
Submitted:
12 April 2023
Posted:
13 April 2023
You are already at the latest version
Abstract
Sheep (Ovis aries) provide a vital source of protein and fibre to human populations. In coming decades, as the pressures associated with rapidly changing climates increase, breeding sheep sustainably as well as producing enough protein to feed a growing human population will pose a considerable challenge for sheep production across the globe. High quality reference genomes and other genomic resources can help to meet these challenges by: 1) informing breeding programmes by adding a priori information about the genome, 2) providing tools such as pangenomes for characterising and conserving genetic diversity, and 3) improving our understanding of fundamental biology using the power of genomic information to link cell, tissue and whole animal scale knowledge. In this review we describe recent advances in the genomic resources available for sheep, discuss how these might help to meet future challenges for sheep production, and provide some insight into what the future might hold.
Keywords:
Sheep
; genome
; transcriptome
; pangenome
; gene editing
Introduction
The domestic sheep (Ovis aries), is an important farmed animal species providing a source of protein and fibre to human populations across the globe. Sheep have excelled over the centuries in a range of production systems and environments (Mignon-Grasteau et al., 2005; Marshall et al., 2014; Alberto et al., 2018). Production systems differ across the globe, often with arable land, breed, environment, and key local and international markets playing a role in the type of production system used. The UK sheep industry, for example, is primarily based on sheep meat production, where the stratified system consists of three sectors: hill, upland and lowland, each utilising different breeds and production systems (Conington et al., 2001). The UK sheep sector currently largely uses traditional breeding practices, with a few exceptions, while in Australia, New Zealand and elsewhere advanced genomics enabled breeding schemes have been widely implemented (Daetwyler et al., 2010; Brito et al., 2017a). Sheep production systems in place in countries that produce a large amount of sheep meat, including the UK, Australia and New Zealand rely on a relatively small number of popular breeds, to support large export markets. In contrast sheep production within low and middle income countries (LMICs) is orientated towards small holder systems that make use of a diverse range of breeds that are adapted to harsh climatic and nutritional conditions (Marshall et al., 2019). In LMICs sheep production is vital to the livelihoods and nutritional needs of both individuals and communities, and often plays a multifaceted role within society (Marshall et al., 2019).
The future of sheep production, and its contributing role in global food production, will become more apparent in coming decades due to predicted extremes of climate and a growing human population that is expected to reach almost 9 billion by 2050 (McKenzie and Williams, 2015). Increases in food production from sheep need to be achieved with societal expectations around animal health and welfare in mind and should be guided through initiatives for responsible animal breeding such as Code EFABAR (EFFAB 2020). Sheep are also a source of greenhouse gases (Marino et al., 2016), and ambitious targets are being set to cut greenhouse gas emissions across the globe by 2030, which will require breeding strategies that reduce environmental impact (Mollenhorst and de Haas 2019). In addition, future breeding programmes will need to maintain genetic diversity to increase performance and strengthen resilience in the face of climatic extremes and other pressures (Dumont et al., 2020). In coming years breeding sheep sustainably using fewer resources, whilst flexibly meeting societal expectations, as well as producing enough protein to feed a growing human population, will pose a considerable challenge for sheep breeders and producers across the globe (Hayes et al., 2013). High quality reference genomes and other genomic tools and resources can help to meet these challenges (Clark et al., 2020). For example, they can: 1) inform breeding programmes including those enabled by genomic selection and genome editing (Georges et al., 2019), 2) provide tools for characterising and conserving genetic diversity (Talenti et al., 2022), and 3) improve our understanding of fundamental biology to link cell, tissue and whole animal scale knowledge (Giuffra and Tuggle, 2019) (Figure 1). Here we describe recent advances in the genomic resources available for sheep, discuss how these might help to meet future challenges for sheep production, and provide some insight into potential future opportunities.
Towards a high quality highly contiguous reference genome for sheep
The genomic resources for sheep have gradually been improving in quality and resolution over the last twenty years in parallel with advances in sequencing technology. This is particularly evident when describing improvements in the quality and contiguity of the reference genome sequence for sheep. The reference genome sequence is the version of the sheep genome accepted by the sheep genomics community as a standard for comparison to sequence information generated in their own studies. A contiguous, high quality, well annotated and assembled reference genome sequence for sheep is a hugely valuable research tool, providing a searchable map of the genome including the locations of expressed and regulatory regions. There have been several versions of the reference genome sequence for sheep and each new version has kept pace with advancements in sequencing technology, starting with the ovine radiation hybrid panel (Cockett 2006). The first true version of the reference genome sequence for sheep (Ovis_aries_1.0; GCA_000005525.1) was a guided assembly using the bovine genome. It was generated from six female sheep of different breeds sequenced at 0.5× coverage by 454 FLX (Dalrymple et al., 2007). Seven years later in 2014 the Texel reference genome sequence Oar_v3.1 (GCA_000298735.1), assembled from two unrelated Texel sheep using Illumina short read sequencing at 150× coverage, was released (Jiang et al., 2014). This assembly offered an improved contiguity (N50 contig length of approximately 40 Kb) and a genome length of 2.6 Gb (Jiang et al., 2014) (Table 1). The Oar_v3.1 genome assembly revealed segmental duplications within Texel sheep, along with a large run of homozygosity that contained the MSTN gene (Jiang et al., 2014). Previously a variant in the 3’ UTR region in the MSTN gene, that disrupted miRNA binding, had been shown to control the muscle hypertrophy (double muscling) phenotype in Texel sheep (Clop et al., 2006). The Oar_v3.1 reference genome provided a resource to interrogate the genomic regions associated with muscling in Texel sheep in more detail including the MSTN gene and the Texel muscling QTL (TM-QTL) on chromosome 18 (Macfarlane et al. 2014).
More recently, long read sequencing technologies capable of generating contiguous reads of up to 10 Kb in length have provided a means to significantly improve the contiguity of a reference genome sequence (Pollard et al., 2018). A combination of Illumina® GAII sequencing, Roche 454 sequencing and PacBio® RSII technologies were used to gap fill Oar_v3.1 generating the more contiguous Texel Oar_v4.0 (GCA_000298735.2) genome (Table 1). Oar_v3.1 and Oar_v4.0 remained the gold standard reference genome sequences for sheep until 2020 when a new reference genome sequence was released that was generated using both Illumina® HiSeq X short reads and PacBio® RS II long read technology. This new reference genome Oar_rambouillet_v1.0 (GCA_002742125.1) was built from the DNA of a single Rambouillet ewe Benz2616 (Liu et al., 2016). Oar_rambouillet_v1.0 had fewer contigs and a considerably greater contig N50 length than Oar_v3.1 and Oar_v4.0, replacing the Texel as the new reference genome sequence for sheep (Table 1).
In 2022 a de novo assembly of the same Rambouillet ewe used to generate the Oar_rambouillet_v1.0 assembly was published, ARS-UI_Ramb_v2.0 (GCA_016772045.1) (Davenport et al., 2022). This new assembly was built using ∼50× coverage Oxford Nanopore® PromethION reads (N50 47 kb) and 75× coverage Pacific Biosciences (PacBio) reads (N50 13 kb), with Hi-C data for scaffolding and Illumina short read data for final polishing (Davenport et al., 2022). The result was a 15-fold improvement in contiguity and increased accuracy over Oar_rambouillet_v1.0 (Table 1). The ARS-UI_Ramb_v2.0 genome is now the community adopted reference genome sequence. It has provided the sheep genomics community with a very high quality reference genome assembled into fewer contigs than even the ARS1 goat genome (Table 1), which it at the time of its release was considered the gold standard of farmed animal genomes (Bickhart et al., 2017; Worley, 2017).
Annotation of regulatory regions in the reference genome sequence by the Ovine FAANG project
High resolution annotation information, that accurately defines gene models and regulatory regions, adds basic functional genomic knowledge to the reference genome sequences for farmed animals increasing their power and utility as research tools (Georges et al., 2019; Giuffra and Tuggle, 2019; Clark et al., 2020). The USDA NIFA funded Ovine FAANG project, led by the University of Idaho, provided the opportunity to annotate regulatory genomic regions in the new Rambouillet genome (Murdoch, 2019). The Functional Annotation of Animal Genomes (FAANG) consortium is a concerted international effort to use molecular assays, developed during the Human ENCODE project (Birney et al., 2007), to annotate the majority of functional elements in the genomes of domesticated animals (Andersson et al., 2015; Giuffra and Tuggle, 2019). By applying a set of core assays defined by the FAANG consortium, including five ChIP-Seq marks, ATAC-Seq, CAGE-Seq, RNA-Seq and methylation information, across a set of 56 tissues from Benz2616, the Ovine FAANG project developed a set of deep and robust expressed elements and regulatory features in the Rambouillet genome (Murdoch, 2019). Some of these datasets are already available, via the FAANG Data Portal (https://data.faang.org) (Harrison et al., 2021), including the CAGE dataset which provides a high resolution annotation of transcription start sites in the Oar_rambouillet_v1.0 genome (Salavati et al., 2020). RefSeq have also provided an annotation of the coding regions for ARS-UI_Ramb_v2.0 (GCF_016772045.1) using the mRNA-Seq, CAGE and Iso-Seq data. Once the ATAC-Seq and ChIP-Seq data become available it will be possible for Ensembl to incorporate them into a regulatory build (Zerbino et al., 2015). This will mean that a genome-wide set of regions that are likely to be involved in gene regulation in sheep will be available in the Ensembl genome browser as a resource for the farmed animal genomics community. The Ovine FAANG project provides a valuable resource to facilitate a deeper understanding of how the regulatory regions of the genome control complex traits in sheep. It also provides a foundation for comparative analysis with other farmed animal species in which similar annotation datasets are available e.g. for cattle, chicken, goat and pig (Foissac et al., 2019; Goszczynski et al., 2021; Kern et al., 2021).
From the human literature we know that as many as 90% of variants underlying complex traits identified in Genome Wide Association Studies (GWAS) are located in non-coding regions of the genome (Tam et al., 2019). In addition to the efforts of the Ovine FAANG project, in annotating the Rambouillet reference genome sequence, there have been a small number of other studies to date that have characterised regulatory regions in the sheep genome. For example, (Davenport et al., 2021) used histone modifications that distinguish active or repressed chromatin states, CTCF binding, and DNA methylation to characterize regulatory elements in liver, spleen, and cerebellum tissues from four yearling sheep to identify the regulatory regions of genes that play key roles in defining health and economically important traits. To evaluate the impact of selection and domestication on regulatory sequences (Naval-Sanchez et al., 2018) used histone modification and gene expression data. Their analyses showed that selective sweeps were significantly enriched for protein coding genes, proximal regulatory elements of genes and genome features associated with active transcription and indicated that remodelling of gene expression is likely to have been one of the evolutionary forces driving phenotypic diversification in domestic sheep (Naval-Sanchez et al., 2018). Both studies demonstrate the value of regulatory annotation information in understanding the genomic processes driving complex traits and shaping the characteristics and genetic diversity of global sheep populations.
Annotating expressed regions in the sheep genome, the sheep gene expression atlas and beyond
Advances in transcriptome sequencing technology and reductions in cost have also led to improvements in annotation of the expressed regions in the reference genome sequence for sheep over the last decade. Coding regions in the Oar_v3.1 reference genome (Jiang et al., 2014) were annotated by Ensembl with their ‘Genebuild’ pipeline (Aken et al., 2016) using RNA-sequencing data from more than 80 tissues collected from a Texel ewe, lamb and ram trio (http://useast.ensembl.org/Ovis_aries/Info/Annotation) When released the Oar_v3.1 annotation was one of the most comprehensive annotations of any of the farmed animal species and was widely used by the community until Oar_rambouillet_v1.0 was annotated by Ensembl in 2020 (http://www.ensembl.org/Ovis_aries_rambouillet/Info/Annotation). Over the last decade a vast amount of RNA-sequencing data for sheep has been generated, capturing global transcriptomic complexity across multiple tissues, cell types and developmental stages (Jiang et al., 2014; Clark et al., 2017). In 2017 a large scale gene expression atlas (http://biogps.org/sheepatlas) was generated from tissues and cells collected from all of the major organ systems from adult Texel x Scottish Blackface sheep and from juvenile, neonatal and prenatal developmental stages (Clark et al., 2017). Of the 20,921 protein coding genes, that were annotated in the Oar v3.1 reference genome, 19,921 (92%) had detectable expression in at least one tissue in the sheep gene expression atlas dataset (Clark et al., 2017). Network-based cluster analysis, using the software package Graphia (Freeman et al., 2022), was used to describe the overall transcriptional signatures present in the sheep gene expression atlas and assign those signatures, where possible, to specific tissues or cell types.
The next frontier for the sheep transcriptome will be to fully resolve the tissue- and cell- type specific transcriptional signatures generated for the sheep atlas from bulk tissue samples, at a single cell resolution. Single-cell sequencing technologies enable the deconvolution of transcriptional and regulatory complexity in tissues comprised of many different cell types e.g. (Schaum et al. 2018). Atlases of gene expression generated using single cell sequencing technologies have already been created for pig (https://dreamapp.biomed.au.dk/pigatlas/) (Wang et al., 2022). Building similar single cell transcriptomic resources for sheep from multiple tissue types and developmental stages and adding regulatory information with single cell ATAC-seq, for example, would provide insights into cell composition, cell-to-cell interactions and the cellular heterogeneity of tissues. As datasets of this type are generated for more species of farmed animals sets of cell specific marker genes that are conserved across species will be revealed. Such markers could be applied as a proxy for a particular cell type e.g. (Herrera-Uribe et al. 2021) and may be useful as a costly but high value intermediate phenotype for complex trait prediction, providing a powerful tool for linking genotype to phenotype in sheep and other farmed animal species.
The Power of PanGenomes – moving beyond a single reference genome sequence
Recent advances in long read sequencing technologies, and reductions in cost, have meant that in addition to a single reference genome per farmed animal species it is now possible to generate chromosome level genomes for many different breeds and populations. Many new chromosome level genomes including, for example, Hu sheep (Li et al., 2021), Dorper (Qiao et al., 2022), and Tibetan sheep (Li et al., 2022b) have recently been deposited in NCBI (Table 2). In addition, recently a pangenome for sheep was generated that included new long-read assemblies for 13 different breeds (Li et al., 2022a). Currently, NCBI reports that there are 55 genome assemblies for sheep (https://www.ncbi.nlm.nih.gov/data-hub/genome/?taxon=9940). Some of these are alternate-pseudohaplotypes, where two pseudohaplotype assemblies of the diploid genome have been generated, and each release of the reference genome sequences for the Rambouillet and Texel are also included in the database. In total on NCBI there are 19 unique breeds of sheep that have chromosome level assemblies (Table 2). These breeds represent 11 different countries (Table 2), and include the Suffolk, a British breed, that is a very popular terminal sire across the globe (https://www.suffolksheep.org/history/), and the Dorper a versatile composite that is used extensively for production in tropical regions (http://agtr.ilri.cgiar.org/dorper). Assembly statistics for the Rambouillet reference genome sequence (ARS-UI_Ramb_v2.0) are included to demonstrate that the majority of these new genome assemblies, generated using different long read sequencing technologies, are close to reference quality in terms of contiguity.
The number of breeds and populations with chromosome level genome assemblies will rise significantly, as global pangenome efforts that aim to capture the global diversity of sheep breeds, gather pace. The concept of a ‘pangenome’ is probably most simply defined as any collection of genomic sequences to be analysed jointly or to be used as a reference (The Computational Pan-Genomics Consortium 2018). The USDA NIFA Ovine Pangenome Project, for example, plans to generate eight new haplotype resolved assemblies from crosses of breeds selected for their divergent characteristics, using the trio-binning approach developed by (Koren et al., 2018). For trio-binning usually an F1 cross of two disparate breeds of sheep, chosen to maximise heterozygosity, is generated. The genome assembly then relies on using short read Illumina data from the two parental genomes to first partition the long reads from the offspring into haplotype-specific sets. Each parental haplotype is then assembled independently, resulting in a complete diploid reconstruction, and effectively two new reference assemblies, one for each of the two parental breeds, as described in (Koren et al., 2018). This strategy has proved very successful in cattle (Koren et al., 2018; Rice et al. 2020) and has been used so far to produce the White Dorper x Romanov assemblies for sheep Oar_ARS-UKY_Romanov_v1.0 (GCA_022244705.1 ) and Oar_ARS-UKY_WhiteDorper_v1.0 (GCA_022244695.1) (Table 2).
These new chromosome level assemblies for sheep will improve our understanding genome diversity and the drivers of breed-specific characteristics. As such global pangenome efforts should aim to capture the genomic diversity of global sheep populations. This genetic diversity provides a foundational resource for breed improvement and for the adaptation of sheep populations to changing environments and changing demands (FAO 2015). It will be important, for example, to include the United Kingdom’s native sheep breeds that have become the mainstay of sheep production across the globe. This is particularly important in the context of breed conservation as many of the UK breeds, including for example the Norfolk Horn the ancestor of the Suffolk, are rare and declining in numbers (https://www.rbst.org.uk/norfolk-horn). Many European rare and indigenous breeds exhibit widespread heterozygote deficit due to declining diversity and are being lost due to introgression into large commercial populations (Lawson Handley et al. 2007). Similarly, in LMICs where small-holder farmers rely on a wide diversity of breeds adapted to local conditions (Marshall et al. 2019), the genomic diversity of indigenous breeds should be represented. For example, West and Central African indigenous breeds, such as the Cameroon sheep, represent a unique reservoir of genetic diversity and have followed the tracks of human migration across the globe contributing to the formation of Caribbean breeds (Spangler et al., 2017; Wiener et al., 2022). The Cameroon sheep is also anecdotally thought to be tolerant to Trypanosomiasis (Geerts et al. 2009).Genomic drivers of adaptation in local indigenous breeds to specific environmental challenges, including resistance or tolerance to specific diseases, needs to be better understood (FAO 2015). Genomic information provided by global pangenome efforts for sheep should help to remedy this through comparative approaches, such as those described in (Dutta et al., 2020) for water buffalo and cattle populations, to identify loci present in one breed, species or population that are missing another.
Reference quality genome sequences representing the global diversity of sheep breeds also provide genomic resources that are relevant in a country or continent specific context. This is important because it can minimise reference mapping bias when working with short read whole genome sequencing data (Chen et al., 2021). For example, for a study investigating population genomics in sheep from the African continent using short read data, the Dorper (Qiao et al., 2022) a South African breed, would be a more appropriate reference assembly than the European Texel or Rambouillet. However, even when reference genome sequences for multiple different breeds are available the use of reference genome sequences that represent only a single individual, for understanding population diversity at genomic level are still limited. There are two main reasons for this (described in (Talenti et al., 2022)); i) because a single reference genome represents one consensus haplotype of a single individual, and as such it would be expected that large sections of the diversity represented in the global pangenome for sheep will be missing from the reference sequence, and ii) reference mapping bias causes downstream analyses to be biased towards the alleles and haplotypes present in the reference sequence. Graph-based genomes, that integrate long read genome sequences for a subset of representative breeds and short read sequence data from hundreds of breeds and individuals to build a pangenome graph, provide an alternative, to capture global diversity. Graph based pangenomes have recently been produced for other ruminants including, cattle (Crysnanto and Pausch, 2020; Crysnanto et al., 2021; Talenti et al., 2022) and goats (Li et al., 2019), and a sheep pangenome graph which includes 13 breeds is also now available (Li et al., 2022a). The graph based pangenomes generated for cattle have been shown to increase read mapping rates, reduce allelic biases and identify structural variants with a high level of accuracy (Talenti et al., 2022). As such a graph-based genome for sheep incorporating many different breeds and populations spanning the depth and breadth of genetic diversity from across the globe, would provide a hugely informative research tool to inform future breeding and conservation strategies.
Characterising global diversity in sheep populations using other genomic resources
Before the development of long read sequencing and pangenomes, sheep benefitted from the availability of several genotyping tools, including the Illumina® 50K Ovine Beadchip, both for the purposes of genomic selection, and for capturing genetic diversity using a set of genetic markers. The Illumina® OvineSNP50 BeadChip was developed by the International Sheep Genomics Consortium (ISGC; www.sheephapmap.org; Kijas et al., 2009). (Kijas et al., 2012) used the Illumina® 50K chip to genotype 49,034 SNPs in 2,819 animals from a diverse collection of 74 sheep breeds, generating the sheep HapMap dataset (https://www.sheephapmap.org/hapmap.php), which provided a global picture of the genetic history of sheep and variation across breeds. More recent studies have added 50K genotyping data from additional geographical locations and local and indigenous breeds not represented in the original HapMap dataset (Kijas et al., 2012), including from, Asia (Wei et al., 2015), Russia (Deniskova et al., 2018) India (Kumar et al., 2021) and Eastern Europe (Machová et al. 2023). Adding 50K genotypes from the African continent e.g. from North and East Africa (Ahbara et al., 2019) and West/Central Africa (Wiener et al. 2022) illustrates the unique diversity represented by these breeds and highlights the importance of including the diversity they represent in new genomic resources for sheep (Figure 2). In addition, characterising the genetics of production breeds is also important to understand genetic relationships between breeds. The 50K chip has been used, for example, to characterise the genetic diversity of terminal sires in the US (Davenport et al., 2020) and the genetic diversity in New Zealand’s composite flocks was characterised using a higher density 600K chip (Brito et al., 2017b). When combined the 50K genotype datasets for sheep now probably capture a considerable amount of the genetic diversity represented by sheep breeds from across the globe.
Genotyping data is also useful for conservation purposes. Many indigenous local breeds are now very rare, including for example, the Cameroon sheep from West/Central Africa. As such zoo populations often provide important reservoirs of genetic diversity that can be characterised using genotyping tools (Woodruff 2001). The three ‘Beale Park’ individuals shown in Figure 2 below are a trio of Cameroon sheep from a wildlife park collection in the UK. Although they are purportedly a “West/Central African” breed, these individuals originated from zoo populations that have been bred in Europe over several generations. Analysis of their 50K genotypes reflect this, as they cluster some distance from the rare Cameroon sheep and closer to the more common Barbados Blackbelly which is similar in appearance. As such their genetics are unlikely to be sufficiently representative of Cameroon sheep populations from West/Central Africa to be useful for conservation purposes.
A wealth of short read whole genome sequencing data also now exists for sheep breeds and populations from across the globe. (Li et al., 2020), for example, performed deep resequencing of 248 sheep, including wild Ovis orientalis landraces and improved breeds, and were able to detect genomic regions containing genetic variation of relevance to domestication, breeding, and selection. With additional whole genome sequencing data they were then able to define chromosomal evolution between wild, hybrid and domestic sheep (Li et al., 2022b). Recently, (Deng et al., 2020) also provided a comprehensive genomic analysis of haplotype diversity in the Y chromosome, mitochondrial DNA, and variants called from whole genome sequence data from 595 sheep representing 118 domestic populations.
Climate change and the pressures it will place on food production will shape future sheep populations and production systems, making characterising and conserving existing genomic diversity increasingly important (Georges et al., 2019). Short read whole genome sequencing data can provide a tool to investigate adaptation in populations of sheep living in diverse and extreme environments at the genomic level e.g. (Yang et al., 2016; Wiener et al., 2021). (Wiener et al., 2021) identified over three million single nucleotide variants across twelve Ethiopian sheep populations and applied landscape genomics approaches to investigate the association between these variants and environmental variables. (Yang et al., 2016) performed whole genome sequencing of 77 sheep living a varying altitude and detected a novel set of candidate genes associated with hypoxia response at high altitudes and water reabsorption in arid environments. These studies illustrate how informative large scale short read whole genome sequencing from diverse populations of sheep can be in identifying the genomic variation driving complex traits such as environmental adaptation and resilience in extreme environments. Harnessing the power of this functional variation will be important in future breeding strategies that aim to select for resilience traits that will help to mitigate the effects of extremes of climate on sheep production systems.
The wealth of short read whole genome sequencing data for sheep provides a rich and diverse set of sequence information from which to call variants. There are several resources available to view and mine this data including iSheep: an integrated resource for sheep variant, phenotype and genome information (Wang et al., 2021). The sheep genomes database (SheepGenomesDB) (https://sheepgenomesdb.org) houses the sequence variants called, using a standardised pipeline, from sheep short read whole genome sequencing data that has been deposited in the public archives. It is a hugely valuable community resource, not least because calling variants against the reference genome sequence takes a considerable amount of time and computational resource. Through the application of a single harmonised pipeline for read quality control, mapping, variant detection, and annotation, SheepGenomesDB makes available variant collections derived in a standardised manner against the reference genome. The recent change from the Texel Oarv3.1 to the Rambouillet ARS-UI_Ramb_v2.0, as the community adopted reference genome sequence, means that a new set of consensus variant calls is currently being generated. The new variant call set will be deposited in the European Variant Archive (EVA). Once in EVA the variant track can be visualised against the Rambouillet ARS-UI_Ramb_v2.0 by using the Ensembl genome browser (Hunt et al., 2018). Generating this new set of consensus calls for sheep will provide a hugely useful set of genetic markers representing global diversity.
Given the amount and diversity of whole genome sequencing data that is publicly available, it would now also be possible to generate a diverse haplotype reference panel for sheep, similar to those available for pig (Nosková et al., 2021) and cattle (Snelling et al., 2020), for imputation purposes. This resource would open-up a host of possibilities for low pass sequencing of many individuals capturing both between and within population diversity and providing the potential to improve genomic prediction by optimising the markers used in evaluation.
Genomic selection in sheep – integrating available genomic resources as a priori information in breeding programmes
A key component of improving profit and production output in sheep, particularly in Australia and New Zealand, has been the use of genomic selection (Daetwyler et al. 2010). Genomic selection is a form of marker-assisted selection in which genetic markers covering the whole genome are used to estimate an animal’s breeding value (Goddard and Hayes, 2007). In sheep causative variants for production relevant traits with large phenotypic effects, have been successfully detected, using quantitative, population and molecular genetics approaches e.g. for carcass traits (Clop et al., 2006; Tellam et al., 2012; Matika et al., 2016). However, the majority of health, welfare and resilience traits, are polygenic and any causative variants are likely to have small effects, which makes detecting them more difficult (Georges et al., 2019). Functional genomic data can help enrich for variance in quantitative traits (reviewed in Johnsson 2023). Since most causal variants for complex traits are likely to be located in regulatory regions of the genome and will impact complex traits by changing gene expression (Tam et al., 2019) improvements in prediction accuracy could be achieved by filtering the genetic marker information, used for genomic selection, based upon whether the genetic variants reside in regulatory regions of the genome and then developing robust prediction models that can accommodate information about genome function (Georges et al., 2019).
Recently, new methods for integrating genomic information, such as gene expression or methylation data, into genomic prediction models have been proposed e.g. (Xiang et al., 2019, 2021). These multi-layered models, which are based on the combination and ranking of many types of functional genomic data from multiple individuals, have been shown for cattle to facilitate further improvements in predicting genetic merit and consequently on genomic selection (Xiang et al., 2019, 2021). (Liu et al., 2022) also recently demonstrated the feasibility of linking variants associated with complex traits from GWAS with gene expression information across tissues and cell types in cattle and the consortia has plans to extend these efforts to the other farmed animal species. There are, however, currently only a handful of datasets for sheep, that are suitable for this purpose, such as a recently published expression QTL study from muscle and liver for carcass traits (Yuan et al., 2021b). However, the potential to generate gene expression information at a population scale now exists due to a reduction in cost of RNA-sequencing and the development of new assays that are deployable at scale such as Illumina 3’-sequencing. The challenge for sheep may be accessing phenotype data as recording in sheep is much less advanced across traits than for cattle, pigs and chicken. However, accurate recording to inform selection strategies will becoming increasingly important as future extremes of climate put pressure on producers to select animals that are more resilient.
New genomic resources can inform genome editing and the use of sheep as biomedical models
While genomic selection is likely to provide the foundation of many future commercial breeding programmes for sheep, it is limited by the genetic pool of the population under selection. If a target trait is not encoded in the genome of a breeding population, then it is not possible to select for it. Genome editing has the potential to offer an effective solution to this problem (McFarlane et al., 2019). Sheep are particularly amenable to genome editing and it has been applied successfully for a small number of production relevant target genes, reviewed in (Proudfoot et al., 2015). Advances in the genomic resources for sheep will provide information to identify new editing targets particularly those that control breed-specific characteristics that may be present in one breeding population but not in another. One example is the ‘polled’ or hornlessness trait that is a distinct characteristic of some breeds such as the Poll Dorset. Horns can cause injury both to the sheep themselves and to their handlers and consequently, particularly in production animals, polledness is desirable. However, some production breeds with desirable resilience and sustainability traits, like the Wiltshire Horn, a wool-shedding breed with a good carcass and high feed efficiency, have undesirable large horns that make them difficult to handle and manage. Gene editing for polledness has been achieved successfully in cattle, reviewed in (Van Eenennaam, 2019), but in sheep is likely to be more complex, reviewed in (Simon et al. 2022). A 1.78Kb insertion in the 3’UTR region of the RXFP2 gene on chromosome 10 has been identified which is strongly associated with polledness in GWAS (Wiedemar and Drögemüller, 2015) however it does not segregate in the same way across all breeds (Lühken et al., 2016). Comparative approaches to analyse breed specific genomic resources for sheep, across individuals and populations, will help to reveal the functional basis of traits present in one breed or population that are desirable in another providing novel targets for selective breeding and/or genome editing (Clark 2022).
In addition to their role as food production animals sheep are also important biomedical models (Banstola and Reynolds, 2022). The new highly contiguous ARS-UI_Ramb_v2.0 reference genome and associated annotation, provides a research tool that can inform studies designed to identify alleles encoding human physiological processes and diseases. One recent example, is the novel sheep model of CLN1 disease, in which gene editing was used to insert a disease-causing PPT1 (R151X) human mutation into the orthologous sheep locus (Eaton et al., 2019; Nelvagal et al., 2022). High-throughput CRISPR/Cas9 knock-out libraries, such as those available for pigs e.g., Yu et al., (2022), will help considerably with identifying novel alleles for genome editing in both human and farmed animal studies but at present a lack of suitable primary cell lines for sheep is a barrier to progress. As the applications of genome editing technologies in the biomedical field expand a high-quality annotated reference genome for sheep on which to base target selection will become even more useful.
The future
In addition to the new genomic resources for sheep described above there are further exciting developments on the horizon (Figure 2). For example, recent improvements in tools and resources for long read sequencing have made assembling fully contiguous assembled telomere to telomere genomes possible. The human telomere-to-telomere assembly is a revolutionary new tool for human research unlocking the complex regions of the genome to study genome function and genetic variation (Nurk et al., 2022). A telomere-to-telomere reference assembly for sheep is currently being generated for the Ruminant Telomere-to-Telomere project which is led by the USDA and University of Idaho.
From a transcriptome perspective, since publication of the sheep gene expression atlas, expanded transcriptomes, that include histological tissue maps and characterisation of all RNA populations, have been published, e.g. for pig (Jin et al., 2021), and similar new resources of this type for sheep will soon follow. Furthermore, long read RNA isoform sequencing technologies, can now capture full-length isoform information, even at single cell level resolution. These technologies make transcript annotation considerably easier and allow for the characterisation of splicing events and prediction of full-length open reading frames. Isoform sequencing (Iso-Seq) data for a small subset of tissues is available for sheep, for the purposes of annotating the Rambouillet genome, and from a small number of published studies that have focused on specific tissues relevant to phenotypes of interest (Yuan et al., 2021a, 2022). New long read isoform sequencing datasets for multiple tissues, cell types and developmental stages, will provide a valuable novel resource for genome annotation and build on the transcriptomic resources already provided by short read RNA-Seq data. Long read sequencing technologies will also facilitate, the generation of breed- specific transcriptomes. These breed-specific transcriptomes based on full-length isoform information, will allow the classification of sets of pan-genes and pan-transcriptomes for sheep providing new insights into how isoform usage can influence key traits across different breeds.
The primary challenge facing the sheep and wider farmed animal genomics community now is harnessing the power of a highly accurate reference genome with functional genomics data at a population scale and from there how to leverage this information to enhance genomic prediction (reviewed in Johnsson 2023). The potential to go ‘beyond the genome’ by using epigenetic modifications to predict genetic merit also shows significant potential (reviewed in Clarke et al., 2021). DNA methylation arrays, for example, have proved to be useful tools for informing breeding programmes for sheep, and provide an opportunity to accelerate the physiological response of breeding populations to environmental pressures (Clarke et al., 2021). Tools to visualise the combination of genetic variation with predicted function will be critical in advancing the sheep genomics field. Functional genomic comparisons of different sheep breeds will become increasingly powerful as haplotype-resolved reference genomes and pangenomes with matched functional annotation data become the new standard for sheep and other economically important farmed animal species.
Conclusions
The field of sheep genomics has undoubtedly moved into a new era. New functional annotation datasets for sheep for many different tissues and cell types provide new resources to link cell, tissue and whole animal scale knowledge. Novel opportunities also now exist for interrogating gene regulation information at single cell resolution providing a much more complete picture of transcriptional complexity in sheep. Affordable long read sequencing technologies have caused an explosion in the number of new genome assemblies that are being generated for many different breeds and populations. Genetic improvement in the future will also almost certainly include the use of pangenomes to understand and visualise the diversity of farmed animal genomes (Hayes and Daetwyler, 2019). For this reason, pangenome efforts should ensure they capture the global genetic diversity of sheep breeds, including those from the global south. Logistical considerations will inevitably arise with the rapid expansion of genomes and genomic resources for sheep. Genome browsers, such as Ensembl, will need to keep pace with how rapidly these new genomic and transcriptomic resources are being generated. This will need to happen quickly in order that the community can maximise the benefit of this new information, and will require resources, effort and funding (Cunningham et al., 2022). The sheep genomics research community will also need to work with stakeholders to decide what the priorities are for the coming decade. These priorities should be centred around providing resources that can inform global sheep breeding systems in a way that will help to accelerate their response to future extremes of climate, produce healthier improved animals and provide enough food for a growing human population.
Funding
This work was supported by Biotechnology and Biological Sciences Research Council (BBSRC) grants “Empowering sheep breeding by identifying variants associated with growth traits using allele-specific expression” (BB/S01540X/1) and “Ensembl in a new era - deep genome annotation of domesticated animal species and breeds” (BB/W018772/1) as well as Institute Strategic Programme Grant “Prediction of genes and regulatory elements in farm animal genomes” (BBS/E/D/10002070) awarded to the Roslin Institute. This work was also supported in part by the Gates Foundation and with UK aid from the UK Foreign, Commonwealth and Development Office (Grant Agreement OPP1127286) under the auspices of the Centre for Tropical Livestock Genetics and Health (CTLGH), established jointly by the University of Edinburgh, SRUC (Scotland’s Rural College), and the International Livestock Research Institute.
Competing Interests
The authors declare that they have no financial or non-financial interests that are directly or indirectly related to the work submitted for publication.
References
- Ahbara, A., Bahbahani, H., Almathen, F., Al Abri, M., Agoub, M. O., Abeba, A., et al. (2019). Genome-Wide Variation, Candidate Regions and Genes Associated With Fat Deposition and Tail Morphology in Ethiopian Indigenous Sheep. Front. Genet.9. https://www.frontiersin.org/articles/10.3389/fgene.2018.00699. [CrossRef]
- Aken, B. L., Ayling, S., Barrell, D., Clarke, L., Curwen, V., Fairley, S., et al. (2016). The Ensembl gene annotation system. Database 2016, baw093. [CrossRef]
- Alberto, F. J., Boyer, F., Orozco-terWengel, P., Streeter, I., Servin, B., de Villemereuil, P., et al. (2018). Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 9, 813. [CrossRef]
- Andersson, L., Archibald, A. L., Bottema, C. D., Brauning, R., Burgess, S. C., Burt, D. W., et al. (2015). Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 16, 57. [CrossRef]
- Animal Genetics Training Resource (AGTR) - Dorper, available via http://agtr.ilri.cgiar.org/dorper, last accessed 11th April 2023.
- Banstola, A., and Reynolds, J. N. J. (2022). The Sheep as a Large Animal Model for the Investigation and Treatment of Human Disorders. Biology. 11. [CrossRef]
- Bickhart, D. M., Rosen, B. D., Koren, S., Sayre, B. L., Hastie, A. R., Chan, S., et al. (2017). Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–650. [CrossRef]
- Birney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigó, R., Gingeras, T. R., Margulies, E. H., et al. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. [CrossRef]
- Brito, L. F., Clarke, S. M., McEwan, J. C., Miller, S. P., Pickering, N. K., Bain, W. E., et al. (2017a). Prediction of genomic breeding values for growth, carcass and meat quality traits in a multi-breed sheep population using a HD SNP chip. BMC Genet. 18, 7. [CrossRef]
- Brito, L.F., McEwan, J.C., Miller, S.P. et al. (2017b) Genetic diversity of a New Zealand multi-breed sheep population and composite breeds’ history revealed by a high-density SNP chip. BMC Genet 18, 25. [CrossRef]
- Chen, N.-C., Solomon, B., Mun, T., Iyer, S., and Langmead, B. (2021). Reference flow: reducing reference bias using multiple population genomes. Genome Biol. 22, 8. [CrossRef]
- Clark, E. L. (2022). “Breeding in an Era of Genome Editing" In: Encyclopedia of Sustainability Science and Technology - Animal Breeding and Genetics ed. R. A. Meyers (New York, NY: Springer New York), 1–16. [CrossRef]
- Clark, E. L., Bush, S. J., McCulloch, M. E. B., Farquhar, I. L., Young, R., Lefevre, L., et al. (2017). A high resolution atlas of gene expression in the domestic sheep (Ovis aries). PLOS Genet. 13, e1006997. [CrossRef]
- Clark, E. L., Archibald, A. L., Daetwyler, H. D., Groenen, M. A. M., Harrison, P. W., Houston, R. D., et al. (2020). From FAANG to fork: application of highly annotated genomes to improve farmed animal production. Genome Biol. 21, 285. [CrossRef]
- Clarke S, Caulton A, McRae K, Brauning R, Couldrey C, Dodds K. Beyond the genome: a perspective on the use of DNA methylation profiles as a tool for the livestock industry. Anim Front. 11(6):90-94. [CrossRef]
- Clop, A., Marcq, F., Takeda, H., Pirottin, D., Tordoir, X., Bibé, B., et al. (2006). A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 38, 813–818. [CrossRef]
- Cockett NE. (2006) The sheep genome. Genome Dyn. 2:79-85. [CrossRef]
- Conington, JE., Bishop, SC., Grundy, B., Waterhouse, A., & Simm, G. (2001). Multi-trait selection indexes for sustainable UK hill sheep production. Animal Science, 73, 413 - 423.
- Crysnanto, D., and Pausch, H. (2020). Bovine breed-specific augmented reference graphs facilitate accurate sequence read mapping and unbiased variant discovery. Genome Biol., 184. [CrossRef]
- Crysnanto, D., Leonard, A. S., Fang, Z.-H., and Pausch, H. (2021). Novel functional sequences uncovered through a bovine multiassembly graph. Proc. Natl. Acad. Sci. 118, e2101056118. [CrossRef]
- Cunningham, F., Allen, J. E., Allen, J., Alvarez-Jarreta, J., Amode, M. R., Armean, I. M., et al. (2022). Ensembl 2022. Nucleic Acids Res. 50, D988–D995. [CrossRef]
- Daetwyler, H. D., Hickey, J. M., Henshall, J. M., Dominik, S., Gredler, B., van der Werf, J. H. J., et al. (2010). Accuracy of estimated genomic breeding values for wool and meat traits in a multi-breed sheep population. Anim. Prod. Sci. 50, 1004–1010. [CrossRef]
- Dalrymple, B. P., Kirkness, E. F., Nefedov, M., McWilliam, S., Ratnakumar, A., Barris, W., et al. (2007). Using comparative genomics to reorder the human genome sequence into a virtual sheep genome. Genome Biol. 8, R152. [CrossRef]
- Davenport, K.M., Hiemke, C., McKay, S.D., Thorne, J.W., Lewis, R.M., Taylor, T. and Murdoch, B.M. (2020), Genetic structure and admixture in sheep from terminal breeds in the United States. Anim Genet, 51: 284-291. [CrossRef]
- Davenport, K. M., Massa, A. T., Bhattarai, S., McKay, S. D., Mousel, M. R., Herndon, M. K., et al. (2021). Characterizing Genetic Regulatory Elements in Ovine Tissues. Front. Genet. 12, 566. https://www.frontiersin.org/article/10.3389/fgene.2021.628849. [CrossRef]
- Davenport, K. M., Bickhart, D. M., Worley, K., Murali, S. C., Salavati, M., Clark, E. L., et al. (2022). An improved ovine reference genome assembly to facilitate in-depth functional annotation of the sheep genome. Gigascience 11, giab096. [CrossRef]
- Deng, J., Xie, X.-L., Wang, D.-F., Zhao, C., Lv, F.-H., Li, X., et al. (2020). Paternal Origins and Migratory Episodes of Domestic Sheep. Curr. Biol. 30, 4085-4095.e6. [CrossRef]
- Deniskova, T. E., Dotsev, A. V, Selionova, M. I., Kunz, E., Medugorac, I., Reyer, H., et al. (2018). Population structure and genetic diversity of 25 Russian sheep breeds based on whole-genome genotyping. Genet. Sel. Evol. 50, 29. [CrossRef]
- Dumont B, Puillet L, Martin G, Savietto D, Aubin J, Ingrand S, Niderkorn V, Steinmetz L and Thomas M (2020) Incorporating Diversity Into Animal Production Systems Can Increase Their Performance and Strengthen Their Resilience. Front. Sustain. Food Syst. 4:109. [CrossRef]
- Dutta, P., Talenti, A., Young, R. et al. Whole genome analysis of water buffalo and global cattle breeds highlights convergent signatures of domestication. Nat Commun 11, 4739 (2020). [CrossRef]
- Eaton, S. L., Proudfoot, C., Lillico, S. G., Skehel, P., Kline, R. A., Hamer, K., et al. (2019). CRISPR/Cas9 mediated generation of an ovine model for infantile neuronal ceroid lipofuscinosis (CLN1 disease). Sci. Rep. 9, 9891. [CrossRef]
- European Forum of Farmed Animal Breeders (EFFAB) (2020) Code of Good Practice for Farm Animal Breeding Organisations (Code EFABAR). (available at: http://www.responsiblebreeding.eu/uploads/2/3/1/3/23133976/01_general_document_2020_final-code_efabar.pdf).
- FAO. (2015) The Second Report on the State of the World’s Animal Genetic Resources for Food and Agriculture, edited by B.D. Scherf & D. Pilling. FAO Commission on Genetic Resources for Food and Agriculture Assessments. Rome (available at http://www.fao.org/3/a-i4787e/index.html).
- Foissac, S., Djebali, S., Munyard, K., Vialaneix, N., Rau, A., Muret, K., et al. (2019). Multi-species annotation of transcriptome and chromatin structure in domesticated animals. BMC Biol. 17, 108. [CrossRef]
- Freeman, T. C., Horsewell, S., Patir, A., Harling-Lee, J., Regan, T., Shih, B. B., et al. (2022). Graphia: A platform for the graph-based visualisation and analysis of high dimensional data. PLOS Comput. Biol. 18, e1010310. [CrossRef]
- Geerts S, Osaer S, Goossens B and Faye D. (2009) Trypanotolerance in small ruminants of sub-Saharan Africa. Trends in Parasitology, 25(3):132-8. [CrossRef]
- Georges, M., Charlier, C., and Hayes, B. (2019). Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 20, 135–156. [CrossRef]
- Giuffra, E., and Tuggle, C. K. (2019). Functional Annotation of Animal Genomes (FAANG): Current Achievements and Roadmap. Annu. Rev. Anim. Biosci. 7, 65–88. [CrossRef]
- Goddard, M. E., and Hayes, B. J. (2007). Genomic selection. J. Anim. Breed. Genet. 124, 323–330. [CrossRef]
- Goszczynski DE, Halstead MM, Islas-Trejo AD, Zhou H, Ross PJ. Transcription initiation mapping in 31 bovine tissues reveals complex promoter activity, pervasive transcription, and tissue-specific promoter usage. Genome Res. 2021 Apr;31(4):732-744. [CrossRef]
- Harrison PW, Sokolov A, Nayak A, Fan J, Zerbino D, Cochrane G and Flicek P (2021) The FAANG Data Portal: Global, Open-Access, “FAIR”, and Richly Validated Genotype to Phenotype Data for High-Quality Functional Annotation of Animal Genomes. Front. Genet. 12:639238. [CrossRef]
- Hayes B. J. and Daetwyler H. D. (2019) 1000 Bull Genomes Project to Map Simple and Complex Genetic Traits in Cattle: Applications and Outcomes. Ann Rev Animal Biosci. 7:1, 89-102. [CrossRef]
- Hayes, B. J., Lewin, H. A., and Goddard, M. E. (2013). The future of livestock breeding: genomic selection for efficiency, reduced emissions intensity, and adaptation. Trends Genet. 29, 206–214. [CrossRef]
- Herrera-Uribe J, Wiarda JE, Sivasankaran SK, Daharsh L, Liu H, Byrne KA, Smith TPL, Lunney JK, Loving CL, Tuggle CK. (2021) Reference Transcriptomes of Porcine Peripheral Immune Cells Created Through Bulk and Single-Cell RNA Sequencing. Front Genet. 23;12:689406. [CrossRef]
- Hunt, S. E., McLaren, W., Gil, L., Thormann, A., Schuilenburg, H., Sheppard, D., et al. (2018). Ensembl variation resources. Database, bay119. [CrossRef]
- International Sheep Genomics Consortium (ISGC), available via https://www.sheephapmap.org/, last accessed 11th April 2023.
- Jiang, Y., Xie, M., Chen, W., Talbot, R., Maddox, J. F., Faraut, T., et al. (2014). The sheep genome illuminates biology of the rumen and lipid metabolism. Science 344, 1168–1173. [CrossRef]
- Jin, L., Tang, Q., Hu, S., Chen, Z., Zhou, X., Zeng, B., et al. (2021). A pig BodyMap transcriptome reveals diverse tissue physiologies and evolutionary dynamics of transcription. Nat. Commun. 12, 3715. [CrossRef]
- Johnsson, M. (2023) The big challenge for livestock genomics is to make sequence data pay, arXiv, 2302.01140. [CrossRef]
- Kern, C., Wang, Y., Xu, X., Pan, Z., Halstead, M., Chanthavixay, G., et al. (2021). Functional annotations of three domestic animal genomes provide vital resources for comparative and agricultural research. Nat. Commun. 12, 1821. [CrossRef]
- Kijas J.W., Townley D., Dalrymple B.P., Heaton M.P., Maddox J.F., McGrath A., Wilson P.et al. (2009). A genome wide survey of SNP variation reveals the genetic structure of sheep breeds. PLoS ONE4(3):E4668. [CrossRef]
- Kijas, J. W., Lenstra, J. A., Hayes, B., Boitard, S., Porto Neto, L. R., San Cristobal, M., et al. (2012). Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 10, e1001258. [CrossRef]
- Koren, S., Rhie, A., Walenz, B. P., Dilthey, A. T., Bickhart, D. M., Kingan, S. B., et al. (2018). De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol., 10.1038/nbt.4277. [CrossRef]
- Kumar, H., Panigrahi, M., Rajawat, D., Panwar, A., Nayak, S. S., Kaisa, K., et al. (2021). Selection of breed-specific SNPs in three Indian sheep breeds using ovine 50 K array. Small Rumin. Res. 205, 106545. [CrossRef]
- Lawson Handley, L.-J., Byrne, K., Santucci, F., Townsend, S., Taylor, M., Bruford, M. W., et al. (2007). Genetic structure of European sheep breeds. Heredity. 99, 620–631. [CrossRef]
- Li, R., Fu, W., Su, R., Tian, X., Du, D., Zhao, Y., et al. (2019). Towards the Complete Goat Pan-Genome by Recovering Missing Genomic Segments From the Reference Genome. Front. Genet. 10, 1169. https://www.frontiersin.org/article/10.3389/fgene.2019.01169. [CrossRef]
- Li, X., Yang, J., Shen, M., Xie, X.-L., Liu, G.-J., Xu, Y.-X., et al. (2020). Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 11, 2815. [CrossRef]
- Li, R., Yang, P., Li, M., Fang, W., Yue, X., Nanaei, H. A., et al. (2021). A Hu sheep genome with the first ovine Y chromosome reveal introgression history after sheep domestication. Sci. China. Life Sci. 64, 1116–1130. [CrossRef]
- Li, R., Gong, M., Zhang, X., Wang, F., Liu, Z., Zhang, L., et al. (2022a). The first sheep graph-based pan-genome reveals the spectrum of structural variations and their effects on tail phenotypes. bioRxiv, 2021.12.22.472709. [CrossRef]
- Li, X., He, S.-G., Li, W.-R., Luo, L.-Y., Yan, Z., Mo, D.-X., et al. (2022b). Genomic analyses of wild argali, domestic sheep, and their hybrids provide insights into chromosome evolution, phenotypic variation, and germplasm innovation. Genome Res. 32, 1669–1684. [CrossRef]
- Liu, S., Gao, Y., Canela-Xandri, O., Wang, S., Yu, Y., Cai, W., et al. (2022). A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 54, 1438–1447. [CrossRef]
- Liu, Y., Murali, S. C., Harris, R. A., English, A. C., Qin, X., Skinner, E., et al. (2016). P1009 Sheep reference genome sequence updates: Texel improvements and Rambouillet progress. J. Anim. Sci. 94, 18–19. [CrossRef]
- Lühken, G., Krebs, S., Rothammer, S., Küpper, J., Mioč, B., Russ, I., et al. (2016). The 1.78-kb insertion in the 3′-untranslated region of RXFP2 does not segregate with horn status in sheep breeds with variable horn status. Genet. Sel. Evol. 48, 78. [CrossRef]
- Macfarlane JM, Lambe NR, Matika O, Johnson PL, Wolf BT, Haresign W, Bishop SC, Bünger L. Effect and mode of action of the Texel muscling QTL (TM-QTL) on carcass traits in purebred Texel lambs. Animal. 2014 Jul;8(7):1053-61. [CrossRef]
- Machová K, Marina H, Arranz JJ, Pelayo R, Rychtářová J, Milerski M, Vostrý L, Suárez-Vega A. (2023) Genetic diversity of two native sheep breeds by genome-wide analysis of single nucleotide polymorphisms. Animal. Jan;17(1):100690. [CrossRef]
- Marino, R., Atzori A.S., D’Andrea, M., Iovane, G., Trabalza-Marinucci, M., and Rinaldi, L. (2016). Climate change: Production performance, health issues, greenhouse gas emissions and mitigation strategies in sheep and goat farming. Small Rumin. Res. 135, 50–59. [CrossRef]
- Marshall, F. B., Dobney, K., Denham, T., and Capriles, J. M. (2014). Evaluating the roles of directed breeding and gene flow in animal domestication. Proc. Natl. Acad. Sci. 111, 6153 LP – 6158. [CrossRef]
- Marshall, K., Gibson, J. P., Mwai, O., Mwacharo, J. M., Haile, A., Getachew, T., et al. (2019). Livestock Genomics for Developing Countries – African Examples in Practice. Front. Genet. 10, 297. [CrossRef]
- Matika, O., Riggio, V., Anselme-Moizan, M., Law, A. S., Pong-Wong, R., Archibald, A. L., et al. (2016). Genome-wide association reveals QTL for growth, bone and in vivo carcass traits as assessed by computed tomography in Scottish Blackface lambs. Genet. Sel. Evol. 48, 11. [CrossRef]
- McFarlane, G. R., Salvesen, H. A., Sternberg, A., and Lillico, S. G. (2019). On-Farm Livestock Genome Editing Using Cutting Edge Reproductive Technologies. Front. Sustain. Food Syst. 3, 106. https://www.frontiersin.org/article/10.3389/fsufs.2019.00106. [CrossRef]
- McKenzie, F. C., and Williams, J. (2015). Sustainable food production: constraints, challenges and choices by 2050. Food Secur. 7, 221–233. [CrossRef]
- Mignon-Grasteau, S., Boissy, A., Bouix, J., Faure, J.-M., Fisher, A. D., Hinch, G. N., et al. (2005). Genetics of adaptation and domestication in livestock. Livest. Prod. Sci. 93, 3–14. [CrossRef]
- Mollenhorst, H., Y. de Haas, 2019. The contribution of breeding to reducing environmental impact of animal production. Wageningen Livestock Research, Report 1156.
- Murdoch, B. M. (2019). The functional annotation of the sheep genome project. J. Anim. Sci. 97, 16. [CrossRef]
- Naval-Sanchez, M., Nguyen, Q., McWilliam, S., Porto-Neto, L. R., Tellam, R., Vuocolo, T., et al. (2018). Sheep genome functional annotation reveals proximal regulatory elements contributed to the evolution of modern breeds. Nat. Commun. 9, 859. [CrossRef]
- Nelvagal, H. R., Eaton, S. L., Wang, S. H., Eultgen, E. M., Takahashi, K., Le, S. Q., et al. (2022). Cross-species efficacy of enzyme replacement therapy for CLN1 disease in mice and sheep. J. Clin. Invest. 132. [CrossRef]
- Nosková, A., Bhati, M., Kadri, N. K., Crysnanto, D., Neuenschwander, S., Hofer, A., et al. (2021). Characterization of a haplotype-reference panel for genotyping by low-pass sequencing in Swiss Large White pigs. BMC Genomics 22, 290. [CrossRef]
- Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V, Mikheenko, A., et al. (2022). The complete sequence of a human genome. Science (80). 376, 44–53. [CrossRef]
- Pollard, M. O., Gurdasani, D., Mentzer, A. J., Porter, T., and Sandhu, M. S. (2018). Long reads: their purpose and place. Hum. Mol. Genet. 27, R234–R241. [CrossRef]
- Proudfoot, C., Carlson, D. F., Huddart, R., Long, C. R., Pryor, J. H., King, T. J., et al. (2015). Genome edited sheep and cattle. Transgenic Res. 24, 147–153. [CrossRef]
- Qiao, G., Xu, P., Guo, T., Wu, Y., Lu, X., Zhang, Q., et al. (2022). Genetic Basis of Dorper 13:846449. [CrossRef]
- Rice, E. S., Koren, S., Rhie, A., Heaton, M. P., Kalbfleisch, T. S., Hardy, T., Hackett, P. H., Derek M Bickhart, Rosen, B. D., Vander Ley, B., Maurer, N. W., Green, R. E., Phillippy, A. M., Petersen, J. L., Smith, T. P. L. (2020) Continuous chromosome-scale haplotypes assembled from a single interspecies F1 hybrid of yak and cattle, GigaScience, Volume 9, Issue 4, giaa029. [CrossRef]
- Salavati, M., Caulton, A., Clark, R., Gazova, I., Smith, T. P. L., Worley, K. C., et al. (2020). Global Analysis of Transcription Start Sites in the New Ovine Reference Genome (Oar rambouillet v1.0). Front. Genet. 11, 1184. [CrossRef]
- Schaum N, Karkanias J, Neff NF, May AP, Quake SR, Wyss-Coray T, et al. (2018) Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 562:367. [CrossRef]
- Simon R, Drögemüller C. and Lühken G. (2022) The Complex and Diverse Genetic Architecture of the Absence of Horns (Polledness) in Domestic Ruminants, including Goats and Sheep. Genes (6;13(5):832. [CrossRef]
- Snelling, W. M., Hoff, J. L., Li, J. H., Kuehn, L. A., Keel, B. N., Lindholm-Perry, A. K., et al. (2020). Assessment of Imputation from Low-Pass Sequencing to Predict Merit of Beef Steers. Genes.. 11. [CrossRef]
- Spangler, G. L., Rosen, B. D., Ilori, M. B., Hanotte, O., Kim, E.-S., Sonstegard, T. S., et al. (2017). Whole genome structural analysis of Caribbean hair sheep reveals quantitative link to West African ancestry. PLoS One 12, e0179021. [CrossRef]
- Talenti, A., Powell, J., Hemmink, J. D., Cook, E. A. J., Wragg, D., Jayaraman, S., et al. (2022). A cattle graph genome incorporating global breed diversity. Nat. Commun. 13, 910. [CrossRef]
- Tam, V., Patel, N., Turcotte, M., Bossé, Y., Paré, G., and Meyre, D. (2019). Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484. [CrossRef]
- Tellam, R. L., Cockett, N. E., Vuocolo, T., and Bidwell, C. A. (2012). Genes contributing to genetic variation of muscling in sheep. Front. Genet. 3, 164. [CrossRef]
- The Computational Pan-Genomics Consortium (2018) Computational pan-genomics: status, promises and challenges, Briefings in Bioinformatics, 19, 1, 118–135. [CrossRef]
- The Rare Breed Survival Trust - Norfolk Horn, available via: https://www.rbst.org.uk/norfolk-horn, last accessed 11th April 2023.
- The Suffolk Sheep Society - History, available via: https://www.suffolksheep.org/history/, last accessed 11th April 2023.
- Van Eenennaam, A. L. (2019). Application of genome editing in farm animals: cattle. Transgenic Res. 28, 93–100. [CrossRef]
- Wang Z-H, Zhu Q-H, Li X, Zhu J-W, Tian D-M, Zhang S-S, Kang H-L, Li C-P, Dong L-L, Zhao W-M and Li M-H (2021) iSheep: an Integrated Resource for Sheep Genome, Variant and Phenotype. Front. Genet. 12:714852. [CrossRef]
- Wang, F., Ding, P., Liang, X., Ding, X., Brandt, C. B., Sjöstedt, E., et al. (2022). Endothelial cell heterogeneity and microglia regulons revealed by a pig cell landscape at single-cell level. Nat. Commun. 13, 6748. [CrossRef]
- Wei, C., Wang, H., Liu, G., Wu, M., Cao, J., Liu, Z., et al. (2015). Genome-wide analysis reveals population structure and selection in Chinese indigenous sheep breeds. BMC Genomics 16, 194. [CrossRef]
- Wiedemar, N., and Drögemüller, C. (2015). A 1.8-kb insertion in the 3’-UTR of RXFP2 is associated with polledness in sheep. Anim. Genet. 46, 457–461. [CrossRef]
- Wiener, P., Robert, C., Ahbara, A., Salavati, M., Abebe, A., Kebede, A., et al. (2021). Whole-Genome Sequence Data Suggest Environmental Adaptation of Ethiopian Sheep Populations. Genome Biol. Evol. 13. [CrossRef]
- Wiener, P., Salavati, S., Djikeng, A., Van Tassell, C. P., Rosen, B. D., Spangler, G. L., et al. (2022). Genetic diversity of the Cameroon Blackbelly sheep, an indigenous sheep from West Africa. in World Congress in Genetics Applied to Livestock Production. pg 1717-1720. https://www.wageningenacademic.com/pb-assets/wagen/WCGALP2022/37_007.pdf.
- Woodruff DS. (2001) Populations, Species, and Conservation Genetics. Encyclopedia of Biodiversity. 811–29. [CrossRef]
- Worley, K. C. (2017). A golden goat genome. Nat. Genet. 49, 485–486. [CrossRef]
- Xiang, R., Berg, I. van den, MacLeod, I. M., Hayes, B. J., Prowse-Wilkins, C. P., Wang, M., et al. (2019). Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits. Proc. Natl. Acad. Sci. 116, 19398 LP – 19408. [CrossRef]
- Xiang, R., MacLeod, I. M., Daetwyler, H. D., de Jong, G., O’Connor, E., Schrooten, C., et al. (2021). Genome-wide fine-mapping identifies pleiotropic and functional variants that predict many traits across global cattle populations. Nat. Commun. 12, 860. [CrossRef]
- Yang, J., Li, W.-R., Lv, F.-H., He, S.-G., Tian, S.-L., Peng, W.-F., et al. (2016). Whole-Genome Sequencing of Native Sheep Provides Insights into Rapid Adaptations to Extreme Environments. Mol. Biol. Evol. 33, 2576–2592. [CrossRef]
- Yu C, Zhong H, Yang X, Li G, Wu Z, Yang H. (2022) Establishment of a pig CRISPR/Cas9 knockout library for functional gene screening in pig cells. Biotechnol J. 17(7):e2100408. [CrossRef]
- Yuan, Z., Ge, L., Sun, J., Zhang, W., Wang, S., Cao, X., et al. (2021a). Integrative analysis of Iso-Seq and RNA-seq data reveals transcriptome complexity and differentially expressed transcripts in sheep tail fat. PeerJ, 9, e12454. [CrossRef]
- Yuan, Z., Sunduimijid, B., Xiang, R., Behrendt, R., Knight, M. I., Mason, B. A., et al. (2021b). Expression quantitative trait loci in sheep liver and muscle contribute to variations in meat traits. Genet. Sel. Evol. 53, 8. [CrossRef]
- Yuan, Z., Ge, L., Zhang, W., Lv, X., Wang, S., Cao, X., et al. (2022). Preliminary Results about Lamb Meat Tenderness Based on the Study of Novel Isoforms and Alternative Splicing Regulation Pathways Using Iso-seq, RNA-seq and CTCF ChIP-seq Data. Foods 11. [CrossRef]
- Zerbino, D. R., Wilder, S. P., Johnson, N., Juettemann, T., and Flicek, P. R. (2015). The Ensembl Regulatory Build. Genome Biol. 16, 56. [CrossRef]
Figure 1.
Schematic describing how the new genomic resources for sheep will help to inform sheep breeding with the goal of providing healthier and improved animals, to meet growing pressures on food production, while maintaining genomic diversity (adapted from Clark et al. 2020).
Figure 1.
Schematic describing how the new genomic resources for sheep will help to inform sheep breeding with the goal of providing healthier and improved animals, to meet growing pressures on food production, while maintaining genomic diversity (adapted from Clark et al. 2020).
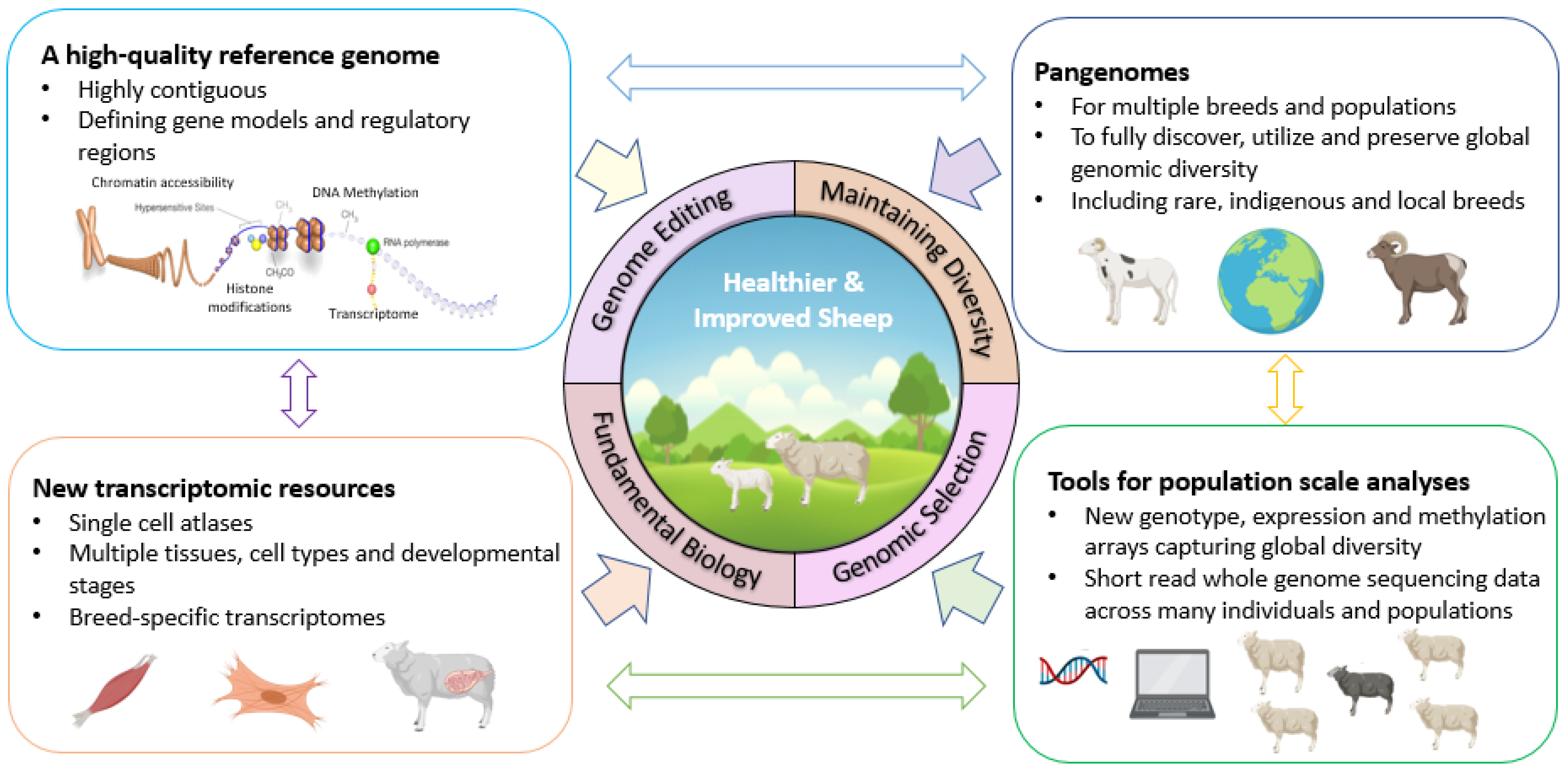
Figure 2.
Principal component analysis illustrating the genetic diversity of sheep breeds from across the globe using 50K genotyping data (PC1 contributed 16% and PC3 7% to the variance). Included in the analysis are 50K genotypes from the HapMap dataset from Kijas et al. 2012, populations of East African sheep from Ahbara et al. 2019 (orange circle) and West and Central African sheep (blue circle) from Wiener et al. 2022. Cameroon sheep from the zoo collection at Beale Park (unpublished) are circled in purple.
Figure 2.
Principal component analysis illustrating the genetic diversity of sheep breeds from across the globe using 50K genotyping data (PC1 contributed 16% and PC3 7% to the variance). Included in the analysis are 50K genotypes from the HapMap dataset from Kijas et al. 2012, populations of East African sheep from Ahbara et al. 2019 (orange circle) and West and Central African sheep (blue circle) from Wiener et al. 2022. Cameroon sheep from the zoo collection at Beale Park (unpublished) are circled in purple.
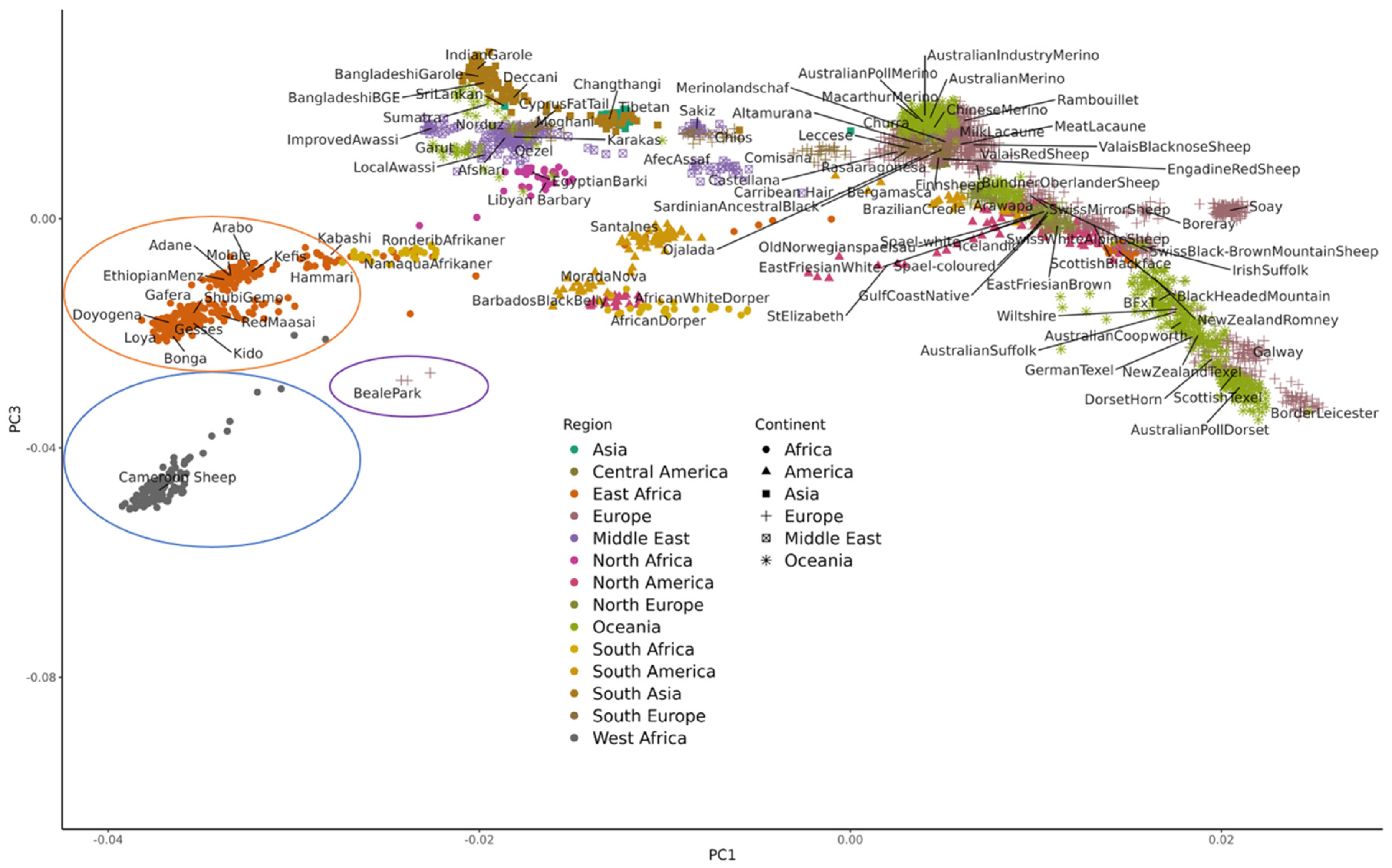

Table 1.
Genome summary statistics for popular sheep reference genome sequence releases based on information from the National Centre for Biotechnology Information (NCBI) genome database.
Table 1.
Genome summary statistics for popular sheep reference genome sequence releases based on information from the National Centre for Biotechnology Information (NCBI) genome database.
| Genome assembly | Breed | Genome size (Mb) | Number of contigs | Contig N50 length | Contig L50 length |
|---|---|---|---|---|---|
| Ovis_aries_1.0 (GCA_000005525.1) | Mixed | 2,861 | 2,352,347 | 685 | 545,914 |
| Oar_v3.1 (GCA_000298735.1) | Texel | 2,619 | 130,764 | 40,376 | 18,404 |
| Oar_v4.0 (GCA_000298735.2) | Texel | 2,616 | 48,481 | 150,472 | 5,008 |
| Oar_rambouillet_v1.0 (GCA_002742125.1) | Rambouillet | 2,870 | 7,486 | 2,572,683 | 313 |
| ARS-UI_Ramb_v2.0 (GCA_016772045.1) | Rambouillet | 2,628 | 226 | 43,178,051 | 24 |
| ARS1 (GCA_001704415.1) | Goat | 2,923 | 30,399 | 26,244,591 | 32 |
Table 2.
Chromosome level assemblies for breeds of sheep listed in NCBI, including basic assembly statistics and GenBank accessions.
Table 2.
Chromosome level assemblies for breeds of sheep listed in NCBI, including basic assembly statistics and GenBank accessions.
| Breed | Country | GenBank Accession | Contig N50 (Mb) | No. of Contigs | Publication |
|---|---|---|---|---|---|
| Yunnan | China | GCA_022416785.1 | 71.9 | 1,354 | Li et al. 2022a |
| Chinese Merino | China | GCA_022432825.1 | 60 | 1,773 | Li et al. 2022a |
| Qaioke | China | GCA_022416685.1 | 75 | 1,654 | Li et al. 2022a |
| Hu | China | GCA_011170295.1 | 8.7 | 4,131 | Li et al. 2021 |
| Tibetan | Tibet | GCA_017524585.1 | 74.6 | 168 | Li et al. 2022b |
| Kermani | Iran | GCA_022432835.1 | 80.3 | 1,678 | Li et al. 2022a |
| Kazak | Kazakhstan | GCA_022432845.1 | 73.4 | 1,851 | Li et al. 2022a |
| Ujimqin | Mongolia | GCA_022416755.1 | 75.7 | 1,539 | Li et al. 2022a |
| Waggir | Afghanistan | GCA_024222265.1 | 73.6 | 843 | - |
| Texel | Netherlands | GCA_022416775.1 | 47.6 | 1,838 | Li et al. 2022a |
| Romney | UK | GCA_022538005.1 | 68.3 | 1,553 | Li et al. 2022a |
| Suffolk | UK | GCA_022416725.1 | 64.5 | 1,520 | Li et al. 2022a |
| Charollais | UK | GCA_022416745.1 | 65.1 | 1,430 | Li et al. 2022a |
| Polled Dorset | UK | GCA_022416915.1 | 92.4 | 1,297 | Li et al. 2022a |
| East Friesian | Germany | GCA_018804185.1 | 85.3 | 972 | Qiao et al. 2022 |
| Romanov | Russia | GCA_024222175.1 | 31.8 | 1,179 | Li et al. 2022a |
| Romanov | Russia | GCA_022244705.1 | 62.3 | 499 | - |
| Dorper | South Africa | GCA_019145175.1 | 73.3 | 142 | Qiao et al. 2022 |
| White Dorper | South Africa | GCA_022416695.1 | 17.9 | 2,133 | Li et al. 2022a |
| White Dorper | South Africa | GCA_022244695.1 | 61.8 | 1,178 | - |
|
Rambouillet (ARS-UI_Ramb_v2.0) |
France | GCA_016772045.1 | 43.2 | 225 | Davenport et al. 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



