Submitted:
17 April 2023
Posted:
18 April 2023
You are already at the latest version
Abstract
Intestinal commensal microbes, called the gut microbiota, play a critical role in host immune homeostasis through active microbial metabolites and innate and adaptive immune responses. Dysbiosis or imbalance of the gut microbiota composition is associated with inflammatory bowel disease (IBD) which includes Crohn’s disease and ulcerative colitis. The incidence and prevalence rates of IBD have been increasing worldwide. It is well recognized that butyrate, a microbial metabolite of diet fibers, is a major energy source for colonocytes and plays a crucial role in regulating immune function and maintaining epithelial barrier function and intestinal homeostasis. Emerging evidence suggests that butyrate might be a potential therapeutic agent to treat IBD. In this review, we discuss about gut microbial metabolites, particularly butyrate, the synthesis and metabolism of butyrate, mechanisms of butyrate in immune and epithelial barrier function. Furthermore, we review the current research on various therapeutic implications of butyrate in IBD.
Keywords:
butyrate
; inflammatory bowel disease
; gut microbiota
; microbial metabolites
; nutrients
; gut homeostasis
; immune responses
; T-cells
1. Introduction
Inflammatory bowel disease (IBD) is characterized by chronic intestinal inflammation in the gastrointestinal (GI) tract that includes Crohn’s disease (CD) and ulcerative colitis (UC). Although both CD and UC present with chronic inflammation, they differ in many aspects such as location, distribution, and depth of inflammation, and complications and rectal involvement (Table 1). The exact cause of IBD is still not well understood, but the pathogenesis is interlinked with genetic factors, abnormal immune reactivity, microbiota dysbiosis, diet, and environmental factors being involved. The dynamic balance between commensal microflora and host defensive responses in the intestine plays a key role in the initiation and chronic progression of IBD [1]. Disturbed immune function and epithelial barrier integrity are the major features of IBD.
While the pathogenic mechanisms of CD and UC remain unknown, IBD is not curable. Current therapies for IBD, including corticosteroids, immunomodulators, and biologics, are designed to induce remission [2]. However, patients’ response to the treatments decreases over time, and relapses occur frequently. Moreover, the side effects of these treatments are significant, and sometimes intolerable to patients. It is important to identify novel therapeutic targets and discover effective and safe treatments for IBD patients. Short chain fatty acids (SCFAs) are the most abundant microbial metabolites in the intestine and provide 60-70% of energy needs for colonocytes [3]. Specifically, butyrate is the major fuel source for the epithelial cells and has gained more attention than any other SCFA as it regulates intestinal homeostasis and maintains epithelial integrity. A reduced number of butyrate-producing bacteria and lowered butyrate concentration have been found in IBD [4,5]. As butyrate is shown to modulate immune function and intestinal barrier function, it is considered a therapeutic target in the treatment of IBD. In this review, we discuss the production and metabolism of butyrate and the therapeutic implications of butyrate in IBD.
2. Gut microbiota and metabolites
The human gastrointestinal tract harbors a complex and diverse microbial population termed gut microbiota. The gut microbiota comprises trillions of microbes including archaea, bacteria, fungi, and viruses. Many bacteria, particularly anaerobic bacteria, colonize the intestinal tract in a symbiotic relationship, which plays a critical role in maintaining the intestinal homeostasis of the host. The high-throughput DNA sequencing technology has enhanced our understanding of gut microbiota without the need for microbial culturing. More than 1,000 bacterial species colonized in the human gastrointestinal tract especially in the colon. Most of these bacterial species belong to two major phyla: Firmicutes and Bacteroidetes [6].
The gut microbiota produces a wide range of metabolites including short chain fatty acids (SCFAs), polyamines, vitamins, tryptophan-derived metabolites, and secondary bile acids using exogenous undigested dietary substrates and endogenous compounds [7]. These metabolites can be classified into three types: (1) Metabolites produced by microbial transformation of dietary components or drugs such as compound K; (2) Metabolites produced from host secretions that are modified by gut microbiota such as secondary bile acids; (3) Metabolites synthesized by gut microbiota from diet components such as SCFAs [8]. The microbial metabolites can be both beneficial and toxic to the host (Figure 1).
Primary bile acids cholic acid and chenodeoxycholic acids are synthesized from cholesterol and conjugated to glycine or taurine in the liver then stored in the gallbladder and released into intestine to facilitate dietary fats emulsification, digestion, and absorption in the small intestine. The remaining bile acids are absorbed in the terminal ileum and reached to the liver through enterohepatic circulation [9]. The escaped bile salts during enterohepatic circulation become substrates for gut microbial metabolism including deconjugation, oxidation, epimerization and dehydroxylation. The bacteria genera including Clostridium, Bifidobacterium, Bacteroides, Listeria and Lactobacillus are involved in the deconjugation of bile acids. Bacteroides, Eggerthella, Escherichia, Clostridium, Ruminococcus and Peptostreptococcus are involved in oxidation and epimerization [10]. The intestinal bacteria Clostridium and Eubacterium genera transforms cholic acid and chenodeoxycholic acid into deoxycholic acid and lithocholic acid, respectively by dihydroxylation using hydroxysteroid dehydrogenase enzymes [10,11,12]. Undigested dietary proteins enter the colon and serve as substrate for gut microbial metabolism. Tryptophan is an essential amino acid consumed from diet. Undigested or escaped tryptophan is fermented by colonic bacteria, producing various metabolites, indole, indoleacetic acid, indole-3-lactate, indole-3-propionate through direct tryptophan transformation pathway [13]. Indole-producing bacteria, such as Acinetobacter oleivorans, Vibrio cholera, Escherichia coli, Pseudomonas chlororaphis, and Synbiobacterium thermophilus, produce indole from tryptophan [14].
SCFAs including acetic, propionic, and butyric acids are a group of carboxylic acids that consist lesser than six carbon atoms. SCFAs are derived from fermentation of non-digestible carbohydrates in the proximal colon and by proteolytic fermentation in the distal colon. SCFAs can be formed from fermentable carbohydrates through the glycolytic pathway and the pentose phosphate pathways by microbial fermentation [15]. Butyrate is mainly produced from species of Firmicutes phylum including Roseburia species, Faecalibacterium prausnitzii, Eubacterium rectale, whereas acetate and propionates are produced from the species of Bacteroidetes phylum [16,17]. The production of SCFAs in the intestine is substrate dependent. About 300 to 600 mmol of SCFAs are produced in the human intestine per day and only a small amount of SCFAs (~ 10 mmol) are excreted through fecal excretion. The remaining SCFAs are rapidly absorbed by the host epithelial cells via passive diffusion or active transport [18,19].
3. Butyrate production, absorption, and metabolism
Butyrate, a four-carbon SCFA, is one of the major energy sources for intestinal epithelial cells. Gut microbiota produces butyrate from acetyl-CoA, lysine, glutarate, or succinate pathways in the colon [20]. Various bacterial species in the human intestine generate enzymes that can synthesize butyrate from complex fermentable substrates. The predominant butyrogenic bacterial species including Faecalibacterium prausnitzii, Clostridium spp., Eubacterium spp., and Roseburia spp. are from two clusters (Clostridium clusters IV and XIVa) in Firmicutes phylum and the Clostridiales order [21,22]. Most of luminal butyrate is synthesized from non-digestible carbohydrates via acetyl-CoA pathway (Figure 2). In first step, non-digestible carbohydrates are catabolized into pyruvate through pentose phosphate pathway or Embden-meyerhof-parnas pathway. Pyruvate can be converted into acetyl-CoA, which is further breakdown into butyryl-CoA. In the final step, butyryl-CoA can be converted into butyrate by butyryl-CoA: acetyl-CoA transferase or phosphorylated to butyryl-phosphate through phospho-transbutyrylase and then subsequently converted to butyrate through butyrate kinase [23,24,25]. Acetate is required to produce butyrate via butyryl-CoA: acetyl-CoA transferase through cross-feeding microbial reactions. Butyrate is produced by cross-feeding interactions between acetate producing Bifidobacterium spp. and acetate utilizing Faecalibacterium prausnitzii [22]. Moreover, the metabolite cross-feeding within microbial community plays a key role in maintaining the diversity of gut microbial ecosystem [26]. In succinate pathway, butyrogenic bacteria convert succinate to crotonyl-CoA, which is subsequently converted into butyrate. Crotonyl-CoA is the common butyrate precursor in L-lysine and glutarate pathways (Figure 2).
Gut microbiome-derived butyrate is taken up rapidly by colonocytes through passive non-ionic diffusion or active carrier-mediated transport [27]. Ionized form of butyrate is transported across the apical surface of intestinal epithelial cells through active transport mediated by H+-monocarboxylate transporter-1 (MCT1) and Na+-coupled monocarboxylate transporter-1 (SMCT1). Solute carrier family 5 member 8 (SLC5A8) is one of the major SMCT1 transporters of butyrate across the colonocytes [28]. The gene expression levels of SLC5A8 are abundant in the apical membrane of the colon and ileum. On the basolateral membrane, butyrate is transported through carrier-mediated bicarbonate exchange system [29]. Butyrate predominantly presents in the anionic form in the colon due to colonic luminal pH conditions. Thus, it requires carrier-mediated transportation for the cellular entry.
The absorbed butyrate is metabolized in the intestinal epithelial cells, liver cells, and other tissues and cells [30]. In the epithelial cells, butyrate is transformed into acetyl-CoA, and enters tricarboxylic acid (TCA) cycle in the mitochondria to produce ATP, which is consumed by the colon epithelial cells. The portion of butyrate which is not utilized by epithelial cells can reach to the liver via portal circulation, where it is metabolized into acetyl-CoA and becomes a substrate for fatty acids, cholesterol, and ketone bodies by hepatocytes [21,31]. The plasma concentration of butyrate is very low compared to colonic levels, only 2% of butyrate enters systemic circulation, being utilized by other tissues and cells [31]. The remaining SCFAs including butyrate are excreted through the lungs and urine.
4. Role and mechanisms of butyrate in the regulation of barrier function and immune response
The single layer of intestinal epithelium serves as a barrier between the host and its external environment that controls the interaction between luminal contents and the internal milieu of the body. The intestinal epithelial monolayer contains several types of specialized cells: (1) enterocytes, for absorption of nutrients; (2) goblet cells, producing secretory and gel forming mucins which are glycosylated proteins that form polymeric nets called mucus layer, a physical barrier between intestinal bacteria and epithelial cells; (3) enteroendocrine cells, secreting various hormones regulating digestive function; (4) Paneth cells, residing at crypt base and secreting antimicrobial peptides such as lysozyme, defensins, and cryptidins; (5) microfold cells (M cells), sampling antigens from lumen to sub-epithelium; and (6) tuft cells, for chemosensing function in the epithelium [32,33]. These epithelial cells are connected by intercellular desmosomes, tight junctions (TJs), and adherent junctions (AJs), which create a physical barrier for luminal contents of the gut and regulate epithelial permeability. TJs are a complex network formed by transmembrane proteins such as claudins, occludin, tricellulin, and junctional adhesion molecules and cytosolic scaffold proteins such as zonulae occludens (ZO) and cingulin [34,35]. Both TJs and AJs are connected to the actin cytoskeleton and form an apical junction complex. On the basal side, epithelial cells are connected by hemidesmosomes.
The intestinal epithelium lies between the commensal organisms in the gut lumen and the immune cells in lamina propria. The complex immune interactions between commensal microflora, the epithelial layer, and the sub-epithelial immune cells maintain homeostasis under normal conditions. Lamina propria contains the gut-associated lymphoid tissue (GALT) which is comprised of Peyer’s patches, a group of lymphoid follicles containing several immune cells, such as specialized M cells, dendritic cells, T cells, B cells, intraepithelial lymphocytes, and macrophages [36]. The dendritic cells (DCs) from lamina propria sample the luminal food and microbial antigens by extending their dendrites between epithelial cells and transport to antigen-presenting cells (APCs) in GALT [37,38]. Upon activation, GALT performs effector immune functions by activating immune cells to produce specific cytokines from T cells and immunoglobulins from B cells. Antigens in the gut lumen can be taken up by specialized M cells and delivered to DCs for effector functions in the Peyer’s patches [39]. Intestinal epithelial cells themselves can also act as dynamic sensors by pattern recognition molecule receptors (PRRs) such as toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD) like receptors (NLRs) to sense pathogen-associated molecular patterns.
Gut microflora and their metabolites play a major role in maintaining epithelial barrier function and immune homeostasis. Among the microbial metabolites, butyrate involves a number of signaling pathways in the gut immune cells and epithelial cells for restoration of impaired colonic barrier function and gut homeostasis (Figure 3). Pathophysiology of IBD involves both epithelial barrier dysfunction and abnormal immune cell activation. Changes in TJs structure, down-regulation of claudin proteins, and up-regulation of pore-forming claudin-2 were observed in both CD and UC conditions [34]. Since 2007, butyrate was found to enhance the intestinal barrier function by facilitating tight junction assembly via activation of AMPK, Akt, and other signaling pathways in a dose-dependent manner as shown in studies with transepithelial electrical resistance (TEER) and fluorescein isothiocyanate-dextran (FITC-dextran) permeability assays in in vitro settings [40,41,42]. Marinelli et al. [43] demonstrated that butyrate regulates the epithelial barrier function by acting as a signaling molecule for cell-surface G-protein-coupled receptors (GPRs) and nuclear factors (NFs). Indeed, butyrate was found to induce T cell-independent IgA secretion in the colon via activation of GPR41 (free fatty acid receptor 3, FFAR3) and GPR109A (hydrocarboxylic acid receptor 2, HCAR2), and inhibition of histone deacetylase (HDAC) to restore epithelial barrier function under inflammatory conditions [44]. Studies also explored the effect of butyrate on claudins expression. Zheng et al. [45] reported that butyrate promotes epithelial barrier function through interleukin-10 receptor α-subunit (IL-10RA) dependent repression of claudin-2 TJ protein. Wang et al. [46] demonstrated that butyrate treatment improved epithelial barrier function via up-regulation of claudin-1 transcription by
facilitating the interaction between specific motifs in the claudin-1 promoter region and SP1 transcription factor. Moreover, butyrate enhances mucin secretion and protects epithelial cells by inducing MUC2 gene expression via AP-1 and acetylation/methylation of histones at the MUC2 promoter in intestinal epithelial goblet cells [47]. Hypoxia-inducible factor 1 (HIF-1) - dependent mechanism may also contribute to butyrate-enhanced epithelial barrier function [48].
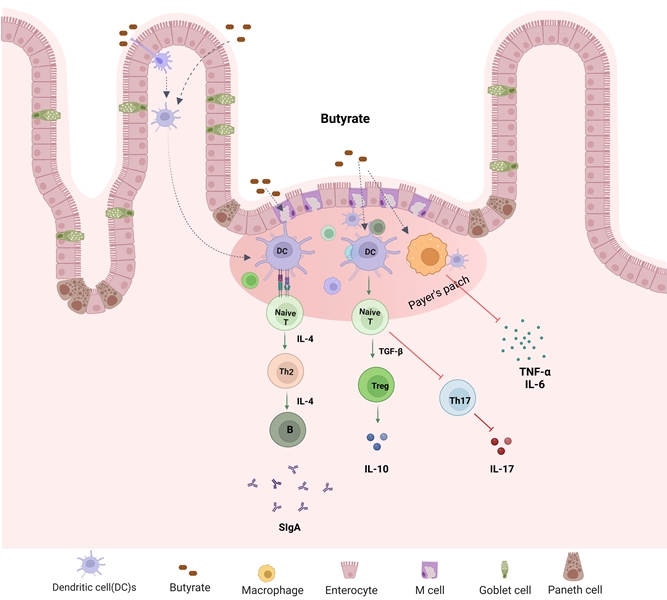
An inappropriate immune response to antigens derived from intestinal components is a key feature in IBD, leading to imbalance of inflammatory cytokines, tissue damage, and disease progression [49,50]. Increased phagocytic activity of macrophages and cytokines’ secretion (for example, IL-1, IL-6, IL-17, and TNF) has been found in IBD patients [51]. T lymphocytes (T-cells) play a crucial role in maintaining immune homeostasis by regulating innate and adaptive immune responses. Upon specific antigen stimulation, naïve CD4+ T-cells differentiate into effector T helper (Th) cells including Th1, Th2, T regulatory (Treg), and Th17 cells [52]. Each Th type secret specific cytokines to perform protective or pathogenic roles. Treg cells have immunosuppressive properties that help to maintain immune homeostasis by secreting anti-inflammatory cytokines including IL-10 [53]. IBD is associated with dysregulated T-cell immune responses such as increased Th1, Th2, Th17 cells function and decreased Treg cells function [54]. Th17 produce inflammatory cytokines such as IL-17A, IL-17F, and IL-21 which are involved in the pathogenesis of IBD. Gut microbial metabolite butyrate regulates differentiation and proliferation of T cells (Figure 4). Butyrate administration enhanced Treg cells function and suppressed IL-17 levels as well as Th17 cells in the peripheral blood and colon tissues of TNBS-induced colitis rats compared to control group [53]. Zimmerman et al. [55] have demonstrated that butyrate inhibits proliferation of both CD4+ and CD8+ T cells in a dose-dependent manner and it induces apoptosis in T cells through the Fas-mediated apoptosis pathway. Butyrate facilitates Treg cell differentiation by increasing histone H3 acetylation at the promoter and CNS3 region of the FOX3 gene locus [56]. Chen et al. [57] found that butyrate enhanced Th1 differentiation by promoting IFN-γ levels and T-bet expression in healthy condition, but inhibited Th1 differentiation through IL-10 production and T-bet expression in colonic inflammation. In addition, butyrate has shown to regulate inflammatory response by influencing NF-κB activity. NF-κB is a transcription factor involved in the regulation of various inflammatory mediators and cytokines expression including TNF-α and IL-6 [58]. Butyrate is shown to reduce inflammatory response by suppressing NF-κB activity. Several studies have demonstrated the ability of butyrate to reduce NF-κB activity in human colon cell lines and in lamina propria mononuclear cells isolated from CD patients [59,60,61]. Butyrate activates transmembrane GPRs and nuclear receptors such as aryl hydrocarbon receptor (AhR) in the intestinal epithelial cells. AhR is a ligand-activated transcription factor that resides in cytosol in activated form, and translocates to nucleus up on activation thereby regulating AhR-dependent gene expression [62,63]. SCFAs including butyrate are shown to enhance AhR ligand interations in mouse and human colon cells [43,64].
5. Therapeutic implications of butyrate for IBD
IBD is characterized with aberrant immune response and barrier dysfunction and associated with reduced number of butyrate-producing bacteria in the gut. As butyrate was found to not only provide energy to colonic epithelial cells but also help maintain intestinal integrity and modulate immune responses [42], numerous studies have investigated the role of various forms of butyrate in reducing gut inflammation [65,66,67]. Many of the studies have demonstrated the efficacy of oral butyrate supplements, butyrate enema, butyrogenic diet and bacterial supplements in the treatment of IBD.
5.1. Butyrate supplements
5.1. a. Oral administration
Dysbiosis of gut microbiota leads to decreased butyrate synthesis and impaired butyrate metabolism as observed in IBD [68]. Although a low concentration of butyric acid is commonly present in our daily regular diet, it may not be sufficient to restore the epithelial function in inflammation in the colon. Many studies have investigated the therapeutic potential of butyrate oral supplements in gut inflammation in both preclinical studies and clinical trials. Table 2 summarizes these results (Table 2). Butyrate has been shown to reduce gut inflammation and ameliorate symptoms in a dose dependent manner. Butyrate at 20 mg/kg/day or lower doses was found to have no significant effect, while at 100 mg/kg it was effective against inflammation in mice [67,69]. Lee et al. [67] reported that the oral supplementation of sodium butyrate at 100 mg/kg of body weight daily decreased colitis scores, prevented body weight loss, and induced histone H3 acetylation in colonic mucosa in mouse models of acute and chronic colitis. Moreover, butyrate treatment restored the microbial community diversity and reduced microbiota dysbiosis in gut inflammation [70].
As orally supplemented butyrate is rapidly absorbed in the duodenum, majority of the orally administered butyrate would not reach the colon. Moreover, the clinical application of oral butyrate is limited due to its unpleasant taste and odor. To address these issues, some studies used colon targeted formulations and encapsulated butyrate to test if butyrate in such formulations has better effects in IBD patients especially UC patients [66,71,72]. Sabatino et al. [71] demonstrated that enteric-coated butyrate tablets administration effectively reduced ileocaecal inflammation and maintained clinical remission in Crohn’s disease patients. Lipophilic microencapsulated-sodium-butyrate treatment showed enrichment of butyrogenic colonic bacteria in IBD patients [66]. Wang et al. [73] developed butyrate micelles so that butyrate is released in the lower gastrointestinal tract. They found that butyrate micelles significantly improved intestinal barrier function and reduced disease severity in DSS-induced colitis and CD45RBhiT-cell transfer colitis in mice.

Table 2.
Impact of oral butyrate supplements on IBD.
| Treatment name | Concentration | Colitis model | Effects | Authors | |
|---|---|---|---|---|---|
| Mice | Sodium butyrate | 0.5% of sodium butyrate | DSS-induced colitis | Decreased mucosal inflammation | Vieira et al. [74] |
| Mice | Butyrate-releasing polysaccharide derivative | 200 mg/kg | DSS-induced colitis | Reduced disease activity index, rebalanced gut microbiota and reversed the imbalance between pro- and anti-inflammatory cytokines | Zha et al. [65] |
| Mice | Balatable butyrate-releasing derivative, N-(1-carbamoyl-2-phenylethyl) butyramide (FBA) | 42.5 mg/kg | DSS-induced colitis | Reduced disease activity index | Simeoli et al. [75] |
| Mice | Sodium butyrate | 200 mM | Citrobacter rodentium infection model | Prevented mice from weight loss and suppressed intestinal inflammation | Zhou et al. [76] |
| Mice | Sodium butyrate | 200 mM | DSS-induced colitis | Suppressed intestinal inflammation and lowered pathology scores | Zhou et al. [76] |
| Mice | Sodium butyrate | 5 g/L | TNBS induced colitis | Decreased disease activity index and suppressed inflammation | Chen et al. [77] |
| Mice | Sodium butyrate | 100 mg/kg/day | DSS-induced acute colitis Piroxicam-induced chronic colitis |
Decreased colitis scores and prevented weight loss | Lee et al. [67] |
| Mice | Sodium butyrate | DSS-induced colitis | Decreased disease activity index, and restored the balance of gut microbial communities | Dou et al. [70] | |
| Mice | Sodium butyrate150 mM | 150 mM sodium butyrate | DSS-induced colitis | No significant difference in histologic scores | Lee et al. [78] |
| Human | Enteric-coated tablets | 4 g/day | Crohn's disease | Induced clinical improvement and reduced disease activity index | Sabatino et al. [71] |
| Human | Sodium butyrate tablets | 4 g/day | Crohn's disease | Induced clinical improvement or remission | Di Sabatino et al. [79] |
| Human | Microencapsulated-sodium-butyrate | 1800 mg/day | IBD-both CD and UC | Increases the growth of bacteria able to produce SCFA with potential anti-inflammatory action | Faccin et al. [66] |
| Human | Microencapsulated sodium butyrate | 1000 mg/day | UC in clinical remission | Helped to maintain clinical remission | Vernero et al. [72] |
| Human | Sodium butyrate | 150 mg/twice a day | IBD-both CD and UC | No significant effects in newly diagnosed children and adolescents | Pietrzak et al. [80] |
5.1. b. Butyrate enemas
Treatment with butyrate enemas had mixed effects in preclinical and clinical studies as summarized in Table 3 (Table 3). Butyrate enema showed inhibition of NF-κB activation in lamina propria macrophages of UC patients, and it also reduced disease activity [81]. Segain et al. [59] observed reduction of TNF-α induced NF-κB in colon tissues in butyrate enema treated colitis rats. However, some clinical studies found that butyrate enema did not show any significant improvement on UC patients in remission and in patients with left-sided UC [82,83].
5.2. Butyrogenic diets
As IBD is associated with decreased butyrate-producing bacteria and butyrate production in the colon, many investigators have tested if intake of butyrate producing fermentable dietary fibers could be beneficial for IBD. Fernandez-Banares et al. [88] observed increased concentrations of fecal butyrate after the intake of fiber rich plantago ovata seeds in UC patients. Moreover, plantago ovata seed supplementation showed effectiveness in maintaining UC remission. Further studies confirmed that butyrogenic diet supplementations attenuated colonic inflammation by regulation of gut microbial balance, increased production of SCFAs, upregulation of anti-inflammatory cytokines and Treg cells, and reduced the mucosal damage (Table 4). Fiber rich diet such as oat bran and germinated barley foodstuff have shown positive effects in IBD, especially in reducing risk of relapse while maintaining prolonged remission in UC patients [89,90]. It was shown that β-glucan derived from oats and barley ameliorates colitis through the regulation of tight junction proteins and inhibition of pro-inflammatory factors by increased SCFAs production via gut microbial fermentation [91,92]. IBD patients showed good tolerability to dietary fiber intake, particularly during clinical remission stage [90,93]. Despite the beneficial effects of fiber, IBD patients are advised to reduce fiber consumption during disease exacerbation period. Thus, the long term effects of high fiber intake in active CD remain uncertain due to limited clinical data [94,95].
5.3. Combination therapies
A combination therapy is a treatment modality that combines two or more therapeutic agents. It is found in most of the studies that combination therapies with butyrate and other agents are more effective than single therapy in the treatment of IBD or colitis models. Please see Table 5 for a summary of the outcomes of the studies (Table 5). Combinations of butyrate with other SCFAs, prebiotics, and probiotics have been investigated. Mixture of butyrate, Pistacia atlantica, and Lactobacillus casei or butyrate, Lactobacillus casei, and L-carnitin showed synergistic effects than single agent on TNBS-induced rat colitis model [101,102]. Combination of SCFAs, mainly acetate, propionate, and butyrate, showed increased effects against colitis [78]. However, treatment of SCFAs rectal enema (sodium acetate, propionate, and butyrate) did not improve histological and clinical state of left-sided UC [103]. Co-administration of sodium butyrate and mesalazine improved the efficacy of oral mesalazine in UC patients [104].
6. Conclusions
In conclusion, butyrate at appropriate concentrations helps to maintain intestinal barrier function and regulate immune response in the gut. Clinical trials and animal studies have shown that butyrate can reduce mucosal inflammation and improve barrier function in UC and CD. Butyrate formulations and butyrogenic compounds may represent alternative therapeutic approaches for IBD. Combination therapies with butyrate and other SCFAs may further increase the efficacy of butyrate in the treatment of IBD.
Author Contributions
All the authors have read and approved the final version of the paper and contributed to the work. NR searched literatures and drafted the manuscript. XZS and RG reviewed and revised it. XZS obtained the funds and helped to organize the review.
Funding
This study was supported in part by the United States Department of Defense (W81XWH2010681 to XZS) and the National Institute of Health (R01 DK124611 to XZS).
Acknowledgments
The authors wish to thank Dr. Yingzi Cong for beneficial discussions. All graphical images were created using BioRender.
Conflicts of Interest statement
The authors declare that this work was completed in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Xavier, R.J.; Podolsky, D.K. Unravelling the Pathogenesis of Inflammatory Bowel Disease. Nature 2007, 448, 427–434. [CrossRef]
- Dassopoulos, T.; Sultan, S.; Falck–Ytter, Y.T.; Inadomi, J.M.; Hanauer, S.B. American Gastroenterological Association Institute Technical Review on the Use of Thiopurines, Methotrexate, and Anti–TNF-α Biologic Drugs for the Induction and Maintenance of Remission in Inflammatory Crohn’s Disease. Gastroenterology 2013, 145, 1464–1478. [CrossRef]
- Ghishan, F.K.; Kiela, P.R. Epithelial Transport in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2014, 20, 1099–1109. [CrossRef]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low Counts of Faecalibacterium Prausnitzii in Colitis Microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K. A Decrease of the Butyrate-Producing Species Roseburia Hominis and Faecalibacterium Prausnitzii Defines Dysbiosis in Patients with Ulcerative Colitis. Gut 2014, 63, 1275–1283. [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature 2012, 489, 220–230. [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut Microbiota, Metabolites and Host Immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [CrossRef]
- Feng, W.; Ao, H.; Peng, C. Gut Microbiota, Short-Chain Fatty Acids, and Herbal Medicines. Front. Pharmacol. 2018, 9, 1354. [CrossRef]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47, 241–259. [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile Acid–Microbiota Crosstalk in Gastrointestinal Inflammation and Carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [CrossRef]
- Ridlon, J.M.; Hylemon, P.B. Identification and Characterization of Two Bile Acid Coenzyme A Transferases from Clostridium Scindens, a Bile Acid 7α-Dehydroxylating Intestinal Bacterium. J. Lipid Res. 2012, 53, 66–76. [CrossRef]
- Pavlović, N.; Goločorbin-Kon, S.; Ðanić, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile Acids and Their Derivatives as Potential Modifiers of Drug Release and Pharmacokinetic Profiles. Front. Pharmacol. 2018, 9, 1283. [CrossRef]
- Su, X.; Gao, Y.; Yang, R. Gut Microbiota-Derived Tryptophan Metabolites Maintain Gut and Systemic Homeostasis. Cells 2022, 11, 2296. [CrossRef]
- Zhang, J.; Zhu, S.; Ma, N.; Johnston, L.J.; Wu, C.; Ma, X. Metabolites of Microbiota Response to Tryptophan and Intestinal Mucosal Immunity: A Therapeutic Target to Control Intestinal Inflammation. Med. Res. Rev. 2021, 41, 1061–1088. [CrossRef]
- Hugenholtz, F.; Mullaney, J.A.; Kleerebezem, M.; Smidt, H.; Rosendale, D.I. Modulation of the Microbial Fermentation in the Gut by Fermentable Carbohydrates. Bioact. Carbohydrates Diet. Fibre 2013, 2, 133–142. [CrossRef]
- Wang, G.; Huang, S.; Wang, Y.; Cai, S.; Yu, H.; Liu, H.; Zeng, X.; Zhang, G.; Qiao, S. Bridging Intestinal Immunity and Gut Microbiota by Metabolites. Cell. Mol. Life Sci. 2019, 76, 3917–3937.
- Louis, P.; Hold, G.L.; Flint, H.J. The Gut Microbiota, Bacterial Metabolites and Colorectal Cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [CrossRef]
- Høverstad, T. Studies of Short-Chain Fatty Acid Absorption in Man. Scand. J. Gastroenterol. 1986, 21, 257–260. [CrossRef]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The Role of Short-Chain Fatty Acids in Microbiota–Gut–Brain Communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [CrossRef]
- Levine, M.; Lohinai, Z.M. Resolving the Contradictory Functions of Lysine Decarboxylase and Butyrate in Periodontal and Intestinal Diseases. J. Clin. Med. 2021, 10, 2360. [CrossRef]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A Double-Edged Sword for Health? Adv. Nutr. 2018, 9, 21–29. [CrossRef]
- Fu, X.; Liu, Z.; Zhu, C.; Mou, H.; Kong, Q. Nondigestible Carbohydrates, Butyrate, and Butyrate-Producing Bacteria. Crit. Rev. Food Sci. Nutr. 2019, 59, S130–S152. [CrossRef]
- Louis, P.; Flint, H.J. Diversity, Metabolism and Microbial Ecology of Butyrate-Producing Bacteria from the Human Large Intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [CrossRef]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the Bacterial Butyrate Synthesis Pathways by Analyzing (Meta) Genomic Data. MBio 2014, 5, e00889-14. [CrossRef]
- Bui, T.P.N.; Ritari, J.; Boeren, S.; De Waard, P.; Plugge, C.M.; De Vos, W.M. Production of Butyrate from Lysine and the Amadori Product Fructoselysine by a Human Gut Commensal. Nat. Commun. 2015, 6, 10062. [CrossRef]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; Van Harsselaar, J. Short Chain Fatty Acids in Human Gut and Metabolic Health. Benef. Microbes 2020, 11, 411–455. [CrossRef]
- Nedjadi, T.; Moran, A.W.; Al-Rammahi, M.A.; Shirazi-Beechey, S.P. Characterization of Butyrate Transport across the Luminal Membranes of Equine Large Intestine. Exp. Physiol. 2014, 99, 1335–1347. [CrossRef]
- Takebe, K.; Nio, J.; Morimatsu, M.; Karaki, S.-I.; Kuwahara, A.; Kato, I.; Iwanaga, T. Histochemical Demonstration of a Na+-Coupled Transporter for Short-Chain Fatty Acids (Slc5a8) in the Intestine and Kidney of the Mouse. Biomed. Res. 2005, 26, 213–221. [CrossRef]
- Guilloteau, P.; Martin, L.; Eeckhaut, V.; Ducatelle, R.; Zabielski, R.; Van Immerseel, F. From the Gut to the Peripheral Tissues: The Multiple Effects of Butyrate. Nutr. Res. Rev. 2010, 23, 366–384. [CrossRef]
- Wong, J.M.W.; De Souza, R.; Kendall, C.W.C.; Emam, A.; Jenkins, D.J.A. Colonic Health: Fermentation and Short Chain Fatty Acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [CrossRef]
- Boets, E.; Gomand, S. V; Deroover, L.; Preston, T.; Vermeulen, K.; De Preter, V.; Hamer, H.M.; Van den Mooter, G.; De Vuyst, L.; Courtin, C.M. Systemic Availability and Metabolism of Colonic-derived Short-chain Fatty Acids in Healthy Subjects: A Stable Isotope Study. J. Physiol. 2017, 595, 541–555. [CrossRef]
- Laukoetter, M.G.; Nava, P.; Nusrat, A. Role of the Intestinal Barrier in Inflammatory Bowel Disease. World J. Gastroenterol. WJG 2008, 14, 401. [CrossRef]
- Vereecke, L.; Beyaert, R.; van Loo, G. Enterocyte Death and Intestinal Barrier Maintenance in Homeostasis and Disease. Trends Mol. Med. 2011, 17, 584–593. [CrossRef]
- Hering, N.A.; Fromm, M.; Schulzke, J. Determinants of Colonic Barrier Function in Inflammatory Bowel Disease and Potential Therapeutics. J. Physiol. 2012, 590, 1035–1044. [CrossRef]
- Sánchez de Medina, F.; Romero-Calvo, I.; Mascaraque, C.; Martínez-Augustin, O. Intestinal Inflammation and Mucosal Barrier Function. Inflamm. Bowel Dis. 2014, 20, 2394–2404. [CrossRef]
- Takiishi, T.; Fenero, C.I.M.; Câmara, N.O.S. Intestinal Barrier and Gut Microbiota: Shaping Our Immune Responses throughout Life. Tissue barriers 2017, 5, e1373208. [CrossRef]
- Foti, M.; Ricciardi-Castagnoli, P. Antigen Sampling by Mucosal Dendritic Cells. Trends Mol. Med. 2005, 11, 394–396. [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal Epithelial Cells: Regulators of Barrier Function and Immune Homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [CrossRef]
- Chehade, M.; Mayer, L. Oral Tolerance and Its Relation to Food Hypersensitivities. J. Allergy Clin. Immunol. 2005, 115, 3–12. [CrossRef]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of Butyrate on Intestinal Barrier Function in a Caco-2 Cell Monolayer Model of Intestinal Barrier. Pediatr. Res. 2007, 61, 37–41. [CrossRef]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco-2 Cell Monolayers. J. Nutr. 2009, 139, 1619–1625. [CrossRef]
- Yan, H.; Ajuwon, K.M. Butyrate Modifies Intestinal Barrier Function in IPEC-J2 Cells through a Selective Upregulation of Tight Junction Proteins and Activation of the Akt Signaling Pathway. PLoS One 2017, 12, e0179586. [CrossRef]
- Marinelli, L.; Martin-Gallausiaux, C.; Bourhis, J.-M.; Béguet-Crespel, F.; Blottière, H.M.; Lapaque, N. Identification of the Novel Role of Butyrate as AhR Ligand in Human Intestinal Epithelial Cells. Sci. Rep. 2019, 9, 643. [CrossRef]
- Isobe, J.; Maeda, S.; Obata, Y.; Iizuka, K.; Nakamura, Y.; Fujimura, Y.; Kimizuka, T.; Hattori, K.; Kim, Y.-G.; Morita, T. Commensal-Bacteria-Derived Butyrate Promotes the T-Cell-Independent IgA Response in the Colon. Int. Immunol. 2020, 32, 243–258. [CrossRef]
- Zheng, L.; Kelly, C.J.; Battista, K.D.; Schaefer, R.; Lanis, J.M.; Alexeev, E.E.; Wang, R.X.; Onyiah, J.C.; Kominsky, D.J.; Colgan, S.P. Microbial-Derived Butyrate Promotes Epithelial Barrier Function through IL-10 Receptor–Dependent Repression of Claudin-2. J. Immunol. 2017, 199, 2976–2984. [CrossRef]
- Wang, H.-B.; Wang, P.-Y.; Wang, X.; Wan, Y.-L.; Liu, Y.-C. Butyrate Enhances Intestinal Epithelial Barrier Function via Up-Regulation of Tight Junction Protein Claudin-1 Transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [CrossRef]
- Burger-van Paassen, N.; Vincent, A.; Puiman, P.J.; van Der Sluis, M.; Bouma, J.; Boehm, G.; Van Goudoever, J.B.; Van Seuningen, I.; Renes, I.B. The Regulation of Intestinal Mucin MUC2 Expression by Short-Chain Fatty Acids: Implications for Epithelial Protection. Biochem. J. 2009, 420, 211–219. [CrossRef]
- Fachi, J.L.; de Souza Felipe, J.; Pral, L.P.; da Silva, B.K.; Corrêa, R.O.; de Andrade, M.C.P.; da Fonseca, D.M.; Basso, P.J.; Câmara, N.O.S.; e Souza, É.L. de S. Butyrate Protects Mice from Clostridium Difficile-Induced Colitis through an HIF-1-Dependent Mechanism. Cell Rep. 2019, 27, 750–761. [CrossRef]
- Brown, S.J.; Mayer, L. The Immune Response in Inflammatory Bowel Disease. Off. J. Am. Coll. Gastroenterol. ACG 2007, 102, 2058–2069. [CrossRef]
- Guan, Q.; Zhang, J. Recent Advances: The Imbalance of Cytokines in the Pathogenesis of Inflammatory Bowel Disease. Mediators Inflamm. 2017, 2017. [CrossRef]
- Maloy, K.J.; Powrie, F. Intestinal Homeostasis and Its Breakdown in Inflammatory Bowel Disease. Nature 2011, 474, 298–306. [CrossRef]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+ T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012. [CrossRef]
- Zhang, M.; Zhou, Q.; Dorfman, R.G.; Huang, X.; Fan, T.; Zhang, H.; Zhang, J.; Yu, C. Butyrate Inhibits Interleukin-17 and Generates Tregs to Ameliorate Colorectal Colitis in Rats. BMC Gastroenterol. 2016, 16, 1–9. [CrossRef]
- Bouma, G.; Strober, W. The Immunological and Genetic Basis of Inflammatory Bowel Disease. Nat. Rev. Immunol. 2003, 3, 521–533. [CrossRef]
- Zimmerman, M.A.; Singh, N.; Martin, P.M.; Thangaraju, M.; Ganapathy, V.; Waller, J.L.; Shi, H.; Robertson, K.D.; Munn, D.H.; Liu, K. Butyrate Suppresses Colonic Inflammation through HDAC1-Dependent Fas Upregulation and Fas-Mediated Apoptosis of T Cells. Am. J. Physiol. Liver Physiol. 2012, 302, G1405–G1415. [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature 2013, 504, 446–450. [CrossRef]
- Chen, L.; Sun, M.; Wu, W.; Yang, W.; Huang, X.; Xiao, Y.; Ma, C.; Xu, L.; Yao, S.; Liu, Z. Microbiota Metabolite Butyrate Differentially Regulates Th1 and Th17 Cells’ Differentiation and Function in Induction of Colitis. Inflamm. Bowel Dis. 2019, 25, 1450–1461. [CrossRef]
- Venkatraman, A.; Ramakrishna, B.S.; Shaji, R. V; Kumar, N.S.N.; Pulimood, A.; Patra, S. Amelioration of Dextran Sulfate Colitis by Butyrate: Role of Heat Shock Protein 70 and NF-ΚB. Am. J. Physiol. Liver Physiol. 2003, 285, G177–G184. [CrossRef]
- Segain, J.P.; De La Blétiere, D.R.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottiere, H.M.; Galmiche, J.P. Butyrate Inhibits Inflammatory Responses through NFκB Inhibition: Implications for Crohn’s Disease. Gut 2000, 47, 397–403. [CrossRef]
- Inan, M.S.; Rasoulpour, R.J.; Yin, L.; Hubbard, A.K.; Rosenberg, D.W.; Giardina, C. The Luminal Short-Chain Fatty Acid Butyrate Modulates NF-ΚB Activity in a Human Colonic Epithelial Cell Line. Gastroenterology 2000, 118, 724–734. [CrossRef]
- Yin, L.; Laevsky, G.; Giardina, C. Butyrate Suppression of Colonocyte NF-ΚB Activation and Cellular Proteasome Activity. J. Biol. Chem. 2001, 276, 44641–44646. [CrossRef]
- Lee, H.U.; McPherson, Z.E.; Tan, B.; Korecka, A.; Pettersson, S. Host-Microbiome Interactions: The Aryl Hydrocarbon Receptor and the Central Nervous System. J. Mol. Med. 2017, 95, 29–39. [CrossRef]
- Yu, M.; Wang, Q.; Ma, Y.; Li, L.; Yu, K.; Zhang, Z.; Chen, G.; Li, X.; Xiao, W.; Xu, P. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int. J. Biol. Sci. 2018, 14, 69. [CrossRef]
- Jin, U.-H.; Cheng, Y.; Park, H.; Davidson, L.A.; Callaway, E.S.; Chapkin, R.S.; Jayaraman, A.; Asante, A.; Allred, C.; Weaver, E.A. Short Chain Fatty Acids Enhance Aryl Hydrocarbon (Ah) Responsiveness in Mouse Colonocytes and Caco-2 Human Colon Cancer Cells. Sci. Rep. 2017, 7, 10163. [CrossRef]
- Zha, Z.; Lv, Y.; Tang, H.; Li, T.; Miao, Y.; Cheng, J.; Wang, G.; Tan, Y.; Zhu, Y.; Xing, X. An Orally Administered Butyrate-Releasing Xylan Derivative Reduces Inflammation in Dextran Sulphate Sodium-Induced Murine Colitis. Int. J. Biol. Macromol. 2020, 156, 1217–1233. [CrossRef]
- Facchin, S.; Vitulo, N.; Calgaro, M.; Buda, A.; Romualdi, C.; Pohl, D.; Perini, B.; Lorenzon, G.; Marinelli, C.; D’Incà, R. Microbiota Changes Induced by Microencapsulated Sodium Butyrate in Patients with Inflammatory Bowel Disease. Neurogastroenterol. Motil. 2020, 32, e13914. [CrossRef]
- Lee, C.; Kim, B.G.; Kim, J.H.; Chun, J.; Im, J.P.; Kim, J.S. Sodium Butyrate Inhibits the NF-Kappa B Signaling Pathway and Histone Deacetylation, and Attenuates Experimental Colitis in an IL-10 Independent Manner. Int. Immunopharmacol. 2017, 51, 47–56. [CrossRef]
- Huda-Faujan, N.; Abdulamir, A.S.; Fatimah, A.B.; Anas, O.M.; Shuhaimi, M.; Yazid, A.M.; Loong, Y.Y. The Impact of the Level of the Intestinal Short Chain Fatty Acids in Inflammatory Bowel Disease Patients versus Healthy Subjects. Open Biochem. J. 2010, 4, 53. [CrossRef]
- Chang, P. V; Hao, L.; Offermanns, S.; Medzhitov, R. The Microbial Metabolite Butyrate Regulates Intestinal Macrophage Function via Histone Deacetylase Inhibition. Proc. Natl. Acad. Sci. 2014, 111, 2247–2252. [CrossRef]
- Dou, X.; Gao, N.; Yan, D.; Shan, A. Sodium Butyrate Alleviates Mouse Colitis by Regulating Gut Microbiota Dysbiosis. Animals 2020, 10, 1154. [CrossRef]
- Sabatino, A. Di; Morera, R.; Ciccocioppo, R.; Cazzola, P.; Gotti, S.; Tinozzi, F.P.; Tinozzi, S.; Corazza, G.R. Oral Butyrate for Mildly to Moderately Active Crohn’s Disease. Aliment. Pharmacol. Ther. 2005, 22, 789–794. [CrossRef]
- Vernero, M.; De Blasio, F.; Ribaldone, D.G.; Bugianesi, E.; Pellicano, R.; Saracco, G.M.; Astegiano, M.; Caviglia, G.P. The Usefulness of Microencapsulated Sodium Butyrate Add-on Therapy in Maintaining Remission in Patients with Ulcerative Colitis: A Prospective Observational Study. J. Clin. Med. 2020, 9, 3941. [CrossRef]
- Wang, R.; Cao, S.; Bashir, M.E.H.; Hesser, L.A.; Su, Y.; Hong, S.M.C.; Thompson, A.; Culleen, E.; Sabados, M.; Dylla, N.P. Treatment of Peanut Allergy and Colitis in Mice via the Intestinal Release of Butyrate from Polymeric Micelles. Nat. Biomed. Eng. 2022, 1–18. [CrossRef]
- Vieira, E.L.M.; Leonel, A.J.; Sad, A.P.; Beltrão, N.R.M.; Costa, T.F.; Ferreira, T.M.R.; Gomes-Santos, A.C.; Faria, A.M.C.; Peluzio, M.C.G.; Cara, D.C. Oral Administration of Sodium Butyrate Attenuates Inflammation and Mucosal Lesion in Experimental Acute Ulcerative Colitis. J. Nutr. Biochem. 2012, 23, 430–436. [CrossRef]
- Simeoli, R.; Mattace Raso, G.; Pirozzi, C.; Lama, A.; Santoro, A.; Russo, R.; Montero-Melendez, T.; Berni Canani, R.; Calignano, A.; Perretti, M. An Orally Administered Butyrate-releasing Derivative Reduces Neutrophil Recruitment and Inflammation in Dextran Sulphate Sodium-induced Murine Colitis. Br. J. Pharmacol. 2017, 174, 1484–1496. [CrossRef]
- Zhou, Z.; Cao, J.; Liu, X.; Li, M. Evidence for the Butyrate Metabolism as Key Pathway Improving Ulcerative Colitis in Both Pediatric and Adult Patients. Bioengineered 2021, 12, 8309–8324. [CrossRef]
- Chen, G.; Ran, X.; Li, B.; Li, Y.; He, D.; Huang, B.; Fu, S.; Liu, J.; Wang, W. Sodium Butyrate Inhibits Inflammation and Maintains Epithelium Barrier Integrity in a TNBS-Induced Inflammatory Bowel Disease Mice Model. EBioMedicine 2018, 30, 317–325. [CrossRef]
- Lee, J.G.; Lee, J.; Lee, A.; Jo, S.V.; Park, C.H.; Han, D.S.; Eun, C.S. Impact of Short-Chain Fatty Acid Supplementation on Gut Inflammation and Microbiota Composition in a Murine Colitis Model. J. Nutr. Biochem. 2022, 101, 108926. [CrossRef]
- Di Sabatino, A.; Cazzola, P.; Ciccocioppo, R.; Morera, R.; Biancheri, P.; Rovedatti, L.; Cantoro, L.; Vanoli, A.; Tinozzi, F.P.; Tinozzi, S. Efficacy of Butyrate in the Treatment of Mild to Moderate Crohn’s Disease. Dig. Liver Dis. Suppl. 2007, 1, 31–35. [CrossRef]
- Pietrzak, A.; Banasiuk, M.; Szczepanik, M.; Borys-Iwanicka, A.; Pytrus, T.; Walkowiak, J.; Banaszkiewicz, A. Sodium Butyrate Effectiveness in Children and Adolescents with Newly Diagnosed Inflammatory Bowel Diseases—Randomized Placebo-Controlled Multicenter Trial. Nutrients 2022, 14, 3283. [CrossRef]
- Lührs, H.; Gerke, T.; Müller, J.G.; Melcher, R.; Schauber, J.; Boxberger, F.; Scheppach, W.; Menzel, T. Butyrate Inhibits NF-ΚB Activation in Lamina Propria Macrophages of Patients with Ulcerative Colitis. Scand. J. Gastroenterol. 2002, 37, 458–466. [CrossRef]
- Hamer, H.M.; Jonkers, D.M.A.E.; Vanhoutvin, S.A.L.W.; Troost, F.J.; Rijkers, G.; de Bruïne, A.; Bast, A.; Venema, K.; Brummer, R.-J.M. Effect of Butyrate Enemas on Inflammation and Antioxidant Status in the Colonic Mucosa of Patients with Ulcerative Colitis in Remission. Clin. Nutr. 2010, 29, 738–744. [CrossRef]
- Steinhart, A.H.; Hiruki, T.; Brzezinski, A.; Baker, J.P. Treatment of Left-sided Ulcerative Colitis with Butyrate Enemas: A Controlled Trial. Aliment. Pharmacol. Ther. 1996, 10, 729–736. [CrossRef]
- Okamoto, T.; Sasaki, M.; Tsujikawa, T.; Fujiyama, Y.; Bamba, T.; Kusunoki, M. Preventive Efficacy of Butyrate Enemas and Oral Administration of Clostridium Butyricum M588 in Dextran Sodium Sulfate-Induced Colitis in Rats. J. Gastroenterol. 2000, 35, 341–346. [CrossRef]
- Butzner, J.D.; Parmar, R.; Bell, C.J.; Dalal, V. Butyrate Enema Therapy Stimulates Mucosal Repair in Experimental Colitis in the Rat. Gut 1996, 38, 568–573. [CrossRef]
- Kanauchi, O.; Iwanaga, T.; Mitsuyama, K.; Saiki, T.; Tsuruta, O.; Noguchi, K.; Toyonaga, A. Butyrate from Bacterial Fermentation of Germinated Barley Foodstuff Preserves Intestinal Barrier Function in Experimental Colitis in the Rat Model. J. Gastroenterol. Hepatol. 1999, 14, 880–888. [CrossRef]
- Scheppach, W.; Sommer, H.; Kirchner, T.; Paganelli, G.-M.; Bartram, P.; Christl, S.; Richter, F.; Dusel, G.; Kasper, H. Effect of Butyrate Enemas on the Colonic Mucosa in Distal Ulcerative Colitis. Gastroenterology 1992, 103, 51–56. [CrossRef]
- Fernandez-Banares, F.; Hinojosa, J.; Sanchez-Lombrana, J.L.; Navarro, E.; Martınez-Salmerón, J.F.; Garcıa-Pugés, A.; González-Huix, F.; Riera, J.; González-Lara, V.; Domınguez-Abascal, F. Randomized Clinical Trial of Plantago Ovata Seeds (Dietary Fiber) as Compared with Mesalamine in Maintaining Remission in Ulcerative Colitis. Am. J. Gastroenterol. 1999, 94, 427–433. [CrossRef]
- Hallert, C.; Björck, I.; Nyman, M.; Pousette, A.; Grännö, C.; Svensson, H. Increasing Fecal Butyrate in Ulcerative Colitis Patients by Diet: Controlled Pilot Study. Inflamm. Bowel Dis. 2003, 9, 116–121. [CrossRef]
- Hanai, H.; Kanauchi, O.; Mitsuyama, K.; Andoh, A.; Takeuchi, K.; Takayuki, I.; Araki, Y.; Fujiyama, Y.; Toyonaga, A.; Sata, M. Germinated Barley Foodstuff Prolongs Remission in Patients with Ulcerative Colitis. Int. J. Mol. Med. 2004, 13, 643–647. [CrossRef]
- Chen, M.; Tian, S.; Li, S.; Pang, X.; Sun, J.; Zhu, X.; Lv, F.; Lu, Z.; Li, X. β-Glucan Extracted from Highland Barley Alleviates Dextran Sulfate Sodium-Induced Ulcerative Colitis in C57BL/6J Mice. Molecules 2021, 26, 5812. [CrossRef]
- Bai, J.; Zhao, J.; Waleed, A.-A.; Wang, J.; Xue, L.; Liu, J.; Wang, Y.; Fan, M.; Qian, H.; Li, Y. Oat β-Glucan Alleviates DSS-Induced Colitis via Regulating Gut Microbiota Metabolism in Mice. Food Funct. 2021, 12, 8976–8993. [CrossRef]
- Nyman, M.; Nguyen, T.D.; Wikman, O.; Hjortswang, H.; Hallert, C. Oat Bran Increased Fecal Butyrate and Prevented Gastrointestinal Symptoms in Patients with Quiescent Ulcerative Colitis—Randomized Controlled Trial. Crohn’s Colitis 360 2020, 2, otaa005. [CrossRef]
- Wedlake, L.; Slack, N.; Andreyev, H.J.N.; Whelan, K. Fiber in the Treatment and Maintenance of Inflammatory Bowel Disease: A Systematic Review of Randomized Controlled Trials. Inflamm. Bowel Dis. 2014, 20, 576–586. [CrossRef]
- Limketkai, B.N.; Iheozor-Ejiofor, Z.; Gjuladin-Hellon, T.; Parian, A.; Matarese, L.E.; Bracewell, K.; MacDonald, J.K.; Gordon, M.; Mullin, G.E. Dietary Interventions for Induction and Maintenance of Remission in Inflammatory Bowel Disease. Cochrane Database Syst. Rev. 2019. [CrossRef]
- Xu, Z.; Chen, W.; Deng, Q.; Huang, Q.; Wang, X.; Yang, C.; Huang, F. Flaxseed Oligosaccharides Alleviate DSS-Induced Colitis through Modulation of Gut Microbiota and Repair of the Intestinal Barrier in Mice. Food Funct. 2020, 11, 8077–8088. [CrossRef]
- Kang, S.; You, H.J.; Ju, Y.; Kim, H.J.; Jeong, Y.J.; Johnston, T. V; Ji, G.E.; Ku, S.; Park, M.S. Butyl-Fructooligosaccharides Modulate Gut Microbiota in Healthy Mice and Ameliorate Ulcerative Colitis in a DSS-Induced Model. Food Funct. 2022, 13, 1834–1845. [CrossRef]
- Liu, J.; Wang, Z.; Mai, P.; Hao, Y.; Wang, Z.; Wang, J. Quinoa Bran Soluble Dietary Fiber Ameliorates Dextran Sodium Sulfate Induced Ulcerative Colitis in BALB/c Mice by Maintaining Intestinal Barrier Function and Modulating Gut Microbiota. Int. J. Biol. Macromol. 2022, 216, 75–85. [CrossRef]
- Wang, N.; Chen, W.; Cui, C.; Zheng, Y.; Yu, Q.; Ren, H.; Liu, Z.; Xu, C.; Zhang, G. The Peanut Skin Procyanidins Attenuate DSS-Induced Ulcerative Colitis in C57BL/6 Mice. Antioxidants 2022, 11, 2098. [CrossRef]
- De Preter, V.; Falony, G.; Windey, K.; Hamer, H.M.; De Vuyst, L.; Verbeke, K. The Prebiotic, Oligofructose-enriched Inulin Modulates the Faecal Metabolite Profile: An in Vitro Analysis. Mol. Nutr. Food Res. 2010, 54, 1791–1801. [CrossRef]
- Gholami, M.; Ghasemi-Niri, S.F.; Maqbool, F.; Baeeri, M.; Memariani, Z.; Pousti, I.; Abdollahi, M. Experimental and Pathalogical Study of Pistacia Atlantica, Butyrate, Lactobacillus Casei and Their Combination on Rat Ulcerative Colitis Model. Pathol. Pract. 2016, 212, 500–508. [CrossRef]
- Moeinian, M.; Ghasemi-Niri, S.F.; Mozaffari, S.; Abdolghaffari, A.H.; Baeeri, M.; Navaea-Nigjeh, M.; Abdollahi, M. Beneficial Effect of Butyrate, Lactobacillus Casei and L-Carnitine Combination in Preference to Each in Experimental Colitis. World J. Gastroenterol. WJG 2014, 20, 10876. [CrossRef]
- Breuer, R.I.; Soergel, K.H.; Lashner, B.A.; Christ, M.L.; Hanauer, S.B.; Vanagunas, A.; Harig, J.M.; Keshavarzian, A.; Robinson, M.; Sellin, J.H. Short Chain Fatty Acid Rectal Irrigation for Left-Sided Ulcerative Colitis: A Randomised, Placebo Controlled Trial. Gut 1997, 40, 485–491. [CrossRef]
- Vernia, P.; Monteleone, G.; Grandinetti, G.; Villotti, G.; Di Giulio, E.; Frieri, G.; Marcheggiano, A.; Pallone, F.; Caprilli, R.; Torsoli, A. Combined Oral Sodium Butyrate and Mesalazine Treatment Compared to Oral Mesalazine Alone in Ulcerative Colitis. Dig. Dis. Sci. 2000, 45, 976–981. [CrossRef]
- Gibbs, B.; Brown, B.I. Butyrate Therapy for Treatment-Resistant Ulcerative Colitis: A Case Study. 2022.
Figure 1.
Synthesis of microbial metabolites in the intestine. Commensal bacteria in the intestine utilize non-digested fermentable carbohydrates and proteins from the host ingested diet and produce SCFAs and vitamins. Likewise, gut bacteria transform non-absorbed primary bile salts into secondary bile acids. These microbial metabolites modulate the host physiological functions and provide health benefits.
Figure 1.
Synthesis of microbial metabolites in the intestine. Commensal bacteria in the intestine utilize non-digested fermentable carbohydrates and proteins from the host ingested diet and produce SCFAs and vitamins. Likewise, gut bacteria transform non-absorbed primary bile salts into secondary bile acids. These microbial metabolites modulate the host physiological functions and provide health benefits.
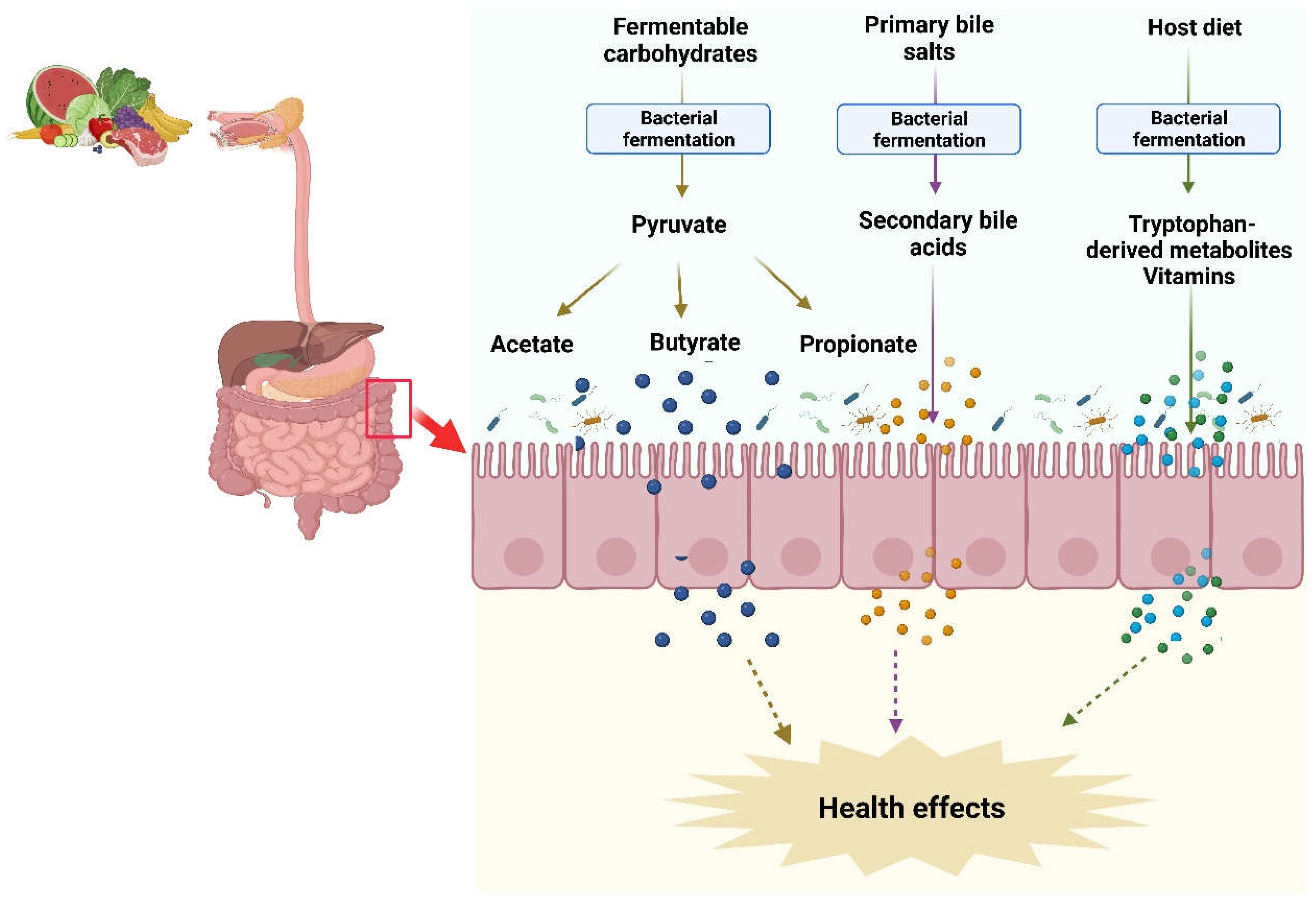
Figure 2.
Schematic representation of bacterial metabolic pathways involved in butyrate production and absorption in the intestine. Butyrate is synthesized by intestinal bacteria via four pathways from non-digestible carbohydrates, succinate, L-lysate, and glutarate. It is taken up and metabolized by the colonic epithelial cells. Low levels of butyrate enter into the liver and regulates fatty acids metabolism. Small amounts of butyrate enter into the systemic circulation and may reach to other tissues.
Figure 2.
Schematic representation of bacterial metabolic pathways involved in butyrate production and absorption in the intestine. Butyrate is synthesized by intestinal bacteria via four pathways from non-digestible carbohydrates, succinate, L-lysate, and glutarate. It is taken up and metabolized by the colonic epithelial cells. Low levels of butyrate enter into the liver and regulates fatty acids metabolism. Small amounts of butyrate enter into the systemic circulation and may reach to other tissues.
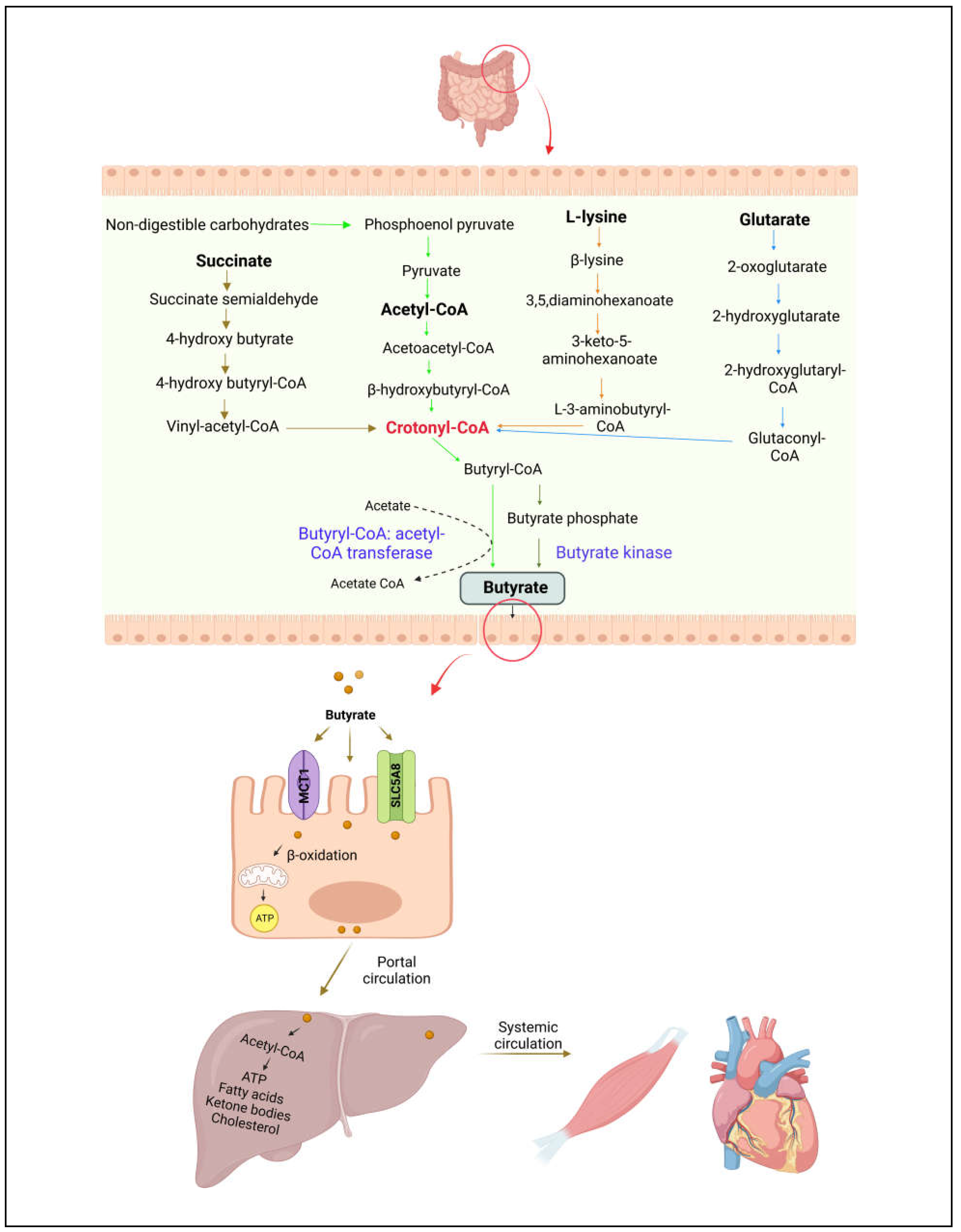
Figure 3.
Schematic overview of butyrate transport and cellular mechanisms. Butyrate is absorbed by intestinal epithelial cells via active transport mediated by MCT1 and SMCT1 transporters or via passive diffusion. Butyrate activates GPRs and couples to G proteins to interact with downstream effectors such as HDACs to reduce inflammation.
Figure 3.
Schematic overview of butyrate transport and cellular mechanisms. Butyrate is absorbed by intestinal epithelial cells via active transport mediated by MCT1 and SMCT1 transporters or via passive diffusion. Butyrate activates GPRs and couples to G proteins to interact with downstream effectors such as HDACs to reduce inflammation.
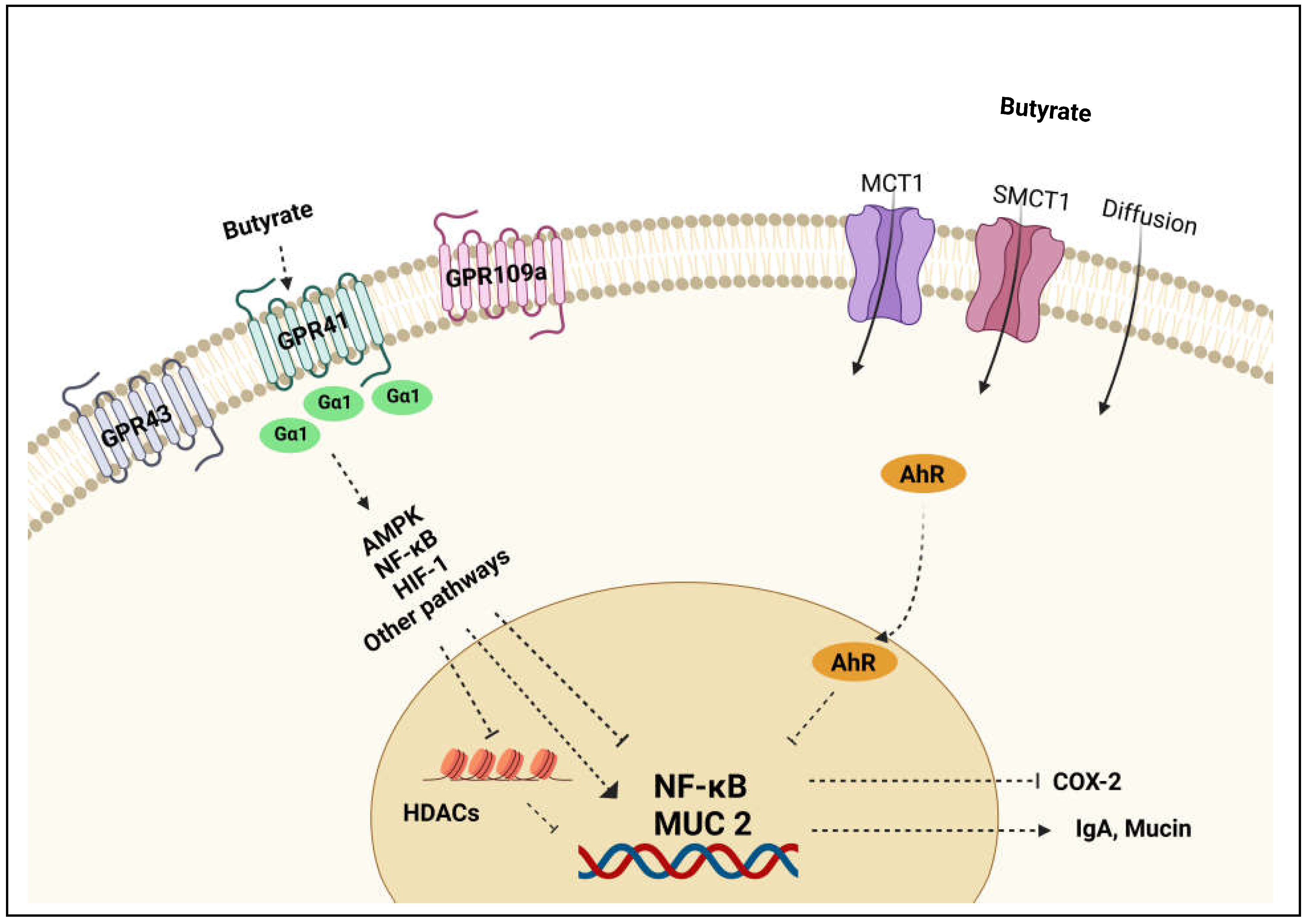
Figure 4.
Microbial metabolite butyrate in the regulation of host immune function. Butyrate plays a key role in maintenance of intestine immune homeostasis. It promotes Treg differentiation and secretion of IL-10 and secretary IgA but inhibits Th17 cell differentiation.
Figure 4.
Microbial metabolite butyrate in the regulation of host immune function. Butyrate plays a key role in maintenance of intestine immune homeostasis. It promotes Treg differentiation and secretion of IL-10 and secretary IgA but inhibits Th17 cell differentiation.
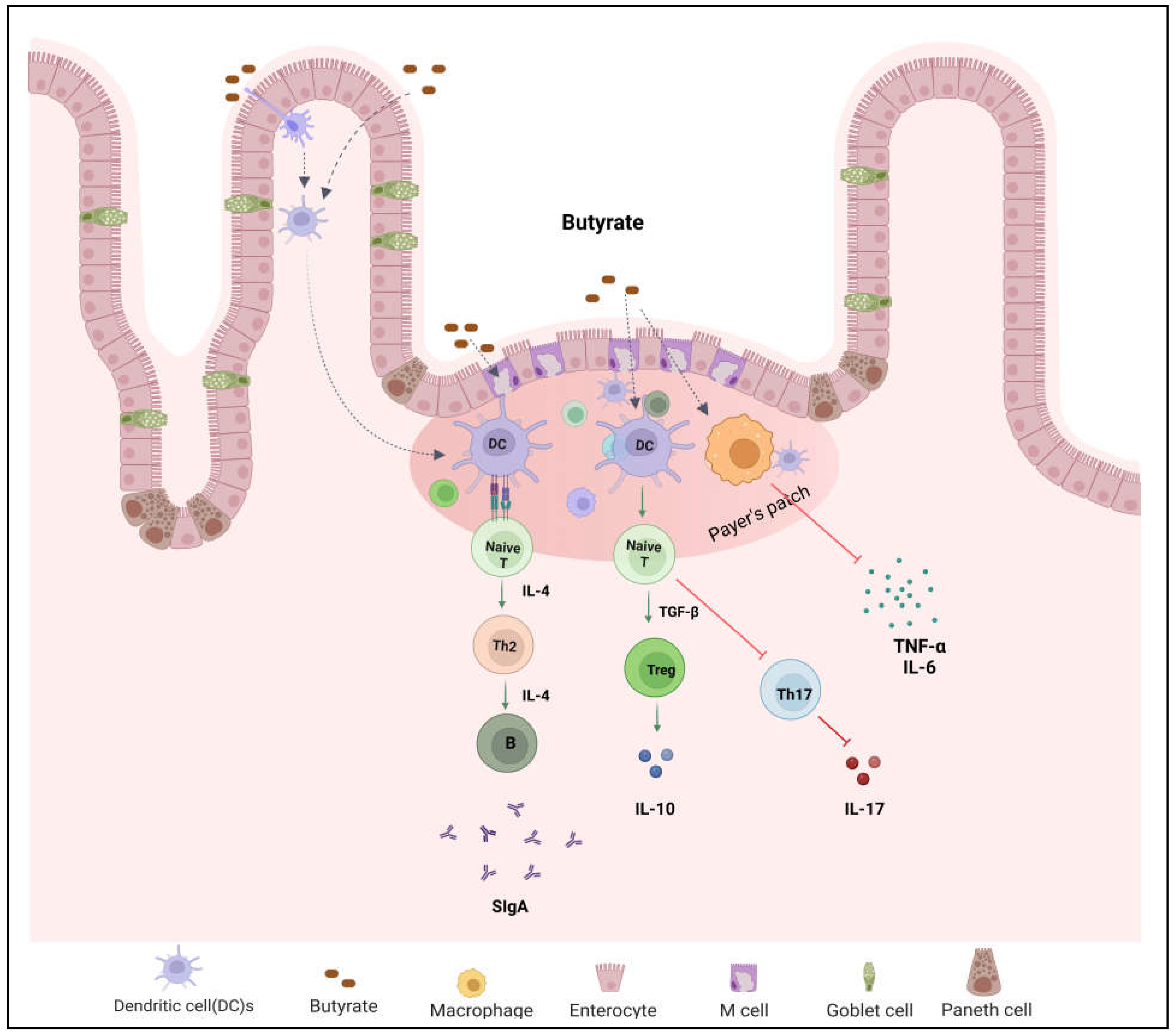
Table 1.
Comparison of Crohn’s disease and ulcerative colitis.
| Features | Crohn’s disease | Ulcerative colitis |
|---|---|---|
| Location | Any part of the GI tract | Large intestine |
| Inflammation | Transmural | Superficial |
| Complications | Fistula development, obstruction | No fistula, Hemorrhage |
| Distribution | Continuous | Discontinuous and patchy |
| Rectal involvement | Common | Occasional |
Table 3.
Impact of butyrate enemas on IBD.
| Concentration | Colitis model | Duration | Effects | Authors | |
|---|---|---|---|---|---|
| Rat | 3 ml of 100 mM | DSS-induced colitis | 17 days | Decreased ulcer index and myeloperoxidase activity | Okamoto et al. [84] |
| Rat | 100 mM sodium butyrate | TNBS induced colitis | Day 5 to 23 | Decreased inflammation and improved clinical recovery | Butzner et al. [85] |
| Rat | 3% of sodium butyrate | DSS-induced colitis | Decreased mucosal damage, no difference in the incidence of diarrhea | Kanauchi et al. [86] | |
| Rat | 100 mM sodium butyrate | TNBS induced colitis | 2 wks | Decreased inflammation and stimulated mucosal repair | Segain et al. [59] |
| Human | 100 mM sodium butyrate | Ulcerative colitis | 2 wks | Decreased disease activity index and inflammation | Scheppach et al. [87] |
| Human | 60 mL of 80 mM sodium butyrate | Ulcerative colitis | 3 and 6 wks | Nightly butyrate enema was not efficacious for distal ulcerative colitis | Steinhart et al. [83] |
| Human | 60 mL of 100 mM sodium butyrate | Ulcerative colitis | 4 and 8 wks | Decreased disease activity index and mucosal inflammation after 8 wks | Luhrs et al. [81] |
| Human | 60 mL of 100 mM sodium butyrate | Ulcerative colitis | 20 days | No significant effects of butyrate administration on parameters of oxidative stress were found | Hamer et al. [82] |
Table 4.
Impact of butyrogenic diet on IBD.
| Treatment | Disease or model | Effects | Authors | |
|---|---|---|---|---|
| Rat | Germinated barley foodstuff | DSS-induced colitis | Bloody diarrhea and mucosal damage were dose dependently decreased | Kanauchi et al. [86] |
| Mice | Flaxseed oligosaccharides | DSS-induced colitis | Decreased disease activity index, improved colon histology, and increased cecal SCFAs levels | Xu et al. [96] |
| Mice | Oat β-glucan | DSS-induced colitis | Suppressed colonic inflammatory infiltration and increased SCFAs concentrations | Bai et al. [92] |
| Mice | Butyl-fructooligosaccharides | DSS-induced colitis | Increased cecal butyrate concentration, increased occludin mRNA expression | Kang et al. [97] |
| Mice | Soluble dietary fiber from quinoa bran | DSS-induced colitis | Decreased disease activity index, increased microbial diversity and SCFAs | Liu et al. [98] |
| Mice | Peanut skin procyanidins extract | DSS-induced colitis | Suppressed inflammatory responses, increased butyrate producing bacterial abundance and colon SCFAs | Wang et al. [99] |
| Human | Plantago ovata seeds | Ulcerative colitis in remission | Increased fecal butyrate levels | Fernandez-Banares et al. [88] |
| Human | Oat bran | Ulcerative colitis | Increased fecal butyrate and maintained the remission phase | Hallert et al. [89] |
| Human | Germinated barley food stuff | Ulcerative colitis in remission | Effective in maintenance of prolonged remission | Hanai et al. [90] |
| Human | Prebiotic oligofructose-enriched inulin | Crohn's disease | The relative levels of butyrate and acetaldehyde increased compared to baseline | De Preter et al. [100] |
| Human | Oat bran | Ulcerative colitis in remission | Increased fecal SCFAs including butyric acid and reduced the risk of relapse | Nyman et al. [93] |
Table 5.
Effects of butyrate combination therapies on IBD.
| Treament name | Concentration | Colitis model | Duration | Effects | Authors | |
|---|---|---|---|---|---|---|
| Mice | SCFAs | 67.5 mM acetate, 40 mM butyrate, 25.9 mM propionate | DSS-induced colitis | No significant difference in histologic scores but IL-17A producing T cells increased | Lee et al. [78] | |
| Rat | Pistacia atlantica, butyrate, Lactobacillus casei | 25 mg/kg atlantica, 0.5% butyrate, and 108 CFU of Lactbacillus | TNBS-induced colitis | 10 days | Reduced the severity of inflammation | Gholami et al. [101] |
| Human | Plantago ovata seeds and mesalamine | 20 g seeds and 1.5 g mesalamine/day | Ulcerative colitis remission | 12 months | Effective in remission maintenance | Fernandez-Banares et al. [88] |
| Human | Sodium butyrate and mesalazine | 4 g/day butyrate and 2.4 g/day mesalamine | Ulcerative colitis | 6 wks | Improved the efficacy of mesalazine | Vernia et al. [104] |
| Human | Calcium magnesium butyrate along with Mezavant treatment | 1.2 g/day magnesium butyrate and 4.8 g/day mezavant |
Ulcerative colitis | Relief of symptoms | Gibbs and Brown. [105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



