
Submitted:
01 April 2023
Posted:
04 April 2023
You are already at the latest version
Abstract
The steady-state dissolution rates of basaltic glass and labradorite have been measured in the presence of 10 to 700 x 10-3 mol kgw-1 aqueous NaCl, KCl, CaCl2, and MgCl2 at 25 oC. All rates were measured in mixed flow reactors, and at pH~3.6 by the addition of HCl to the reactive fluids. Steady-state basaltic glass dissolution rates based on Si release increased by ~0.3 log units in the presence of 1 x 10-2 mol kgw-1 of CaCl2 or MgCl2, compared to their rates in 10 x 10-3 mol kgw-1 of NaCl, and KCl. In contrast, the steady-state dissolution rates of labradorite decreased by ~0.4 log units in the presence of 10 x 10-3mol kgw-1 of CaCl2 or MgCl2, compared to their rates in 10 x 10-3 mol kgw-1 of NaCl, and KCl. These contrasting behaviours likely reflect the varying effects of these cations on the stability of rate controlling Si-rich activated complexes on the surface of the solids. On average, the Si release rates of these solids are similar to each other and increase slightly with increasing ionic strength. As the pH of water charged with 10 to 30 bars CO2 is ~3.6, the results of this study indicate that both basaltic glass and labradorite dissolution will likely be effective at increasing pH and adding Ca to the aqueous phase in saline fluids. This observation supports potential efforts to store carbon through its mineralization in saline aquifers containing Ca-bearing feldspar and in submarine basalts.
Keywords:
Labradorite
; basaltic glass
; mineral carbonation
; dissolution rates
1. Introduction
This study is focused on quantifying the effect of the ionic strength and the concentration of some common aqueous chloride salts on the dissolution rates of basaltic glass and Ca-rich plagioclase. The dissolution rates of these solids have received significant recent attention due to their potential use as the feedstock for carbon mineral storage in the subsurface (McGrail et al., 2014, 2006; Matter et al., 2007; 2011, 2016; Alfredsson et al., 2008; Goldberg et al., 2008; Oelkers et al., 2008; Goldberg and Salagle, 2009; Matter and Kelemen, 2009; Schaef et al., 2009; Shikazono et al., 2009; Gislason et al., 2010; Kampman et al. 2009; Schaef and McGrail, 2009; Gysi and Stefansson, 2011; Pham et al., 2011; Wolff-Boenisch et al., 2011; Munz et al., 2012; Hellevang et al., 2013; Gislason and Oelkers, 2014; Wolff-Boenisch and Galeczka, 2018; Tutolo et al., 2021; Snæbjörnsdóttir et al., 2018, 2020; Kelemen et al., 2019; 2020; Galeczka et al., 2022; Oelkers et al., 2023). Notably, there have been a large number of studies reporting the measured dissolution rates of basaltic glass (e.g. Crovisier et al., 1985, 1987; Gislason and, Eugster,1987; Guy, 1989; Oelkers and Gislason, 2001; Wolff-Boenisch et al., 2004a, b, 2006; 2011; Stockmann et al., 2011; Parruzot et al., 2015) as well as of the Ca-rich feldspar labradorite (Sjoberg, 1985; Casey et al., 1988, 1989; Cygan et al., 1989; Nesbitt et al., 1991, Welch and Ullman, 1996; Taylor et al., 2000; Caroll and Knauss; 2005; Gudbrandsson et al., 2014; Wild et al., 2016; Gadikota et al., 2020; de Obeso et al., 2023). These past studies have explored in detail the effects of temperature, presence of aqueous organic species, pH, distance from equilibrium and crystallographic orientation on measured dissolution rates. The present study builds on these past efforts by measuring the dissolution rates of basaltic glass and labradorite as a function of reactive aqueous concentrations of NaCl, KCl, CaCl2, and MgCl2.
The motivation for this study is to better quantify the effect of major cations on the dissolution kinetics of basaltic glass and Ca-rich plagioclase in an attempt to better constrain the potential applicability of saline fluids in carbon mineralization efforts. Notably, carbon dioxide storage in the subsurface is increasingly targeted for injection into saline reservoirs, in an attempt to preserve freshwater resources (e.g. Celia et al., 2015; Kumar et al., 2020; Ringrose et al, 2021). Some of these efforts will be aimed at injecting CO2 into reactive basaltic or ultramafic rocks containing saline fluids or seawater to enhance the long-term security of the carbon storage (e.g., Luo et al. 2012; Marieni et al., 2021; Voigt et al., 2021). Towards the improved quantification of such efforts, the dissolution rates of basaltic glass and labradorite have been determined at steady-state, far-from-equilibrium conditions in flow through reactors at pH of 3.6 and 25°C in the presence of aqueous metal chloride solutions having ionic strengths up to 0.7 mol kgw-1. The purpose of this study is to report the results of these measurements and to use them to assess the effect of water salinity on subsurface mineral carbonation.
2. Theoretical Background
The standard state adopted in this study is that of unit activity of pure minerals and H2O at any temperature and pressure. For aqueous species other than H2O, the standard state is unit activity of the species in a hypothetical one molal solution referenced to infinite dilution at any temperature and pressure. All thermodynamic calculations reported in this study were performed using the geochemical modelling code PHREEQC3 (Parkhurst and Appelo, 2013), together with its carbfix.dat database (Voigt et al., 2018).
This study is focused on the characterization of dissolution rates of basaltic glass and labradorite. The dissolution of these solids is controlled by the detachment of metals from the surface of these solids (Brantley et al., 2008). Within the context of Transition State Theory, surface reaction-controlled dissolution rates, r, can be considered to be the difference between the forward rate (r+) and the reverse rate (r-) such that
Taking account of the law of detailed balancing, it can be shown that Eqn. (1) is equivalent to (Aagaard and Helgeson, 1977, 1982; Lasaga, 1981; Schott and Oelkers, 1995; Oelkers, 2001)
where A refers to the chemical affinity of the reaction, σ stands for Temkin's average stoichiometric number equal to the ratio of the rate of destruction of the activated or precursor complex relative to the overall rate, R designates the gas constant and T denoted the temperature. Experimental evidence suggests that the value of σ in Eqn. (2) is 3 for basaltic glass and plagioclase (Oelkers et al., 1994; Oelkers, 2001). The form of Eq. (1) is such that overall rates (r) equal forward rates (r+) when A >> σRT. All dissolution rates in the present study were measured at far-from-equilibrium conditions, such that A >> σRT. At these conditions and thus r ≈ r+.
According to Transition State Theory applied to surface-controlled dissolution reactions, the forward rate, r+, is proportional to the concentration of a rate controlling surface complex such that (Eyring, 1935; Schott et al., 2009):
where k+ refers to a rate constant and [
. enotes the concentration of the rate controlling surface complex. Previous work on the basaltic glass and plagioclases having less than ~75% Ca in their cation site (<An75) suggests that their far-from-equilibrium dissolution rates are proportional to the concentration of a Si-rich surface complex formed by the Al-proton exchange reactions leading to the following rate equation (Oelkers et al., 1994; Oelkers and Schott, 1995; Oelkers, 2001; Oelkers and Gislason, 2001; Gislason and Oelkers, 2003; Carroll and Knauss, 2005, Schott et al., 2009). This observation leads to an equation describing basaltic glass and labradorite dissolution rates of the form:
where represents a rate constant, s refers to the specific surface area of the mineral, and n denotes an exponential factor equal to ~0.33. Although monovalent and divalent cations, including Na, K, Mg, and Ca, also need to be removed from the structure of basaltic glass and labradorite to create their dissolution rate controlling Si-rich surface complex, the degree to which these cations influence the dissolution rate of these solids has yet to be quantified. The role of these cations in the dissolution rate of these solids will be explored in detail in the present study.
Rates measured in the present study are normalized to the geometric surface areas of the basaltic glass and labradorite samples. These surface areas were calculated using (Tester et al., 1994):
where refers to the rock density of the rock and deff to the effective particle diameter. This latter term, deff , was calculated using:
where dmax and dmin correspond to the minimum and maximum grain size of the prepared powders, respectively.
3. Materials and Methods
Solids
The basaltic glass used in this study was originally collected from Stapafell Mountain in SW Iceland. This basaltic glass has been previously used in a large number of different experimental studies (e.g. Oelkers and Gislason, 2001; Gislason and Oelkers, 2003; Flaathen et al., 2010; Stockmann et al., 2011, 2012; Wolff-Boenisch et al., 2011; Prikryl et al., 2017; Saetre et al., 2018; Wolff-Boenisch and Galeczka, 2018; Clark et al., 2019; Ralston et al., 2023). The chemical composition of this glass, determined by X-ray fluorescence spectrometry, is shown in Table 1. Back scattering images obtained using scanning electron microscope confirm that while some minor microcrystalline phases are present in this glass, they are confined within the glass shards (Wolff-Boenisch, 2004).
The labradorite used in this study was collected from an anorthosite intrusion located on the Hrappsey Islands in Breidafjördur, Western Iceland (Kirstmannsdottir, 1971). Its chemical composition, provided in Table 1, was determined using standard wavelength dispersive techniques using the JEOL Superprobe JSL 8200 electron microprobe located at the GET/CNRS in Toulouse, France. These analyses were performed using an acceleration voltage of 15 kV, a beam current of 15 nA, and a beam diameter of 2 µm.
The solid materials were first dried at room temperature for several days before being crushed in plastic bags using a plastic hammer. The solids were then further ground with an agate mortar and dry sieved to obtain the 45–125 µm size fraction. This size fraction was gravitationally settled to remove fine particles, and subsequently cleaned ultrasonically five times in acetone. The resulting powder was oven-dried overnight at 60 oC. The geometric surface areas of these powders were calculated using Eqns. (5) and (6) and equal to 284 cm2/g for both solids.
Reactive Fluids
Reactive inlet fluids were created by dissolving Merck/Sigma-Aldrich analytical grade NaCl, KCl, CaCl2·2H2O, or MgCl2·6H2O in deionized Millipore™ water with ionic strengths ranging from 0.01 to 2.1 mol kg−1. The concentration of these chloride bearing inlet solutions was selected to cover a broad range of ionic strengths. Reagent grade HCl was added to each fluid to adjust the pH to 3.6. This reactive fluid pH was selected so that it would have a similar pH to likely CO2-charged injection waters and to avoid the precipitation of most secondary phases. The pH of pure water in equilibrium with 25 bars of CO2 pressure at 25°C is 3.2 (Gislason et al., 2010).
Figure 1.
Design of the mixed-flow reactor system used to perform the experiments reported in the present study.
Figure 1.
Design of the mixed-flow reactor system used to perform the experiments reported in the present study.
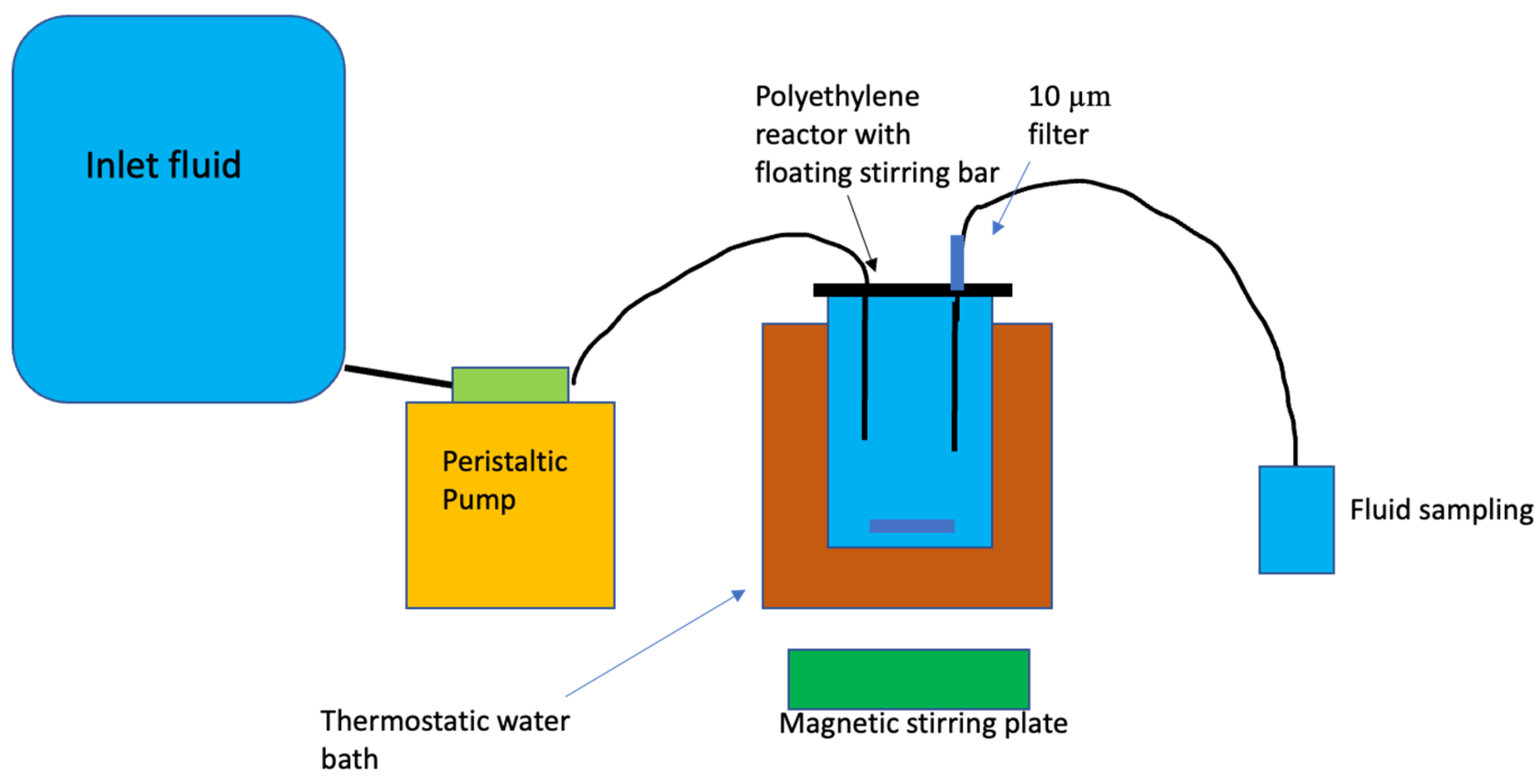
Experimental Design
Basaltic glass and labradorite dissolution rates were performed in mixed-flow reactors. This reactor system is similar to those used in past dissolution rate studies (c.f. Stockmann et al., 2011, Gudbrandsson et al., 2014). This system consisted of 300 mL acid washed high density polyethylene reactors maintained at constant temperature in a thermostat-controlled water bath. Temperature was kept constant during the experiment at 25°C. All the reactor components were made of polyethylene to avoid corrosion. At the beginning of the experiment all reactors, connectors, and tubing were cleaned with a 0.1M HCl solution for 24h and rinsed with Millipore™ water prior to each experiment. All outlet fluid sample bottles went through the same cleaning procedure prior to sampling to prevent contamination. These reactors were stirred using floating stir bars located on the bottom of the reactors. The stirring bars were rotated by a magnetic stirrer located underneath the water bath. Basaltic glass and labradorite dissolution experiments were initiated by placing 4g of cleaned powder and the selected initial reactive fluid into the reactor. The reactors were then sealed and a Masterflex™ cartridge pump delivered the inlet fluid into the polyethylene mixed-flow reactors at a rate of 0.85–1.0 g/min. The reactive inlet fluids were stored in 12L compressible plastic bags during the experiments. Inlet solutions of selected concentrations of NaCl, KCl, CaCl2 and MgCl2 were sequentially pumped into the reactor. The reactor fluid passed through a 10µm filter when exiting the reactor and was further filtered through a 0.2 µm cellulose acetate filter prior to chemical analysis. The outlet fluid pH was measured at 25° C using a Eutech Instruments© pH meter coupled to a Eutech Instruments© electrode with a 3 M KCl outer filling solution. The electrode was calibrated with NBS standards at pH 4.01, and 7, with an average error less than 0.03 pH units. Fluid samples were collected and acidified with 0.5% concentrated supra-pure HNO3 prior to their analysis. The Si concentrations of all inlet and outlet fluids were measured by inductively coupled argon plasma using a Spectro Vision optical emission spectrometer (ICP-OES). Analytical uncertainties on ICP-OES analyses are estimated to be 3–5% based on repeated analyses.
As was the case for a number of past mineral dissolution rate studies (e.g Flaathen et al., 2010; Saldi et al., 2010; Oelkers et al., 2011), experiments in this study were run in several distinct series on individual solid powders. At the start of each experimental series, the initial reactive fluid was injected into the rector at a constant flow rate. The reactors were then operated for at least 48 h before the first outlet fluid sample was collected. Subsequent fluid sampling was timed to allow at least 3 residence times to pass between each sampling. The residence time is defined as the reactor volume divided by the fluid flow rate and ranged from 5-6 h. Steady-state was assumed when four consecutive rate determinations from Si outlet concentrations were constant with analytical uncertainty. Steady state was usually attained after 7-8 days. After reaching steady-state with the initial NaCl-bearing fluid, the inlet fluid was replaced with KCl of the same cation concentration and the procedure repeated. On attainment of this second steady-state, the inlet fluid was then replaced with a CaCl2 bearing inlet fluid of similar ionic concentration resulting in a third steady-state over time. Finally, the CaCl2 inlet fluid was replaced with a MgCl2 bearing inlet solution of similar ionic concentration until a fourth and last steady-state was attained. In total, each solid was dissolved in the presence of four different cation-chloride salts in the order of NaCl, KCl, CaCl2, MgCl2. Running several distinct salts in each series facilitated identification of the effects of each on measured rates.
Basaltic glass and labradorite dissolution rates reported in this present study are based on Si release. Silicon release rates are commonly used as a measure of the dissolution rate of silicate minerals as this metal is critical for maintaining their structure (cf. Oelkers, 2001; Brantley, 2003; Brantley et al., 2008; Hermanska et al., 2022). These rates (rSi,i) were calculated from steady state Si concentrations in the outlet fluids of the reactors using:
where F represents the fluid flow rate, CSi stands for the concentration of Si in the outlet fluid at steady state, νi signifies the stoichiometric number of Si in one mole of the solid (1 for basaltic glass and 3.26 for labradorite), Ai denotes the specific geometric surface area of ith solid, and mi represents the initial mass of the ith solid in the reactor.
rSi,i = (F CSi)/(νi Ai, mi)
4. Results
A representative variation of the measured outlet fluid Si concentration as a function of time is shown in Figure 2. The measured Si concentrations of all samples collected during this study is provided in Table S1 of the electronic supplement. The Si release rates rapidly attain a near constant value in all experiments during each reactive series although some scatter is evident. The steady-state Si concentrations for each experiment are reported in Table 2. Integration of the temporal Si release was used to estimate the total mass of solid dissolved during each experimental series. In each case no more than 1% of the total mass of the solid was dissolved during any experimental series.
Measured Si concentrations were used to determine the steady-state dissolution rate of the solids. These rates are reported in Table 2 and illustrated as a function of the concentration of each added chloride salt in Figure 3. Rates are relatively unaffected by the addition of the selected chloride salts to the reactive fluids. Geometrically measured dissolution rates vary from 10-9.0 to 10-8.3 mol m-2 s-1 for basaltic glass and from 10-9.5 to 10-8.5 mol m-2 s-1 for labradorite. Nevertheless, some trends are apparent. The addition of divalent metal chlorides tend to increase the steady-state dissolution rate of basaltic glass compared to that of the monovalent chloride salts. Steady state basaltic glass dissolution rates measured in the presence of 0.01 mol kg-1 NaCl and 0.01 mol kg-1 KCl are approximately 0.3 orders of magnitude slower than rates measured in either 0.01 mol kg-1 CaCl2 or MgCl2. These differences appear to decrease somewhat with increasing salt concentrations. In contrast, the addition of divalent metal chlorides tends to decrease the steady-state dissolution rate of labradorite compared to that of the monovalent chloride salts. Steady state labradorite dissolution rates measured in the presence of 0.01 mol kg-1 NaCl and 0.01 mol kg-1 KCl are approximately 0.3 orders of magnitude slower than rates measured in either 0.01 mol kg-1 CaCl2 or MgCl2. Similar to the dissolution behaviour of basaltic glass, the differences in these rates appear to decrease with increasing salt concentration.
The variation of measured steady-state rates with the ionic strength of the reactive fluid is depicted in Figure 4. The steady-state dissolution rates of both basaltic glass and labradorite increase slightly in response to the ionic strength of the reactive fluid, as indicated by the trend line in this figure. Nevertheless, a systematic scatter is evident in the variation of labradorite rates with increasing ionic strength reflecting an apparent small inhibitory effect of the presence of divalent metal cations on the rates.
5. Discussion
5.1. Comparison of Basaltic Glass and Labradorite Si Release Rates
The results of this study allow the direct comparison of the dissolution rates of basaltic glass and an intermediate feldspar. These solids are both commonly present in basaltic rocks and have similar atomic Ca/Si ratios. The atomic Ca/Si ratio of Stapafell basaltic glass and of Hrappsey Islands feldspar are 0.263 and 0.286, respectively. The availability of Ca is essential for the mineralization of CO2 as solid calcium carbonate during carbon storage efforts. The measured Si release rates of these two solids are compared directly in Figure 5. As can be seen in this figure, the Si release rate of these solids are identical within uncertainty. This concurrence suggests that glassy and crystalline basalts might be equally effective at capturing and storing CO2 through calcium carbonate precipitation.
5.2. Comparison to Previously Measured Rates
A comparison of measured steady-state Stapafell basaltic glass dissolution rates with those of past studies is presented in Figure 6A. This comparison is confounded by the variety of conditions and fluid compositions considered in the various experiments. Oelkers and Gislason (2001) measured the dissolution rates at pH 3 and pH 11 in the presence of added aqueous Al, Si and organic acids and found the rates depended on the aqueous Al activity of the reactive fluids. Gislason and Oelkers (2003) reported the dissolution rates of Stapafell basaltic glass as a function of pH and temperature. Stockmann et al. (2011) measured the dissolution rates of this glass in the presence of carbonate precipitates on its surface. They found little effect of precipitated carbonate minerals on the dissolution rates of the basaltic glass surfaces. Stockmann et al. (2012) measured dissolution rates in the presence of dead and alive bacteria and concluded that the nutrients and the presence of bacteria slowed substantially dissolution. Wolff-Boenisch et al. (2004, 2011) reported the dissolution rates of this basaltic glass in the presence of inorganic cations, aqueous fluoride, and organic acids. They found that fluoride and aqueous organic acids substantially accelerated basaltic glass dissolution.
The rates summarised in Figure 6A suggest that the presence of increased concentrations of NaCl, KCl, CaCl2, and MgCl2 do not strongly influence the rates of basaltic glass dissolution. This behaviour contrasts to that of aqueous Al, F, and organic acid anions. The effect of these aqueous species on basaltic glass dissolution rates were attributed to a dissolution mechanism that required the removal of aqueous Al to form the rate limiting Si-rich activated complex on the basaltic glass surface (e.g. Oelkers, 2001; Oelkers and Gislason, 2001; Schott et al., 2009).
Gudbrandsson et al. (2014) reported dissolution rates of the identical plagioclase considered in this study. These past dissolution rates were measured in a mixed flow reactor system in the presence of an aqueous solution having a 0.01 mol kg-1 ionic strength using NH4Cl as a background electrolyte at 22 oC. These previously reported rates are compared with those measured in the present study as a function of pH in Figure 6B. It can be seen that the rates measured in the present study are consistent with those of past efforts. The rates of Gudbrandsson et al., 2014), which were normalized to BET surface area, were recalculated to geometrically normalized dissolution rates using Eqn. 5 and 6 and the grain sizes reported in the original publication. The previously reported rate measured at pH=3.76 is directly comparable to that of the present study, and equal to 10-9.23 mol m-2 s-1. The rates measured at this pH and in the presence of 0.01 mol kg-1 NaCl and 0.01 mol kg-1 KCl in the present study are 10-9.02 and 10-9.09 mol m-2 s-1 respectively. Although these rates are similar, they suggest that the presence of Na and K in the reactive solution may have a slight accelerating effect on the rate of bytownite dissolution at this pH.
5.3. Implications for the Dissolution Mechanism of Basaltic Glass and Feldspars
Past studies have reported that the far-from-equilibrium dissolution rates of basaltic glass and the feldspars having an anorthite content less than 75% slow significantly and systematically as a function of increasing aqueous aluminium activity. This observation was attributed to the dissolution mechanism of these solids. In both cases, the dissolution rate variation with aqueous Al activity was attributed to the control of these rates by a Si-rich activated complex formed from the removal of Al from the aluminosilicate structure.
The results of the present study suggest that the presence of CaCl2 and MgCl2 decreases labradorite dissolution rates by as much as 0.3 log units compared to corresponding rates in the presence of aqueous NaCl and KCl solutions. This observation, which contrasts to the results of Oelkers and Schott (1995), suggest that these divalent metals may limit the formation of the Si-rich activated complex at the surface of this initially Ca-rich feldspar. A potential pathway for this effect is that the presence of these divalent metals in solution could disfavour the removal of divalent metals from the near surface of the dissolving felspar. This possibility could be further tested through a detailed study of the surface chemistry of Ca-rich feldspars after their interacton with compositionally distinct divalent metal bearing fluids.
In contrast to the inhibitory effect of solute CaCl2 and MgCl2 on labradorite dissolution, these salts are observed to increase basaltic glass dissolution rates. It seems likely that this observation is due to the role of these salts in increasing the ionic strength of the aqueous solutions. This interpretation is supported by the distribution of rates illustrated in Figure 4, where glass rates appear to plot as a linear function of ionic strength, in contrast to behaviour of the feldspar dissolution rates, appear to plot as a linear function of ionic strength. An increase in silica glass dissolution rates was reported by Icenhower and Dove (2000); the increase of silicate dissolution rates with increasing ionic strength was attributed to the increased charging of the mineral surface (Brady and Walther, 1990).
5.4. Consequences for Carbon Mineral Storage in Saline Aquifers
This study considered the dissolution rates of basaltic glass and labradorite. The dissolution of these minerals will favour the mineralisation of CO2 through the combination of adding Ca to the aqueous phase and increasing the pH of mildly acidic waters. The results presented in this study indicate that the dissolution rates of neither basaltic glass nor labradorite are inhibited by the presence of elevated concentrations of NaCl, KCl, CaCl2 or MgCl2 in the aqueous phase at pH~3.5. In fact, rates appear to increase to some extent as the ionic strength of the aqueous phase increases. The pH of the reactive fluids considered in this study is similar to that of water in equilibrium with 10 to 50 bars of CO2. It therefore seems likely that both basaltic glass and labradorite dissolution will be at least equally able to promote the mineral storage of CO2 in saline as in fresh water aquifers.
6. Conclusions
The rates generated in the present study indicate that the dissolution of basaltic glass and plagioclase are equally efficient at releasing Ca to the aqueous solution and adding alkalinity to the aqueous phase at pH 3.6. This pH is close to that expected in waters charged with CO2 at pressures of 10 to 30 bars. Notably, the dissolution rates of these minerals are found to increase mildly with increasing fluid ionic strength to as much as 2.1 mol kg H2O-1. Taken together these results support the likely success of mineral carbonation of basaltic rocks located in saline aquifers.
Conflict of Interests
The authors declare that they have no conflict of interest regarding t content of this manuscript.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
K.G. Mesfin: Performed the experiments; D. Wolff-Boenisch: Designed experiments, S.R Gislason: Designed Experiments; E.H. Oelkers: Interpreted data and wrote manuscript.
Acknowledgments
We thank our friends and colleagues at University of Iceland, Iwona M. Galeczka, Snorri Gudbrandsson and Eydis Eiriksdottir for their help during this study. This project was funded by the Icelandic Science Foundation RANNÍS Geothermal Research Group GEORG 09-02-001. EHO and SRG would like to thank professors Hussein A. Hoteit, and Abdulkader M. Alafifi for their hospitality while EHO and SRG stayed at KAUST University Saudi Arabia in 2023 where much of this manuscript was revised.
References
- Aagaard, P.; Helgeson, H.C. Thermodynamic and kinetic constraints on the dissolution of feldspars. Geol. Soc. Am. Abstr. Progr. 1977, 9, 873. [Google Scholar]
- Aagaard, P.; Helgeson, H.C. Thermodynamic and kinetic constraints on reaction-rates among minerals and aqueous-Solutions.1. Theoretical Considerations. Am. J. Sci. 1982, 282, 237–285. [Google Scholar] [CrossRef]
- Alfredsson, H.A.; Hardarson, B.S.; Franzson, H.; Gislason, S.R. CO2 sequestration in basaltic rock at the Hellisheidi site in SW Iceland: Stratigraphy and chemical composition of the rocks at the injection site. Min. Mag. 2008, 72, 1–5. [Google Scholar] [CrossRef]
- Brady, P.V.; Walther, J.V. Kinetics of quartz dissolution at low temperatures. Chem. Geol. 1990, 82, 253–264. [Google Scholar] [CrossRef]
- Brantley, S.L. 5.03 - Reaction Kinetics of Primary Rock-forming Minerals under Ambient Conditions, in: Holland, H.D., Turekian, K.K. (Eds.), Treatise on Geochemistry. Pergamon, Oxford, 2003; pp. 73–117.
- Brantley, S.L.; Kubicki, J.; White, A. (Eds.) , 2008. Kinetics of Water-Rock Interaction. Springer-Verlag, New York, 2008; 833 p.
- Carroll, S.A.; Knauss, K.G. Dependence of labradorite dissolution kinetics on CO2(aq), Al(aq), and temperature. Chem. Geol. 2005, 217, 213–225. [Google Scholar] [CrossRef]
- Casey, W.H.; Westrich, H.R.; Arnold, G.W. Surface chemistry of labradorite feldspar reacted with aqueous solutions at pH 2, 3, and 12. Geochim. Cosmochim. Acta 1988, 52, 2795–2807. [Google Scholar] [CrossRef]
- Casey, W.H.; Westrich, H.R.; Arnold, G.W.; Banfield, J.F. The surface chemistry of dissolving labradorite feldspar. Geochim. Cosmochim. Acta 1989, 53, 821–832. [Google Scholar] [CrossRef]
- Celia, M.A.; Bachu, S.; Nordbotten, J.M.; Bandilla, K.W. Status of CO2 storage in deep saline aquifers with emphasis on modeling approaches and practical simulations. Water Resour. Res. 2015, 51, 6846–6892. [Google Scholar] [CrossRef]
- Clark, D.E.; Galeczka, I.M.; Dideriksen, K.; Voigt, M.J.; Wolff-Boenisch; Gislason, S. R. Experimental observations of CO2-water-basaltic glass interaction in a large column reactor experimental 50 oC. Int. J. Greenhouse Gas Cont. 2019, 89, 9–19. [Google Scholar] [CrossRef]
- Crovisier, J.L.; Fritz, B.; Grambow, B. , Eberhart, J.P. Dissolution of basaltic glass in seawater: Experiments and thermodynamic modelling. MRS Online Proceedings Library 1985, 50, 273–282. [Google Scholar] [CrossRef]
- Crovisier, J.L.; Honnorez, J.; Eberhart, J.P. Dissolution of basaltic glass in seawater: Mechanism and rate. Geochim. Cosmochim. Acta 1987, 51, 2977–2990. [Google Scholar] [CrossRef]
- Cygan, R.T.; Casey, W.H.; Boslough, M.B.; Westrich, H.R.; Carr, M.J.; Holdren, G.R., Jr. Dissolution kinetics of experimentally shocked silicate minerals. Chem. Geol. 1989, 78, 229–244. [Google Scholar] [CrossRef]
- de Obeso, J.C.; Awolayo, A.N.; Nightingale, M.J.; Tan, C.; Tutolo, B.M. Experimental study on plagioclase dissolution rates at conditions relevant to mineral carbonation of seafloor basalts. Chem. Geol. 2023, 620, 121348. [Google Scholar] [CrossRef]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 3, 107. [Google Scholar] [CrossRef]
- Flaathen, T.K.; Gislason, S.R.; Oelkers, E.H. The effect of aqueous sulphate on basaltic glass dissolution rates. Chem. Geol. 2010, 277, 345–354. [Google Scholar] [CrossRef]
- Gadikota, G.; Matter, J.; Kelemen, P.; Brady, P.V.; Park, A.-H. Elucidating the differences in the carbon mineralization behaviours of calcium and magnesium bearing alumino-silicates and magnesium silicates for CO2 storage. Fuel 2020, 277, 117900. [Google Scholar] [CrossRef]
- Galeczka, I.M.; Stefánsson, A.; Kleine, B.I.; Gunnarsson-Robin, J.; Ó Snæbjörnsdóttir, S.Ó.; Sigfússon, B.; Gunnarsdóttir, S.H.; Weisenberger,T. B.; Oelkers, E.H. A pre-injection assessment of CO2 and H2S mineralization reactions at the Nesjavellir (Iceland) geothermal storage site. Int. J. Greenhouse Gas Cont. 2022, 115, 103610. [Google Scholar] [CrossRef]
- Gislason, S.R.; Eugster, H.P. Meteoric water-basalt interactions: I. A laboratory study. Geochim. Cosmochim. Acta 1987, 51, 2827–2840. [Google Scholar] [CrossRef]
- Gislason, S.R.; Oelkers, E.H. Mechanism, rates, and consequences of basaltic glass dissolution: II. An experimental study of the dissolution rates of basaltic glass as a function of pH and temperature. Geochim. Cosmochim. Acta 2003, 67, 3817–3832. [Google Scholar] [CrossRef]
- Gislason, S.R.; Oelkers, E.H. Carbon Storage in Basalt. Science 2014, 344, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Gislason, S. R.; Wolff-Boenisch, D.; Stefansson, A.; Oelkers, E. H.; Gunnlaugsson, E.; Sigurdardottir, H.; Sigfusson, B.; Broecker, W.S.; Matter, J. M.; Stute, M.; et al. Mineral sequestration of carbon dioxide in basalt: A preinjection overview of the CarbFix project. Int. J. Greenhouse Gas Control 2010, 4, 537–545. [Google Scholar] [CrossRef]
- Goldberg, D.S.; Slagle, A.L. A global assessmnet of deep-sea basalt sites for carbon storage. Energy Proced. 2009, 1, 3675–3682. [Google Scholar] [CrossRef]
- Goldberg, D.S.; Takahashi, T.; Slagel, A. Carbon dioxide sequestration in deep-sea basalt. Proc. Nat. Acad. Sci. USA 2008, 105, 9920–9925. [Google Scholar] [CrossRef]
- Gudbrandsson, S.; Wolff-Boenisch, D.; Gislason, S.R.; Oelkers, E.H. Experimental determination of plagioclase dissolution rates as a function of its composition and pH at 22 oC. Geochim. Cosmochim. Acta 2014, 139, 154–172. [Google Scholar] [CrossRef]
- Guy, C. Mécanismes de dissolution des solides dans les solutions hydrothermales deduits du comportement des verres basaltiques et de calcites deformees. Universite Paul Sabatier, Toulouse, France., 1989.
- Gysi, A.P.; Stefansson, A. CO2–water–basalt interaction. Numerical simulation of low temperature CO2 sequestration into basalts. Geochim. Cosmochim. Acta 2011, 75, 4728–4751. [Google Scholar] [CrossRef]
- Hellevang, H.; Pham, V.T.H.; Aagaard, P. Kinetic modelling of CO2–water–rock interactions. Int. J. Greenhouse Gas Contr. 2013, 15, 3–15. [Google Scholar] [CrossRef]
- Heřmanská, M.; Voigt, M.J.; Marieni, C.; Declercq, J.; Oelkers, E.H. A comprehensive and internally consistent mineral dissolution rate database: Part I: Primary silicate minerals and glasses. Chem. Geol. 2022, 597, 120807. [Google Scholar] [CrossRef]
- Icenhower, J.P.; Dove, P.M. Solution kinetic effects of amorphous silica into sodium chloride solutions: Effects of temperature and ionic strength. Geochim. Cosmochim. Acta 2000, 64, 4193–4203. [Google Scholar] [CrossRef]
- Kampman, N.; Bickle, K.; Becker, J.; Assayag, N.; Chapman, H. Feldspar dissolution kinetics and Gibbs free energy dependence in a CO2-enriched groundwater system, Green River Utah. Earth Planet. Sci. Lett. 2009, 284, 473–488. [Google Scholar]
- Kelemen, P.B.; Benson, S.M.; Pilorge, H.; Psarras, P.; Wilcox, J. An overview of the status and challenges of CO2 storage in minerals and geological formations. Front. Clim. 2019, 2019, 00009. [Google Scholar] [CrossRef]
- Kelemen, P.B.; McQueen, N.; Wilcox, J.; Renforth, P.; Dipple, G.; Vankeuren, A.P. Engineered carbon mineralization in ultramafic rocks for CO2 removal from air: Review and new insights. Chem. Geol. 2020, 550, 119628. [Google Scholar] [CrossRef]
- Kristmannsdottir, H. Anorthosite inclusions in tertiary dolerite from the island groups Hrappsey and Purkey, West Iceland. J. Geol. 1971, 79, 741–748. [Google Scholar] [CrossRef]
- Kumar, S.; Foroozesh, J.; Edlmann, K.; Rezk, M.G.; Lim, C.Y. A comprehensive review of value-added CO2 sequestration in subsurface saline aquifers. J. Natural Gas Sci. Eng. 2020, 81, 103437. [Google Scholar] [CrossRef]
- Lasaga, A.C. Transition state theory. Rev. Min. 1981, 8, 135–169. [Google Scholar]
- Luo, S.; Xu, R.; Jiang, P. Effect of reactive surface area of minerals on mineralization trapping of CO2 in saline aquifers. Pet. Sci. 2012, 9, 400–407. [Google Scholar] [CrossRef]
- Marieni, C.; Voigt, M. , Clark, D.E., Gíslason, S.R., Oelkers, E.H. Mineralization potential of water-dissolved CO2 and H2S injected into basalts as function of temperature: Freshwater versus Seawater. Int. J. Greenhouse Gas Cont. 2021, 109, 103357. [Google Scholar] [CrossRef]
- Matter, J.M.; Broecker, W.S.; Gislason, S.R.; Gunnlaugsson, E.; Oelkers, E.H.; Stute, M.; Sigurdardottir, H.; Stefansson, A.; Alfreðsson, H.A.; Aradóttir, E.S.; et al. The CarbFix Pilot Project-Storing carbon dioxide in basalt. Energy Procedia 2011, 4, 5579–5585. [Google Scholar] [CrossRef]
- Matter, J.M.; Kelemen, P.B. Enhanced in situ carbonation of peridotite for permanent CO2 storage. Geochim. Cosmochim. Acta 2009, 73, A848. [Google Scholar]
- Matter, J.M.; Takahashi, T.; Goldberg, D. Experimental evaluation of in-situ CO2–water–rock reactions during CO2 injection in basaltic rocks: Implicationsfor geological CO2 sequestration. Geochem. Geophys. Geosyst. 2007, 8, Q02001. [Google Scholar] [CrossRef]
- Matter, J.M.; Stute, M.; Snæbjörnsdottir, S.Ó.; Oelkers, E.H.; Gislason, S.R.; Aradottir, E.S.; Sigfusson, B.; Gunnarsson, I.; Sigurdardottir, H.; Gunnlaugsson, E.; et al. Rapid carbon mineralization for permanent disposal of anthropogenic carbon dioxide emissions. Science 2016, 352, 1312–1314. [Google Scholar] [CrossRef]
- McGrail, B.P.; Schaef, H.T.; Ho, A.M.; Chien, Y.-J.; Dooley, J.J.; Davidson, C. L. Potential for carbon dioxide sequestration in flood basalts. J. Geophys. Res. 2006, 111, B12201. [Google Scholar] [CrossRef]
- McGrail, B.P.; Spane, F.A.; Amonette, J.E.; Thompson, C.R.; Brown, C.F. Injection and Monitoring at the Wallula Basalt Pilot Project. Energy Proced. 2014, 63, 2939–2948. [Google Scholar] [CrossRef]
- Munz, I.A.; Brandvoll, Ø.; Haug, T.A.; Iden, K.; Smeets, R.; Kihle, J.; Johansen, H. Mechanisms and rates of plagioclase carbonation reactions. Geochim. Cosmochim. Acta 2012, 77, 27–51. [Google Scholar] [CrossRef]
- Nesbitt, H.; Macrae, N.; Shotyk, W. Congruent and incongruent dissolution of labradorite in dilute, acidic, salt solutions. J. Geol. 1991, 99, 429–442. [Google Scholar] [CrossRef]
- Oelkers, E.H. General kinetic description of multioxide silicate mineral and glass dissolution. Geochim. Cosmochim. Acta 2001, 65, 3703–3719. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Gislason, S.R. The mechanism, rates and consequences of basaltic glass dissolution: I. An experimental study of the dissolution rates of basaltic glass as a function of aqueous Al, Si and oxalic acid concentration at 25 _C and pH = 3 and 11. Geochim. Cosmochim. Acta 2001, 65, 3671–3681. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Gislason, S.R.; Matter, J. Mineral carbonation of CO2. Elements 2008, 4, 333–337. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Gislason, S.R.; Kelemen, P.B. Moving Subsurface Carbon Mineral Storage Forward. Carbon Capture Sci. Tech. 2023, 7, 100098. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Golubev, S.V.; Pokrovsky, O.S.; Bénézeth., P. Do organic ligands affect calcite dissolution rates? Geochim. Cosmochim. Acta 2011, 75, 1799–1813. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Schott, J. Experimental study of anorthite dissolution and the relative mechanism of feldspar hydrolysis. Geochim. Cosmochim. Acta 1995, 59, 5039–5053. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Schott, J.; Devidal, J.-L. The effect of aluminium, pH, and chemical affinity on the rates of aluminosilicate dissolution reactions. Geochim. Cosmochim. Acta 1994, 58, 2011–2024. [Google Scholar] [CrossRef]
- Parruzot, B.; Jollivet, P.; Rebiscoul, D.; Gin, S. Long-term alteration of basaltic glass: Mechanisms and rates. Geochim. Cosmochim. Acta 2015, 154, 28–48. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3 — A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations, in: U.S. Geological Survey Techniques and Methods, 2013, Book 6, Chapter A43. pp. 6–43A.
- Pham, V.T.H.; Lu, P.; Aagaard, P.; Zhu, C.; Hellevang, H. On the potential of CO2–water–rock interactions for CO2 storage using a modified kinetic model. Int. J. Greenhouse Gas Contr. 2011, 5, 1002–1015. [Google Scholar] [CrossRef]
- Prikryl, J.; Jha, D.; Stefansson, A.; Stipp, S.L.S. Mineral dissolution in porous media: An experimental and modelling study on kinetics, porosity and surface area evolution. App. Geochem. 2017, 87, 57–70. [Google Scholar] [CrossRef]
- Ralston, S.J.; Peretyazhko, T.S.; Sutter, B.; Ming, D.W.; Morris, R.V.; Garcia, A.; Ostwald, A. Phyllosilicate formation on early Mars via open-system acid alteration of basaltic glass. Earth Planet. Sci. Let. 2023, 603, 117987. [Google Scholar] [CrossRef]
- Ringrose, P.S.; Furre, A.-K.; Gilfillan, S.M.V.; Krevor, S.; Landrø, M.; Leslie, R.; Meckel,T. ; Nazarian, B., Zahid, A. Storage of carbon dioxide in saline aquifers: Physicochemical processes, key constraints, and scale-up potential. Ann. Rev. Chem. Biomolecular 2021, 12, 471–494. [Google Scholar] [CrossRef]
- Saetre, C.; Hellevang, H.; Riu, L.; Dypvik, H.; Pilorget, C.; Poulet, F.; Werner, S.C. Experimental hydrothermal alteration of basaltic glass with relevance to Mars. Meteoritics Planet. Sci. 2018, 54, 357–378. [Google Scholar] [CrossRef]
- Saldi, G.D.; Schott, J.; Pokrovsky, O.S.; Oelkers, E.H. An experimental study of magnesite dissolution rates at neutral to alkaline conditions and 150 and 200 oC as a function of pH, total dissolved carbonate concentration, and chemical affinity. Geochim. Cosmochim. Acta 2010, 74, 6344–6356. [Google Scholar] [CrossRef]
- Schott, J.; Oelkers, E. H. Dissolution and crystallization rates of silicate minerals as a function of chemical affinity. Pure Appl. Chem. 1995, 67, 903–910. [Google Scholar] [CrossRef]
- Schott, J.; Pokrovsky, O.S.; Oelkers, E.H. The link between mineral dissolution/ precipitation kinetics and solution chemistry. Rev. Mineral. Geochem. 2009, 70, 207–258. [Google Scholar] [CrossRef]
- Schaef, H.T.; McGrail, B.P. Dissolution of Columbia River Basalt under mildly acidic conditions as a function of temperature: Experimental results relevant to the geological sequestration of carbon dioxide. Appl. Geochem. 2009, 24, 980–987. [Google Scholar] [CrossRef]
- Schaef, H.T.; McGrail, B.P.; Owen, A.T. Basalt–CO2–H2O interactions and variability in carbonate mineralization rates. Energy Procedia 2009, 1, 4899–4906. [Google Scholar] [CrossRef]
- Shikazono, N.; Harada, H.; Ikeda, N.; Kashiwagi, H. Dissolution of basaltic rocks and its application to underground sequestration of CO2 – estimate of mineral trapping by dissolution–precipitation simulation. Jpn. Mag. Mineral. Petrol. Sci. 2009, 38, 149–160. [Google Scholar]
- Sjoberg, L. the effect of pH and phthalic acid on labradorite dissolution kinetoics. Geol. Foren. I Stockhom Forh. 1985, 107, 311–313. [Google Scholar] [CrossRef]
- Snæbjörnsdóttir, S.Ó.; Gislason, S.R.; Galeczka, I.M.; Oelkers, E.H. Reaction path modelling of in-situ mineralisation of CO2 at the CarbFix site at Hellisheidi, SW-Iceland. Geochimica et Cosmochimica Acta 2018, 220, 348–366. [Google Scholar] [CrossRef]
- Snæbjörnsdóttir, S.Ó.; Sigfússon, B.; Marieni, C.; Goldberg, D.; Gislason, S.R.; Oelkers, E.H. Carbon dioxide storage through mineral carbonation. Nat. Rev. Earth Environ. 2020, 1, 90–102. [Google Scholar] [CrossRef]
- Stockmann, G.J.; Wolff-Boenisch, D.; Gislason, S.R.; Oelkers, E.H. Do carbonate precipitates affect dissolution kinetics? 1: Basaltic glass. Chem. Geol. 2011, 284, 306–316. [Google Scholar] [CrossRef]
- Stockmann, G.J.; Shirokova, L.S.; Pokrovsky, O.S.; Bénézeth, P.; Bovet, N.; Gislason, S. R.; Oelkers, E.H. Does the presence of heterotrophic bacterium Pseudomonas reactans affect basaltic glass dissolution rates? Chem. Geo. 2012, 296–297, 1–18. [Google Scholar] [CrossRef]
- Taylor, A.S.; Blum, J.D.; Lasaga, A.C. The dependence of labradorite dissolution and Sr isotope release rates on solution saturation state. Geochim. Cosmochim. Acta 2000, 64, 2389–2400. [Google Scholar] [CrossRef]
- Tester, J.W.; Worley, W.G.; Robinson, B.A.; Grigsby, C.O.; Feerer, J.L. Correlating quartz dissolution kinetics in pure water from 25 to 625°C. Geochim. Cosmochim. Acta 1994, 58, 2407–2420. [Google Scholar] [CrossRef]
- Tutolo, B.M.; Aaolayo, A.; Brown, C. Alkalinity generation constraints on basalt carbonation for carbon dioxide removal at the gigaton-per-year scale. Environ. Sci. Technol. 2021, 55, 906–915. [Google Scholar] [CrossRef]
- Voigt, M.; Marieni, C.; Clark, D.E.; Gíslason, S.R.; Oelkers, E.H. Evaluation and refinement of thermodynamic databases for mineral carbonation. Energy Procedia 2018, 146, 81–91. [Google Scholar] [CrossRef]
- Voigt, M.; Marieni, C.; Baldermann, A.; Galeczka, I.M.; Wolff-Boenisch, D.; Oelkers, E.H.; Gislason, S.R. An experimental study of basalt–seawater–CO2 interaction at 130 °C. Geochim. Cosmochim, Acta 2021, 308, 21–41. [Google Scholar] [CrossRef]
- Welch, S.A.; Ullman, W.J. Feldspar dissolution in acidic and organic solutions. Compositional and pH dependence of dissolution rate. Geochim. Cosmochim. Acta 1996, 60, 2939–2948. [Google Scholar] [CrossRef]
- Wild, B.; Daval, D.; Guyot, F.; Knauss, K.G.; Pollet-Villard, M.; Imfeld, G. pH-dependent control of feldspar dissolution rate by altered surface layers. Chem. Geol. 2016, 442, 148–159. [Google Scholar] [CrossRef]
- Wolff-Boenisch, D.; Gislason, S.R.; Oelkers, E.H. The effect of fluoride on the dissolution rates of natural glasses at pH 4 and 25 oC. Geochim. Cosmochim. Acta 2004, 68, 4571–4582. [Google Scholar] [CrossRef]
- Wolff-Boenisch, D.; Gislason, S.R.; Oelkers, E.H.; Putnis, C.V. The dissolution rates of natural glasses as a function of their composition at pH 4 and 10.6, and temperatures from 25 to 74 oC. Geochim. Cosmochim. Acta 2004, 68, 4843–4858. [Google Scholar] [CrossRef]
- Wolff-Boenisch, D.; Gislason, S.R.; Oelkers, E.H. The effect of crystallinity on dissolution rates and CO2 consumption capacity of silicates. Geochim. Cosmochim. Acta 2006, 70, 858–870. [Google Scholar] [CrossRef]
- Wolff-Boenisch, D.; Galeczka, I.M. Flow-through reactor experiments on basalt-(sea)water-CO2 reactions at 90°C and neutral pH. What happens to the basalt pore space under post-injection conditions? Int. J. Greenhouse Gas Cont. 2018, 68, 176–190. [Google Scholar] [CrossRef]
- Wolff-Boenisch, D. On the buffer capacity of CO2-charged seawater used for carbonation and subsequent mineral sequestration. Energy Procedia 2021, 4, 3738–3745. [Google Scholar] [CrossRef]
- Wolff-Boenisch, D.; Wenau, S.; Gislason, S.R.; Oelkers, E.H. Dissolution of basalts and peridotite in seawater, in the presence of ligands, and CO2: Implications for mineral sequestration of carbon dioxide. Geochim. Cosmochim. Acta 2011, 75, 5510–5525. [Google Scholar] [CrossRef]
Figure 2.
Temporal evolution of silica concentrations is the outlet fluids during the basaltic glass dissolution experimental series G-100. This series consisted of a suite of four distinct experiments performed on a single basaltic glass powder, where the inlet solution composition was fixed by the addition to the aqueous inlet solution of HCl and 0.1 mol kg-1 of either NaCl, KCl, CaCl2 or MgCl2. The vertical lines denote times at which the intel fluid was changed, whereas the dashed horizontal lines correspond to steady-state outlet Si concentrations.
Figure 2.
Temporal evolution of silica concentrations is the outlet fluids during the basaltic glass dissolution experimental series G-100. This series consisted of a suite of four distinct experiments performed on a single basaltic glass powder, where the inlet solution composition was fixed by the addition to the aqueous inlet solution of HCl and 0.1 mol kg-1 of either NaCl, KCl, CaCl2 or MgCl2. The vertical lines denote times at which the intel fluid was changed, whereas the dashed horizontal lines correspond to steady-state outlet Si concentrations.
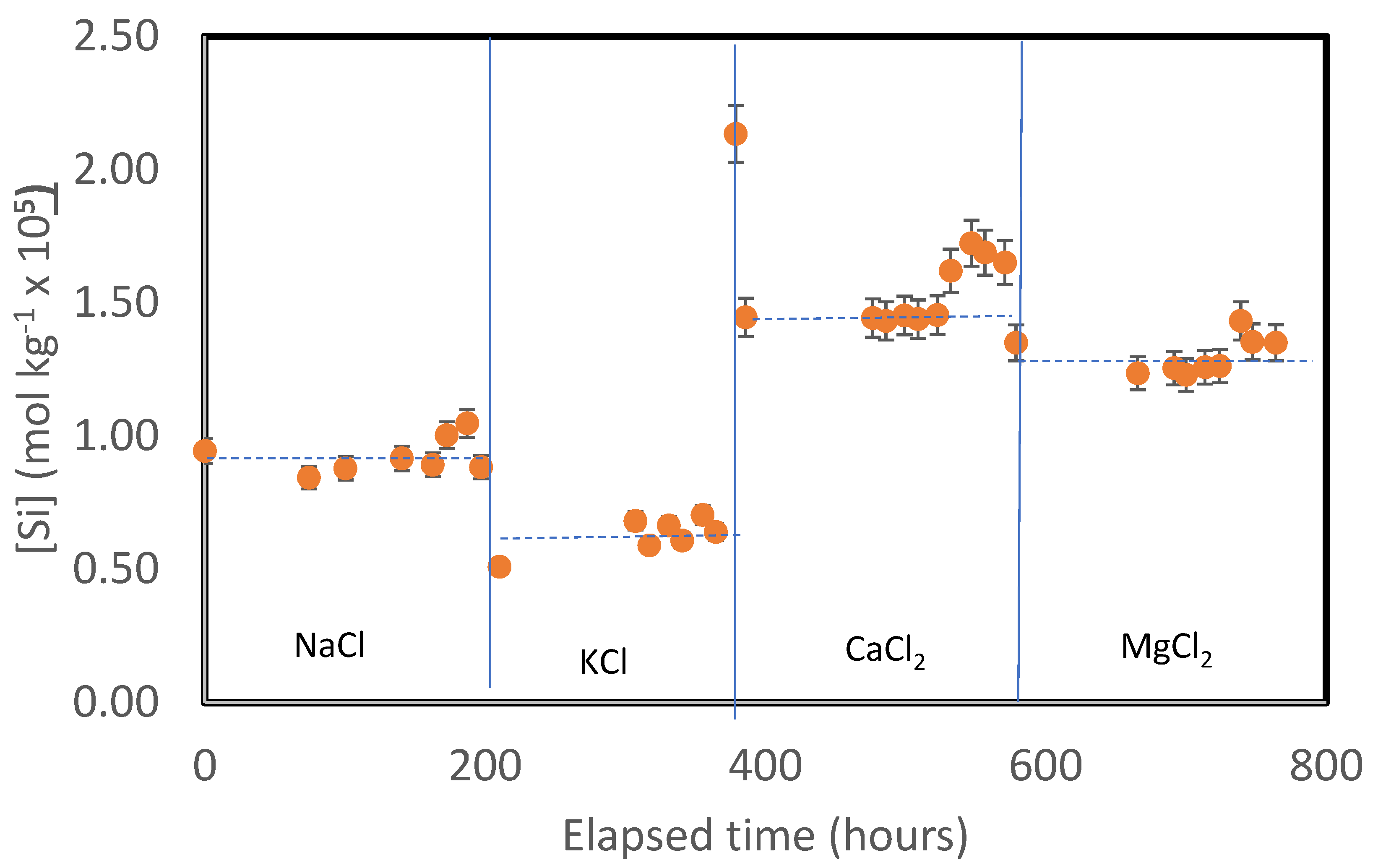
Figure 3.
Variation of measured steady state dissolution rates for a) basaltic glass and b) plagioclase as a function of the concentration of salt added to the inlet fluid. The open squares, open diamonds, filled circles and filled triangles represent rates measured in the presence of NaCl, KCl, CaCl2, and MgCl2, respectively.
Figure 3.
Variation of measured steady state dissolution rates for a) basaltic glass and b) plagioclase as a function of the concentration of salt added to the inlet fluid. The open squares, open diamonds, filled circles and filled triangles represent rates measured in the presence of NaCl, KCl, CaCl2, and MgCl2, respectively.
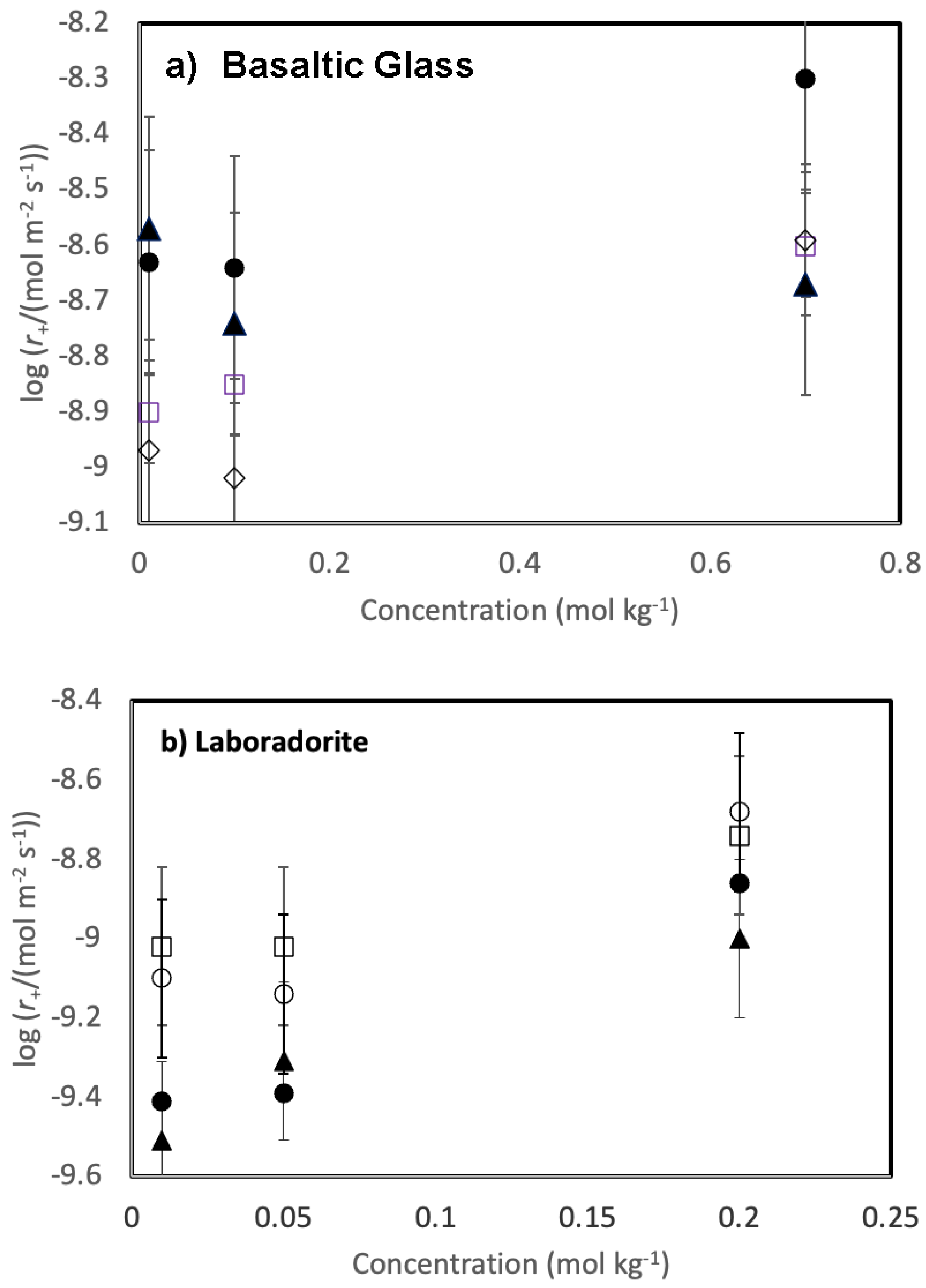
Figure 4.
Variation of measured steady state dissolution rates for a) basaltic glass and b) plagioclase as a function of the logarithm of the inlet fluid ionic strength. The dashed lines in this figure correspond to a least squares fit of the measured rates, where the equation of this line is provided in the figure.
Figure 4.
Variation of measured steady state dissolution rates for a) basaltic glass and b) plagioclase as a function of the logarithm of the inlet fluid ionic strength. The dashed lines in this figure correspond to a least squares fit of the measured rates, where the equation of this line is provided in the figure.
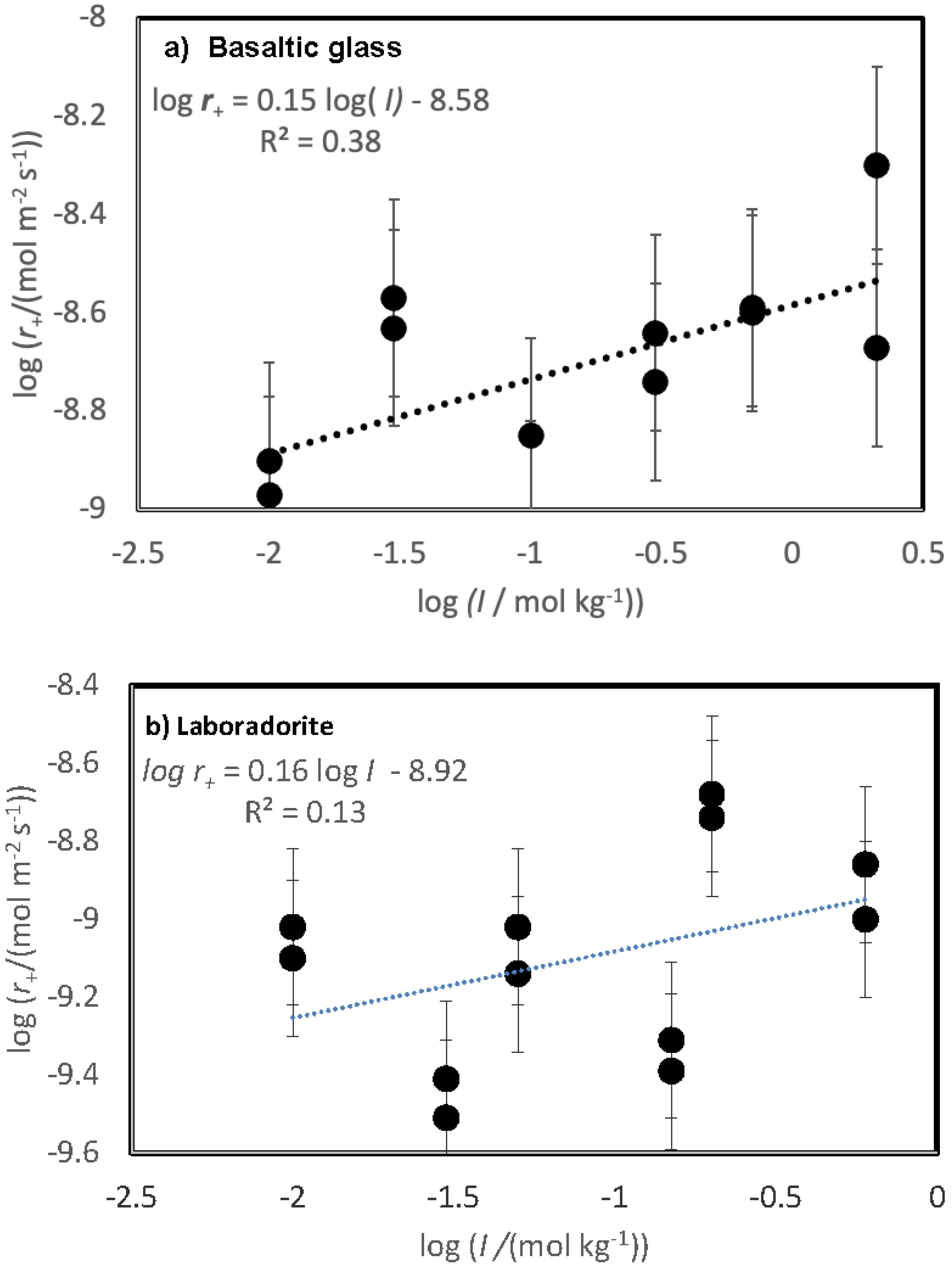
Figure 5.
Comparison between the logarithm of the Si release rate of basaltic glass and labradorite as a function of the logarithm of ionic strength. The open squares and filled circles represent the measured dissolution rates of basaltic glass and labradorite, respectively.
Figure 5.
Comparison between the logarithm of the Si release rate of basaltic glass and labradorite as a function of the logarithm of ionic strength. The open squares and filled circles represent the measured dissolution rates of basaltic glass and labradorite, respectively.
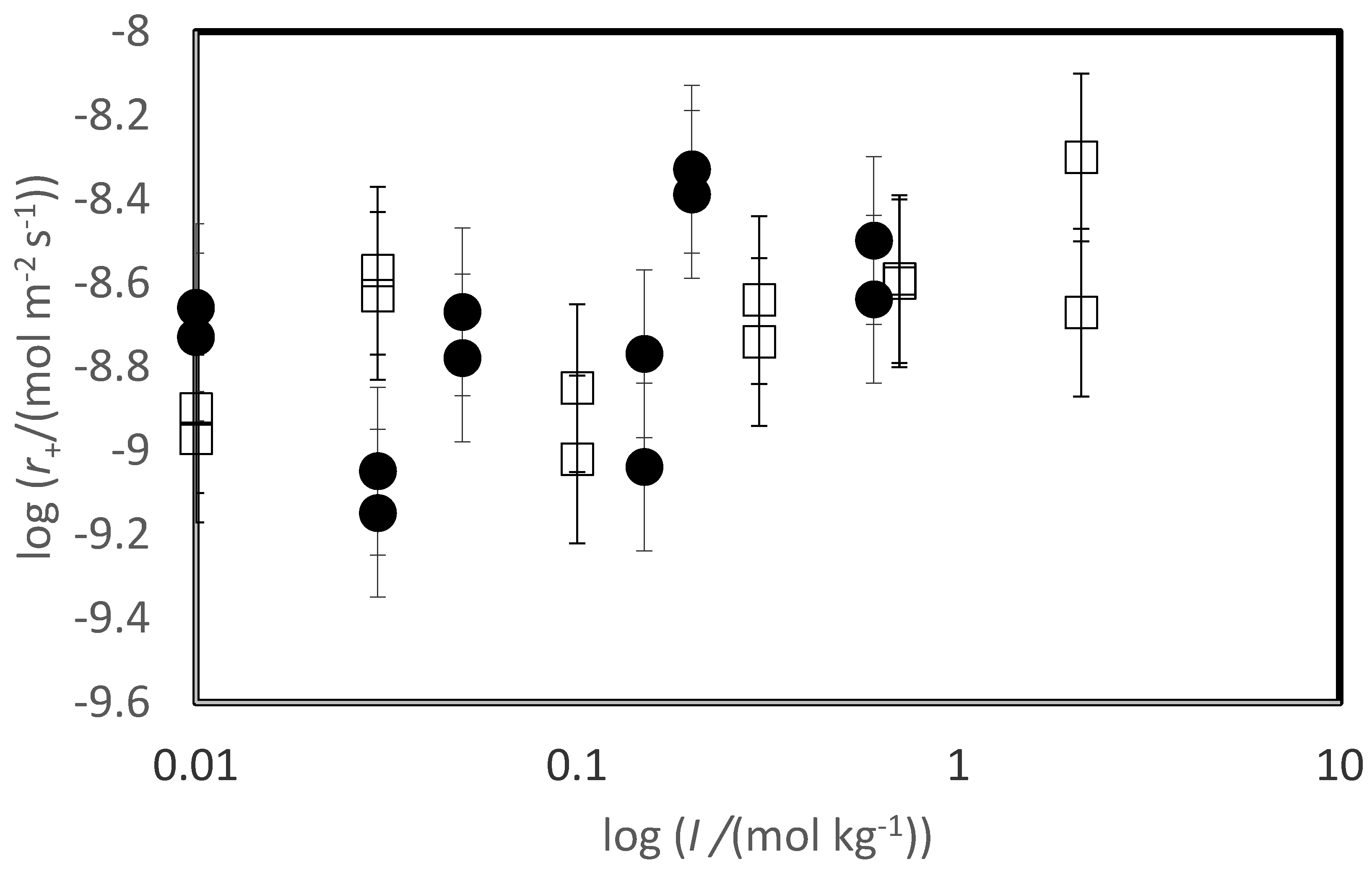
Figure 6.
Comparison of measured geometric surface area normalized steady state far-from-equilibrium dissolution rates measured in the present study with those reported in the literature as a function of pH at ~25 oC: a) Stapafell basaltic glass b) plagioclase with a composition of ~An70. The source of the data represented by the symbols are defined in the plot. The Gudbrandsson et al. (2014)* rates were performed using the same plagioclase sample as in the present study. Error bars correspond to a 0.2 uncertainty in measured rates.
Figure 6.
Comparison of measured geometric surface area normalized steady state far-from-equilibrium dissolution rates measured in the present study with those reported in the literature as a function of pH at ~25 oC: a) Stapafell basaltic glass b) plagioclase with a composition of ~An70. The source of the data represented by the symbols are defined in the plot. The Gudbrandsson et al. (2014)* rates were performed using the same plagioclase sample as in the present study. Error bars correspond to a 0.2 uncertainty in measured rates.
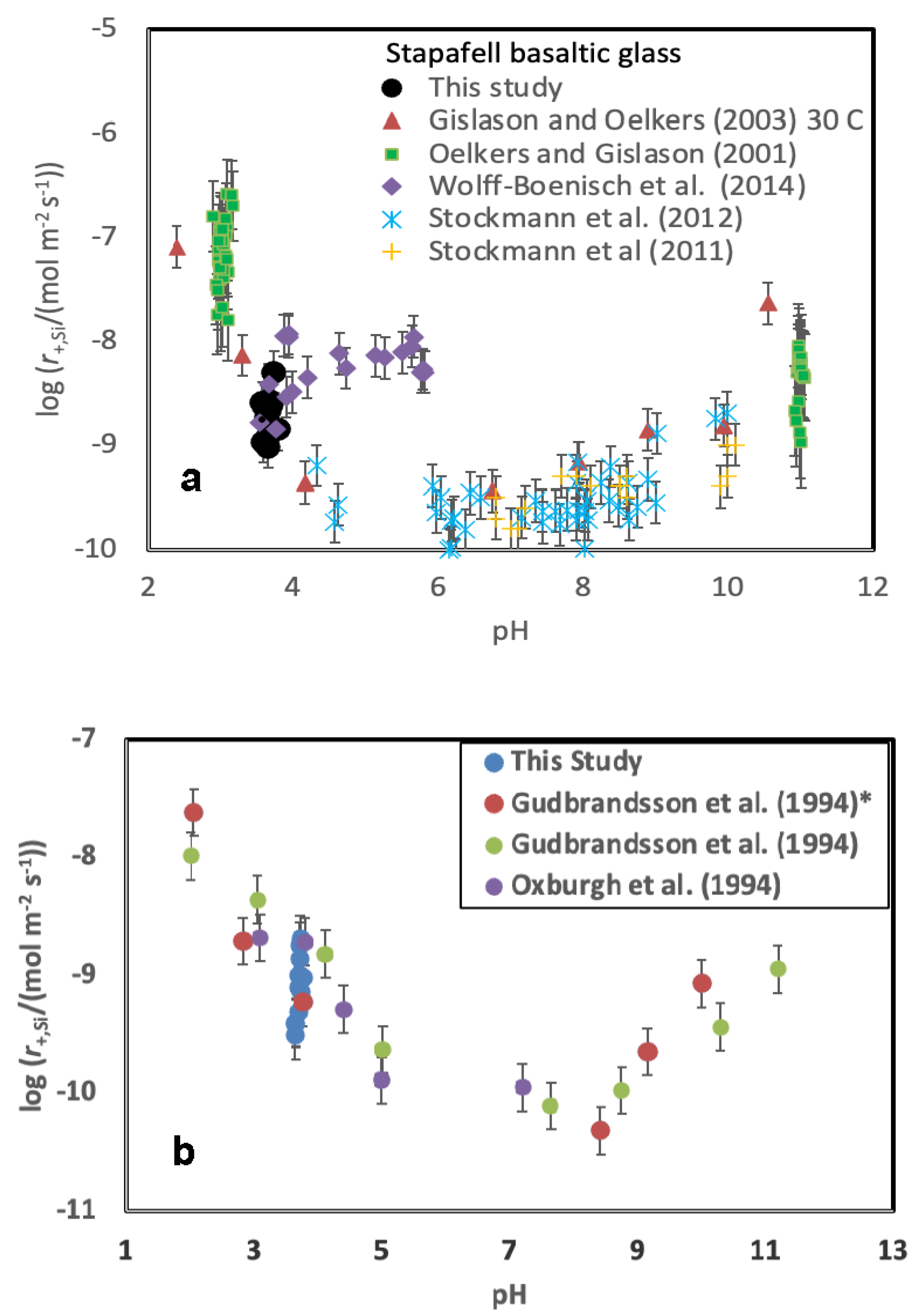

Table 1.
Chemical composition and surface areas of the basaltic glass and labradorite used in the study normalized to one silicon and to 8 oxygens, respectively.
Table 1.
Chemical composition and surface areas of the basaltic glass and labradorite used in the study normalized to one silicon and to 8 oxygens, respectively.
| Solid | Chemical composition | Ageo(m2/g) |
|---|---|---|
| Normalized to 1 silicon | ||
| Basaltic glass | Si1.000Al0.365Fe0.191Mn0.003 Mg0.294 Ca0.263 Na0.081 K0.008 Ti0.025 P0.004 O3.405 | 0.0284 |
| Labradorite | Si1.000Al0.684Ca0.286Na0.139K0.002Fe0.008O3.107Normalized to 8 Oxygen | 0.0284 |
| Basaltic glass | Si2.281 Al0.833 Fe0.436 Mn0.007 Mg0.669 Ca0.6 Na0.184 K0.019 Ti0.058 P0.008 O8 | 0.0284 |
| Labradorite | Si2.359Al1.612Ca0.674Na0.327K0.006Fe0.018O8 | 0.0284 |
Table 2.
Summary of measured steady-state dissolution rates generated in the present study. All experiments were performed at atmospheric PCO2.
Table 2.
Summary of measured steady-state dissolution rates generated in the present study. All experiments were performed at atmospheric PCO2.
| Experiment ID | Inlet Fluid Composition |
Ionic strength (mol kgw-1) | Exp. Duration (h) | pH in |
pH out |
[Si] out (mol x 105) |
Log r+si Geo1 |
|---|---|---|---|---|---|---|---|
| G-NaCl-10 | 0.01m NaCl, HCl | 0.01 | 210 | 3.59 | 3.67 | 0.95 | -8.90 |
| G-KCl-10 | 0.01m KCl, HCl | 0.01 | 176 | 3.57 | 3.60 | 0.81 | -8.97 |
| G-CaCl2-10 | 0.01m CaCl2.2H20, HCl | 0.03 | 193 | 3.55 | 3.61 | 1.77 | -8.63 |
| G-MgCl2-10 | 0.01m MgCl2.6H20, HCl | 0.03 | 209 | 3.64 | 3.67 | 2.00 | -8.57 |
| G-NaCl-100 | 0.1m NaCl, HCl | 0.1 | 210 | 3.86 | 3.80 | 1.06 | -8.85 |
| G-KCl-100 | 0.1mm KCl, HCl | 0.1 | 176 | 3.65 | 3.66 | 0.71 | -9.02 |
| G-CaCl2-100 | 0.1mm CaCl2.2H20, HCl | 0.3 | 193 | 3.53 | 3.66 | 1.75 | -8.64 |
| G-MgCl2-100 | 0.1mm MgCl2.6H20, HCl | 0.3 | 209 | 3.59 | 3.64 | 1.37 | -8.74 |
| G-NaCl-700 | 0.7mm NaCl, HCl | 0.7 | 210 | 3.57 | 3.71 | 1.92 | -8.60 |
| G-KCl-700 | 0.7m KCl, HCl | 0.7 | 176 | 3.64 | 3.58 | 1.94 | -8.59 |
| G-CaCl2-700 | 0.7m CaCl2.2H20, HCl | 2.1 | 193 | 3.56 | 3.74 | 3.77 | -8.30 |
| G-MgCl2-700 | 0.7m MgCl2.6H20, HCl | 2.1 | 209 | 3.63 | 3.67 | 1.62 | -8.67 |
| L-NaCl-10 | 0.01m NaCl, HCl | 0.01 | 208 | 3.68 | 3.76 | 1.80 | -9.02 |
| L-KCl-10 | 0.01m KCl, HCl | 0.01 | 223 | 3.62 | 3.73 | 1.38 | -9.09 |
| L-CaCl2-10 | 0.01m CaCl2.2H20, HCl | 0.03 | 236 | 3.64 | 3.72 | 0.67 | -9.41 |
| L-MgCl2-10 | 0.01m MgCl2.6H20, HCl | 0.03 | 237 | 3.64 | 3.70 | 0.53 | -9.51 |
| L-NaCl-50 | 0.05m NaCl, HCl | 0.05 | 208 | 3.64 | 3.74 | 1.64 | -9.03 |
| L-KCl-50 | 0.05m KCl, HCl | 0.05 | 223 | 3.66 | 3.73 | 1.26 | -9.14 |
| L-CaCl2-50 | 0.05m CaCl2.2H20, HCl | 0.15 | 236 | 3.61 | 3.65 | 0.70 | -9.40 |
| L-MgCl2-50 | 0.05m MgCl2.6H20, HCl | 0.15 | 237 | 3.65 | 3.65 | 0.84 | -9.13 |
| L-NaCl-200 | 0.2m NaCl, HCl | 0.2 | 208 | 3.63 | 3.71 | 3.12 | -8.75 |
| L-KCl-200 | 0.2m KCl, HCl | 0.2 | 223 | 3.66 | 3.65 | 3.57 | -8.69 |
| L-CaCl2-200 | 0.2m CaCl2.2H20, HCl | 0.6 | 236 | 3.66 | 3.69 | 2.37 | -8.86 |
| L-MgCl2-200 | 0.2m MgCl2.6H20, HCl | 0.6 | 237 | 3.68 | 3.70 | 1.73 | -9.00 |
1 Rates for basaltic glass are based on 1 Si per formula unit, whereas rates for labradorite are based on 2.36 Si per formula unit.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



