovery rates of 98.8% and 94.8% for DHLc and Lc, respectively). In this way, the crude extract to be purified was poor in sugars and rich in the STLs of interest.
3) Reversed-chromatography. As for the analytic scale, a Phenyl Butyl (PHC4) reversed-phase was used for the purification step. This functionalized silica is very selective for mid-polar compounds and can tolerate up to 100% of water. Its availability also as a flash column allowed an easy transfer of the separation method. The two targeted molecules, together with a mixture of less polar STLs, were obtained. After a freeze-drying step, 642.3 ± 76,3 mg of DHLc and 175.3 ± 32.9 mg of Lc were obtained (means of three extraction-purification cycles), corresponding to a yield of 0.86 ± 0.10 and 0.23 ± 0.04 mg/g dry matter.

Table 4.
Content of DHLc and Lc after three ethyl-acetate extractions. Values are means (± standard deviations) of three samples.
Table 4.
Content of DHLc and Lc after three ethyl-acetate extractions. Values are means (± standard deviations) of three samples.
| STLs Concentration (mM) ± SD | ||
| DHLc | Lc | |
| Initial H2O | 0.571 ± 0.066 | 0.134 ± 0.005 |
| EtOAc 1 | 0.345 ± 0.010 | 0.081 ± 0.002 |
| EtOAc 2 | 0.153 ± 0.004 | 0.033 ± 0.003 |
| EtOAc 3 | 0.066 ± 0.000 | 0.013 ± 0.011 |
| FinalH2O | 0.004 ± 0.016 | 0.005 ± 0.003 |
2.4. Analytical Validation of STLs
After flash-chromatography purification, one-dimensional (1H-NMR and 13C-NMR) and two-dimensional (HMBC, HSQC-DEPT) NMR experiments in DMSO confirmed the structures of the two isolated STLs. Our chemical shifts and assignments are reported in Table 5 and were consistent with the ones reported in the literature[19,20].
Table 5.
1H and 13C NMR data for DHLc and Lc.
| DHLc: (CD3)2SO | Lc: (CD3)2SO | |||
| Position | 1H (mult, J Hz) | 13C | 1H (mult, J Hz) | 13C |
| 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 |
- - 6.27 (s) - 3.76–3.64 (m) 3.76–3.64 (m) 2.17–2.05 (m) 3.61–3.49 (m) 9: 2.74–2.55 (m) 9’: 2.24 (dd, 13.6, 1.8) - 2.74–2.55 (m) - 1.25 (d, 7.0) 2.33 (s 15: 4.64 (ddd, 18.8, 5.8, 1.9) 15’: 4.22 (ddd, 18.9, 4.9, 1.5) |
132.0 194.9 132.5 175.8 48.4 80.9 60.5 68.4 48.7 147.5 40.9 178.3 15.7 21.5 61.8 |
- - 6.28 (d, 1.0) - 3.83–3.67 (m) 3.83–3.67 (m) 3.12–3.03 (m) 3.83–3.67 (m) 9: 2.74–2.55 (m) 9’: 2.24 (dd, 13.6, 1.8) - - - 13: 6.13 (dd, 3.0, 1.5) 13’: 6.01 (dd, 3.2, 1.5) 2.33 (s 15: 4.66 (d, 18.7) 15’: 4.25 (d, 18.8) |
132.3 194.8 132.7 169.2 48.3 81.1 56.8 66.9 48.8 147.0 138.4 175.4 121.9 21.5 61.8 |
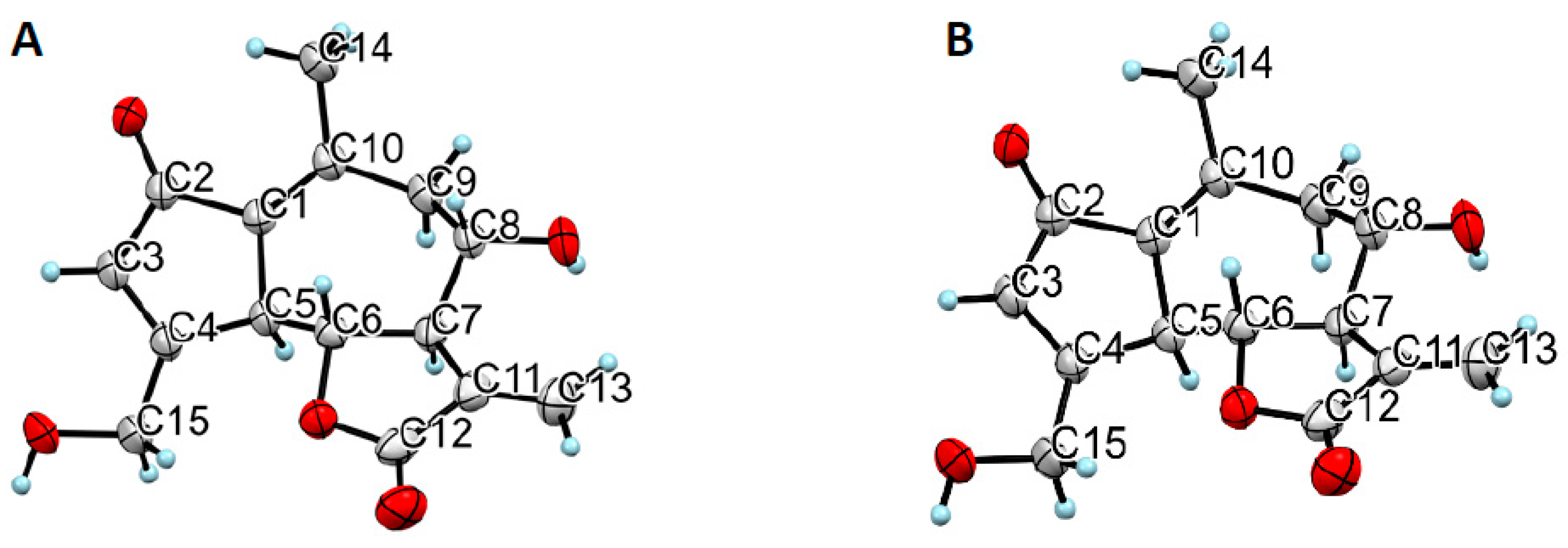
Moreover, crystallographic studies were performed on single crystals to establish the configuration of the chiral centres and to assure that no racemization occurred during the extraction-purification process. For both DHLc and Lc, the configurations found were consistent with the ones reported in literature[21,22]. The absolute configurations were assigned to be 5S,6R,7R,8S,11S for DHLc and 5S,6R,7R,8S for Lc. In the case of DHLc, one molecule of water of crystallization was present.
Figure 5.
ORTEP drawing of both 11β,13-dihydrolactucin·H2O (A) and lactucin (B).
2.5. Isolation of 11β,13-dihydrolactucin-glycoside
As mentioned above, to determine the content of the main STLs involved in our study after different extraction conditions, we planned to have the five standards available. To the best of our knowledge, 11β,13-dihydrolactucin-glycoside is not commercially available. Therefore, an extraction-purification process was developed. Contrary to the conditions to isolate DHLc and Lc, a short extraction time was chosen to limit the hydrolysis of conjugated STLs, including DHLc-gly, to the corresponding free forms. In addition, to limit the presence of sugars, a MeOH/H2O (70/30, v/v) mixture was chosen. Moreover, to reduce the presence of inulin in the water phase, a precipitation step was introduced after the extraction. Ethanol, well known in the literature, was used[23]. This time, after the liquid-liquid extraction step, the water phase, rich in conjugated STLs, was kept and freeze-dried. After a reversed-phase chromatography, 11β,13-dihydrolactucin-glycoside was obtained as a white powder (13 mg, corresponding to a yield of 3.07 mg/g dry matter).
2.6. Synthesis of STLs standards
Given the low extraction yields and the instability of the conjugated STLs during the extraction process, we thought that a synthetic strategy towards DHLc-ox and Lc-ox would be interesting. Again, to the best of our knowledge, these two conjugated STLs are not commercially available. Starting from extracted DHLc and Lc, 11β,13-dihydrolactucin-oxalate and lactucin-oxalate were synthesized in two steps (Figure 6).
The first one consists in the acylation of the primary alcohol of DHLc/Lc using equimolar quantities of methyl 2-chloro-2-oxo-acetate in the presence of a base (Et3N) in acetonitrile (ACN) for DHLc (64% yield) and in N,N-Dimethylformamide (DMF) for Lc (80% yield). After optimization, to limit the formation of the di-acylated undesired product, no catalyst was used (i.e., DMAP) and the acyl chloride was diluted and added dropwise at 0°C to the reaction mixture. In this way, the more reactive primary alcohol can selectively react. For the synthesis of Lc methyl-oxalate, DMF was chosen as the reaction solvent given the poor solubility of Lc in ACN. The second step consists in the hydrolysis of the obtained methyl-oxalate derivative with LiOH, leading to the formation of the two lithium salts of DHLc-ox and Lc-ox which were subsequently purified by reversed-phase preparative HPLC (DHLc: 21% yield and Lc: 16% yield). Unfortunately, the yields of this second step were low because of the concomitant hydrolysis of the oxalate group and the formation of the starting DHLc and Lc. Therefore, to improve the reaction yield, the optimization of this second step would be required.
3. Discussion
In our study, we developed a three-step process for the large-scale extraction and purification of 11β,13-dihydrolactucin and lactucin, starting from a chicory genotype rich in their corresponding conjugated forms. As previously mentioned, these molecules have a pharmacological interest, together with a nutritional (“bittering agents”) and analytical interest (HPLC-UV standards).
Our work addresses the need of producing, at low cost and in a simple manner, these expensive commercially available molecules (40,000 € for one gram of 11β,13-dihydrolactucin[24]). Indeed, to date, there are no publications or patents describing an extraction-purification process that simultaneously targets the two sesquiterpene lactones with a significant productive capacity. The oldest works focus on the discovery and identification of STLs[2,25], while the most recent works look at the biological activity of isolated molecules and/or chicory extracts[7,26]. Overall, these studies do not prioritize the STLs quantity produced. A few patents focus on lactucin isolation; however, these processes involve a large number of extractions and fractionation steps and a multitude of solvents[27,28,29,30].
In our three-step process, starting from freeze-dried and grounded chicory roots, an extraction by aqueous maceration is carried out under conditions favourable to the hydrolysis of conjugated STLs towards the two targeted molecules. Eventually, the two molecules and the less polar STLs fraction are obtained simultaneously after a liquid-liquid extraction step followed flash chromatography purification (purity greater than 95%).
Given the existence of more efficient techniques, such as supercritical fluid extractions (SFEs) or continuous extractions, we are aware that the water maceration can be seen as the limiting step of the process, not leading to the best productive capacity. Nevertheless, we think that it represents a really simple and convenient technique to extract important quantities of DHLc and Lc at a laboratory scale which eventually allows a semi-synthetic approach and further structure-activity relationship studies. However, it would be interesting to try to optimize this step by assisting the maceration with ultrasounds.
The strength of the process is represented by the long extraction time, which leads to the hydrolysis of the conjugated STLs and higher concentrations of targeted DHLc and Lc. Interestingly, in the literature, previous studies suggested the instability of oxalyl and gyclosyl STLs. For instance, Graziani et al. stated that the conversion of glycosides and oxalates to their free forms can be induced by treating a chicory extract with enzymes or acidified methanol[3]. Moreover, Sessa et al. showed that the principal conjugates in Lactuca species were not glycosides as frequently reported in earlier studies, but oxalates. The group suggested that in earlier research these conjugate forms may not have been recovered because of their degradation during the isolation[14]. However, to the best of our knowledge, our study represents the first case where the hydrolysis of conjugated STLs is achieved and observed without the use of enzymes or acidified solvents.
Thanks to the large quantities isolated, the two STLs were subsequently used in the context of semi-synthesis to generate other chicory STLs which are not commercially available (DHLc-ox and Lc-ox). Moreover, several 11β,13-dihydrolactucin and lactucin analogues were synthesized and will be initially tested for their antibacterial properties. Given this global health challenge[31], our study can open the way to the discovery of new promising hits to fight against antibacterial resistance. In addition, our work can open the way to the cultivation of industrial chicory as a source of interesting bioactive molecules.
4. Materials and Methods
4.1. Plant Material
Field-grown chicory roots selected for their high-STLs-content genotype were provided by the company Florimond-Desprez (Cappelle-en-Pévèle, France). The corresponding plants were cultivated in 2020 (Coutiches, France) and the roots were harvested in November from field plot as a pool of 60 roots. They were washed with cold water, cut into slices (robot coupe CL-52, Quiétalis, Tourcoing, France) and freeze-dried (SP VirTis 25L Genesis, BTF, Diemoz, France). The dried slices were then ground in two steps, with the use of a speed blender in a 1L stainless steel container (Waring blender, Fisher Scientific, Illkirch, France) followed by an ultra-centrifugal mill (ZM 200 Retsch™, Eragny sur Oise, France) to obtain a fine and homogeneous particle size. The resulting powder was stored at -20°C.
4.2. Reagents and Chemicals
The reagents for the synthesis and the solvents (LC-MS grade) were purchased from commercial suppliers (Fisher Scientific, Sigma Aldrich) and used without further purification. Ultra-pure water was obtained from a Milli-Q system (Milli-Q® Advantage A10). The standards, 11β,13-dihydrolactucin and lactucin, were provided by Extrasynthèse (Genay, France).
4.3. Method Development
4.3.1. Extraction of STLs From Freeze-dried Chicory Roots
Extractions were carried out by maceration with the desired solvent (solid-liquid ratio of 1/10 (g Powder/mL solvent)) on 100 mg of chicory roots using a laboratory shaker (Eppendorf ThermoMixer C, Hamburg, Germany) and an agitation speed of 1000 rpm. The effect of different solvents, temperatures and times was studied. The solvents used were ultra-pure water, water/methanol mixtures (50/50 and 80/20, v/v), ethanol and methanol. For this first investigation, an extraction temperature of 30°C and an extraction time of 15 min were chosen. Then, the effect of the extraction temperature was studied in the range from 30°C to 100°C using ultra-pure water and an extraction time of 15 min. Ultimately, the extraction time was studied in the range from 15 min to 17 h. For each studied parameter, three repetitions were done. At the end of each extraction, following a centrifugation step (3 min, RCF = 48), the supernatant was separated from the powder deposit and filtered through a 45 µm suitable filter: 0.45 µm PP Whatman UNIFILTER microplate (Cytiva, VWR, Rosny-sous-Bois, France) for aqueous filtration, and a syringe-driven filter unit with a polytetrafluoroethylene membrane for organic solvents. Samples were taken to be injected into a UPLC-DAD system to determine STLs content.
4.3.2. Scaling-up
4.3.2.1. Selective Fractionation of Free STLs and Removal of Sugars by Liquid-liquid Extraction
100 mg of chicory root powder were extracted by water maceration as previously mentioned. Three repetitions were done. After the centrifugation step (3 min, RCF = 48), 100 µL of the supernatant were kept aside for the following HPLC-DAD analysis, while 500 µL of the supernatant were extracted with ethyl acetate (500 µL). The liquid-liquid extraction step was carried out using a laboratory shaker (Eppendorf ThermoMixer C, Hamburg, Germany), for 10 min at 30°C and 1000 rpm. The two layers were separated and the ethyl acetate phase (450 µL) was transferred into another tube and evaporated under vacuum. The residue obtained was then re-dissolved in the same volume of a water/methanol mixture (50/50, v/v) and kept aside for UPLC-DAD analysis. The remaining water layer was extracted two more times with ethyl acetate following the same procedure. At the end of the experiment, five samples for each repetition were analysed: the initial filtered water phase, the three ethyl acetate phases and the final filtered water phase (after the three extractions).
4.3.2.2. The Standardized Big-scale Process
Extractions were carried out in a temperature-controlled shaker (Gerhardt THO 500/1, Les Essarts Le Roi, France) and three plastic bins of 5 L were used. 250 g of chicory powder were weighted in each bin, suspended in 2.5 L of ultra-pure water and agitated for 17 h at 30°C. After the extraction, the liquid was combined and centrifuged (5 min, RCF = 20335). 6 L of supernatant were collected and concentrated using an industrial rotavapor (BUCHI Rotavapor® R-220 Pro). The remaining liquid (~ 1 L) was then extracted three times with ethyl acetate. The liquid-liquid extraction was carried out in centrifuge bottles of 1 L. After shaking, a centrifugation step was performed (5 minutes, 20335*g). The ethyl acetate phases were then combined (~ 3 L), dried over anhydrous magnesium sulphate and concentrated under reduced pressure. Finally, a crude extract of 3-4 grams was obtained. Two equivalents of celite were then added to prepare the solid deposit for the following reversed-phase chromatography. The obtained powder was loaded into an 80 g PHC4 15µm F0080 flash column and purified using an Interchim® PuriFlash 4250 system (Interchim, Montluçon France) with 0.1% (v/v) formic acid in acetonitrile (B) and 0.1% (v/v) formic acid in water (A) as the mobile phase. The gradient elution was as follows: isocratic at 10% B for 40 minutes; linear from 10% to 100% B in 2 min; isocratic at 100% B for 3 min; linear from 100% to 50% B in 2 min and isocratic at 50% B for 3 min. After a freeze-drying step, pure 11β,13-dihydrolactucin and lactucin were obtained as white powders. DHLc: HRMS (TOF, ES+) m/z [M+H]+: calcd. for C15H18O5 279.1232, found 279.1226. Lc: HRMS (TOF, ES+) m/z [M+H]+: calcd. for C15H16O5 277.1076, found 277.1068.
4.4.1. 11β,13-. dihydrolactucin-glycoside Isolation
40 g of dried chicory root powder were weighted in a glass bottle of 1 L and extracted with a mixture of methanol/water (70/30, v/v, solid-liquid ratio of 1/10 (g Powder/mL solvent)) in a temperature-controlled shaker (Eppendorf ThermoMixer C, Hamburg, Germany) at 30°C for 1 hour. After the extraction, the liquid was centrifuged (5 min, RCF = 20335*g). Ethanol (40%) was added to the supernatant and the mixture was left to stand overnight at a temperature of 4°C. A liquid-liquid extraction step with ethyl acetate was then carried out as previously described. The water phase was recovered, freeze-dried and a solid deposit was prepared as already indicated. 11β,13-dihydrolactucin-glycoside was obtained after reversed-phase chromatography using a 25 g PHC4 15µm F0025 flash column and an Interchim® PuriFlash 4250 system with 0.1% (v/v) formic acid in acetonitrile (B) and 0.1% (v/v) formic acid in water (A) as the mobile phase. The gradient elution was as follows: isocratic at 1% B for 7 min; linear from 1 to 10% B in 11 min; isocratic at 10% B for 17 min; linear from 10 to 37% B in 15 min; linear from 37 to 100% B in 5 min; isocratic at 100% B for 3 min; linear from 100 to 50% B in 2 min and isocratic at 50% B for 3 min. 1H NMR (300 MHz, DMSO) δ 6.57 (s, 1H), 5.22 (m, 2H), 4.97-4.91 (m, 2H), 4.67-4.57 (m, 2H), 4.53-4.48 (m, 1H), 4.23 (d, J = 7.7 Hz, 1H), 3.76-3.72 (m, 2H), 3.69-3.64 (m, 1H), 3.59-3.53 (m, 1H), 3.48-3.41 (m, 1H), 3.19-2.99 (m, 4H), 2.73-2.59 (m, 2H), 2.34 (s, 3H), 2.25 (dd, J = 13.0, 1.3 Hz, 1H), 2.16-2.09 (m, 1H), 1.25 (d, J = 6.9 Hz, 3H). 13C NMR (75 MHz, DMSO) δ 195.01, 178.28, 170.56, 147.84, 133.50, 132.20, 103.28, 80.85, 77.29, 76.85, 74.03, 70.33, 68.39, 68.32, 61.35, 60.43, 48.80, 48.54, 40.99, 21.55, 15.72. HRMS (TOF, ES+) m/z [M+H]+: calcd. for C21H28O10 441.1761, found 441.1758.
4.5. STLs Analysis
4.5.1. Quantification of STLs by UPLC-DAD
STLs content was determined using an Ultimate 3000RS system equipped with an LPG-3400RS pump, a WPS-3000TRS autosampler and a DAD-3000RS (Thermo Fisher Scientific, Waltham, MA, USA). Chromatographic separation was achieved on an Uptisphere Strategy PHC4 column (3 µm, 150 x 3 mm: Interchim, Montluçon, France) at 45 °C with 0.1% (v/v) formic acid in acetonitrile (B) and 0.1% (v/v) formic acid in water (A) as mobile phases at flow rate of 0,700 mL/min. The gradient elution was as follows: linear from 6 to 17.5% B in 14 min; linear from 17.5 to 85% B in 1 min; isocratic at 85% B for 2 min; linear from 85 to 6% B in 1 min; and isocratic at 6% B for 6 min. The injection volume was 5.0 μL. The quantification was performed by external calibration using standards (DHLc-gly, DHLc-ox, DHLc, Lc-ox, Lc). The respective calibration curves were constructed by linear regression plotting signal area versus compound concentration.
4.5.2. LC-QTOF-HRMS Analysis of STLs
STLs were identified using an Ultimate 3000RS system equipped with a DAD-3000RS (Thermo Fisher Scientific, Waltham, MA, USA), interfaced with a high-resolution quadrupole time-of-flight mass spectrometer and equipped with an ESI source (Impact II, Bruker Daltonik GmbH, Bremen, Germany). 100 mg of freeze-dried chicory root powder were extracted with 1 mL of ultra-pure water for 1 h at 30°C at a speed of 1000 rpm (Eppendorf ThermoMixer C, Hamburg, Germany). Following a centrifugation step (3 min, RCF = 48*g), the supernatant was filtered through a 0.45 µm PP Whatman UNIFILTER microplate (Cytiva, VWR, Rosny-sous-Bois, France) and transferred to a vial for LC-MS analysis. Chromatographic separation was achieved on an Uptisphere Strategy PHC4 column (2.2 µm, 150 x 3 mm; Interchim, Montluçon, France) at 45°C with 0.1% (v/v) formic acid in acetonitrile (B) and 0.1% (v/v) formic acid in water (A) as the mobile phase at a flow rate of 0.5 mL/min. The gradient elution was as follows: isocratic at 2% B for 1 min; linear from 2 to 50% B in 32 min; linear from 50 to 100% B in 5 min; isocratic at 100% B for 7 min; linear from 100 to 2% B in 5 min; and isocratic at 2% B for 7 min. The injection volume was 2.0 μL. The mass spectrometer was operated in full spectral acquisition mode, in the positive mode at 4,5 kV with the following parameters: nebulizer gas (N2), 2.4 Bar; dry gas (N2), 10 mL/min; dry heater, 250 °C; collision cell energy, 11.0 eV; end plate offset, 500 V. The acquisition was performed in full scan mode in the 90–1200 m/z range with an acquisition rate of 1 Hz and with mass accuracy < 5 ppm. Software Compass OtofControl 4.1 was used for the operation of the mass spectrometer and for data acquisition. Data were processed by the software Compass DataAnalysis 4.4 (Bruker Daltonik GmbH, Bremen, Germany).
4.5.3. NMR analysis
NMR spectra were recorded on a Bruker® Avance-300 spectrometer. The results were calibrated to signals from the solvent as an internal reference [e.g., 2.50 (residual DMSO d6) and 39.52 (DMSO d6) ppm for 1H and 13C NMR spectra, respectively]. Chemical shifts (δ) are in parts per million (ppm) downfield from tetramethylsilane (TMS). The assignments were made using one-dimensional (1D) 1H and 13C spectra and two-dimensional (2D) HSQC-DEPT and HMBC spectra. NMR coupling constants (J) are reported in Hertz (Hz) and splitting patterns are indicated as follows: s (singlet), brs (broad singlet), d (doublet), dd (doublet of doublet), ddd (double of doublet of doublet), m (multiplet).
4.5.4. X-ray Structural Determination
Single-crystal X-ray crystallographic analyses were conducted on 11β,13-dihydrolactucin and lactucin. Diffraction data were obtained using a combination of phi and omega -scans on a Bruker Apex DUO diffractometer equipped with a Photon 3 hybrid pixel area detector, mounted on a four-circle D8 goniometer. Cu Kα radiation (λ = 1.54178 Å) was obtained using an ImuS Incoatec Cu microfocus sealed tube. Data solution was found using SHELXT[32] and refined using SHELXL[33], as implemented in the Olex 2 crystallographic suite for small molecules[34].
4.6. Chemistry
Progress of the chemical reactions was monitored by thin layer chromatography (TLC) and/or by Ultra Performance Liquid Chromatography—Mass Spectrometry (UPLC-MS). TLC was performed using Merck® commercial aluminium sheets coated with silica gel 60 F254. Visualization was achieved by fluorescence quenching under UV light at 254 nm, 215 nm or stained by potassium permanganate and bromocresol green. Purifications were performed by reversed-phase flash chromatography on prepacked columns (Macherey-Nagel® Chromabond) under pressure using a Combiflash® C18 Rf200 instrument and by preparative HPLC Buchi Pure C-380 on an OmniSphere C18 Dynamax (10 µm, 250 mm × 41.4 mm) column. Products were detected by UV absorption at 215 nm and 254 nm. UPLC-MS analysis was performed on LC-MS Waters ACQUITY UPLC I-Class system equipped with a UPLC I BIN SOL MGR solvent manager, a UPLC I SMP MGR-FTN sample manager, an ACQUITY UPLC I-Class eK PDA Detector photodiode array detector (210–400 nm) and an ACQUITY QDa (Performance) as mass detector (full scan ESI+/- in the range 30–1250). Acquity BEH C18 column (1.7 μm, 50 mm × 2.1 mm) was used for UPLC analysis. The injection volume was 0.5 μL. For a 5 min analysis, the elution was done at pH 3.8 from 100% H2O/0.1% ammonium formate to 2% H2O/98% ACN/0.1% ammonium formate over 3.5 min. A flow rate at 600 µL/min was used. For a 30 min analysis, the elution was done at pH 3.8 from 100% H2O/0.1% ammonium formate to 100% ACN/0.1% ammonium formate over 25 min. A flow rate at 600 µL/min was used. HRMS analysis were performed on a LCT Premier XE Micromass, using a Waters C18 X-Bridge (3.5 μm, 50 mm x 4.6 mm). A gradient starting from 98% H2O 5 mM ammonium formate pH = 3.8 and reaching 100% ACN 5 mM ammonium formate pH = 3.8 within 3 min at a flow rate of 1 mL/min was used.
4.6.1. Chemical Synthesis
4.6.1.1. General Procedure for the Synthesis of DHLc-Me-oxalate and Lc-Me-oxalate
In a 25 mL round bottom flask, 1 eq. of STL was dissolved either in dry ACN (for DHLc) or DMF (for Lc) (0.18 M). Then, 2 eq. of Et3N were added. After cooling to 0°C, a solution of methyl 2-chloro-2-oxo-acetate (1 eq.) in dry ACN or DMF (0.18 M) was added dropwise over 5 minutes under argon atmosphere. The mixture was stirred for 2 h at 0°C under argon. Then, 0.5 eq. of Et3N were added, followed by a solution of methyl 2-chloro-2-oxo-acetate (0.25 eq.) in dry ACN or DMF (0.18 M). After completion, the reaction mixture was evaporated under reduced pressure. The following purification by reversed-phase chromatography afforded the desired compounds.
DHLc-Me-oxalate was obtained as a white powder after purification by reversed-phase chromatography on a 15 g-cartridge, with a gradient from 90% of H2O to 30/70 of ACN/H2O (210 mg, 64% yield). 1H NMR (300 MHz, DMSO) δ 6.35 (d, J = 1.1 Hz, 1H, H-3), 5.42 (dd, J = 17.4, 1.6 Hz, 1H, H-15), 5.25 (d, J = 5.3 Hz, 1H, OH-8), 5.05 (d, J = 16.7 Hz, 1H, H-15’), 3.89–3.73 (m, 5H, H-5, H-6, H-18), 3.63–3.51 (m, 1H, H-8), 2.76–2.60 (m, 2H, H-11), 2.34 (s, 3H, H-14), 2.28 (dd, J = 13.6, 1.8 Hz, 1H, H-9), 2.20–2.08 (m, 1H, H-7), 1.26 (d, J = 7.0 Hz, 3H, H-13). 13C NMR (75 MHz, DMSO) δ 193.81 (Cq, C-2), 177.54 (Cq, C-12), 165.73 (Cq, C-4), 157.07 (Cq, C-17), 156.26 (Cq, C-16), 148.52 (Cq, C-10), 132.69 (CH, C-3), 131.17 (Cq, C-1), 80.07 (CH, C-6), 67.72 (CH, C-8), 64.83 (CH2, C-15), 59.84 (CH, C-7), 53.38 (CH3-C18), 48.34 (CH2, C-9), 48.11 (CH, C-5), 40.42 (CH, C-11), 21.18 (CH3, C-14), 15.17 (CH3, C-13).
Lc-Me-oxalate was obtained as a white powder after purification by reversed-phase flash chromatography on a 4 g-cartridge, with a gradient from 90% of H2O to 30/70 of ACN/H2O (53 mg, 80%). 1H NMR (300 MHz, DMSO) δ 6.37 (d, J = 1.3 Hz, 1H, H-3), 6.15 (dd, J = 3.0, 1.4 Hz, 1H, H-13), 6.04 (dd, J = 3.2, 1.4 Hz, 1H, H-13’), 5.51 (d, J = 5.4 Hz, 1H, OH-8), 5.43 (dd, J = 17.3, 1.6 Hz, 1H, H-15), 5.09 (d, J = 16.8 Hz, 1H, H-15’), 3.98 (d, J = 10.0 Hz, 1H, H-5), 3.87–3.70 (m, 5H, H-6, H-8, H-18), 3.16–3.06 (m, 1H, H-7), 2.78 (dd, J = 13.5, 10.7 Hz, 1H, H-9), 2.34 (s, 3H, H-14), 2.35–2.28 (m, 1H, H-9). 13C NMR (75 MHz, DMSO) δ 194.26 (Cq, C-2), 169.03 (Cq, C-12), 165.86 (Cq, C-4), 157.59 (Cq, C-17), 156.78 (Cq, C-16), 148.62 (Cq, C-10), 138.24 (Cq, C-11), 133.48 (CH, C-3), 131.94 (Cq, C-1), 122.25 (CH2, C-13), 80.88 (CH, C-6), 66.83 (CH, C-8), 65.30 (CH2, C-15), 56.63 (CH, C-7), 53.90 (CH3-C18), 48.92 (CH2, C-9), 48.58 (CH, C-5), 21.74 (CH3, C-14).
4.6.1.2. General Procedure for the Synthesis of DHLc-oxalate and Lc-oxalate
A 10 ml round bottom flask was charged with 1 eq. of STL-methyl-oxalate, 1:1 THF:H2O (1M) and a magnetic stir bar under ambient atmosphere. After cooling the biphasic mixture to 0°C, 1 eq. of LiOH (aq.) 0.5 M was added dropwise. The mixture was then vigorously stirred at 0°C for 10 minutes. The presence of the acid was confirmed by LC-MS and TLC staining with bromocresol green (blue spot). The crude was then extracted six times with ethyl acetate. The water layer was freeze-dried and the pure oxalates were obtained after purification by reversed-phase preparative HPLC.
DHLc-oxalate was obtained as a white powder after purification by reversed-phase preparative HPLC, with a gradient from 100% H2O (0.1% HCOOH) to 50% ACN (0.1% HCOOH) in 25 min (30mg, 21%). 1H NMR (300 MHz, DMSO) δ 6.27 (d, J = 1.2 Hz, 1H, H-3), 5.37 (dd, J = 5.4, 1.6 Hz, 1H, H-15), 5.23 (bs, 1H, OH-8), 5.02 (d, J = 17.3 Hz, 1H, H-15), 3.84 (d, J = 10.0 Hz, 1H, H-5), 3.80–3.75 (m, 1H, H-6), 3.60–3.54 (m, 1H, H-8), 2.72 (dd, J = 13.5, 10.9 Hz, 1H, H-9’), 2.68–2.62 (m, 1H, H-11), 2.34 (s, 3H, H-14), 2.27 (dd, J = 13.6, 1.8 Hz, 1H, H-9), 2.20–2.12 (m, 1H, H-7), 1.26 (d, J = 7.0 Hz, 3H, H-13). 13C NMR (75 MHz, DMSO) δ 194.28 (Cq, C-2), 178.06 (Cq, C-12), 166.61 (Cq, C-4), 159.27 (Cq, C-16/17), 149.04 (Cq, C-10), 133.04 (CH, C-3), 131.69 (Cq, C-1), 80.60 (CH, C-6), 68.25 (CH, C-8), 64.75 (CH2, C-15), 60.37 (CH, C-7), 48.86 (CH2, C-9), 48.66 (CH, C-5), 40.94 (CH, C-11), 21.70 (CH3, C-14), 15.70, (CH3, C-13). HRMS (TOF, ES+) m/z [M+H]+: calcd. for C17H18O8 351.1080, found 351.1074.
Lc-oxalate was obtained as a white powder after purification by reversed-phase preparative HPLC, with a gradient from 100% H2O (0.1% HCOOH) to 30% ACN (0.1% HCOOH) in 25 min (12.3 mg, 16%). 1H N(300 MHz, DMSO) δ 6.30 (s, 1H, H-3), 6.16 (d, J = 1.5 Hz, 1H, H-13), 6.04 (d, J = 1.8 Hz, 1H, H-13’), 5.53 (bs, 1H, OH-8), 5.37 (d, J = 17.1 Hz, 1H, H-15), 5.03 (d, J = 17.4 Hz, 1H, H-15’), 3.98 (d, J = 10.0 Hz, 1H, H-5), 3.85–3.80 (m, 1H, H-6), 3.78–3.73 (m, 1H, H-8), 3.16-3.09 (m, 1H, H-7), 2.79 (dd, J = 13.5, 10.7 Hz, 1H, H-9’), 2.36 (s, 3H, H-14), 2.33 (dd, J = 13.8, 1.8 Hz, 1H, H-9). 13C NMR (75 MHz, DMSO) δ 194.25 (Cq, C-2), 169.06 (Cq, C-12), 166.54 (Cq, C-4), 160.32 (Cq, C-17), 159.76 (Cq, C-16), 148.58 (Cq, C-10), 138.26 (Cq, C-11), 133.25 (CH, C-3), 131.96 (Cq, C-1), 122.23 (CH2, C-13), 80.90 (CH, C-6), 66.85 (CH, C-8), 64.33 (CH2, C-15), 56.66 (CH, C-7), 48.90 (CH2, C-9), 48.60 (CH, C-5), 21.75 (CH3, C-14). HRMS (TOF, ES+) m/z [M+H]+: calcd. for C17H16O8 349.0923, found 349.0918.
4.7. Statistical Assays
All extractions were performed in triplicate. Data are expressed as the means ± standard deviations of each extraction condition. The assumptions of normality, homogeneity of variance and independence were checked by Shapiro-Wilk test, Bartlett test, and Durbin-Watson test, respectively. Significant differences of different extraction conditions on total free and conjugated STLs concentrations were determined using ANOVA followed by Tukey’s or based on the marginal means post-hoc test with adjustment by Holm’s or Bonferroni method. The significance threshold was set to 0.05 for all statistical tests. All analyses were done with R Statistics 4.2.2. (R Core Team (2017). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/.)
5. Conclusions
The experiments carried out in this paper have shown that the optimal extraction operating conditions to increase the content of 11β,13-dihydrolactucin and lactucin were obtained with a 17h water maceration at a temperature of 30°C. With these conditions, starting from a chicory genotype rich in conjugated STLs, we managed to decrease their concentration, favouring the liberation of the corresponding free STLs. This result was obtained without employing enzymes or acidified solvents as previously reported in the literature[3,25]. On this basis, a big scale process was developed and allowed us to obtain 642.3 ± 76.3 mg of DHLc and 175.3 ± 32.9 mg of Lc (means of three extraction-purification cycles). To date, these two STLs can be bought only from two providers (Extrasynthese and Phytolab) and are really expensive (€40,000 for one gram of 11β,13-dihydrolactucin). Our process is simple, economical and characterized by a limited number of steps (3 main) and solvents involved (ultra-pure water, ethyl acetate, acetonitrile). The two extracted molecules were used in the context of semi-synthesis to generate potential antibacterial analogues. Moreover, 11β,13-dihydrolactucin-oxalate and lactucin-oxalate were synthesized and used as analytical standards. 11β,13-dihydrolactucin-glycoside was extracted from chicory roots and also used as analytical standard. Additional studies considering the nature of the hydrolysis of conjugated STLs (enzymatic or chemical) and the peculiar behaviour of STLs towards different extraction temperatures would be interesting.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, NMR spectra of DHLc, Lc, DHLc-Me-oxalate, Lc-Me-oxalate, DHLc-oxalate, Lc-oxalate, DHLc-glycoside; Figure S1: HPLC profiles (254 and 320 nm) of a water/methanol 50/50 and pure water extracts and different content in 3-CQA; Figure S2: Base peak chromatogram (BPC, All − MS) of a water chicory extract obtained in the positive mode; diagnostic fragment ions in the positive mode for DHLc-gly, DHLc-ox, DHLc, Lc-ox, Lc.
Author Contributions
Conceptualization, F.R., P.H., N.W., J.L.H.; methodology, F.R., P.H., A.B., B.G., P.R.; validation, F.R., P.H.; writing—original draft preparation, F.R., P.H., N.W., J.L.H.; writing—review and editing, all authors; supervision, P.H., N.W. and J.L.H. All authors have read and agreed to the published version of the manuscript.
Funding
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 847568, was supported by the French government through the Programme Investissement d’Avenir (I-SITE ULNE/ANR-16-IDEX-0004 ULNE) managed by the Agence Nationale de la Recherche, by the Programme d’Investissement d’Avenir “Mustart ANR-20-PAMR-0005”, by Institut National de la Santé et de la Recherche Médicale, by Université de Lille and by Institut Pasteur de Lille. The NMR facilities were funded by the Région Hauts-de-France, CNRS, Institut Pasteur de Lille, the Fonds Européens de Développement Régional (FEDER), Ministère de l’Enseignement supérieur, de la Recherche et de l’Innovation (MESRI) and Université de Lille.
Acknowledgments
We thank the Chevreul Institute (FR CNRS 2638) for providing X-ray facility access. It is funded by “Ministère de l’Enseignement Supérieur de la Recherche et de l’Innovation”, region “Hauts-de-France”, ERDF program of the European Union and “Métropole Européenne de Lille”. We thank Mathieu Beaumesnil for having run the Q-TOF experiments. We would like to thank Nicolas Henry, Marine Cordonnier and the team from Florimond Desprez for providing chicory root materials.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
ACN, acetonitrile; DHLc, 11,13-dihydrolactucin; DHLc-ox, 11,13-dihydrolactucin-oxalate; DHLc-gly, 11,13-dihydrolactucin-glycosyde; DHLp, 11,13-dihydrolactucopicrin; DMF, dimethylformamide; DMSO, dimethylsulfoxide; DMAP, 4-dimethylaminopyridine; Et3N, triethylamine; EtOAc, ethyl acetate; eq, equivalent; LiOH, lithium hydroxide; MeOH, methanol; SFEs, supercritical fluid extractions; STLs, sesquiterpene lactones; THF, tetrahydrofuran.
Bibliographic References
- Padilla-Gonzalez:, G.F.; dos Santos, F.A.; Da Costa, F.B. Sesquiterpene Lactones: More Than Protective Plant Compounds With High Toxicity. Critical Reviews in Plant Sciences 2016, 35, 18–37. [Google Scholar] [CrossRef]
- Van Beek, T.A.; Maas, P.; King, B.M.; Leclercq, E.; Voragen, A.G.J.; De Groot, A. Bitter Sesquiterpene Lactones from Chicory Roots. J. Agric. Food Chem. 1990, 38, 1035–1038. [Google Scholar] [CrossRef]
- Graziani, G.; Ferracane, R.; Sambo, P.; Santagata, S.; Nicoletto, C.; Fogliano, V. Profiling Chicory Sesquiterpene Lactones by High Resolution Mass Spectrometry. Food Research International 2015, 67, 193–198. [Google Scholar] [CrossRef]
- Padilla-Gonzalez, G.F. Sesquiterpene Lactones: More Than Protective Plant Compounds With High Toxicity. 21.
- Ivanescu, B.; Miron, A.; Corciova, A. Sesquiterpene Lactones from Artemisia Genus: Biological Activities and Methods of Analysis. Journal of Analytical Methods in Chemistry 2015, 2015, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J. Structure-Activity Relationships of Sesquiterpene Lactones. In Studies in Natural Products Chemistry; Elsevier, 2006; Vol. 33, pp. 309–392 ISBN 978-0-444-52717-2.
- Häkkinen, S.T.; Nohynek, L.; Ivanov, M.; Tsitko, I.; Matos, M.; Baixinho, J.P.; Ivasiv, V.; Fernández, N. Chicory Extracts and Sesquiterpene Lactones Show Potent Activity against Bacterial and Fungal Pathogens. 2021, 23.
- Wang, X.; Liu, M.; Cai, G.H.; Chen, Y.; Shi, X.C.; Zhang, C.C.; Xia, B.; Xie, B.C.; Liu, H.; Zhang, R.X.; et al. A Potential Nutraceutical Candidate Lactucin Inhibits Adipogenesis through Downregulation of JAK2/STAT3 Signaling Pathway-Mediated Mitotic Clonal Expansion. Cells 2020, 9, 331. [Google Scholar] [CrossRef]
- Imam, K.M.S.U.; Tian, Y.; Xin, F.; Xie, Y.; Wen, B. Lactucin, a Bitter Sesquiterpene from Cichorium Intybus, Inhibits Cancer Cell Proliferation by Downregulating the MAPK and Central Carbon Metabolism Pathway. Molecules 2022, 27, 7358. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, T.A.; Kelley, C.J.; Karchesy, Y.; Laurantos, M.; Nguyen-Dinh, P.; Arefi, A.G. Antimalarial Activity of Lactucin and Lactucopicrin: Sesquiterpene Lactones Isolated from Cichorium Intybus L. Journal of Ethnopharmacology 2004, 95, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Wesołowska, A.; Nikiforuk, A.; Michalska, K.; Kisiel, W.; Chojnacka-Wójcik, E. Analgesic and Sedative Activities of Lactucin and Some Lactucin-like Guaianolides in Mice. Journal of Ethnopharmacology 2006, 107, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Silva, P. LEISHMANICIDAL, ANTI-INFLAMMATORY AND ANTI-OBESITY PROPERTIES OF NATURAL PRODUCTS FROM COMMON MEDICINAL AND EDIBLE PLANTS. 135. [CrossRef]
- Jaśkiewicz, A.; Budryn, G.; Carmena-Bargueño, M.; Pérez-Sánchez, H. Evaluation of Activity of Sesquiterpene Lactones and Chicory Extracts as Acetylcholinesterase Inhibitors Assayed in Calorimetric and Docking Simulation Studies. Nutrients 2022, 14, 3633. [Google Scholar] [CrossRef]
- Sessa, R.A.; Bennett, M.H.; Lewis, M.J.; Mansfield, J.W.; Beale, M.H. Metabolite Profiling of Sesquiterpene Lactones from Lactuca Species. Journal of Biological Chemistry 2000, 275, 26877–26884. [Google Scholar] [CrossRef]
- Willeman, H.; Hance, P.; Fertin, A.; Voedts, N.; Duhal, N.; Goossens, J.-F.; Hilbert, J.-L. A Method for the Simultaneous Determination of Chlorogenic Acid and Sesquiterpene Lactone Content in Industrial Chicory Root Foodstuffs. The Scientific World Journal 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chadni, M.; Isidore, E.; Diemer, E.; Ouguir, O.; Brunois, F.; Catteau, R.; Cassan, L.; Ioannou, I. Optimization of Extraction Conditions to Improve Chlorogenic Acid Content and Antioxidant Activity of Extracts from Forced Witloof Chicory Roots. Foods 2022, 11, 1217. [Google Scholar] [CrossRef] [PubMed]
- Milala, J.; Grzelak, K.; Król, B.; Juśkiewicz, J.; Zduńczyk, Z. COMPOSITION AND PROPERTIES OF CHICORY EXTRACTS RICH IN FRUCTANS AND POLYPHENOLS.
- Agriculturae Conspectus Scientificus. 2014, 79.
- Deng, Y.; Scott, L.; Swanson, D.; Snyder, J.K.; Sari, N.; Dogan, H. Guaianolide Sesquiterpene Lactones From Cichorium Intybus (Asteraceae) [1]. Zeitschrift für Naturforschung B 2001, 56, 787–796. [Google Scholar] [CrossRef]
- Michalska, K.; Szneler, E.; Kisiel, W. Complete NMR Spectral Assignments of Two Lactucin-Type Sesquiterpene Lactone Glycosides from Picris Conyzoides: Sesquiterpene Lactone Glycosides from Picris Conyzoides. Magn. Reson. Chem. 2011, 49, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.; Zheng, G.; Zhang, Q.; Jin, P.; Zhang, H.; Su, L.; Qin, D.; Yao, G. Sesquiterpenoids with Diverse Carbon Skeletons from the Roots of Cichorium Glandulosum and Their Anti-Inflammatory Activities. Fitoterapia 2019, 136, 104170. [Google Scholar] [CrossRef] [PubMed]
- Ruban, G.; Zabel, V.; Gensch, K.H.; Smalla, H. The Crystal Structure and Absolute Configuration of Lactucin. Acta Crystallogr B Struct Sci 1978, 34, 1163–1167. [Google Scholar] [CrossRef]
- Escobar-Ledesma, F.R.; Sánchez-Moreno, V.E.; Vera, E.; Ciobotă, V.; Jentzsch, P.V.; Jaramillo, L.I. Extraction of Inulin from Andean Plants: An Approach to Non-Traditional Crops of Ecuador. Molecules 2020, 25, 5067. [Google Scholar] [CrossRef] [PubMed]
- 11beta,13-Dihydrolactucin 3810, CAS 83117-63-9 - SESQUITERPENE. Available online: https://www.extrasynthese.com/sesquiterpene/290-11beta13-dihydrolactucin.html (accessed on 5 March 2023).
- Leclercq, E.; Netjes, J.J. Release of Sesquiterpene Lactones by Enzymatic Liquefaction of Chicory Roots. Z Lebensm Unters Forch 1985, 181, 475–477. [Google Scholar] [CrossRef]
- Baixinho, J.P.; Anastácio, J.D.; Ivasiv, V.; Cankar, K.; Bosch, D.; Menezes, R.; de Roode, M.; dos Santos, C.N.; Matias, A.A.; Fernández, N. Supercritical CO2 Extraction as a Tool to Isolate Anti-Inflammatory Sesquiterpene Lactones from Cichorium Intybus L. Roots. Molecules 2021, 26, 2583. [Google Scholar] [CrossRef]
- Ajiaikebaier, A.; Wei, Z.; Sun, G. Preparation Method of Lactucin, CN111689935A, 2020.
- Preparation Method and Application of Lactucin Extracted from Cichorium Intybus, CN112939912A, 2021.
- Lactucin and Its Preparation Method and Application, CN101099566A, 2008.
- Qin, D.; Zou, N.; Cai, G.; Wang, S.; Su, L.; Dang, T. Lactucin and Application Thereof as Anti-Inflammatory Component, CN112079804A, 2020.
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin Microbiol Rev 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT – Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr A Found Adv 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr A Found Crystallogr 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2 : A Complete Structure Solution, Refinement and Analysis Program. J Appl Crystallogr 2009, 42, 339–341. [Google Scholar] [CrossRef]
Figure 1.
Three main steps of the extraction-purification process.
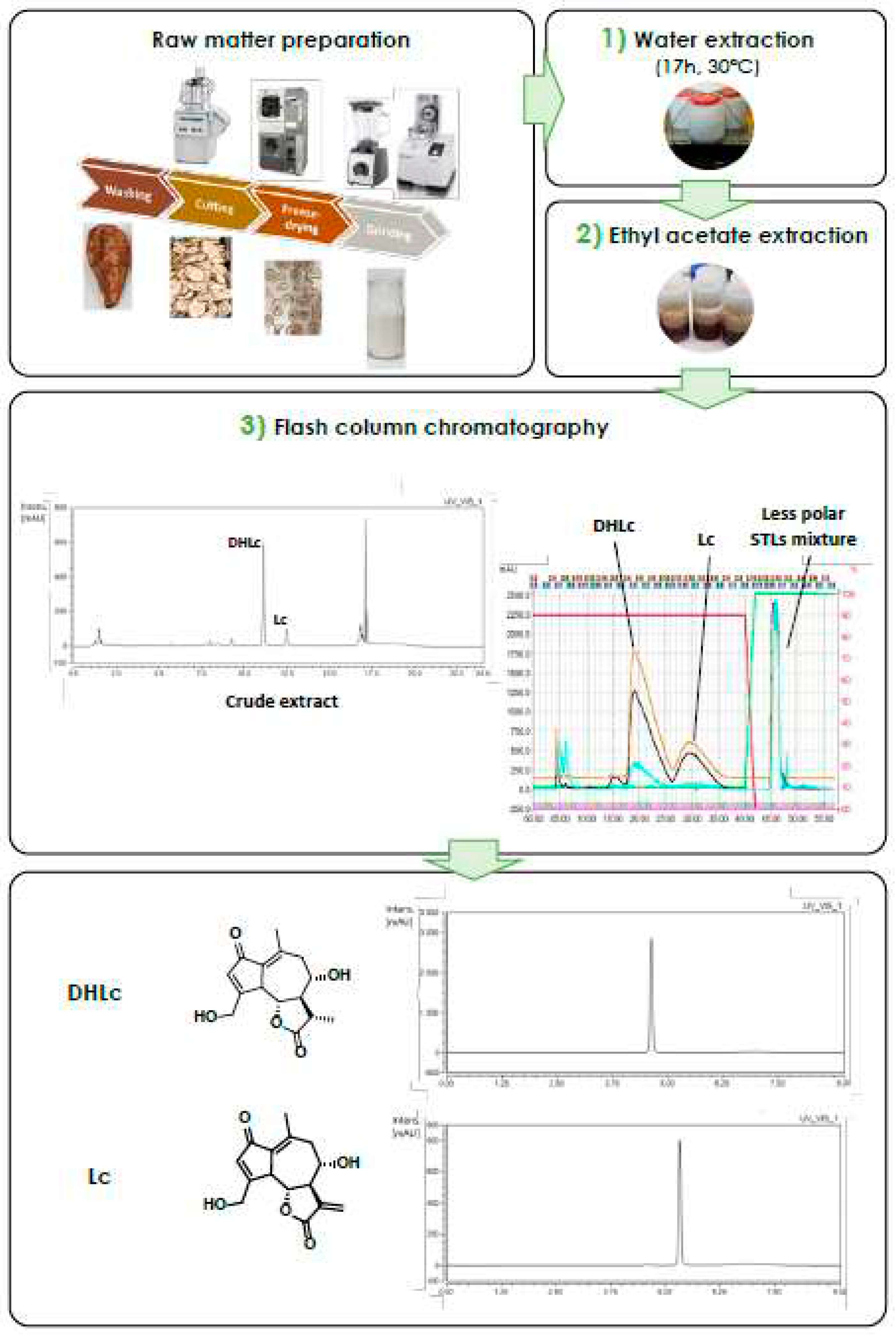
Figure 2.
HPLC profile of a water chicory extract (A) and zoom on DHLc, Lc and their conjugated forms (B).
Figure 2.
HPLC profile of a water chicory extract (A) and zoom on DHLc, Lc and their conjugated forms (B).
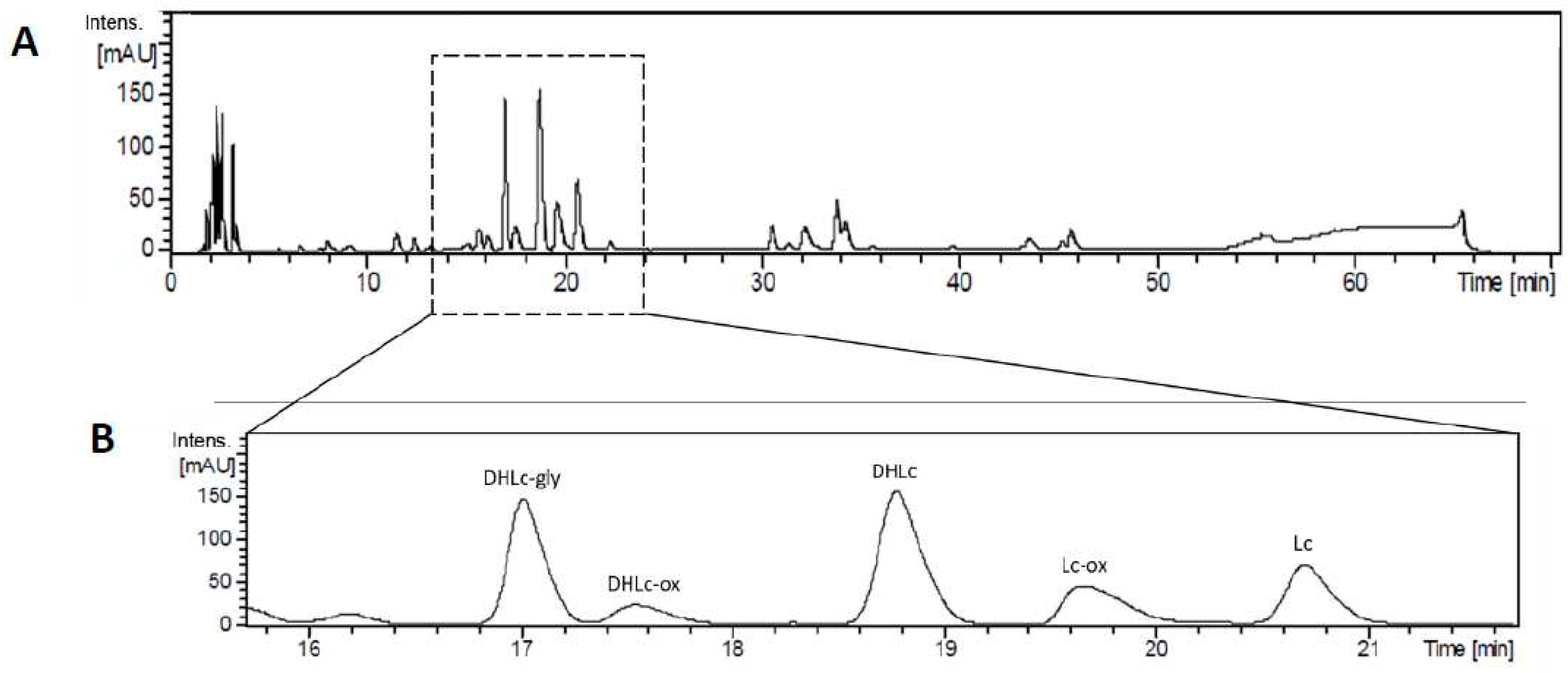
Figure 3.
Chemical structures of DHLc, Lc and their conjugated forms and aimed hydrolysis.
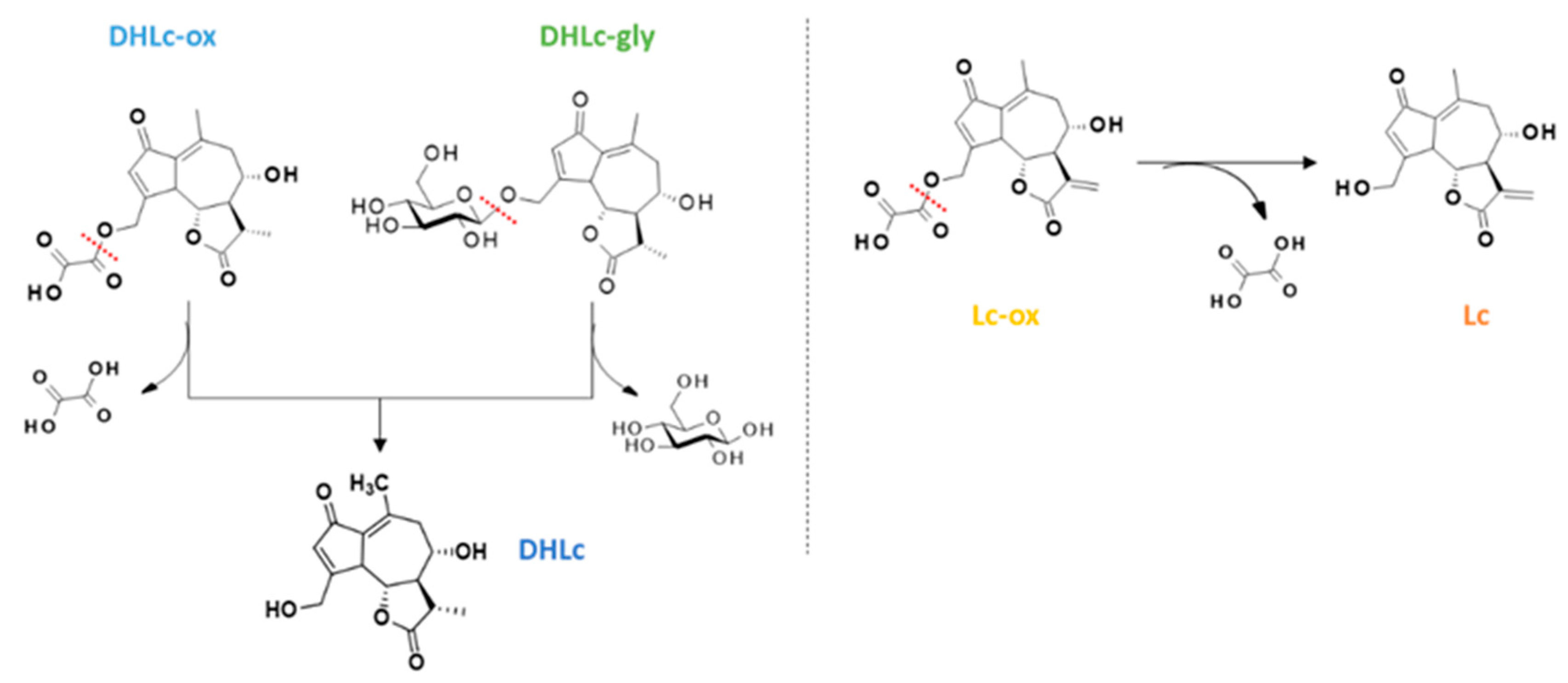
Figure 4.
Effect of the extraction solvent on the levels of free (A) and conjugated (B) STLs. Effect of the extraction temperature on the levels of free (C) and conjugated (D) STLs. Effect of the extraction time at 50°C on the levels of free (E) and conjugated (F) STLs. Effect of the extraction time at 30°C on the levels of free (G) and conjugated (H) STLs.
Figure 4.
Effect of the extraction solvent on the levels of free (A) and conjugated (B) STLs. Effect of the extraction temperature on the levels of free (C) and conjugated (D) STLs. Effect of the extraction time at 50°C on the levels of free (E) and conjugated (F) STLs. Effect of the extraction time at 30°C on the levels of free (G) and conjugated (H) STLs.
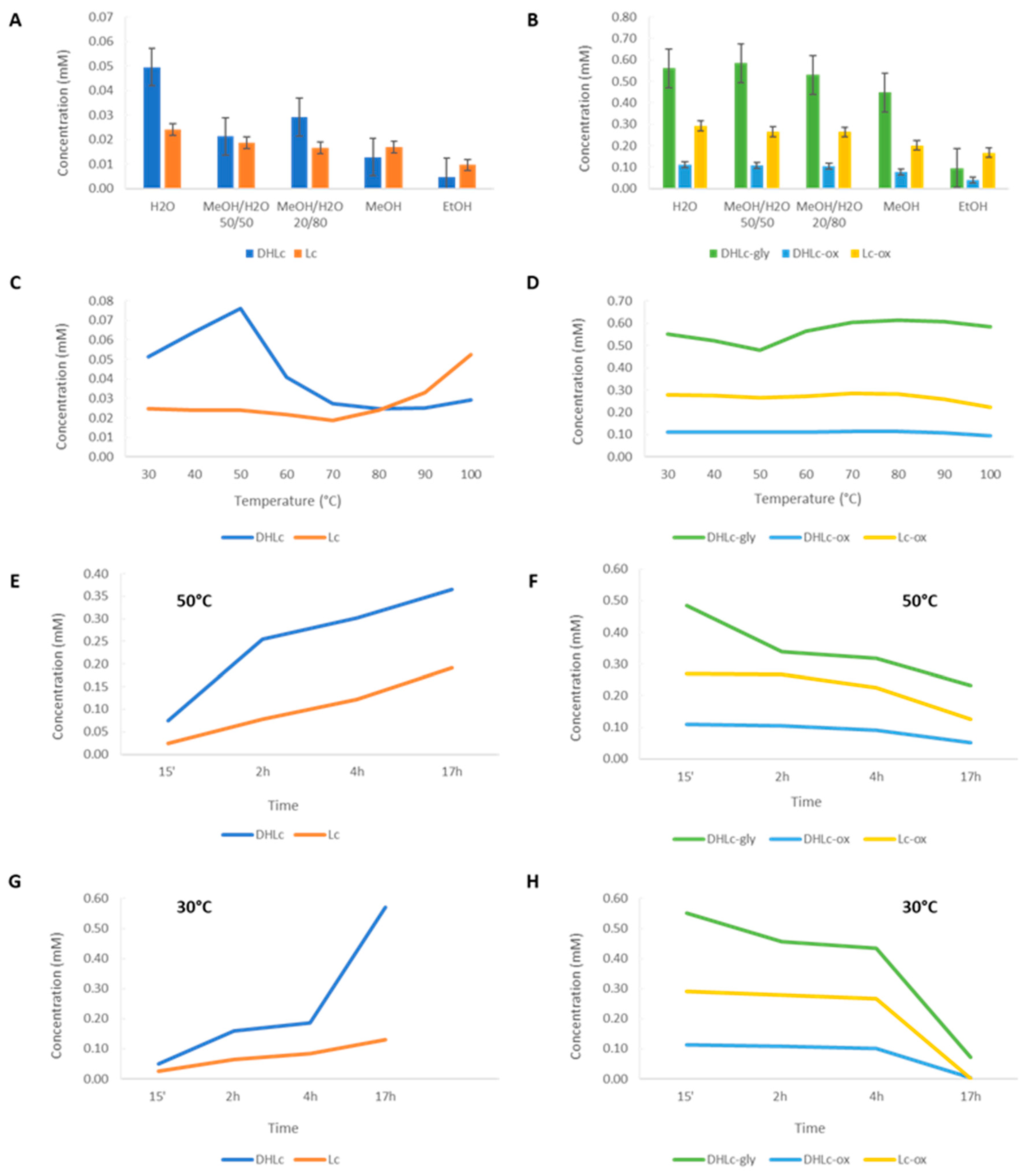
Figure 6.
Synthetic strategy towards DHLc-ox and Lc-ox.
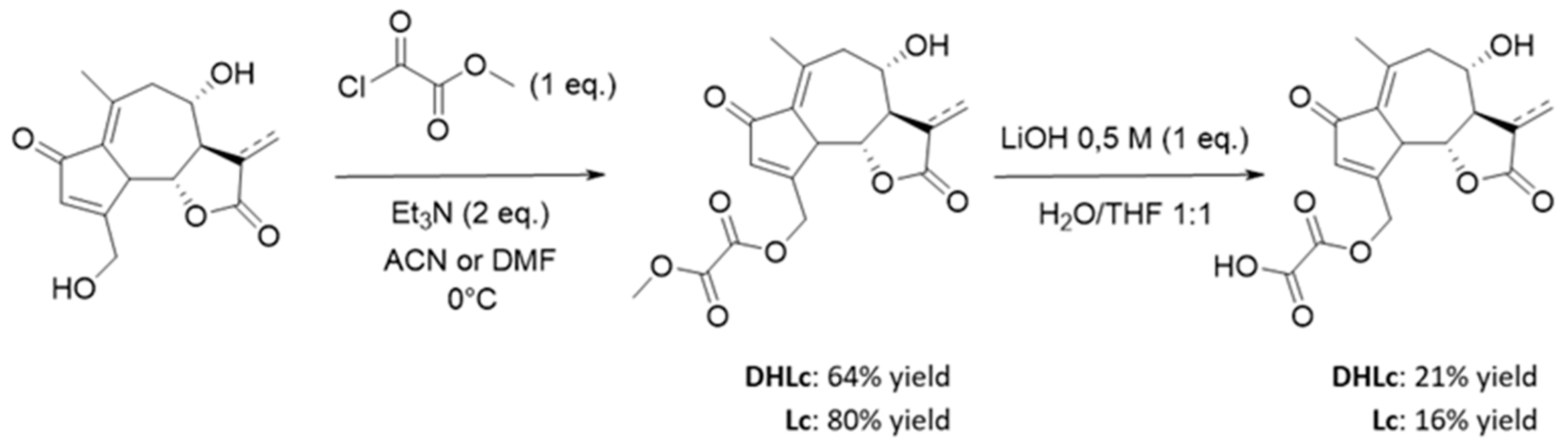
Table 1.
Main STLs found in chicory roots.
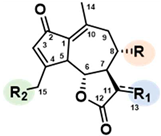 |
R | R1 | R2 | |
| Lactucin | OH | CH2 | OH | |
| Lactucin-15-oxalate | OH | CH2 | OCOCOOH | |
| Lactucin-15-glycoside | OH | CH2 | OGlucose | |
| 11β,13-dihydrolactucin | OH | CH3 | OH | |
| 11β,13-dihydrolactucin-15-oxalate | OH | CH3 | OCOCOOH | |
| 11β,13-dihydrolactucin-15-glycoside | OH | CH3 | OGlucose | |
| 8-deoxylactucin | H | CH2 | OH | |
| 8-deoxylactucin-15-oxalate | H | CH2 | OCOCOOH | |
| 8-deoxylactucin-15-glycoside | H | CH2 | OGlucose | |
| 11β,13-dihydro-8-deoxylactucin | H | CH3 | OH | |
| 11β,13-dihydro-8-deoxylactucin-oxalate | H | CH3 | OCOCOOH | |
| 11β,13-dihydro-8-deoxylactucin-glycoside | H | CH3 | OGlucose | |
| Lactucopicrin |  |
CH2 | OH | |
| Lactucopicrin-15-oxalate | CH2 | OCOCOOH | ||
| Lactucopicrin-15-glycoside | CH2 | OGlucose | ||
| 11β,13-dihydrolactucopicrin | CH3 | OH | ||
| 11β,13-dihydrolactucopicrin-15-oxalate | 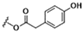 |
CH3 | OCOCOOH | |
| 11β,13-dihydrolactucopicrin-15-glycoside | CH3 | OGlucose |
Table 2.
Molecular fragments of STLs determined by positive analysis.
| Compound | Molecular weight | Retention time (min) | ESI major fragment ions (m/z) |
|---|---|---|---|
| DHLc-gly | 440 | 16.9 | 463 (100) [M+Na]+; 441 (13) [M+H]+; 279 (18) [M-glycosyl+H]+; 261 (9) [M-glycosyl- H2O+H]+; 243 (10) [M-glycosyl-2H2O+H]+; 215 (8) [M-glycosyl- H2O-HCO2H+H]+ |
| DHLc-ox | 350 | 17.7 | 373 (57) [M+Na]+; 351 (100) [M+H]+; 261 (94) [M-oxaloyl- H2O+H]+; 243 (56) [M-oxaloyl-2H2O+H]+; 215 (39) [M-oxaloyl-H2O-HCO2H+H]+ |
| DHLc | 278 | 18.8 | 301 (100) [M+Na]+; 279 (59) [M+H]+; 261 (7) [M- H2O+H]+; 243 (8) [M-2H2O+H]+; 215 (17) [M-HCO2H-H2O+H]+ |
| Lc-ox | 348 | 19.7 | 371 (73) [M+Na]+; 349 (86) [M+H]+; 259 (100) [M-oxaloyl-H2O+H]+; 241 (96) [M-oxaloyl-2H2O+H]+; 213 (60) [M-oxaloyl-H2O-HCO2H+H]+ |
| Lc | 276 | 20.7 | 299 (100) [M+Na]+; 277 (44) [M+H]+; 259 (8) [M– H2O+H]+; 241 (17) [M–2H2O+H]+; 213 (18) [M-HCO2H-H2O+H]+ |
Table 3.
Content of DHLc-gly, DHLc-ox, DHLc, Lc-ox and Lc extracted under different conditions. Values are means (± standard deviations) of three samples.
Table 3.
Content of DHLc-gly, DHLc-ox, DHLc, Lc-ox and Lc extracted under different conditions. Values are means (± standard deviations) of three samples.
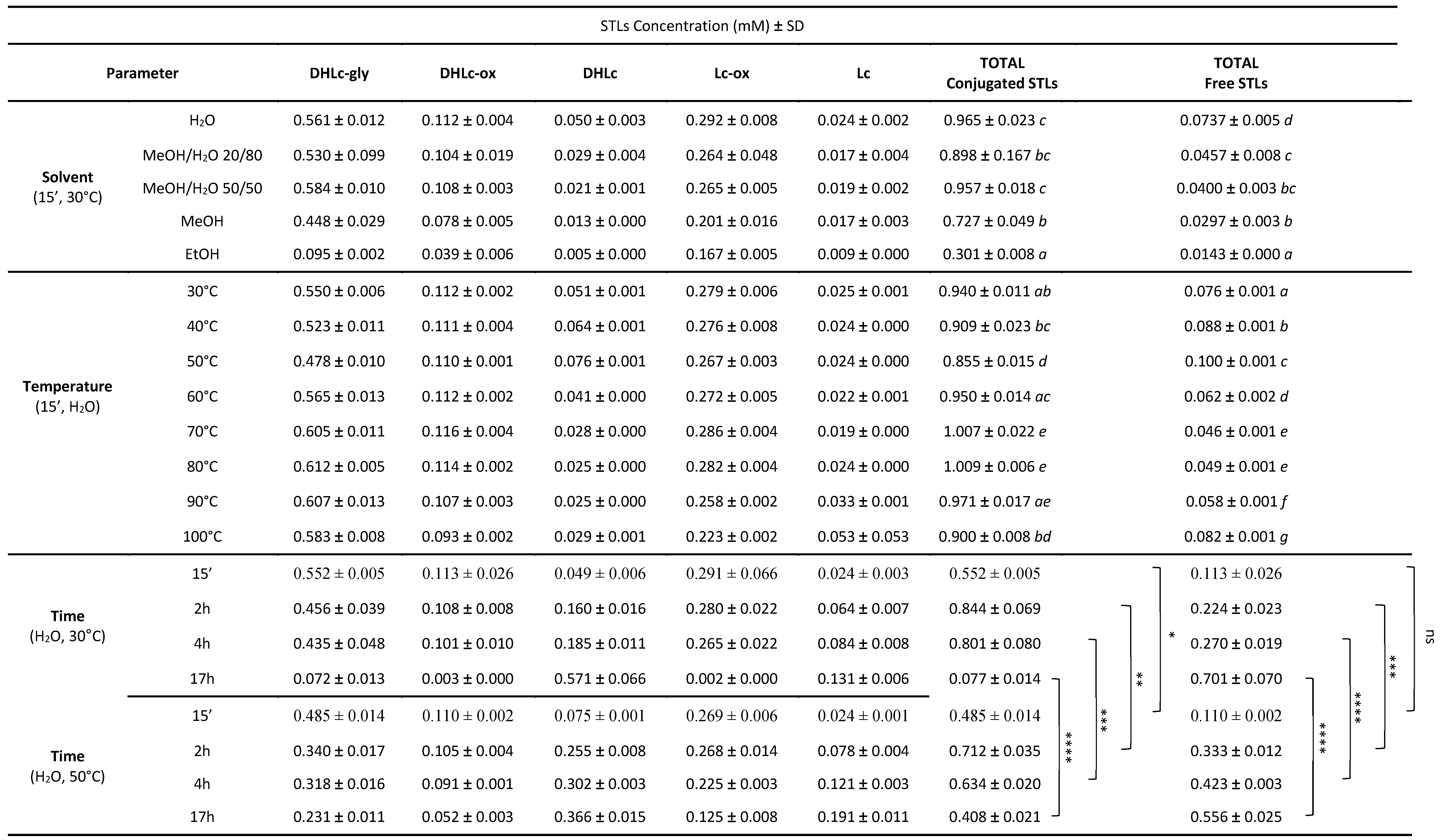
For solvent and temperature, different letters in the same column within the same parameters indicate significant differences (P < 0.05). For time, statistical differences between 30°C and 50°C are noted as ns not significant, * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



