
Submitted:
06 April 2023
Posted:
10 April 2023
You are already at the latest version
Abstract
The Burkholderia cepacia complex (Bcc) are a group of increasingly multi-drug resistant opportunistic bacteria that can cause severe pulmonary infections. This resistance is driven through a combination of intrinsic factors and the carriage of a broad range of conjugative plasmids harbouring virulence determinants. Therefore, novel treatments are required to not only treat Bcc infection but also to prevent further spread of these virulence determinants. In the search for phages infective for two clinical Bcc isolates, CSP1 phage, a PRD1-like phage was isolated. CSP1 phage was found to require pilus machinery commonly encoded on conjugative plasmids to facilitate infection of multiple Gram-negative bacteria genera including Escherichia and Pseudomonas. Whole genome sequencing and characterisation of one of the clinical Burkholderia isolates revealed it to be Burkholderia contaminans. B. contaminans 5080 was found to contain a genome of over 8 Mbp encoding multiple intrinsic resistance factors, such as efflux pump systems, but more interestingly, carried three novel plasmids encoding multiple putative virulence factors for increased host fitness, including antimicrobial resistance. Even though PRD1-like phages are broad host range, their use in novel antimicrobial treatments shouldn’t be dismissed, as the dissemination potential of conjugative plasmids is extensive. Continued survey of clinical bacterial strains is
Keywords:
Burkholderia cepacia complex
; bacteriophage
; PRD1-like phage
; conjugative plasmid
1. Introduction
The rising occurrence and spread of antibiotic resistance are of great concern to public health. It is predicted that by 2050, antibiotic resistance will cause 10 million global premature fatalities and cost approximately 100 trillion USD in damages (1). Gram-negative bacteria are of particular concern due to their innate cell wall structure, incidence of horizontal gene transfer and acquisition of resistance determinants (2). One such group of organisms are the Burkholderia cepacia complex (Bcc), containing over 24 species that cause severe and chronic pulmonary infections in immunocompromised individuals, such as those with cystic fibrosis (3). The Bcc are renowned for their innate resistance to a wide range of antibiotics, including aminoglycosides, β-lactams, chloramphenicol, quinolones, tetracyclines and trimethoprim. They can also persist within nutrient limited environments and are often found to contaminate sterile products such as intravenous drugs, saline and other medical solutions (4). The Bcc ability to persist in unfavourable conditions and possess resistance to antimicrobials is mainly attributed to their innate cellular resistance mechanisms but also can be acquired through the uptake of mobile genetic elements (MGEs).
In addition to the intrinsically encoded antibiotic resistance determinants, including at least 19 known antibiotic extrusions efflux pumps providing resistance to chloramphenicol, tetracyclines, trimethoprim and some quinolone antibiotics in addition to resistance conferred by enzymatic modification of such as inducible β- lactamase enzymes including PenA1 that possess a broad hydrolytic spectrum, degrading ampicillin, aztreonam and ceftazidime (5), the Bcc also gain various antimicrobial resistance and metabolism factors from the horizontal gene transfer of MGEs. Plasmids, transposons, integrons and genomic islands have been found within Bcc strains that encode not only for antimicrobial resistance but also addition carbon and lipid metabolism, arsenic and toluene resistance and other virulence factors (6, 7). Dissemination of these resistance determinants from environmental into clinical settings is of concern, as environmental reservoirs are hotspots for antibiotic resistance genes and opportunistic bacteria such as Burkholderia sp. and Pseudomonas sp. (8). The spread of these determinants through MGEs such as plasmids is becoming more apparent, and the discovery of novel antimicrobials is now lagging the prevalence of antibiotic resistant bacteria. Last resort treatments are quickly becoming futile. However, one promising pipeline for novel antimicrobials are bacteriophages (also known as phages), bacterial specific viruses that rapidly infect and lyse their host.
Phages are inherently bactericidal in nature, and it is thought that they can be harnessed to treat multi-drug resistant bacterial infections. Since phages are also very selective in their host, they are harmless to commensal microbiome species or eukaryotic cells (4). Phage therapy research has regained momentum in the recent decades as a result of the increase in antibiotic resistant bacteria, with a focus on isolating and characterising phages that can be of suitable use for alternative therapies. However, a drawback of using phages is that they can be lysogenic, integrating into the host genome and in some cases can encode additional host advantages (9). Therefore, suitable phages need to be purely lytic to avoid any chance of in efficient bacterial lysis. Despite effort, the pool of phages infective against Bcc organisms is not well developed compared to other pathogenic bacteria. Therefore, this study aimed to isolate and characterise phages infective for two clinical Bcc isolates.
Here we report the isolation of a PRD1-like phage, CSP1 phage, which is a part of a genus of phages that utilise pilus machinery encoded by conjugative plasmids to infect multiple genera of Gram-negative bacteria. Whole genome sequencing of one clinical Burkholderia isolate identified it as Burkholderia contaminans and was found to carry three conjugative plasmids. One of the plasmids was related to previously isolated Incompatibility (Inc) group P found within Gram-negative bacteria including Burkholderia, Pseudomonas and Acidovorax. These plasmids contained putative antimicrobial resistance genes amongst other fitness determinants. While CSP1 phage (and other PRD1-like phages) are broad host range and often overlooked compared to strain specific phages, they could provide additional utility to phage therapies by specifically targeting pathogenic bacteria carrying and spreading antimicrobial resistance via conjugative plasmids.
2. Results and Discussion
2.1. Isolation of PRD1-like phage that infects clinical Burkholderia cepacia complex strains
Screening of pond water samples from Victoria, Australia resulted in the visualisation of plaques on two clinical isolates of Burkholderia cepacia complex (as determined by MALDI-TOF) isolated from a patient with Bcc bacteraemia in a hospital in metropolitan Melbourne, Victoria, Australia. Whole genome sequencing and assembly of the phage (CSP1 phage) using Illumina technology revealed a 14,942 bp linear dsDNA genome with a G+C content of 48.4% (Figure 1A). Whole genome BLASTn against the GenBank database indicated CSP1 phage displayed ~97.85% identity (and 100% coverage) to PRD1 phage, and similarly high identity to group of previously characterised phages known as PRD1-like phages (10). Intergenomic similarities and a phylogenetic tree were generated to compare the homology between CSP1, PRD1 and the other PRD1-like phages indicating CSP1 shared between between 93.35% and 97.87% intergenomic identity with PRD1 and other PRD1-like phages including PR5, PR772, PR3, L17 and PR4 (Figure 1B). Generation of a DNA polymerase phylogenetic tree also showed agreement with the intergenomic similarity analysis (Figure 1C). CSP1 phage forms a distinct clade with L17, PR3, PR772, PR5, PR4 and PRD1 phages. CSP1 phage DNA polymerase also shares homology to a group of Vibrio phages. PRD1 and PR4 phages represent two individual species currently classified by the International Committee on Taxonomy of Viruses (ICTV) in the Alphatectivirus genus. The other phages (L17, PR3, PR5, PR772 and LNA9) are greater than 95% similar to PRD1 phage and are known as PRD1-like phages. Since CSP1 is also >95% identical to PRD1 phage, CSP1 phage is also classified as a PRD1-like phage.
Annotation of the CSP1 phage genome revealed it is flanked by 110 bp inverted terminal repeats and encodes 30 putative open reading frames (ORFs) organised in characteristic modules (Figure 1A; Table 1). The gene products were assigned putative functions based on similarity to known domains based on the conserved domain database (CDD) and the Pfam database (11). Flanking the genome next to the inverted repeats are orf1, orf2, and orf29 and orf30. These genes encode products essential for phage replication including a terminal protein, DNA polymerase, and two single-stranded DNA binding proteins, respectively. PRD1 phage utilises a unique protein-primed DNA replication mechanism where the terminal protein is covalently bound to the 5’ end of the genome and initiates replication of the rest of the genome, with the inverted repeats serving as replication origins (12).
The virion structure of PRD1 phage has been extensively characterised (13-17). The capsid consists of 240 copies of the major capsid protein (orf14) arranged in a pseudo T=25 triangulation (15) with the minor capsid protein (orf21) stretching the capsid and cementing the edges of the icosahedron (13). On each capsid vertex but the twelfth are receptor binding proteins (orf4) and spike proteins (orf6) (13). The receptor binding and spike proteins attach to the penton base protein (orf5), protruding from capsid vertices and are responsible for host receptor recognition (14). On the twelfth vertex is the packaging complex that consists of the DNA packaging ATPase (orf11) and DNA packaging proteins (orf9, orf13 and orf15). This complex spans from the inner membrane to the outer capsid and is involved in both packaging of newly synthesised virus particles and injection of the viral genome into the host cell by formation of a tubular structure (15, 16). The tubular structure is derived from the inner membrane and consists of numerous proteins encoded by orf7, orf11, orf14 and orf18 (17).
Finally, CSP1 phage contains a predicted four gene lysis cluster comprising of endolysin (orf3), holin (orf24) and Rz/Rz1 (orf25/orf26) transglycosylases (18). The endolysin (orf3) has 1,4-β- N-acetylmuramidase activity while the Rz/Rz1 transglycosylases form a complex that spans the cytoplasmic membrane to outer membrane and aids in holin function. Holin proteins accumulate and form lesions in the cytoplasmic membrane that allow for the endolysin to reach the periplasm and degrade peptidoglycan, ultimately causing host cell lysis (18).
2.2. CSP1 phage relies on the sex pilus for successful infection
PRD1 phage has been shown to infect Gram-negative bacteria (i.e. Pseudomonas, Escherichia, Klebsiella) that harbour incompatibility (Inc) group P, W, or N plasmids (10). Given the genomic similarity between CSP1 and PRD1 phages, we reasoned CSP1 phage would share this property. To test this, we examined the ability of CSP1 phage to lyse strains of E. coli DH5α and P. aeruginosa PAO1 carrying derivatives of the conjugative IncP plasmid RP1 (ie., pUB307 and pUB1601). CSP1 phage could lyse E. coli DH5α and P. aeruginosa PAO1 only if they carried a conjugative IncP plasmid (Table 2). We further explored the ability of CSP1 phage to lyse the strains when they carried conjugation defective IncP plasmids due to transposon deletions. When the strains with the conjugation defective plasmids were challenged by the addition of CSP1 phage no lysis was observed suggesting that CSP1 phage requires an intact and fully functional mating complex to infect the Gram-negative bacteria.
2.3. Whole genome sequence confirms Bcc isolate as Burkholderia contaminans
Lysis of Burkholderia by a PRD1-like phage has not been described previously. As CSP1 phage appeared to rely on the conjugation machinery encoded by a conjugative plasmid for infection, this suggested our clinical Burkholderia sp. isolates were carrying a conjugative plasmid(s). To investigate we performed whole genome sequencing on Burkholderia sp. 5080 utilising both Illumina short-read and Oxford Nanopore long-read sequencing. The complete genome assembly was represented by six circularised contigs; three chromosomes and three plasmids (Table 3). Bcc typically contain multi-chromosome (also known as replicons) genomes as seen with this isolate, and are hypothesised to be essential for their fitness and persistence in a wide range of environments (19).
Preliminary attempts as species-level classification using 16S rRNA sequence identity, like previous MALDI-TOF identification, could not distinguish Burkholderia sp. 5080 from other members of the Bcc with matches of >99.8% identity to B. cepacia, B. cenocepacia and B. contaminans isolates, among others (Figure 2A). The Bcc are genetically very similar (and almost biochemically identical) and therefore 16S rRNA comparison has limitations in resolving species classification (19). To determine the species more definitively, we utilised whole-genome similarity analyses including average nucleotide identity (ANI) and genome DNA-DNA hybridisation (GDDH). ANI analysis determined that our isolate, Burkholderia sp. 5080, was most closely related to B. contaminans SK875 and B. contaminans ZCC, sharing 99.98% and 99.96% average nucleotide identity, respectively. The ANI results also better defined the groupings, with clearer clustering of each species group and fewer outliers (Figure 2B). Further comparison of the B. contaminans cluster showed that Burkholderia sp. 5080 shared 97.7% and 96.2% GDDH with B. contaminans SK875 and B. contaminans ZCC, respectively. Whole genome phylogenetic analysis ultimately agreed with the ANI and GDDH analyses, resolving each clade or species cluster and showed little substitutions between the genomes (Supplementary Figure 1). Furthermore, analysis with the GTDB-Tk pipeline correlated the identification as B. contaminans. Combined, these results indicate that the Burkholderia sp. 5080 isolate can be classified as Burkholderia contaminans and will be referred to as such moving forward.
B. contaminans 5080 encodes 7,655 predicted coding-sequences (CDS) across the three replicons. Functional categorisation of these using cluster of orthologous groups (COGs) resulted in ~88% CDS’s with at least one COG prediction, with some proteins containing multiple COG functions (Supplementary Figure 2). Given the role of efflux pumps in antimicrobial resistance within the Bcc (23) we inspected the genome and identified 117 CDS with predicted efflux pump-related function (Supplementary Table 1). Overall, the three B. contaminans 5080 replicons were found to encode at least 35 putative efflux pump systems: 17 in replicon 1, 10 in replicon 2 and 8 in replicon 3. Most of these efflux systems fell into the RND family efflux pump family, however there were also MATE, ABC and MFS family efflux pumps present (Figure 8). Some examples of these pumps included the OqxAB and CeoAB multidrug efflux pumps able to provide resistance to fluroquinolones and trimethoprim (20-22), the AcrAB-TolC efflux pump that gives rise to β-lactams (23), MexAB-OprM efflux pump that confers resistance to meropenem and ciprofloxacin (24) and ArpABC efflux pump involved in tetracycline, carbenicillin, chloramphenicol and streptomycin resistance (25). Other genes associated with antimicrobial resistance were also present including tetAR encoding for tetracycline resistance, penA and ampC that are β-lactamases that hydrolyse carbapenem antibiotics (26) and omp38 that encodes a porin protein able to lower cell membrane permeability to gain resistance to carbapenems and cephalosporins (27). Since Bcc organisms can occupy a large range of ecological niches, it is no surprise a large component of the genome has beneficial functions for the bacteria to adapt and persist in these environments and tolerate various antimicrobial compounds (28).
2.4. B. contaminans contains three conjugative plasmids
B. contaminans 5080 contained three unique DNA plasmids of 95.8 kb, 70.78 kb and 49.76 kb in size here named pBCO-1, pBCO-2 and pBCO-3, respectively (Table 4). Preliminary analysis revealed plasmid pBCO-3 belongs to the IncPβ family whereas pBCO-1 and pBCO-2 are uncharacterised. IncP plasmids are broad host range, conjugative plasmids that have been encountered in a wide range of Gram-negative bacteria (29). pBCO-1 and pBCO-2 are both also suspected to be conjugative as they encode the virB operons (30).
The first plasmid, pBCO-1, was found to share high identity (~99%) across a 71-kb region, the plasmid backbone, with three previously uncharacterised plasmid sequences from strains B. contaminans ZCC, B. contaminans XL73 and Burkholderia sp. FXe9 (Table 4, Figure 3). These strains were isolated from environmental locations (soil or plant-associated rhizosphere) in China between 2017-2021 (Table 4). pBCO-1 was found to contain an additional 25,471 bp sequence containing a nested transposon, here named Tn6611. Tn6611 is flanked by 38 bp inverted repeat sequences which are further flanked by 5 bp target site duplication sequences (TGGAA), a characteristic feature of a transposition event. This transposon contains a transposition module related to the Tn3-family and an accessory module of unknown function. Within the accessory module is an additional insertion of IS256 and appears to flank a remnant of another transposase, suggesting multiple insertions that have generated the structure seen in Tn6611.
Adjacent to the transposition module is an insertion of another intact transposon here named Tn6610 (Figure 3). Tn6610 belongs to the Tn5053 res-site hunter transposon family and is flanked a 25 bp inverted repeats. This transposon has a disrupted transposition module as an insertion of IS21 occurs in tniQ, that is essential for transposition (31, 32). Nonetheless Tn6610 appears to have inserted in a res site of the carrier transposon (Tn6611) and is flanked by 5 bp target site duplications (GTCAG) suggesting its placement within Tn6611 occurred prior to the acquisition of IS21 or transposed via tniQ being supplied in trans. The accessory module of Tn6610 is unique as res-site hunter family transposons traditionally contain either an integron or mercury(II) resistance module (31). In this case, Tn6610 instead contained a cluster of genes encoding an ABC type phosphonate transporter system (phnCDE), for phosphonate uptake and transport. Downstream of phnCDE is ptxD, a phosphonate dehydrogenase that degrades phosphonate to phosphate (33). Phosphonates are used in antibiotics such as fosfomycin (34) and fungicides and herbicides containing glyphosate (35). While phnCDE are typically of chromosomal origin (36), it is seen that phnCDE and ptxD were transferred through a transposon and inserted within pBCO-1.
The second plasmid, pBCO-2, shared >99% sequence similarity with five uncharacterised plasmids in GenBank (Table 4, Figure 3). Annotation of the pBCO-2 plasmid genes revealed that majority encode proteins of unknown function. Genes encoding proteins of known function appeared to be involved in plasmid maintenance and stability as well as type II and type IV secretion systems responsible for conjugative pilus and secretion machinery. Comparison of pBCO-2 with its close relatives suggests that the plasmids are conserved with no insertions of transposable elements (Figure 3).
The third plasmid, pBCO-3, belongs to the well characterised IncPβ group, sharing >99% homology with other IncPβ plasmids (Table 4, Figure 3). IncP plasmids are broad host range and have been isolated from a range of Gram-negative bacteria. The plasmids differ based on insertions of MGEs integrated between the plasmid modules (37). A truncated IS91 is present that is missing a portion of the 5’ end of the element, probably due to an intermolecular deletion. Adjacent to the IS91 are three genes, one that encodes a hydroxyacylglutathione hydrolase family protein, GloB, one that encodes a sterol desaturase family protein (SDFP) and one gene with an unknown function. There are also an additional two genes, one that encodes a vicinal oxygen chelate (VOC) family protein that is a putative GloA protein and an uroporphyrin-III methyltransferase that is involved in the cobalamin and siroheme biosynthetic pathway (38). GloA and GloB are known to detoxify the glycolysis by-product methylglyoxal and are associated with antibiotic resistance, biofilm formation and cell growth (39, 40). Their placement within this region is unknown but could have been part of a larger transposon with IS91. Analysis of the regions flanking the Tn6612 revealed that to the left was the IncP backbone whereas the right had a remnant tniQ gene and a complete tniR gene from a res-site hunter transposon. Immediately adjacent to tniR was a deleted form of parA which is commonly encountered in IncP plasmids and is known to be required for transposition of res-site hunter transposons (32). The remainder of the res hunter transposon has been deleted.
Based on the finding of the three plasmids, and the data available in Genbank, it can be presumed that pBCO-1 and pBCO-2 are narrow host range and replicate in Burkholderia species as they have only been detected in Burkholderia sp. (Table 4). The plasmids in GenBank homologous to pBCO-1 were isolated from environmental sources whereas those related to pBCO-2 were from clinical environments (Table 4). The third plasmid pBCO-3 belongs to the IncPβ group which are well established broad host range plasmids that can replicate in a diverse range of Gram-negative bacteria. All three plasmids identified in B. contaminans are presumed to be conjugative and have acquired genes from environmental and medical settings via MGEs such as transposons. These plasmids can further disseminate these genes in other Burkholderia strains and even more broadly to other Gram-negative bacteria.
3. Conclusions
This study describes the isolation of a PRD1-like phage, CSP1, the first time this phage has been shown to infect two members of the Burkholderia cepacia complex. CSP1, like PRD1, was shown to require the pilus machinery of a conjugative IncP plasmid for infection. Further investigation of one the clinical Burkholderia isolates revealed it B. contaminans harbouring three conjugative plasmids, one of which was identified as an IncP plasmid. We believe it is this conjugative plasmid, pBCO-3, that is providing the CSP1 receptor for infection. Both the chromosomes and plasmids of Burkholderia contaminans contained over 35 efflux pump systems and genes encoding for multidrug resistance, metal ion transport and host virulence, providing fitness advantages within both environmental and clinical settings. The core regions of the plasmids were highly conserved within the Burkholderia genus and other Gram-negative bacteria such as Pseudomonas and Acidovorax, highlighting the broad spread of these plasmids. The dissemination of these plasmids and their unique insertion regions containing virulence genes are of great concern with the rise of multi-drug resistant bacteria. While phages such as PRD1 and CSP1 can be very broad host range, they provide a novel treatment alternative, particularly in use with other strain specific phages to target virulent clinical pathogens.
4. Materials and methods
4.1. Bacterial strains and media
Bacterial cultures were grown in LB media (1% tryptone, 0.5% yeast extract and 1% sodium chloride ± 1.2% agar) from Oxoid (ThermoFisher Scientific, Australia). All cultures were grown aerobically at 37°C, and liquid cultures were incubated with shaking. The transposon mutagenesis strains used in this study were previously characterised in Petrovski and Stanisich (31).
4.2. Isolation and purification of phages
Pond water samples (1 ml) from Victoria, Australia were filtered using a 0.22-μm pore size cellulose acetate membrane filter. 1 ml of these samples were then incubated with the three Burkholderia strains (Table 1) in 10 ml cultures overnight. Following incubations, the cultures were pelleted by centrifugation at 6,000 x g for 10 min and filtration through a 0.22-μm cellulose acetate membrane filter. Fresh lawn plates of the strains were prepared and 100 μl aliquots of the filtrates were applied onto the lawn plates and allowed to dry. Plates were incubated and visually inspected for the presence of the plaques the following day. Single plaques were purified through three rounds of dilution and re-isolation to ensure purity.
4.3. Phage DNA isolation, genome sequencing and analysis
Purified phage particles were polyethylene glycol (PEG) precipitated and followed by a proteinase K treatment to extract DNA as described previously (41). Isolated phage DNA (100 ng) was prepared using the NEBNext® Ultra™ II DNA Library Prep Kit (NEB) followed by sequencing on an Illumina MiSeq v3 600-cycle kit with 300 bp paired-end reads. Raw data were filtered with fastp v0.23.2 (42), with default settings,. The phage genome was assembled with SPAdes v3.15.4 (43). Raw sequence reads were manually inspected in CLC Genomics WorkBench v9.5.5 (Qiagen) using the duplicated sequence’s function to detect regions of high starting position coverage to determine the genome termini (44). The phage genomes were manually screened for putative open reading frames (ORFs) with a minimum size of 100 bp using Geneious Prime 2022.2.1 and Glimmer (45). Sequence similarity searches were conducted using the predicted amino acid sequences against the GenBank database and the BLASTp algorithm was used (46, 47). Conserved domains and motifs were identified using the conserved domain database (CDD) (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) and the Pfam database (http://pfam.sanger.ac.uk) (11). For phage comparisons, VIRIDIC (48) was used to predict intergenomic similarities with the heatmap generated by GraphPad Prism 9.
4.4. Bacterial and plasmid DNA isolation and genome sequencing
DNA was extracted from 5 ml of log-phage bacterial culture using the Monarch® HMW DNA Extraction Kit (New England Biolabs) according to manufacturer’s instructions. For short-read Illumina sequencing, DNA (100 ng) was prepared using the NEBNext® UltraTM II DNA Library Prep Kit (New England Biolabs) and sequenced on an Illumina MiSeq V3 600-cycle kits (Illumina) to generate 300 bp paired-end reads. For long-read Oxford Nanopore sequencing, DNA (1 μg) was prepared using the Ligation Sequencing Kit (LSK-109) with Native Barcoding Expansion (NBD104), loaded onto a MinION/GridION SpotON flow cell (R9.4.1) and sequenced on a GridION (Oxford Nanopore). Basecalling was performed on-device in super accurate mode. Long-read data was assembled using Flye v2.9 (49) and then polished with short-read data using Polypolish v0.5.0 (50).
4.5. Genome annotation
For whole genome characterisation, gene annotation was completed by Prokka 1.14.5 and the genome was visualised using ClicO FS (51). For comparison to other species, the Burkholderia Genome DB (https://www.burkholderia.com/) was used to collate other Bcc genomes and 16s rRNA genes. For 16s rRNA phylogenetic analysis, A BLASTn search was conducted using Burkholderia sp. 5080 first 16s rRNA gene within chromosome 1. The top BLASTn hits were collated and the first 16s rRNA gene from the chromosome 1 was extracted for analysis. The collated 16s rRNA genes were then aligned with Geneious Prime (45) plug-in MUSCLE v3.8.425 (52) and a maximum-likelihood tree using the alignment was generated in MEGA11 (53). Average nucleotide identities (ANI) and a Neighbour-joining whole genome phylogenetic tree were generated using Kostas Lab tools (54), the heatmap of the ANI matrix was generated in GraphPad Prism 9. DNA:DNA hybridization (DDH) was calculated using the Genome-to-Genome Distance Calculator (55). Classification was also performed using the Type Strain Genome Server (TYGS) (55) and the classify-wf workflow with GTDB-Tk v.2.2.6 using the TDB-Tk reference data version r207 (56). Core gene analysis of B. contaminans 5080 was completed using Roary Galaxy v3.11.2 (57) with default parameters and EggNOG-mapper Galaxy v2.1.9 (58) using eggnog v5.0 database was used to assign COGs functions. ARBicate Galaxy v1.0.1 (https://github.com/tseemann/ABRicate) and the CARD database (https://card.mcmaster.ca/) were used to screen for virulence factors. The plasmids were annotated using both Prokka and BLASTp for determination of CDS function. Plasmid map comparisons were generated using Clinker v0.0.25 (59). EggNOG-mapper Galaxy v2.1.9 (58) using eggnog v5.0 database was used to assign COGs functions. ARBicate Galaxy v1.0.1 (https://github.com/tseemann/ABRicate) and the CARD database (https://card.mcmaster.ca/) were used to screen for virulence factors. The plasmids were annotated using both Prokka and BLASTp for determination of CDS function. Plasmid map comparisons were generated using Clinker v0.0.25 (59).
Author Contributions
Conceptualization, C.R.S., S.P. and S.B.; formal analysis, C.R.S., S.P. and S.B.; investigation, C.R.S.; data curation, C.R.S., S.P. and S.B.; writing—original draft preparation, C.R.S., S.P. and S.B.; writing—review and editing, C.R.S., S.P. and S.B.; supervision, S.P. and S.B.; resources, S.P. All authors have read and agreed to the published version of the manuscript.
Funding
C.R.S. was supported by a La Trobe University postgraduate award and a Defence Science Institute (DSI) RHD student grant.
Data Availability Statement
The nucleotide sequences for the bacteria and phage sequenced and assembled in this study are available on NCBI Genbank under the following accession numbers: Burkholderia phage vB-Bco-CSP1, OQ674210; Burkholderia contaminans 5080, CP120947- CP120952.
Acknowledgements
We thank Dr Mark Chan for providing the Burkholderia cepacia complex strains. We thank the La Trobe University Genomics Platform for their 2200 TapeStation service.
References
- O'Neill J. Review on antimicrobial resistance: tackling drug-resistant infections globally: final report and recommendations. London: Wellcome Trust; 2016. p. 80 pp.
- Breijyeh Z, Jubeh B, Karaman R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules. 2020, 25. [Google Scholar]
- Tavares M, Kozak M, Balola A, Sá-Correia I. Burkholderia cepacia Complex Bacteria: a Feared Contamination Risk in Water-Based Pharmaceutical Products. Clin Microbiol Rev. 2020, 33.
- Lauman P, Dennis JJ. Advances in Phage Therapy: Targeting the Burkholderia cepacia Complex. Viruses. 2021, 13, 1331. [Google Scholar] [CrossRef] [PubMed]
- Scoffone VC, Trespidi G, Barbieri G, Irudal S, Perrin E, Buroni S. Role of RND Efflux Pumps in Drug Resistance of Cystic Fibrosis Pathogens. Antibiotics. 2021, 10, 863. [Google Scholar]
- Holden MTG, Seth-Smith HMB, Crossman LC, Sebaihia M, Bentley SD, Cerdeño-Tárraga AM, et al. The Genome of Burkholderia cenocepacia J2315, an Epidemic Pathogen of Cystic Fibrosis Patients. Journal of Bacteriology. 2009, 191, 261–277. [Google Scholar] [CrossRef]
- Shields MS, Reagin MJ, Gerger RR, Campbell R, Somerville C. TOM, a new aromatic degradative plasmid from Burkholderia (Pseudomonas) cepacia G4. Applied and Environmental Microbiology. 1995, 61, 1352–1356.
- Esiobu N, Armenta L, Ike J. Antibiotic resistance in soil and water environments. International Journal of Environmental Health Research. 2002, 12, 133–144. [Google Scholar] [CrossRef]
- Lin DM, Koskella B, Lin HC. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J Gastrointest Pharmacol Ther. 2017, 8, 162–173. [Google Scholar] [CrossRef]
- Olsen RH, Siak JS, Gray RH. Characteristics of PRD1, a plasmid-dependent broad host range DNA bacteriophage. J Virol. 1974, 14, 689–699. [Google Scholar] [CrossRef]
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic acids research. 2015; 44(D1), D279–D285.
- Savilahti H, Bamford DH. Protein-primed DNA replication: role of inverted terminal repeats in the Escherichia coli bacteriophage PRD1 life cycle. Journal of Virology. 1993, 67, 4696–4703. [Google Scholar] [CrossRef]
- Oksanen HM, Abrescia NGA. Membrane-Containing Icosahedral Bacteriophage PRD1: The Dawn of Viral Lineages. In: Greber UF, editor. Physical Virology: Virus Structure and Mechanics. Cham: Springer International Publishing; 2019; 85–109.
- Huiskonen JT, Manole V, Butcher SJ. Tale of two spikes in bacteriophage PRD1. Proceedings of the National Academy of Sciences. 2007, 104, 6666–6671. [Google Scholar] [CrossRef] [PubMed]
- Gowen B, Bamford JK, Bamford DH, Fuller SD. The tailless icosahedral membrane virus PRD1 localizes the proteins involved in genome packaging and injection at a unique vertex. J Virol. 2003, 77, 7863–7871. [Google Scholar] [CrossRef] [PubMed]
- Bamford D, Mindich L. Structure of the lipid-containing bacteriophage PRD1: disruption of wild-type and nonsense mutant phage particles with guanidine hydrochloride. J Virol. 1982, 44, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Peralta B, Gil-Carton D, Castaño-Díez D, Bertin A, Boulogne C, Oksanen HM, et al. Mechanism of Membranous Tunnelling Nanotube Formation in Viral Genome Delivery. PLOS Biology. 2013, 11, e1001667. [Google Scholar]
- Krupovič M, Cvirkaitė-Krupovič V, Bamford DH. Identification and functional analysis of the Rz/Rz1-like accessory lysis genes in the membrane-containing bacteriophage PRD1. Molecular Microbiology. 2008, 68, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Mahenthiralingam E, Urban TA, Goldberg JB. The multifarious, multireplicon Burkholderia cepacia complex. Nat Rev Microbiol. 2005, 3, 144–156. [Google Scholar] [CrossRef]
- Podnecky N, Rhodes K, Schweizer H. Efflux Pump-mediated Drug Resistance in Burkholderia. Frontiers in Microbiology. 2015, 6. [Google Scholar]
- Kim HB, Wang M, Park CH, Kim EC, Jacoby GA, Hooper DC. oqxAB encoding a multidrug efflux pump in human clinical isolates of Enterobacteriaceae. Antimicrob Agents Chemother. 2009, 53, 3582–3584.
- Nair BM, Cheung KJ, Jr. , Griffith A, Burns JL. Salicylate induces an antibiotic efflux pump in Burkholderia cepacia complex genomovar III (B. cenocepacia). J Clin Invest. 2004, 113, 464–473.
- Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol. 1995, 16, 45–55. [Google Scholar] [CrossRef]
- Choudhury D, Ghose A, Dhar Chanda D, Das Talukdar A, Dutta Choudhury M, Paul D, et al. Premature Termination of MexR Leads to Overexpression of MexAB-OprM Efflux Pump in Pseudomonas aeruginosa in a Tertiary Referral Hospital in India. PLoS One. 2016, 11, e0149156. [Google Scholar]
- Kieboom J, de Bont J. Identification and molecular characterization of an efflux system involved in Pseudomonas putida S12 multidrug resistance. Microbiology (Reading). 2001;147(Pt 1):43-51.
- Becka SA, Zeiser ET, LiPuma JJ, Papp-Wallace KM. Activity of Imipenem-Relebactam against Multidrug- and Extensively Drug-Resistant Burkholderia cepacia Complex and Burkholderia gladioli. Antimicrobial Agents and Chemotherapy. 2021, 65, e01332–21. [Google Scholar]
- Aunkham A, Schulte A, Winterhalter M, Suginta W. Porin involvement in cephalosporin and carbapenem resistance of Burkholderia pseudomallei. PLoS One. 2014, 9, e95918. [Google Scholar]
- Lewis ER, Torres AG. The art of persistence-the secrets to Burkholderia chronic infections. Pathog Dis. 2016, 74. [Google Scholar]
- Popowska M, Krawczyk-Balska A. Broad-host-range IncP-1 plasmids and their resistance potential. Frontiers in Microbiology. 2013, 4. [Google Scholar]
- Christie, PJ. Type IV secretion: the Agrobacterium VirB/D4 and related conjugation systems. Biochim Biophys Acta. 2004, 1694, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Petrovski S, Stanisich VA. Tn502 and Tn512 are res site hunters that provide evidence of resolvase-independent transposition to random sites. J Bacteriol. 2010, 192, 1865–1874.
- Minakhina S, Kholodii G, Mindlin S, Yurieva O, Nikiforov V. Tn5053 family transposons are res site hunters sensing plasmidal res sites occupied by cognate resolvases. Molecular Microbiology. 1999, 33, 1059–1068.
- Relyea HA, van der Donk WA. Mechanism and applications of phosphite dehydrogenase. Bioorganic Chemistry. 2005, 33, 171–189. [Google Scholar] [CrossRef]
- Martínez A, Osburne MS, Sharma AK, DeLong EF, Chisholm SW. Phosphite utilization by the marine picocyanobacterium Prochlorococcus MIT9301. Environmental Microbiology. 2012, 14, 1363–1377.
- Dann E, McLeod A. Phosphonic acid: a long-standing and versatile crop protectant. Pest Management Science. 2021, 77, 2197–2208. [Google Scholar] [CrossRef]
- Poehlein A, Daniel R, Schink B, Simeonova DD. Life based on phosphite: a genome-guided analysis of Desulfotignum phosphitoxidans. BMC Genomics. 2013, 14, 753. [Google Scholar]
- Schlüter A, Szczepanowski R, Pühler A, Top EM. Genomics of IncP-1 antibiotic resistance plasmids isolated from wastewater treatment plants provides evidence for a widely accessible drug resistance gene pool. FEMS Microbiology Reviews. 2007, 31, 449–477. [Google Scholar] [CrossRef] [PubMed]
- Vévodová J, Graham RM, Raux E, Schubert HL, Roper DI, Brindley AA, et al. Structure/Function Studies on a S-Adenosyl-l-methionine-dependent Uroporphyrinogen III C Methyltransferase (SUMT), a Key Regulatory Enzyme of Tetrapyrrole Biosynthesis. Journal of Molecular Biology. 2004, 344, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Sanchez A, Feng Y, Berude JC, Portnoy DA. Detoxification of methylglyoxal by the glyoxalase system is required for glutathione availability and virulence activation in Listeria monocytogenes. PLoS Pathog. 2021, 17, e1009819. [Google Scholar]
- Haris M, Chen C, Wu J, Ramzan MN, Taj A, Sha S, et al. Inducible knockdown of Mycobacterium smegmatis MSMEG_2975 (glyoxalase II) affected bacterial growth, antibiotic susceptibility, biofilm, and transcriptome. Archives of Microbiology. 2021, 204, 97. [Google Scholar]
- Petrovski S, Seviour RJ, Tillett D. Genome sequence and characterization of the Tsukamurella bacteriophage TPA2. Appl Environ Microbiol. 2011, 77, 1389–1398.
- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018, 34, i884–i90. [Google Scholar] [CrossRef]
- Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. Using SPAdes De Novo Assembler. Current Protocols in Bioinformatics. 2020, 70, e102. [Google Scholar]
- Batinovic S, Stanton CR, Rice DTF, Rowe B, Beer M, Petrovski S. Tyroviruses are a new group of temperate phages that infect Bacillus species in soil environments worldwide. BMC Genomics. 2022, 23, 777. [Google Scholar]
- Kearse M, Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., Buxton, S., Cooper, A., Markowitz, S., Duran, C., Thierer, T., Ashton, B., Mentjies, P., & Drummond, A. . Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012, 1647–1649.
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Moraru C, Varsani A, Kropinski AM. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses. 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nature Biotechnology. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Wick RR, Holt KE. Polypolish: Short-read polishing of long-read bacterial genome assemblies. PLOS Computational Biology. 2022, 18, e1009802. [Google Scholar]
- Cheong W-H, Tan Y-C, Yap S-J, Ng K-P. ClicO FS: an interactive web-based service of Circos. Bioinformatics. 2015, 31, 3685–3687. [Google Scholar] [CrossRef]
- Edgar, RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura K, Stecher G, Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol. 2021, 38, 3022–3027.
- Rodriguez-R L, Konstantinidis K. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Preprints; 2016.
- Meier-Kolthoff JP, Carbasse JS, Peinado-Olarte RL, Göker M. TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Research. 2021, 50(D1), D801–D807.
- Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics. 2022, 38, 5315–5316. [Google Scholar] [CrossRef]
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Rodríguez del Río Á, Giner-Lamia J, Cantalapiedra CP, Botas J, Deng Z, Hernández-Plaza A, et al. Functional and evolutionary significance of unknown genes from uncultivated taxa. bioRxiv. 2022:2022.01.26.477801.
- Gilchrist CLM, Chooi Y-H. clinker & clustermap.js: automatic generation of gene cluster comparison figures. Bioinformatics. 2021, 37, 2473–2475. [Google Scholar]
Figure 1.
Characterisation of Burkholderia phage CSP1. A) Genome map of CSP1 phage. Features are colour coded based on predicted function. B) Whole genome comparison of CSP1 phage with other Alphatectivirus phages. C) Maximum likelihood phylogenetic tree tree of CSP1 phage with top GenBank matches using the DNA polymerase gene. The percentage of trees in which the associated taxa clustered together is shown next to the branches (of 100 bootstraps).
Figure 1.
Characterisation of Burkholderia phage CSP1. A) Genome map of CSP1 phage. Features are colour coded based on predicted function. B) Whole genome comparison of CSP1 phage with other Alphatectivirus phages. C) Maximum likelihood phylogenetic tree tree of CSP1 phage with top GenBank matches using the DNA polymerase gene. The percentage of trees in which the associated taxa clustered together is shown next to the branches (of 100 bootstraps).

Figure 2.
Figure 2. Whole genome and phylogenetic comparisons of Bcc strains. A) 16s rRNA maximum-likelihood phylogenetic tree of the top GenBank hits against B. contaminans 5080. Bootstrap values are shown next to the branches. B) Average nucleotide identity heatmap of whole Bcc genomes. In both A and B, B. contaminans 5080 isolated from this study is highlighted by blue shading.
Figure 2.
Figure 2. Whole genome and phylogenetic comparisons of Bcc strains. A) 16s rRNA maximum-likelihood phylogenetic tree of the top GenBank hits against B. contaminans 5080. Bootstrap values are shown next to the branches. B) Average nucleotide identity heatmap of whole Bcc genomes. In both A and B, B. contaminans 5080 isolated from this study is highlighted by blue shading.
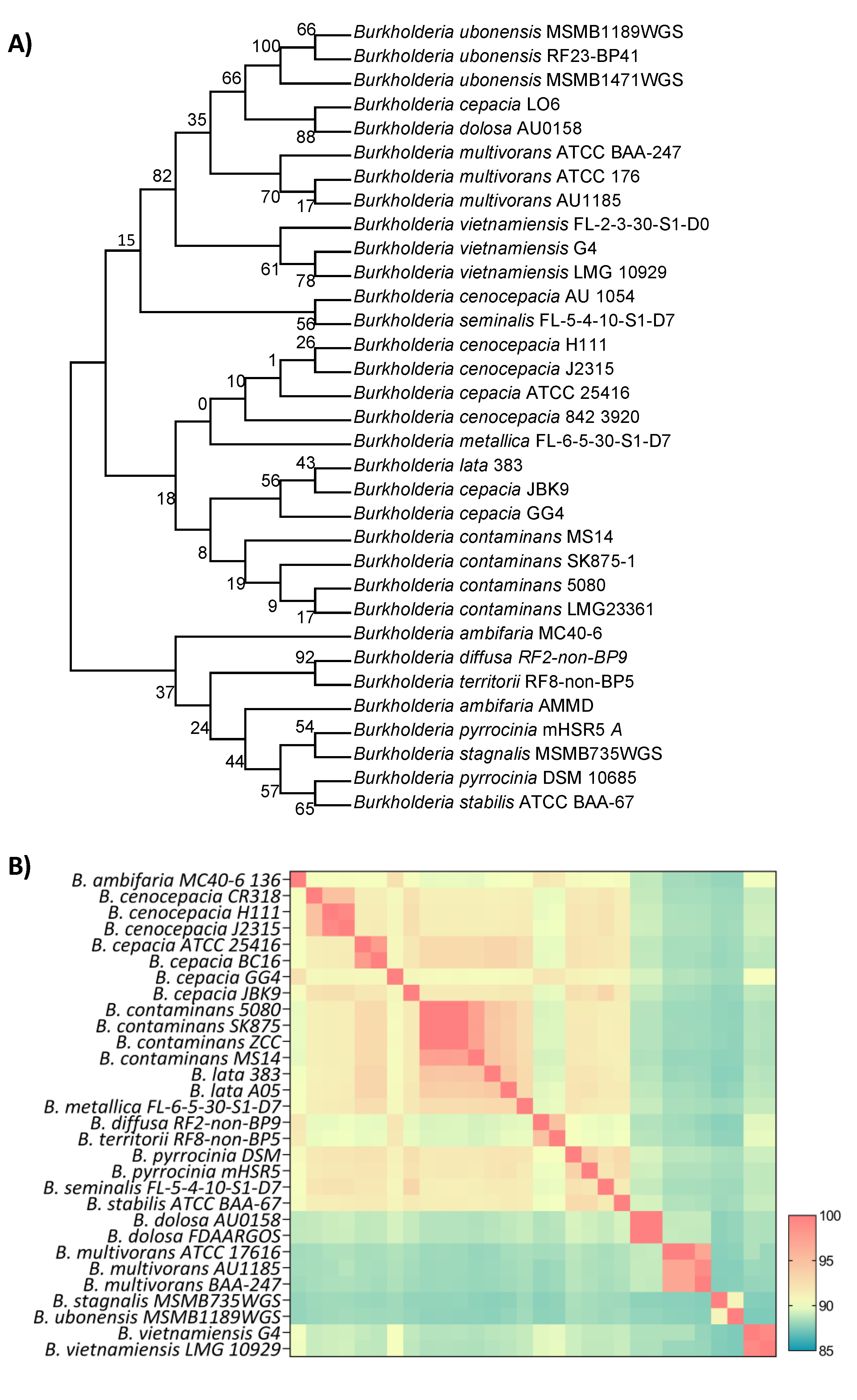
Figure 3.
Plasmid map comparisons of B. contaminans 5080 plasmids A) pBCO-1 , B) pBCO-2 and 3) pBCO-3 with closest GenBank matches. Unique transposon insertions in pBCO-1 and pBCO-3 are shown in expanded form. CDS in wholeplasmid comparisons are displayed by various colours depending on their homology and nucleotide identities are showed by a grey-scale key and CDS in transposon insertion sequences and represented named arrows, remnant genes are denoted by prime notations (e.g., tniQ’). Inverted repeats are shown by vertical lines with small arrows indicating the orientation of the repeat and 5 bp target duplication sequences are shown adjacent to the inverted repeat. All putative mobile elements are indicated with a bracket with the name of the element. The plasmid from B. contaminans FXe9 (unnamed2) was omitted due to the presence of a large duplication in the plasmid sequence.
Figure 3.
Plasmid map comparisons of B. contaminans 5080 plasmids A) pBCO-1 , B) pBCO-2 and 3) pBCO-3 with closest GenBank matches. Unique transposon insertions in pBCO-1 and pBCO-3 are shown in expanded form. CDS in wholeplasmid comparisons are displayed by various colours depending on their homology and nucleotide identities are showed by a grey-scale key and CDS in transposon insertion sequences and represented named arrows, remnant genes are denoted by prime notations (e.g., tniQ’). Inverted repeats are shown by vertical lines with small arrows indicating the orientation of the repeat and 5 bp target duplication sequences are shown adjacent to the inverted repeat. All putative mobile elements are indicated with a bracket with the name of the element. The plasmid from B. contaminans FXe9 (unnamed2) was omitted due to the presence of a large duplication in the plasmid sequence.
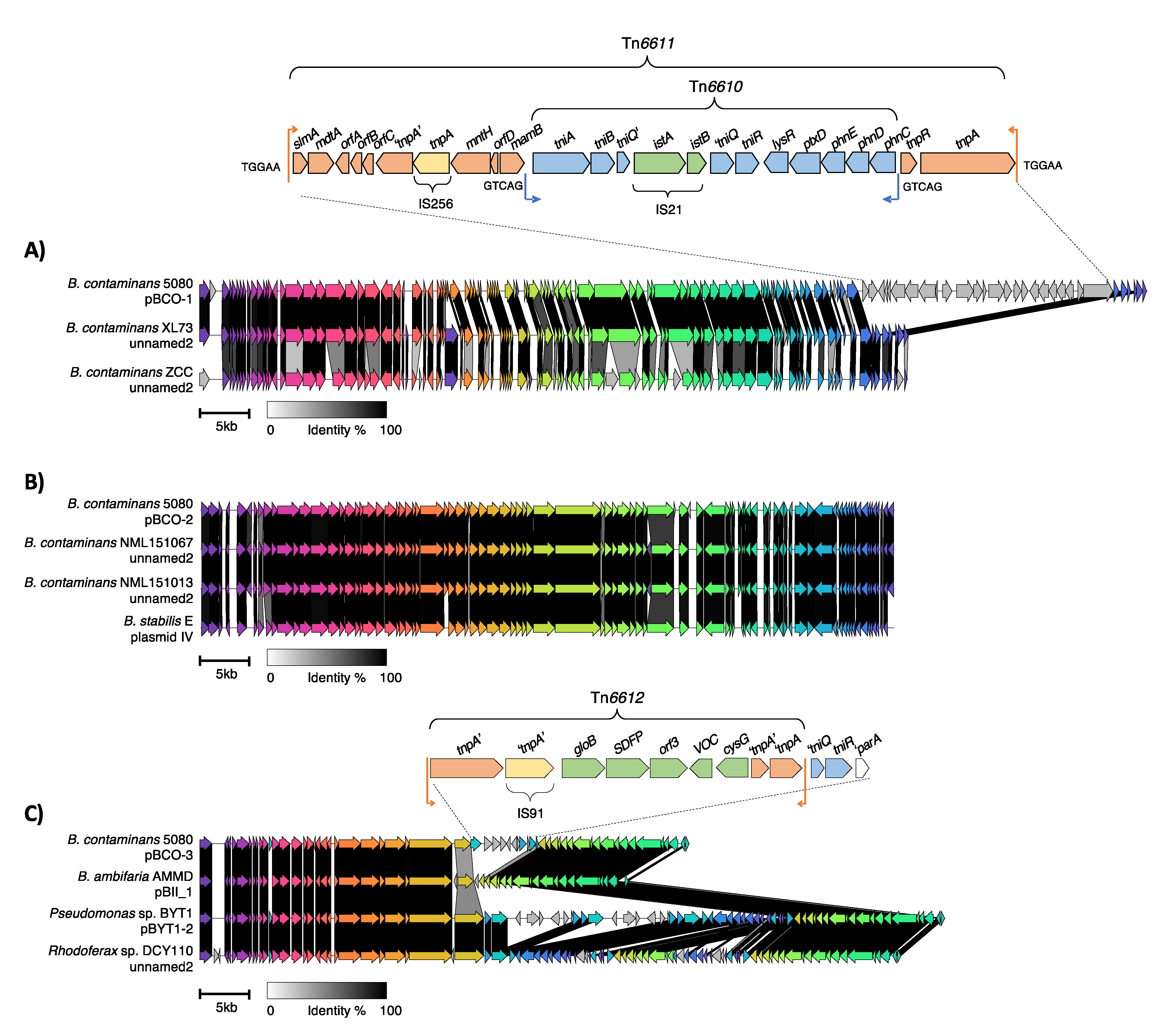

Table 1.
ORF annotations of CSP1 genome with predicted functions.
| ORF | Predicted function | Size (bp) |
Coordinates | Conserved protein domain |
E-value |
|---|---|---|---|---|---|
| 1 | Terminal protein | 780 | 233 – 1,012 | - | 0.0 |
| 2 | DNA polymerase | 1662 | 1,016 – 2,677 | pfam03175 | 0.0 |
| 3 | N-acetylmuramidase endolysin | 450 | 2,680 – 3,129 | pfam11860 | 2e-118 |
| 4 | Receptor binding protein | 1776 | 3,128 – 4,903 | - | 0.0 |
| 5 | Penton protein | 381 | 4,907 – 5,287 | pfam08948 | 6e-89 |
| 6 | Spike protein | 1026 | 5,287 – 6,312 | pfam08949; pfam08948 | 0.0 |
| 7 | Assembly protein | 261 | 6,331 – 6,591 | - | 2e-56 |
| 8 | Assembly protein | 207 | 6,581 – 6,787 | - | 2e-38 |
| 9 | DNA packaging efficiency factor |
501 | 6,787 – 7,287 | - | 4-104 |
| 10 | Assembly protein | 324 | 7,321 – 7,644 | - | 1e-70 |
| 11 | DNA packaging ATPase | 645 | 7,680 – 8,323 | cd01127 | 6e-168 |
| 12 | Hypothetical protein | 129 | 8,335 – 8,463 | - | 9e-20 |
| 13 | DNA packaging protein | 129 | 8,463 – 8,591 | - | 4e-21 |
| 14 | Major capsid protein | 1188 | 8,598 – 9,785 | pfam09018 | 0.0 |
| 15 | DNA packaging protein | 144 | 9,804 – 9,947 | - | 1e-24 |
| 16 | Hypothetical protein | 84 | 9,959 – 10,042 | ||
| 17 | Hypothetical protein | 123 | 10,047 – 10,169 | - | 3e-19 |
| 18 | DNA delivery protein | 273 | 10,171 – 10,443 | - | 2e-55 |
| 19 | DNA delivery protein | 165 | 10,443 – 10,607 | pfam11087 | 6e-27 |
| 20 | DNA delivery protein | 206 | 10,620 – 10,825 | pfam11087 | 1e-04 |
| 21 | Minor capsid protein | 255 | 10,835 – 11,089 | pfam09300 | 1e-52 |
| 22 | DNA delivery protein | 624 | 11,204 – 11,827 | - | 4e-145 |
| 23 | Hypothetical protein | 354 | 11,838 – 12,191 | pfam09301 | 5e-79 |
| 24 | Transglycosylase (Rz) | 810 | 12,192 – 13,001 | pfam01464; cd00254 | 0.0 |
| 25 | Putative Rz1 | 465 | 12,539 – 13,003 | ||
| 26 | Holin | 354 | 12,998 – 13,351 | pfam16083 | 4e-80 |
| 27 | Hypothetical protein | 363 | 13,345 – 13,707 | - | 5e-86 |
| 28 | Hypothetical protein | 234 | 13,670 – 13,903 | - | 1e-58 |
| 29 | ssDNA-binding protein | 285 | 13,862 – 14,146 | - | 1e-62 |
| 30 | ssDNA-binding protein | 483 | 14,219 – 14,701 | - | 4e-108 |
Table 2.
List of bacterial strains tested for CSP1 phage infection.
| Strain | Description | CSP1 lysis a | Conjugation b |
|---|---|---|---|
| Burkholderia sp. 5080 | Wild-type strain | + | NA |
| Burkholderia sp. 3720 | Wild-type strain | + | NA |
| E. coli | Lab strain | - | - |
| E. coli (pUB307) | Lab strain with an IncP plasmid pUB307 | + | + |
| E. coli (pUB1601::Tn502) | pUB1601 Tn502 deletant mutant | - | - |
| E. coli (pUB307::Tn502) | pUB307 Tn502 deletant mutant | - | - |
| P. aeruginosa | Lab strain | - | - |
| P. aeruginosa (pUB307) | Lab strain with an IncP plasmid | + | + |
a + indicates lysis; - indicates no lysis. b + indicates ability to conjugate; - indicates inability to conjugate. E. coli strains were DH5α; P. aeruginosa were PAO1
Table 3.
Properties of Burkholderia sp. 5080 genome.
| Replicon 1 | Replicon 2 | Replicon 3 | Plasmid 1 | Plasmid 2 | Plasmid 3 | |
|---|---|---|---|---|---|---|
| Size (bp) | 3,799,460 | 3,244,835 | 1,529,354 | 95,791 | 70,779 | 49,756 |
| G+C content (%) | 66.2 | 66.7 | 65.8 | 58.9 | 62.2 | 65.6 |
| CDS | 3, 429 | 2, 853 | 1, 304 | 124 | 85 | 67 |
| rRNA | 12 | 3 | 3 | - | - | - |
| tRNA | 64 | 11 | 6 | - | 1 | - |
Table 4.
Plasmids pBCO-1 and pBCO-2 and their close relatives.
| Plasmid | Strain | Size (bp) | Country | Source | Year | Accession |
|---|---|---|---|---|---|---|
| pBCO-1 | Burkholderia contaminans 5080 | 95,791 | Australia | Clinical | 2018 | CP120950 |
| unnamed2 | Burkholderia contaminans ZCC | 71,607 | China | Soil | 2017 | CP042168 |
| unnamed2 | Burkholderia contaminans XL73 | 71,703 | China | Cucumber rhizosphere | 2018 | CP046611 |
| unnamed2 | Burkholderia sp FXe9 | 10,5779 | China | Soil | 2021 | CP101282 |
| pBCO-2 | Burkholderia contaminans 5080 | 70,779 | Australia | Clinical | 2018 | CP120951 |
| plasmid IV | Burkholderia stabilis E | 70,766 | Switzerland* | Contaminated wipes | 2017 | LR025745 |
| unnamed2 | Burkholderia contaminans NML151067 | 70,795 | Canada | Sputum | 2015 | CP102471 |
| unnamed2 | Burkholderia contaminans NML151013 | 70,795 | Canada | Sputum | 2015 | CP102466 |
| unnamed2 | Burkholderia contaminans toggle1 | 77,157 | Taiwan | Blood -human | 2018 | CP073666 |
| unnamed | Burkholderia aenigmatica CMCC(B)23010 | 448,767 | China | Unknown | 2019 | CP091649 |
* Genbank entry does not indicate the country of isolation, country of the research group is displayed.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



