
Submitted:
12 April 2023
Posted:
13 April 2023
You are already at the latest version
Abstract
Plant extracts have been evaluated to determine their bioactivities and their potential use in wound healing. In this study, a conductive composite material, based on graphene oxide (GO), nanocellulose (CNF) and tannins (TA) from pine bark, reduced using polydopamine (PDA), was developed for wound dressing. The amount of CNF and TA was varied in the composite material and a complete characterization including SEM, FTIR, XRD, XPS, TGA was performed. Also, the conductivity, mechanical properties, cytotoxicity, and in vitro wound healing of the materials were evaluated. The results showed a successful physical interaction between CNF, TA and GO. In-creasing the CNF amount in the composite reduced the thermal properties, surface charge and conductivity of the material, but its strength, cytotoxicity and wound healing performance were improved. The TA incorporation slightly reduced the cell viability and migration, which may be associated with the doses used and the chemical composition of the extracts. However, the in vitro obtained results demonstrated that these composite materials can be suitable for wound healing.
Keywords:
graphene oxide
; nanocellulose
; reduced graphene oxide
; polydopamine
; wound healing
1. Introduction
Skin is the largest body organ and comprises approximately one-sixth of the body mass with the primary role of protecting the inner organs from the external environment [1] Extreme loss of skin function and structure due to injury and illness will result in substantial physiological imbalance and may ultimately lead to major disability or even death [2]. To remediate diseased or damaged tissue, surgical intervention can be accomplished by anatomically localizing the wound borders, thereby closing the wound, and reducing infection or contamination from the external environment [3]. For wounds that are not large-surface or deep, a combination of new strategies, using advanced biomaterials science, cells, and biochemical cues (proteins/growth factors (GFs)), are currently available [4].
In particular, with the development of advanced biomaterials many forms of dressings, such as nanofibers, membranes, foams, hydrocolloids and, hydrogels have been created [5]. Ideally, wound dressing should promote healing and prevent contamination. To accelerate healing, the dressings should provide a moist wound environment, remove excess wound exudates, provide thermal insulation, allow gaseous exchange, be impermeable to microorganisms, allow current circulation and have low adherence to the wound site [6].
A wound electrical signal at the wound site regulates cell behaviors, promoting wound healing [7]. Previous studies have reported 4 μA/cm2 wound current measurement immediately after trauma, which gradually increased to 10 μA/cm2 after 60 min. These wound potentials can promote wound regeneration by inducing cell behaviors through electrical stimulation [8]. Unfortunately, most wound dressings are non-electroactive and are therefore unable to respond to physiological electrical signals at wound sites during healing.
Nevertheless, the incorporation of electroactive materials such as reduced graphene oxide (rGO) into the wound dressing could be accelerated, distributing endogenous electrical stimulation more effectively to tissues in the wound [9]. Carbon materials are difficult to disperse in the matrix, restricting the functionality of materials, and they also lack cell affinity [10]. Both problems can be overcome using a biocompatible matrix that supports the electroactive material.
Cellulose nanofibers (CNF) are made from wood-derived fiber (pulp) that has been micro-refined to the nano level. One of the most important characteristics of nanocellulose is that it can be colloidally stable in an aqueous solution for a wide range of salt concentrations and pH [11]. Moreover, it can assemble with other nanoparticles in a colloidal suspension in order to be transformed into a gel that is utilized to create multifunctional composites [12]. Nevertheless, CNF are poor electrochemically and unstable, which can be overcome by adding rGO to develop CNF-rGO composites. The interaction in the material is formed through a hydrogen bond between the unreacted oxygen-containing groups existing on rGO surface and CNF hydrophilic polymer matrix, endowing the composite with superior properties through a conjugate structure [13].
rGO has a multipotential to produce advanced materials for tissue engineering that require electrical signal transmission to strengthen cell–cell interactions [14]. rGO is produced from graphene oxide (GO), a graphene derivative carrying functional group [12], which can interact with CNF and simultaneously be partially reduced by Dopamine (DA) [15]. DA is generally considered to be a type of cell-compatible molecule, which can be strongly adsorbed into a majority of substrate materials through covalent conjugation or physical interactions [16]. In a weak alkaline pH, DA will simultaneously undergo self-polymerization to generate a hydrophilic polydopamine (PDA) coating on the surfaces of rGO to enhance its water dispersity [15]. Simultaneously, after the self-polymerization, PDA can be anchored on the GO nanosheets through Schiff or Michael‘s addition reaction, and GO can be partially reduced [17]. Thus, the obtained rGO not only presents good dispersity and good electrical conductivity but also possesses excellent photothermal, antibacterial properties and bioactivity from PDA, which contribute to producing a bioactive wound dressing.
Condensed tannins also reduce GO similarly to DA [18] and elicit several bioactivities [19], which can be transferred to a dressing. Phenolic groups in tannins also aid anchoring to the rGO surface through π–π interactions [20].
Thus, this study developed CNF/rGO composites loaded with TA from pine bark, reduced with PDA, to be used as a wound dressing. To this purpose, the effect of different CNF concentrations (5, 15, 25, 50 % w/w) on the CNF/rGO was evaluated and the materials were analyzed in terms of surface morphology, spectroscopy characterization, electric conductivity, swelling, traction and deformation, cell biocompatibility and in vitro wound healing.
2. Materials and Methods
2.1. Production of Cellulose Nanofibrils (CNF)
The bleached Kraft hardwood pulp (80% Eucalyptus globulus and 20% Eucalyptus nitens) used to produce CNFs was provided by the company CMPC pulp S.A. (Santa Fe Mill, Chile). The pulp was rehydrated, filtered, and pelleted according to ISO 5263. The chemical characterization of cellulosic fibers was performed through acid hydrolysis based on the method described by Zeng et al. [21] and Andrade et al. [22].
The enzymatic hydrolysis was performed using the commercial enzyme complex of cellulases Quimizime B, which had a higher content of endoglucanases and an activity of 7.75 U/mL enzyme. Enzyme activity was determined according to the methodology of Ghose [23].
2.2. Mechanical-Enzymatic Pretreatment
Thirty grams of the oven-dried pulp of Kraft bleached eucalyptus pulp (BHKP) at 10% (w/w) were refined in a PFI mill at 4000 revolutions to increase the accessibility of the enzyme in the substrate. Then, 0.05% of Quimizime B enzyme was added to the dry weight of the pulp. The enzymatic pretreatment conditions were: 60 min of reaction, temperature of 48 °C, 5% consistency, pH 5 (adjusted with 0.1M HCl), and constant stirring at 800 rpm with a Stirrer Type BS. After the reaction time, the enzyme was denatured at 80 °C for 20 min. Next, the pulp was refined at 46,000 revolutions in a PFI mill at 10% consistency. Then, 0.05% of Quimizime B enzyme was added in relation to the dry weight of the pulp. The enzymatic pretreatment conditions were: 60 min of reaction, temperature of 48 °C, 5% consistency, pH 5 (adjusted with 0.1M HCl), and constant stirring at 800 rpm with a Stirrer Type BS. After the reaction time, the enzyme was denatured at 80◦C for 20 min. Next, the pulp was refined at a pressure of 700 bar using Gea Niro Soavi Panda Plus 2000 homogenizing equipment (Dusseldorf, Germany) provided with an S-type impact head. The fiber suspension was passed 15 times through the equipment to produce the CNFs; the final consistency was between 0.5-1%.
2.3. Synthesis of Graphene Oxide (GO)
GO was synthesized from natural graphite powder using the modified Hummers method [24]. Briefly, H2SO4 (270 mL) and H2PO3 (30 mL) were mixed in a beaker under stirring in an ice bath. Next, 2.25 g of graphite powder and 13.5 g of KMnO4 were slowly added. The temperature was controlled not to exceed 45 °C, and the mixture was stirred for 1 h. The reaction was then stopped with H2O2 (60 mL) until a color change from brown to dark green was observed. The final mixture was centrifuged (Rotina 380R; Hettich, Germany) for 20 min at 5000 rpm, and the supernatant was removed. Subsequently, the oxidation product was continuously washed and centrifuged with ethanol (100 mL) and HCl (20 mL) to remove the metal ions (using AgNO3 as an indicator). The mixture was then washed four times with Milli-Q® water and the supernatant was removed in each wash. The final mixture had a pH value of around 4. Next, the product was dialyzed in dialysis membranes (12000 MWCO) for three days to remove any remaining impurities and lyophilized for 72 h (Labconco freeze-dry system; Germany) to obtain a solid dispersion of graphite oxide (GpO). Finally, the GpO was exfoliated for 30 min to obtain a graphene oxide suspension, which was lyophilized and kept in a sealed flask for further use.
2.4. Pinus radiata Bark Extract Production
Pinus radiata bark extracts were produced through a pilot-scale extraction process, as described by Bocalandro et al. [25]. For this purpose, a reactor volume of 4 m3 and a vapor heating system composed of a shell and a tube heat exchanger with 6 m2 heat transference area were used. In addition, a recirculation circuit for the extracted solution was implemented. Briefly, the Pinus radiata bark was ground with a double-knife mill to an average size lower than 20 mm. Then, the bark was dried at room temperature to a humidity of 24.5% (dry weight), and 100 kg (dry weight) of bark was soaked in an ethanol/water solution at a 1:20 ratio (w/v) for 120 min at 120°C. Subsequently, the ethanol was evaporated in a vacuum (absolute pressure 0.05 bar) at room temperature. Thus, the water-insoluble particulate material after decanting and the water-soluble polyphenol fraction were obtained. Finally, the water-soluble polyphenols were lyophilized at room temperature and the obtained extracts were stored in sealed amber glass containers for further analysis.
2.5. Synthesis of Reduced Graphene Oxide/Nanocellulose (rGO/CNF) and Reduced Graphene Oxide/Nanocellulose/Tannin (rGO/CNF/TA) Composites
The composite material was synthesized by dissolving 100 mg of GO in 20 mL of MilliQ® water, stirring at 250 rpm for 15 min at room temperature. After the GO dissolution, it was put in an ultrasonic bath (Digital Ultrasonic cleaner model CD 4820, 42 kHz, 160 W, Shenzhen Codyson Electrical Co. Ltd., China) with ice for 30 min. Twenty-five mL of CNF were centrifuged to 1500 rpm for 10 min to 5 °C, eliminating the supernatant. Subsequently, in a buffer Tris solution (200 mL MilliQ® water, 24.05 mg Tris), 100 mg of DA were added plus the GO solution previously prepared and the CNF, in different amounts (5, 15, 25, 50 mg), with stirring to 250 rpm and adjusting to 8.5 pH. The mix was left to react for 24 h at 60 °C and 750 rpm. The samples were cooled to 30°C and centrifuged at 11000 rpm for 30 min to 15 °C, eliminating the supernatant. Next, they were washed twice with MilliQ® water at 11000 rpm for 15 min at 15 °C. The resulting samples were sonicated for 10 min and deposited in a Petri dish to dry in a vacuum oven (Huanghua Faithful Instrument Co. Ltd., China) at 45 °C. The samples were named rGO/CNF5, rGO/CNF15, rGO/CNF25, and rGO/CNF50.
In the case of the samples with bark tannin, the tannin was incorporated after the washing step in two proportions (12.5 and 25 mg), the mix being sonicated for 10 min and then dried as previously described. The samples were named rGO/CNF25/TA5, rGO/CNF25/TA10.
2.6. Physicochemical Characterization of rGO/CNF and rGO/CNF/TA Composites
The morphology and physicochemical aspects of rGO/CNF and rGO/CNF/TA composites were studied through scanning electron microscopy (SEM), Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), and by examining the surface charge for dynamic light scattering (DLS). A detailed characterization of the materials is included in the Supplementary Materials (SM).
2.7. Conductivity Measurements
The resistance values were measured with an LF meter 4192A (Keysight, California, USA). The resistivity could be calculated using the resistance by the following formula: ρ = RS/L, where R is the resistance of the sample, and S and L represent the cross-sectional area and length of the sample, respectively [26]. Thus, the conductivity (σ) was calculated through σ = 1/ρ.
2.8. Swelling Behavior of rGO/CNF and rGO/CNF/TA Composites
The swelling ratio (SR) of the materials was determined by placing samples in contact with water and phosphate-buffered saline (PBS, pH=7.4) at room temperature for 5, 15, 30, 60, 90, 120 and 240 minutes. First, the samples were dried at 105°C for 24 h and then the dry samples were cut (1.0 cm2), and 10 mL of liquid medium were dripped onto the surface of the samples. Each time, the excess liquid was eliminated, and the SR of the rGO/CNF and rGO/CNF/TA composites was determined using Equation (1):
where Wdry is the dry sample weight, and Wwet is the wet sample weight (after contact with the liquid medium).
2.9. Mechanical Properties
The mechanical properties of the rGO/CNF and rGO/CNF/TA composites were measured using a Universal Testing Machine (Shimadzu EZ-XS, Japan) equipped with a 20 N load cell at a temperature of 20 °C and relative humidity of 50%. All samples were cut following the shape template of 13 mm width, 19 mm length and 0.18 thickness. The samples were held between two clamps and pulled by the top clamp at 0.1 mm/s. The elongation and breaking force were measured when the material tore apart. The elongation at break, tensile strength and elastic modulus were calculated using Equations (2), (3) and (4):
2.10. Cytotoxicity Assay
Cytotoxicity was determined using the 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide (MTT) assay. Human dermal fibroblast cells were used to evaluate the in vitro cytotoxicity of all synthesized materials.
These experiments were conducted using a cell density of 104 cells/mL. First, 1 mL of DMEM medium was added to 10 mg of material and also to the individual materials (rGO, and TA) to promote full contact. After 24 h of incubation at 37 °C, the supernatant was recovered and mixed with 5% (v/v) fetal bovine serum (FBS), 1% (v/v) antibiotics (100 units/mL of penicillin and 100 units/mL of streptomycin). The supernatant from each sample was added to the cells and incubated for 48 h at 37 °C under humidified air with 5% (v/v) CO2. At the end of the incubations, the supernatants were removed, and the cells were washed with PBS, pH 7.4. Then, 100 μL of fresh DMEM medium was added to the cells and 5 mg/mL MTT solution was added for the determination of cell viability. The plates were incubated for 4 h at 37 °C with CO2, then 25 μL of the medium were removed and 50 μL of dimethyl sulfoxide (DMSO) were added to the wells. After 10 min, the supernatant was removed by aspiration, and the formazan crystals were dissolved in DMSO (100 μL per well), followed by shaking for 5 min. The absorbance was determined using a microplate reader (Biotek synergy 2, Algilent, CA, USA) at a wavelength of 540 nm. The cell viability (%), relative to control cells, was calculated from Atest/(Acontrol)×100 %, where Atest and Acontrol are the absorbance values of the wells (with the material) and control wells (without the material), respectively. DMEM medium was used as a positive control.
2.11. In Vitro Wound Healing Assay (Scratch Test)
An in vitro wound healing assay was performed according to previously described experimental procedures [27], with slight modifications. Briefly, human dermal fibroblast cells (50.000 cells/well) were seeded into a 24-well plate and incubated at 37 °C for 48 h in a humidified atmosphere with 5% CO2. Subsequently, a vertical scratch was manually created in the middle of the human dermal fibroblast monolayer, using a 200 µL sterile pipet tip. Then, each material was fixed on CellCrown 24 inserts (Corning Incorporated, PA, USA) and placed on the 24-well plate without touching the surface. The wound closure rate and the cell migration were monitored over time (0, 12, 24, 36, 42, and 48 h) using a light microscope (MOTIC AE31, Richmond, Canada). Finally, the images were analyzed using ImageJ® software. The wound closure rates were calculated according to Equation (5):
where A0 is the initial wound area and At is the wound area after each time interval.
2.12. Statistical Analysis
The spectra obtained in the chemical characterization of the rGO/CNF and rGO/CNF/TA composites were acquired in triplicate and processed using OriginPro8.5® software. All experiments were carried out in triplicate, and the results are expressed as the mean ± standard deviation value. The studies of means were performed using multifactorial analysis of variance (ANOVA), whose accepted significance was p-value ≤ 0.05, and an analysis of multiple ranges (Duncan test, 95% confidence) in Statgraphics Centurion XVII® software. The mean values and the error bars are reported in each figure.
3. Results and Discussion
3.1. Morphological Characterization of rGO/CNF and rGO/CNF/TA Composites
The raw and the composite materials formulated are presented in Figure 1. After the reduction with DA, rGO (Figure 1c) presented a homogeneous aspect, black in color, shapeable and easy to mold, different to GO (Figure 1b). After the addition of CNF (Figure 1a), no visual changes were observed (comparing Figure 1c vs 1d) or with TA addition (comparing Figure 1c vs 1d vs 1e) in the micromorphology.
Figure 1.
Images of a) CNF, b) GO, c) rGO, d) rGO/CNF25, e) rGO/CNF25/TA10.

The morphology of the samples was characterized by scanning electron microscopy (SEM) measurements. As shown in SM, Figure S1, the GO presented an unordered sheets distribution exhibiting a wrinkled structure which was due to the freeze-drying process during which the stacked GO layers were further separated from each other, promoting a significant increase in the specific surface area. After the conformation of the composite, the GO wrinkled structure disappeared and the surface presented an increased number of fibers, which is concordant with the amount of CNF added (Figure S1b–e). The TA inclusion softened the surface even more, making it more homogeneous, coating and connecting with CNF and rGO. A structural comparison with other composite materials based on rGO/CNF led to a similar conformation: the GO wrinkles softness with the CNF addition [28,29].
3.2. Spectroscopical Characterization of rGO/CNF and rGO/CNF/TA Composites
The FTIR GO spectrum (Figure 2a) shows a characteristic band at 1635 cm−1, which arises from C=C stretching vibration of the sp2 carbon skeletal network (from unoxidized sp2 C=C bonds)[30,31]. The bands related to oxygen-containing functional groups were visible, such as the C−OH stretching vibration at ∼3335 cm−1, carbonyl groups –C=O at 1730 cm−1, C−O−C epoxy groups at 1194 cm−1and C−O flexion vibrational mode at 1050 cm−1[31,32]. The CNF spectrum presented a band at 3333 cm−1related to O-H stretching vibration, at 1635 cm−1 for -OH bending of absorbed water and at 1031 cm-1 for C-O stretching and at 885 cm−1 for -CH alkane bending [33]. The TA spectrum presented a band at 3200 cm−1 for the aromatic and non-aromatic hydroxyl groups, at 1615 cm−1 the C=O stretching vibration, at 1260 and 1060 cm-1 the C-O aromatic stretching, and at 1030 cm−1 the aliphatic C-O stretching [34,35].
The FTIR demonstrated the successful reduction of GO by DA (Figure 2b). The manifold peaks within the range of 800 to 1900 cm−1 were attributed to various oxygen-containing groups on the surface of the GO sheet and CNF, which after DA reduction treatment were modified. In the composite rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50 the C=O stretching vibration of the carboxyl group and epoxy deformation peak present at 1730 cm−1 and 1194 cm−1, respectively; almost disappeared and the band at 1050 cm−1 was reduced, indicating that the oxygen-containing functional groups of GO were successfully removed. During the grafting of CNF to the GO, the intensity of the absorbed water (at 1635 cm−1) become substantially reduced, while the C=C stretching at 1559 cm−1 emerged [31]. The FTIR spectra for rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50 seem identical, suggesting analogous functional groups constituents. The TA inclusion reduced even more main functional groups of rGO/CNF25; in the samples rGO/CNF25/TA5, rGO/CNF25/TA10 the vibrations 1732, 1559, 1030 cm−1 almost disappear (Figure 2c).
Figure 2.
FTIR Spectra a) GO, CNF, TA, b) rGO and rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50 samples, c) TA, rGO/CNF25, rGO/CNF25/TA5, rGO/CNF25/TA10, d) X-ray diffraction patterns of a rGO, CNF, composite materials rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50 and rGO/CNF25/TA5, rGO/CNF25/TA10.
Figure 2.
FTIR Spectra a) GO, CNF, TA, b) rGO and rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50 samples, c) TA, rGO/CNF25, rGO/CNF25/TA5, rGO/CNF25/TA10, d) X-ray diffraction patterns of a rGO, CNF, composite materials rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50 and rGO/CNF25/TA5, rGO/CNF25/TA10.
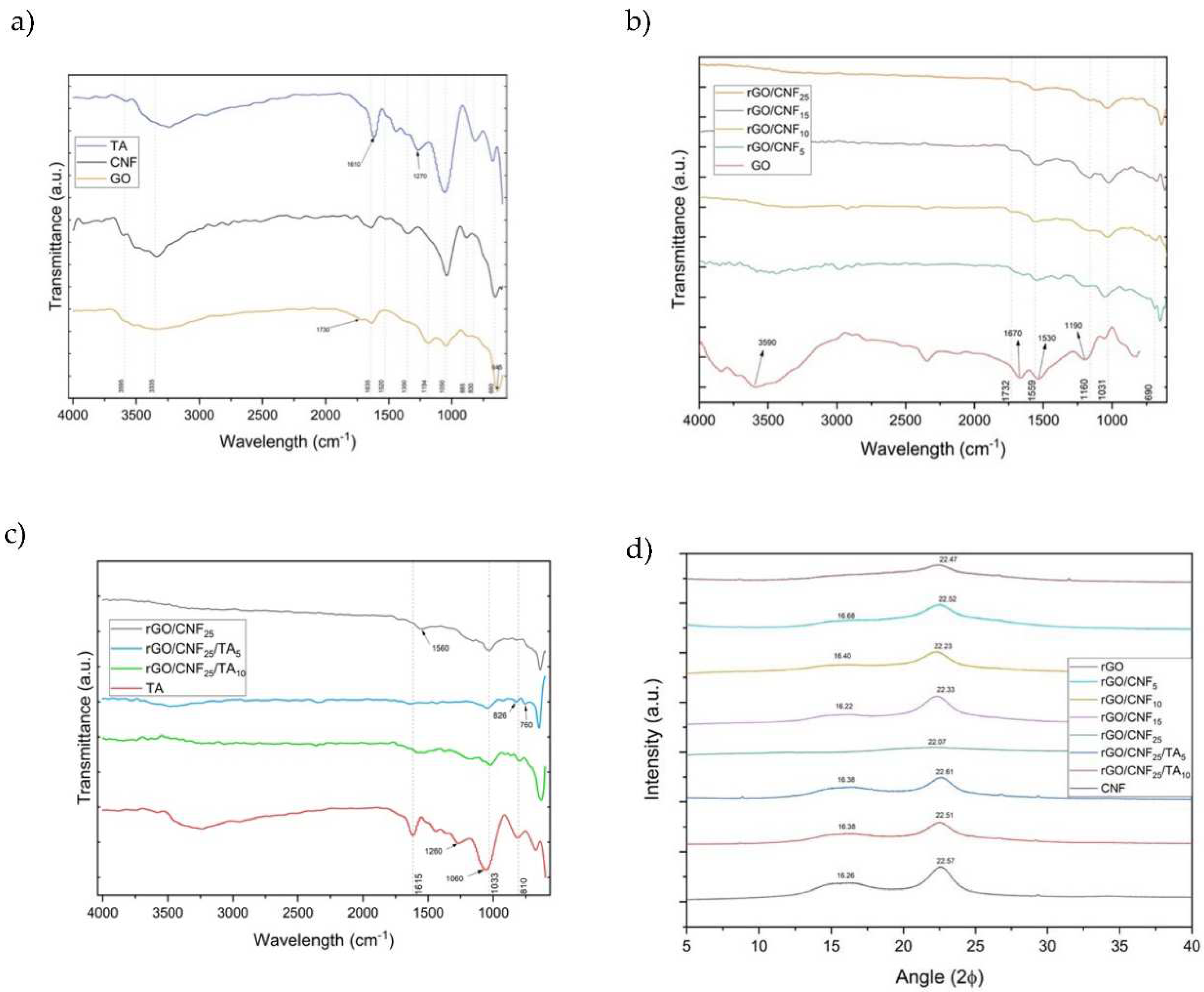
The reduction of GO and the effect of grafting CNF and TA was also verified by XRD and XPS spectroscopy. Figure 2d presents the XRD patterns of GO, CNF and the composite materials, including the samples with TA. GO presented a single peak at 2θ = 8.68° which corresponds to an interlayer d-spacing of 10.2 Å. This larger d-spacing may be due to the presence of oxygen functionalities such as epoxy and hydroxyl on the basal planes and carboxyl on the edges of the graphene sheets (FTIR, Figure 2a). After the GO reduction by DA, a new broad pick at 2θ = 22.47° was obtained, reducing the inter-planar distance to 3.95 Å, which may be attributed to the π–π stacking interactions of the aromatic rings of the coated PDA and/or rGO [36], indicating the successful GO reduction [37]. CNF presented a typical signature of cellulose I structure, with peaks at 16.26° and 22.57° [38]. The CNF inclusion on the rGO sheet preserved the picks, but the sample crystallinity increased, and this value rose even more with TA addition.
The surface compositions of GO, CNF, TA, rGO/CNF25, GO/CNF25/TA10 were studied by XPS (Figure 3). As can be seen in Figure 3a, all samples exhibited two characteristic picks in 284.8 eV corresponding to the C 1s and 533.6 eV corresponding to the O 1s coinciding with data reported by Liu et al. [39]. The characteristic pick 401 eV corresponding to the N 1s appeared in the spectrum of TA, rGO/CNF25 and GO/CNF25/TA10. Also, for the samples rGO/CNF25 and rGO/CNF25/TA10, the C/O ratio increased markedly due to the elimination of oxygen-containing functional groups, indicating the successful reduction of GO.
Deconvolution spectra at the basic C 1 s level of all samples shown in Figure 3b–f resulted in four maximum components with binding energies at 284.8, 285.6, 286.6 and 287.6 eV for the C–C, C–OH, C=O and O–C=O species, respectively. The samples containing CNF at 286.1 eV evidenced the species C-N (Figure 3d–f), increasing the intensity of a C-N bond in the GO presence (Figure 3e), indicating that the GO has reacted with the DA to form a new covalent bond. This showed that the CNF was successfully functionalized with catechol groups through PDA coating. Similar results were reported by other authors working with similar materials [40,41]. The TA inclusion decreases the C-N intensity, which can be attributed to a higher C-OH interaction from the TA compound’s structure.
Figure 3.
a) XPS survey spectrum of GO, TA, CNF, rGO/CNF25, rGO/CNF25/TA10; deconvoluted C 1S XPS spectra of b) TA, c) rGO, d) CNF, e) rGO/CNF25, f) rGO/CNF25/TA10.
Figure 3.
a) XPS survey spectrum of GO, TA, CNF, rGO/CNF25, rGO/CNF25/TA10; deconvoluted C 1S XPS spectra of b) TA, c) rGO, d) CNF, e) rGO/CNF25, f) rGO/CNF25/TA10.
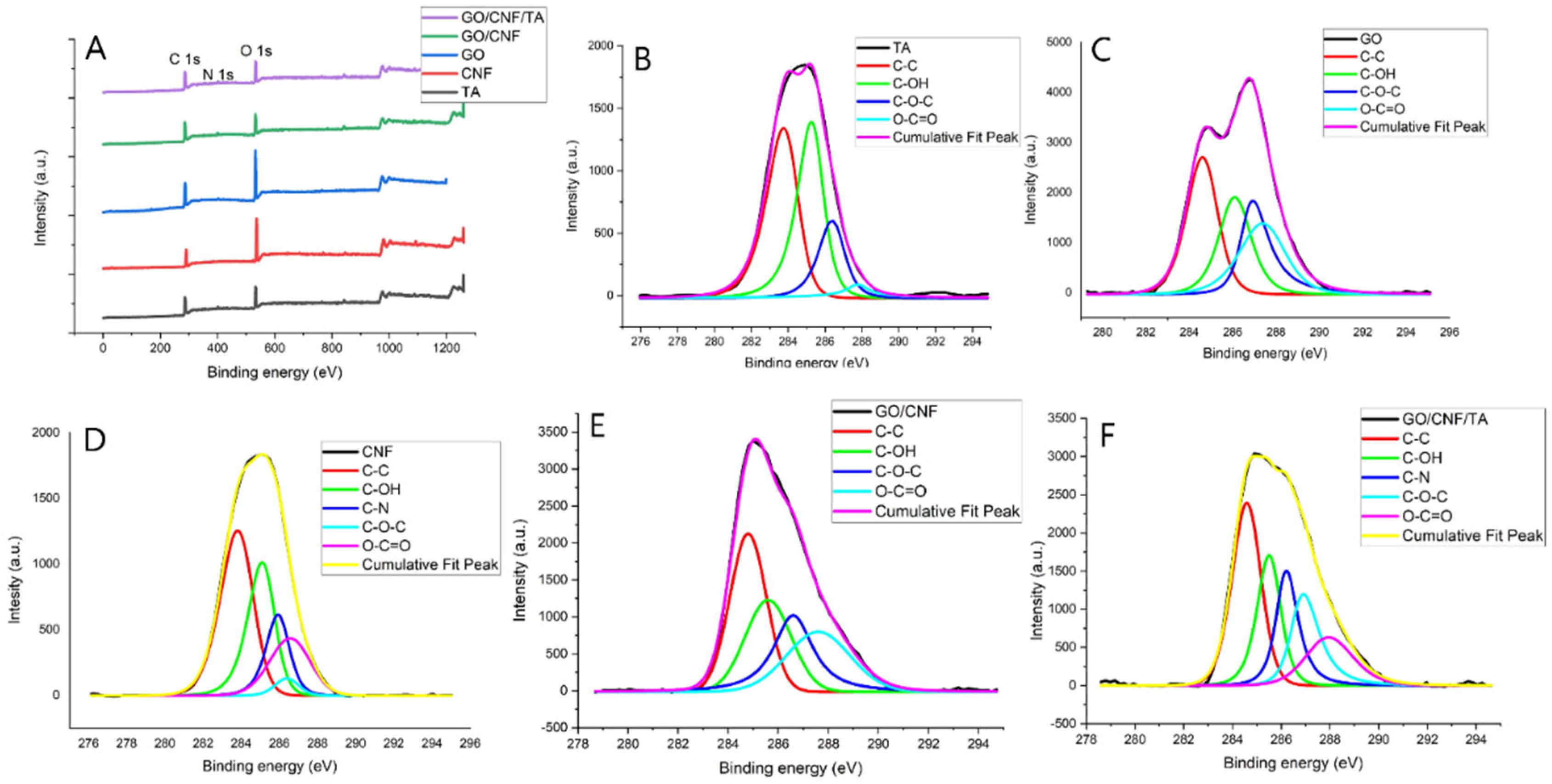
The percentage in area of the maximum components with binding energies of all the samples in the deconvolution of the spectra at basic level C 1 s is presented in Table 1. All samples had the aliphatic group (C-C%) with high percentages, this being the case of TA, CNF and GO/CNF25/TA10, with more than 45% of the area. Comparing the behavior of the alcohol, amine and ester groups of CNF and GO with rGO/CNF25, there is a clear reduction in their % of the area observed, caused by the formation of a new carbonyl group.
The rGO/CNF25 loaded with the TA increases the C-OH bond until reaching values close to 30% in the peak area. The amide, carboxyl, and C-N groups exhibit similar behavior, which may be because these groups are attached to the carbon skeleton of CNF or have been able to form hydrogen bonds with GO.

Table 1.
Percentage in area of maximum components with binding energies of all samples.
| Sample | C-C% | C-OH% | C-O-C% | O-C=O% | C-N% |
|---|---|---|---|---|---|
| TA | 47 | 33 | 16 | 3 | N.D. |
| CNF | 46 | 32 | 17 | 3 | 19 |
| GO | 30 | 23 | 22 | 23 | N.D. |
| rGO/CNF25 | 38 | 2 | 37 | 21 | 13 |
| GO/CNF25/TA10 | 46 | 28 | 25 | 25 | 20 |
3.3. Thermal Stability of the Composite Materials
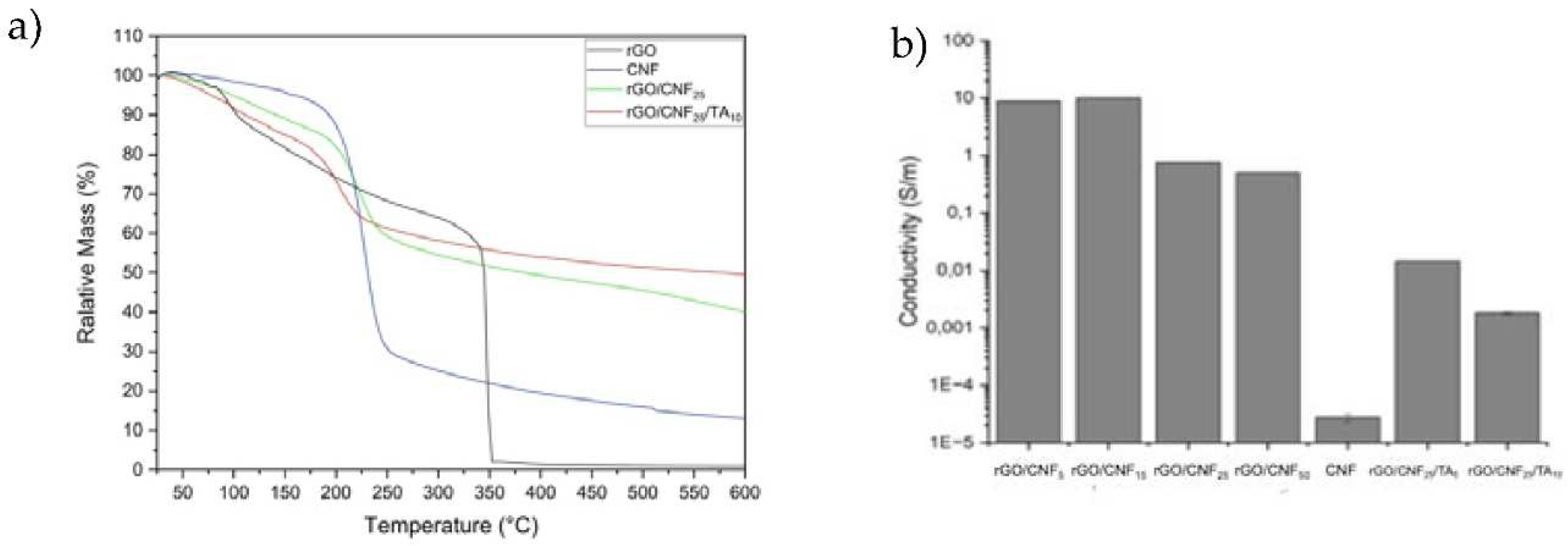
The thermal stability of CNF, rGO and the rGO/CNF25, GO/CNF25/TA10 composite film was evaluated via thermogravimetric analysis (TGA) and differential thermogravimetric analysis (DTG) (Figure 4). CNF showed a main weight loss at 227 °C, which was attributed to the decomposition of cellulose [42]. The rGO presented two stages in the process of thermal weight loss: the first stage ended at 100°C with 10 wt% weight loss, due to the moisture, and the second stage, at 345 °C, was mainly attributed to the decomposition of the labile residual oxygen-containing groups on the surface of GO [43]. The addition of 25% CNF to rGO decreases the temperature of degradation to 223 °C. Other studies have shown a similar effect on cellulose/graphene composites: the inclusion of more CNF content lowering the thermal decomposition temperature [44]. The further addition of TA to the composite reduces the thermal stability even more, to 202 °C.
Figure 4.
a) Thermogravimetric analysis (TGA) of rGO, CNF, rGO/CNF25, rGO/CNF25/TA10 and b) Conductivity of rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50, CNF, rGO/CNF25/TA5 and rGO/CNF25/TA10.
Figure 4.
a) Thermogravimetric analysis (TGA) of rGO, CNF, rGO/CNF25, rGO/CNF25/TA10 and b) Conductivity of rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50, CNF, rGO/CNF25/TA5 and rGO/CNF25/TA10.
3.4. Conductivity and Surface Charge of the Composite Materials
A four-probe tester was used to evaluate the surface electrical resistance of the composite materials. In Figure 4b the conductivity of rGO with different amounts of CNF is shown, also rGO/CNF25 with different amounts of TA. The GO after the reduction with PDA was a fragile material -as will be discussed further-making it impossible to measure it (data not shown). The incorporation of increasing amounts of CNF decreased the electrical activity of the materials, which can be mainly attributed to the insulating nature of the CNF [45]. Neat CNF was not electrically conductive. When the amount of CNF was increased, their absorbance on the surface of rGO increased, which leads to the thickening of the CNF stacking structure units, reducing the connection of rGO sheets and eliminating the conductive channels, thus diminishing the electrical activity. The TA addition resulted in an inverse effect on the material‘s conductivity, which can be attributed to the fact that TA exert a kind of blocking of the electrical charges. The TAs have several alcohol groups in their structure that do not allow the molecule to polarize, thus preventing the movement of electrons and the conductivity per se.
The surface charge of the CNF and GO was -53.3 mV and -96.9 mV, respectively. When the CNF was added to the GO to form the composite materials, this value was reduced to -31.95 mV on average, and no significant difference was observed among samples. The value diminution may be caused by the assembly of the functional compounds after the DA reduction. The addition of TA increases the z-value of composite material, making it more negative (-85.2 mV), which may be caused by the contribution of -OH groups of flavan-3-ols, which is in concordance with the conductivity behavior of TA samples. The z-values found for CNF and GO were comparable to those reported by other authors [46,47]. Reports of the z-potential of the other materials CNF/go/TA were not found.
3.5. Swelling Behavior of Composite Materials
The swelling percentage of the materials was carried out using PBS as a model fluid in order to determine the absorption capacity over time (Figure S2). The material initial humidity was on average 15%. The CNF by their hydrophilic nature, absorbed the fluid instantaneously, reaching 100% of swelling capacity in the first minutes (data not shown). The rGO had a slow absorption rate, reaching a maximum capacity of 48 g/g in close to 80 min. The CNF incorporation into rGO helped to augment the velocity of PBS absorption in the first minutes, but the equilibrium values were maintained, close to 50 g/g in less time than rGO. The TA inclusion helped even more to improve the material‘s swelling velocity (25 g/g in 5 min), which may be caused by the availability of OH groups, contributing to the interaction with the fluid, since PBS has a high hydrogen bond acceptor value.
3.6. Tension and Deformation of Composite Materials
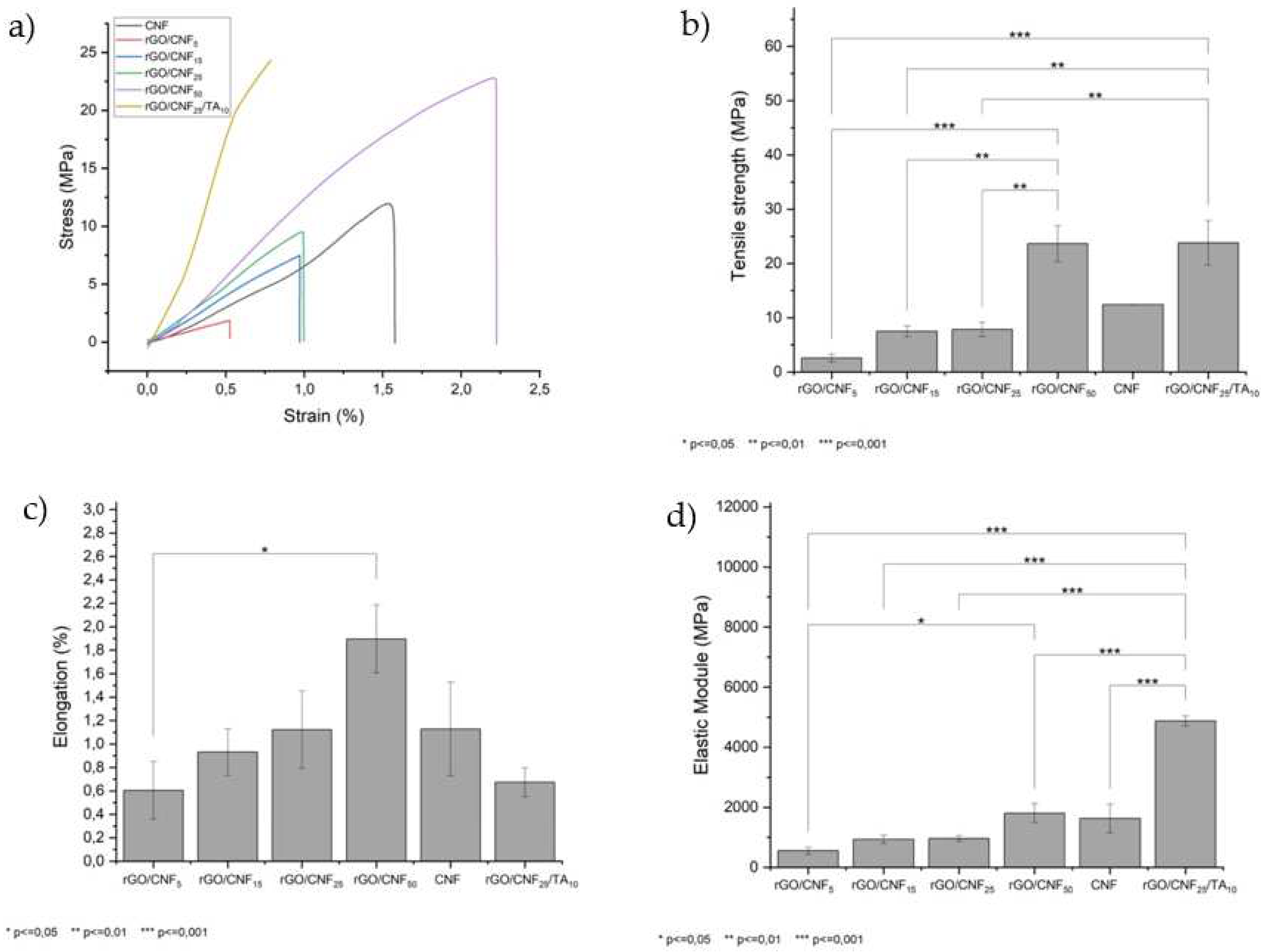
Figure 5a presents the effect of adding different CNF concentrations on the mechanical properties of the material. The CNF presented a tensile strength of 12.4 MPa, a maximum deformation of 1.13% and an elastic module of 1624 MPa. The rGO when synthesized was extremally fragile and it was not possible to analyze it mechanically; nevertheless, the CNF inclusion increased its tensile strength by 90% compared with the native CNF in the sample with CNF 50% (rGO/CNF50) (Figure 5b). The same behavior was observed for the elongation and elastic module of the samples (Figure 5c,d), reaching in the most favorable case an increase of 11% and 68%, respectively. Also, the TA inclusion in the sample with CNF 25% (rGO/CNF25/TA10) showed even higher tensile strength, being 23.8 MPa, as well as elongation at a break of 0.6% and an elastic modulus of 4879 MPa. These results are probably caused by CNF interactions with the oxygenated groups of GO through hydrogen bonds, promoting the interfacial joint and improving the mechanical properties [45], while the addition of TA would produce π–π interactions with the graphene sheets [48].
Figure 5.
Mechanical properties of materials including a) Stress vs Strain curves (b) tensile strength, (b) elongation at break, and (c) elastic modulus of the neat CNF, rGO and rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50, rGO/CNF25/TA10.
Figure 5.
Mechanical properties of materials including a) Stress vs Strain curves (b) tensile strength, (b) elongation at break, and (c) elastic modulus of the neat CNF, rGO and rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50, rGO/CNF25/TA10.
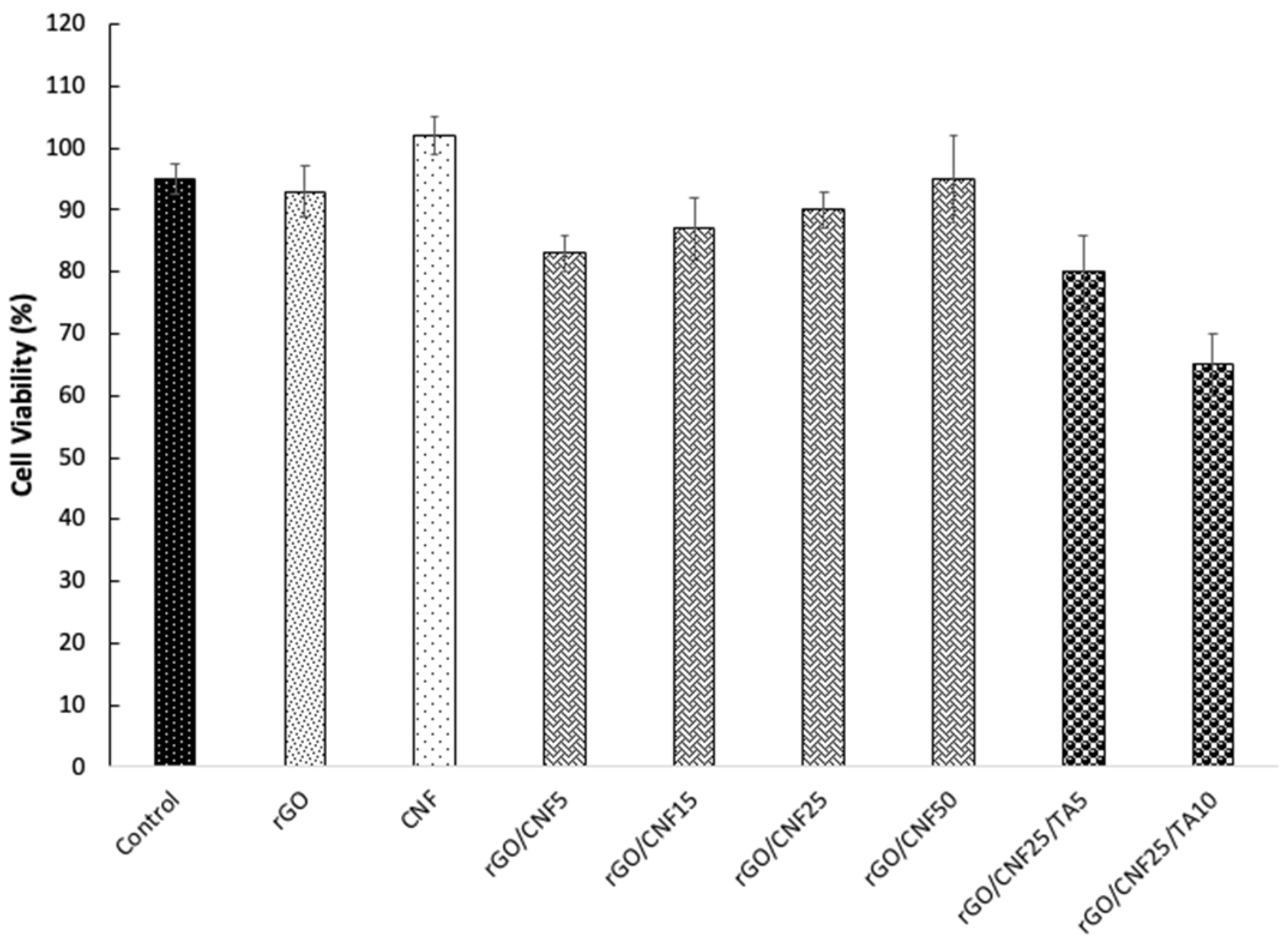
3.7. Cytotoxicity Assays
An ideal wound dressing should exhibit biocompatible properties and not produce intolerable toxic effects on the body. Biocompatibility of composite materials was examined through cell viability using human fibroblast as a model cell (Figure 6). The raw CNF viability was higher than that of rGO, and the addition of CNF to the composite increased the compatibility, but the inclusion of TA was not favorable, decreasing the cell viability, especially for the higher concentration studied. However, the cell viability for the highest TA concentration was higher than 65% whereas an optimal cellular viability of 80% was determined for the sample rGO/CNF25/TA5. It is well known that TA exert several bioactivities such as anti-inflammatory, antimicrobial, antioxidant and anticancer activities, as well as their involvement in cardiovascular, neuroprotective and general metabolic disease prevention [19]. Thus, we believe that the TA inclusion in the wound dressing will contribute to improving healing. Nevertheless, TA have also been reported to be toxic for the cells, depending on their chemical structure [49]. The TA studied here were obtained from pine bark and their chemical composition is mainly condensed tannins with high molecular weight (see Tables S1 and S2). Studies focusing on pine radiata and human fibroblast to evaluate cytotoxicity were not found, but other authors working with Enzogenol®, also a pine radiata bark extract, evaluated the influence of different concentrations of extracts (30-5000 ng/mL) on human neuroblastomas cells, to determine cell survival, did not find significant changes [50]. In our cytotoxicity test after 24 h of material incubation, the released TA reached values of 53500 ng/ml for 5% TA and 93500 ng/mL for 10% TA, which was almost twenty times superior to the concentration assayed in that report.
Figure 6.
Human fibroblast cell viability in presence of neat CNF, rGO and rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50, rGO/CNF25/TA5, rGO/CNF25/TA10.
Figure 6.
Human fibroblast cell viability in presence of neat CNF, rGO and rGO/CNF5, rGO/CNF15, rGO/CNF25, rGO/CNF50, rGO/CNF25/TA5, rGO/CNF25/TA10.
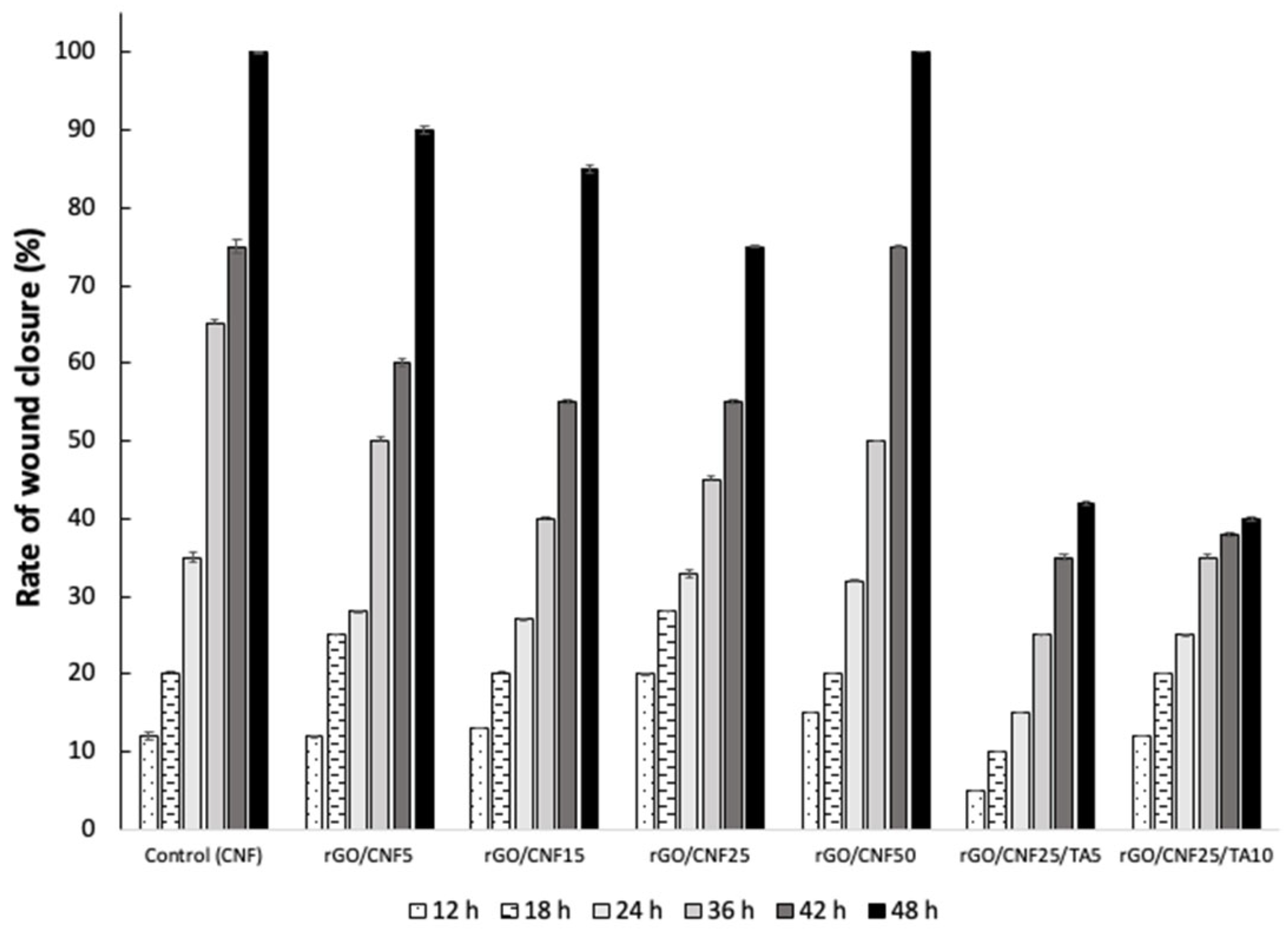
3.8. In Vitro Wound Healing Assay (Scratch Test)
An in vitro wound healing assay (scratch) using human fibroblast cells was performed. In this test, it was assumed that fibroblast cells would attempt to migrate along the edges of the scratch zone to establish cell–cell contact, leading to the closure of the wound. Both cell migration and wound closure rates were monitored over time. The optical microscopy images show that fibroblast cells migrated to the scratch zone after 48 h incubation in contact with the materials, which was confirmed by the wound closure rates (Figure 7). The samples rGO/CNF5 and rGO/CNF15 had a superior performance to that of rGO/CNF25, which could be influenced by the higher conductivity. The wound closure of the sample rGO/CNF50 was similar to the control, maybe influenced by the higher amount of CNF. The TA inclusion in the composite was not favorable for the migration of cells, obtaining values lower than those of the samples without TA, which agrees with cell viability results. Schmit et al.[51], studied different bark fractions (rich in catechins) of P. rigida. Preparations from this bark are used in traditional medicine because of their anti-inflammatory, astringent, expectorant, antidiarrheic, antihemorrhagic, antimicrobial, and wound-healing properties. They found through the scratch assay that the migratory and proliferative activities of mouse fibroblast after 12 h of incubation were dose dependent. Most samples showed enhanced cell numbers when concentrations of 1 and 10 μM were studied, whereas 20 μM concentrations led mostly to a reduction. Also, from the eleven compounds tested, only epicatechin-3-O-gallate and 4′-O-methylepicatechin-3-O-gallate had a favorable effect on wound healing. If the chemical composition of our bark pine extracts it is compared with those results, in their composition gallate moieties were not found (Table S1), which could explain the unfavorable results observed.
Figure 7.
In vitro wound healing over time for raw CNF, rGO/CNF5, rGO/CNF15, rGO/CNF25 rGO/CNF50 rGO/CNF15, rGO/CNF50.
Figure 7.
In vitro wound healing over time for raw CNF, rGO/CNF5, rGO/CNF15, rGO/CNF25 rGO/CNF50 rGO/CNF15, rGO/CNF50.
4. Conclusions
A new composite material based on rGO/CNF/TA was developed under physical crosslinks and reduction with PDA. The chemical characterization showed an effective reduction and crosslinking among the components. The TA incorporation was favorable for the composite, increasing the interaction with PDA. The increasing incorporation of CNF into rGO led to a reduction in thermal stability, surface charge, and conductivity, but the mechanical properties were improved. The cytotoxicity and wound healing of the composite materials in human fibroblast improved with the incorporation of CNF, but the addition of TA resulted in a slight diminution of the cell viability and reduced cell migration, which may be associated with the doses used and the chemical nature of pine extracts. However, the sample rGO/CNF25/TA5 showed a cell viability of 80%. Based on the obtained results, it can be concluded that these composite materials might be used as efficient materials for wound healing. Further in vivo tests will be carried out in order to confirm these in vitro results.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1. SEM images a) GO, b) rGO/CNF5, c) rGO/CNF15, d) rGO/CNF25, e) rGO/CNF50 f) rGO/CNF25/TA; Figure S2. Swelling behavior of neat materials and composite samples on PBS fluid on time; Table S1. Phenol composition and content of the Pinus Radiata bark extract; Table S2. Average molecular weight number (Mn) of pine extracts at different percentages of sample development, determinate by GPC.
Author Contributions
Conceptualization, K.F. and C.A.; methodology, M.F.M, M.P., A.LL., M.B., S.C., V.A. and M.T.; formal analysis, C.A., S.C., I.R., M.T. and V.A.; investigation, S.C., I.R., and V.A.; resources, K.F.; data curation, S.C., I.R., V.A., M.T., and M.B.; writing—original draft preparation, K. F.; writing—review and editing, K.F. and L.I.A.; visualization, K.F. and L.I.A; supervision, K.F and C.A.; project administration, K.F.; funding acquisition, K.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fondo Nacional de Desarrollo Científico y Tecnológico (Fondecyt), Project nº 1210770, Agencia Nacional de Investigación y Desarrollo (ANID), Chile.
Institutional Review Board Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
K.F. thanks to Fondecyt Project nº 1210770, Agencia Nacional de Investigación y Desarrollo (ANID), Chile.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Payne, P.A., Measurement of properties and function of skin. Clinical Physics and Physiological Measurement, 1991. 12(2): p. 105-129. [CrossRef]
- Chua, A.W., et al., Skin tissue engineering advances in severe burns: review and therapeutic applications. Burns Trauma, 2016. 4: p. 3. [CrossRef]
- Zhong, S.P., Y.Z. Zhang, and C.T. Lim, Tissue scaffolds for skin wound healing and dermal reconstruction. Wiley Interdiscip Rev Nanomed Nanobiotechnol, 2010. 2(5): p. 510-25. [CrossRef]
- Balasubramani, M., T.R. Kumar, and M. Babu, Skin substitutes: a review. Burns, 2001. 27(5): p. 534-44. [CrossRef]
- Boateng, J. and O. Catanzano, Advanced Therapeutic Dressings for Effective Wound HealingA Review. Journal of Pharmaceutical Sciences, 2015. 104(11): p. 3653-3680. [CrossRef]
- Agrawal, P., et al., Role of polymeric biomaterials as wound healing agents. Int J Low Extrem Wounds, 2014. 13(3): p. 180-90. [CrossRef]
- Farber, P.L., F.C. Isoldi, and L.M. Ferreira, Electric Factors in Wound Healing. Advances in Wound Care, 2021. 10(8): p. 461-476. [CrossRef]
- Zhao, M., et al., Electrical signals control wound healing through phosphatidylinositol-3-OH kinase-gamma and PTEN. Nature, 2006. 442(7101): p. 457-60.
- Fan, Z., et al., A Novel Wound Dressing Based on Ag/Graphene Polymer Hydrogel: Effectively Kill Bacteria and Accelerate Wound Healing. Advanced Functional Materials, 2014. 24(25): p. 3933-3943. [CrossRef]
- Tang, P., et al., Mussel-Inspired Electroactive and Antioxidative Scaffolds with Incorporation of Polydopamine-Reduced Graphene Oxide for Enhancing Skin Wound Healing. ACS Applied Materials & Interfaces, 2019. 11(8): p. 7703-7714. [CrossRef]
- Fall, A.B., et al., Colloidal stability of aqueous nanofibrillated cellulose dispersions. Langmuir, 2011. 27(18): p. 11332-8. [CrossRef]
- Brakat, A. and H. Zhu, Nanocellulose-Graphene Derivative Hybrids: Advanced Structure-Based Functionality from Top-down Synthesis to Bottom-up Assembly. ACS Applied Bio Materials, 2021. 4(10): p. 7366-7401. [CrossRef]
- Wang, Y., et al., Stretchable, Conductive, and Self-Healing Hydrogel with Super Metal Adhesion. Chemistry of Materials, 2018. 30(13): p. 4289-4297. [CrossRef]
- Shin, S.R., et al., Graphene-based materials for tissue engineering. Adv Drug Deliv Rev, 2016. 105(Pt B): p. 255-274. [CrossRef]
- Huang, N., et al., Multifunctional Electrochemical Platforms Based on the Michael Addition/Schiff Base Reaction of Polydopamine Modified Reduced Graphene Oxide: Construction and Application. ACS Appl Mater Interfaces, 2015. 7(32): p. 17935-46. [CrossRef]
- Zhou, P., et al., Rapidly-deposited polydopamine coating via high temperature and vigorous stirring: formation, characterization and biofunctional evaluation. PLoS One, 2014. 9(11): p. e113087. [CrossRef]
- Huang, N., et al., Multifunctional Electrochemical Platforms Based on the Michael Addition/Schiff Base Reaction of Polydopamine Modified Reduced Graphene Oxide: Construction and Application. ACS Applied Materials & Interfaces, 2015. 7(32): p. 17935-17946. [CrossRef]
- Sileika, T.S., et al., Colorless Multifunctional Coatings Inspired by Polyphenols Found in Tea, Chocolate, and Wine. Angewandte Chemie International Edition, 2013. 52(41): p. 10766-10770. [CrossRef]
- Maugeri, A., et al., Pharmacology and toxicology of tannins. Archives of Toxicology, 2022. 96(5): p. 1257-1277. [CrossRef]
- Wang, Y., Z. Shi, and J. Yin, Facile Synthesis of Soluble Graphene via a Green Reduction of Graphene Oxide in Tea Solution and Its Biocomposites. ACS Applied Materials & Interfaces, 2011. 3(4): p. 1127-1133. [CrossRef]
- Zeng, J., et al., Cellulose nanofibrils manufactured by various methods with application as paper strength additives. Scientific Reports, 2021. 11(1): p. 11918. [CrossRef]
- Andrade, A., et al., Effect of the chemical and structural characteristics of pulps of Eucalyptus and Pinus on the deconstruction of the cell wall during the production of cellulose nanofibrils. Cellulose, 2021. 28(9): p. 5387-5399. [CrossRef]
- Ghose, T.K., Measurement of cellulase activities. Pure and Applied Chemistry, 1987. 59(2): p. 257-268. [CrossRef]
- Marcano, D.C., et al., Improved Synthesis of Graphene Oxide (vol 4, pg 4806, 2010). Acs Nano, 2018. 12(2): p. 2078-2078.
- Bocalandro, C., et al., Comparison of the composition of Pinus radiata bark extracts obtained at bench- and pilot-scales. Industrial Crops and Products, 2012. 38: p. 21-26. [CrossRef]
- Wei, J., et al., Bioinspired 3D Printable, Self-Healable, and Stretchable Hydrogels with Multiple Conductivities for Skin-like Wearable Strain Sensors. ACS Applied Materials & Interfaces, 2021. 13(2): p. 2952-2960. [CrossRef]
- Schuhladen, K., et al., Production of a novel poly(ɛ-caprolactone)-methylcellulose electrospun wound dressing by incorporating bioactive glass and Manuka honey. J Biomed Mater Res B Appl Biomater, 2021. 109(2): p. 180-192. [CrossRef]
- Li, M.Z., et al., A novel graphene-based micro/nano architecture with high strength and conductivity inspired by multiple creatures. Scientific Reports, 2021. 11(1). [CrossRef]
- Hsu, H.H., et al., An Eco-Friendly, Nanocellulose/RGO/in Situ Formed Polyaniline for Flexible and Free-Standing Supercapacitors. Acs Sustainable Chemistry & Engineering, 2019. 7(5): p. 4766-4776. [CrossRef]
- Faniyi, I.O., et al., The comparative analyses of reduced graphene oxide (RGO) prepared via green, mild and chemical approaches. SN Applied Sciences, 2019. 1(10): p. 1181. [CrossRef]
- Wang, J., et al., Nanocellulose-assisted low-temperature synthesis and supercapacitor performance of reduced graphene oxide aerogels. Journal of Power Sources, 2017. 347: p. 259-269. [CrossRef]
- Song, J., X. Wang, and C.-T. Chang, Preparation and Characterization of Graphene Oxide. Journal of Nanomaterials, 2014. 2014: p. 276143. [CrossRef]
- Hsu, H.H., et al., An Eco-Friendly, Nanocellulose/RGO/in Situ Formed Polyaniline for Flexible and Free-Standing Supercapacitors. ACS Sustainable Chemistry & Engineering, 2019. 7(5): p. 4766-4776. [CrossRef]
- Soto, R., J. Freer, and J. Baeza, Evidence of chemical reactions between di- and poly-glycidyl ether resins and tannins isolated from Pinus radiata D. Don bark. Bioresource Technology, 2005. 96(1): p. 95-101. [CrossRef]
- Zhu, Y.H., et al., The synthesis of tannin-based graphene aerogel by hydrothermal treatment for removal of heavy metal ions. Industrial Crops and Products, 2022. 176. [CrossRef]
- Gu, R.P., W.Z. Xu, and P.A. Charpentier, Synthesis of polydopamine-coated graphene-polymer nanocomposites via RAFT polymerization. Journal of Polymer Science Part a-Polymer Chemistry, 2013. 51(18): p. 3941-3949. [CrossRef]
- Liao, J.M., et al., Antibacterial Performance of a Mussel-Inspired Polydopamine-Treated Ag/Graphene Nanocomposite Material. Materials, 2019. 12(20). [CrossRef]
- Mohaiyiddin, M.S., et al., Characterization of nanocellulose recovery from Elaeis guineensis frond for sustainable development. Clean Technologies and Environmental Policy, 2016. 18(8): p. 2503-2512. [CrossRef]
- Liu, X.R., et al., Thiol-branched graphene oxide and polydopamine-induced nanofibrillated cellulose to strengthen protein-based nanocomposite films. Cellulose, 2019. 26(12): p. 7223-7236. [CrossRef]
- Liu, X., et al., Thiol-branched graphene oxide and polydopamine-induced nanofibrillated cellulose to strengthen protein-based nanocomposite films. Cellulose, 2019. 26(12): p. 7223-7236. [CrossRef]
- Li, M., et al., Preparation of and research on bioinspired graphene oxide/nanocellulose/polydopamine ternary artificial nacre. Materials & Design, 2019. 181. [CrossRef]
- Gan, P.G., et al., Thermal properties of nanocellulose-reinforced composites: A review. Journal of Applied Polymer Science, 2020. 137(11): p. 48544. [CrossRef]
- Zhao, Y., et al., Mechanical Reinforcement in Nylon 6 Nanocomposite Fiber Incorporated with Dopamine Reduced Graphene Oxide. Materials, 2022. 15(15): p. 5095. [CrossRef]
- Nguyen Dang, L. and J. Seppälä, Electrically conductive nanocellulose/graphene composites exhibiting improved mechanical properties in high-moisture condition. Cellulose, 2015. 22(3): p. 1799-1812. [CrossRef]
- Ding, Z.J., Y.J. Tang, and P. Zhu, Reduced graphene oxide/cellulose nanocrystal composite films with high specific capacitance and tensile strength. International Journal of Biological Macromolecules, 2022. 200: p. 574-582. [CrossRef]
- Anikushin, B.M., et al., Zeta Potential of Nanosized Particles of Cellulose as a Function of pH. Chemistry and Technology of Fuels and Oils, 2022. 57(6): p. 913-916. [CrossRef]
- Simsek, B., et al., Improvement of the Graphene Oxide Dispersion Properties with the Use of TOPSIS Based Taguchi Application. Periodica Polytechnica-Chemical Engineering, 2018. 62(3): p. 323-335. [CrossRef]
- Guo, Q., et al., Fabrication of Super Extensible and Highly Tough Graphene Composite Hydrogels by Thermal Treatment Strategy for the Mixture of Tannin and Graphene Oxide. Macromolecular Chemistry and Physics, 2017. 218(6): p. 1600549. [CrossRef]
- He, F., et al., Structure-Dependent Eco-Toxicity of Vegetable Tannin. Processes, 2022. 10(5): p. 816. [CrossRef]
- Schmidt, A.J., et al., Impact of Plant Extracts Tested in Attention-Deficit/Hyperactivity Disorder Treatment on Cell Survival and Energy Metabolism in Human Neuroblastoma SH-SY5Y Cells. Phytotherapy Research, 2010. 24(10): p. 1549-1553. [CrossRef]
- Schmidt, C.A., et al., Catechin Derivatives from Parapiptadenia rigida with in Vitro Wound-Healing Properties. Journal of Natural Products, 2010. 73(12): p. 2035-2041. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



