
Submitted:
12 April 2023
Posted:
12 April 2023
Read the latest preprint version here
Abstract
Adrenarche is a developmental phase that occurs only in humans and chimpanzees. It is characterized by the maturation of the adrenal zona reticularis, culminating in the synthesis and secretion of enormous amounts of dehydroepiandro-sterone sulfate (DHEAS) into the blood stream. The purpose of adrenarche, and the flooding of the circulation with DHEAS in the period preceding the onset of adult human body size, has been described as “enigmatic.” This laboratory has uncovered a previously unknown, human-specific “kill-switch” tumor suppression mechanism in which circulating DHEAS is pumped into cells that have undergone tumorigenic mutation(s)— primarily in the TP53 tumor suppressor gene. We refer to such tumorigenic single cells as singularities. The advantage of eliminating tumorigenic cells while they are still in their single cell state, before they have evolved into the heterogeneous tumor cell populations that have proven refractory to chemotherapeutic sterilization, is obvious. This is accomplished by activation of the kill-switch in singularities by de-sulfating imported DHEAS to dehydroepiandrosterone (DHEA), a potent uncompetitive inhibitor of the enzyme Glucose-6-phosphate Dehydrogenase (G6PD). G6PD is responsible for the production of most of the NADPH required for the synthesis and maintenance of activity of anti-oxidant selenoproteins, and for the ubiquinol that prevents ferroptosis. In singularities, uncompetitive inhibition of G6PD by DHEA rapidly becomes irreversible, leading to ROS/ferroptosis-mediated cell death.
Keywords:
p53
; adrenarche
; dehydroepiandrosterone
; DHEA
; DHEAS
Introduction
Adrenarche is a developmental stage distinct from and preceding gonadarche and pubarche (Figure 1). It is characterized by the maturation of the zona reticularis in the adrenal gland, and subsequent secretion into the circulation of enormous amounts of dehydroepiandrosterone sulfate (DHEAS). Accordingly, the word adrenarche literally means “the awakening of the adrenal gland.” Despite extensive research, the purpose of adrenarche remains unknown, and it has been called “enigmatic” by several authors [1,2]. Among primates, true adrenarche occurs only in the Hominini lineage— humans, chimpanzees and bonobos— and the absence of adrenarche in other species typically used as animal models has hindered its study. The levels of circulating DHEAS in humans, for example, are some 10,000-fold higher than in mice.
Adrenarche also precedes the pubertal growth spurt, which averages approximately 8 centimeters (3 inches)/year in Western children. Peak growth for girls is typically 6-12 months before the onset of menarche, after which growth slows, such that only an additional 5-7.6 cm (2-3 inches) of growth occurs during adulthood, on average. Boys tend to have their growth spurt later than girls. Peak growth for boys is right before spermarche and averages approximately 9 centimeters (3.5 inches)/year [3]. Thus, adrenarche occurs immediately preceding the onset of the most dramatic increases in body stature that occur in our species.
In adults, height/stature is associated with lifetime risk of cancer. The ‘million women study’ followed 1,297,124 women for a median time of 9.4 years each and reported an overall 16% increase in cancer risk for every 10 cm (4 inches) in height above average [4]. This finding was reproduced in a cohort of 144,701 Canadian women (median follow-up 12 years) by Kabat et al. [5], in 310,000 male and female UK Biobank participants [6], and in 22 million Koreans [7]. The increased lifetime risk of cancer for men vs. women (Figure 2) is also driven by body size [8]. Further support for the association between stature and cancer comes from studies of dwarf humans with Laron Syndrome— one of which studies lasted 57 years. This work demonstrated a near total absence of cancer in these long-lived, small-bodied humans [9,10].
This laboratory has recently identified a human-specific, “kill switch” tumor suppression mechanism that is dependent upon the DHEAS that is synthesized and secreted into the circulation at adrenarche [11,12,13]. In brief, circulating DHEAS is specifically imported into cells that have suffered a tumorigenic lesion— typically in the p53 tumor suppressor gene. Such cells— which we refer to as singularities— would typically proceed to a neoplastic, tumorigenic state were it not for this kill switch mechanism. DHEAS that has been specifically imported into singularities is then de-sulfated to DHEA, where DHEA’s unique uncompetitive inhibition of Glucose-6-phosphate dehydrogenase (G6PD) leads to depletion of the NADPH that is required for the synthesis and maintenance of activity of ROS-detoxifying selenoproteins; and for the synthesis of ubiquinone, and its reduction to ubiquinol, that is required to prevent ferroptosis. Loss of control of intracellular ROS accumulation and ferroptosis extinguishes the singularity while it is still in its single cell state, before it has had the opportunity to evolve into the heterogeneous tumor cell populations that are so difficult to eradicate. Adrenarche thus triggers the kill switch immediately preceding the dramatic increase in stature— and therefore cancer risk— that occurs during the human adolescent growth spurt (Figure 1).
In the remainder of this article, we will discuss the components of the human-specific kill switch tumor suppression mechanism, demonstrating their evolution through the course of primate speciation, culminating in the advent of adrenarche in the Hominini. We shall show how the evolution of the kill switch answers many long-standing questions in human biology, such as the purpose of adrenarche, and how it evolved despite the fact that the proximal metabolite of DHEAS, i.e., DHEA, is a powerful, rapidly irreversible inhibitor of so fundamental an enzyme as G6PD; why anthropoid primates, including humans, underwent selection for the loss of the ability to synthesize vitamin C; why hominoid primates, including humans, underwent selection for dramatically high levels of circulating uric acid, precipitating risk for the development of the debilitating disease of gout; and many more such long-standing, here-to-fore unanswered questions in human biology.
The lex naturalis equation links speciation to lifetime cancer risk
Recent results now indicate that every vertebrate species has evolved its own, unique, species-specific mechanism of tumor suppression [11,12,13,14,15]. As is evident by comparative cancer research, evolution by natural selection has evolved a general rule that a certain low amount of neoplastic transformation is enabled in vertebrate animals, probably because a certain low amount of genomic instability is required so that the process of speciation can respond to changes in the environment, or exploit new opportunities. The low, lifetime risk of cancer in the vast majority of larger, longer-lived vertebrate animals is fairly rigidly set at ≤ 5% [14]. Not only is the lifetime cancer risk of most vertebrates set at this low rate, but their cancer risk as a function of time remains essentially flat throughout their lifespans. This is in sharp contrast to lifetime cancer risk in humans, which begins to dramatically rise once we have exceeded the lifespan that has characterized our species for more than 99% of its existence— averaging 25 years, with 88% mortality by age 30— and reaches a value tenfold higher than the vertebrate norm [3,4,5,6]. The low, flat cancer risk of other vertebrates with lifespans and body size equal to or exceeding that of our species— for example elephants [14] and whales [15]— demonstrates that cancer risk is aberrant in humans (Figure 2). This disparity recently helped to uncover an equation linking lifetime cancer risk (R) to evolutionary variables associated with speciation, including body size (S), lifespan (Li), species-specific mechanism of tumor suppression (T), and carcinogen exposure (E), such that in order for the process of speciation to include an increase in body size, an equilibrating “improvement” in tumor suppression mechanism for the new species must occur, in order to keep R at the value of the ancestral species, ≤ 5% (Figure 3, and graphical abstract). Natural evolution enforces this lex naturalis equation (lex naturalis, “natural law), never permitting an increase in body size during speciation without such an equilibrating change in one of the other variables, generally an improvement in T [12].
Evolution of the human-specific kill-switch tumor suppression mechanism
Stage 1. Alu transposon-mediated origin of primates by the saltatory evolution of an adrenal gland uniquely capable of secretion of enormous amounts of DHEA and DHEAS
It is commonly believed that primates arose as a result of the demise of the dinosaurs, which gave early primates the ecological “space” to evolve. However, new evidence suggests that primates arose as a direct result of the same asteroid impact that led to the demise of the dinosaurs. Thus, it is now established that the Chicxulub impact occurred at a rare site on the Earth’s surface (the Gulf of Mexico) where enormous natural concentrations of hydrocarbons exist(ed) [16]. The intense heat associated with this impact converted these hydrocarbons into polycyclic aromatic hydrocarbons (PAH) [17], creating worldwide PAH contamination (increasing E in the lex naturalis equation).
As instigators of genomic plasticity, PAH activate transposons [18,19,20,21,22,23,24], the movement of which within the genome have been responsible for many speciation events. In particular, the Alu family of transposons originated with— and is specific for— the primate lineage. At the origin of primates, PAH-stimulated mobilization of Alu transposons induced abrupt evolutionary modification of the embryonic adrenogonadal anlage such that, instead of androgens being produced primarily by the gonads, as in other vertebrates, the novel primate adrenal was capable of synthesizing and secreting dehydroepiandrosterone (DHEA). This modification is thought to have enabled the proto primate to exploit the PAH-contaminated environment that it found itself in, by detoxifying PAH [11,12,13]. Thus, DHEA directly inhibits CYP1A1 and CYP1A2, cytochrome p450 enzymes that activate PAH into DNA-reactive species [25,26,27]. DHEA is also a powerful uncompetitive inhibitor of Glucose-6-phosphate Dehydrogenase (G6PD), the enzyme responsible for producing most of the NADPH required for the synthesis and activity maintenance of the selenoproteins involved in the detoxification of Reactive Oxygen Species (ROS), and for the synthesis and reduction of ubiquinone to ubiquinol, required for the prevention of ferroptosis. Uncompetitive enzyme inhibition is unique in that, unlike other forms of enzyme inhibition, in the presence of substrate (in this case, Glucose-6-phosphate, G6P) and inhibitor (DHEA), it rapidly becomes irreversible (Figure 4). From British biochemist Athel Cornish-Bowden [28]:
“Cases of uncompetitive inhibition by species that are not involved in the reaction are virtually unknown…Uncompetitive effects may not merely be mechanistically implausible but may be so detrimental to organisms that display them that there has been evolutionary selection against such inhibition by naturally occurring metabolites. It may therefore be worthwhile to point out that any metabolic pathway in which uncompetitive inhibition can occur can potentially respond catastrophically to the presence of inhibitor.”
It has long been a puzzlement as to why humans have evolved such enormous levels of circulating DHEAS— the highest by far in the Animal Kingdom— when its proximate metabolite, DHEA, is a potentially irreversible inhibitor of so critical an enzyme as G6PD. This question is now resolved with the discovery that circulating DHEAS is the core component of the kill-switch tumor suppression mechanism that evolved in our species. This, and the other components of the kill switch mechanism that will be discussed below, are presented collectively in Figure 5.
Stage 2. Vitamin C auxotrophy as a kill-switch improvement in haplorrhine primates, including humans
It has also been a long-standing puzzlement as to why anthropoid primates, including humans, underwent selection for vitamin C auxotrophy, creating the risk of the debilitating disease of scurvy. This question, too, is answered by the discovery that, in addition to DHEA, high G6P concentrations are also required in order for uncompetitive inhibition of G6PD to become irreversible. That selection occurred for the ability of primate cells to accumulate intracellular G6P inside of singularities— and thereby enhance the effectiveness of DHEA-mediated inhibition of G6PD— is made clear by the retention of Alu transposon-mediated inactivation of the Gulanolactone Oxidase gene (GLO) over vast stretches of evolutionary time. The initiating substrate for the vitamin C pathway is G6P, and thus active GLO acts as a “sink” for G6P, preventing its accumulation inside of singularities. While Alu insertion-mediated inactivation of GLO made anthropoid primates, including humans, auxotrophic for vitamin C, it also, and more importantly from the perspective of kill switch evolution, eliminated this sink for G6P (12). This finding finally presents an answer to the long-standing question of why vitamin C auxotrophy was selected for in anthropoid primates, including humans.
Stage 3. G6PC promoter modification as a kill-switch improvement in anthropoid primates, including humans
In a similar fashion to GLO, Glucose-6-phosphatase (G6PC) activity also represented a sink for G6P, particularly in the presence of DHEA, which induces its transcription factors, PGC-1α and HNF-4α. G to T transversion mutations— the signature mutations of PAH (GLO deletion also occurred in a PAH-contaminated environment, the Paleocene-Eocene Thermal Maximum, which occurred about 56 million years ago [12])— in the G6PC gene promoter Tetrad occurred exclusively in the anthropoid lineage, disabling the ability of PGC-1α and HNF-4α to activate G6PC activity [Figure 6]. Thus, in a lineage-specific manner, intracellular accumulation of G6P was enabled by this promoter modification. Together, the inactivation of GLO and the modification of the G6PC promoter during primate evolution enhanced the function of the primate kill-switch by disabling these sinks for G6P, enabling the accumulation of G6P to drive uncompetitive inhibition of G6PD by DHEA to irreversibility within singularities.
Stage 5.11Incorporation of uric acid into the primate kill switch replaced the antioxidant properties of vitamin C lost with GLO deletion
In our search for an antioxidant that might have compensated for the loss of vitamin C, we identified uric acid as a likely candidate. In fact, uric acid has been shown to be superior to vitamin C with respect to the detoxification of some metabolites of PAH. For example, Uric acid was found to completely suppress benzo[a]pyrene quinone formation at a concentration significantly less than vitamin C [42].
Just as the purpose of adrenarche has been called “enigmatic,” the purpose of high uric acid levels in higher primates has been termed “elusive” [43]. Thus, the question has been repeatedly asked, why would natural selection allow the accumulation of uric acid despite the physiological consequences of gout and liver and kidney damage? Bruce Ames proposed that the high uric acid levels in humans was associated with increased lifespan in our species [44]. Indeed, among mammals there is a positive correlation between serum uric acid (UA) levels and life span [45]. A study from the National Institute on Aging published data supporting this observation in which female mice genetically engineered to possess high uric acid levels experienced significantly increased longevity compared to normal female mice [46].
That the lex naturalis equation was an effective guide in dissecting the evolution of the components of the primate kill switch is demonstrated by the fact that when we examined the association of uric acid biology with lifespan in primates, we discovered a published literature that corroborated such an association. For example, in a study examining the role of uric acid as a determinant of lifespan in 22 primate species, a highly significant positive correlation was observed between uric acid concentrations and maximum lifespan [47]. In particular, beginning in catarrhine primates, multiple selection events occurred that successively increased the levels of circulating uric acid, enabling the exploitation of the different variables within the lex naturalis equation described above. (Normal circulating levels of uric acid levels in humans are 4.9-7.0 mg/dL for men and 3.9-6.0 mg/dL for women [106]. Hominoid apes have circulating levels of uric acid similar to those of humans, while in non-primate mammals, such levels range from 10-20 ug/ml [107].) This succession of selection events driving increased uric acid concentrations began with an alteration in the uric acid transporter (URAT1), in which it sharply diverged mechanistically from the high affinity, high capacity kinetics of URAT1 in basal anthropoids, and the low affinity, high capacity kinetics of URAT1 in Strepsirrhines, to a high affinity, low capacity version of URAT1 that was ideal for maintaining high, constant levels of circulating uric acid [48].
Subsequently, a series of mutations in the uric acid oxidase (UOX) gene drove extraordinary increases in circulating uric acid [49], and further mutations in SLC2A9, the uric acid transporter, put uric acid import into the cell under the control of the p53 tumor suppressor. Thus, in a normal cell with intact p53, uric acid SLC2A9 pumps uric acid into cells in response to increases in intracellular ROS, neutralizing those ROS. But in terms of the kill-switch, this would be detrimental as it would neutralize the ROS generated by DHEA’s uncompetitive inhibition of G6PD. Therefore, as Itahana and coworkers discovered [50], when p53 is inactivated— as it would be in the creation of a singularity— SLC2A9 stops pumping antioxidant uric acid, allowing ROS levels to climb in the p53-affected cell. These two mechanisms thus work in concert to extinguish singularities. When p53 is inactivated, creating a singularity, DHEAS and G6P from the circulation are both pumped into the singularity, driving uncompetitive inhibition of G6PD toward irreversibility, raising ROS to toxic levels. Simultaneously, the import of uric acid is terminated, preventing its antioxidant properties from countering DHEA-mediated increase in intracellular ROS, enabling the extinguishing of singularities while they are still in their single cell state.
Stage 6.1DHEAS vs. DHEA
Based upon data obtained in mice and rats, it has often been proposed that supplementation with DHEA might prevent cancer in our species [51,52,53,54,55,56,57,58,59,60,61,62,63,64,65]. But, as was shown in Figure 5, above, the evolution of the human-specific kill-switch tumor suppression mechanism depended not only on extremely high levels of circulating DHEAS, but also on an extremely low ratio of circulating DHEA/DHEAS, an important finding that has never here-to-fore been reported. Thus, from the Old-World Monkeys to humans, while there has been a ten-fold increase in the concentrations of circulating DHEAS during speciation, there has been a simultaneous 56-fold decrease in the DHEA/DHEAS ratio.
Why have constant, successive decreases in the DHEA/DHEAS ratio been selected for during primate evolution? Unlike DHEAS, which is hydrophilic and requires active transport to enter cells, DHEA is lipophilic, and crosses cell membranes by passive diffusion— which could trigger the kill-switch tumor suppression mechanism in normal, healthy cells. Evolutionary selection for lower DHEA/DHEAS ratios enabled fine tuning of the kill-switch tumor suppression mechanism by preventing DHEA from entering normal cells “uninvited,” thereby focusing the kill-switch upon singularities, enabling our species and those immediately preceding it to harness fire. In humans, circulating DHEA is therefore maintained at concentrations 10,000-fold lower than circulating DHEAS [75]. Cancer suppression by DHEA in mice is meaningless as regards cancer prevention in humans because mice did not evolve a species-specific kill-switch based on DHEA/DHEAS. Administration of DHEA to humans bypasses 66 million years of evolutionary selection for minimization of its serum concentration. It is thus difficult to find justification for the fact that, in sharp contrast to the regulatory authorities in virtually every other developed nation, those in the United States permit oral DHEA to be sold over-the-counter and online, without regulation, and without clinical trials to assess safety [76].
Stage 7.1Adrenarche powers up the human-specific kill-switch immediately prior to the human adolescent growth spurt, and the attainment of near-adult size, when cancer risk would otherwise increase because of the increase in stature
Using National Cancer Institute (NCI) statistics for modern humans, Figure 8 illustrates that the combined strategies of small body size during adolescence, followed by the activation of our species-specific kill-switch tumor suppression mechanism immediately prior to the adolescent growth spurt, and continuing through young adulthood (≤ 25 years of age)— effectively limits lifetime cancer risk to the ≤ 5% that is the rule for vertebrate animals that was illustrated in Figure 2. Note that the low cancer incidence in modern “young adulthood” corresponds to the primitive lifespan of about 25 years, with 88% mortality occurring before age 30 [11], and references therein. Thus, levels of circulating DHEAS reach their peak at about age 25, and thereafter undergo steady decline (Figure 9). This indicates that, in contrast to other vertebrate species (e.g., elephants, whales) that evolved genetic kill-switch tumor suppression mechanisms that remain active throughout their lifespans, humans, with their epigenetic kill-switch mechanism based on DHEAS, proceed through the extended modern human lifespan unprotected by their species-specific tumor suppression mechanism. These data indicate that the synthesis and secretion of DHEAS came at some cost, such that its elimination was “writ” into our natural, species-specific lex naturalis equation.
DHEAS is a small molecule and is therefore pharmacologically tractable
As noted above, DHEAS naturally circulates at an extremely high concentration in humans (11.5 uM; compare to DHEA, which circulates at a concentration 10,000-fold lower). But once the primitive lifespan is surpassed, a steady decline in circulating DHEAS occurs such that modern humans enter their extended lifespan dramatically deficient in this core component of the human-specific kill-switch tumor suppression mechanism. However, since DHEAS is a small molecule, it is pharmacologically tractable, such that peak levels of circulating DHEAS can be maintained throughout the extended modern human lifespan (Figure 10). In fact, the lex naturalis equation predicts that we may be able to anticipate future evolution of our species-specific kill-switch tumor suppression mechanism, and pharmacologically induce still further decreases in the DHEA/DHEAS ratio (Figure 11). This can be accomplished not only by increasing the administered dose of DHEAS to achieve supra-peak levels in the circulation, but also by inhibiting the synthesis of DHEA by any of a number of different methods, e.g., the pharmacological application of statins [77] simultaneously with DHEAS. While this strategy of anticipating future evolution of the DHEA/DHEAS ratio may enhance the process of “normalization” of lifetime cancer risk for humans across the board, it may be a particularly useful strategy to prevent the high risk of cancer in persons harboring mutations in various tumor suppressive genes that are associated with extremely high cancer risk. These include, for example, the association of BRCA mutations with breast cancer [78,79], and mismatch repair gene mutations in Lynch syndrome, the most common inherited cancer which in the United States alone causes ≈ 4,200 colorectal cancers and 1,800 endometrial cancers per year [80,81].
Emergence of transposons within the primate genome coincides with emergence of the components of the primate kill switch tumor suppression mechanism
Only ≈ 1–2% of the ≈ 3 billion bases in the human genome code for proteins, while an astonishing 40–50% is derived from mobile repetitive sequences collectively known as transposons. Transposons, nick-named “jumping genes” for their ability to incorporate copies of themselves throughout a genome, were first discovered by Barbara McClintock in her seminal studies in maize [82]. DNA transposons like Mariner were intensely active from about 66 million years ago to about 37 million years ago (Figure 12), at which time their ability to transpose was extinguished in the germline DNA of primates. While Mariner and other DNA transposons are no longer actively “jumping” within our germline DNA, their legacy in establishing primate regulatory networks remains. But there is a downside to this legacy. Thus, DNA transposons such as RAG1/RAG2 and piggybac can become active during early development and cause pediatric cancers by inactivating tumor suppressor genes [83].
The transposons that concern us here have the potential to be transcribed into RNA, reverse transcribed back into DNA, and then re-inserted into the genome. These are known as retrotransposons. Many retrotransposons are enriched in the dinucleotide CpG, which functions as a binding domain for transcription modulating proteins. This enables retrotransposons to wire new transcriptional networks by providing binding motifs in genes that were previously not targets of a particular transcription factor, making domestication of transposons into functional cis-regulatory elements a widespread phenomenon across both plants and animals.
Primate genomes have been modified by waves of transposon insertions during the course of their evolution, beginning, as we have noted, with the Alu-mediated creation of a DHEA/DHEAS-secreting adrenal gland which marked the origin of primates. SINE (Short Interspersed Nuclear Elements) transposons are typically lineage specific, making them good markers of evolutionary divergence. The Alu family of transposons are SINE transposons. In addition to Alu retrotransposons, there are LINE1 and SVR retrotransposons in primate genomes. Measured by bulk, LINE-1 is the largest transposon family in humans with an estimated 500,000 copies representing 17% of the human genome. It is also the sole autonomous transposon in humans— that is, it can complete the entire process of retrotransposition without help. The reverse transcriptase encoded by LINE-1 is the key component required for its transposition process, and for the retro-transposition of Alu and SVA transposons, which depend upon this enzyme,e for their retro-transposition.
SVA are composite retrotransposons that uniquely arose in hominoids by stepwise fusions of three genetic elements: SINE-R, a variable number of tandem repeat (VNTR) sequence, and Alu. There are six main SVA subfamilies known (SVA-A through F) and nearly half of the annotated ≈ 3,700 copies are human-specific, including all of the SVA-Es and SVA-Fs. Because of their density of CpG dinucleotides, SVA retrotransposons have been called “mobile CpG islands.” As such, they have the capacity to effect dramatic changes in the genes near to which they insert.
The Alu family of transposons are most numerous, with more than a million copies representing 10.7% of the human genome. Because they, too, are rich in CpG dinucleotides, Alu elements have played a major role in the regulation of tissue-specific genes. Su et al. [84] showed that Alu elements are bound by nucleosomes that contain histones bearing marks of active chromatin, and they show tissue-specific enrichment for the enhancer mark H3K4me1. These authors demonstrated that a proportion of Alu elements were indeed bona fide active enhancers, showing long-range interactions with gene promoters. Their function as enhancers increases with their age in the human genome, and in addition to their real time role as active enhancers, there appear to be a large number that can be considered to be proto-enhancers ready to serve as starting material for the de novo birth of future enhancers. Alu elements have also been shown to induce an array of alternative mRNA splicing events, termed “Aluternative splicing” [85].
The successive waves of new retrotransposon insertions have been met in human cells by the evolution of new tools to silence those invading elements. Although almost 50% of the human genome consists of transposons and transposon-like repetitive elements— the signature left behind of past insertion events— only a small proportion (<0.05%) of these remain active today, comprising some 30-45 subfamilies of Alu, Line1 and SVA transposons. In real time, transposon insertions are deleterious and are associated with a plethora of diseases, including cancer. But over evolutionary periods of time (i.e., tens of thousands to hundreds of thousands of years), transposon insertions provide a large part of the genomic plasticity required for speciation to respond to changes in the environment, or to exploit new opportunities. In the human genome, the most recently active have been the Alu-Y and Alu-S elements [86]. These exist within a sea of inactive transposons resulting from a number of processes that have evolved to suppress them. DNA methylation of transposon CpG dinucleotides, for example, attracts repressive histone modifications, inactivating transposon transcription. Since the 5-methylcytsine of methylated CpGs spontaneously deaminates to thymidine at a high rate, DNA methylation also leads to mutational degradation of transposons. Chemically-mediated modification can also occur at other bases in transposon DNA, and the heavy CpG methylation pattern that characterizes transposons denies repair enzymes access to these damaged DNA bases, degrading transposon structure and hence their mobility. There are a number of additional ways that primate cells suppress transposons. Thus, Tiwari et al. [87] showed that the p53 tumor suppressor protein constitutively restrains human LINE1s by cooperatively engaging sites in the 5'UTR and stimulating local deposition of repressive histone marks at these transposons. Consistent with this, the elimination of p53 or the removal of corresponding binding sites in LINE1s, can stimulate these retroelements to become hyperactive. Since p53 is inactivated by one means or another in virtually all human tumors, LINE1s jumping about the genome have been described as an important mechanism of genome instability leading to cancer [88]. And in reference to immune strategies to treat cancer, activation of LINE1 transcription has been shown to be a major mechanism of T cell exhaustion [89]. In response to transposon infection, particularly that of LINE1, primates also underwent a rapid expansion of the APOBEC3 family of cytosine deaminases which degrade transposons by inducing mutations in them [90]. Yet another mechanism of transposon suppression involves a family of zinc finger proteins that bind as transcription factors to methylated CpGs and play a critical role in gene regulation. KRAB Zinc Finger (KZNF) genes are one of the fastest growing gene families in primates because they represent a primary defense mechanism against newly emergent retrotransposons. KZNF gene expansion limits the activity of newly emergent retrotransposon classes, and this is followed, in a sort of evolutionary arms race, by selection for mutations in these retrotransposons that enable them to evade repression by eliminating the binding site for KZNF proteins; then, in tit-for-tat response, KZNF genes undergo selection to enable re-repression of transposons, and so on. Jacobs et al. [91] reported that two primate-specific KZNF genes rapidly evolved to repress SVA and LINE1 retrotransposon families shortly after these transposons began to spread in our ancestral genome. ZNF91 underwent a series of structural changes 8–12 million years ago that enabled it to repress SVA elements. ZNF93 evolved earlier to repress the primate LINE1 lineage, until ∼12.5 million years ago when the LINE1PA3-subfamily of retrotransposons escaped ZNF93’s restriction by removing code for the ZNF93-binding site from its DNA.
It is of great interest that transposon insertions into the primate genome coincide with alterations in gene expression producing “improvements” in the primate kill switch tumor suppression mechanism, culminating in the advent of adrenarche in the Hominini, with dramatically increased levels of circulating DHEAS and decreased levels of DHEA relative to DHEAS (Figure 12).
The harnessing of fire coincided with a dramatic increase in transposon activity in the human genome, as compared to chimpanzees
As noted above, chimpanzees have been shown to forage for food in fire-combusted landscapes [37,38], and to prefer cooked to raw food [92]. Thus, their exposure to PAH was much higher than that of non-hominin primates, a fact that correlates with their much higher levels of circulating DHEAS, and their much lower DHEA/DHEAS ratios (Figure 12). But humans harnessed fire and used it in every aspect of their existence— to warm their habitats and ward off predators, and to heat-process their food, all of which dramatically increased E in their lex naturalis equation. This increase in E required equilibration during speciation within the genus Homo. Although a steady increase in body size did occur from the Australopithecines to and through Homo species, the intense increase in E experienced by the Homo lineage as a result of the harnessing of fire dominated the equilibration of the Homo lex naturalis equation, and was only made possible by still further improvements in T, species-specific mechanism of tumor suppression, as depicted in Figure 5, above.
There is evidence that the harnessing of fire potentiated human evolution by PAH-mediated activation of transposons. As noted above, Alu and other transposons are activated by DNA damage, including the type induced by PAH [93,94,95,96]. PAH specifically target methylated CpG dinucleotides, inducing DNA hypomethylation that allows transposons to jump [96,97,98,99]. Across the primate lineages, the rate of Alu transposon insertion has differed by 15-fold, with human, chimpanzees and bonobos showing the highest rate, and among these hominins, humans showing by far the highest rate (3.5 times more Alu insertions in humans than in chimpanzees [100]. In terms of total transposon insertions, there have been nearly 15,000 that are exclusive to the human genome [101] (Figure 12). Thus, there is a linear correlation between Alu activity and increasing exposure to PAH, culminating in the primate genus that harnessed fire as a tool. As we noted above, insertion of Alu transposons alter the expression of genes near to which they have inserted [102,103], providing a mechanism of action for the increased levels of circulating DHEAS in humans compared to chimpanzees, and in chimpanzees compared to the rest of the Hominidae.
The existence of species-specific mechanisms of tumor suppression precludes the pre-testing of our proposed DHEAS maintenance protocol in non-human animals. Neither is it possible to test it in vitro, since cancer cells have already achieved the genetic and epigenetic modifications that enabled their escape from the kill switch. Nor can it be tested in normal cells, because the kill switch evolved to be triggered in response to inactivation of TP53. Single cell DNA/mRNA sequencing techniques are not capable of analyzing such a phenomenon that occurs at a frequency of one cell in 10-7. From what, then, can we derive assurance that the expense of a clinical trial involving a large number of subjects for a long period of time is warranted? We argue that such risk is dramatically reduced by the fact that the human-specific kill-switch tumor suppression mechanism provides answers for a list of longstanding questions in human biology. For example, why do humans have such enormous concentrations of circulating DHEAS when this androgen is the immediate precursor of DHEA, a powerful inhibitor of so critical an enzyme as G6PD, and with uncompetitive reaction kinetics that can rapidly become irreversible? This longstanding question is finally explained by the fact that circulating DHEAS is the core component of the human-specific kill-switch tumor suppression mechanism.
Another long-standing question in human biology is, “What is the purpose of adrenarche, a developmental period the function of which has long been called ‘enigmatic?’” This question, too, finds resolution in the human-specific kill-switch tumor suppression mechanism. Thus, adrenarche focuses the powering up of the kill-switch exactly when it is needed and not before— in the developmental period immediately preceding the onset of the adolescent growth phase, when 80% of adult body size is achieved, with a corresponding increase in the number of cells at risk for neoplastic transformation. The purpose of adrenarche, thus, is tumor suppression.
Another long unanswered question: “Why has the loss of GLO activity been retained by natural selection, making humans (and other haplorrhine primates) auxotrophic for vitamin C?” As discussed above, in order for uncompetitive inhibition of G6PD by DHEA to become irreversible, and catastrophic to singularities, not only inhibitor (DHEA) is required, but also equal amounts of substrate, G6P. The GLO pathway was a “sink” for G6P, working against the evolving primate kill-switch by withdrawing this necessary co-factor for uncompetitive inhibition. Primates therefore underwent selection for inactivation of GLO, enabling the accumulation of vitamin C within singularities.
Why has an anthropoid primate-specific GAAT sequence motif evolved in the G6PC promoter? Like GLO, the G6PC pathway was a sink for G6P. And thus, like the second impact of a one-two punch, modification of the G6PC promoter eliminated this additional “sink” for G6P, enhancing the ability of anthropoid primates to drive uncompetitive inhibition of G6PD to irreversibility in singularities.
And why the successive deletions within the Uric Acid Oxidase (UOX) gene, and the modification of URAT1 retention kinetics, both of which lead to accumulation of circulating uric acid, and the painful disease of gout? Here again, this longstanding question is finally answered. UOX deletion/URAT1 modification enabled uric acid to take the place of vitamin C as an antioxidant, and selection for the placing of its transport protein SLC2A9 under p53 control made it an integral component of the primate kill-switch.
We are left with a situation that may be unique in the history of cancer research— but in line with recent identification of species-specific mechanisms of tumor suppression— where justification for a large-scale clinical trial in humans must be based on indirect evidence consisting of the large number of long-standing questions in human biology that are answered by the kill switch mechanism. It is extremely unlikely that so many longstanding, unresolved questions in human biology all intersect at kill-switch function, and obtain compelling explanation by such intersection, if that is not where they derive their evolutionary significance. Reliance upon such data has a high probability of being superior to that derived from animal models, as animal models have been shown to be unreliable predictors of cancer drug efficacy in humans [104,105]. We therefore argue that the probability that the human-specific kill switch tumor suppression mechanism described in this manuscript— a system focused by adrenarche upon the developmental period immediately preceding the attainment of near-adult body size and the attendant increase in cancer risk— represents a fundamental means of tumor suppression in our species, deserving of large-scale clinical trials to test this hypothesis.
Funding
This research was based upon the following funding: NCI 5R29CA04217-03.
Conflicts of Interest
The author reports no conflict of interest.
References
- Auchus, R.J. The physiology and biochemistry of adrenarche. Endocrine development 2011, 20, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Cumberland, A.L.; Hirst, J.J.; Badoer, E.; Wudy, S.A.; Greaves, R.F.; Zacharin, M.; Walker, D.W. The Enigma of the Adrenarche: Identifying the Early Life Mechanisms and Possible Role in Postnatal Brain Development. Int. J. Mol. Sci. 2021, 22, 4296. [Google Scholar] [CrossRef] [PubMed]
- Limony, Y.; Kozieł, S.; Friger, M. Age of onset of a normally timed pubertal growth spurt affects the final height of children. Pediatr. Res. 2015, 78, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Cairns, B.J.; Casabonne, D.; Wright, F.L.; Reeves, G.; Beral, V. Height and cancer incidence in the Million Women Study: prospective cohort, and meta-analysis of prospective studies of height and total cancer risk. Lancet Oncol. 2011, 12, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Kabat, G.C.; Heo, M.; Kamensky, V.; Miller, A.B.; Rohan, T.E. Adult height in relation to risk of cancer in a cohort of Canadian women. Int. J. Cancer 2012, 132, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.-S.; An, J.; Law, M.H.; Whiteman, D.C.; Neale, R.E.; Gharahkhani, P.; MacGregor, S. Height and overall cancer risk and mortality: evidence from a Mendelian randomisation study on 310,000 UK Biobank participants. Br. J. Cancer 2018, 118, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Lee, D.H.; Han, K.-D.; Yoon, H.; Shin, C.M.; Park, Y.S.; Kim, N. Adult height in relation to risk of cancer in a cohort of 22,809,722 Korean adults. Br. J. Cancer 2019, 120, 668–674. [Google Scholar] [CrossRef]
- Fu, B.; Song, M.; Li, X.; Han, J.; Adami, H.; Giovannucci, E.; Mucci, L. Height as a mediator of sex differences in cancer risk. Ann. Oncol. 2020, 31, 634–640. [Google Scholar] [CrossRef]
- Janecka, A.; Kołodziej-Rzepa, M.; Biesaga, B. Clinical and Molecular Features of Laron Syndrome, A Genetic Disorder Protecting from Cancer. In vivo 2016, 30, 375–381. [Google Scholar]
- Laron, Z.; Kauli, R. Fifty seven years of follow-up of the Israeli cohort of Laron Syndrome patients—From discovery to treatment. Growth Horm. IGF Res. 2015, 28, 53–56. [Google Scholar] [CrossRef]
- Nyce, J.W. Detection of a novel, primate-specific ‘kill switch’ tumor suppression mechanism that may fundamentally control cancer risk in humans: an unexpected twist in the basic biology of TP53. Endocrine-Related Cancer 2018, 25, R497–R517. [Google Scholar] [CrossRef] [PubMed]
- Nyce, J.W. A lex naturalis delineates components of a human-specific, adrenal androgen-dependent, p53-mediated ‘kill switch’ tumor suppression mechanism. Endocrine-Related Cancer 2020, 27, R51–R65. [Google Scholar] [CrossRef] [PubMed]
- Nyce, J.W. Species-specific mechanisms of tumor suppression are fundamental drivers of vertebrate speciation: critical implications for the ‘war on cancer’. Endocrine-Related Cancer 2019, 26, C1–C5. [Google Scholar] [CrossRef]
- Abegglen, L.M.; Caulin, A.F.; Chan, A.; Lee, K.; Robinson, R.; Campbell, M.S.; Kiso, W.K.; Schmitt, D.L.; Waddell, P.J.; Bhaskara, S.; et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA 2015, 314, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Chai, S.; Huang, X.; Wang, Y.; Xiao, L.; Xu, S.; Yang, G. Novel Genomic Insights into Body Size Evolution in Cetaceans and a Resolution of Peto’s Paradox. Am. Nat. 2022, 199, E28–E42. [Google Scholar] [CrossRef]
- Kaiho, K.; Oshima, N. Site of asteroid impact changed the history of life on Earth: the low probability of mass extinction. Sci. Rep. 2017, 7, 14855. [Google Scholar] [CrossRef] [PubMed]
- Lyons, S.L.; Karp, A.T.; Bralower, T.J.; Grice, K.; Schaefer, B.; Gulick, S.P.S.; Morgan, J.V.; Freeman, K.H. Organic matter from the Chicxulub crater exacerbated the K–Pg impact winter. Proc. Natl. Acad. Sci. USA 2020, 117, 25327–25334. [Google Scholar] [CrossRef]
- Hassanin, A.A.I.; Tavera-Garcia, M.; Moorthy, B.; Zhou, G.D.; Ramos, K.S. Lung genotoxicity of benzo(a)pyrene in vivo involves reactivation of LINE-1 retrotransposon and early reprogramming of oncogenic regulatory networks. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, L816–L822. [Google Scholar] [CrossRef]
- Lu, K.; Hallberg, L.; Tomlinson, J.; Ramos, K. Benzo(a)pyrene activates L1Md retrotransposon and inhibits DNA repair in vascular smooth muscle cells. Mutat. Res. Mol. Mech. Mutagen. 2000, 454, 35–44. [Google Scholar] [CrossRef]
- Ramos, K.S.; Montoya-Durango, D.E.; Teneng, I.; Nanez, A.; Stribinskis, V. Epigenetic control of embryonic renal cell differentiation by L1 retrotransposon. Birth Defects Res. Part A: Clin. Mol. Teratol. 2011, 91, 693–702. [Google Scholar] [CrossRef]
- Rudin, C.M.; Thompson, C.B. Transcriptional activation of short interspersed elements by DNA-damaging agents. Genes chromosomes cancer 2001, 30, 64–71. Available online: https://pubmed.ncbi.nlm.gov/11107177/. [CrossRef] [PubMed]
- Stribinskis, V.; Ramos, K.S. Activation of Human Long Interspersed Nuclear Element 1 Retrotransposition by Benzo(a)pyrene, an Ubiquitous Environmental Carcinogen. Cancer Res 2006, 66, 2616–2620. [Google Scholar] [CrossRef] [PubMed]
- Teneng, I.; Stribinskis, V.; Ramos, K.S. Context-specific regulation of LINE-1. Genes Cells 2007, 12, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Teneng, I.; Montoya-Durango, D.E.; Quertermous, J.L.; Lacy, M.E.; Ramos, K.S. Reactivation of L1 retrotransposon by benzo(a)pyrene involves complex genetic and epigenetic regulation. Epigenetics 2011, 6, 355–367. [Google Scholar] [CrossRef]
- Ciolino, H.P.; Yeh, G.C. The Steroid Hormone Dehydroepiandrosterone InhibitsCYP1A1 Expression in Vitro By a Post-transcriptional Mechanism. J. Biol. Chem. 1999, 274, 35186–35190. [Google Scholar] [CrossRef] [PubMed]
- Belic, A.; Tóth, K.; Vrzal, R.; Temesvári, M.; Porrogi, P.; Orbán, E.; Rozman, D.; Dvorak, Z.; Monostory, K. Dehydroepiandrosterone post-transcriptionally modifies CYP1A2 induction involving androgen receptor. Chem. Interactions 2013, 203, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Ciolino, H.; MacDonald, C.; Memon, O.; Dankwah, M.; Yeh, G.C. Dehydroepiandrosterone inhibits the expression of carcinogen-activating enzymesin vivo. Int. J. Cancer 2003, 105, 321–325. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Why is uncompetitive inhibition so rare? FEBS Lett. 1986, 203, 3–6. [Google Scholar] [CrossRef]
- Huang, N.; Dardis, A.; Miller, W.L. Regulation of Cytochrome b5 Gene Transcription by Sp3, GATA-6, and Steroidogenic Factor 1 in Human Adrenal NCI-H295A Cells. Mol. Endocrinol. 2005, 19, 2020–2034. [Google Scholar] [CrossRef]
- Abbott, D.H.; Bird, I.M. Nonhuman primates as models for human adrenal androgen production: Function and dysfunction. Rev. Endocr. Metab. Disord. 2008, 10, 33–42. [Google Scholar] [CrossRef]
- Jimenez, P.; Saner, K.; Mayhew, B.; Rainey, W.E. GATA-6 Is Expressed in the Human Adrenal and Regulates Transcription of Genes Required for Adrenal Androgen Biosynthesis. Endocrinology 2003, 144, 4285–4288. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Loomis, D.; Guyton, K.Z.; Grosse, Y.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef] [PubMed]
- Diallo, A.; Deschasaux, M.; Latino-Martel, P.; Hercberg, S.; Galan, P.; Fassier, P.; Allès, B.; Guéraud, F.; Pierre, F.H.; Touvier, M. Red and processed meat intake and cancer risk: Results from the prospective NutriNet-Santé cohort study. Int. J. Cancer 2017, 142, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Turesky, R.J. Mechanistic Evidence for Red Meat and Processed Meat Intake and Cancer Risk: A Follow-up on the International Agency for Research on Cancer Evaluation of 2015. Chim. Int. J. Chem. 2018, 72, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Parra-Soto, S.; Ahumada, D.; Petermann-Rocha, F.; Boonpoor, J.; Gallegos, J.L.; Anderson, J.; Sharp, L.; Malcomson, F.C.; Livingstone, K.M.; Mathers, J.C.; et al. Association of meat, vegetarian, pescatarian and fish-poultry diets with risk of 19 cancer sites and all cancer: findings from the UK Biobank prospective cohort study and meta-analysis. BMC Med. 2022, 20, 79. [Google Scholar] [CrossRef] [PubMed]
- Key, T.J.; Appleby, P.N.; Crowe, F.L.; E Bradbury, K.; Schmidt, J.A.; Travis, R.C. Cancer in British vegetarians: updated analyses of 4998 incident cancers in a cohort of 32,491 meat eaters, 8612 fish eaters, 18,298 vegetarians, and 2246 vegans. Am. J. Clin. Nutr. 2014, 100 (Suppl. 1), 378S–385S. [Google Scholar] [CrossRef]
- Herzog, N.M.; Pruetz, J.D.; Hawkes, K. Investigating foundations for hominin fire exploitation: Savanna-dwelling chimpanzees (Pan troglodytes verus) in fire-altered landscapes. J. Hum. Evol. 2022, 167, 103193. [Google Scholar] [CrossRef]
- Pruetz, J.D.; LaDuke, T.C. Brief communication: Reaction to fire by savanna chimpanzees (Pan troglodytes verus) at Fongoli, Senegal: Conceptualization of “fire behavior†and the case for a chimpanzee model. Am. J. Phys. Anthr. 2009, 141, 646–650. [Google Scholar] [CrossRef]
- Gowlett, J.A. The discovery of fire by humans: a long and convoluted process. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 2016, 371, 20150164. [Google Scholar] [CrossRef]
- Aarts, J.M.; Alink, G.M.; Scherjon, F.; MacDonald, K.; Smith, A.C.; Nijveen, H.; Roebroeks, W. Fire Usage and Ancient Hominin Detoxification Genes: Protective Ancestral Variants Dominate While Additional Derived Risk Variants Appear in Modern Humans. PLOS ONE 2016, 11, e0161102. [Google Scholar] [CrossRef]
- Vangenot, C.; Gagneux, P.; de Groot, N.G.; Baumeyer, A.; Mouterde, M.; Crouau-Roy, B.; Darlu, P.; Sanchez-Mazas, A.; Sabbagh, A.; Poloni, E.S. Humans and Chimpanzees Display Opposite Patterns of Diversity in Arylamine N-Acetyltransferase Genes. G3 Genes|Genomes|Genetics 2019, 9, 2199–2224. [Google Scholar] [CrossRef] [PubMed]
- Terao, J.; Matsushita, S. Quinone formation from benzo[a]pyrene by free radicals: effects of antioxidants. Free Radical Biology Medicine 1988, 4, 205–208. Available online: https://europepmcorg/article/MED/3360379. [CrossRef] [PubMed]
- Li, Z.; Hoshino, Y.; Tran, L.; A Gaucher, E. Phylogenetic Articulation of Uric Acid Evolution in Mammals and How It Informs a Therapeutic Uricase. Mol. Biol. Evol. 2021, 39, msab312. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G. Antioxidants and longevity of mammalian species. Basic life sciences 1985, 35, 15–73. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Camandola, S.; Feldman, N.H.; Yoon, J.S.; Haran, J.B.; Arguelles, S.; Mattson, M.P. Uric acid enhances longevity and endurance and protects the brain against ischemia. Neurobiol. Aging 2018, 75, 159–168. [Google Scholar] [CrossRef]
- Cutler, R.G. Urate and ascorbate: their possible roles as antioxidants in determining longevity of mammalian species. Arch. Gerontol. Geriatr. 1984, 3, 321–348. [Google Scholar] [CrossRef]
- Tan, P.K.; Farrar, J.E.; Gaucher, E.A.; Miner, J.N. Coevolution of URAT1 and Uricase during Primate Evolution: Implications for Serum Urate Homeostasis and Gout. Mol. Biol. Evol. 2016, 33, 2193–2200. [Google Scholar] [CrossRef]
- Oda, M.; Satta, Y.; Takenaka, O.; Takahata, N. Loss of Urate Oxidase Activity in Hominoids and its Evolutionary Implications. Mol. Biol. Evol. 2002, 19, 640–653. [Google Scholar] [CrossRef]
- Itahana, Y.; Han, R.; Barbier, S.; Lei, Z.; Rozen, S.; Itahana, K. The uric acid transporter SLC2A9 is a direct target gene of the tumor suppressor p53 contributing to antioxidant defense. Oncogene 2014, 34, 1799–1810. [Google Scholar] [CrossRef]
- Kelloff, G.J.; Crowell, J.A.; Hawk, E.T.; Steele, V.E.; Lubet, R.A.; Boone, C.W.; Covey, J.M.; Doody, L.A.; Omenn, G.S.; Greenwald, P.; et al. Strategy and planning for chemopreventive drug development: Clinical development plans II. J. Cell. Biochem. 1996, 63, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Keiloff, G.J.; Boone, C.W.; Crowell, J.A.; Steele, V.E.; Lubet, R.A.; Doody, L.A.; Malone, W.F.; Hawk, E.T.; Sigman, C.C. New agents for cancer chemoprevention. J. Cell. Biochem. 1996, 63, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Kelloff, G.J.; Boone, C.W.; Steele, V.E.; Fay, J.R.; Lubet, R.A.; Crowell, J.A.; Sigman, C.C. Mechanistic considerations in chemopreventive drug development. J. Cell. Biochem. 1994, 56, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kelloff, G.J.; Crowell, J.A.; Boone, C.W.; Steele, V.E.; Lubet, R.A.; Greenwald, P.; Alberts, D.S.; Covey, J.M.; Doody, L.A.; Knapp, G.G. Clinical development plan: DHEA analog 8354. Journal of cellular biochemistry. Supplement 1994, 20, 141–146. [Google Scholar] [PubMed]
- Schwartz, A.G.; Pashko, L.L. Cancer prevention with dehydroepiandrosterone and non-androgenic structural analogs. J. Cell. Biochem. 1995, 59, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Sundar, J.; Gnanasekar, M. Can dehydroepiandrostenedione (DHEA) target PRL-3 to prevent colon cancer metastasis? Med Hypotheses 2013, 80, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Osawa, E.; Nakajima, A.; Yoshida, S.; Omura, M.; Nagase, H.; Ueno, N.; Wada, K.; Matsuhashi, N.; Ochiai, M.; Nakagama, H.; et al. Chemoprevention of precursors to colon cancer by dehydroepiandrosterone (DHEA). Life Sci. 2002, 70, 2623–2630. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Calderón, Y.N.; López-Marure, R. Dehydroepiandrosterone inhibits proliferation and suppresses migration of human cervical cancer cell lines. Anticancer research 2014, 34, 4039–4044. [Google Scholar]
- Pietri, E.; Massa, I.; Bravaccini, S.; Ravaioli, S.; Tumedei, M.M.; Petracci, E.; Donati, C.; Schirone, A.; Piacentini, F.; Gianni, L.; et al. Phase II Study of Dehydroepiandrosterone in Androgen Receptor-Positive Metastatic Breast Cancer. Oncologist 2018, 24, 743-e205. [Google Scholar] [CrossRef]
- Schwartz, A.G.; Whitcomb, J.M.; Nyce, J.W.; Lewbart, M.L.; Pashko, L.L. Dehydroepiandrosterone and structural analogs: a new class of cancer chemopreventive agents. Advances in cancer research 1988, 51, 391–424. [Google Scholar] [CrossRef]
- Nyce, J.W.; Magee, P.N.; Hard, G.C.; Schwartz, A.G. Inhibition of 1,2-dimethylhydrazine-induced colon tumorigenesis in Balb/c mice by dehydroepiandrosterone. Carcinog. 1984, 5, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Honda, A. Dehydroepiandrosterone and Its Derivatives: Potentially Novel Anti-Proliferative and Chemopreventive Agents. Curr. Pharm. Des. 2006, 12, 3411–3421. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.B.; Shantz, L.M.; Talalay, P. Modulation of growth, differentiation and carcinogenesis by dehydroepiandrosterone. Adv. Enzym. Regul. 1987, 26, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Mayer, D.; Forstner, K.; Kopplow, K. Induction and Modulation of Hepatic Preneoplasia and Neoplasia in the Rat by Dehydroepiandrosterone. Toxicol. Pathol. 2003, 31, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Pashko, L.L.; Lewbart, M.L.; Schwartz, A.G. Inhibition of 12-O-tetradecanoylphorbol-13-acetate-promoted skin tumor formation in mice by 16α-fluoro-5-androsten-17-one and its reversal by deoxyribonucleosides. Carcinogenesis 1991, 12, 2189–2192. [Google Scholar] [CrossRef] [PubMed]
- Edes, A.N. Dehydroepiandrosterone-sulfate (DHEA-S), sex, and age in zoo-housed western lowland gorillas (Gorilla gorilla gorilla). Primates 2017, 58, 385–392. [Google Scholar] [CrossRef]
- Smail, P.J.; Faiman, C.; Hobson, W.C.; Fuller, G.B.; Winter, J.S. Further Studies on Adrenarche in Nonhuman Primates*. Endocrinology 1982, 111, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, R.M. Hormones and Human and Nonhuman Primate Growth. Horm. Res. Paediatr. 2017, 88, 15–21. [Google Scholar] [CrossRef]
- Conley, A.J.; Pattison, J.C.; Bird, I.M. Variations in Adrenal Androgen Production Among (Nonhuman) Primates. Semin. Reprod. Med. 2004, 22, 311–326. [Google Scholar] [CrossRef]
- Bernstein, R.M.; Sterner, K.N.; Wildman, D.E. Adrenal androgen production in catarrhine primates and the evolution of adrenarche. Am. J. Phys. Anthr. 2012, 147, 389–400. [Google Scholar] [CrossRef]
- Sabbi, K.H.; Muller, M.N.; Machanda, Z.P.; Otali, E.; Fox, S.A.; Wrangham, R.W.; Thompson, M.E. Human-like adrenal development in wild chimpanzees: A longitudinal study of urinary dehydroepiandrosterone-sulfate and cortisol. Am. J. Primatol. 2019, 82, e23064–e23064. [Google Scholar] [CrossRef] [PubMed]
- Blevins, J.K.; Coxworth, J.E.; Herndon, J.G.; Hawkes, K. Brief communication: Adrenal androgens and aging: Female chimpanzees (Pan troglodytes) compared with women. Am. J. Phys. Anthr. 2013, 151, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Behringer, V.; Hohmann, G.; Stevens, J.M.G.; Weltring, A.; Deschner, T. Adrenarche in bonobos (Pan paniscus): evidence from ontogenetic changes in urinary dehydroepiandrosterone-sulfate levels. J. Endocrinol. 2012, 214, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Grizzle, W.; Blevins, J.; Hawkes, K. Development of adrenal cortical zonation and expression of key elements of adrenal androgen production in the chimpanzee (Pan troglodytes) from birth to adulthood. Mol. Cell. Endocrinol. 2014, 387, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Bélanger, A.; Cusan, L.; Gomez, J.-L.; Candas, B. Marked Decline in Serum Concentrations of Adrenal C19 Sex Steroid Precursors and Conjugated Androgen Metabolites During Aging. J. Clin. Endocrinol. Metab. 1997, 82, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Nyce, J. Alert to US physicians: DHEA, widely used as an OTC androgen supplement, may exacerbate COVID-19. Endocrine-Related Cancer 2021, 28, R47–R53. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gu, Y.-Y.; Jing, F.; Yu, C.-X.; Guan, Q.-B. The Effect of Statins on Levels of Dehydroepiandrosterone (DHEA) in Women with Polycystic Ovary Syndrome: A Systematic Review and Meta-Analysis. J. Pharmacol. Exp. Ther. 2019, 25, 590–597. [Google Scholar] [CrossRef]
- Zheng, H.; Siddharth, S.; Parida, S.; Wu, X.; Sharma, D. Tumor Microenvironment: Key Players in Triple Negative Breast Cancer Immunomodulation. Cancers 2021, 13, 3357. [Google Scholar] [CrossRef]
- Wu, Q.; Siddharth, S.; Sharma, D. Triple Negative Breast Cancer: A Mountain Yet to Be Scaled Despite the Triumphs. Cancers 2021, 13, 3697. [Google Scholar] [CrossRef]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef]
- Boland, P.M.; Yurgelun, M.B.; Boland, C.R. Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA: A Cancer J. Clin. 2018, 68, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S. Barbara McClintock and the discovery of jumping genes. Proc. Natl. Acad. Sci. USA 2012, 109, 20198–20199. [Google Scholar] [CrossRef] [PubMed]
- Henssen, A.G.; Kentsis, A. Emerging functions of DNA transposases and oncogenic mutators in childhood cancer development. J. Clin. Investig. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Han, D.; Boyd-Kirkup, J.; Yu, X.; Han, J.-D.J. Evolution of Alu Elements toward Enhancers. Cell Rep. 2014, 7, 376–385. [Google Scholar] [CrossRef]
- Chen, L.-L.; Yang, L. ALU ternative Regulation for Gene Expression. Trends Cell Biol. 2017, 27, 480–490. [Google Scholar] [CrossRef]
- Mills, R.E.; Bennett, E.A.; Iskow, R.C.; Devine, S.E. Which transposable elements are active in the human genome? Trends Genet. 2007, 23, 183–191. [Google Scholar] [CrossRef]
- Tiwari, B.; Jones, A.E.; Caillet, C.J.; Das, S.; Royer, S.K.; Abrams, J.M. p53 directly represses human LINE1 transposons. Minerva Anestesiol. 2020, 34, 1439–1451. [Google Scholar] [CrossRef]
- McKerrow, W.; Wang, X.; Mendez-Dorantes, C.; Mita, P.; Cao, S.; Grivainis, M.; Ding, L.; LaCava, J.; Burns, K.H.; Boeke, J.D.; et al. LINE-1 expression in cancer correlates with p53 mutation, copy number alteration, and S phase checkpoint. Proc. Natl. Acad. Sci. USA 2022, 119, e2115999119. [Google Scholar] [CrossRef] [PubMed]
- Marasca, F.; Sinha, S.; Vadalà, R.; Polimeni, B.; Ranzani, V.; Paraboschi, E.M.; Burattin, F.V.; Ghilotti, M.; Crosti, M.; Negri, M.L.; et al. LINE1 are spliced in non-canonical transcript variants to regulate T cell quiescence and exhaustion. Nat. Genet. 2022, 54, 180–193. [Google Scholar] [CrossRef]
- Modenini, G.; Abondio, P.; Boattini, A. The coevolution between APOBEC3 and retrotransposons in primates. Mob. DNA 2022, 13, 27. [Google Scholar] [CrossRef]
- Jacobs, F.M.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1. 2014.
- Warneken, F.; Rosati, A.G. Cognitive capacities for cooking in chimpanzees. Proc. R. Soc. B: Biol. Sci. 2015, 282, 20150229. [Google Scholar] [CrossRef] [PubMed]
- Rota, F.; Conti, A.; Campo, L.; Favero, C.; Cantone, L.; Motta, V.; Polledri, E.; Mercadante, R.; Dieci, G.; Bollati, V.; et al. Epigenetic and Transcriptional Modifications in Repetitive Elements in Petrol Station Workers Exposed to Benzene and MTBE. Int. J. Environ. Res. Public Health 2018, 15, 735. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Thompson, C.B. Transcriptional activation of short interspersed elements by DNA-damaging agents. Genes, chromosomes cancer 2001, 30, 64–71. Available online: https://pubmedncbinlmnihgov/11107177/. [CrossRef] [PubMed]
- Okudaira, N.; Okamura, T.; Tamura, M.; Iijma, K.; Goto, M.; Matsunaga, A.; Ochiai, M.; Nakagama, H.; Kano, S.; Fujii-Kuriyama, Y.; et al. Long interspersed element-1 is differentially regulated by food-borne carcinogens via the aryl hydrocarbon receptor. Oncogene 2012, 32, 4903–4912. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kalia, V.; Perera, F.; Herbstman, J.; Li, T.; Nie, J.; Qu, L.; Yu, J.; Tang, D. Prenatal airborne polycyclic aromatic hydrocarbon exposure, LINE1 methylation and child development in a Chinese cohort. Environ. Int. 2017, 99, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Smith, L.E.; Feng, Z.; Tang, M.; Lee, C.S.; Pfeifer, G.P. Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer research 2001, 61, 7110–7117. [Google Scholar] [PubMed]
- Herbstman, J.B.; Tang, D.; Zhu, D.; Qu, L.; Sjödin, A.; Li, Z.; Camann, D.; Perera, F.P. Prenatal Exposure to Polycyclic Aromatic Hydrocarbons, Benzo[ a ]pyrene–DNA Adducts, and Genomic DNA Methylation in Cord Blood. Environ. Health Perspect. 2012, 120, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Li, G.; Wei, W.; Bai, Y.; Feng, Y.; Fu, M.; Guan, X.; Li, M.; Li, H.; Wang, C.; et al. Epigenome-wide DNA methylation signature of benzo[a]pyrene exposure and their mediation roles in benzo[a]pyrene-associated lung cancer development. J. Hazard. Mater. 2021, 416, 125839. [Google Scholar] [CrossRef]
- Mills, R.E.; Bennett, E.A.; Iskow, R.C.; Luttig, C.T.; Tsui, C.; Pittard, W.S.; Devine, S.E. Recently Mobilized Transposons in the Human and Chimpanzee Genomes. Am. J. Hum. Genet. 2006, 78, 671–679. [Google Scholar] [CrossRef]
- Tang, W.; Mun, S.; Joshi, A.; Han, K.; Liang, P. Mobile elements contribute to the uniqueness of human genome with 15,000 human-specific insertions and 14 Mbp sequence increase. DNA Res. 2018, 25, 521–533. [Google Scholar] [CrossRef]
- Häsler, J.; Strub, K. Alu elements as regulators of gene expression. Nucleic Acids Res. 2006, 34, 5491–5497. [Google Scholar] [CrossRef]
- Häsler, J.; Strub, K. Alu elements as regulators of gene expression. Nucleic Acids Res. 2006, 34, 5491–5497. [Google Scholar] [CrossRef] [PubMed]
- Shanks, N.; Greek, R.; Greek, J. Are animal models predictive for humans? Philos. Ethic- Humanit. Med. 2009, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. American journal of translational research 2014, 6, 114–118. [Google Scholar] [PubMed]
- Huang, Y.; Zhang, S.; Shen, J.; Yang, J.; Chen, X.; Li, W.; Wang, J.; Xu, X.; Xu, X.; Liu, Z.; et al. Association of plasma uric acid levels with cognitive function among non-hyperuricemia adults: A prospective study. Clin. Nutr. 2021, 41, 645–652. [Google Scholar] [CrossRef]
- Durgapal, S.; Jantwal, A.; Upadhyay, J.; Joshi, T.; Kumar, A. Antioxidants Effects in Health: The Bright and the Dark Side. Chapter 4.19, URIC ACID. PP 505-516, 2022, Elsevier. [CrossRef]
Figure 1.
Adrenarche triggers the human-specific kill switch tumor suppression mechanism exactly when it is needed— immediately preceding the dramatic increase in body height that would otherwise cause an increase in cancer risk.
Figure 1.
Adrenarche triggers the human-specific kill switch tumor suppression mechanism exactly when it is needed— immediately preceding the dramatic increase in body height that would otherwise cause an increase in cancer risk.
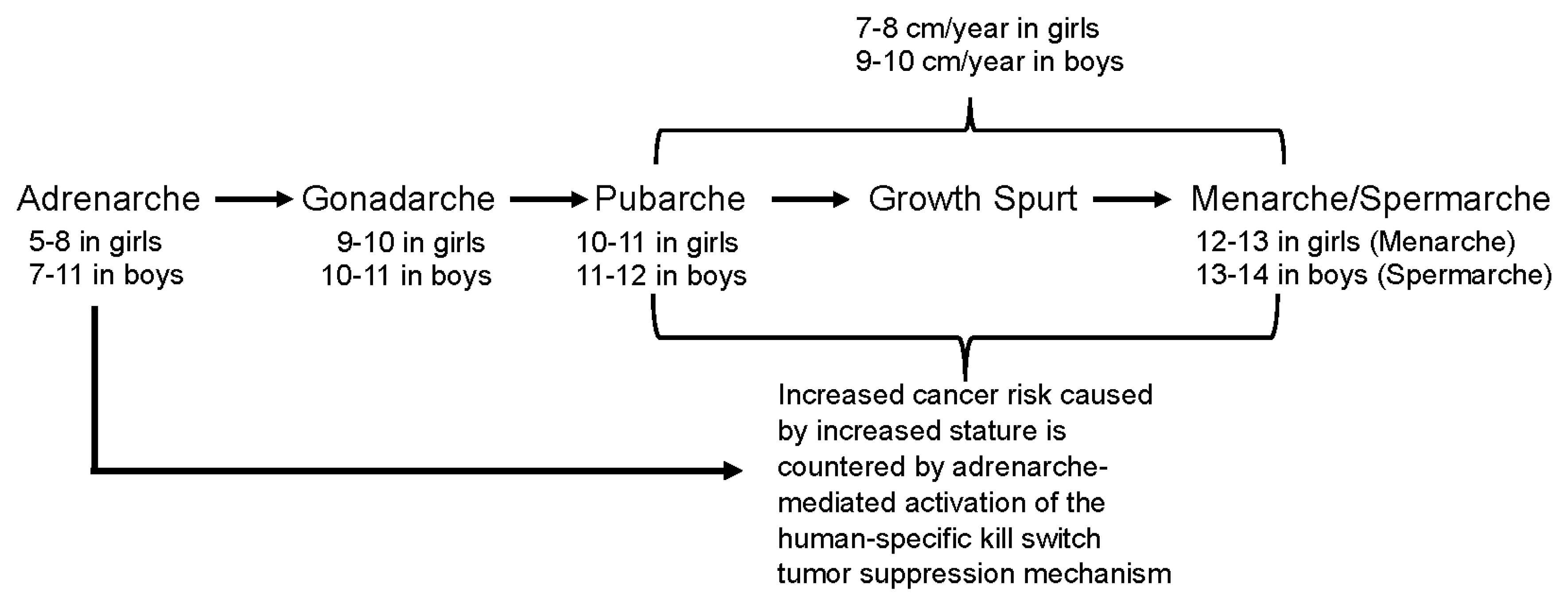
Figure 2.
The evolutionary process of speciation maintains lifetime cancer risk in vertebrates at less than or equal to 5% [1,2,3,4,5,6]. Human cancer incidence data [5,6].
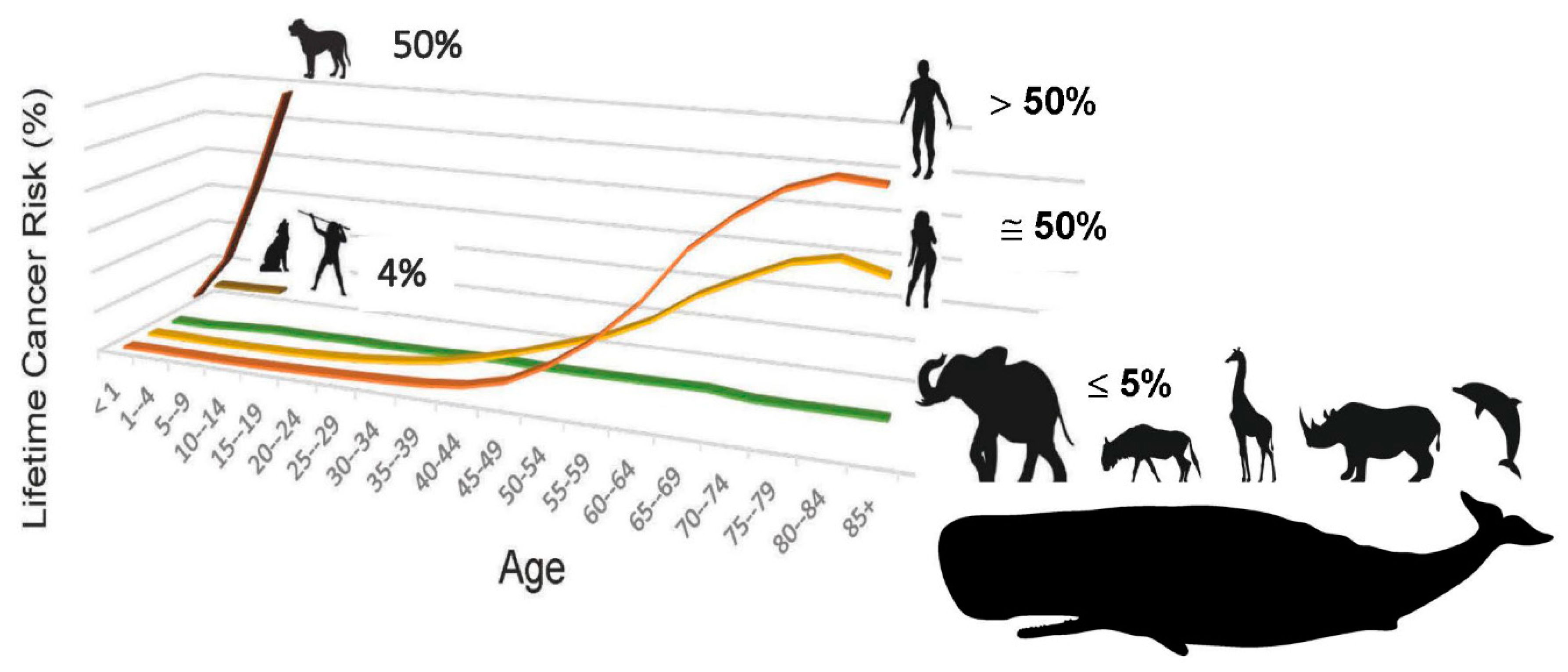
Figure 3.
(A) The lex naturalis equation links the speciation variables of body size (S), lifespan (Li), species-specific mechanism of tumor suppression (T-1), and carcinogen exposure (E) to lifetime cancer risk (R-1) [7]. (B) During speciation, any increase in one of the variables, for example body size, must be offset by an equilibrating change in one of the other variables, for example species-specific mechanism of tumor suppression, in order to maintain lifetime cancer risk at a level no greater than that of the ancestral species, ≤ 5%. T is the probability that a species-specific mechanism of tumor suppression will extinguish all singularities, and R is the probability of incurring cancer within the species’ lifetime. They are thus represented as their reciprocals, thereby allowing manipulation of the equation, e.g., Li = T/[S][E][R].
Figure 3.
(A) The lex naturalis equation links the speciation variables of body size (S), lifespan (Li), species-specific mechanism of tumor suppression (T-1), and carcinogen exposure (E) to lifetime cancer risk (R-1) [7]. (B) During speciation, any increase in one of the variables, for example body size, must be offset by an equilibrating change in one of the other variables, for example species-specific mechanism of tumor suppression, in order to maintain lifetime cancer risk at a level no greater than that of the ancestral species, ≤ 5%. T is the probability that a species-specific mechanism of tumor suppression will extinguish all singularities, and R is the probability of incurring cancer within the species’ lifetime. They are thus represented as their reciprocals, thereby allowing manipulation of the equation, e.g., Li = T/[S][E][R].
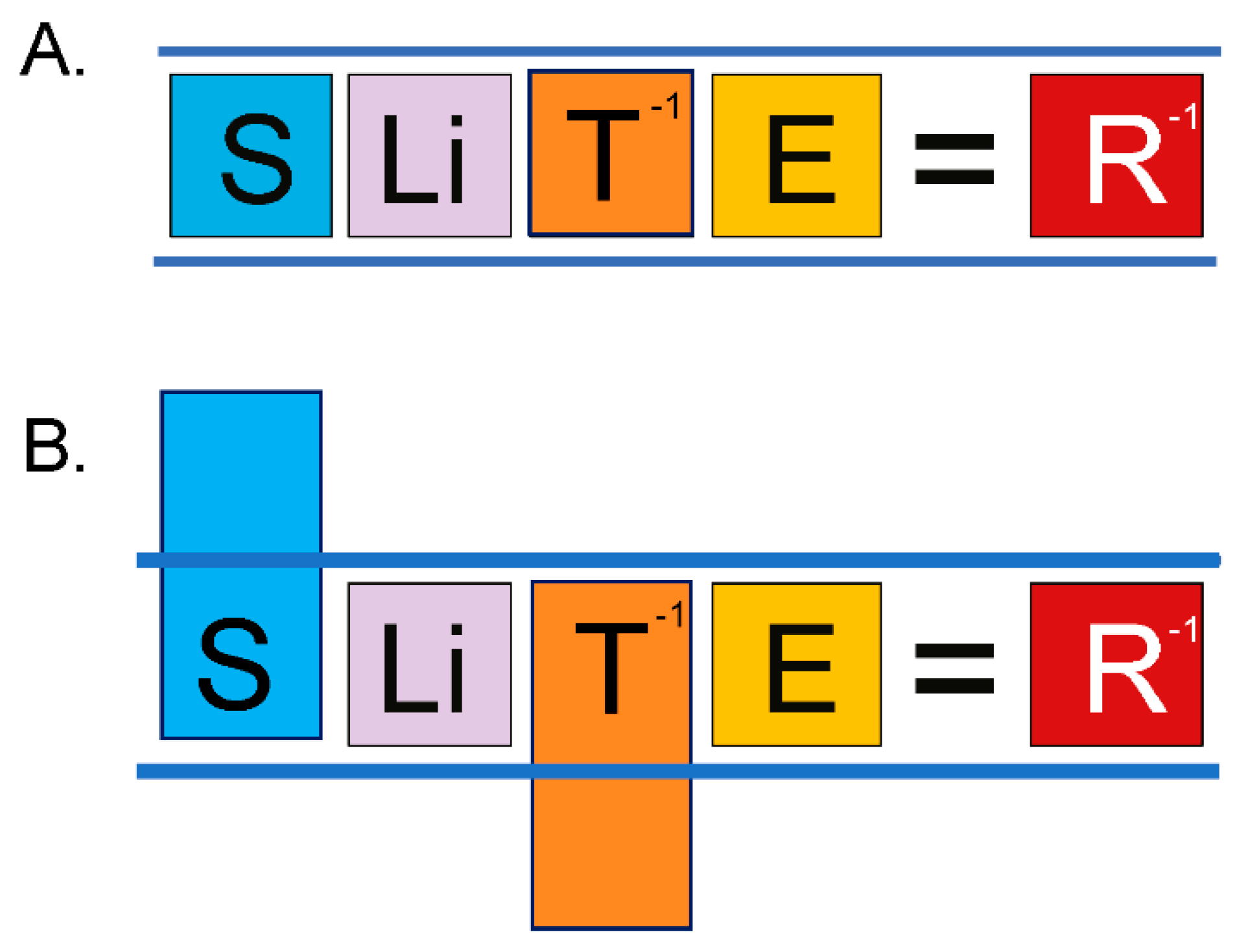
Figure 4.
Uncompetitive enzyme inhibition is unique because (1) it requires substrate [S] to bind to the target enzyme before inhibitor [I] can bind; (2) its is not inhibited by increasing [S]; and (3) it therefore rapidly becomes irreversible in the presence of high intracellular concentrations of substrate and inhibitor.
Figure 4.
Uncompetitive enzyme inhibition is unique because (1) it requires substrate [S] to bind to the target enzyme before inhibitor [I] can bind; (2) its is not inhibited by increasing [S]; and (3) it therefore rapidly becomes irreversible in the presence of high intracellular concentrations of substrate and inhibitor.

Figure 5.
The primate lineage underwent selection for a succession of “improvements” in the kill switch tumor suppression mechanism that marked its origin, culminating in the evolution of adrenarche in the hominins, and in Homo sapiens with the highest levels of circulating DHEAS, and lowest DHEA to DHEAS ratios [66,67,68,69,70,71,72,73,74].
Figure 5.
The primate lineage underwent selection for a succession of “improvements” in the kill switch tumor suppression mechanism that marked its origin, culminating in the evolution of adrenarche in the hominins, and in Homo sapiens with the highest levels of circulating DHEAS, and lowest DHEA to DHEAS ratios [66,67,68,69,70,71,72,73,74].
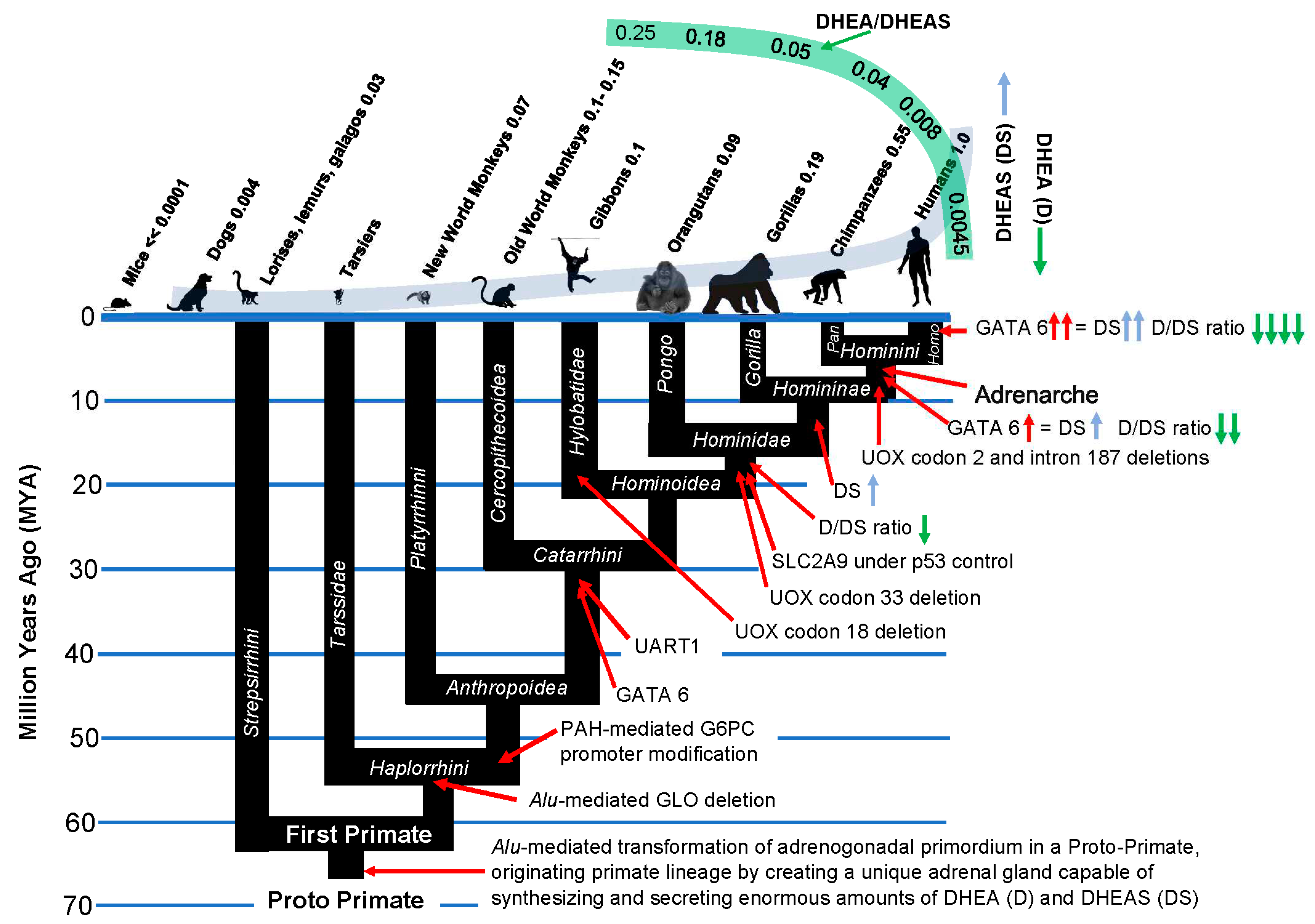
Figure 6.
Mutations in the G6PC promoter that occurred exclusively in the anthropoid primate lineage disabled the DHEA-mediated induction of PGC-1α and HNF-4α, the transcription factor activators of G6PC. Along with GLO deletion, this enabled the accumulation of G6P in singularities.
Figure 6.
Mutations in the G6PC promoter that occurred exclusively in the anthropoid primate lineage disabled the DHEA-mediated induction of PGC-1α and HNF-4α, the transcription factor activators of G6PC. Along with GLO deletion, this enabled the accumulation of G6P in singularities.
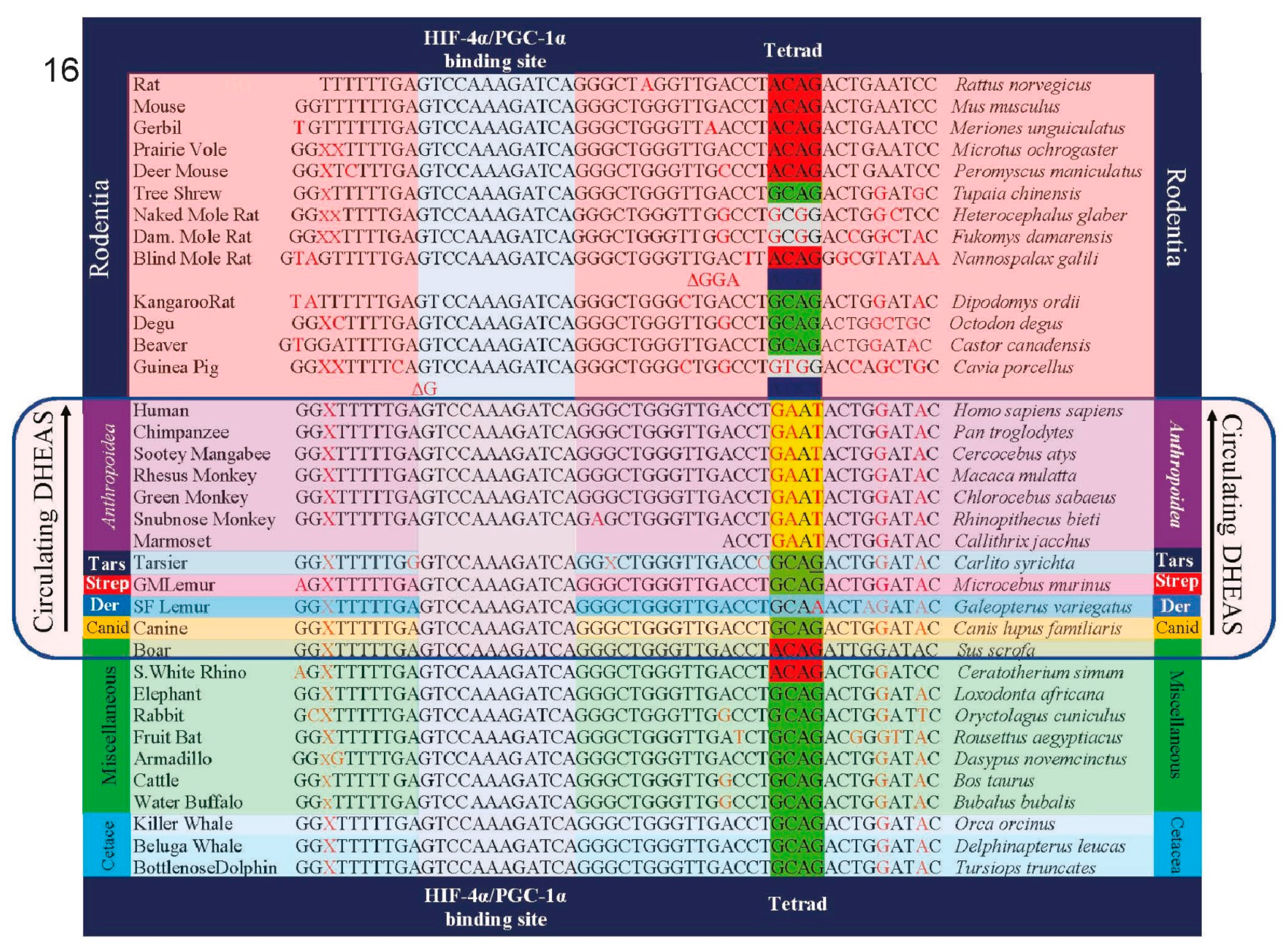
Figure 7.
Lex naturalis equations for hominid species. Gorilla speciation emphasized body size, enabled by both a vegetarian diet (decreasing E) and a doubling of levels of circulating DHEAS compared to Pongo— also an obligate vegetarian species— which attained less body size, but increased lifespan as compared to Gorilla. Chimpanzees exploited fire-combusted sites to forage for PAH-contaminated food, but their increase in E was a fraction of that experienced by humans. Accordingly, higher DHEAS levels enabled the harnessing of fire.
Figure 7.
Lex naturalis equations for hominid species. Gorilla speciation emphasized body size, enabled by both a vegetarian diet (decreasing E) and a doubling of levels of circulating DHEAS compared to Pongo— also an obligate vegetarian species— which attained less body size, but increased lifespan as compared to Gorilla. Chimpanzees exploited fire-combusted sites to forage for PAH-contaminated food, but their increase in E was a fraction of that experienced by humans. Accordingly, higher DHEAS levels enabled the harnessing of fire.
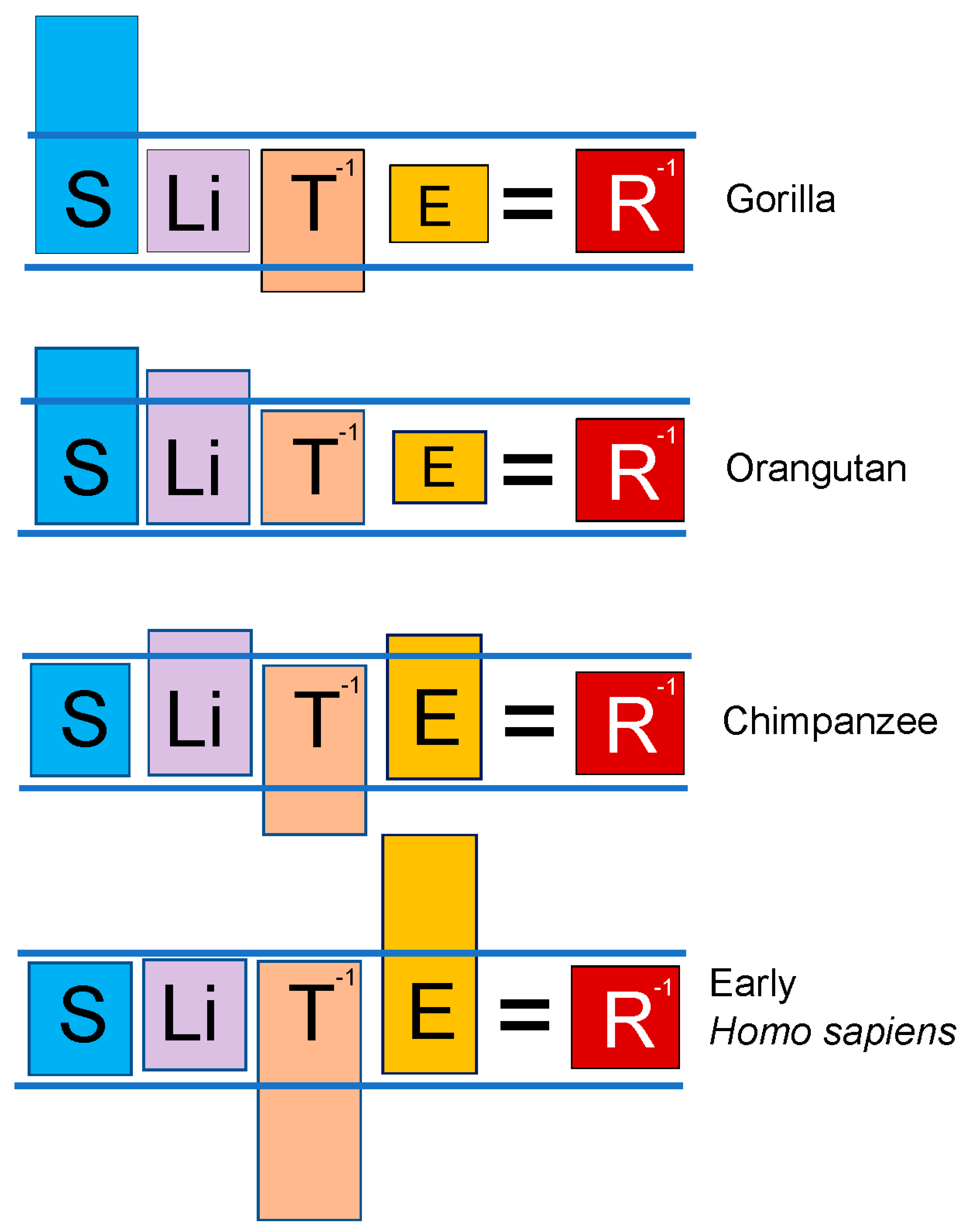
Figure 8.
Lifetime cancer risk in humans during the natural lifespan of our species was <5%, in keeping with other vertebrate animals, and the strictures of natural selection in the lex naturalis equation. Cancer incidence data from the National Cancer Institute (NCI) at https://www.cancer.gov/about-cancer/causes-prevention/risk/age.
Figure 8.
Lifetime cancer risk in humans during the natural lifespan of our species was <5%, in keeping with other vertebrate animals, and the strictures of natural selection in the lex naturalis equation. Cancer incidence data from the National Cancer Institute (NCI) at https://www.cancer.gov/about-cancer/causes-prevention/risk/age.
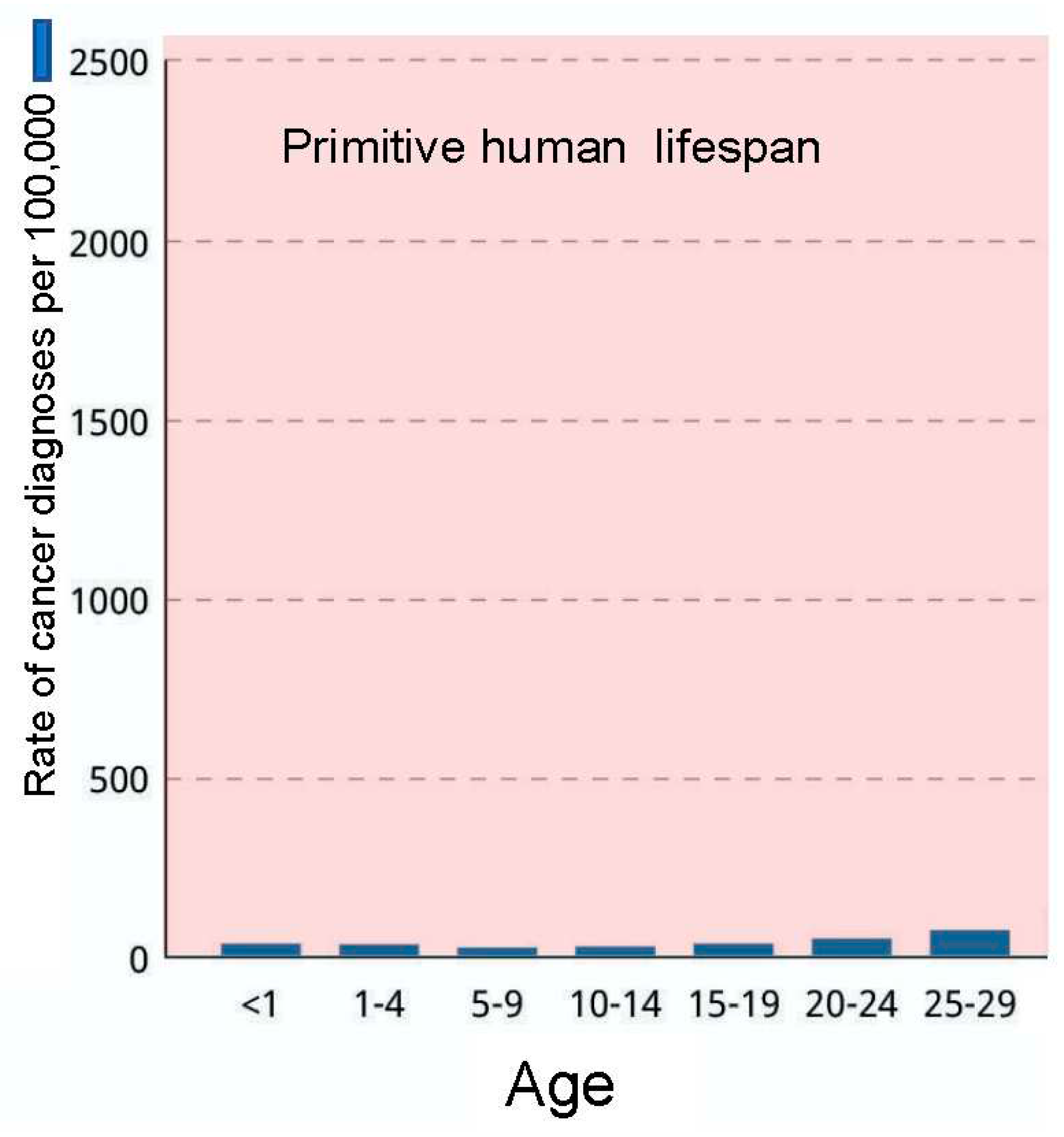
Figure 9.
Circulating levels of DHEAS— the core component of our human-specific “kill-switch” tumor suppression mechanism— reach their peak in humans at about age 25, and rapidly decrease thereafter. Thus, the “kill-switch” evolved to protect only during the primitive lifespan. Cancer statistics from: https://www.cancer.gov/about-cancer/causes-prevention/risk/age.
Figure 9.
Circulating levels of DHEAS— the core component of our human-specific “kill-switch” tumor suppression mechanism— reach their peak in humans at about age 25, and rapidly decrease thereafter. Thus, the “kill-switch” evolved to protect only during the primitive lifespan. Cancer statistics from: https://www.cancer.gov/about-cancer/causes-prevention/risk/age.
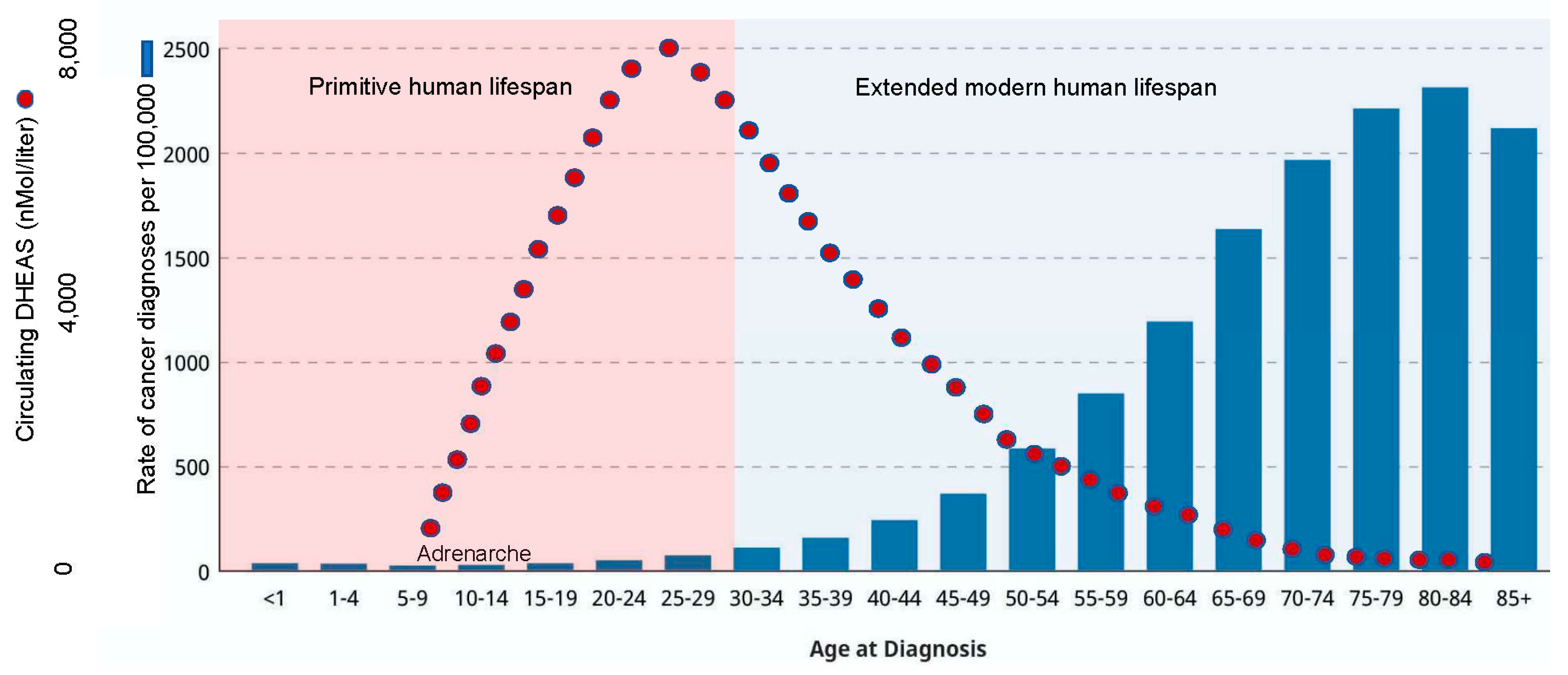
Figure 10.
DHEAS is a small molecule and is therefore pharmacologically tractable. Will this “normalize” cancer risk in our species from its current aberrant rate to the ≤ 5% of other vertebrate species, as depicted above? Only studies in humans of pharmacological maintenance of peak levels of DHEAS throughout the modern lifespan will answer this question.
Figure 10.
DHEAS is a small molecule and is therefore pharmacologically tractable. Will this “normalize” cancer risk in our species from its current aberrant rate to the ≤ 5% of other vertebrate species, as depicted above? Only studies in humans of pharmacological maintenance of peak levels of DHEAS throughout the modern lifespan will answer this question.
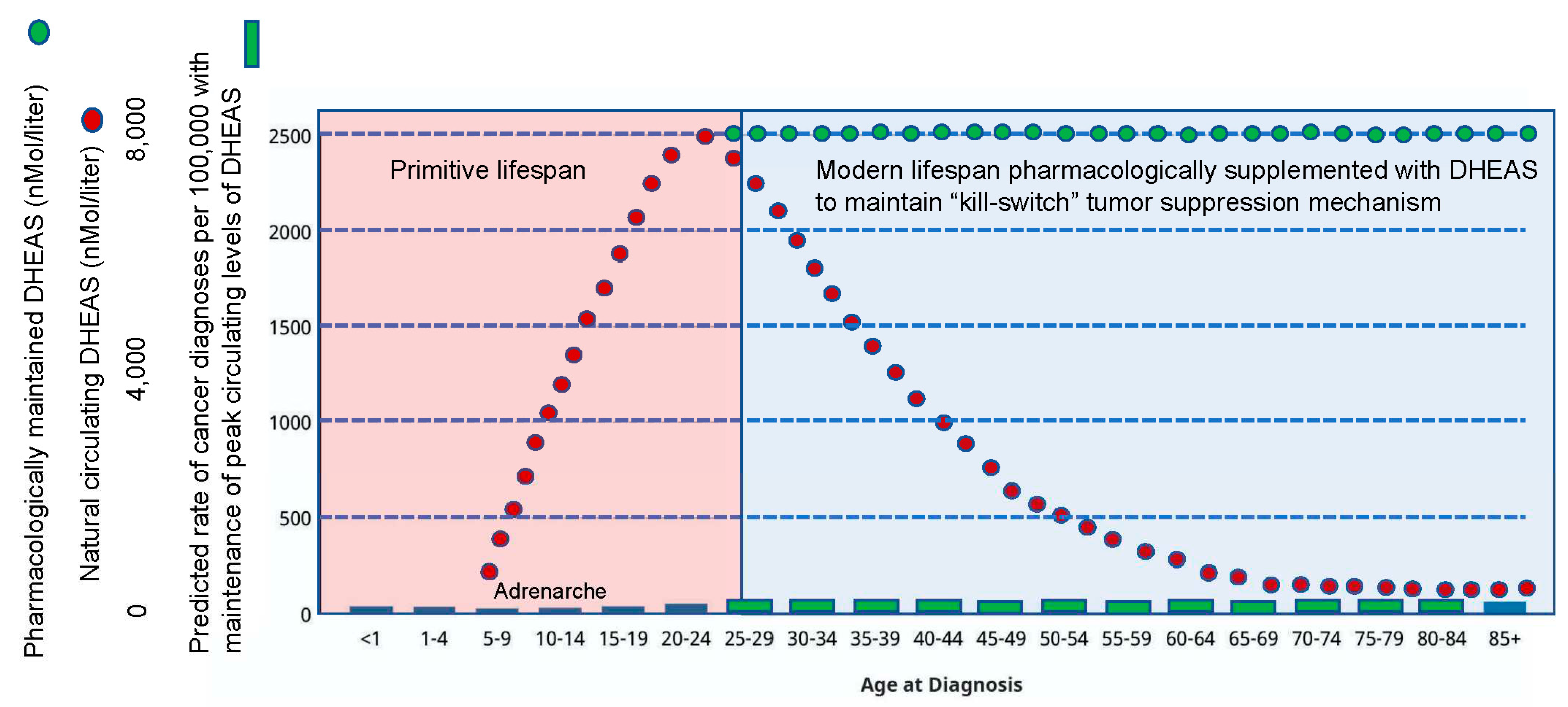
Figure 11.
Anticipating the future evolution of the human-specific “kill-switch” tumor suppression mechanism (increasing DHEAS, decreasing DHEA/DHEAS ratio), suggests a strategy to prevent cancer in persons with mutations that strongly predispose for malignancy, e.g., BRCA-positive women with strong predilection for breast cancer.
Figure 11.
Anticipating the future evolution of the human-specific “kill-switch” tumor suppression mechanism (increasing DHEAS, decreasing DHEA/DHEAS ratio), suggests a strategy to prevent cancer in persons with mutations that strongly predispose for malignancy, e.g., BRCA-positive women with strong predilection for breast cancer.
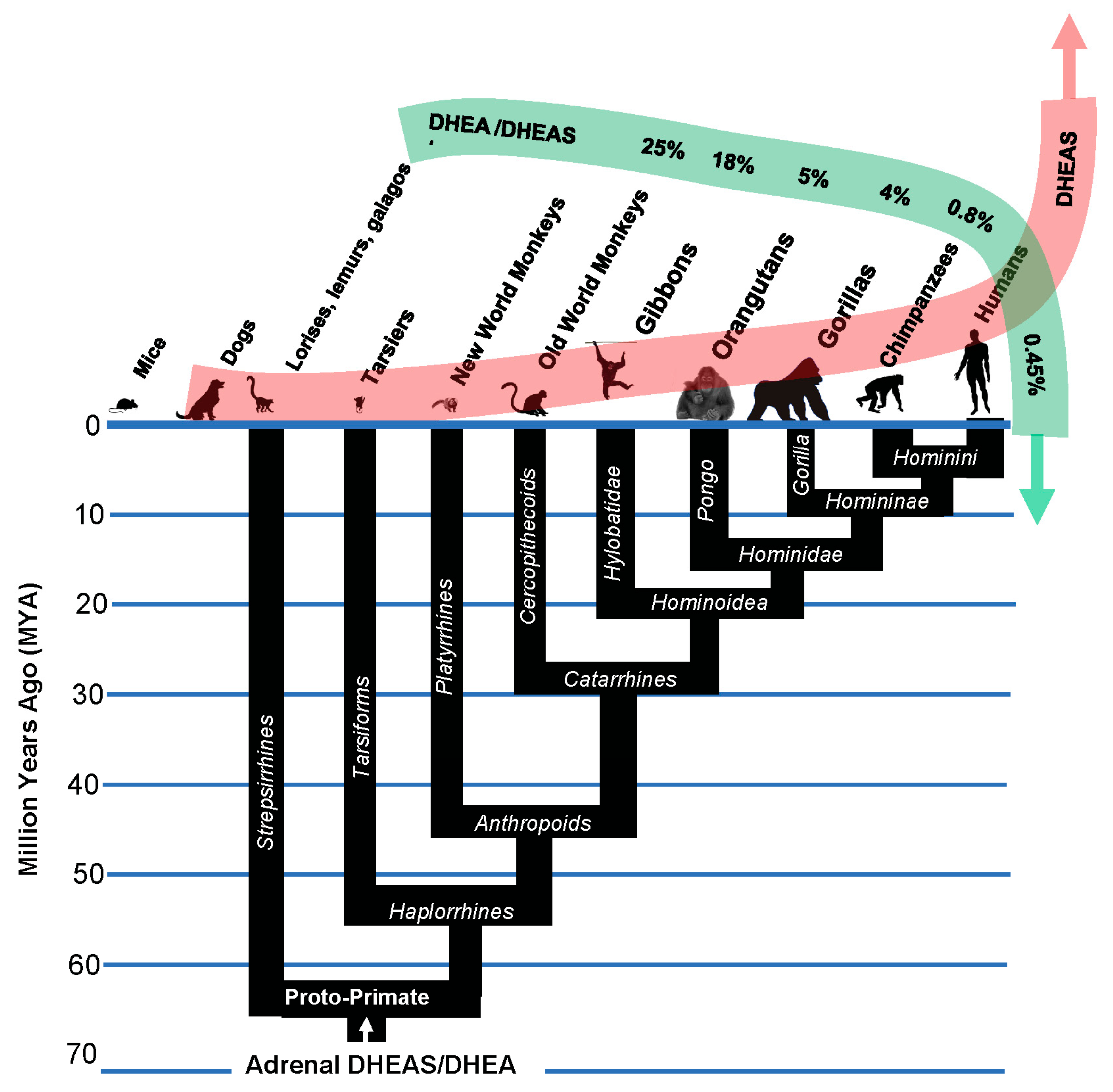
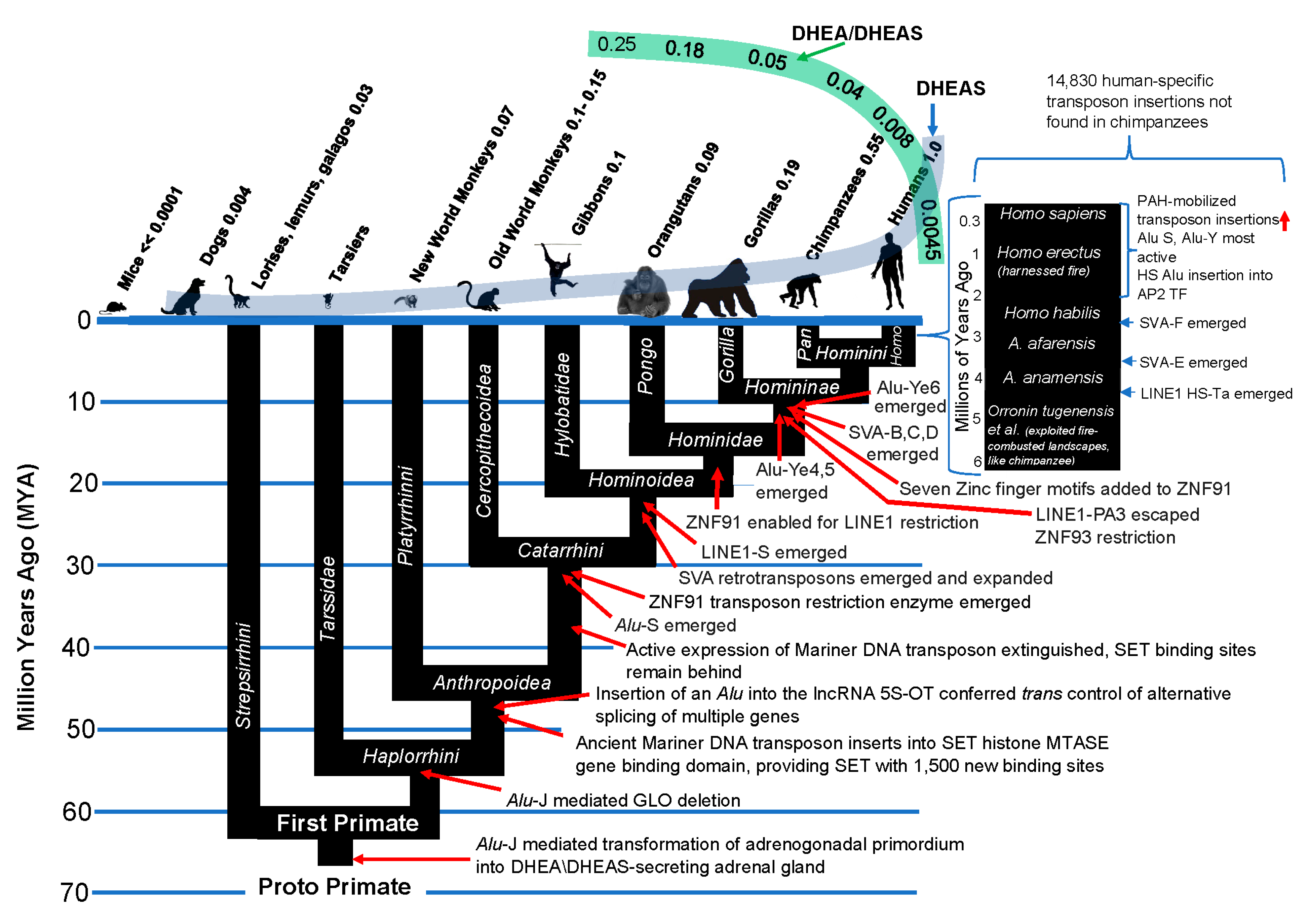
Figure 12.
Transposon insertions into the primate genome correlate with “improvements” in the primate kill switch tumor suppression mechanism. LINE1 HS-Ta, LINE1 human-specific, transcriptionally active; HS Alu, Human-specific Alu; TF, transcription factor.Conclusion.
Figure 12.
Transposon insertions into the primate genome correlate with “improvements” in the primate kill switch tumor suppression mechanism. LINE1 HS-Ta, LINE1 human-specific, transcriptionally active; HS Alu, Human-specific Alu; TF, transcription factor.Conclusion.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



