
Submitted:
12 April 2023
Posted:
13 April 2023
You are already at the latest version
Abstract
The androgen/androgen receptor (AR)-signaling axis plays a central role in the development and growth of prostate cancer (PCa) cells. Upon androgen-binding the AR dimerizes with another AR, translocates into the nucleus where the AR-dimer activates/inactivates androgen-dependent genes. In consequence treatments for locally advanced or metastatic PCa are commonly based on androgen deprivation therapies (ADT). Unfortunately, the clinical benefits of ADT are only transitory and most tumors develop mechanisms allowing the AR to bypass its need for physiological levels of circulating androgens. In the clinic failure of ADT is often characterized by the synthesis of a C-terminally truncated, constitutively active AR splice variant, termed AR-V7. In contrast to AR, the constitutively active AR-V7 does no longer need androgenic stimuli for nuclear entry and/or dimerization. The goal of the present study was to mechanistically decipher the interaction between full-length AR (AR-FL) and AR-V7 in AR-null HEK-293 cells using the NanoLuc Binary Technology (NanoBiT) structural complementation assay under androgen stimulation and deprivation conditions. Our data point toward a hypothesis that AR-FL/AR-FL homodimers form in the cytoplasm, whereas AR-V7/AR-V7 localize in the nucleus. However, after 15 min of androgen stimulation, all AR-FL/AR-FL, AR-FL/AR-V7 and AR-V7/AR-V7 dimers localized in the nucleus. In this way, we can show an androgen-regulated interaction between AR-FL and AR-V7 at forming heterodimers that localize in the nucleus, whereas AR-V7/AR-V7 dimers were found to localize in the absence of androgens in the nucleus. Treatment with enzalutamide diminished the luminescence of AR-FL homodimers and AR-FL/AR-V7 heterodimers but not AR-V7/AR-V7 homodimers.
Keywords:
Androgen receptor
; androgen receptor splice variant 7
; prostate cancer
; localization
; NanoBiT
1. Introduction
The AR gene is located on Xq11-12 and encodes a 110 kDa steroid hormone receptor with four distinct functional motifs. The first exon codes for the amino N-terminal domain (NTD), which is the transcriptional regulatory region of the protein. Exons 2 and 3 code for the central DNA-binding domain (DBD), exon 4 codes for the hinge region harboring parts of the bipartite nuclear localization signal (NLS) and exons 5 to 8 code for the ligand binding domain (LBD) [1]. In the absence of the ligand (inactive state), the cytoplasmic receptor is bound by heat-shock proteins (predominantly HSP90). Androgen binding, specifically dihydrotestosterone (DHT) or testosterone to the LBD, causes a conformational change that releases AR from the HSP complex, facilitating AR dimerization, exposure of NLS rapid nuclear translocation and binding to androgen-response elements (AREs) in the promoter region of androgen-regulated genes. AREs serve as a platform for recruiting coactivators and basal transcription machinery, initiating gene transcription that results in diverse biological outcomes depending on the cell context [2,3]. AR intra- and intermolecular dimerization and the localization of AR have been studied in detail [4,5]. Intramolecular dimerization occurs via NTD-LBD interactions in the cytoplasm, whereas intermolecular dimerization by DBD-DBD and NTD-LBD interactions emerge after localization to the nucleus, which is ligand dependent [4]. However, whether AR intermolecular dimerization starts in the cytoplasm, in the nucleus or possibly in both compartments remains unknown.
Alternative splicing of the AR is a specific mechanism that has gained attention in several contexts, including the progression to castration-resistant prostate cancer (CRPC) and in resistance against second-generation endocrine treatments [6,7]. To date, more than 30 AR splice variants (AR-Vs) have been identified in prostate cancer cell lines and xenograft tumors at the mRNA level [reviewed in 8].
Most AR-Vs share a common structural feature, as they exhibit a truncated LBD in both clinical samples and prostate cancer cell lines. These structural similarities in AR-Vs may result in androgen-independent transcriptional activity. Most transcripts coding for truncated AR-Vs may arise due to incorporation of cryptic exons, exon skipping or genetic rearrangements.
Two of the best characterized AR-Vs, AR-V7 and AR-V567es, have demonstrated constitutive activity in different cell systems. AR-V7 was originally discovered in the androgen-independent 22Rv1 cell line, whereas AR-V567es was identified in LuCaP 86.2 and 136 xenografts and found to increase proliferation in androgen-dependent LNCaP cells in the absence of androgens [9,10]. AR-V7 is expressed at low levels in primary PCa but increases in CRPC [11,12]. Androgen receptor splice variants, more specifically AR-V7, have been described to mediate resistance to second-generation drugs targeting the AR pathway, i.e., enzalutamide and abiraterone [6,13]. However, the molecular mechanisms by which AR-Vs in castration-resistant PCa (CRPC) are regulated are not fully understood [14,15].
In contrast to the canonical AR signaling pathway, in which NLSs are necessary for nuclear localization, the exact mechanism enabling AR-Vs to enter the nucleus and exert their function is less clear. Since the bipartite AR-NLS is located in exons 3 and 4, most AR-Vs are expected to be predominantly cytoplasmic because of the truncated exon 3; there are a few exceptions, including AR-V567es, which retains both exons. Surprisingly, AR-V7, without a full NLS, exhibits constitutive nuclear localization and transcriptional activity [14]. However, a potential NLS is presumed in the cryptic exon CE3 of AR-V7 [16,17]. Chan and colleagues described that truncated AR-Vs with the AR NTD/DBD core and diverse COOH terminal extensions exhibit a basal level of nuclear localization sufficient for androgen-independent transcriptional activity regardless of whether they harbor the exon 4-enconded NLS or NLS-like COOH-terminal extensions. They concluded that the canonical NLS in the AR hinge region is not the only determinant of AR nuclear access and transcriptional activity [18]. Recently, it has been demonstrated that AR-Vs form heterodimers with transcription factors, such as ZFX, which harbors intact nuclear localization sequences and could enable the nuclear localization of AR-Vs [19].
Cao et al. studied the interaction of AR-FL, AR-V7 homodimers and AR-FL/AR-V7 heterodimers in COS-7 cells, an African green monkey kidney fibroblast-like cell line without endogenous AR expression [13]. AR-V7 facilitated AR-FL nuclear localization in the absence of androgens. In addition, enzalutamide, an AR signaling inhibitor, inhibited AR-FL translocation to the nucleus, but this effect was mitigated in the presence of AR-V7. In addition, nuclear localization of AR-V7/AR-V7 was not affected by androgens or enzalutamide [13].
Xu and colleagues studied the ability of AR-V7 and AR-V567es to interact with AR-FL to form heterodimers and homodimers in the absence of androgens in PC3 cells [5]. Their study indicates that AR-V/AR-FL dimerization is mediated by both a DBD/DBD and an N/C (NTD-LBD) interaction. However, since AR-Vs have lost their LBD, the LBD is provided by AR-FL and the NTD from AR-V in the heterodimer [5]. AR-V7/AR-V7 homodimers show only the DBD-DBD interaction. Xu and coworkers detected AR-V7/AR-FL heterodimers and AR-V7/AR-V7 homodimers primarily in the nucleus of PC-3 cells. AR-FL/AR-FL homodimers were observed only after DHT treatment and mostly occurred in the nucleus of PC-3 cells.
Özgün et al. investigated the DNA binding of AR-FL, AR-V7 homodimers and AR-V7/AR-FL heterodimers. AR-FL/AR-V7 heterodimers readily form in the nucleus via intermolecular N/C (NTD-LBD) interactions. However, DNA binding occupancy is determined by protein monomers, not homodimer or heterodimer complexes [20].
However, it remains unclear when AR-FL/AR-FL homodimers enter the nucleus after DHT treatment. Furthermore, we do not know if AR-V7/AR-FL heterodimers form in the cytoplasm or if they enter the nucleus as AR-V7/AR-V7 and AR-FL/AR-FL homodimers and perform a partner swap in the nucleus.
We used human embryonic kidney cells (HEK-293) that do not express endogenous AR protein and transfected them with plasmids coding for AR-FL/AR-FL homodimers, AR-FL/AR-V7 heterodimers or AR-V7/AR-V7 homodimers under androgen treatment (DHT) or androgen deprivation conditions (enzalutamide). We applied NanoLuc Binary Technology (NanoBiT) for highly sensitive intracellular detection of protein:protein interactions [21] for AR-FL/AR-FL and AR-V7/AR-V7 homodimers and AR-FL/AR-V7 heterodimers. AR-FL/AR-FL homodimers formed in the cytoplasm. The localization of AR-FL/AR-FL homodimers shifted into the nucleus within 15 min after DHT treatment. AR-V7/AR-V7 homodimers were constitutively located in the nucleus, and neither DHT nor enzalutamide affected the localization of AR-V7/AR-V7 or its status as a dimer. However, our data indicate that AR-V7/AR-FL heterodimers form in the nucleus after AR-FL homodimers are translocated to the nucleus.
2. Materials and Methods
2.1. Cloning of tagged androgen receptor constructs
To study the interaction of androgen receptor and splice variant AR-V7 homo-or heterodimers, we used the NanoBiT PPI MCS Starter System (Promega, Madison, WI, USA). Primers were designed to PCR amplify the coding regions of AR-FL and AR-V7 and at the same time to add specific restriction sites. After agarose gel purification, the PCR fragments and target vectors were restriction digested to allow an in-frame insertion of the coding regions into the expression vectors containing the large BiT (LgBiT) and small BiT (SmBiT) fragments (Figure 1). The sequences of the amplification primers are listed in Table 1. All expression vectors were verified by sequencing. The SmBiT-PRKACA:LgBiT-PRKAR2A pair served as a positive control, in which fusion partners interact without adding a compound. PRKACA is the catalytic subunit α of protein kinase A, and PRKARS2A is the cAMP-dependent protein kinase type II-alpha regulatory subunit.
2.2. Cell culture conditions HEK293
Cells were cultured in DMEM (Sigma‒Aldrich, Darmstadt, Germany) supplemented with 10% charcoal-stripped fetal bovine serum (Sigma‒Aldrich), 1% penicillin‒streptomycin and 20 mM HEPES (Pan Biontech, Aidenbach, Germany). For expression of tagged AR-FL and AR-V7 constructs, 2 µg (1 µg of each LgBit and SmBit interaction partner) was transfected into subconfluent grown HEK293 cells using the jetPRIME transfection system according to the manufacturer’s recommendations (Polyplus transfection, Illkirch, France). Twenty-four hours after transfection, cells were used for short- and long-term stimulation experiments.
2.3. Luminescence in plates
Luminescence measurements were carried out in a TECAN Infinite M200 Pro (Tecan, Männedorf, Switzerland) plate luminometer. Briefly, HEK293 cells were seeded in white plates at a density of 10,000 cells/well, incubated overnight and transfected with AR expression constructs or control constructs. After 24 hours, Nano-Glo Live Cell substrate (Promega) was added, and protein‒protein interactions were induced by adding DHT (1 nM final concentration). An intact NanoLuc luciferase protein tag was formed through direct protein‒protein interaction. Luminescence was measured in the luminometer, preheated to 37 °C, over a period of 2 seconds and normalized as counts per second (CPS).
2.4. Confocal microscopy
We used confocal microscopy to assess the subcellular localization of tagged AR proteins and the localization shift following stimulation with DHT. Therefore, HEK-293 cells were seeded on coverslips coated with poly-L-lysine and placed in 6-well plates. HEK-293 cells were cotransfected with different androgen receptor full-length (AR-FL) and androgen receptor splice variant-7 (AR-V7)-tagged constructs (Suppl. Table 1). Twenty-four hours after transfection, the cells were stimulated with 1 nM DHT for 0, 15, 30 or 60 min. Immediately after, cells were fixed with 4 % paraformaldehyde (PFA) for 20 min at room temperature and washed three times each for 5 min with phosphate-buffered saline (PBS). Then, the cells were permeabilized with 0.2% Triton X-100 solution for 10 min at room temperature. The samples were blocked with 1% BSA+ 0.1% Triton X-100 solution for 20 min at room temperature. To detect the tagged AR protein, we used a NanoLuc Luciferase Antibody (R&D Systems, Clone 965853; 1:500), which detects both the LgBit component as well as the complemented NanoLuc, overnight at 4 °C.
After overnight incubation, samples were washed with PBS and incubated with the secondary Alexa Fluor 488 anti-mouse IgG antibody (Invitrogen, Darmstadt, Germany; A-11001; 1:1000) diluted in blocking solution (1% BSA+ 0.1% Triton X-100) for 60 min at room temperature. Again, samples were washed with PBS and preserved in one single step by using Mounting Medium (Carl Roth, Karlsruhe, Germany) containing DAPI (4’,6-diamidino-2-phenylindole) for staining cell nuclei. The samples were analyzed by fluorescence microscopy with a Leica SP5 II (Leica, Wetzlar, Germany). Suppl. Table 2 shows an overview of the timeline from seeding to mounting of the HEK-293 cell line.
2.5. Live cell luminescent imaging
For live cell luminescence imaging, HEK-293 cells were plated in chambered coverslips coated with poly-L-lysine as described previously. Following 24 h of incubation, cells were cotransfected with AR-FL and AR-V7 NanoBiT constructs (Suppl. Table 1) and incubated overnight. Immediately before imaging, cells were stimulated with 1 nM DHT and NanoGlo live-cell substrate (Promega). Luminescence was recorded and integrated for a total of 90 min with an interval of 10 min and an exposure time of 10 seconds using a Leica DMi8 TIRF Widefield Fluorescence Microscope (Leica) equipped with an EMCCD camera (Andor, Oxford instruments, iXon Ultra, Abingdon, UK).
3. Results
3.1. Characterization of AR-FL and AR-V7 in the NanoBiT protein‒protein interaction assay
As it is known that the orientation of the NanoLuc Binary protein tag components influences the results of live cell luminescence reactions, we first generated AR-FL- and AR-V7-tagged protein expression constructs to include all possible combinations of homodimers and heterodimers. There were four different combinations each for AR-FL and AR-V7 homodimers (Ho 1-8) and eight different AR-FL/AR-V7 heterodimer combinations (He 1-8) (Figure 1, Figure 2A). HEK-293 cells were transiently transfected with 1 µg plasmid combinations (0.5 µg each plasmid construct). Twenty-four hours after transfection, the cell culture medium was replaced, and the cells were stimulated with 1 nM DHT or 10 µM enzalutamide for another 24 hours. Control cells were transfected with a negative control vector encoding HaloTag-SmBiT, a structurally stable protein that is expressed throughout the cell and coexpressed with a respective LgBiT fusion construct.
Without stimulation, the HEK-293 cells expressing tagged AR-FL (Ho 1-4) did not show any luminescence signal increase compared to the negative control, confirming the lack of AR-FL dimerization in the absence of androgens. However, cells expressing tagged AR-V7 (Ho 5-8) displayed constitutive luminescence activity, which was elevated by 3.7-, 3.5-, 3.8- and 3.7-fold compared to that of the negative control. This confirmed the ability of AR-V7 to form functionally active homodimers in the absence of androgens (Cao et al. 2014; Xu et al. 2015).
When stimulated with 1 nM DHT, all AR-FL combinations displayed a marked fold increase in luminescence by 7.4-, 10-, 13.8- and 10-fold compared to that of negative control cells, whereas AR-V7 combinations showed identical 3.9-, 3.8-, 4.0- and 3.8-fold luminescence to that of control cells. This again confirms that AR-FL dimers only form in the presence of androgens, whereas the extent of AR-V7 dimerization cannot be further enhanced.
Anti-androgen treatment with 10 µM enzalutamide, on the other hand, did not induce dimerization of AR-FL monomers, nor did it inhibit the dimer formation of AR-V7, as these constructs consistently showed 4.3-, 4.1-, 4.2- and 3.8-fold higher luminescence compared to that of negative control-transfected cells (Figure 2B).
Next, we tested the capabilities to assess the formation of AR-FL/AR-V7 heterodimers when the binary NanoLuc components were split between AR-FL and AR-V7. Without stimulation, none of the eight distinct combinations showed any increased luminescent signal compared to negative control cells. This demonstrates that in the absence of androgens, no heterodimer formation was present. After stimulation with 1 nM DHT, all 8 combinations displayed an increase in luminescence by 5-, 4.9-, 16.5-, 7.8-, 10.8-, 11-, 4.2- and 5.5-fold compared to that of control cells (Figure 2C).
In summary, we confirmed that AR-FL resides as a monomer in the cytoplasm in the absence of androgens and that dimer formation can be readily stimulated by DHT. AR-V7 forms a homodimer in the absence of androgen, and this dimer formation can neither be further stimulated by androgens nor inhibited by enzalutamide. When both AR-FL and AR-V7 were present, no trace of heterodimer formation was observed in the absence of androgens.
For all further experiments, we selected the homodimer 3 (N-LgBit AR/C-SmBit AR), homodimer 5 (N-LgBit AR-V7/N-SmBit AR-V7) and heterodimer 3 (C-LgBit AR/C-SmBit AR-V7) plasmid combinations that exhibited the most pronounced increase in luminescence after stimulation of protein‒protein interaction (Figure 2 B and C).
3.2. Localization of recombinant AR and AR-V7 proteins
Next, we were interested in assessing the subcellular localization of AR-FL and AR-V7 proteins upon dihydrotestosterone stimulation. For this, we used immunofluorescence (IF) and confocal microscopy. For detection purposes, we used an antibody specific to the NanoLuc luciferase protein. Importantly, the detection antibody binds to both the complemented NanoLuc luciferase and the LargeBiT fragment but not to the SmBiT fragment, so the subcellular localization information does not provide any information about possible dimer formation.
Without androgen stimulation, we detected AR-FL monomers exclusively in the cytoplasm, whereas AR-V7 homodimers were exclusively located in the nucleus (Figure 3). Upon stimulation with DHT, we observed the translocation of AR-FL from the cytoplasm into the nucleus. This translocation was almost complete in all cells observed after 15 min, and only small residues of AR-FL were detectable in the cytoplasm. This translocation was stable over the observed period of 60 min. However, although this technique is suitable to track the subcellular location of tagged AR constructs, it cannot pinpoint the exact location where AR-FL dimerization takes place.
Regardless of whether AR-FL enters the nucleus as a monomer or as a dimer after DHT stimulation, we detected the presence of AR-FL/AR-V7 heterodimers. Considering the observation that AR-V7 is located exclusively in the nucleus and that this localization is independent of any stimulation with DHT, the results indicate that the formation of heterodimers occurs in the nucleus after AR-FL monomers or homodimers translocate into the nucleus. (Figure 3).
In summary, AR-FL is initially located in the cytoplasm before previous DHT treatment, whereas AR-V7 is located in the nucleus. Upon androgen stimulation, AR-FL translocates into the nucleus, where it interacts with AR-V7.
3.3. Bioluminescence imaging
After 30 min of androgen stimulation, the integrated luminescence signal for the AR-FL homodimers crossed the detection threshold and was clearly detectable. AR-FL/AR-V7 dimers were detecTable 50 min after stimulation. For both construct combinations, time course imaging allowed us to observe changes in luminescence intensity, but it was not possible to visualize translocation of the luminescent signals. (Figure 4 A-B). Unfortunately, with this method, we reached the technical limit of the visual detection threshold. Therefore, to overcome these technical limitations, future experiments in an Olympus LV200 bioluminescence imager would be necessary, which has been described to perform bioluminescence imaging with sufficient resolution to clearly detect and localize luminescence signals [22].
3.4. Kinetics
We recorded the luminescence kinetics using AR-FL homodimer and AR-FL/AR-V7 heterodimer formation. Immediately after androgen stimulation, luminescence was measured in 3 min intervals with a TECAN Infinite M200 PRO (Figure 5). Therefore, the AR-FL homodimer showed a fast-detectable luminescence signal after 3-6 min that continuously increased until 24 min after androgen stimulation, reached saturation and remained stable for the following measured time points.
A different picture was seen for the AR-FL/AR-V7 heterodimer. Luminescence, as a marker for dimerization, increased slower in a linear fashion until 15 min and then remained at a constant level. However, the level of luminescence was lower for the AR-FL/AR-V7 heterodimer than for the AR-FL homodimer.
We suggest that interaction partners interact faster when close, as observed for AR-FL. However, when the interaction partners can only meet after translocation to the nucleus, as for AR-FL and AR-V7, they interact slower. This kinetics is seen directly (Figure 5A) and when the delta luminescence (change in luminescence per 3 min interval) is considered (Figure 5B).
4. Discussion
Based on the very strict dependency of PCa on AR signaling, most systemic therapies directly target the androgen receptor, androgen biosynthesis and/or interaction with androgens. As a consequence, tumors develop resistance to AR-targeted therapies. These resistance mechanisms can include AR overexpression, AR gene amplification, mutations in the ligand binding site of the AR, intracrine androgen synthesis and expression of constitutive splice variants [7,23]. In particular, alternative splicing of AR is a specific mechanism that has gained attention, as it is relevant in the progression to CRPC [7,8]. The most prominent AR splice variant, AR-V7, has been described to mediate resistance to the second generation anti-androgens enzalutamide and abiraterone [6]. However, nuclear AR-V7 expression can be detected in primary prostate cancer prior to long-term androgen deprivation and castration resistance [24]. A meta-analysis recently showed that the AR-V7-positive proportion was significantly higher in CRPC than in newly diagnosed prostate cancer [25]. In addition, especially for hormone-sensitive PCa patients, AR-V7 positivity reveals a worse prognosis of first-line hormonal therapy and prostatectomy, as shown by shorter PFS and OS [25]. The expression of AR-V7 indicates a poor prognosis and is an independent risk factor for reduced overall survival in mCRPC patients treated with endocrine therapy [26].
In our study, we focused on AR and AR-V7. In particular, in the kinetics of their homodimerizations and heterodimerization. We studied these interactions by using NanoLuc Binary Technology (NanoBiT), which can characterize protein–protein interactions in live cells, allowing real-time detection of complex formation [21].
We confirmed that AR-V7 homodimerization occurs in the absence of androgen and that its interaction cannot be further stimulated (dihydrotestosterone/DHT) or inhibited with anti-androgens (enzalutamide). In contrast to other reports, which described AR-FL/AR-V7 dimerization in PC3 cells that does not require androgen stimulation [5,27]), we observed that the formation of AR-FL/AR-V7 heterodimers strictly occurs upon androgen stimulation with DHT (Figure 3). A plausible theory for this discrepancy is that Streicher et al. and Xu et al. performed their localization studies in prostate cancer PC3 cells, whereas our study was performed in HEK-293 cells. Xu and colleagues found that similar to AR-FL/AR-V7 dimerization, AR-V7/AR-V7 dimers were detected primarily in the nucleus. Our data point to the possibility that AR-FL forms dimers in the cytoplasm upon androgen stimulation and translocates into the nucleus, in which AR-FL may interact with AR-V7 to form heterodimers. The differences in dimerization and luminescence kinetics further support our theory that for heterodimer formation, translocation of the AR-FL partner into the nucleus is necessary, and therefore, this luminescence kinetics is slower.
AR-V7 resides constitutively in the nucleus [16,18]. As stimulation with DHT induces AR-FL and AR-V7 proteins to form heterodimers, we further assessed the effect of androgen stimulation on AR-FL and AR-V7 subcellular localization by immunofluorescence (IF) staining in AR protein-null HEK-293 cells. Consistent with previous reports, AR-V7 was found exclusively in the nucleus, whereas AR-FL localized predominantly in the cytoplasm under androgen-deprived conditions. However, as early as 15 mins after androgen stimulation, AR-FL homodimers were found mainly in the nucleus (Figure 3). Concordantly, when measuring the luminescence of AR-FL/AR-V7, luminescent signals needed approximately 15 min to reach their maximum after the substrate was added (Figure 5). We can hypothesize that the AR-FL/AR-V7 interaction takes place in the nucleus and not the cytoplasm. Nevertheless, we could not confirm this result by bioluminescent imaging due to the technical limitations of our approach.
Cao et al. found that AR-V7 can co-occupy the promoter of the PSA gene with AR-FL [13]. Our theory that AR-FL and AR-V7 interact in the nucleus upon androgen stimulation considers the following possibilities: 1. AR-FL and AR-V7 dimers may reside together at the androgen response elements (AREs), and their close interaction produces the detected NanoBiT luminescent signals or 2. AR-FL and AR-V7 once in the nucleus form heterodimers to promote gene transcription.
To gain a better resolution of the time course of AR-FL/AR-FL homodimers and AR-FL/AR-V7 heterodimers, we measured luminescence after androgen stimulation in a 3 min interval.
As expected, the AR-FL/AR-FL homodimer showed a luminescence signal after 3 min that continuously increased until 24 min after androgen stimulation and then remained stable for the following measured time points. Because of the early detection of the luminescence signal, we suggest that AR-FL/AR-FL homodimerization starts in the cytoplasm and that the homodimers are transported into the nucleus. This finding is in accordance with the model presented by Feldman and Feldman [3]. However, others suggest that homodimerization of AR-FL/AR-FL starts in the nucleus [4]. A possible explanation for this discrepancy is that AR-FL/AR-FL homodimerization can start in the cytoplasm, but enrichment of the homodimers is observed in the nucleus. However, AR-FL/AR-V7 heterodimer formation may occur differently. Luminescence as a marker for dimerization increased slower and in a linear fashion until 15 min and then remained at a constant level. We suggest that within 15 min after stimulation, AR-FL translocates into the nucleus, in which it encounters AR-V7 homodimers to form AR-FL/AR-V7 heterodimers. The luminescence levels of AR-FL/AR-V7 are lower than those of AR-FL/AR-FL. This is in accordance with reduced fluorescence levels for AR-FL/AR-V7 constructs compared with AR-FL/AR-FL constructs [5].
However, we have to consider that we and others applied in vitro model systems, but AR-V7 in the clinical setting is a dynamic marker that can change according to treatment conditions [28] and can also heterodimerize with other AR-Vs [29]. Interestingly, our research group observed general cytoplasmic and granular cytoplasmic staining patterns for AR-V7 in immunohistochemical staining on a tissue microarray with 410 primary PCa specimens, i.e., patients were not yet treated with ADT. However, AR-V7 nuclear staining occurred only in 25 cases (6.2%). AR-V7 granular staining was unexpectedly associated with longer relapse-free survival (RFS), whereas staining of the cytoplasm was associated with shorter RFS. More importantly, the granular staining pattern was similar to that of GOLGB1 (synonymous: giantin), a major protein of the Golgi apparatus. The coinciding staining pattern suggests that AR-V7 is localized in the Golgi apparatus [30]. When looking carefully at the AR-V7 IF-staining pictures presented in this work, a granular fluorescent pattern could be distinguished around the nucleus, but due to the close proximity of the Golgi apparatus to the nucleus, it is difficult to determine the exact location. Considering the longer RFS associated with granular staining, we suggest that AR-V7 is not functionally active in these cases and may play a role in the protein degradation process in the Golgi apparatus. Li et al. described proteasomal degradation of AR-V7 in prostate cancer cells controlled by protein phosphatase 1 [31].
There are different possibilities for the constitutive expression of AR-V7 in the nucleus. At first, amino acids or their changes in the cryptic exon CE3 could be responsible for AR-V7 expression and localization. Chan et al. showed that K629A and R631A mutations in CE3 shifted AR-V7 expression from predominantly nuclear to a mixed nuclear/cytoplasmic pattern [18]. Nuclear import of AR-V7 is not mediated by the microtubule pathway but possibly by the importin α/β machinery [32]. Furthermore, Src family kinases have been identified as potential regulators of AR-V7 expression and AR-V7 localization [33]. Altogether, the regulation of AR-V7 still appears to be a complex process.
AR-V7 has been detected in many different cancer and normal cell lines and in normal tissues, such as the liver, spleen, testis, skeletal muscle, small intestine, adipose tissue, and cervix [34]. AR-V7 may regulate wound repair via tenascin c [35]. Cai and coworkers identified several transcriptional targets uniquely activated by AR-V7, e.g., ZNF32, FZD6, HDAC3, PHF21B, and SKP2 [19]. SKP2 plays an active role in the G1/S phase transition via p27Kip1 degradation in quiescent cells [36], and SKP2 protein expression is elevated in PCa [37]. ZNF32 promotes the self-renewal of colorectal cancer cells by regulating the LEPR-STAT3 signaling pathway [38]. FDR6, a member of the Wnt pathway, is expressed at the RNA level in normal prostate tissue, but its expression is elevated in PCa [39]. HDAC3 belongs to the commonly overexpressed genes in many solid tumors, including PCa [40]. PHF21B constitutively activates Wnt/β-catenin signaling, which promotes a PCa stem cell-like phenotype [41]. Furthermore, expression of AR-V7, but not AR-FL, was positively correlated with the cell cycle gene UBE2C in clinical CRPC specimens and in PCa cells following treatment with enzalutamide or abiraterone [42]. UBE2C is also a commonly overexpressed gene in many solid tumors, including PCa [40]. Altogether, AR-V7 can induce a specific transcriptional program of genes functioning mostly as oncogenes, which aggravate PCa. Therefore, AR-V7 may still receive special consideration as a future therapeutic target in CRPC beyond AR.
5. Conclusions
In summary, AR-FL is initially located as a monomer in the cytoplasm before DHT treatment and possibly as an AR-FL/AR-FL homodimer shortly after DHT treatment. The localization of AR-FL/AR-FL homodimers shifted into the nucleus within 15 min after DHT treatment. AR-V7/AR-V7 homodimers were constitutively located in the nucleus, and neither DHT nor enzalutamide affected the localization of AR-V7/AR-V7 or its status as a dimer. AR-FL/AR-V7 heterodimers form only after DHT stimulation. Our data indicate that AR-V7/AR-FL heterodimers form in the nucleus after translocation of AR-FL homodimers to the nucleus.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org: Table S1: List of plasmids used and cloned in this study; Table S2: Workflow of immunofluorescence detection.
Author Contributions
S.W., J.G., and H.T. designed the study and were responsible for the conceptualization. J.G., S.W., K.W., A.N., P.T., B.S., Z.W. and P. M. performed the experiments and microscopic work. S.W., J.G. and H.T. performed statistical analysis. J.G., S.W., P.T., and B.S. prepared the tables and figures. H.T., S.W., J.G., R.P., Z.C., MVC, and B.W. were responsible for the original draft preparation, wrote and edited the manuscript, R.P. and B.W. did the project supervision, HT and S.W. were responsible for funding acquisition. All authors reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
HT and ZC were supported in a D-A-CH project by the Deutsche Forschungsgemeinschaft/Fonds für wissenschaftliche Forschung, Österreich (TA 145/17-1 and I 4859). MVC was supported by a research grant of the Fondtion Cancer, Luxembourg (FC2020/01A). HT and SW were supported by the Wilhelm-Sander Stiftung (Förderungs Nr. 2015.171.1). ZW was supported by Deutsche Forschungsgemeinschaft CRC1181-Z02 – project 261193037. Microscopic detection of luminescent signals was enabled by an ANDOR IXON ULTRA EMCCD camera funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) —project 261193037. Confocal microscopy was enabled on a Leica SP5 laser scanning microscope funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) — project 52732026. The funding organizations were not involved in any aspects of study design, implementation, data analysis or interpretation.
Acknowledgments
We would like to thank PD Dr. Frédéric R. Santer/Department of Urology, Medical University of Innsbruck for kindly supplying the AR and AR-V7 plasmids and helpful discussion. The authors also acknowledge support by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg within the funding program Open Access Publishing.
Conflicts of Interest
Paul Muschler is employee of Promega GmbH. The other authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Gelmann, E.P. Molecular biology of the androgen receptor. J Clin Oncol 2002, 20, 3001–3015. [Google Scholar] [CrossRef] [PubMed]
- Trapman, J.; Cleutjens, K.B. Androgen-regulated gene expression in prostate cancer. Semin Cancer Biol 1997, 8, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat Rev Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- van Royen, M.E.; van Cappellen, W.A.; de Vos, C.; Houtsmuller, A.B.; Trapman, J. Stepwise androgen receptor dimerization. J Cell Sci 2012, 125, 1970–1979. [Google Scholar] [CrossRef]
- Xu, D.; Zhan, Y.; Qi, Y.; Cao, B.; Bai, S.; Xu, W.; Gambhir, S.S.; Lee, P.; Sartor, O.; Flemington, E.K. , et al. Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Res 2015, 75, 3663–3671. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L. , et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Azoitei, A.; Merseburger, A.S.; Godau, B.; Hoda, M.R.; Schmid, E.; Cronauer, M.V. C-terminally truncated constitutively active androgen receptor variants and their biologic and clinical significance in castration-resistant prostate cancer. J Steroid Biochem Mol Biol 2017, 166, 38–44. [Google Scholar] [CrossRef]
- Wach, S.; Taubert, H.; Cronauer, M. Role of androgen receptor splice variants, their clinical relevance and treatment options. World J Urol 2020, 38, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 2008, 68, 5469–5477. [Google Scholar] [CrossRef]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H. , et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest 2010, 120, 2715–2730. [Google Scholar] [CrossRef]
- Li, H.; Wang, Z.; Xiao, W.; Yan, L.; Guan, W.; Hu, Z.; Wu, L.; Huang, Q.; Wang, J.; Xu, H. , et al. Androgen-receptor splice variant-7-positive prostate cancer: a novel molecular subtype with markedly worse androgen-deprivation therapy outcomes in newly diagnosed patients. Mod Pathol 2018, 31, 198–208. [Google Scholar] [CrossRef]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G. , et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest 2019, 129, 192–208. [Google Scholar] [CrossRef]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Alvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O. , et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014, 5, 1646–1656. [Google Scholar] [CrossRef]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A 2010, 107, 16759–16765. [Google Scholar] [CrossRef]
- Djusberg, E.; Jernberg, E.; Thysell, E.; Golovleva, I.; Lundberg, P.; Crnalic, S.; Widmark, A.; Bergh, A.; Brattsand, M.; Wikstrom, P. High levels of the AR-V7 Splice Variant and Co-Amplification of the Golgi Protein Coding YIPF6 in AR Amplified Prostate Cancer Bone Metastases. Prostate 2017, 77, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B. , et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Hornberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One 2011, 6, e19059. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Li, Y.; Dehm, S.M. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem 2012, 287, 19736–19749. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Tsai, Y.H.; Wang, P.; Wang, J.; Li, D.; Fan, H.; Zhao, Y.; Bareja, R.; Lu, R.; Wilson, E.M. , et al. ZFX Mediates Non-canonical Oncogenic Functions of the Androgen Receptor Splice Variant 7 in Castrate-Resistant Prostate Cancer. Mol Cell 2018, 72, 341–354 e346. [Google Scholar] [CrossRef] [PubMed]
- Ozgun, F.; Kaya, Z.; Morova, T.; Geverts, B.; Abraham, T.E.; Houtsmuller, A.B.; van Royen, M.E.; Lack, N.A. DNA binding alters ARv7 dimer interactions. J Cell Sci 2021, 134. [Google Scholar] [CrossRef]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A. , et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem Biol 2016, 11, 400–408. [Google Scholar] [CrossRef]
- Goda, K.; Takahashi, T.; Suzuki, H. Combining Fluorescence and Bioluminescence Microscopy to Study the Series of Events from Cellular Signal Transduction to Gene Expression. Curr Protoc Cell Biol 2017, 77, 4 35 31–34 35 16. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.E.; Penning, T.M. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab 2010, 21, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, A.; Chen, X.; Herpel, E.; Merseburger, A.S.; Kristiansen, G.; Bernemann, C.; Hohenfellner, M.; Cronauer, M.V.; Duensing, S. Antibody selection influences the detection of AR-V7 in primary prostate cancer. Cancer Treat Res Commun 2020, 24, 100186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, H.; Liang, Z.; Mao, Y.; Wang, C.; Xie, L. The characteristics of androgen receptor splice variant 7 in the treatment of hormonal sensitive prostate cancer: a systematic review and meta-analysis. Cancer Cell Int 2020, 20, 149. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.J.; Hu, Q.; Li, S.Y.; Mao, W.P.; Xu, B.; Chen, M. The Role of Androgen Receptor Splicing Variant 7 in Predicting the Prognosis of Metastatic Castration-Resistant Prostate Cancer: Systematic Review and Meta-Analysis. Technol Cancer Res Treat 2021, 20, 15330338211035260. [Google Scholar] [CrossRef] [PubMed]
- Streicher, W.; Zengerling, F.; Laschak, M.; Weidemann, W.; Hopfner, M.; Schrader, A.J.; Jentzmik, F.; Schrader, M.; Cronauer, M.V. AR-Q640X, a model to study the effects of constitutively active C-terminally truncated AR variants in prostate cancer cells. World J Urol 2012, 30, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.; Lu, C.; Chen, Y.; Paller, C.J.; Carducci, M.A.; Eisenberger, M.A.; Luo, J.; Antonarakis, E.S. Serial blood-based analysis of AR-V7 in men with advanced prostate cancer. Ann Oncol 2015, 26, 1859–1865. [Google Scholar] [CrossRef]
- Zhan, Y.; Zhang, G.; Wang, X.; Qi, Y.; Bai, S.; Li, D.; Ma, T.; Sartor, O.; Flemington, E.K.; Zhang, H. , et al. Interplay between Cytoplasmic and Nuclear Androgen Receptor Splice Variants Mediates Castration Resistance. Mol Cancer Res 2017, 15, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Konig, P.; Eckstein, M.; Jung, R.; Abdulrahman, A.; Guzman, J.; Weigelt, K.; Serrero, G.; Hayashi, J.; Geppert, C.; Stohr, R. , et al. Expression of AR-V7 (Androgen Receptor Variant 7) Protein in Granular Cytoplasmic Structures Is an Independent Prognostic Factor in Prostate Cancer Patients. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Li, Y.; Xie, N.; Gleave, M.E.; Rennie, P.S.; Dong, X. AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget 2015, 6, 33743–33754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, X.; Li, J.; Ledet, E.; Alvarez, X.; Qi, Y.; Fu, X.; Sartor, O.; Dong, Y.; Zhang, H. Androgen receptor splice variants circumvent AR blockade by microtubule-targeting agents. Oncotarget 2015, 6, 23358–23371. [Google Scholar] [CrossRef] [PubMed]
- Szafran, A.T.; Stephan, C.; Bolt, M.; Mancini, M.G.; Marcelli, M.; Mancini, M.A. High-Content Screening Identifies Src Family Kinases as Potential Regulators of AR-V7 Expression and Androgen-Independent Cell Growth. Prostate 2017, 77, 82–93. [Google Scholar] [CrossRef]
- Hu, D.G.; Hickey, T.E.; Irvine, C.; Wijayakumara, D.D.; Lu, L.; Tilley, W.D.; Selth, L.A.; Mackenzie, P.I. Identification of androgen receptor splice variant transcripts in breast cancer cell lines and human tissues. Horm Cancer 2014, 5, 61–71. [Google Scholar] [CrossRef]
- Thomas, R.; Jerome, J.M.; Dang, T.D.; Souto, E.P.; Mallam, J.N.; Rowley, D.R. Androgen receptor variant-7 regulation by tenascin-c induced src activation. Cell Commun Signal 2022, 20, 119. [Google Scholar] [CrossRef] [PubMed]
- Sutterluty, H.; Chatelain, E.; Marti, A.; Wirbelauer, C.; Senften, M.; Muller, U.; Krek, W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol 1999, 1, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Ayala, G.; De Marzo, A.; Tian, W.; Frolov, A.; Wheeler, T.M.; Thompson, T.C.; Harper, J.W. Elevated Skp2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p27 and PTEN and with reduced recurrence-free survival. Clin Cancer Res 2002, 8, 3419–3426. [Google Scholar]
- Li, J.; Li, X.; Lan, L.; Sun, L.; Li, X.; Li, Y.; Tian, Y.; Zhang, T.; Zhou, Y.; Mo, C. , et al. ZNF32 promotes the self-renewal of colorectal cancer cells by regulating the LEPR-STAT3 signaling pathway. Cell Death Dis 2022, 13, 108. [Google Scholar] [CrossRef]
- Wissmann, C.; Wild, P.J.; Kaiser, S.; Roepcke, S.; Stoehr, R.; Woenckhaus, M.; Kristiansen, G.; Hsieh, J.C.; Hofstaedter, F.; Hartmann, A. , et al. WIF1, a component of the Wnt pathway, is down-regulated in prostate, breast, lung, and bladder cancer. J Pathol 2003, 201, 204–212. [Google Scholar] [CrossRef]
- Pilarsky, C.; Wenzig, M.; Specht, T.; Saeger, H.D.; Grutzmann, R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia 2004, 6, 744–750. [Google Scholar] [CrossRef]
- Li, Q.; Ye, L.; Guo, W.; Wang, M.; Huang, S.; Peng, X. PHF21B overexpression promotes cancer stem cell-like traits in prostate cancer cells by activating the Wnt/beta-catenin signaling pathway. J Exp Clin Cancer Res 2017, 36, 85. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E. , et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Principle of the NanoBiT Protein:Protein Interaction system. Each protein of interest is fused at either N or C terminus with LgBiT and SmBiT (A) This creates up to eight different expression constructs. Here the androgen receptor and the androgen receptor splice variant AR-V7 are the proteins of interest. (The figure is copied with permission from the Promega Corporation: NanoBiT® Protein:Protein Interaction System Technical Manual).
Figure 1.
Principle of the NanoBiT Protein:Protein Interaction system. Each protein of interest is fused at either N or C terminus with LgBiT and SmBiT (A) This creates up to eight different expression constructs. Here the androgen receptor and the androgen receptor splice variant AR-V7 are the proteins of interest. (The figure is copied with permission from the Promega Corporation: NanoBiT® Protein:Protein Interaction System Technical Manual).
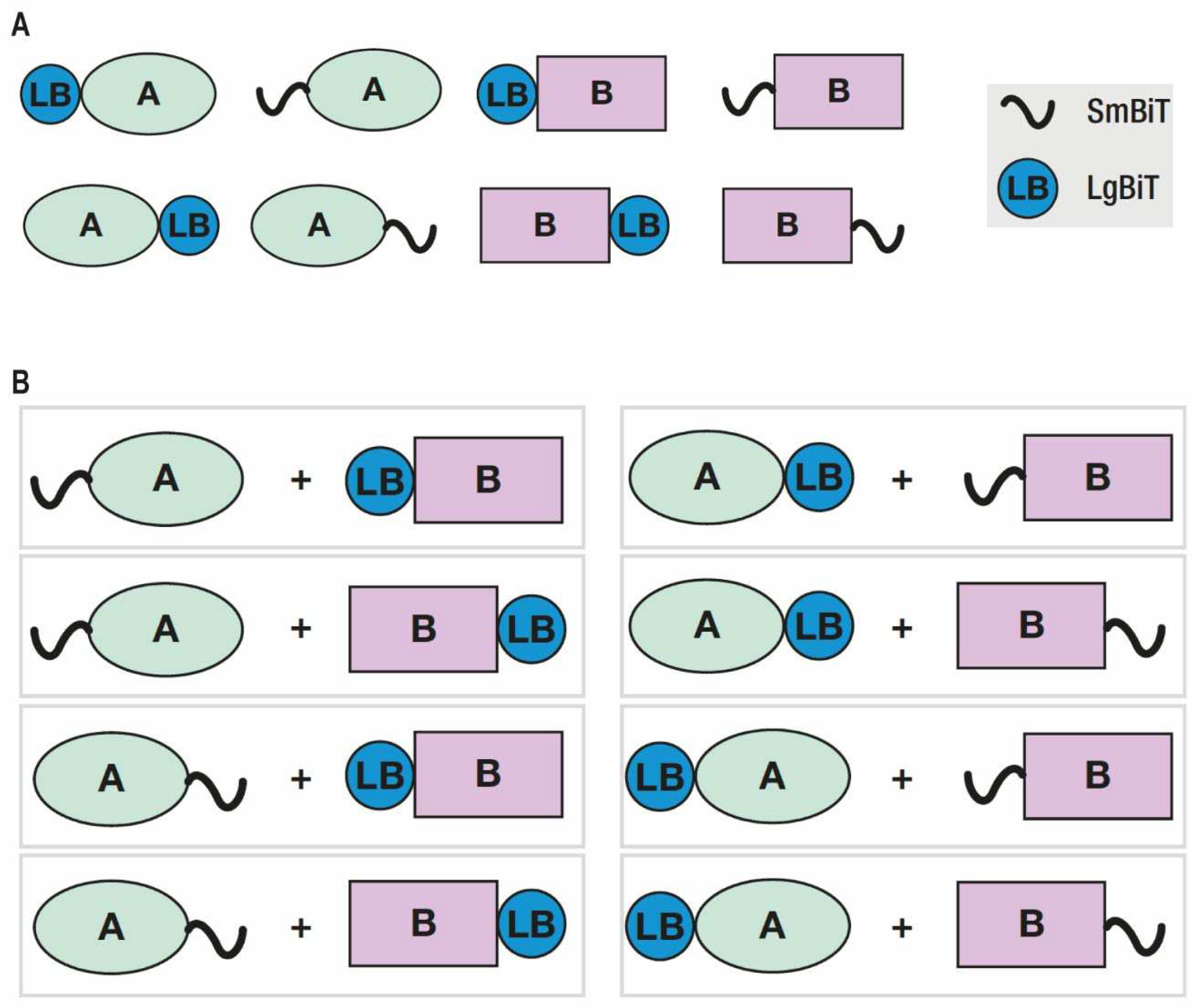
Figure 2.
Nano-Glo® Live Cell assay. (A) HEK-293 cells were transfected with all possible combinations for homo-and-heterodimers (Ho and He) encoding the LargeBiT (LgBiT) and SmallBiT (SmBiT) subunits in the N-and-C terminal of the androgen receptor full length (AR) and androgen receptor splice variant 7 (AR-V7) proteins. (B-C) Luminescence measurements in HEK-293 cells transfected with AR-FL and AR-V7 homo-and-heterodimers after treatment with 1nM DHT or 10 µM enzalutamide. Luminescence in non-stimulated HEK-293 cells were set as base line. Date represents the mean ± SEM (n=3).
Figure 2.
Nano-Glo® Live Cell assay. (A) HEK-293 cells were transfected with all possible combinations for homo-and-heterodimers (Ho and He) encoding the LargeBiT (LgBiT) and SmallBiT (SmBiT) subunits in the N-and-C terminal of the androgen receptor full length (AR) and androgen receptor splice variant 7 (AR-V7) proteins. (B-C) Luminescence measurements in HEK-293 cells transfected with AR-FL and AR-V7 homo-and-heterodimers after treatment with 1nM DHT or 10 µM enzalutamide. Luminescence in non-stimulated HEK-293 cells were set as base line. Date represents the mean ± SEM (n=3).
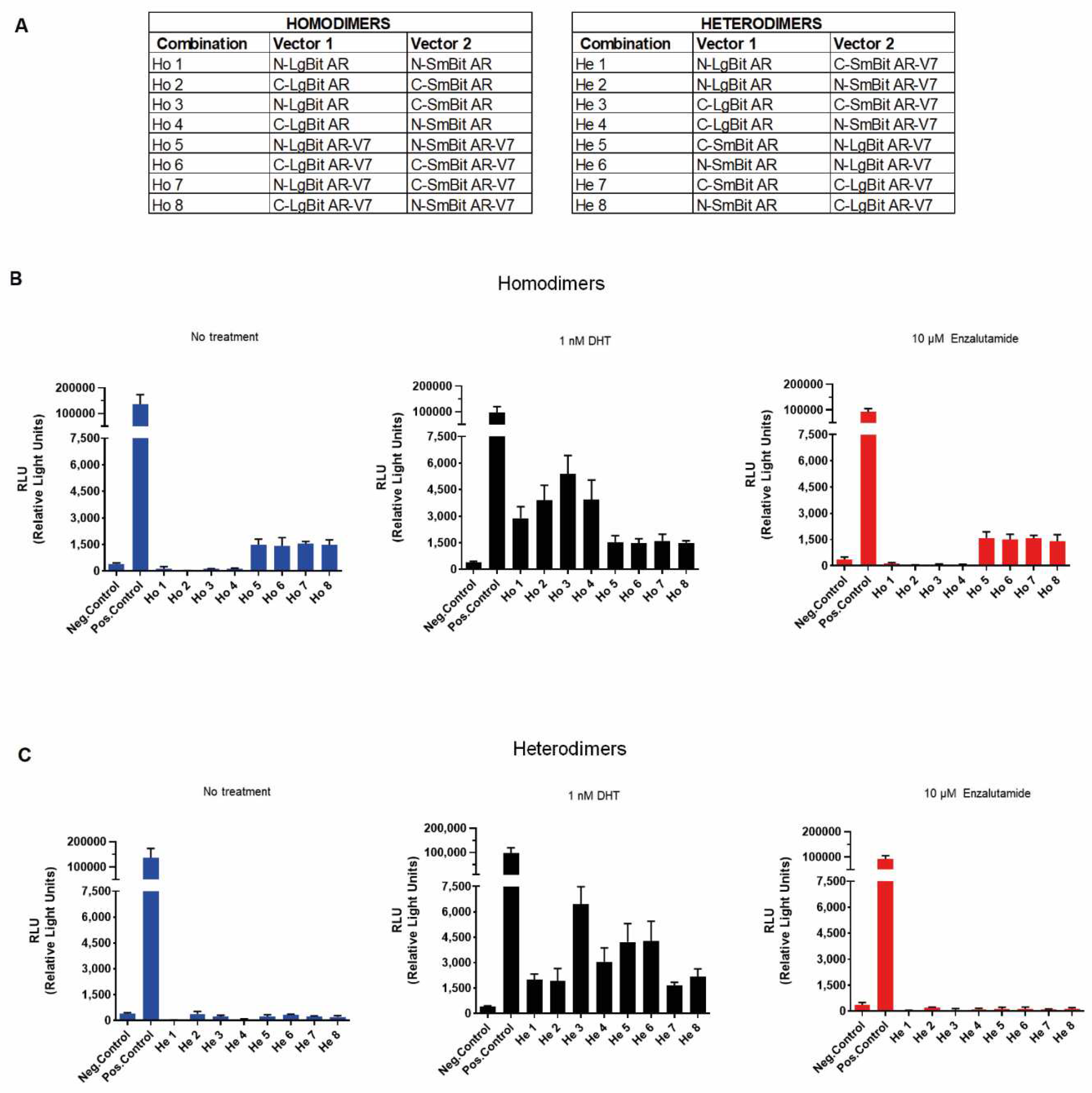
Figure 3.
Immunofluorescence staining of NanoLuc® luciferase. HEK-293 cells transfected with the indicated combinations of plasmid vectors. Immunofluorescence (IF) staining using NanoLuc® and Alexa Fluor™ 488 antibodies. IF staining showing subcellular localization of AR-FL and AR-V7 proteins before and after androgen stimulation. The indicated combination constructs were transfected into HEK-293 cells, and IF staining was conducted 48h after transfection. Cells were stimulated with 1 nM DHT for 0, 15, 30 and 60 min. DAPI, nuclear staining. Only merged pictures shown.
Figure 3.
Immunofluorescence staining of NanoLuc® luciferase. HEK-293 cells transfected with the indicated combinations of plasmid vectors. Immunofluorescence (IF) staining using NanoLuc® and Alexa Fluor™ 488 antibodies. IF staining showing subcellular localization of AR-FL and AR-V7 proteins before and after androgen stimulation. The indicated combination constructs were transfected into HEK-293 cells, and IF staining was conducted 48h after transfection. Cells were stimulated with 1 nM DHT for 0, 15, 30 and 60 min. DAPI, nuclear staining. Only merged pictures shown.
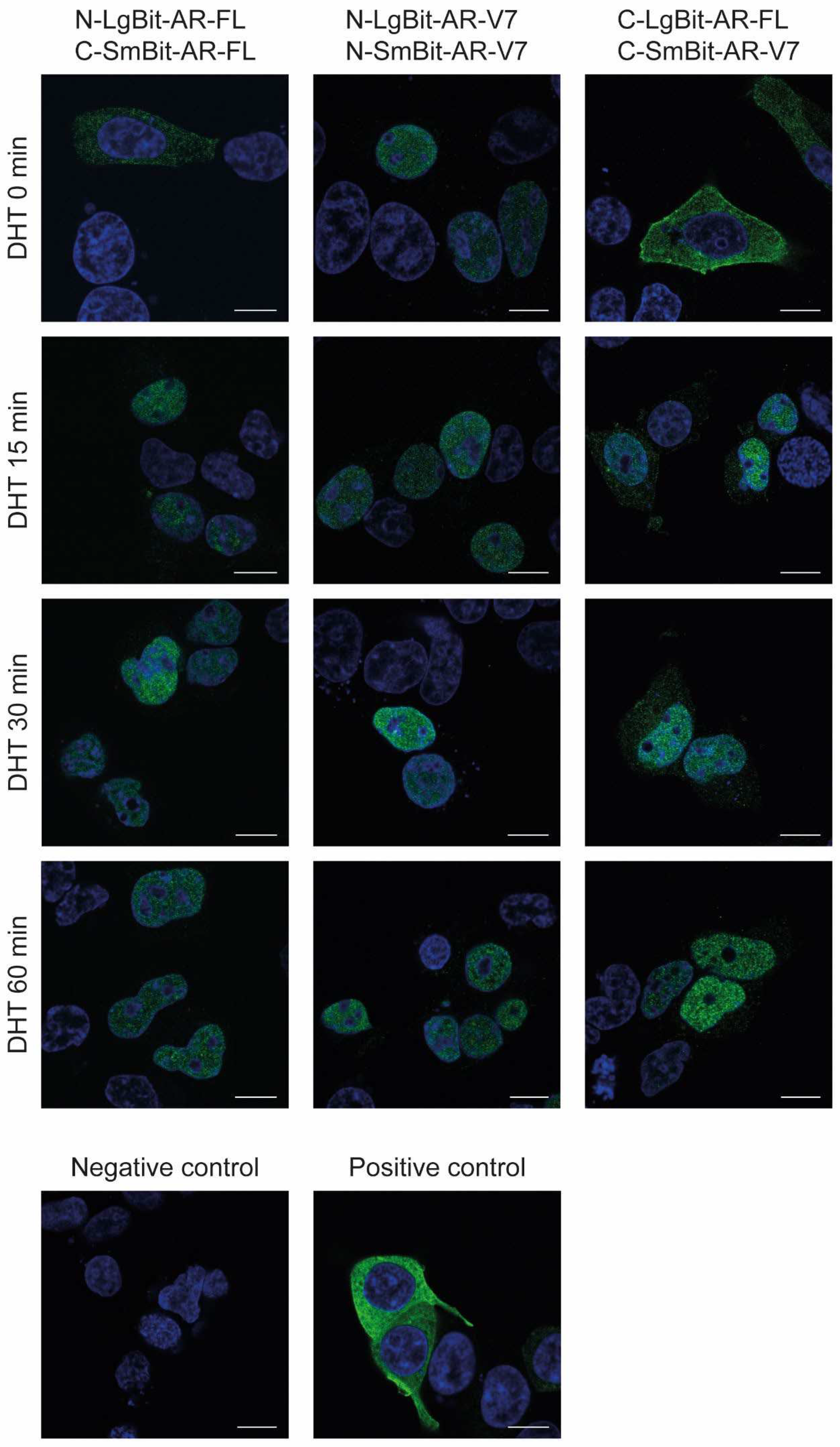
Figure 4.
Bioluminescence imaging. (A-B) HEK-293 cells transfected with homodimer 3 and heterodimer 3 plasmid combinations. Representative images of time course changes in luminescence every 10 min for 90 min after 1 nM DHT stimulation.
Figure 4.
Bioluminescence imaging. (A-B) HEK-293 cells transfected with homodimer 3 and heterodimer 3 plasmid combinations. Representative images of time course changes in luminescence every 10 min for 90 min after 1 nM DHT stimulation.
Figure 5.
Formation of AR-FL homodimers and AR-FL/AR-V7 heterodimers after DHT stimulation. (A) Luminescence measurement AR-FL/AR-FL (red) and AR-FL/AR-V7 (black); (B) Kinetics of luminescence development. Changes in luminescence are displayed in a sliding 3 min window.
Figure 5.
Formation of AR-FL homodimers and AR-FL/AR-V7 heterodimers after DHT stimulation. (A) Luminescence measurement AR-FL/AR-FL (red) and AR-FL/AR-V7 (black); (B) Kinetics of luminescence development. Changes in luminescence are displayed in a sliding 3 min window.
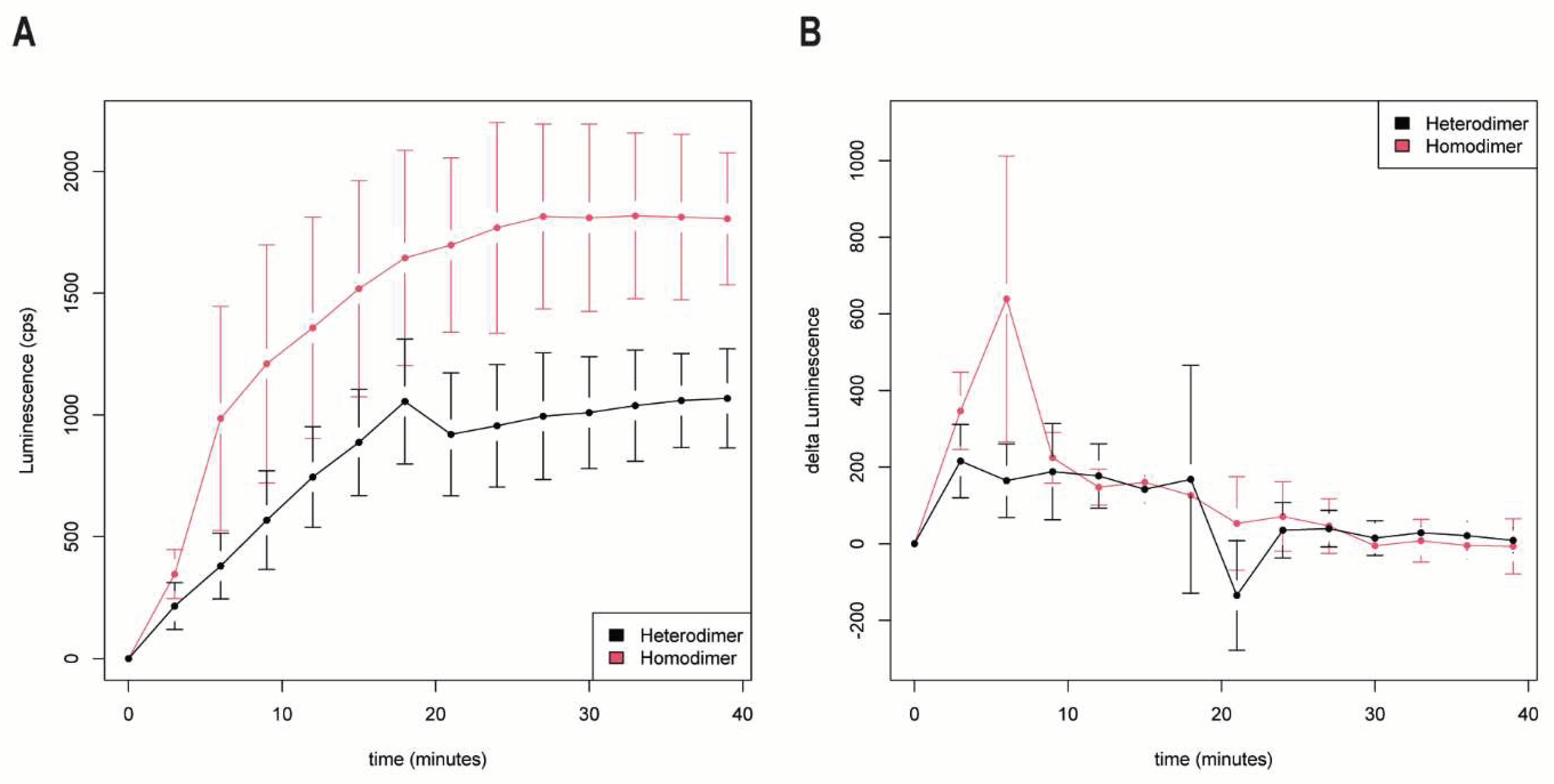

Table 1.
PCR primers.
| Cloning Primer | MCS Restriction site | Sequence |
|---|---|---|
| N-Terminal-AR/ARV7-FW | Xhol | 5′-CTCGAGATGGAAGTGCAGTTAGGGCTGG-3′ |
| N-Terminal-AR-RV | Bglll | 5′-AGATCTGCTTCACTGGGTGTGGAAATAGATGG-3′ |
| N-Terminal-AR-V7-RV | Bglll | 5′-AGATCTTCTTCAGGGTCTGGTCATTTTGAGAT-3′ |
| C-Terminal-AR/ARV7-FW | Bglll | 5′-AGATCTATGGAAGTGCAGTTAGGGCTGG-3′ |
| C-Terminal-AR-RV | Xhol | 5′-CTCGAGCCCTGGGTGTGGAAATAGATGGGCTTG-3′ |
| C-Terminal-AR-V7-RV | Xhol | 5′CTCGAGCCGGGTCTGGTCATTTTGAGATGCTTGCA-3′ |
All primers were obtained from Biomers.net (Ulm, Germany). FW is the forward primer and RV the reverse primer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



