
Submitted:
04 April 2023
Posted:
06 April 2023
You are already at the latest version
Abstract
Small, Ubiquitin-like Modifier (SUMO) is a post-translational modifier with a profound influence on several key biological processes including the mammalian stress response. Of particular interest is its neuroprotective effects, first recognized in the 13-lined ground squirrel (Ictidomys tridecemlineatus), in the context of hibernation torpor. Although the full scope of the SUMO pathway is yet to be elucidated, observations of its importance in managing neuronal responses to ischemia, maintaining ion gradients, and preconditioning of neural stem cells, make it a promising therapeutic target for acute cerebral ischemia. Recent advances in high-throughput screening have enabled the identification of small molecules that can upregulate SUMOylation, some of which have been validated in pertinent preclinical models of cerebral ischemia. Accordingly, the present review aims to summarize current knowledge and highlight the translational potential of the SUMOylation pathway in brain ischemia.
Keywords:
stroke
; ischemia
; neuroprotection
; SUMOylation
; experimental therapeutics
1. Introduction
Despite landmark developments in chemical thrombolysis and mechanical thrombectomy in the past decades, ischemic stroke remains one of the greatest drivers of global disease burden [1-4]. Effective management options are often not available due to temporality of presentation and lack of access to specialist medical facilities, even in high income settings. Eligibility for alteplase/tenecteplase is dependent on patient presentation within 4.5-9 hours and absence of disqualifying comorbidities [5]. Endovascular thrombectomy is an effective method for revascularization, but not available to many patients as they are either not candidates for intervention, do not have access to a skilled operator at a stroke capable center, or may not present within the time window for intervention. In fact, a significant fraction of patients with ischemic stroke do not benefit from rapid recanalization—for example, between 2012-2018 in the United States, only 3.5% of stroke patients received thrombectomy, 9.3% received tPA, and 1% received both [6]. While the positive results reported in the DAWN and DEFUSE-3 trials have likely spurred wider utilization of these methods, the fact remains that the treatment paradigm remains the same: existing care for stroke focuses on the physiologic effects of flow and perfusion on bulk tissue and does not account for the cellular or molecular biological mechanisms of ischemic stroke. Even with advances in imaging to identify patients with less tissue injury and persistent at risk tissue, about half of patients taken for stroke intervention still have unfavorable outcomes (54% [7], 56% [2], 51% [3]). On the contrary, RESCUE-Japan LIMIT [8] , SELECT2 [9], and ANGEL-ASPECT [10] show that patients with significant tissue injury can still benefit from reperfusion.
While structural vessel occlusion is the initiating event in clinical stroke, the pathobiological sequelae that unfolds as a result of such ischemia must ultimately also be targeted and as such the expansion of treatment paradigms to include novel neuroprotetctive therapies will be required both for patients that are successfully treated (e.g., subjective to reperfusion injury) and those that are not. Accordingly, herein, we provide a clinically oriented summary of the current understanding of the role of the Small, Ubiquitin-like Modifier (SUMO) protein in endogenous neuroprotective mechanisms, recapitulate known therapeutic candidates acting on SUMO pathways (SUMOtherapeutics), and conclude by exploring potential future directions and approaches within the context of ischemic stroke.
1.1. Biological Significance of the SUMO Pathway
SUMO is a protein intrinsically involved in orchestrating physiological responses to hypoxia, hypothermia, and DNA damage [11]. SUMO conjugation of proteins (SUMOylation) was first implicated in the torpor conditioning of the 13-lined ground squirrel (Ictidomys tridecemlineatus). The analogous nature of this physiological mechanism of hibernation to the pathological mechanism of ischemic stroke and arousal to reperfusion has made it a direction for inquiry in the context of neuroprotection [12-14]. Postmortem observation of increased SUMOylation within the penumbra of ischemic stroke in human victims has made the potential exploitation of this pathway of clinical interest [15].
The SUMO pathway exerts its effects by way of post-translation modification akin to ubiquitinylation. However, while only 29 variants of ubiquitin binding domains have been identified in humans at the time of writing, over 14,000 SUMO binding domains have been found in human cells [16]. Conversely, whereas ubiquitinylation involves one of almost three dozen forms of the E2 conjugase, SUMOylation relies solely on one—UBC9 [17]. This combination of a vast number of SUMO binding domains coupled with a universal reliance on a single E2 conjugase has significant potential as a wide-reaching yet approachable therapeutic target.
Although 4 SUMO paralogs have been identified in humans, SUMO-1 and SUMO-2/3 (SUMO-2 and SUMO-3 being almost identical) represent the prevalent isotypes [17, 18]. The act of conjugating/deconjugating SUMO, SUMOylation and de-SUMOylation, is primarily carried out by seven Sentrin-specific proteases known as SENP1-7. Of this family, SENP1 (key for maturing translated SUMO) and SENP2 display the broadest substrate affinity for SUMO isotypes and have been found preferentially within the nucleus and nuclear pore complex. The other SENPs are either selectively active on a particular SUMO isoform or are not involved in SUMO maturation, making them of lesser interest for SUMOtherapeutics engineered for global upregulation. Other SUMO-involved enzymes such as DeSI-1, DeSI-2, and USPL1 have been identified but have been found to have weak endopeptidase activity and are not thought to be involved in global SUMOylation to the extent of SENP1 and SENP2 [11, 19, 20].
The exact extent of control exerted by SUMO and the bounds of its physiological interplay remain to be fully elucidated. SUMOylation is involved in numerous processes spanning most organ systems, with downstream effects occasionally resulting in simultaneous upregulation and downregulation of protein machinery like HIF-1a [11]. Knockout experiments have demonstrated that SUMO has a role, albeit undefined, in emotion, cognition, anxiety, and episodic memory [21, 22]. Preclinical work has shown that SUMO serves as a regulator of critical cellular processes including DNA repair, axonal trafficking, brain development, as well as neuronal plasticity and neurotransmission [19, 20, 23-26]. Furthermore, SUMOylation and de-SUMOylation have been identified as potential drivers of various CNS pathologies. Glioblastomas have been found to overexpress SUMO[22]; increased SUMOylation has been associated with Alzheimer’s Disease and Huntington’s Disease; and both increased SUMO1ylation and decreased SUMO2/3ylation have been implicated with alpha-synuclein aggregation in Parkinson’s disease [27]. Moreover, SUMOylation is known to play a role in cardiac [28, 29], renal [30], pulmonary, hepatic and mesenteric ischemic disease processes [14]. These observations, coupled with reports on the critical role of SUMO in cell survival during hyperacute ischemic pathologies such as myocardial infarction[31] make it of particular interest for the management of acute ischemic stroke.
1.2. Role of SUMO in Neuroprotection
The neuroprotective effects of SUMOylation have been widely observed both in vitro and in vivo. Due to the role of SUMO as the “modifier of modifiers” and the great physiological distance between the act of SUMOylation and its ultimate effect; a comprehensive pattern of association between end-effects and neuroprotection remains to be clearly demonstrated. The importance of SUMO in preserving neuronal integrity first observed in natura has been replicated in vitro and in vivo through key preclinical work [19, 21, 23, 25, 26, 32]. Furthermore, recent proteomics-based approaches continue to produce new mechanistic insights into the downstream effects of SUMO in contexts such as the response to cellular stress, development, and differentiation. Despite that, a clear and comprehensive awareness of causal relationships is yet to be defined.
The preclinical literature offers a variety of downstream effects of SUMOylation that may explain the observed impacts. As with other advances in systems biology, greater awareness of the panoply of interactions governed by SUMO represents an expanding list of potentially druggable nodes. A large portion of studies to date have demonstrated the importance of both SUMO1 [33] and SUMO2/3 [32] in neuroprotection. As SUMO proteases have a central role in neuroprotection, factors regulating the activity of these should be considered. For example, SENP1 and 3 have been associated with reversible modulation of activity dependent on levels of reactive oxygen species or genotoxic stress, suggesting a possible role as intracellular sentinels for impending insult [20, 34]. The interested reader is directed to excellent reviews by Droescher et al. and Filippopoulou et al. covering the variety of both physiologic and protective roles SUMO plays within the cell [11, 20].
1.2.1. SUMO and the Ischemia Response
The neuroprotective effects of SUMO regulation are most apparent in the context of the response to physiologic stressors, including hypoxic and ischemic insults. As with other aspects of SUMO-related research, the complete breadth of control exerted by SUMO over the ischemic stress response remains to be comprehensively defined. There are increasing reports of SUMO-interactions with first- and second-order stress pathways integral to neuronal survival. As such, SUMO is believed to be a critical intermediary in optimizing and balancing the various endogenous stress responses to favor regeneration and repair as opposed to apoptosis and degeneration.
Hypoxia-inducible factor (HIF) is a family of transcription modifier proteins that play a central role in the hypoxic response, wound healing, and angiogenesis. In the context of ischemic stroke, HIF activation is associated with increased protection and recovery after insult [35]. Although HIF expression varies based on ambient oxygen tension and interactions with other pathways such as Nuclear Factor κB (NF-κB), prolonged activation has been associated with non-scarring tissue regeneration and is a therapeutic target for several other pathologies [36]. Unsurprisingly, both SUMOylation and the SUMO pathway enzymes have been observed to play a role in both positive and negative regulation of the HIF pathway, with effects varying depending on both the agent and the target of SUMOylation [11]. The differing effects of the family of SUMO-associated E3 ligases is an example of the former—depending on which ligase is involved, HIF transcription may be positively regulated, negatively regulated, both, or unaffected. The E3 ligases Cbx4 and PIAS3 positively impact HIF-1α stability and transcription with and without SUMOylation, respectively [37-39]. Alternatively, PIAS1 SUMOylates and inhibits HIF-1B (also known as ARNT) without an appreciable effect on overall HIF transcription [40]. In some cases, opposing effects can be produced by the same E3 ligase depending on the target of SUMOylation—PIASγ SUMOylation of HIF-1α negatively impacts HIF stability and transcription, but PIASγ SUMOylation of pVHL results in increased ubiquitinylation of the same, ultimately resulting in increased HIF stability [41, 42]. Similar modulating SUMO interactions with the prolyl and asparaginyl hydroxylases that serve as negative feedback to HIF activation have also been reported: SUMOylation of PHD3 and FIH results in downregulation (via potentiation of extant negative feedback) and upregulation of HIF transcription [43, 44]. Finally, experiments in HeLa cultures subjected to oxygen-glucose deprivation (OGD) have demonstrated that changes in SUMOylation in more generalized transcription factors can inflect the activity of HIF-1—specifically, reduction in SUMO2/3ylation of TFAP2 results in an increase in the transcriptional activity of the same while also enhancing that of HIF-1 [45].
Whereas activation of HIF-1 is associated with neuroprotection, upregulation of NF-κB has been observed to correspond with inflammation and neuronal death [46]. Similar to its impact on HIF, SUMO pathways affect multiple nodes within the NF-κB pathway, occasionally with contradicting impacts. The known SUMO influences within the pathway relate primarily to IκBα and the IKK complex [36]. IκBα is a constitutively active inhibitor that functions by preventing nuclear localization of NF-κB. Experiments with over-expressed dominant negative murine homologues of Ubc9 observed delays in IκBα degradation and thus activation of the pathway after in vitro exposure to TNFα, implicating the SUMO E2 in the endogenous activation of NF-κB [47]. At the same time, SUMO1ylated IκBα was discovered in COS7 monkey kidney, HEK-293, and Jurkat human T lymphocyte cultures and was found to be resistant against ubiquitinoylation. Furthermore, the same study found that ubiquitin and SUMO1 competed for the same lysine residue (K21) within IκBα, suggesting competing influences of SUMO and Ubiquitin on IκBα stability and NF-κB activation [36, 48]. Similarly, SUMO has been found to exert indirect influence over IκBα by way of the IKK complex. IKK serves to enable nuclear localization of NF-κB by phosphorylating IκBα. SUMOylation of Annexin-A1 was found to suppress the NF-κB pathway by increasing IKKα degradation in a microglial model of oxygen-glucose deprivation [49]. On the other hand, SUMO1ylation of IKK-γ (also known as NEMO) resulted in greater nuclear trafficking and thus activation of NF-κB [50]. The proinflammatory impact of SUMO1ylating NEMO was demonstrated in reverse by the observation that in vitro overexpression of SENP1 resulted in increased de-SUMOylation of NEMO and concomitant reduction in NF-κB activation [51].
In addition to HIF-1 and NF-κB , SUMO has been found to act as a modulator, albeit less characterized, of many other cellular pathways. STAT, a signaling pathway involved in the inflammatory response, was suppressed by SUMOylation of synthetic liver X receptors (LXR) [52]. SUMOylation of the GluR6 subunit of the Kaianate Receptor (KAR) has been observed to downregulate the JNK cell-death pathway. Similarly, increased SUMOylation of the GluR2 subunit of the AMPA receptor has been observed in a murine model of middle cerebral artery occlusion [53]. Finally, OGD-induced degradation of SENP3 by PERK has been found to result in increased SUMO2/3ylation of Drp1, resulting in greater survival during ischemia but increased death during reperfusion [54-56]. As such, SUMO is integrally linked with the physiological response to ischemia.
1.2.2. Maintenance of Ion Homeostasis
Ion homeostasis is a critical factor in neuronal survival following an ischemic insult. SUMOylation directly or indirectly impacts the balance of calcium, sodium, and potassium within the neuron [55]. Hypoxic-ischemic injury to neurons is known to lead to massive disruption of Ca2+ and Na+ homeostasis, uncontrolled neurotransmitter release and subsequent excitotoxicity [57]. Artificial increases in synaptosomal SUMO1 and SENP1 have been shown to modulate calcium intake and glutamate release with opposing results observed with Kainate or KCl stimuli [58, 59]. In neurons of the dorsal root ganglia, increased SUMOylation has been found to reduce calcium influx and increase membrane migration of sodium channels [60]. The impact of SUMOylation on calcium homeostasis is thought to be due to regulatory interplay with the NCX3 sodium-calcium exchanger at the LYS590 f-loop: experiments with siRNA-induced knockdown of SUMO1 demonstrated a corresponding downregulation of NCX3 and thus exacerbation of ischemic damage in a rat model of MCAO [33].
In addition to direct impact on ion transporters themselves, SUMOylation has been observed modulating receptor expression, possibly as a means of mitigating excitotoxicity and thereby blunting the aforementioned calcium influx. SUMOylation of GluR6 subunit of the Kaianate receptor (KAR) has been found to cause a reduction in end-synaptic potential [61]. In line with these observations, SUMOylation has been shown to modulate synapse formation, neurotransmitter release via SNARE interactions, and synaptic plasticity, suggesting a fundamental role within neuronal synaptic architecture [62].
1.2.3. Preservation of Neural Stem Cell Populations
The brain maintains a limited ability to repair and regenerate itself by way of its small host of native neural stem cells (NSCs). However, its inability to fully compensate for the overwhelming damage caused by ischemic insults is evidenced by the profound morbidity that may follow ischemic stroke [6, 63] . Furthermore, advances in regenerative medicine brought about by exogenous stem cell therapies in other contexts are yet to be replicated for neuropathological indications [64, 65]. Preclinical successes have been observed with graft preconditioning and use of scaffolding but there are considerable challenges associated with such clinical translation.
The SUMO pathway also plays a critical role in the aforementioned quotidian homeostatic processes and responses to stress. Moreover, its role in cellular development and differentiation has been established by the discovery that SUMOylation of chromatin is integral in the maintenance of cellular identity—particularly that of the pluripotent stem cell [17]. Tahmasebi et al. reported not only an increase in Ubc9 expression during the transformation of murine embryonic fibroblasts into induced pluripotent stem cells (iPSCs), but also a profound decrease in iPSC transformation when Ubc9 functionality was exogenously suppressed [66]. Indeed, murine experiments found that knockout of either SUMO (in particular SUMO2) or associated proteases were lethal in utero [17, 67]. In this way, SUMOylation may represent a means of facilitating the survival and possibly expansion of endogenous NSC populations.
In the context of ischemic insult, use of SUMOylation to prepare exogenous NSCs for infarct-like conditions may serve as a form of in vitro preconditioning to enhance chances of exogenous graft survival and expansion [63]. This concept has been demonstrated both in vitro and in vivo: cultured NSCs with induced SUMO upregulation were found to have greater survivability against OGD and increased differentiation both within and without the murine brain [68]. Although the technology remains in its infancy, it has the potential to cause a paradigm shift in ischemic stroke - from damage control to anatomical and functional repair.
2. SUMOtherapeutics for Cerebral Ischemia
As the awareness of SUMO’s regulatory function expands, efforts to identify potentially druggable nodes and small molecule agents to exploit these are already underway. These SUMOtherapeutics can be broadly classified as either targeting a step within the SUMO pathways or as a direct upregulator of SUMOylation (Fig. 1). The former class is unsurprisingly comprised of compounds antagonizing endogenous SUMO inhibitors such as miRNA-182/183 and inhibiting deSUMOylating proteases such as SENP-2. Many of these so-called “inhibitor-inhibitors” have been identified through novel screening assays and are currently undergoing evaluation.
Figure 1.
SUMOylation in cerebral ischemia.
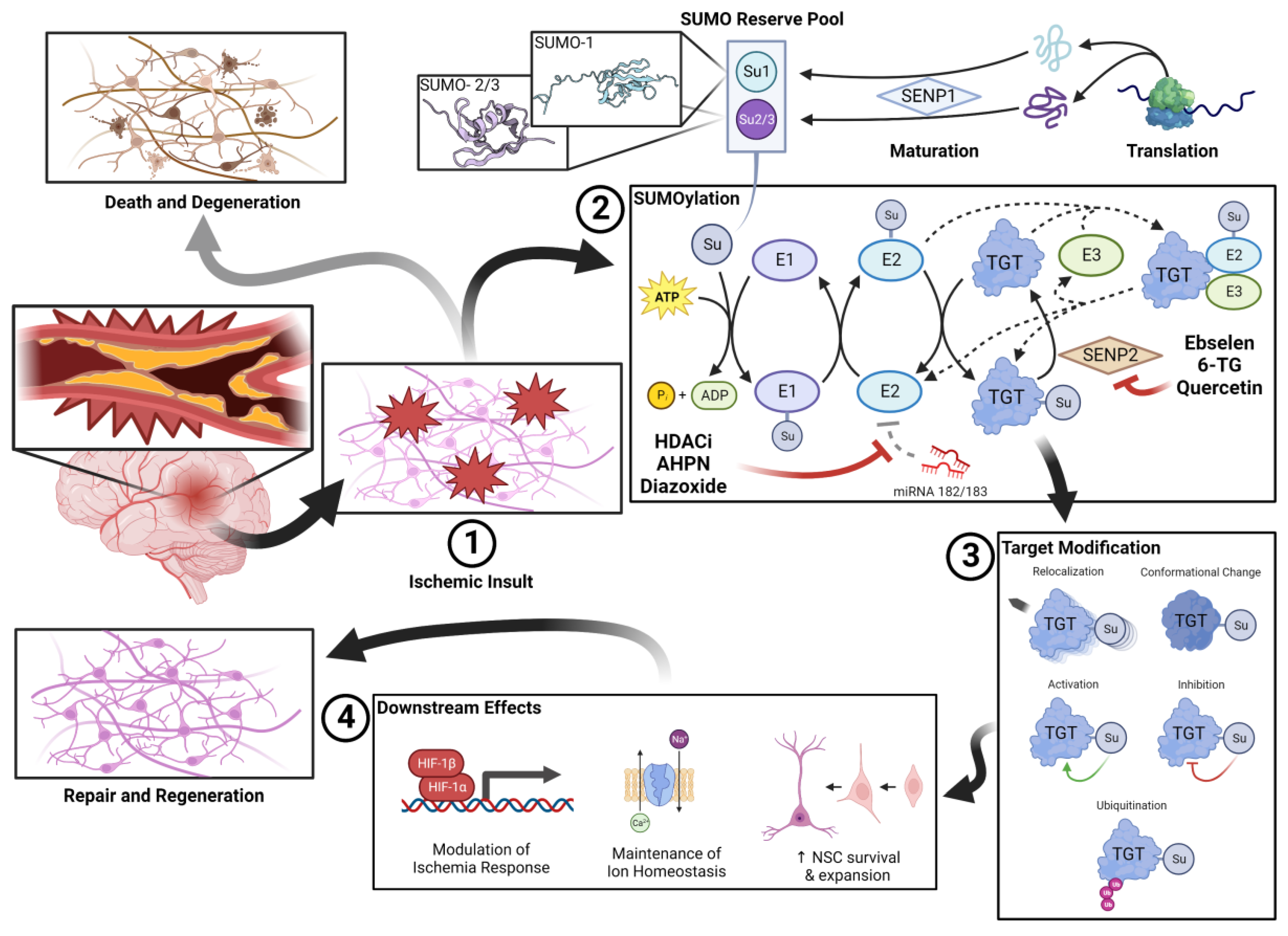
SUMOylation in cerebral ischemia. A pool of unconjugated SUMO is kept in reserve during normoxic conditions. After vascular occlusion, (1) the lack of perfusion results in an ischemic insult. When stressors are detected within the microenvironment, conjugation of the SUMO reserve is rapidly upregulated. SUMOylation (2) is a process analogous to that of ubiquitination. The process of SUMOylation and de-SUMOylation is highly dynamic and the subject of significant regulatory interplay. Endogenous deSUMOylators such as SENP2 and miRNA-182/183 thus represent therapeutic targets. SUMOylated target proteins have a variety of fates (3) that may impact their functionality, conformation, or location within the cellular milieu. Ultimately, SUMOylated proteins effect a variety of (4) pro-survival and pro-regeneration changes including managing the response to ischemia, managing ion homeostasis, and encouraging survival of endogenous/exogenous neural stem cell (NSC) populations. To this end, the regenerative nature of the SUMO response to ischemia stands in stark contrast against the apoptotic and degenerative response that is prevalent within the ischemic brain.
2.1. miRNA-182/183 Inhibitors
Micro-RNA is a negative regulation system that functions by silencing or degrading target mRNA. Based on experiments on hibernating ground squirrels, two major miRNA families have been identified in global SUMO suppression—miRNA-182 (including miR-182, miR-183, and miR-96) and miRNA-200 (including miR-200a/b/c, miR-141, miR-429) [69, 70]. As such, therapeutic upregulation and downregulation are simultaneously of interest due to the implication of SUMO in other pathologies [71, 72]. Advances in quantitative high-throughput screening have enabled the identification of miRNA-182 antagonists with druggable properties [73] (Table 1).
Despite many of the drugs returned by the screen being in preclinical stages of investigation, the histone deacetylase inhibitors (HDACi) romidepsin, panabinostat, entinostat, belinostat, and pracinostat are either currently clinically utilized or ongoing clinical evaluation. Panabinostat is of particular interest as it was found to be one of the most effective miRNA inhibitors when evaluated for both SUMO upregulation and protection against OGD [73]. Given that HDAC activity has been positively correlated with increased penumbra diameter in recent transcriptomics studies [74] and the encouraging in vivo performance of other HDACi (albeit ones with unknown interactions with SUMO) in models of stroke [75-77], the frequency of HDACi amongst the candidates output by the screen is of interest. However, in vivo evaluations of panabinostat and entinostat have been mixed; a study by Al Shoyaib et al. studying the effects of both drugs (oral formulations, dosed on alternate days from post-stroke day 5 to 15) on motor recovery in a photothrombotic model of ischemic stroke in CD-1 mice did not find statistically significant differences in motor recovery or infarct volume when compared to vehicle [78]. Another study in a CD-1 mouse model of intracerebral hemorrhage by Bonsack and Sukumari-Ramesh evaluated the effect of entinostat (delivered intraperitoneally 1 hour after insult at 10mg/kg in PBS) on sensorimotor deficit, neuroinflammation, acute neurodegeneration, and lesion volume and found significant reductions in all measures compared to control [79]. It is important to note that the relative paucity of other in vivo data on these specific drugs for this indication and extremely varied experimental parameters of the aforementioned studies makes comparing the results challenging.
The selective mitochondrial potassium-channel agonist diazoxide has also been identified as an inhibitor of miRNA-182/183. Observations of its protective effects on myocardium in ischemic heart disease inspired studies of its use for cerebral ischemia. A study evaluating infarct volume in a Wistar rat model of middle cerebral artery occlusion (MCAO) found an over two-fold reduction after diazoxide pretreatment [80]. Another study measured cerebral concentrations of heat-shock protein 25 and 70 (HSP25, HSP70) in the brains of Sprague-Dawley rats subject to cerebral ischemia as a consequence of hemorrhagic shock and observed significant increases in concentration with both pretreatment and posttreatment with diazoxide [81].
2.2. SENP Inhibitors
In addition to the significant research effort that has been directed towards understanding of the physiological role of SUMO, other efforts have focused on identifying potential therapeutic candidates by way of recent advances in high-throughput screening. Bernstock et al. developed AlphaScreen (PerkinElmer, MA, USA) as a facile and efficient means of identifying SENP-2 inhibitors (measured by an overall increase in SUMOylation). The primary screening batch consisted of over four thousand compounds from the Sigma LOPAC1280 (Library of Pharmaceutically Active Compounds) and NCATS NPC (National Center for Advancing Translational Science Pharmaceutical Collection), of which less than two hundred remained after the screen. After further in vitro and in vivo testing, eight drugs were identified as lead candidates, of which a final four significantly increased SUMO conjugation (Table 2) [82]. Of these candidates, antioxidant ethyl protocatechuate, and nonselective beta antagonist isoprenaline HCl have not been studied in the context of stroke. The nucleoside analogue 6-Thioguanine has also not been evaluated in the context of stroke, but its parent nucleoside has been identified as a SUMOylator and neuroprotectant [83].
Quercetin, a flavonoid commonly found in citrus fruits, onions, and broccoli, is a natural antioxidant found to have SENP-inhibiting properties [84]. Although successes at stroke prevention or neuroprotection enhancement have been widely reported in the preclinical literature, no consensus exists on mechanism of action. The preclinical literature suggests several possible mechanisms, including reduction of thrombotic inflammation, antithrombotic effects, prevention of oxidative stress, promotion of autophagy, and inhibition of apoptosis [85]. Although a recent meta-analysis of in vivo studies reported neuroprotective effects of quercetin for ischemic stroke, it found evidence of publication bias and methodological issues such as lack of blinding [86]. At the time of writing there has been no clinical advancement of quercetin as a therapeutic.
Another example of a known neuroprotectant that remains mired in translation, ebselen is a selenium-containing glutathione peroxidase mimic that was a known neuroprotective agent even before its SUMOylating properties were discovered. Furthermore, at least one mechanism for its neuroprotective properties other than its capacity as a SUMOylator has been elucidated—it is able to significantly mitigate the deleterious effects of glutamate excitotoxicity [87] and reperfusion injury [88]. At the turn of the 21st century it was a promising candidate for a putative protective agent for stroke, going so far as entering clinical trials in Japan. Although trial data were encouraging, showing statistically significant improvements in infarct volume and functional status [89, 90], these trial data did not approach the efficacy observed in animal models and in vitro studies. This, coupled with concerns over selenium toxicity caused regulatory approval and clinical implementation to stall [91, 92].
As such, the corpus of small animal and tissue culture research on ebselen and quercetin depicts a pattern of encouraging results hampered by difficulties in clinical translation and lack of standardization and may merit revisiting. For example, clinical trials utilized oral suspensions of ebselen, significantly increasing off-target exposure due to its lipophilic nature. Recent advances in catheter technology and mechanical thrombectomy could enable precise and controlled dosing of the compound. Such an approach would have the benefit of limiting off-target exposure and enabling dosing as soon as the thrombus is retrieved and reperfusion injury begins to occur—a characteristic correlated with increased neuronal survival [93, 94].
In addition to revisiting older compounds such as ebselen or quercetin with recently discovered SENP inhibitor characteristics, the potential for design and discovery of novel agents is increasingly at play. With recent advances in high-throughput screening (such as the previously mentioned AlphaScreen), AI-assisted drug design, and other in silico methods, investigating SENP inhibition is of great interest in a variety of clinical contexts. The interested reader is directed to an excellent review by Chen et al. for further information about the development of SENP inhibitors for a variety of other clinical indications [95].

Table 2.
SENP inhibitors evaluated for neuroprotection
| Drug | Tissue/ Animal |
Ischemia Model | Intervention | Measured Outcome |
Results Summary | Study | |
|---|---|---|---|---|---|---|---|
| Quercetin | SHSY5Y, B35, E18 PCN | SHSY5Y, B35: 16hr OGD + 5hr recoveryE18 PCN: 5hr OGD + 16hr recovery | Drug treatment ± pre-treatment | SENP activity, SUMOylation, cell survival, LDH release | Decrease in SENP expression*, Increases in SUMOylation*; increase in cell survival (in SHSY5Y and E18 PCN)*; Decrease in LDH release with co-treatment alone and with pre-treatment* | Lee et al., 2016 [84] | |
| Isoprenaline HCl | B35 | 20hr OGD | 4hr pretreatment + treatment | SUMO-1 expression, cell survival | Increase in SUMO-2/3 conjugation*, no significant OGD protection | Bernstock et al., 2018 [82] | |
| Ethyl protocatechuate | |||||||
| 6-thioguanine | SUMO-1 upregulation*, OGD protection* | ||||||
| Ebselen | |||||||
| C57BL6 mice | N/A | 12mg/kg IP bolus treatment | SUMOylation 1hr after bolus | Increases in SUMO-1 and SUMO-2/3 conjugation* | |||
| SHRSP, WKY rat PDN | 24hr OGD + 3hr recovery | treatment + 3hr posttreatment | Cell survival, LDH activity | OGD protection*, no significant difference in LDH activity | Yamagata et al., 2008 [96] | ||
| SHRSP, WKY rat | 0.5 hr BCCO | 30 mg/kg/day pretreatment for 7 days then 30mg/kg/day posttreatment for 3 days | Apoptotic neurons in CA1 subfield of hippocampus | Almost complete inhibition of apoptosis† | |||
| N/A | 60 mg/kg/day treatment for 6 wks | Oxidative stress (via cortical NO and MDA concentrations); iNOS expression | Reduction in NO and MDA concentrations*; reduction in iNOS expression* | Sui et al., 2005 [97] | |||
| Human | MCAO | 150mg PO BID post-treatment within 12 hours of onset for 2 weeks. Placebo controlled, double blind trial. | Infarct volume 1mo post stroke, GOS 3mo post stroke | Reduction in infarct volume*; superior GOS if administered within 6 hours*; no significant difference in GOS vs. negative control overall | Ogawa et al., 1999 [89] | ||
| AIS | 150mg PO BID posttreatment within 48hrs of onset for 2 weeks. Placebo controlled, double blind trial. | GOS (1- and 3-month), neurological status (2wk, modified Mathew Scale), functional status (2 wk, Barthel Index) | Improvement in 1-month GOS* but no significant difference in 3-month GOS; superior GOS if administered ≤ 24 hrs*; reduction in both impairment (Mathew)* and disability (Barthel)* | Yamaguchi et al., 1998 [90] | |||
| SD rats |
Permanent MCAO | 1mg/kg/hr pretreatment 45min pre-stroke to 4hrs post-stroke | Extent of ischemic damage, oxidative stress (via IHC) | 28% reduction in cortical ischemic damage vs. control†; reduction in oxidative stress markers vs. control† | Imai et al., 2003 [98] | ||
| 2hr FCI | 1mg/kg IV bolus + 1mg/kg/hr IV post-treatment for 24hrs | 24 hr neurological deficit, gray matter damage, Axonal damage and oxidative stress (via IHC) | 40.7% reduced neurological deficit at 24hrs vs control*; 53.6% reduction in gray matter damage*; 46.8% reduction in axonal damage IHC markers* | Imai et al., 2001 [99] | |||
| Wistar rats |
45 min BCCO | 30mg/kg PO bolus pretreatment 2hrs prior to stroke | cortical EAA and NO concentrations, 24hr hippocampal CA1 subfield integrity | Increase in intact CA1 neurons*; No difference in EAA or NO concentrations vs control. | Koizumi et al., 2011 [87] | ||
| 2hr FCI | 10mg/kg and 100mg/kg PO bolus pretreatment 1hr prior to FCI | Reduced glutathione concentration; plasma Selenium; 1wk post stroke infarct size | Increase in perfusion with 100mg/kg*; increase in plasma Selenium* | Salom et al., 2004 [100] | |||
| Wistar rat cerebellar neurons | Glutamate exposure | 25min treatment ± posttreatment; posttreatment | 24hr cell survival, 48hr cell survival | Increase in survival with treatment* and posttreatment* comparable to negative control | Porciúncula et al., 2001 [93] | ||
| Wistar rat hippocampal neurons | 45min OGD | Pretreatment and posttreatment | 3hr cell survival | Increase in survival with treatment* and post-treatment* comparable to negative control | Porciúncula et al., 2003 [94] |
* Results are statistically significant. † No measurement of statistical significance presented. Abbreviations: AIS, Acute Ischemic Stroke; BCCO, bilateral common carotid occlusion; EAA, Excitatory Amino Acid; FCI, Focal Cerebral Ischemia; GOS, Glasgow Outcome Scale; IHC, Immunohistochemistry; iNOS, induced Nitric Oxide Synthase; MCAO, middle cerebral artery occlusion; MDA, malonaldehyde; NO, Nitrous oxide; OGD, Oxygen-glucose deprivation; PCN, primary cortical neuron; PDN, primary dissociated neuron; PO, per os; SD, Sprague-Dawley; WKY, Wistar Kyoto.
2.3. Direct SUMO Upregulators
The increased accessibility and prevalence of high-throughput screening methodologies has enabled the rapid development of libraries of possible SUMOtherapeutic agents. Krajnak et al. and Neurodon LLC (Schererville, IN, USA) utilized a Förster Resonance Energy Transfer (FRET)-based high-throughput screening tool to identify prospective SUMOylators which were further screened in orthogonal assays and filtered based on physicochemical criteria indicating druggability. While the exact compounds utilized remain proprietary, the authors identified eleven small-molecule SUMOylators belonging to the quinoline, benzothiazole, and aminothiazole families. When evaluated in an in vitro model of endoplasmic reticulum stress (via thapsigargin exposure) in CSM14.1 striatal neuroprogenitor and Buffalo Green Monkey Kidney cells, nine out of the eleven identified compounds demonstrated statistically significant SUMO upregulation compared to control [101]. Despite these compounds being developed for clinical indications other than ischemic stroke, the relatively facile nature of such screening tools represents a promising avenue towards an ever-expanding library of SUMOylators.
3. Near Future Innovation in SUMOtherapeutics
The expanding understanding of the SUMO pathway is generating an increasing amount of potential therapeutic indications. In turn, compounds that modulate the pathway are increasingly the subject of research efforts for a variety of conditions including ischemic disease of the heart, kidney, and transplanted organs [16, 102]. With viable solutions to the problems of substrate availability [103] and screening methods [82], preclinical investigation of the SUMO pathway is increasingly facile. In fact, the first clinical trials of a SUMOtherapeutic compound, TAK-981 (Takeda Pharmaceuticals, Cambridge, MA, USA), are ongoing at the time of writing (NCT03648372, NCT04074330, and NCT04381650) [104]. While it is a chemotherapeutic agent, the lessons learned in its development will undoubtedly aid in the development of other SUMOtherapeutics.
Due to the gross similarities between the SUMO pathway and ubiquitinoylation, it is not unreasonable that the development of SUMOtherapeutics would follow the trajectory of the latter’s therapeutic exploitation. Advances in high throughput screening and in proteomics methods along with greater availability of equipment enable SUMOylation research to be conducted at an increased pace. Unfortunately, unlike agents targeted to other indications, SUMOtherapeutics for cerebral ischemia remain closer to the bench than the bedside.
Even so, in a new ‘era of neuroprotection’, advancements in other therapeutic technologies may represent a catalyst for stroke SUMOtherapeutics. To accelerate the development and clinical implementation of these compounds, several hurdles must be overcome such as the lack of standardization in study design, and the distance between preclinical investigations and potential clinical applications. Extant literature on neuroprotection utilizes a variety of animal and tissue models alongside ischemia models that range from global ischemia to focal occlusion on widely varying timescales. The lack of standardization impedes comparisons and meta-analyses of different methods. As such, the guidelines for clinical trial design posited by Tymianski may be adapted to preclinical evaluations of SUMOtherapeutics: study designs should emulate those of prior methodologically sound studies; subject heterogeneity should be minimized while treatment cohort size is maximized; and models should incorporate clinically relevant treatment windows (i.e. emphasis on co-treatment and post-treatment vs. pre-treatment) [105].
The difference between preclinical study design and putative clinical use case makes identifying knowledge gaps and pitfalls more difficult—small animal studies may provide translational utility if care is taken to replicate the clinical picture of ischemic stroke. Finally, promising neuroprotectants that were abandoned due to difficulties in translation may merit revisiting in forms that leverage the newer capabilities available today. In particular, perithrombectomy delivery at the time of recanalization and advances in targeted drug delivery to enhance blood-brain barrier penetration. Provided that these factors are considered, SUMOtherapeutics may represent the vanguard of an entirely new approach in the management of ischemic stroke.
4. Conclusions
The SUMO pathway represents a promising avenue for neuroprotection in the context of ischemic cerebral injury. The variety of druggable nodes and steadily increasing awareness of the scope of SUMO-mediated processes makes it an attractive target for therapeutic exploitation. SUMOtherapeutics for stroke may represent the first truly novel treatment paradigm in stroke since the discovery of tPA and development of thrombectomy techniques. As such, a greater breadth and depth of preclinical inquiry with standardized methodologies and clinically-congruent experimental design that utilizes recent advances in technology and technique is warranted.
References
- "Global, Regional, and National Burden of Stroke and Its Risk Factors, 1990-2019: A Systematic Analysis for the Global Burden of Disease Study 2019." Lancet Neurol 20, no. 10 (2021): 795-820.
- Albers, G. W., M. P. Marks, S. Kemp, S. Christensen, J. P. Tsai, S. Ortega-Gutierrez, R. A. McTaggart, M. T. Torbey, M. Kim-Tenser, T. Leslie-Mazwi, A. Sarraj, S. E. Kasner, S. A. Ansari, S. D. Yeatts, S. Hamilton, M. Mlynash, J. J. Heit, G. Zaharchuk, S. Kim, J. Carrozzella, Y. Y. Palesch, A. M. Demchuk, R. Bammer, P. W. Lavori, J. P. Broderick, M. G. Lansberg, and Defuse Investigators. "Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging." N Engl J Med 378, no. 8 (2018): 708-18. [CrossRef]
- Nogueira, R. G., A. P. Jadhav, D. C. Haussen, A. Bonafe, R. F. Budzik, P. Bhuva, D. R. Yavagal, M. Ribo, C. Cognard, R. A. Hanel, C. A. Sila, A. E. Hassan, M. Millan, E. I. Levy, P. Mitchell, M. Chen, J. D. English, Q. A. Shah, F. L. Silver, V. M. Pereira, B. P. Mehta, B. W. Baxter, M. G. Abraham, P. Cardona, E. Veznedaroglu, F. R. Hellinger, L. Feng, J. F. Kirmani, D. K. Lopes, B. T. Jankowitz, M. R. Frankel, V. Costalat, N. A. Vora, A. J. Yoo, A. M. Malik, A. J. Furlan, M. Rubiera, A. Aghaebrahim, J. M. Olivot, W. G. Tekle, R. Shields, T. Graves, R. J. Lewis, W. S. Smith, D. S. Liebeskind, J. L. Saver, T. G. Jovin, and Dawn Trial Investigators. "Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct." N Engl J Med 378, no. 1 (2018): 11-21. [CrossRef]
- Berkhemer, O. A., P. S. Fransen, D. Beumer, L. A. van den Berg, H. F. Lingsma, A. J. Yoo, W. J. Schonewille, J. A. Vos, P. J. Nederkoorn, M. J. Wermer, M. A. van Walderveen, J. Staals, J. Hofmeijer, J. A. van Oostayen, G. J. Lycklama a Nijeholt, J. Boiten, P. A. Brouwer, B. J. Emmer, S. F. de Bruijn, L. C. van Dijk, L. J. Kappelle, R. H. Lo, E. J. van Dijk, J. de Vries, P. L. de Kort, W. J. van Rooij, J. S. van den Berg, B. A. van Hasselt, L. A. Aerden, R. J. Dallinga, M. C. Visser, J. C. Bot, P. C. Vroomen, O. Eshghi, T. H. Schreuder, R. J. Heijboer, K. Keizer, A. V. Tielbeek, H. M. den Hertog, D. G. Gerrits, R. M. van den Berg-Vos, G. B. Karas, E. W. Steyerberg, H. Z. Flach, H. A. Marquering, M. E. Sprengers, S. F. Jenniskens, L. F. Beenen, R. van den Berg, P. J. Koudstaal, W. H. van Zwam, Y. B. Roos, A. van der Lugt, R. J. van Oostenbrugge, C. B. Majoie, D. W. Dippel, and Mr Clean Investigators. "A Randomized Trial of Intraarterial Treatment for Acute Ischemic Stroke." N Engl J Med 372, no. 1 (2015): 11-20. [CrossRef]
- Ma, H., B. C. V. Campbell, M. W. Parsons, L. Churilov, C. R. Levi, C. Hsu, T. J. Kleinig, T. Wijeratne, S. Curtze, H. M. Dewey, F. Miteff, C. H. Tsai, J. T. Lee, T. G. Phan, N. Mahant, M. C. Sun, M. Krause, J. Sturm, R. Grimley, C. H. Chen, C. J. Hu, A. A. Wong, D. Field, Y. Sun, P. A. Barber, A. Sabet, J. Jannes, J. S. Jeng, B. Clissold, R. Markus, C. H. Lin, L. M. Lien, C. F. Bladin, S. Christensen, N. Yassi, G. Sharma, A. Bivard, P. M. Desmond, B. Yan, P. J. Mitchell, V. Thijs, L. Carey, A. Meretoja, S. M. Davis, G. A. Donnan, and Extend Investigators. "Thrombolysis Guided by Perfusion Imaging up to 9 Hours after Onset of Stroke." N Engl J Med 380, no. 19 (2019): 1795-803. [CrossRef]
- Anand, S. K., W. J. Benjamin, A. R. Adapa, J. V. Park, D. A. Wilkinson, B. J. Daou, J. F. Burke, and A. S. Pandey. "Trends in Acute Ischemic Stroke Treatments and Mortality in the United States from 2012 to 2018." Neurosurg Focus 51, no. 1 (2021): E2. [CrossRef]
- Goyal, M., B. K. Menon, W. H. van Zwam, D. W. Dippel, P. J. Mitchell, A. M. Demchuk, A. Davalos, C. B. Majoie, A. van der Lugt, M. A. de Miquel, G. A. Donnan, Y. B. Roos, A. Bonafe, R. Jahan, H. C. Diener, L. A. van den Berg, E. I. Levy, O. A. Berkhemer, V. M. Pereira, J. Rempel, M. Millan, S. M. Davis, D. Roy, J. Thornton, L. S. Roman, M. Ribo, D. Beumer, B. Stouch, S. Brown, B. C. Campbell, R. J. van Oostenbrugge, J. L. Saver, M. D. Hill, T. G. Jovin, and Hermes collaborators. "Endovascular Thrombectomy after Large-Vessel Ischaemic Stroke: A Meta-Analysis of Individual Patient Data from Five Randomised Trials." Lancet 387, no. 10029 (2016): 1723-31. [CrossRef]
- Yoshimura, S., N. Sakai, H. Yamagami, K. Uchida, M. Beppu, K. Toyoda, Y. Matsumaru, Y. Matsumoto, K. Kimura, M. Takeuchi, Y. Yazawa, N. Kimura, K. Shigeta, H. Imamura, I. Suzuki, Y. Enomoto, S. Tokunaga, K. Morita, F. Sakakibara, N. Kinjo, T. Saito, R. Ishikura, M. Inoue, and T. Morimoto. "Endovascular Therapy for Acute Stroke with a Large Ischemic Region." N Engl J Med 386, no. 14 (2022): 1303-13. [CrossRef]
- Sarraj, A., A. E. Hassan, M. G. Abraham, S. Ortega-Gutierrez, S. E. Kasner, M. S. Hussain, M. Chen, S. Blackburn, C. W. Sitton, L. Churilov, S. Sundararajan, Y. C. Hu, N. A. Herial, P. Jabbour, D. Gibson, A. N. Wallace, J. F. Arenillas, J. P. Tsai, R. F. Budzik, W. J. Hicks, O. Kozak, B. Yan, D. J. Cordato, N. W. Manning, M. W. Parsons, R. A. Hanel, A. N. Aghaebrahim, T. Y. Wu, P. Cardona-Portela, N. Perez de la Ossa, J. D. Schaafsma, J. Blasco, N. Sangha, S. Warach, C. D. Gandhi, T. J. Kleinig, D. Sahlein, L. Elijovich, W. Tekle, E. A. Samaniego, L. Maali, M. A. Abdulrazzak, M. N. Psychogios, A. Shuaib, D. K. Pujara, F. Shaker, H. Johns, G. Sharma, V. Yogendrakumar, F. C. Ng, M. H. Rahbar, C. Cai, P. Lavori, S. Hamilton, T. Nguyen, J. T. Fifi, S. Davis, L. Wechsler, V. M. Pereira, M. G. Lansberg, M. D. Hill, J. C. Grotta, M. Ribo, B. C. Campbell, G. W. Albers, and Select Investigators. "Trial of Endovascular Thrombectomy for Large Ischemic Strokes." N Engl J Med (2023). [CrossRef]
- Huo, X., G. Ma, X. Tong, X. Zhang, Y. Pan, T. N. Nguyen, G. Yuan, H. Han, W. Chen, M. Wei, J. Zhang, Z. Zhou, X. Yao, G. Wang, W. Song, X. Cai, G. Nan, D. Li, A. Y. Wang, W. Ling, C. Cai, C. Wen, E. Wang, L. Zhang, C. Jiang, Y. Liu, G. Liao, X. Chen, T. Li, S. Liu, J. Li, F. Gao, N. Ma, D. Mo, L. Song, X. Sun, X. Li, Y. Deng, G. Luo, M. Lv, H. He, A. Liu, J. Zhang, S. Mu, L. Liu, J. Jing, X. Nie, Z. Ding, W. Du, X. Zhao, P. Yang, L. Liu, Y. Wang, D. S. Liebeskind, V. M. Pereira, Z. Ren, Y. Wang, Z. Miao, and Angel-Aspect Investigators. "Trial of Endovascular Therapy for Acute Ischemic Stroke with Large Infarct." N Engl J Med (2023). [CrossRef]
- Filippopoulou, C., G. Simos, and G. Chachami. "The Role of Sumoylation in the Response to Hypoxia: An Overview." Cells 9, no. 11 (2020). [CrossRef]
- Lee, Y. J., S. Miyake, H. Wakita, D. C. McMullen, Y. Azuma, S. Auh, and J. M. Hallenbeck. "Protein Sumoylation Is Massively Increased in Hibernation Torpor and Is Critical for the Cytoprotection Provided by Ischemic Preconditioning and Hypothermia in Shsy5y Cells." J Cereb Blood Flow Metab 27, no. 5 (2007): 950-62. [CrossRef]
- Lee, Y. J., P. Castri, J. Bembry, D. Maric, S. Auh, and J. M. Hallenbeck. "Sumoylation Participates in Induction of Ischemic Tolerance." J Neurochem 109, no. 1 (2009): 257-67. [CrossRef]
- Oliveira, Frmb, E. S. Soares, C. Harms, H. I. Cimarosti, and R. Sordi. "Sumoylation in Peripheral Tissues under Low Perfusion-Related Pathological States." J Cell Biochem 123, no. 7 (2022): 1133-47. [CrossRef]
- Bernstock, J. D., D. G. Ye, A. Griffin, Y. J. Lee, J. Lynch, L. L. Latour, G. K. Friedman, D. Maric, and J. M. Hallenbeck. "Cerebral Ischemia Increases Small Ubiquitin-Like Modifier Conjugation within Human Penumbral Tissue: Radiological-Pathological Correlation." Front Neurol 8 (2017): 738. [CrossRef]
- Hendriks, I. A., D. Lyon, D. Su, N. H. Skotte, J. A. Daniel, L. J. Jensen, and M. L. Nielsen. "Site-Specific Characterization of Endogenous Sumoylation across Species and Organs." Nat Commun 9, no. 1 (2018): 2456. [CrossRef]
- Sahin, U., H. de The, and V. Lallemand-Breitenbach. "Sumoylation in Physiology, Pathology and Therapy." Cells 11, no. 5 (2022).
- Flotho, A., and F. Melchior. "Sumoylation: A Regulatory Protein Modification in Health and Disease." Annu Rev Biochem 82 (2013): 357-85. [CrossRef]
- Yang, W., H. Sheng, and H. Wang. "Targeting the Sumo Pathway for Neuroprotection in Brain Ischaemia." Stroke Vasc Neurol 1, no. 3 (2016): 101-07. [CrossRef]
- Droescher, M., V. K. Chaugule, and A. Pichler. "Sumo Rules: Regulatory Concepts and Their Implication in Neurologic Functions." Neuromolecular Med 15, no. 4 (2013): 639-60. [CrossRef]
- Zhang, L., X. Liu, H. Sheng, S. Liu, Y. Li, J. Q. Zhao, D. S. Warner, W. Paschen, and W. Yang. "Neuron-Specific Sumo Knockdown Suppresses Global Gene Expression Response and Worsens Functional Outcome after Transient Forebrain Ischemia in Mice." Neuroscience 343 (2017): 190-212. [CrossRef]
- Bernstock, J. D., D. Ye, F. A. Gessler, Y. J. Lee, L. Peruzzotti-Jametti, P. Baumgarten, K. R. Johnson, D. Maric, W. Yang, D. Kogel, S. Pluchino, and J. M. Hallenbeck. "Topotecan Is a Potent Inhibitor of Sumoylation in Glioblastoma Multiforme and Alters Both Cellular Replication and Metabolic Programming." Sci Rep 7, no. 1 (2017): 7425. [CrossRef]
- Yang, W., Q. Ma, G. B. Mackensen, and W. Paschen. "Deep Hypothermia Markedly Activates the Small Ubiquitin-Like Modifier Conjugation Pathway; Implications for the Fate of Cells Exposed to Transient Deep Hypothermic Cardiopulmonary Bypass." J Cereb Blood Flow Metab 29, no. 5 (2009): 886-90. [CrossRef]
- Yang, W., H. Sheng, J. W. Thompson, S. Zhao, L. Wang, P. Miao, X. Liu, M. A. Moseley, and W. Paschen. "Small Ubiquitin-Like Modifier 3-Modified Proteome Regulated by Brain Ischemia in Novel Small Ubiquitin-Like Modifier Transgenic Mice: Putative Protective Proteins/Pathways." Stroke 45, no. 4 (2014): 1115-22.
- Yang, W., H. Sheng, D. S. Warner, and W. Paschen. "Transient Global Cerebral Ischemia Induces a Massive Increase in Protein Sumoylation." J Cereb Blood Flow Metab 28, no. 2 (2008): 269-79. [CrossRef]
- ———. "Transient Focal Cerebral Ischemia Induces a Dramatic Activation of Small Ubiquitin-Like Modifier Conjugation." J Cereb Blood Flow Metab 28, no. 5 (2008): 892-6.
- Chen, Xu, Yuhong Zhang, Qiqi Wang, Yuanyuan Qin, Xinyi Yang, Zhengcao Xing, Yajie Shen, Hongmei Wu, and Yitao Qi. "The Function of Sumoylation and Its Crucial Roles in the Development of Neurological Diseases." The FASEB Journal 35, no. 4 (2021): e21510. [CrossRef]
- Mendler, L., T. Braun, and S. Muller. "The Ubiquitin-Like Sumo System and Heart Function: From Development to Disease." Circ Res 118, no. 1 (2016): 132-44.
- Kho, C., A. Lee, D. Jeong, J. G. Oh, A. H. Chaanine, E. Kizana, W. J. Park, and R. J. Hajjar. "Sumo1-Dependent Modulation of Serca2a in Heart Failure." Nature 477, no. 7366 (2011): 601-5. [CrossRef]
- Guo, C., Q. Wei, Y. Su, and Z. Dong. "Sumoylation Occurs in Acute Kidney Injury and Plays a Cytoprotective Role." Biochim Biophys Acta 1852, no. 3 (2015): 482-9. [CrossRef]
- Zhao, W., X. Zhang, and J. Rong. "Sumoylation as a Therapeutic Target for Myocardial Infarction." Front Cardiovasc Med 8 (2021): 701583. [CrossRef]
- Datwyler, A. L., G. Lattig-Tunnemann, W. Yang, W. Paschen, S. L. Lee, U. Dirnagl, M. Endres, and C. Harms. "Sumo2/3 Conjugation Is an Endogenous Neuroprotective Mechanism." J Cereb Blood Flow Metab 31, no. 11 (2011): 2152-9.
- Cuomo, O., G. Pignataro, R. Sirabella, P. Molinaro, S. Anzilotti, A. Scorziello, M. J. Sisalli, G. Di Renzo, and L. Annunziato. "Sumoylation of Lys590 of Ncx3 F-Loop by Sumo1 Participates in Brain Neuroprotection Induced by Ischemic Preconditioning." Stroke 47, no. 4 (2016): 1085-93. [CrossRef]
- Kunz, K., K. Wagner, L. Mendler, S. Holper, N. Dehne, and S. Muller. "Sumo Signaling by Hypoxic Inactivation of Sumo-Specific Isopeptidases." Cell Rep 16, no. 11 (2016): 3075-86. [CrossRef]
- Dong, P., Q. Li, and H. Han. "Hif-1alpha in Cerebral Ischemia (Review)." Mol Med Rep 25, no. 2 (2022).
- Mabb, A. M., and S. Miyamoto. "Sumo and Nf-Kappab Ties." Cell Mol Life Sci 64, no. 15 (2007): 1979-96.
- Li, J., Y. Xu, X. D. Long, W. Wang, H. K. Jiao, Z. Mei, Q. Q. Yin, L. N. Ma, A. W. Zhou, L. S. Wang, M. Yao, Q. Xia, and G. Q. Chen. "Cbx4 Governs Hif-1alpha to Potentiate Angiogenesis of Hepatocellular Carcinoma by Its Sumo E3 Ligase Activity." Cancer Cell 25, no. 1 (2014): 118-31.
- Pan, Y., Q. Li, Z. Cao, and S. Zhao. "The Sumo E3 Ligase Cbx4 Is Identified as a Poor Prognostic Marker of Gastric Cancer through Multipronged Omic Analyses." Genes Dis 8, no. 6 (2021): 827-37. [CrossRef]
- Nakagawa, K., T. Kohara, Y. Uehata, Y. Miyakawa, M. Sato-Ueshima, N. Okubo, M. Asaka, H. Takeda, and M. Kobayashi. "Pias3 Enhances the Transcriptional Activity of Hif-1alpha by Increasing Its Protein Stability." Biochem Biophys Res Commun 469, no. 3 (2016): 470-6. [CrossRef]
- Tojo, M., K. Matsuzaki, T. Minami, Y. Honda, H. Yasuda, T. Chiba, H. Saya, Y. Fujii-Kuriyama, and M. Nakao. "The Aryl Hydrocarbon Receptor Nuclear Transporter Is Modulated by the Sumo-1 Conjugation System." J Biol Chem 277, no. 48 (2002): 46576-85. [CrossRef]
- Cai, Q., S. C. Verma, P. Kumar, M. Ma, and E. S. Robertson. "Hypoxia Inactivates the Vhl Tumor Suppressor through Piasy-Mediated Sumo Modification." PLoS One 5, no. 3 (2010): e9720. [CrossRef]
- Kang, X., J. Li, Y. Zou, J. Yi, H. Zhang, M. Cao, E. T. Yeh, and J. Cheng. "Piasy Stimulates Hif1alpha Sumoylation and Negatively Regulates Hif1alpha Activity in Response to Hypoxia." Oncogene 29, no. 41 (2010): 5568-78. [CrossRef]
- Nunez-O'Mara, A., A. Gerpe-Pita, S. Pozo, O. Carlevaris, B. Urzelai, F. Lopitz-Otsoa, M. S. Rodriguez, and E. Berra. "Phd3-Sumo Conjugation Represses Hif1 Transcriptional Activity Independently of Phd3 Catalytic Activity." J Cell Sci 128, no. 1 (2015): 40-9.
- Sallais, J., S. Alahari, A. Tagliaferro, J. Bhattacharjee, M. Post, and I. Caniggia. "Factor Inhibiting Hif1-a Novel Target of Sumoylation in the Human Placenta." Oncotarget 8, no. 69 (2017): 114002-18. [CrossRef]
- Chachami, G., N. Stankovic-Valentin, A. Karagiota, A. Basagianni, U. Plessmann, H. Urlaub, F. Melchior, and G. Simos. "Hypoxia-Induced Changes in Sumo Conjugation Affect Transcriptional Regulation under Low Oxygen." Mol Cell Proteomics 18, no. 6 (2019): 1197-209. [CrossRef]
- Zhang, W., I. Potrovita, V. Tarabin, O. Herrmann, V. Beer, F. Weih, A. Schneider, and M. Schwaninger. "Neuronal Activation of Nf-Kappab Contributes to Cell Death in Cerebral Ischemia." J Cereb Blood Flow Metab 25, no. 1 (2005): 30-40.
- Tashiro, K., M. P. Pando, Y. Kanegae, P. M. Wamsley, S. Inoue, and I. M. Verma. "Direct Involvement of the Ubiquitin-Conjugating Enzyme Ubc9/Hus5 in the Degradation of Ikappabalpha." Proc Natl Acad Sci U S A 94, no. 15 (1997): 7862-7.
- Desterro, J. M., M. S. Rodriguez, and R. T. Hay. "Sumo-1 Modification of Ikappabalpha Inhibits Nf-Kappab Activation." Mol Cell 2, no. 2 (1998): 233-9.
- Li, X., Q. Xia, M. Mao, H. Zhou, L. Zheng, Y. Wang, Z. Zeng, L. Yan, Y. Zhao, and J. Shi. "Annexin-A1 Sumoylation Regulates Microglial Polarization after Cerebral Ischemia by Modulating Ikkalpha Stability Via Selective Autophagy." Sci Adv 7, no. 4 (2021).
- Huang, T. T., S. M. Wuerzberger-Davis, Z. H. Wu, and S. Miyamoto. "Sequential Modification of Nemo/Ikkgamma by Sumo-1 and Ubiquitin Mediates Nf-Kappab Activation by Genotoxic Stress." Cell 115, no. 5 (2003): 565-76.
- Yang, T., J. Sun, B. Wei, and S. Liu. "Senp1-Mediated Nemo De-Sumoylation Inhibits Intermittent Hypoxia Induced Inflammatory Response of Microglia in Vitro." J Cell Physiol 235, no. 4 (2020): 3529-38. [CrossRef]
- Lee, J. H., S. M. Park, O. S. Kim, C. S. Lee, J. H. Woo, S. J. Park, E. H. Joe, and I. Jou. "Differential Sumoylation of Lxralpha and Lxrbeta Mediates Transrepression of Stat1 Inflammatory Signaling in Ifn-Gamma-Stimulated Brain Astrocytes." Mol Cell 35, no. 6 (2009): 806-17.
- Cimarosti, H., C. Lindberg, S. F. Bomholt, L. C. Ronn, and J. M. Henley. "Increased Protein Sumoylation Following Focal Cerebral Ischemia." Neuropharmacology 54, no. 2 (2008): 280-9. [CrossRef]
- Guo, C., and J. M. Henley. "Wrestling with Stress: Roles of Protein Sumoylation and Desumoylation in Cell Stress Response." IUBMB Life 66, no. 2 (2014): 71-7. [CrossRef]
- Henley, J. M., T. J. Craig, and K. A. Wilkinson. "Neuronal Sumoylation: Mechanisms, Physiology, and Roles in Neuronal Dysfunction." Physiol Rev 94, no. 4 (2014): 1249-85.
- Guo, C., K. L. Hildick, J. Luo, L. Dearden, K. A. Wilkinson, and J. M. Henley. "Senp3-Mediated Desumoylation of Dynamin-Related Protein 1 Promotes Cell Death Following Ischaemia." EMBO J 32, no. 11 (2013): 1514-28. [CrossRef]
- Neumar, R. W. "Molecular Mechanisms of Ischemic Neuronal Injury." Ann Emerg Med 36, no. 5 (2000): 483-506.
- Feligioni, M., M. P. Mattson, and R. Nistico. "Sumoylation in Neuroplasticity and Neurological Disorders." Neuromolecular Med 15, no. 4 (2013): 637-8. [CrossRef]
- Feligioni, M., A. Nishimune, and J. M. Henley. "Protein Sumoylation Modulates Calcium Influx and Glutamate Release from Presynaptic Terminals." Eur J Neurosci 29, no. 7 (2009): 1348-56. [CrossRef]
- Dustrude, E. T., S. M. Wilson, W. Ju, Y. Xiao, and R. Khanna. "Crmp2 Protein Sumoylation Modulates Nav1.7 Channel Trafficking." J Biol Chem 288, no. 34 (2013): 24316-31.
- Martin, S., A. Nishimune, J. R. Mellor, and J. M. Henley. "Sumoylation Regulates Kainate-Receptor-Mediated Synaptic Transmission." Nature 447, no. 7142 (2007): 321-5. [CrossRef]
- Coelho-Silva, L., G. J. Stephens, and H. Cimarosti. "Sumoylation and Calcium Signalling: Potential Roles in the Brain and Beyond." Neuronal Signal 1, no. 3 (2017): NS20160010. [CrossRef]
- Bernstock, J. D., L. Peruzzotti-Jametti, D. Ye, F. A. Gessler, D. Maric, N. Vicario, Y. J. Lee, S. Pluchino, and J. M. Hallenbeck. "Neural Stem Cell Transplantation in Ischemic Stroke: A Role for Preconditioning and Cellular Engineering." J Cereb Blood Flow Metab 37, no. 7 (2017): 2314-19. [CrossRef]
- Ding, D. C., C. H. Lin, W. C. Shyu, and S. Z. Lin. "Neural Stem Cells and Stroke." Cell Transplant 22, no. 4 (2013): 619-30. [CrossRef]
- Baker, E. W., H. A. Kinder, and F. D. West. "Neural Stem Cell Therapy for Stroke: A Multimechanistic Approach to Restoring Neurological Function." Brain Behav 9, no. 3 (2019): e01214. [CrossRef]
- Tahmasebi, S., M. Ghorbani, P. Savage, G. Gocevski, and X. J. Yang. "The Sumo Conjugating Enzyme Ubc9 Is Required for Inducing and Maintaining Stem Cell Pluripotency." Stem Cells 32, no. 4 (2014): 1012-20. [CrossRef]
- Yamaguchi, T., P. Sharma, M. Athanasiou, A. Kumar, S. Yamada, and M. R. Kuehn. "Mutation of Senp1/Supr-2 Reveals an Essential Role for Desumoylation in Mouse Development." Mol Cell Biol 25, no. 12 (2005): 5171-82.
- Bernstock, J. D., L. Peruzzotti-Jametti, T. Leonardi, N. Vicario, D. Ye, Y. J. Lee, D. Maric, K. R. Johnson, Y. Mou, A. Van Den Bosch, M. Winterbone, G. K. Friedman, R. J. M. Franklin, J. M. Hallenbeck, and S. Pluchino. "Sumoylation Promotes Survival and Integration of Neural Stem Cell Grafts in Ischemic Stroke." EBioMedicine 42 (2019): 214-24. [CrossRef]
- Lee, Y. J., K. R. Johnson, and J. M. Hallenbeck. "Global Protein Conjugation by Ubiquitin-Like-Modifiers During Ischemic Stress Is Regulated by Micrornas and Confers Robust Tolerance to Ischemia." PLoS One 7, no. 10 (2012): e47787. [CrossRef]
- Lee, Y. J., and J. M. Hallenbeck. "Sumo and Ischemic Tolerance." Neuromolecular Med 15, no. 4 (2013): 771-81. [CrossRef]
- Tokarz, P., and K. Wozniak. "Senp Proteases as Potential Targets for Cancer Therapy." Cancers (Basel) 13, no. 9 (2021). [CrossRef]
- Bernstock, J. D., D. G. Ye, Y. J. Lee, F. Gessler, G. K. Friedman, W. Zheng, and J. M. Hallenbeck. "Drugging Sumoylation for Neuroprotection and Oncotherapy." Neural Regen Res 13, no. 3 (2018): 415-16. [CrossRef]
- Bernstock, J. D., Y. J. Lee, L. Peruzzotti-Jametti, N. Southall, K. R. Johnson, D. Maric, G. Volpe, J. Kouznetsova, W. Zheng, S. Pluchino, and J. M. Hallenbeck. "A Novel Quantitative High-Throughput Screen Identifies Drugs That Both Activate Sumo Conjugation Via the Inhibition of Micrornas 182 and 183 and Facilitate Neuroprotection in a Model of Oxygen and Glucose Deprivation." J Cereb Blood Flow Metab 36, no. 2 (2016): 426-41. [CrossRef]
- Nadareishvili, Z., D. Kelley, M. Luby, A. N. Simpkins, R. Leigh, J. K. Lynch, A. W. Hsia, R. T. Benson, K. R. Johnson, J. M. Hallenbeck, and L. L. Latour. "Molecular Signature of Penumbra in Acute Ischemic Stroke: A Pilot Transcriptomics Study." Ann Clin Transl Neurol 6, no. 4 (2019): 817-20. [CrossRef]
- Kim, H. J., M. Rowe, M. Ren, J. S. Hong, P. S. Chen, and D. M. Chuang. "Histone Deacetylase Inhibitors Exhibit Anti-Inflammatory and Neuroprotective Effects in a Rat Permanent Ischemic Model of Stroke: Multiple Mechanisms of Action." J Pharmacol Exp Ther 321, no. 3 (2007): 892-901. [CrossRef]
- Brookes, R. L., S. Crichton, C. D. A. Wolfe, Q. Yi, L. Li, G. J. Hankey, P. M. Rothwell, and H. S. Markus. "Sodium Valproate, a Histone Deacetylase Inhibitor, Is Associated with Reduced Stroke Risk after Previous Ischemic Stroke or Transient Ischemic Attack." Stroke 49, no. 1 (2018): 54-61. [CrossRef]
- Park, M. J., and F. Sohrabji. "The Histone Deacetylase Inhibitor, Sodium Butyrate, Exhibits Neuroprotective Effects for Ischemic Stroke in Middle-Aged Female Rats." J Neuroinflammation 13, no. 1 (2016): 300. [CrossRef]
- Al Shoyaib, A., F. F. Alamri, N. Syeara, S. Jayaraman, S. T. Karamyan, T. V. Arumugam, and V. T. Karamyan. "The Effect of Histone Deacetylase Inhibitors Panobinostat or Entinostat on Motor Recovery in Mice after Ischemic Stroke." Neuromolecular Med 23, no. 4 (2021): 471-84. [CrossRef]
- Bonsack, F., and S. Sukumari-Ramesh. "Entinostat Improves Acute Neurological Outcomes and Attenuates Hematoma Volume after Intracerebral Hemorrhage." Brain Res 1752 (2021): 147222. [CrossRef]
- Shimizu, K., Z. Lacza, N. Rajapakse, T. Horiguchi, J. Snipes, and D. W. Busija. "Mitok(Atp) Opener, Diazoxide, Reduces Neuronal Damage after Middle Cerebral Artery Occlusion in the Rat." Am J Physiol Heart Circ Physiol 283, no. 3 (2002): H1005-11. [CrossRef]
- O'Sullivan, J. C., X. L. Yao, H. Alam, and J. T. McCabe. "Diazoxide, as a Postconditioning and Delayed Preconditioning Trigger, Increases Hsp25 and Hsp70 in the Central Nervous System Following Combined Cerebral Stroke and Hemorrhagic Shock." J Neurotrauma 24, no. 3 (2007): 532-46. [CrossRef]
- Bernstock, J. D., D. Ye, J. A. Smith, Y. J. Lee, F. A. Gessler, A. Yasgar, J. Kouznetsova, A. Jadhav, Z. Wang, S. Pluchino, W. Zheng, A. Simeonov, J. M. Hallenbeck, and W. Yang. "Quantitative High-Throughput Screening Identifies Cytoprotective Molecules That Enhance Sumo Conjugation Via the Inhibition of Sumo-Specific Protease (Senp)2." FASEB J 32, no. 3 (2018): 1677-91. [CrossRef]
- Chojnowski, K., M. Opielka, W. Nazar, P. Kowianski, and R. T. Smolenski. "Neuroprotective Effects of Guanosine in Ischemic Stroke-Small Steps Towards Effective Therapy." Int J Mol Sci 22, no. 13 (2021). [CrossRef]
- Lee, Y. J., J. D. Bernstock, N. Nagaraja, B. Ko, and J. M. Hallenbeck. "Global Sumoylation Facilitates the Multimodal Neuroprotection Afforded by Quercetin against the Deleterious Effects of Oxygen/Glucose Deprivation and the Restoration of Oxygen/Glucose." J Neurochem 138, no. 1 (2016): 101-16. [CrossRef]
- Zhang, L., J. Ma, F. Yang, S. Li, W. Ma, X. Chang, and L. Yang. "Neuroprotective Effects of Quercetin on Ischemic Stroke: A Literature Review." Front Pharmacol 13 (2022): 854249. [CrossRef]
- Guo, C., W. J. Wang, Y. C. Liao, C. Zhao, Y. Yin, M. N. Yao, Y. Ding, and J. W. Wang. "Effect and Mechanisms of Quercetin for Experimental Focal Cerebral Ischemia: A Systematic Review and Meta-Analysis." Oxid Med Cell Longev 2022 (2022): 9749461. [CrossRef]
- Koizumi, H., H. Fujisawa, E. Suehiro, S. Shirao, and M. Suzuki. "Neuroprotective Effects of Ebselen Following Forebrain Ischemia: Involvement of Glutamate and Nitric Oxide." Neurol Med Chir (Tokyo) 51, no. 5 (2011): 337-43. [CrossRef]
- Park, S., S. Kang, D. S. Kim, B. K. Shin, N. R. Moon, and J. W. Daily, 3rd. "Ebselen Pretreatment Attenuates Ischemia/Reperfusion Injury and Prevents Hyperglycemia by Improving Hepatic Insulin Signaling and Beta-Cell Survival in Gerbils." Free Radic Res 48, no. 8 (2014): 864-74.
- Ogawa, A., T. Yoshimoto, H. Kikuchi, K. Sano, I. Saito, T. Yamaguchi, and H. Yasuhara. "Ebselen in Acute Middle Cerebral Artery Occlusion: A Placebo-Controlled, Double-Blind Clinical Trial." Cerebrovasc Dis 9, no. 2 (1999): 112-8.
- Yamaguchi, T., K. Sano, K. Takakura, I. Saito, Y. Shinohara, T. Asano, and H. Yasuhara. "Ebselen in Acute Ischemic Stroke: A Placebo-Controlled, Double-Blind Clinical Trial. Ebselen Study Group." Stroke 29, no. 1 (1998): 12-7.
- Mulder, I. A., E. T. van Bavel, H. E. de Vries, and J. M. Coutinho. "Adjunctive Cytoprotective Therapies in Acute Ischemic Stroke: A Systematic Review." Fluids Barriers CNS 18, no. 1 (2021): 46. [CrossRef]
- Parnham, M. J., and H. Sies. "The Early Research and Development of Ebselen." Biochem Pharmacol 86, no. 9 (2013): 1248-53. [CrossRef]
- Porciuncula, L. O., J. B. Rocha, C. R. Boeck, D. Vendite, and D. O. Souza. "Ebselen Prevents Excitotoxicity Provoked by Glutamate in Rat Cerebellar Granule Neurons." Neurosci Lett 299, no. 3 (2001): 217-20. [CrossRef]
- Porciuncula, L. O., J. B. Rocha, H. Cimarosti, L. Vinade, G. Ghisleni, C. G. Salbego, and D. O. Souza. "Neuroprotective Effect of Ebselen on Rat Hippocampal Slices Submitted to Oxygen-Glucose Deprivation: Correlation with Immunocontent of Inducible Nitric Oxide Synthase." Neurosci Lett 346, no. 1-2 (2003): 101-4. [CrossRef]
- Chen, S., D. Dong, W. Xin, and H. Zhou. "Progress in the Discovery of Small Molecule Modulators of Desumoylation." Curr Issues Mol Biol 35 (2020): 17-34. [CrossRef]
- Yamagata, K., S. Ichinose, A. Miyashita, and M. Tagami. "Protective Effects of Ebselen, a Seleno-Organic Antioxidant on Neurodegeneration Induced by Hypoxia and Reperfusion in Stroke-Prone Spontaneously Hypertensive Rat." Neuroscience 153, no. 2 (2008): 428-35. [CrossRef]
- Sui, H., W. Wang, P. H. Wang, and L. S. Liu. "Protective Effect of Antioxidant Ebselen (Pz51) on the Cerebral Cortex of Stroke-Prone Spontaneously Hypertensive Rats." Hypertens Res 28, no. 3 (2005): 249-54. [CrossRef]
- Imai, H., D. I. Graham, H. Masayasu, and I. M. Macrae. "Antioxidant Ebselen Reduces Oxidative Damage in Focal Cerebral Ischemia." Free Radic Biol Med 34, no. 1 (2003): 56-63. [CrossRef]
- Imai, H., H. Masayasu, D. Dewar, D. I. Graham, and I. M. Macrae. "Ebselen Protects Both Gray and White Matter in a Rodent Model of Focal Cerebral Ischemia." Stroke 32, no. 9 (2001): 2149-54. [CrossRef]
- Salom, J. B., F. J. Perez-Asensio, M. C. Burguete, N. Marin, C. Pitarch, G. Torregrosa, F. J. Romero, and E. Alborch. "Single-Dose Ebselen Does Not Afford Sustained Neuroprotection to Rats Subjected to Severe Focal Cerebral Ischemia." Eur J Pharmacol 495, no. 1 (2004): 55-62. [CrossRef]
- Krajnak, K., and R. Dahl. "Small Molecule Sumoylation Activators Are Novel Neuroprotective Agents." Bioorg Med Chem Lett 28, no. 3 (2018): 405-09. [CrossRef]
- Chang, H. M., and E. T. H. Yeh. "Sumo: From Bench to Bedside." Physiol Rev 100, no. 4 (2020): 1599-619. [CrossRef]
- Melnyk, Oleg, and Jérôme Vicogne. "Total Chemical Synthesis of Sumo Proteins." Tetrahedron Letters 57, no. 39 (2016): 4319-24. [CrossRef]
- Langston, S. P., S. Grossman, D. England, R. Afroze, N. Bence, D. Bowman, N. Bump, R. Chau, B. C. Chuang, C. Claiborne, L. Cohen, K. Connolly, M. Duffey, N. Durvasula, S. Freeze, M. Gallery, K. Galvin, J. Gaulin, R. Gershman, P. Greenspan, J. Grieves, J. Guo, N. Gulavita, S. Hailu, X. He, K. Hoar, Y. Hu, Z. Hu, M. Ito, M. S. Kim, S. W. Lane, D. Lok, A. Lublinsky, W. Mallender, C. McIntyre, J. Minissale, H. Mizutani, M. Mizutani, N. Molchinova, K. Ono, A. Patil, M. Qian, J. Riceberg, V. Shindi, M. D. Sintchak, K. Song, T. Soucy, Y. Wang, H. Xu, X. Yang, A. Zawadzka, J. Zhang, and S. M. Pulukuri. "Discovery of Tak-981, a First-in-Class Inhibitor of Sumo-Activating Enzyme for the Treatment of Cancer." J Med Chem 64, no. 5 (2021): 2501-20. [CrossRef]
- Tymianski, M. "Combining Neuroprotection with Endovascular Treatment of Acute Stroke: Is There Hope?" Stroke 48, no. 6 (2017): 1700-05.
Table 1.
miRNA-182/183 inhibitors evaluated for cerebral ischemia.
| Drug | Tissue/ Animal |
Ischemia Model | Intervention | Measured Outcome |
Results Summary | Study | |
|---|---|---|---|---|---|---|---|
| Orotic Acid | SHSY5Y, E18 PCN | SHSY5Y: 15hr OGD + 6hr recoveryE18 PCN: 5hr OGD + 16hr recovery | Drug co-treatment | Cell survival, SUMO concentration | SUMO upregulation*, OGD protection* | Bernstock et al., 2016 [73] | |
| AHPN | |||||||
| Telmisartan | |||||||
| TW-37 | |||||||
| Dianiline | |||||||
| Diazoxide | |||||||
| NCGC00185916 | SUMO upregulation* | ||||||
| Romidepsin | |||||||
| VX-702 | |||||||
| Lenalidomide | |||||||
| Belinostat | |||||||
| Pracinostat | |||||||
| Licofelone | |||||||
| Fosmidomycin | |||||||
| JWH-015 | |||||||
| Motesanib | |||||||
| Vatalanib | |||||||
| Entinostat | |||||||
| Panobinostat | |||||||
| CD-1 mice | Photothrombotic stroke at PMCA | Drug post-treatment (post stroke day 5-15) | Motor recovery, infarct volumes | No difference vs. negative control for both measures | Al Shoyaib et al., 2016 [78] | ||
| Entinostat | |||||||
| Collagenase-induced ICH | Drug post-treatment (1 hr post stroke, 10 mg/kg IP in PBS) | Sensorimotor deficit score, CD16/32 expression, neurodegeneration (via TUNEL staining neurons), infarct volume | Reduction in day 1 and day 3 post-ICH sensorimotor deficit*. Reduction in CD16/32 expression*, neurodegeneration, and infarct volume* | Bonsack and Sukumari-Ramesh, 2021 [79] | |||
| Diazoxide |
Wistar rats | 1.5 hr MCAO | Drug pretreatment (15min prior to stroke, 30 uL 0.4mM or 2mM ICV bolus) | 24hr post-stroke neurological score, infarct volume |
Increase in neurological score*; reduction in infarct volume* | Shimizu et al., 2002 [80] | |
| SD rats | 1hr RCCA ligation + hemorrhagic shock | Drug pre- and post-treatment: 5mg/kg IP bolus 24hr pre-stroke; 2.3 mg/kg/10min IV infusion 10min or 60min posttreatment | HSP25 and 70 concentrations | Pretreatment: Upregulation of HSP25 and 70*; 60min Posttreatment: Upregulation of HSP25 and 70* | O'Sullivan et al., 2007 [81] |
* Results statistically significant. Abbreviations: HSP, heat shock protein; ICH, intracerebral hemorrhage; IP, intraperitoneal; MCAO, middle cerebral artery occlusion; OGD, Oxygen-glucose deprivation; PCN, primary cortical neuron; PMCA, primary motor cortical area; RCCA, right common carotid artery; SD, Sprague-Dawley
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



