Submitted:
19 April 2023
Posted:
20 April 2023
You are already at the latest version
Abstract
Antiretroviral therapy has effectively suppressed HIV infection and replication and prolonged the lifespan of HIV-infected individuals. In the meantime, various complications including type 2 diabetes associated with long-term antiviral therapy have shown steady increases. Metformin has been the front-line anti-hyperglycemic drug of choice and the most widely prescribed medication for the treatment of type 2 diabetes. However, little is known about the effects of Metformin on HIV infection and replication. In this study, we showed that Metformin treatment enhanced HIV gene expression and transcription in HIV-transfected 293T and HIV-infected Jurkat and human PBMC. Moreover, we demonstrated that Metformin treatment resulted in increased CREB expression and phosphorylation, and TBP expression. Furthermore, we showed that Metformin treatment increased the recruitment of phosphorylated CREB and TBP to the HIV LTR promoter. Lastly, we showed that inhibition of CREB activation significantly abrogated Metformin-enhanced HIV gene expression. Taken together, these results demonstrated that Metformin treatment increased HIV transcription, gene expression, and production though increased CREB phosphorylation and recruitment to the HIV LTR promoter. These findings may help design the clinical management plan and HIV cure strategy of using metformin to treat type 2 diabetes, a comorbidity with an increasing prevalence, in people living with HIV.
Keywords:
Metformin
; HIV transcription and replication
; transactivation
; gene expression
; CREB phosphorylation
; HIV LTR promoter
Introduction
Antiretroviral therapy has effectively suppressed human Immunodeficiency virus (HIV) replication and significantly prolonged the lifespan of HIV-infected population [1,2]. However, this population has still faced other health-relevant disorders and complications [3,4]. These include hypertension and cardiovascular diseases [5-8], renal impairment [9,10], lipodystrophy [11-15], dyslipidemia [16,17], premature/rapid aging [5,18,19], insulin resistance, and diabetes mellitus [20-22], many of which are associated with antiretroviral therapy [23]. The incidence of type 2 diabetes is higher among HIV-infected individuals, as antiretroviral therapy, specifically its component protease inhibitors, is most commonly linked to insulin resistance [20,24-27].
Metformin is the front-line medicine of choice for treating type 2 diabetes and is being prescribed for approximately 120 million individuals worldwide [28]. Its anti-hyperglycemic action is primarily an outcome of decreased glucose production (without overt hypoglycemia) from the liver though inhibition of gluconeogenesis [28,29] and increase in glucose uptake by voluntary/skeletal muscles, although to a smaller extent [30]. It has also been shown to improve insulin sensitivity by increasing the activity of insulin receptor though enhancing and lengthening the tyrosine phosphorylation of β-subunit of this receptor [31]. The mitochondrion is the primary target of Metformin, where it inhibits complex I of the mitochondrial electron transport chain, resulting in decreases in ATP and increases in 5′-adenosine monophosphate (AMP) and subsequent activation of AMP-activated protein kinase (AMPK). AMPK acts as a sensor of cellular energy and a principal organizer of signaling pathways to maintain the balance between metabolic (lipid/glucose) and growth pathways and to restore cellular energy once the energy level is low [30,32-36].
The pleiotropic effects of Metformin have recently gained more attention, from improving lipid profile [37,38] and regulating inflammatory markers in cardiovascular diseases [39,40] to ameliorating tumor establishment, progression, and cancer-related mortality rate [41,42]. Metformin treatment has also been shown to impact HIV comorbidities such as lipodystrophy [11-15], cardiovascular diseases [43-45], and gut microbiota diversity [46,47] in HIV-infected individuals with type 2 diabetes. Several small clinical trials indicate possible effects of Metformin on HIV reservoirs in non-diabetic HIV-infected individuals who are suppressed by antiretroviral therapy [48-50]. However, whether and how Metformin itself affects HIV gene expression and replication is not known.
In the current study, we aimed to investigate effects of Metformin on HIV gene expression and replication and the underlying molecular mechanisms. We began with introduction of HIV into the cells by transfection and infection, treated the cells with Metformin, and determined intracellular HIV gene expression and extracellular HIV production. We then determined effects of Metformin on HIV gene transcription, and expression of several major transcription factors and the recruitment of these transcription factors to the HIV LTR promoter. We also determined effects of Metformin on HIV latency using latent cell lines. Lastly, we validated and substantiated the findings in HIV-infected human peripheral blood mononuclear cells (PBMC). All the results together demonstrated that Metformin enhanced HIV gene expression, transcription, and production and re-activated HIV from latency and that increased cAMP response element-binding protein (CREB) phosphorylation and expression and TATA-binding protein (TBP) expression and their recruitment to the HIV LTR promoter were likely involved in these processes.
Materials and Methods
Cells, plasmids, transfection, and metformin treatment. Human embryonic kidney epithelial cell line 293T was purchased from American Type Culture Collection (Manassas, VA). Jurkat clone E6-1 (#ARP-177) [51], HIV-1 lymphadenopathy-associated virus (LAV)-infected Jurkat E6 clone J1.1 (#ARP-1340), HIV-1 chronically infected U937 clone U1 (#APR-165) and ACH-2 (#ARP-349) [52-54], HIV LTR promoter-driven luciferase reporter cell line TZM-bl (#ARP-8129) [55-57], HIV-1 NL-4-3 LTR-driven luciferase reporter (#ARP-4788) [58,59] were obtained though the NIH HIV Reagent Program. NLGi latently infected Jurkat were generated by infecting Jurkat with NLGi and culturing the infected cells for over 63 days, changing the media every thee days, and monitoring GFP expression in these cells. Buffy coat was purchased from Versiti (Indianapolis, IN). 293T and TZM-bl cells were cultured in Dulbecco’s modified Eagle’s medium (Corning, Manassas, VA). Jurkat, PBMC, ACH-2, J1.1, U1, and NLGi latent Jurkat were cultured in RPMI 1640 medium (Corning). Both DMEM and RPMI1640 were supplemented with 10% fetal bovine serum (R&D Systems, Minneapolis, MN), 100 U/ml penicillin/100 μg/ml streptomycin (Cat # P4333, Sigma-Aldrich, St. Louis, MO) in a 37°C, 5% CO2 incubator. 293T were transfected using the standard calcium phosphate precipitation method [60,61], which often gives rise to 100% transfection efficiency. pcDNA3, pNL4-3, pAP-1-Luc, and pNF-κB-Luc were purchased from Clontech (Mountain View, CA). pGL3.TATA-Luc was constructed using the synthetic adenovirus E1b TATA sequence (TATATAAT) inserted in the pGL3 backbone (Promega, Madison, WI) [62]. pNL4-3-Luc-E- was described elsewhere [63]. HIV reporter virus vector NLGi, a derivative from the pNL4-3 HIV vector with the green fluorescent protein (GFP) reporter gene inserted preceding the Nef gene, was a gift from Dr. B. K. Chen of Mount Sinai School of Medicine [64]. HIV-LTR Core reporter was constructed by insertion of LTR-Core (from pNL4-3 construct) into the pGL3 backbone (Promega, Madison, WI) using restriction digestion and ligation protocol, and primers 5′-TAG AGA GCT CTC TAC AAG GGA CTT TCC G-3′ and 5′-GAG ACA AGC TTT GCT TAT ATG CAG CAT CTG-3′. Lipofectamine™ 3000 Transfection Reagent (# L3000001, ThermoFisher Scientific, Waltham, MA) was used for transfection of 293T cells with pAP-1-Luc, pNF-κB-Luc, and p-TATA-Luc. Metformin was purchased from Cayman Chemical (#13118, Ann Arbor, Michigan), freshly prepared in phosphate-buffered buffer (PBS) and added to the cells as indicated. 666-15 was purchased from Millipore Sigma (# 5383410001, Burlington, MA), dissolved in dimethyl sulfoxide (DMSO), and added to the cells as indicated according to the effective concentration reported in literature [65,66].
HIV production and infection. 293T were plated in a 10 cm dish at a density of 2 x 106 cells per dish and transfected with 20 μg pNL4-3 or 3.3 μg pVSV-G plus 16.7 μg pNL4-3-Luc-E- using the standard calcium phosphate precipitation method. The cells were cultured for 16 h, the culture medium was replaced with fresh medium, and continued to culture for 48 h. The culture medium was collected and briefly centrifuged to remove the cell debris. The cleared supernatant was passed though a syringe filter 0.45 µm (SIMSII, Issaquah, WA) and layered to 20% sucrose solution, followed by ultracentrifugation at 100,000 x g at 4ºC for 2 h. The virus pellet was suspended in PBS, and the suspended viruses were aliquoted, stored at -80oC, and used as virus stock. The virus titer was determined using the reverse transcriptase assay (see below). Jurkat were infected with NL4-3 in the presence of 8 µg/ml polybrene by spinoculation at 850 x g and room temperature for 2 h or transduced by VSV-G-pseudotyped NL4-3-Luc-E-. The infected cells were washed with fresh culture media after infection/transduction and processed for the subsequent experiments.
Cell lysate preparation and Western blotting. Cells were washed twice with ice-cold PBS and lysed in RIPA buffer [10 mM Tris.HCl, pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 1% Triton X-100, 1 mM PMSF, and 1X protease and phosphatase inhibitor cocktail (ThermoFisher Scientific)] on ice for 20 min. The cell debris was removed by centrifugation at 12,000 x g for 15 min to obtain the clear cell lysates. The protein concentration in the lysates was determined using a Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA) and a Bio-Rad iMark microplate reader. The proteins in the lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto Polyvinylidene Fluoride (PVDF) membrane probed with a primary antibody and an appropriate secondary antibody. The probed WB membranes were visualized and imaged using Enhanced Chemiluminescence substrates (#32106, ThermoFisher Scientific), and a Chemidoc MP imaging system (Bio-Rad). The primary antibodies were rabbit monoclonal antibodies AMPKα (#2532), phospho-AMPKα (Th172) (#2535), CREB (#9197), phospho-CREB (Ser133) (#9198), CBP (#7389), p300 (#54062), TBP/TFIID (#44059) from Cell Signaling (Danvers, MA); mouse monoclonal antibodies GAPDH (#sc-32233) from Santa Cruz Biotechnology (Dallas, TX); Anti-HIV-1 p24 Hybridoma (183-H12-5C, #ARP-1513) [67] and rabbit polyclonal anti-HIV-1 Nef protein (#ARP-2949) [68] were obtained though NIH AIDS Reagent Program; and mouse monoclonal antibody β-actin (#A1978) from Sigma-Aldrich (St. Louis, MO); the secondary antibodies were HP-conjugated sheep anti-mouse IgG (#NA931V) and HP-linked donkey anti-rabbit IgG (#NA9340V) from Millipore Sigma (Burlington, MA).
Reverse transcriptase (RTase) assay. The RTase activity assay was performed as previously described [69]. The supernatants containing viruses were collected 48 hours post-transfection and spun down at 500 x g for 5 min to remove the cell debris. The supernatants were then transferred to the new tubes and centrifuged at 21,300 x g at 4°C for 1 h. The virus pellets were suspended and lysed in 10 µl dissociation buffer (50 mM Tris-HCl, pH 7.5, 0.25 M KCl, 0.25% Triton X-100, 20% glycerol, and 1 mM DTT), followed by subjecting the suspended pellets to thee quick freeze-thaw cycles. Subsequently, a 40 µl reaction mixture including 34 µl RT assay buffer (50 mM Tris-HCl pH 7.5, 7.5 mM MgC12, and 0.05 % Triton X-100, 0.5 mM DTT), 5 µl 1 mg/ml poly(A) x (dT)15 (Roche Diagnostics, Indianapolis, IN), and 1 µl [Methyl3H]-deoxythymidine 5′-triphosphate tetrasodium salt (Perkin Elmer, Waltham, MA) was added to the virus lysate and incubated at 37°C for 1 h. The reaction mixtures were then deposited onto nitrocellulose membrane (Bio-Rad, Hercules, CA). The spotted membranes were washed with 2X SSC buffer (0.3 M sodium chloride, 30 mM sodium citrate) thee times, 5 min each, followed by rinsing and dehydrating the membranes in 100% ethanol. The membranes were then air-dried and analyzed utilizing a scintillation counting fluid (#6013631, Perkin Elmer, Waltham, MA) and a microβeta2 scintillation counter (#2450, Perkin Elmer). The recorded RT activity was expressed as counts of incorporated 3H per minute (CPM) per milliliter of the supernatant.
Cell viability and proliferation assays. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to measure the cell number and viability. 293T were seeded in a 24-well plate at a density of 0.6 x 105 per well and cultured for 24 h. The cells were then transfected with cDNA 3 or pNL4-3. Media was replaced with fresh DMEM medium 16 h after transfection, and cells were treated with either PBS or various concentrations of metformin for 48 h. TZM-bl and HIV-infected Jurkat cells were seeded at the same density as 293T above and cultured for 24 h and treated with PBS/metformin for 48 h. The medium was removed and replaced with a complete DMEM medium for 293T and TZM-bl cells or RPMI 1640 medium for Jurkat. MTT (#298-93-1, Biosynth International, Inc, Itasca, IL) was added to each well with the final concentration of 1 mg/ml per well. The plates were incubated in the dark at 37°C for 4 h. The media containing MTT was removed, and DMSO was added to completely dissolve the formazan crystals by shaking the culture plates wrapped with aluminum foil at room temperature for 20-30 min. The plates were shortly centrifuged. The supernatants (100 μl each) were transferred to a 96-well plate for optical density reading. The solvent DMSO was used as the background control and subtracted from the samples’ reading. The absorbance reading was performed at a wavelength of 595 nm with the reference wavelength of 655 nm using an iMark plate reader (Bio-Rad). A fluorometric cell proliferation assay kit (#K307-1000, BioVision, Waltham, MA) was used to determine the cell number according to the manufacturer’s instructions. Briefly, HIV-infected Jurkat were seeded in a 96-well white tissue culture plate with a clear bottom at a density of 1 x 104 per well and cultured for 24 h. The cells were treated and incubated with PBS or various metformin concentrations for 48 h and lysed in 1X cell lysis buffer. Cell lysates (25 µl) was mixed with 25 µl 1X nuclear dye in each well. The plate was gently shaken at room temperature for 15 min on a shaker while protected from light. The fluorescence of treated and untreated cells was measured using a microplate reader (Biotek Synergy HT) at Ex/Em = 480/538 nm. The cell number was determined by serial dilutions of Jurkat to create the standard curve, which was used to calculate the cell number.
RNA isolation, semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR), and quantitative real-time PCR (qRT-PCR). Total RNA was extracted using the TRIzol RNA isolation reagent (#15596018, Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions except for the inclusion of additional step of acidic phenol extraction (#AM9722, Invitrogen) to prevent transfected plasmid DNA from being PCR amplified. Total RNA was used to synthesize cDNA using an iScript™ Reverse Transcription Supermix (#1708890, Bio-Rad). The cDNA was either subjected to the semi-quantitative PCR with the program of 1 cycle of 95°C for 3 min, 35 cycles of 95°C for 1 min, 55°C for 1 min, and 72°C for 2 min, and 1 cycle of 72°C for 8 min, or the qRT-PCR using SYBR Green mix (#1725270, Bio-Rad) with the program of 1 cycle of 95°C for 3 min and 40 cycles at 95°C for 30s, and 60°C for 1 min. The primers were P9501: 5′-CAG ATG CTG CAT ATA AGC AGC TG-3′ and 5T25: 5′-TTT TTT TTT TTT TTT TTT TTT TTT TTG AAG-3′ for unspliced and spliced HIV viral RNA [70]; SK145: 5′-AGT GGG GGG ACA TCA AGC AGC CAT GCA AAT-3′ and SK39: 5′-TTT GGT CCT TGT CTT ATG TCC AGA ATG C-3′ for Gag-Pol RNA [71,72]; 5′-GAA ACT GTG GCG TGA TGG C-3′ and 5′- CCA GTG AGC TTC CCG TTC AG-3′ for GAPDH, which was used as an internal control for normalization. For semi-quantitative RT-PCR, we titrated and optimized the amount of input RNA for reverse transcription and cDNA for PCR to ensure that the amplification was in the linear range.
Luciferase reporter gene assay. Cells were washed with ice-cold PBS and lysed with 1X passive lysis buffer (#E4030, Promega, Madison, WI) for 15 min with intermittent mixing. The cleared cell lysates were obtained by brief centrifugation and added with firefly luciferase assay substrate (#E1500, Promega, Madison, WI) at the ratio of 1:4 (5 µl sample plus 20 µl substrate). The luciferase activity was measured using a Lumat LB 9501 Single Tube Luminometer (Berthold, Hartford, CT). The protein concentration of the cleared cell lysates was also determined using a Bio-Rad DC protein assay kit (Bio-Rad) and used to normalize the luciferase activity, which was expressed as relative luminescence/light unit (rlu).
Chromatin immunoprecipitation (ChIP) assay. The ChIP assay was performed according to the protocol established by Rockland Immunochemicals (Pottstown, PA) with some modifications. Briefly, Cells were washed with ice-cold PBS twice, added freshly made 5 mM Dimethyl 3,3′-dithio-bis (propionimidate) dihydrochloride (#38285-78-8, Sigma-Aldrich), incubated on ice for 30 min, added ice-cold quenching buffer (100 mM Tris.HCl pH 8.0, 150 mM NaCl), and then washed with ice-cold PBS twice. The cells were then added 1% formaldehyde (#410730050 ThermoFisher Scientific), incubated at room temperature for 10 min, added the second quenching buffer (0.125 M glycine), incubated at room temperature for 5 min, and washed with PBS twice. The cross-linked cells were suspended in the swelling buffer [25 mM Hepes, pH 7.8, 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40, 1 mM DTT, 0.5 mM PMSF, 1X protease and phosphatase inhibitor cocktail (#A32959, ThomoFisher Scientific)], followed by several times pipetting and incubation in ice for 15 min. Nuclei were then pelleted and suspended in the nuclear lysis buffer [50 mM Hepes, pH 7.9, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 0.5 mM PMSF, 1X protease and phosphatase inhibitor cocktail (#A32959, ThermoFisher Scientific)]. The nuclear lysates were then subjected to 0.5 unit of micrococcal nuclease (MN) digestion (#88216, ThermoFisher Scientific) per 200 µl sample diluted in MN reaction buffer (50 mM Tris-HCl pH 8.0, 5 mM CaCl2) at room temperature for 10 min. The reaction was stopped by 20 mM EGTA (pH 8.0). The samples were then further sonicated using Sonic Dismembrator (Fisherbrand™ Model 505, FisherScientifc, Pittsburgh, PA) on ice with 5-7 pulses, each for 10 sec with the 10-sec intervals on ice between pulses to complete the break of the nuclear membrane and facilitate the release of all the fragmented chromatin. Subsequently, the lysates were pre-cleared by adding 30 µl protein A agarose beads (#20333, ThermoFisher Scientific)/ml lysate and rotating the samples for 1.5 h at 4°C, followed by centrifugation of samples at 2000 x g for 5 min at 4°C to pellet and discard the beads, while the cleared nuclear lysates were saved and transferred to the new tubes. The primary antibodies of 2-4 µg rabbit monoclonal antibodies for desired targets that were used for Western blotting above (Cell Signaling) or normal rabbit IgG (#J2909, Sigma), was added to the samples and incubated at 4ºC overnight on a rotator, followed by adding protein A agarose beads that were pre-incubated with BSA/Salmon Sperm DNA, and continued to incubate at 4ºC for 4 h. The retrieved beads were washed sequentially twice with each of the following buffers: Low-salt buffer (50 mM Tris.HCl, pH 8.0,150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1 mM EDTA), High-salt buffer (50 mM Tris.HCl, pH 8.0, 500 mM NaCl, 0.1% SDS, 1% Triton X-100, 1 mM EDTA), LiCl buffer [50 mM Tris.HCl, pH 8.0, 250 mM LiCl, 1% sodium deoxycholate, 1% Nonidet P-40, 1 mM EDTA, 0.5 mM PMSF, protease inhibitor cocktail (#A32953, ThermoFisher Scientific)], and TE buffer (10 mM Tris.HCl, pH 8.0, 0.25 mM EDTA pH 8.0) [73]. Each wash was performed by rotating at 4ºC for 10 min. The agarose beads were retrieved by brief centrifugation, and the DNA-protein complexes were eluted in the elution buffer (50 mM Tris.HCl, pH 8.0, 1 mM EDTA pH 8.0, 1% SDS, 50 mM NaHCO3), followed by adding 200 mM NaCl and treatment with 0.1 µg/µl DNase- and protease-free RNase A (#EN0531, ThermoFisher Scientific) at 65ºC overnight and then treatment with 0.2 µg/µl proteinase K (#MC5005, Promega, Madison, WI) at 42ºC for 2 h. The input enzymes and remaining cellular proteins were removed by phenol-chloroform extraction, and the DNA was recovered by ethanol precipitation [1 µl 20 µg/µl Glycogen (#R0561, ThermoFisher Scientific), 7.5 M NH4OAc in the amount of 0.5 X volume of sample and 100% ethanol in the amount of 2.5 X volume of sample] and used as the template for qPCR. The primers were 5′-CAT CCG GAG TAC TTC AAG AAC TGC-3′ and 5′-GGC TTA AGC AGT GGG TTC CCT AG-3 spanning 3′ LTR-promoter region (nt 8984-9202) [73]; 5′-GAG CTT TCT ACA AGG GAC TTT C-3′ and 5′-AGA CCC AGT ACA GGC AAA-3′ for the 5′ LTR promoter region (nt 337-459) [74]; and 5′-CTA GCA TTT CGT CAC ATG GCC-3′ and 5′-GTG GGT TCC CTA GTT AGC CAG-3′ for the larger portion of 5′ LTR region (nt 276-514) targeting CBP, p300 along with TBP, CREB, and its phosphorylated from [75]. qPCR was also performed with U6 primers: 5′-GTG CTC GCT TCG GCA GCA CA-3′ and 5′-AAA ATA TGG AAC GCT TCA CGA-3′ to determine the input DNA and used for normalization.
Human PBMC isolation, activation, and infection. Human PBMC was isolated from fresh buffy coat collected from healthy donors using the density gradient centrifugation method. Briefly, buffy coat was diluted with 2X volume of PBS and was gently layered over an equal volume of Ficoll-Paque PLUS (#17144003, Cytiva, Marlborough, MA), followed by centrifugation for 40 min at 400 x g without break. The PBMC layer was removed and transferred to a new 50 ml falcon tube. PMBC was washed in 25 ml DPBS buffer by two times suspension followed by centrifugation for 10 min at 350 x g to pellet the cells. Isolated PBMC was counted and cultured for 72 h in the presence of 1 µg/ml anti-human CD3 antibody (#317302, BioLegend, San Diego, CA) and 2 µg/ml anti-human CD28 antibody (#302902, BioLegend, , San Diego, CA). The cells were then infected with NL4-3 at a MOI as indicated by spinoculation at 850 x g at room temperature for 2 h in the presence of 8 µg/ml polybrene. The cells were recovered by centrifugation, washed with fresh media, and continued to culture in the presence of 100 IU/ml human IL-2 (#21-8029-U050, Tonbo Bioscience, San Diego, CA) and designated PBS/metformin treatments.
Statistical analysis. All data except for ChIP assay results were analyzed by one-way ANOVA followed by either Bonferroni or Dunnett post hoc tests unless stated otherwise. The results obtained from ChIP assay were analyzed using Two-way ANOVA *: p < 0.05, **: p < 0.01, ***: p < 0.001.
Results
Metformin treatment increased HIV production and intracellular HIV gene expression.
To determine if Metformin treatment would alter HIV production, we first transfected 293T with NL4-3 and treated the cells with Metformin, collected the culture supernatant, and determined the HIV level in the supernatant. We observed that Metformin increased HIV production beginning at a concentration of 0.5 mM and up to 8 mM (Figure 1A). We also noticed significantly fewer cells when Metformin reached to 8 mM (Figure S1A). Thus, we chose to use Metformin at the concentration of 0-4 mM for the subsequent experiments. To determine if Metformin would also increase intracellular HIV gene expression, we harvested the same transfected cells, prepared cell lysates, and performed Western blotting using an anti-p24 antibody as a marker for late gene expression and an anti-Nef antibody for early gene expression. There were parallel increases of p24 and its precursors p55/41 with more Metformin (Figure 1B) and parallel increases of Nef expression with more Metformin (Figure S1B). We next determined Metformin effects on HIV production and intracellular HIV gene expression in the context of HIV infection. To this end, we infected Jurkat with NL4-3, treated the cells with Metformin, collected the supernatant for HIV production, and harvested the cells for intracellular gene expression. Similar to the transfection, more HIV and more p24 and its precursor p55/41 were detected with more Metformin in the context of HIV infection (Figure 1C,D). Similarly, fewer cells were detected with more Metformin, determined by the fluorometric method or direct cell counting (Figure S2A,B). To further confirm Metformin effects on HIV gene expression, we infected Jurkat with replication-defective VSV-G-pseudotyped HIV-Luc, treated the cells with Metformin, and performed Western blotting. Comparably, more p24 and its precursor p55 were detected in these cells with more Metformin (Figure 1E). Taken together, these results demonstrated that Metformin treatment increased HIV production and intracellular HIV gene expression.
Metformin treatment increased HIV RNA expression and transcription.
To determine if Metformin-enhanced HIV gene expression and production resulted from increases in HIV transcription, we determined the level of unspliced HIV RNA in HIV-transfected Metformin-treated 293T using the conventional semi-quantitative RT-PCR and a pair of gag-pol specific primers [71,72]. More Metformin led to higher levels of unspliced HIV RNA (Figure 2A & B). We also determined the total of unspliced and spliced HIV RNA in these transfected cells using qRT-PCR and a pair of primers that were designed to allow detection of both unspliced and spliced HIV RNA [76]. Consistent with the unspliced HIV RNA, more Metformin led to increases in total HIV RNA (Figure 2C). We performed similar qRT-PCR experiments to determine the total HIV RNA in HIV-infected and Metformin-treated Jurkat. Similar results were obtained (Figure 2D). To ascertain that Metformin treatment indeed led to increased HIV transcription, we first treated HIV LTR promoter-driven luciferase (Luc) reporter cell line TZM-bl with Metformin and determined the Luc activity. Higher Luc activities were detected with higher concentrations of Metformin (Figure 3A). We then transfected 293T with HIV LTR-driven Luc reporter plasmid, treated these cells with Metformin, and determined the Luc activity. Similar results were obtained (Figure 3B). All these results together demonstrated that Metformin treatment led to activation of the HIV LTR promoter transcription. The HIV LTR promoter is comprised of thee distinct regions U3, R, and U5. U3 is further divided into the core promoter, the enhancer, and the modulatory region [77]. R contains trans-activation response element TAR region responsible for Tat-enhanced transcription of full-length of HIV RNA. A number of DNA binding sites have been identified within U3 for cellular transcription factors, including multiple DNA binding sites for transcription factors activating protein 1 (AP1) and nuclear factor kappa B (NF-κB). Thus, we first took advantage of AP1 DNA binding site-driven Luc reporter gene (AP1-Luc), NF-κB DNA binding site-driven Luc reporter gene (NF-κB-Luc), and TATA DNA binding site-driven Luc reporter gene (TATA-Luc) and determined the Luc reporter gene activities in response to Metformin treatment. Lower Luc activities were detected with higher Metformin with AP1-Luc (Figure S3A) and NF-κB-Luc (Figure S3B). In comparison, higher Luc activities were detected with higher Metformin with TATA-Luc (Figure S3C). To determine the combined effect of Metformin on the HIV LTR core promoter that contains all these responsive elements, we performed the HIV LTR core promoter-driven Luc reporter gene assay in the presence of Metformin and found the direct activation of metformin on the HIV LTR core promoter (Figure S4), suggesting a net enhancement effect of metformin though CREB and TBP.
Increased CREB expression and phosphorylation and TBP expression by Metformin and HIV
The similar enhancement effects of Metformin on HIV gene expression and production and the TATA-Luc reporter gene activities prompted us to focus on the transcription factors that target the TATA-box of the HIV LTR. TATA-binding protein (TBP)/TFIID are known to bind to the TATA-box region, recruit other basal transcription factors to the promoter to form the RNA polymerase II transcription complex, and facilitate transcription initiation and elongation [78]. TBP also binds to cellular transcription factors and viral proteins to activate transcription [79]. Among these transcription factors is CREB, which functions as a dimer upon phosphorylation [80-82] and has multiple cAMP response elements within the HIV LTR promoter and promotes HIV transcription though the cAMP pathway and CREB binding [83]. Thus, we next determined effects of Metformin on CREB expression and phosphorylation and TBP expression using Western blotting. Compared to the cDNA3 transfection control (Figure 4A), NL4-3 transfection showed trends of increases in CREB expression and phosphorylation and TBP expression at each concentration of Metformin treatment (Figure 4B). Consistent with previous studies [30,32,33,35,36,84], Metformin treatment led to AMPK phosphorylation in both cDNA3 and NL4-3 transfections, albeit with no significant differences between these two transfections. To determine if these changes would occur in the context of HIV infection, we infected Jurkat with NL4-3, treated them with Metformin, and performed Western blotting. Similar results were obtained except for more pronounced increased of CREB phosphorylation (Figure 5A). To further validate these findings, we also performed single-round infection of Jurkat with VSV-G-pseudotyped HIV, treated them with Metformin, and performed Western blotting. Similar to NL4-3 infection, VSV-G-pseudotyped HIV infection showed comparable trends of increased CREB expression and phosphorylation and TBP expression (Figure 5B).
Increased recruitment of phosphorylated CREB and TBP to the HIV LTR promoter by Metformin
We next determined if Metformin-enhanced CREB phosphorylation and TBP expression would result in their recruitment to the HIV LTR promoter. To this end, we transfected 293T with NL4-3, treated them with PBS or Metformin, and performed the chromatin immunoprecipitation (ChIP) assay using specific primers covering the TATA-box region for TBP and the potential DNA binding sites that involve phosphorylated CREB. Corroborated with our previous results (Figure 4 and Figure 5), Metformin treatment led to detection of more phosphorylated CREB and TBP and less total CREB on the LTR promoter than the PBS treatment control (Figure 6). In addition, we performed the ChIP assay for transcription co-factors CREB-binding proteins CBP and p300, which interact with both basal transcription factors and transcription activators [85-87]. Metformin treatment led to more recruitment of CBP but less p300 to the HIV LTR promoter compared to the PBS treatment control (Figure 6).
Metformin treatment increased HIV gene expression, transcription, and production in human PBMC.
To validate and substantiate our findings obtained from cell lines, we isolated peripheral blood mononuclear cells (PBMC) from healthy donors, cultured them in the presence of anti-human CD3/CD28 antibodies for 3 days, infected them with NL4-3, treated them with Metformin, continued to culture for 3 days, and collected the cells for Western blotting and culture supernatant for the RTase assay. Metformin treatment led to increased p24 expression (Figure 7A), increased unspliced HIV RNA (Figure 7B), and higher HIV total RNA levels (Figure 7C), with more HIV production (Figure 7D) in human PBMC. We also performed the similar experiments with concurrent infection and Metformin treatment in human PBMC. Similar results were obtained except for that more pronounced increase of HIV total RNA was noted with Metformin treatment (Figure 7E-H).
Metformin treatment increased CREB expression and phosphorylation and TBP expression and their recruitment to the HIV LTR promoter in human PBMC.
We next determined if Metformin treatment would have similar effects on CREB expression and phosphorylation and TBP expression in HIV-infected human PBMC (Figure 8A-D). Metformin treatment resulted in increased CREB phosphorylation and TBP expression in HIV-infected human PBMC but little changes in CREB expression (Figure 8A). We included AMPK and phosphorylated AMPK as the controls in these experiments and confirmed Metformin-induced AMPK phosphorylation. Then, we performed the ChIP assay and determined the recruitment of phosphorylated CREB, and TBP. Metformin treatment led to increased recruitment of phosphorylated CREB and TBP onto the HIV LTR promoter in these cells (Figure 8B). In addition, we also performed Western blotting and the ChIP assay using the HIV-infected human PBMC derived from Figure 7E-H and obtained similar results in this infection setting (Figure 8C & D). The enhancement effects of Metformin on the HIV LTR promoter and transcription prompted us to investigate whether Metformin would increase HIV transcription and production from several previously characterized HIV latent cells with a low level and persistent HIV replication. We first treated HIV-infected latent CD4+ T lymphocytic cell line J1.1 with Metformin and determined HIV p24 expression using Western blotting and HIV RNA expression by qRT-PCR. More p24 expression was detected in the cells treated with more Metformin (Figure S5A). In the meantime, more unspliced HIV RNA and HIV total RNA were detected in the cells treated with more Metformin (Figure S5B & C). Moreover, more phosphorylated CREB was detected in the cells treated with more Metformin (Figure S5D). We also performed the similar experiments with HIV-infected latent promonocytic cell line U1. A general trend of increased p55 expression, HIV RNA expression, and phosphorylated CREB was observed, albeit in a slightly different kinetics from J1.1 (Figure S6A-D). Similar results were obtained from another HIV-infected latent CD4+ T lymphocytic cell line ACH-2 (Figure S7A,B) and an HIV-infected latent Jurkat cell line we established using HIV reporter viruses NLGi (Figure S7C). These results together indicate that Metformin treatment enhanced HIV transcription and gene expression and was associated with increased CREB phosphorylation.
666-15. treatment abrogated Metformin-enhanced HIV gene expression
To substantiate these findings and ascertain the mechanisms responsible for Metformin-enhanced HIV gene expression, we took advantage of 666-15, a potent and selective inhibitor of CREB activation [88-90] and investigated the effects of this inhibitor on HIV gene expression in the presence of Metformin. As expected, 666-15 treatment led to inhibition of CREB phosphorylation (Figure 9A). In the meantime, it also led to a significantly lower level of p24 expression in 293T treated with Metformin than those treated with Metformin only (Figure 9 B,C), further ascertaining that CREB activation is the major mechanism responsible for metformin-enhanced HIV gene expression.
Discussion
In this study, we first transfected 293T with NL4-3 or infected Jurkat and human PBMC with NL4-3 and treated the cells with Metformin. We showed that Metformin treatment increased HIV gene expression and transcription in these cells. Metformin and its subsequent activation of AMPK have been shown to have diverse effects on infection of various pathogens including viruses, bacteria, and parasites [91]. The effects could be positive or negative, depending on the pathogens. Metformin/AMPK activation promotes replication of viruses such as rotavirus [92], herpes simplex virus type 1 [93], and Epstein-Barr virus [94] or inhibits replication of viruses such as hepatitis C virus [95,96], Zika virus [97], and Dengue virus [98-100]. In case of HIV, AMPK activation is involved in epigallocatechin-3-O-gallate-induced inhibition of Tat transactivation activity on the HIV LTR promoter [101]. AMPK activation is also involved in HIV infection-induced energy deficit and metabolic dysfunction in the context of cocaine use [102]. In contrast to our findings, Metformin has recently been shown to inhibit HIV replication in primary human CD4 T cells and Jurkat [103]. The main differences between our study and the above-mentioned study appears to be that we observed apparent anti-proliferative effects of Metformin on all the cells tested at the same concentrations at which the other study did not notice the anti-proliferative effects of Metformin, and that we normalized all our readouts to the cell number, while the other study did not (personal communication between Drs. He and Guo). Interestingly, the very same study also showed a strong positive correlation between HIV replication and the nucleotide-binding domain and leucine-rich repeat containing receptor X1 transcript levels, which was significantly increased in primary CD4+ T cells when treated with Metformin. These apparent counterintuitive results were interpreted as a compensatory mechanism [103].
The discordance between pharmacological Metformin concentrations in human plasma and in vivo animal studies (0.5-30 μM) and supra pharmacological Metformin concentrations (> 1 mM) in vitro cell culture studies has been a common subject of debate in the field of Metformin research since Metformin was discovered about 70 years ago. Metformin is known to inhibit gluconeogenesis in vivo though inhibition of mitochondrial respiratory chain complex I and activation of AMPK signaling. However, none of these mechanisms have consistently been reproduced in cell cultures when Metformin concentrations are < 1 mM. A number of possibilities have been attributed for this discordance. Among them are lack of Metformin-binding (retaining) proteins in plasma and subsequent high excretion of Metformin though kidney because of highly positive charge of Metformin, selective uptake and accumulation of Metformin in different organs, tissues and cells leading to much higher Metformin working concentrations (than those detected in plasma), different expression of Metformin transporters such as organic cation transporter 1 (OCT1) between cells in organs and tissues in vivo and cell lines in vitro, and different glucose concentrations between cells in vivo and cell cultures in vitro. Thus, the selection of Metformin concentrations in our experiments was based on and consistent with those in the literature [33,84,104-107]. It is also important to note that the accumulation of Metformin in tissues is several times higher than the reported concentrations in the blood [36,108-110], and that the accumulation of Metformin in cell lines are only 10-15% of Metformin in the culture medium [111].
In the study, we also showed that Metformin re-activated HIV in thee lymphocytic cell lines, and one promonocytic cell line. In a 12-week clinical trial involving 13 non-diabetic antiretroviral-suppressed HIV patients (HIV viral load in the blood < 40 copies/ml), Metformin did not show any effects on the reservoir size of HIV-infected latent CD4+ T cells but decreased residual HIV transcription in CD4+ T cells within the colons of 8 HIV-infected individuals [48], while increased HIV transcription in CD4+ T cells within the colons of 4 other HIV-infected individuals [49]. Memory CD4+ T cells constitute the major HIV reservoir in HIV-infected individuals under antiretroviral therapy [112,113]. Metformin has been shown to have fewer HIV-infected memory CD4+ T cells in HIV-infected individuals [50]. Consistent with increased HIV transcription and re-activation of HIV from latency, we showed that Metformin treatment led to increases of the HIV LTR promoter activity. A variety of cellular mechanisms are involved in the establishment and maintenance of HIV latency [114]. Among them is stable repression of the chromatin at the HIV LTR promoter region [114,115], which is primarily controlled by the activities of histone acetylases/deacetylases and lack or sequestration of transcription factors and co-activators/co-repressors [116]. The findings from all these clinical trial studies as well as from the current study indicate that Metformin treatment could help decrease the size of HIV reservoirs in HIV-infected individuals with antiretroviral therapy.
We then determined the underlying molecular mechanisms whereas Metformin treatment augmented HIV transcription by investigating the direct effects of Metformin on the activities of the promoters containing individual DNA-binding sites of transcription factors within the HIV LTR promoter. Consistent with previous studies [70,117-119], we showed that Metformin treatment inhibited NF-κB activity. In addition, we showed that Metformin treatment inhibited AP-1 activity in human embryonic kidney epithelial cell line 293T, while Metformin has been found to enrich AP-1 transcription factor and its regulatory gene network in normal human fibroblasts [120]. This discrepancy may be attributed to the cell-type difference of AP-1 expression and activity between normal human fibroblasts and 293T that were used in our study. Furthermore, we noticed that Metformin treatment led to increased TATA-box transcription activity, which we believe may account for, at least in part, the augmentative effects of HIV transcription. Nevertheless, we performed Western blotting and determined effects of Metformin on CREB expression and phosphorylation and TBP expression. We showed that Metformin treatment resulted in increased CREB expression and phosphorylation and TBP expression in the context of HIV. There are several cAMP-responsive element (CRE) sequences within the HIV LTR promoter, which is the binding site for phosphorylated and dimerized CREB [121-123]. One CRE is located immediately upstream of the transcription start site (+1) of the HIV LTR promoter. Metformin has been shown to decrease CREB phosphorylation and the CRE activity in epithelial cell line MCF-7 [124] but to increase CREB phosphorylation in neuroblastoma cell line SH-SY5Y [125,126], suggesting again that Metformin effects on CREB phosphorylation is cell type-dependent.
Lastly, we demonstrated that Metformin treatment led to increased recruitment of CREB and TBP to the HIV LTR promoter. Phosphorylation of CREB at Ser-133 leads to the recruitment of CBP to CRE though direct interaction and complex formation between phosphorylated CREB and CBP [127]. The tripartite interactions between phosphorylated CREB, CBP, and RNA Pol II as well as bipartite nexus between CREB and TFIID complex have been well demonstrated [128]. The recruitment of CBP by phosphorylated CREB followed by RNA Pol II engagement does not suffice to trigger transcription, and that activated CREB further mediates the recruitment of TFIID as a requirement for transcription induction of the signal-reliant target genes [128]. Interestingly, Metformin in our study did not change TBP expression in non-HIV transfected cells, but slightly increased TBP expression in HIV transfected/infected cells. Besides the increased expression of phosphorylated CREB and TBP, the significant recruitment of these factors alongside CBP to the HIV LTR by Metformin in the context of HIV infection was quite noteworthy. A recent study has indeed shown that activation of the cAMP-PKA-CREB signaling pathway results in enhanced HIV LTR promoter transcription and HIV replication [122]. Importantly, we showed that the inhibition of Metformin-enhanced CREB activation by CREB activation inhibitor 666-15 resulted in a marked decrease in HIV gene expression. These data provide additional mechanistic evidence to support the important roles of CREB activation in Metformin-enhanced HIV transcription and gene expression. Nevertheless, the molecular mechanisms by which Metformin treatment leads to AMPK-independent CREB phosphorylation remains to be investigated.
Author Contributions
Conceptualization: S.R. and J.J.H.; Methodology, S.R. and K.A.T.; Validation, S.R.; Formal Analysis, S.R.; Investigation, S.R. and K.A.T.; Resources, J.J.H.; Writing – Original Draft Preparation, S.R.; Writing – Review & Editing, S.R. and J.J.H.; Supervision, J.J.H.; Project Administration, J.J.H.; Funding Acquisition, J.J.H.
Funding
This work was supported in part by grants R01DA043162 and R01NS094108 (JJH) from the National Institutes of Health and the startup funds from Rosalind Franklin University.
Institutional Review Board Statement
n/a
Informed Consent Statement
n/a
Data Availability Statement
All the data and detailed methodologies in the study will be made available upon request.
Acknowledgments
n/a.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies. Lancet 2008, 372, 293–299. [CrossRef] [PubMed]
- Thaker, H.K.; Snow, M.H. HIV viral suppression in the era of antiretroviral therapy. Postgraduate Medical Journal 2003, 79, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Cirión, A.; Sereti, I. Immunometabolism and HIV-1 pathogenesis: food for thought. Nature Reviews Immunology 2021, 21, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.C.; Sereti, I. Serious Non-AIDS Events: Therapeutic Targets of Immune Activation and Chronic Inflammation in HIV Infection. Drugs 2016, 76, 533–549. [Google Scholar] [CrossRef] [PubMed]
- Hsue, P.Y.; Waters, D.D. HIV infection and coronary heart disease: mechanisms and management. Nature Reviews Cardiology 2019, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Protogerou, A.D.; Fransen, J.; Zampeli, E.; Argyris, A.A.; Aissopou, E.; Arida, A.; Konstantonis, G.D.; Tentolouris, N.; Makrilakis, K.; Psichogiou, M.; et al. The Additive Value of Femoral Ultrasound for Subclinical Atherosclerosis Assessment in a Single Center Cohort of 962 Adults, Including High Risk Patients with Rheumatoid Arthritis, Human Immunodeficiency Virus Infection and Type 2 Diabetes Mellitus. PLoS One 2015, 10, e0132307. [Google Scholar] [CrossRef] [PubMed]
- Savès, M.; Chêne, G.; Ducimetière, P.; Leport, C.; Le Moal, G.; Amouyel, P.; Arveiler, D.; Ruidavets, J.B.; Reynes, J.; Bingham, A.; et al. Risk factors for coronary heart disease in patients treated for human immunodeficiency virus infection compared with the general population. Clin Infect Dis 2003, 37, 292–298. [Google Scholar] [CrossRef]
- Triant, V.A.; Lee, H.; Hadigan, C.; Grinspoon, S.K. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. The Journal of clinical endocrinology and metabolism 2007, 92, 2506–2512. [Google Scholar] [CrossRef]
- Overton, E.T.; Nurutdinova, D.; Freeman, J.; Seyfried, W.; Mondy, K.E. Factors associated with renal dysfunction within an urban HIV-infected cohort in the era of highly active antiretroviral therapy. HIV Med 2009, 10, 343–350. [Google Scholar] [CrossRef]
- Mocroft, A.; Lundgren, J.D.; Ross, M.; Fux, C.A.; Reiss, P.; Moranne, O.; Morlat, P.; Monforte, A.; Kirk, O.; Ryom, L. Cumulative and current exposure to potentially nephrotoxic antiretrovirals and development of chronic kidney disease in HIV-positive individuals with a normal baseline estimated glomerular filtration rate: a prospective international cohort study. Lancet HIV 2016, 3, e23–e32. [Google Scholar] [CrossRef]
- Bonnet, E. New and emerging agents in the management of lipodystrophy in HIV-infected patients. HIV AIDS (Auckl) 2010, 2, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Miralles, C.; Negredo, E.; Domingo, P.; Estrada, V.; Gutierrez, F.; Lozano, F.; Martinez, E. Disorders of body fat distribution in HIV-1-infected patients. AIDS Rev 2009, 11, 126–134. [Google Scholar] [PubMed]
- Brown, T.T. Approach to the human immunodeficiency virus-infected patient with lipodystrophy. J Clin Endocrinol Metab 2008, 93, 2937–2945. [Google Scholar] [CrossRef] [PubMed]
- Coll, B.; van Wijk, J.P.; Parra, S.; Castro Cabezas, M.; Hoepelman, I.M.; Alonso-Villaverde, C.; de Koning, E.J.; Camps, J.; Ferre, N.; Rabelink, T.J.; et al. Effects of rosiglitazone and metformin on postprandial paraoxonase-1 and monocyte chemoattractant protein-1 in human immunodeficiency virus-infected patients with lipodystrophy. Eur J Pharmacol 2006, 544, 104–110. [Google Scholar] [CrossRef] [PubMed]
- van Wijk, J.P.; de Koning, E.J.; Cabezas, M.C.; op’t Roodt, J.; Joven, J.; Rabelink, T.J.; Hoepelman, A.I. Comparison of rosiglitazone and metformin for treating HIV lipodystrophy: a randomized trial. Ann Intern Med 2005, 143, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Riddler, S.A.; Smit, E.; Cole, S.R.; Li, R.; Chmiel, J.S.; Dobs, A.; Palella, F.; Visscher, B.; Evans, R.; Kingsley, L.A. Impact of HIV infection and HAART on serum lipids in men. Jama 2003, 289, 2978–2982. [Google Scholar] [CrossRef] [PubMed]
- Fourie, C.M.; Van Rooyen, J.M.; Kruger, A.; Schutte, A.E. Lipid abnormalities in a never-treated HIV-1 subtype C-infected African population. Lipids 2010, 45, 73–80. [Google Scholar] [CrossRef]
- Gross, Andrew M. ; Jaeger, Philipp A.; Kreisberg, Jason F.; Licon, K.; Jepsen, Kristen L.; Khosroheidari, M.; Morsey, Brenda M.; Swindells, S.; Shen, H.; Ng, Cherie T.; et al. Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Molecular Cell 2016, 62, 157–168. [Google Scholar] [CrossRef]
- De Francesco, D.; Wit, F.W.; Bürkle, A.; Oehlke, S.; Kootstra, N.A.; Winston, A.; Franceschi, C.; Garagnani, P.; Pirazzini, C.; Libert, C.; et al. Do people living with HIV experience greater age advancement than their HIV-negative counterparts? AIDS 2019, 33. [Google Scholar] [CrossRef]
- Han, J.H.; Gordon, K.; Womack, J.A.; Gibert, C.L.; Leaf, D.A.; Rimland, D.; Rodriguez-Barradas, M.C.; Bisson, G.P. Comparative Effectiveness of Diabetic Oral Medications Among HIV-Infected and HIV-Uninfected Veterans. Diabetes Care 2017, 40, 218–225. [Google Scholar] [CrossRef]
- Mathabire Rücker, S.C.; Tayea, A.; Bitilinyu-Bangoh, J.; Bermúdez-Aza, E.H.; Salumu, L.; Quiles, I.A.; Szumilin, E.; Chirwa, Z.; Rick, F.; Maman, D. High rates of hypertension, diabetes, elevated low-density lipoprotein cholesterol, and cardiovascular disease risk factors in HIV-infected patients in Malawi. Aids 2018, 32, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Kalra, B.; Agrawal, N.; Unnikrishnan, A.G. Understanding diabetes in patients with HIV/AIDS. Diabetology & Metabolic Syndrome 2011, 3, 2. [Google Scholar] [CrossRef]
- da Cunha, J.; Maselli, L.M.F.; Stern, A.C.B.; Spada, C.; Bydlowski, S.P. Impact of antiretroviral therapy on lipid metabolism of human immunodeficiency virus-infected patients: Old and new drugs. World J Virol 2015, 4, 56–77. [Google Scholar] [CrossRef] [PubMed]
- Avari, P.; Devendra, S. Human immunodeficiency virus and type 2 diabetes. London J Prim Care (Abingdon) 2017, 9, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Monroe, A.K.; Glesby, M.J.; Brown, T.T. Diagnosing and Managing Diabetes in HIV-Infected Patients: Current Concepts. Clinical Infectious Diseases 2014, 60, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.T.; Cole, S.R.; Li, X.; Kingsley, L.A.; Palella, F.J.; Riddler, S.A.; Visscher, B.R.; Margolick, J.B.; Dobs, A.S. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005, 165, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Galli, L.; Salpietro, S.; Pellicciotta, G.; Galliani, A.; Piatti, P.; Hasson, H.; Guffanti, M.; Gianotti, N.; Bigoloni, A.; Lazzarin, A.; et al. Risk of type 2 diabetes among HIV-infected and healthy subjects in Italy. Eur J Epidemiol 2012, 27, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: an overview. Clinical Science 2011, 122, 253–270. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science (New York, N.Y.) 2005, 310, 1642–1646. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin—mode of action and clinical implications for diabetes and cancer. Nature Reviews Endocrinology 2014, 10, 143–156. [Google Scholar] [CrossRef]
- Gunton, J.E.; Delhanty, P.J.; Takahashi, S.; Baxter, R.C. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2. J Clin Endocrinol Metab 2003, 88, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Detaille, D.; Guigas, B.; Leverve, X.; Wiernsperger, N.; Devos, P. Obligatory role of membrane events in the regulatory effect of metformin on the respiratory chain function. Biochem Pharmacol 2002, 63, 1259–1272. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, G.M.; Leclerc, G.J.; Kuznetsov, J.N.; DeSalvo, J.; Barredo, J.C. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS One 2013, 8, e74420. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- Salpeter, S.R.; Buckley, N.S.; Kahn, J.A.; Salpeter, E.E. Meta-analysis: metformin treatment in persons at risk for diabetes mellitus. Am J Med 2008, 121, 149–157. [Google Scholar] [CrossRef]
- Gokcel, A.; Gumurdulu, Y.; Karakose, H.; Melek Ertorer, E.; Tanaci, N.; BascilTutuncu, N.; Guvener, N. Evaluation of the safety and efficacy of sibutramine, orlistat and metformin in the treatment of obesity. Diabetes Obes Metab 2002, 4, 49–55. [Google Scholar] [CrossRef]
- Andrews, M.; Soto, N.; Arredondo, M. [Effect of metformin on the expression of tumor necrosis factor-alpha, Toll like receptors 2/4 and C reactive protein in obese type-2 diabetic patients]. Rev Med Chil 2012, 140, 1377–1382. [Google Scholar] [CrossRef]
- Nath, N.; Khan, M.; Paintlia, M.K.; Singh, I.; Hoda, M.N.; Giri, S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol 2009, 182, 8005–8014. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, T.I.; Jeon, S.M.; Hong, S.P.; Cheon, J.H.; Kim, W.H. The effects of metformin on the survival of colorectal cancer patients with diabetes mellitus. Int J Cancer 2012, 131, 752–759. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Esteva, F.J.; Ensor, J.; Hortobagyi, G.N.; Lee, M.H.; Yeung, S.C. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann Oncol 2012, 23, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, P.; Blanc, M. [Metabolic abnormalities, lipodystrophy and cardiovascular risk in HIV-infected patients]. Rev Prat 2006, 56, 987–994. [Google Scholar] [PubMed]
- Fitch, K.; Abbara, S.; Lee, H.; Stavrou, E.; Sacks, R.; Michel, T.; Hemphill, L.; Torriani, M.; Grinspoon, S. Effects of lifestyle modification and metformin on atherosclerotic indices among HIV-infected patients with the metabolic syndrome. Aids 2012, 26, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, S.D.; Meininger, G.E.; Lareau, M.T.; Dolan, S.E.; Killilea, K.M.; Hadigan, C.M.; Lloyd-Jones, D.M.; Klibanski, A.; Frontera, W.R.; Grinspoon, S.K. Effects of exercise training and metformin on body composition and cardiovascular indices in HIV-infected patients. Aids 2004, 18, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Hoel, H.; Hove-Skovsgaard, M.; Hov, J.R.; Gaardbo, J.C.; Holm, K.; Kummen, M.; Rudi, K.; Nwosu, F.; Valeur, J.; Gelpi, M.; et al. Impact of HIV and Type 2 diabetes on Gut Microbiota Diversity, Tryptophan Catabolism and Endothelial Dysfunction. Sci Rep 2018, 8, 6725. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Corrigendum: Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2017, 545, 116. [Google Scholar] [CrossRef] [PubMed]
- Routy, J.P.; Isnard, S.; Mehraj, V.; Ostrowski, M.; Chomont, N.; Ancuta, P.; Ponte, R.; Planas, D.; Dupuy, F.P.; Angel, J.B. Effect of metformin on the size of the HIV reservoir in non-diabetic ART-treated individuals: single-arm non-randomised Lilac pilot study protocol. BMJ Open 2019, 9, e028444. [Google Scholar] [CrossRef]
- Planas, D.; Pagliuzza, A.; Ponte, R.; Fert, A.; Marchand, L.R.; Massanella, M.; Gosselin, A.; Mehraj, V.; Dupuy, F.P.; Isnard, S.; et al. LILAC pilot study: Effects of metformin on mTOR activation and HIV reservoir persistence during antiretroviral therapy. EBioMedicine 2021, 65, 103270. [Google Scholar] [CrossRef]
- Shikuma, C.M.; Chew, G.M.; Kohorn, L.; Souza, S.A.; Chow, D.; SahBandar, I.N.; Park, E.Y.; Hanks, N.; Gangcuangco, L.M.A.; Gerschenson, M.; et al. Short Communication: Metformin Reduces CD4 T Cell Exhaustion in HIV-Infected Adults on Suppressive Antiretroviral Therapy. AIDS Res Hum Retroviruses 2020, 36, 303–305. [Google Scholar] [CrossRef]
- Weiss, A.; Wiskocil, R.L.; Stobo, J.D. The role of T3 surface molecules in the activation of human T cells: a two-stimulus requirement for IL 2 production reflects events occurring at a pre-translational level. J Immunol 1984, 133, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.L.; Rowe, T.; Justement, J.S.; Butera, S.T.; June, C.H.; Folks, T.M. An HIV-1-infected T cell clone defective in IL-2 production and Ca2+ mobilization after CD3 stimulation. J Immunol 1991, 147, 3145–3148. [Google Scholar] [CrossRef] [PubMed]
- Folks, T.M.; Justement, J.; Kinter, A.; Dinarello, C.A.; Fauci, A.S. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 1987, 238, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Folks, T.M.; Clouse, K.A.; Justement, J.; Rabson, A.; Duh, E.; Kehrl, J.H.; Fauci, A.S. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc Natl Acad Sci U S A 1989, 86, 2365–2368. [Google Scholar] [CrossRef] [PubMed]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Wu, X.; O’Brien, W.A.; Ratner, L.; Kappes, J.C.; Shaw, G.M.; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol 2000, 74, 8358–8367. [Google Scholar] [CrossRef]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 1998, 72, 2855–2864. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, Q.; Chen, X.; Luo, R.H.; Li, Y.; Liu, X.; Yang, L.M.; Zheng, Y.T.; Wang, P. Modification of N-terminal α-amine of proteins via biomimetic ortho-quinone-mediated oxidation. Nat Commun 2021, 12, 2257. [Google Scholar] [CrossRef]
- Jeeninga, R.E.; Hoogenkamp, M.; Armand-Ugon, M.; de Baar, M.; Verhoef, K.; Berkhout, B. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J Virol 2000, 74, 3740–3751. [Google Scholar] [CrossRef]
- Klaver, B.; Berkhout, B. Comparison of 5’ and 3’ long terminal repeat promoter function in human immunodeficiency virus. J Virol 1994, 68, 3830–3840. [Google Scholar] [CrossRef]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol 1995, 69, 6705–6711. [Google Scholar] [CrossRef]
- Chen, C.; Okayama, H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 1987, 7, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kim, B.O.; Kao, C.; Jung, C.; Dalton, J.T.; He, J.J. Tip110, the human immunodeficiency virus type 1 (HIV-1) Tat-interacting protein of 110 kDa as a negative regulator of androgen receptor (AR) transcriptional activation. J Biol Chem 2004, 279, 21766–21773. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, Y.; Farzan, M.; Choe, H.; Ohagen, A.; Gartner, S.; Busciglio, J.; Yang, X.; Hofmann, W.; Newman, W.; et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 1997, 385, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The Selective Downregulation of Class I Major Histocompatibility Complex Proteins by HIV-1 Protects HIV-Infected Cells from NK Cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Guo, J.; Lu, X.; Qiao, Y.; Liu, D.; Pan, S.; Liang, L.; Liu, C.; Zhu, H.; Liu, Z.; et al. cAMP-response element binding protein mediates podocyte injury in diabetic nephropathy by targeting lncRNA DLX6-AS1. Metabolism 2022, 129, 155155. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Qiang, G.; Wang, K.; Dai, J.; McCann, M.; Munoz, M.D.; Gil, V.; Yu, Y.; Li, S.; et al. Secreted EMC10 is upregulated in human obesity and its neutralizing antibody prevents diet-induced obesity in mice. Nature Communications 2022, 13, 7323. [Google Scholar] [CrossRef] [PubMed]
- Chesebro, B.; Wehrly, K.; Nishio, J.; Perryman, S. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol 1992, 66, 6547–6554. [Google Scholar] [CrossRef]
- Shugars, D.C.; Smith, M.S.; Glueck, D.H.; Nantermet, P.V.; Seillier-Moiseiwitsch, F.; Swanstrom, R. Analysis of human immunodeficiency virus type 1 nef gene sequences present in vivo. J Virol 1993, 67, 4639–4650. [Google Scholar] [CrossRef]
- Rahimian, P.; He, J.J. Exosome-associated release, uptake, and neurotoxicity of HIV-1 Tat protein. J Neurovirol 2016, 22, 774–788. [Google Scholar] [CrossRef]
- Zheng, L.; Yang, W.; Wu, F.; Wang, C.; Yu, L.; Tang, L.; Qiu, B.; Li, Y.; Guo, L.; Wu, M.; et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res 2013, 19, 5372–5380. [Google Scholar] [CrossRef]
- Naitou, H.; Mimaya, J.-i.; Horikoshi, Y.; Morita, T. Quantitative Detection of Human Immunodeficiency Virus Type 1 (HIV-1) RNA by PCR and Use as a Prognostic Marker and for Evaluating Antiretroviral Therapy. Biological & Pharmaceutical Bulletin 1997, 20, 1317–1320. [Google Scholar] [CrossRef]
- ten Haaft, P.; Cornelissen, M.; Goudsmit, J.; Koornstra, W.; Dubbes, R.; Niphuis, H.; Peeters, M.; Thiriart, C.; Bruck, C.; Heeney, J.L. Virus load in chimpanzees infected with human immunodeficiency virus type 1: effect of pre-exposure vaccination. J Gen Virol 1995, 76 ( Pt 4) Pt 4, 1015–1020. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, Y.; Timani, K.A.; He, J.J. Tip110 Protein Binds to Unphosphorylated RNA Polymerase II and Promotes Its Phosphorylation and HIV-1 Long Terminal Repeat Transcription *. Journal of Biological Chemistry 2014, 289, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Pedro, K.D.; Agosto, L.M.; Sewell, J.A.; Eberenz, K.A.; He, X.; Bass, J.I.F.; Henderson, A.J. A functional screen identifies transcriptional networks that regulate HIV-1 and HIV-2. Proceedings of the National Academy of Sciences 2021, 118, e2012835118. [Google Scholar] [CrossRef] [PubMed]
- Elbezanti, W.; Lin, A.; Schirling, A.; Jackson, A.; Marshall, M.; Duyne, R.V.; Maldarelli, F.; Sardo, L.; Klase, Z. Benzodiazepines Drive Alteration of Chromatin at the Integrated HIV-1 LTR. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Rabi, S.A.; Laird, G.M.; Eisele, E.E.; Zhang, H.; Margolick, J.B.; Siliciano, R.F. A Novel PCR Assay for Quantification of HIV-1 RNA. Journal of Virology 2013, 87, 6521. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.A.; Bentley, K.; Peeters, A.; Churchill, M.J.; Deacon, N.J. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res 2000, 28, 663–668. [Google Scholar] [CrossRef]
- Li, L.; Dahiya, S.; Kortagere, S.; Aiamkitsumrit, B.; Cunningham, D.; Pirrone, V.; Nonnemacher, M.R.; Wigdahl, B. Impact of Tat Genetic Variation on HIV-1 Disease. Adv Virol 2012, 2012, 123605–123605. [Google Scholar] [CrossRef]
- Chiang, C.M.; Ge, H.; Wang, Z.; Hoffmann, A.; Roeder, R.G. Unique TATA-binding protein-containing complexes and cofactors involved in transcription by RNA polymerases II and III. The EMBO Journal 1993, 12, 2749–2762. [Google Scholar] [CrossRef]
- Ferreri, K.; Gill, G.; Montminy, M. The cAMP-regulated transcription factor CREB interacts with a component of the TFIID complex. Proc Natl Acad Sci U S A 1994, 91, 1210–1213. [Google Scholar] [CrossRef]
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 2011, 12, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Saluja, D.; Vassallo, M.F.; Tanese, N. Distinct subdomains of human TAFII130 are required for interactions with glutamine-rich transcriptional activators. Molecular and cellular biology 1998, 18, 5734–5743. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, S.; Wang, Z.; Guo, S.; Zhao, J.; Yi, D.; Li, Q.; Liu, Z.; Guo, F.; Li, X.; et al. The CREB Regulated Transcription Coactivator 2 Suppresses HIV-1 Transcription by Preventing RNA Pol II from Binding to HIV-1 LTR. Virol Sin 2021, 36, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256. [Google Scholar] [CrossRef] [PubMed]
- Song, C.Z.; Keller, K.; Chen, Y.; Murata, K.; Stamatoyannopoulos, G. Transcription coactivator CBP has direct DNA binding activity and stimulates transcription factor DNA binding through small domains. Biochemical and biophysical research communications 2002, 296, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Janknecht, R.; Hunter, T. Transcription. A growing coactivator network. Nature 1996, 383, 22–23. [Google Scholar] [CrossRef] [PubMed]
- He, Z.-X.; Wei, B.-F.; Zhang, X.; Gong, Y.-P.; Ma, L.-Y.; Zhao, W. Current development of CBP/p300 inhibitors in the last decade. European Journal of Medicinal Chemistry 2021, 209, 112861. [Google Scholar] [CrossRef]
- Xie, F.; Li, B.X.; Kassenbrock, A.; Xue, C.; Wang, X.; Qian, D.Z.; Sears, R.C.; Xiao, X. Identification of a Potent Inhibitor of CREB-Mediated Gene Transcription with Efficacious in Vivo Anticancer Activity. J Med Chem 2015, 58, 5075–5087. [Google Scholar] [CrossRef]
- Qin, Y.; Chen, W.; Jiang, G.; Zhou, L.; Yang, X.; Li, H.; He, X.; Wang, H.L.; Zhou, Y.B.; Huang, S.; et al. Interfering MSN-NONO complex-activated CREB signaling serves as a therapeutic strategy for triple-negative breast cancer. Sci Adv 2020, 6, eaaw9960. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, P.; Tan, Y.; Feng, P.; Zhang, Z.; Liang, H.; Duan, W.; Jin, Z.; Wang, X.; Liu, J.; et al. C1q-TNF-related protein-3 attenuates pressure overload-induced cardiac hypertrophy by suppressing the p38/CREB pathway and p38-induced ER stress. Cell Death Dis 2019, 10, 520. [Google Scholar] [CrossRef]
- Silwal, P.; Kim, J.K.; Yuk, J.-M.; Jo, E.-K. AMP-Activated Protein Kinase and Host Defense against Infection. Int J Mol Sci 2018, 19, 3495. [Google Scholar] [CrossRef] [PubMed]
- Green, V.A.; Pelkmans, L. A Systems Survey of Progressive Host-Cell Reorganization during Rotavirus Infection. Cell Host Microbe 2016, 20, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Leyton, L.; Arancibia, Y.; Cuevas, A.; Zambrano, A.; Concha, M.I.; Otth, C. Modulation of the AMPK/Sirt1 axis during neuronal infection by herpes simplex virus type 1. J Alzheimers Dis 2014, 42, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; Lo, K.W.; Ko, C.W.; Young, L.S.; Dawson, C.W. Inhibition of the LKB1-AMPK pathway by the Epstein-Barr virus-encoded LMP1 promotes proliferation and transformation of human nasopharyngeal epithelial cells. J Pathol 2013, 230, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gomez, M.; Diago, M.; Andrade, R.J.; Calleja, J.L.; Salmeron, J.; Fernandez-Rodriguez, C.M.; Sola, R.; Garcia-Samaniego, J.; Herrerias, J.M.; De la Mata, M.; et al. Treatment of insulin resistance with metformin in naive genotype 1 chronic hepatitis C patients receiving peginterferon alfa-2a plus ribavirin. Hepatology 2009, 50, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gu, F.; Guan, J.-L. Metformin Might Inhibit Virus through Increasing Insulin Sensitivity. Chin Med J (Engl) 2018, 131, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Ramos da Silva, S.; Huang, I.C.; Jung, J.U.; Gao, S.-J. Suppression of Zika Virus Infection and Replication in Endothelial Cells and Astrocytes by PKA Inhibitor PKI 14-22. Journal of Virology 2018, 92, e02019–e02017. [Google Scholar] [CrossRef] [PubMed]
- Htun, H.L.; Yeo, T.W.; Tam, C.C.; Pang, J.; Leo, Y.S.; Lye, D.C. Metformin Use and Severe Dengue in Diabetic Adults. Sci Rep 2018, 8, 3344. [Google Scholar] [CrossRef]
- Osuna-Ramos, J.F.; Reyes-Ruiz, J.M.; del Ángel, R.M. The Role of Host Cholesterol During Flavivirus Infection. Frontiers in Cellular and Infection Microbiology 2018, 8. [Google Scholar] [CrossRef]
- Soto-Acosta, R.; Bautista-Carbajal, P.; Cervantes-Salazar, M.; Angel-Ambrocio, A.H.; Del Angel, R.M. DENV up-regulates the HMG-CoA reductase activity through the impairment of AMPK phosphorylation: A potential antiviral target. PLoS Pathog 2017, 13, e1006257. [Google Scholar] [CrossRef]
- Zhang, H.S.; Wu, T.C.; Sang, W.W.; Ruan, Z. EGCG inhibits Tat-induced LTR transactivation: role of Nrf2, AKT, AMPK signaling pathway. Life Sci 2012, 90, 747–754. [Google Scholar] [CrossRef]
- Samikkannu, T.; Atluri, V.S.; Nair, M.P. HIV and Cocaine Impact Glial Metabolism: Energy Sensor AMP-activated protein kinase Role in Mitochondrial Biogenesis and Epigenetic Remodeling. Sci Rep 2016, 6, 31784. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, Q.; Ghneim, K.; Wang, L.; Rampanelli, E.; Holley-Guthrie, E.; Cheng, L.; Garrido, C.; Margolis, D.M.; Eller, L.A.; et al. Multi-omics analyses reveal that HIV-1 alters CD4+ T cell immunometabolism to fuel virus replication. Nature Immunology 2021, 22, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Ko, Y.; Choi, A.; Lee, M.; Lee, J.A. Metformin displays in vitro and in vivo antitumor effect against osteosarcoma. Korean J Pediatr 2016, 59, 374–380. [Google Scholar] [CrossRef]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002, 51, 2420–2425. [Google Scholar] [CrossRef]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Sundelin, E.I.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Hougaard Christensen, M.M.; Brøsen, K.; Frøkiær, J.; Jessen, N. In Vivo Imaging of Human 11C-Metformin in Peripheral Organs: Dosimetry, Biodistribution, and Kinetic Analyses. J Nucl Med 2016, 57, 1920–1926. [Google Scholar] [CrossRef]
- Wilcock, C.; Wyre, N.D.; Bailey, C.J. Subcellular distribution of metformin in rat liver. J Pharm Pharmacol 1991, 43, 442–444. [Google Scholar] [CrossRef]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Scientific Reports 2017, 7, 15992. [Google Scholar] [CrossRef] [PubMed]
- Fromentin, R.; Bakeman, W.; Lawani, M.B.; Khoury, G.; Hartogensis, W.; DaFonseca, S.; Killian, M.; Epling, L.; Hoh, R.; Sinclair, E.; et al. CD4+ T Cells Expressing PD-1, TIGIT and LAG-3 Contribute to HIV Persistence during ART. PLoS pathogens 2016, 12, e1005761. [Google Scholar] [CrossRef] [PubMed]
- Chew, G.M.; Fujita, T.; Webb, G.M.; Burwitz, B.J.; Wu, H.L.; Reed, J.S.; Hammond, K.B.; Clayton, K.L.; Ishii, N.; Abdel-Mohsen, M.; et al. TIGIT Marks Exhausted T Cells, Correlates with Disease Progression, and Serves as a Target for Immune Restoration in HIV and SIV Infection. PLoS Pathog 2016, 12, e1005349. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef]
- Coiras, M.; López-Huertas, M.R.; Pérez-Olmeda, M.; Alcamí, J. Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nature Reviews Microbiology 2009, 7, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Lewin, S.R.; Ross, A.L.; Ananworanich, J.; Benkirane, M.; Cannon, P.; Chomont, N.; Douek, D.; Lifson, J.D.; Lo, Y.-R.; et al. International AIDS Society global scientific strategy: towards an HIV cure 2016. Nature Medicine 2016, 22, 839. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Yi, Y.; Liu, Y.; Liu, X.; Keller, E.T.; Qian, C.-N.; Zhang, J.; Lu, Y. Metformin targets multiple signaling pathways in cancer. Chin J Cancer 2017, 36, 17–17. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, T.; Hsu, A.; Edwards, P.A.; Gao, H.; Qiao, X. Metformin reduces inflammation in diabetic human vitreous by activating AMPK and inhibiting NFκB signaling pathway. Investigative Ophthalmology & Visual Science 2019, 60, 6548–6548. [Google Scholar]
- Salminen, A.; Hyttinen, J.M.T.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 2011, 89, 667–676. [Google Scholar] [CrossRef]
- Gillespie, Z.E.; Wang, C.; Vadan, F.; Yu, T.Y.; Ausió, J.; Kusalik, A.; Eskiw, C.H. Metformin induces the AP-1 transcription factor network in normal dermal fibroblasts. Scientific Reports 2019, 9, 5369. [Google Scholar] [CrossRef]
- Cristillo, A.D.; Highbarger, H.C.; Dewar, R.L.; Dimitrov, D.S.; Golding, H.; Bierer, B.E. Up-regulation of HIV coreceptor CXCR4 expression in human T lymphocytes is mediated in part by a cAMP-responsive element. Faseb j 2002, 16, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Luna, L.; Pirrone, V.; Krebs, F.C.; Wigdahl, B.; Nonnemacher, M.R. cAMP Signaling Enhances HIV-1 Long Terminal Repeat (LTR)-directed Transcription and Viral Replication in Bone Marrow Progenitor Cells. Clinical Medicine Insights: Pathology, 1177. [Google Scholar] [CrossRef]
- Krebs, F.C.; Goodenow, M.M.; Wigdahl, B. Neuroglial ATF/CREB factors interact with the human immunodeficiency virus type 1 long terminal repeat. J Neurovirol 1997, 3 Suppl 1, S28–32. [Google Scholar]
- Kim, H.G.; Hien, T.T.; Han, E.H.; Hwang, Y.P.; Choi, J.H.; Kang, K.W.; Kwon, K.-i.; Kim, B.-H.; Kim, S.K.; Song, G.Y.; et al. Metformin inhibits P-glycoprotein expression via the NF-κB pathway and CRE transcriptional activity through AMPK activation. Br J Pharmacol 2011, 162, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Khang, R.; Ham, S.; Jeong, G.R.; Kim, H.; Jo, M.; Lee, B.D.; Lee, Y.I.; Jo, A.; Park, C.; et al. Activation of the ATF2/CREB-PGC-1α pathway by metformin leads to dopaminergic neuroprotection. Oncotarget 2017, 8, 48603–48618. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Park, P.H.; Hong, J.T.; Choi, D.Y. Activation of AMPK/aPKCζ/CREB pathway by metformin is associated with upregulation of GDNF and dopamine. Biochem Pharmacol 2020, 180, 114193. [Google Scholar] [CrossRef]
- Parker, D.; Ferreri, K.; Nakajima, T.; LaMorte, V.J.; Evans, R.; Koerber, S.C.; Hoeger, C.; Montminy, M.R. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol Cell Biol 1996, 16, 694–703. [Google Scholar] [CrossRef]
- Nakajima, T.; Uchida, C.; Anderson, S.F.; Parvin, J.D.; Montminy, M. Analysis of a cAMP-responsive activator reveals a two-component mechanism for transcriptional induction via signal-dependent factors. Genes Dev 1997, 11, 738–747. [Google Scholar] [CrossRef]
Figure 1.
Effects of Metformin on HIV production. A & B. 293T were plated in a 6-well cell culture plate at a density of 2 x 105 per well, transfected with 3.5 μg pNL4-3, cultured for 16 h, changed medium, added Metformin, and continued to culture for 48 h. The culture medium was collected to determine HIV production by the reverse transcriptase (RTase) activity assay (A), while the cells were harvested to determine HIV intracellular gene expression by Western blotting against an anti-p24 antibody or anti-β-actin antibody (B). p24 expression was quantitated by densitometry, normalized to the loading control, β-actin, and expressed by fold-change in reference to the first sample without Metformin treatment. C & D. Jurkat (1 x 106) were infected with 10,000 cpm RT equivalent HIV NL4-3 viruses via spinoculation in the presence of 1x polybrene, washed with PBS, cultured for 5 days with changes of fresh medium every 48 h to reach the maximum number of infected cells, added Metformin, and continued to culture for 72 h. The culture medium was collected to determine HIV production by the RTase assay (C), while the cells were harvested to determine HIV intracellular gene expression by Western blotting against an anti-p24 antibody or anti-GAPDH antibody (D). p24 expression was quantitated as above. E. 1 x 105 Jurkat cells were seeded in 12-well plate and infected with 100,000 cpm RT equivalent VSV-G-pseudotyped HIV-Luc, added Metformin, and cultured for 72 h. The cells were washed with PBS and collected for Western blotting against an anti-p24 antibody or anti-GAPDH antibody. p24 expression was quantitated as above. The RTase activities were normalized to the cell counts (A & C). The data were Mean ± SD of multiple replicates (12 replicates for A, 9 replicates for C) or representative of thee to four independent experiments (B, D, & E).
Figure 1.
Effects of Metformin on HIV production. A & B. 293T were plated in a 6-well cell culture plate at a density of 2 x 105 per well, transfected with 3.5 μg pNL4-3, cultured for 16 h, changed medium, added Metformin, and continued to culture for 48 h. The culture medium was collected to determine HIV production by the reverse transcriptase (RTase) activity assay (A), while the cells were harvested to determine HIV intracellular gene expression by Western blotting against an anti-p24 antibody or anti-β-actin antibody (B). p24 expression was quantitated by densitometry, normalized to the loading control, β-actin, and expressed by fold-change in reference to the first sample without Metformin treatment. C & D. Jurkat (1 x 106) were infected with 10,000 cpm RT equivalent HIV NL4-3 viruses via spinoculation in the presence of 1x polybrene, washed with PBS, cultured for 5 days with changes of fresh medium every 48 h to reach the maximum number of infected cells, added Metformin, and continued to culture for 72 h. The culture medium was collected to determine HIV production by the RTase assay (C), while the cells were harvested to determine HIV intracellular gene expression by Western blotting against an anti-p24 antibody or anti-GAPDH antibody (D). p24 expression was quantitated as above. E. 1 x 105 Jurkat cells were seeded in 12-well plate and infected with 100,000 cpm RT equivalent VSV-G-pseudotyped HIV-Luc, added Metformin, and cultured for 72 h. The cells were washed with PBS and collected for Western blotting against an anti-p24 antibody or anti-GAPDH antibody. p24 expression was quantitated as above. The RTase activities were normalized to the cell counts (A & C). The data were Mean ± SD of multiple replicates (12 replicates for A, 9 replicates for C) or representative of thee to four independent experiments (B, D, & E).
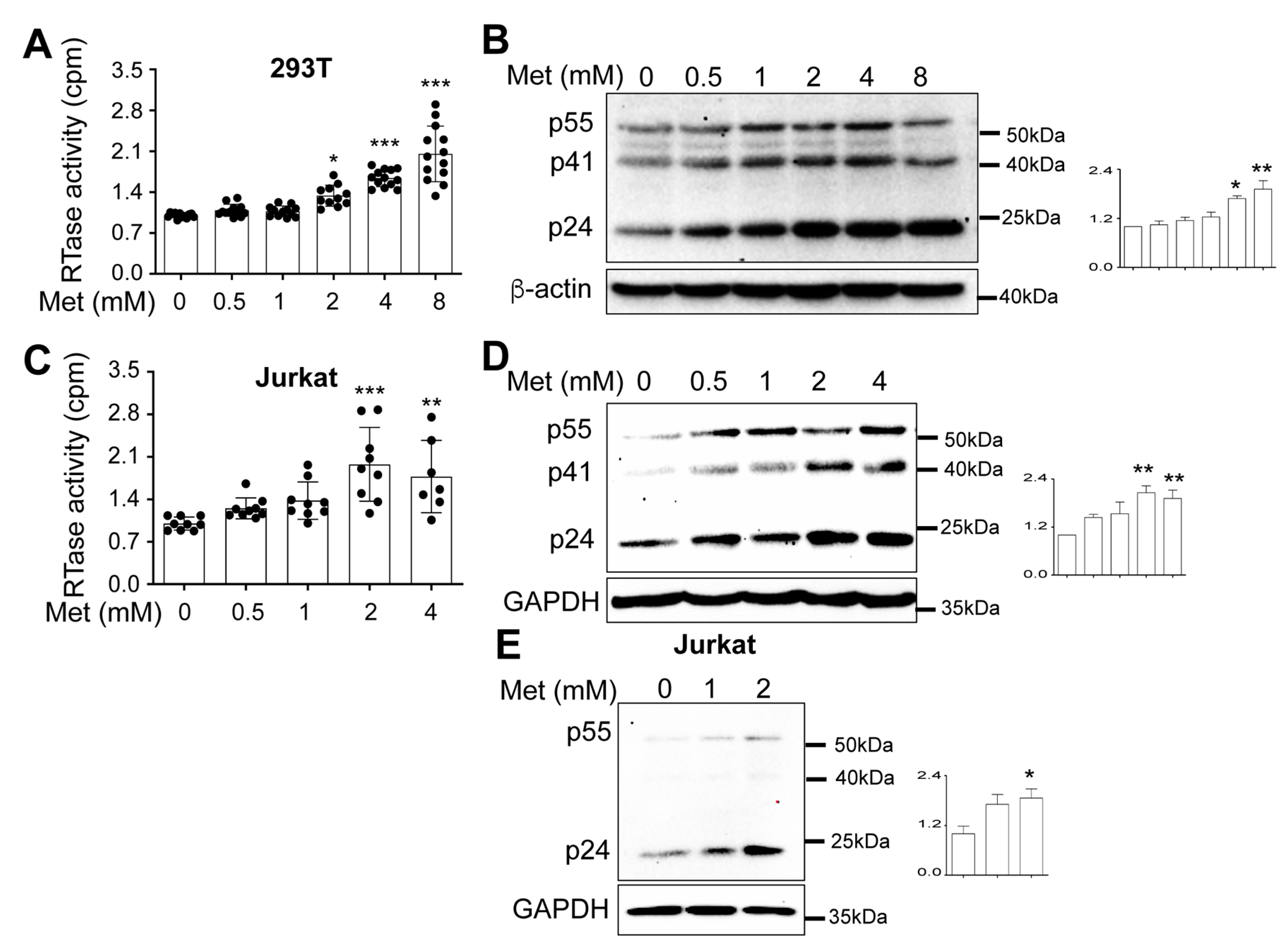
Figure 2.
Effects of Metformin on HIV RNA expression. A-C. 293T were plated in a 6-well cell culture plate at a density of 2 x 105 per well, transfected with 3.5 μg pcDNA3, or pNL4-3, cultured for 16 h, changed medium, added Metformin, and continued to culture for 48 h. The cells were harvested for total RNA isolation and determined for gag-pol RNA by the conventional RT-PCR followed by agarose gel electrophoresis (A) and densitometry quantitation for p24 (B) or determined for total viral RNA by real-time qRT-PCR (C). D. Jurkat were infected with HIV NL4-3 viruses for 5 days as above, added Metformin, and cultured for 72 h. The infected cells were harvested for total RNA isolation and determined for total viral RNA by real-time qRT-PCR. The data were Mean ± SD of 2 replicates for A & B and multiple replicates for C & D.
Figure 2.
Effects of Metformin on HIV RNA expression. A-C. 293T were plated in a 6-well cell culture plate at a density of 2 x 105 per well, transfected with 3.5 μg pcDNA3, or pNL4-3, cultured for 16 h, changed medium, added Metformin, and continued to culture for 48 h. The cells were harvested for total RNA isolation and determined for gag-pol RNA by the conventional RT-PCR followed by agarose gel electrophoresis (A) and densitometry quantitation for p24 (B) or determined for total viral RNA by real-time qRT-PCR (C). D. Jurkat were infected with HIV NL4-3 viruses for 5 days as above, added Metformin, and cultured for 72 h. The infected cells were harvested for total RNA isolation and determined for total viral RNA by real-time qRT-PCR. The data were Mean ± SD of 2 replicates for A & B and multiple replicates for C & D.

Figure 3.
Effects of Metformin on the HIV LTR promoter activity. A. TZM-bl were plated in a 12-well plate at a density of 1 x 105 cells/well, added Metformin, cultured for 48 h, and harvested for the Luc reporter gene assay. B. 293T were plated in a 12-well plate at a density of 1 x 105 cells/well, transfected with 10 ng of HIV-LTR-Luc adjusted to 1.5 µg DNA per well using pcDNA3, cultured for 16 h, changed with fresh medium, added Metformin, continued to culture for 48 h, and harvested for the Luc reporter gene assay. The Luc activity was normalized to the corresponding cellular protein concentration or cell counts. The data were Mean ± SD of 6 replicates (A & B).
Figure 3.
Effects of Metformin on the HIV LTR promoter activity. A. TZM-bl were plated in a 12-well plate at a density of 1 x 105 cells/well, added Metformin, cultured for 48 h, and harvested for the Luc reporter gene assay. B. 293T were plated in a 12-well plate at a density of 1 x 105 cells/well, transfected with 10 ng of HIV-LTR-Luc adjusted to 1.5 µg DNA per well using pcDNA3, cultured for 16 h, changed with fresh medium, added Metformin, continued to culture for 48 h, and harvested for the Luc reporter gene assay. The Luc activity was normalized to the corresponding cellular protein concentration or cell counts. The data were Mean ± SD of 6 replicates (A & B).
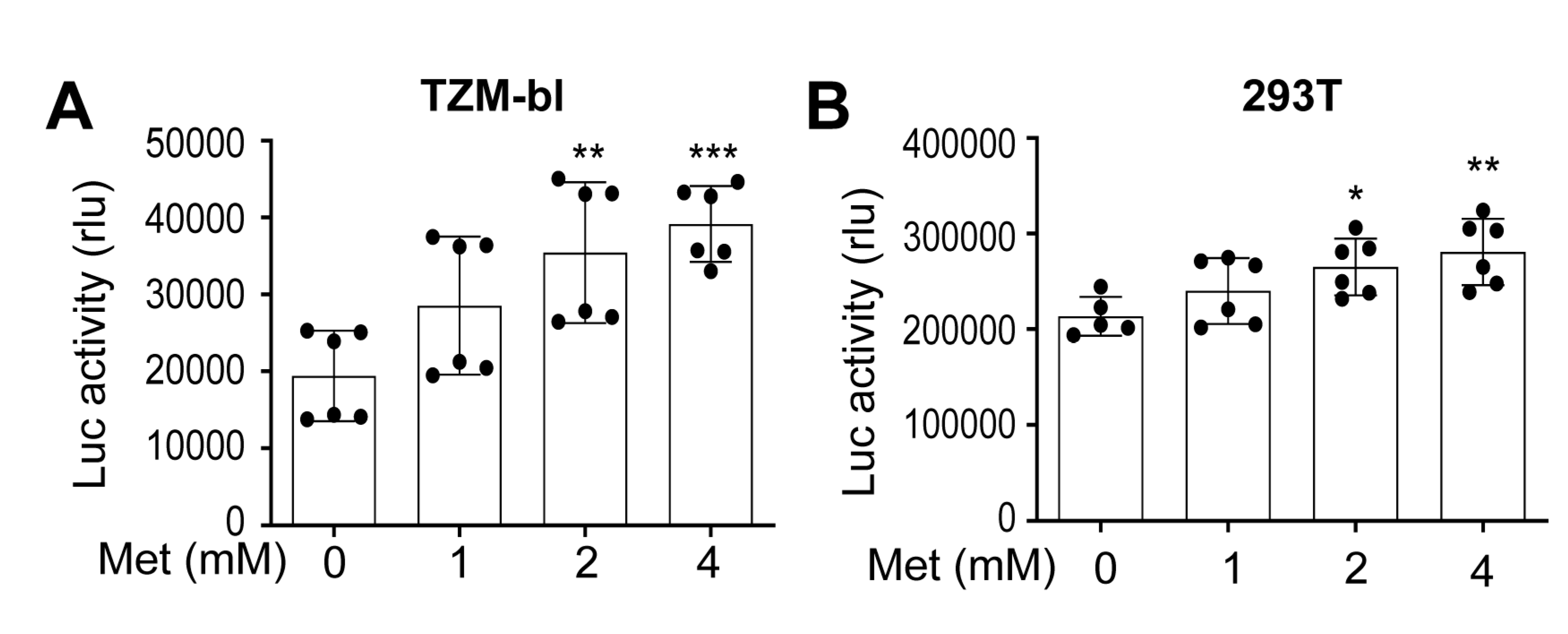
Figure 4.
Effects of Metformin on expression of transcription factors CREB and TBP. 293T were plated in a 6-well plate at a density of 2 x 105 per well, transfected with 3.5 μg pcDNA3 (A), or pNL4-3 (B), cultured for 16 h, changed fresh medium, added Metformin, and continued to culture for 48 h. The cells were harvested for Western blotting against an anti-p24, p-AMPK, AMPK, p-CREB, CREB, TBP, GAPDH, or β-actin antibody. p-CREB was normalized to CREB, while CREB and TBP expression were normalized to the loading control GAPDH. The data were representative of thee independent experiments and Mean ± SD of triplicate (CREB & TBP) or duplicate (p-CREB) samples.
Figure 4.
Effects of Metformin on expression of transcription factors CREB and TBP. 293T were plated in a 6-well plate at a density of 2 x 105 per well, transfected with 3.5 μg pcDNA3 (A), or pNL4-3 (B), cultured for 16 h, changed fresh medium, added Metformin, and continued to culture for 48 h. The cells were harvested for Western blotting against an anti-p24, p-AMPK, AMPK, p-CREB, CREB, TBP, GAPDH, or β-actin antibody. p-CREB was normalized to CREB, while CREB and TBP expression were normalized to the loading control GAPDH. The data were representative of thee independent experiments and Mean ± SD of triplicate (CREB & TBP) or duplicate (p-CREB) samples.
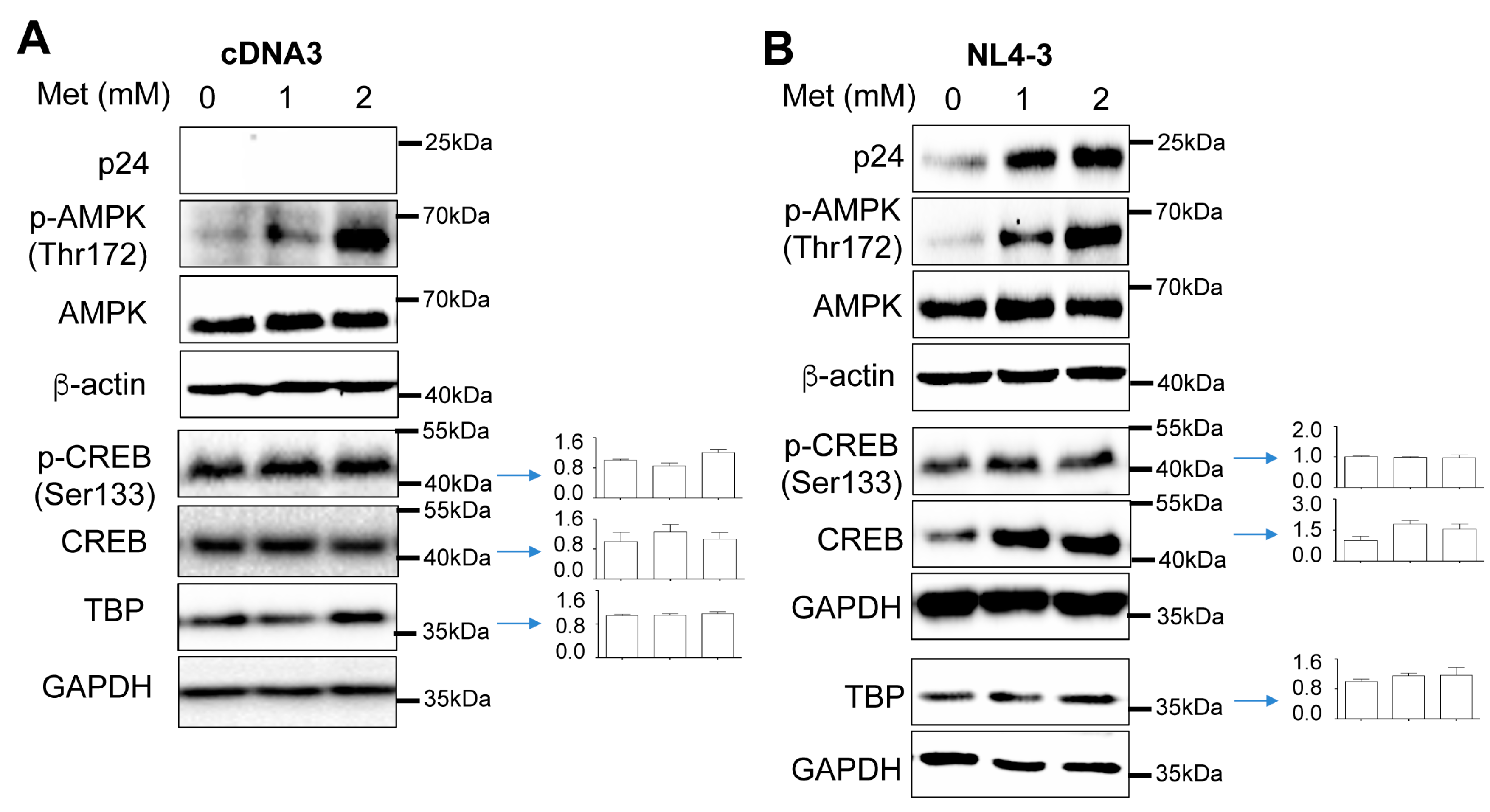
Figure 5.
Effects of Metformin on expression of transcription factors CREB and TBP in the context of HIV infection. A. Jurkat (1 x 106) were infected with 10,000 cpm-equivalent HIV NL4-3 viruses via spinoculation in the presence of 1x polybrene, washed with PBS, cultured for 5 days with changes of fresh medium every 48 h, added Metformin, and continued to culture for 72 h. The cells were harvested for Western blotting. B. Jurkat (1 x 105) were infected with 100,000 cpm-equivalent VSV-G-pseudotyped HIV-Luc, added Metformin, and cultured for 72 h. The cells were washed with PBS and collected for Western blotting. p-CREB was normalized to CREB, while CREB, and TBP expression were normalized to the loading control GAPDH. The data represent two (A) and thee (B) independent experiments.
Figure 5.
Effects of Metformin on expression of transcription factors CREB and TBP in the context of HIV infection. A. Jurkat (1 x 106) were infected with 10,000 cpm-equivalent HIV NL4-3 viruses via spinoculation in the presence of 1x polybrene, washed with PBS, cultured for 5 days with changes of fresh medium every 48 h, added Metformin, and continued to culture for 72 h. The cells were harvested for Western blotting. B. Jurkat (1 x 105) were infected with 100,000 cpm-equivalent VSV-G-pseudotyped HIV-Luc, added Metformin, and cultured for 72 h. The cells were washed with PBS and collected for Western blotting. p-CREB was normalized to CREB, while CREB, and TBP expression were normalized to the loading control GAPDH. The data represent two (A) and thee (B) independent experiments.
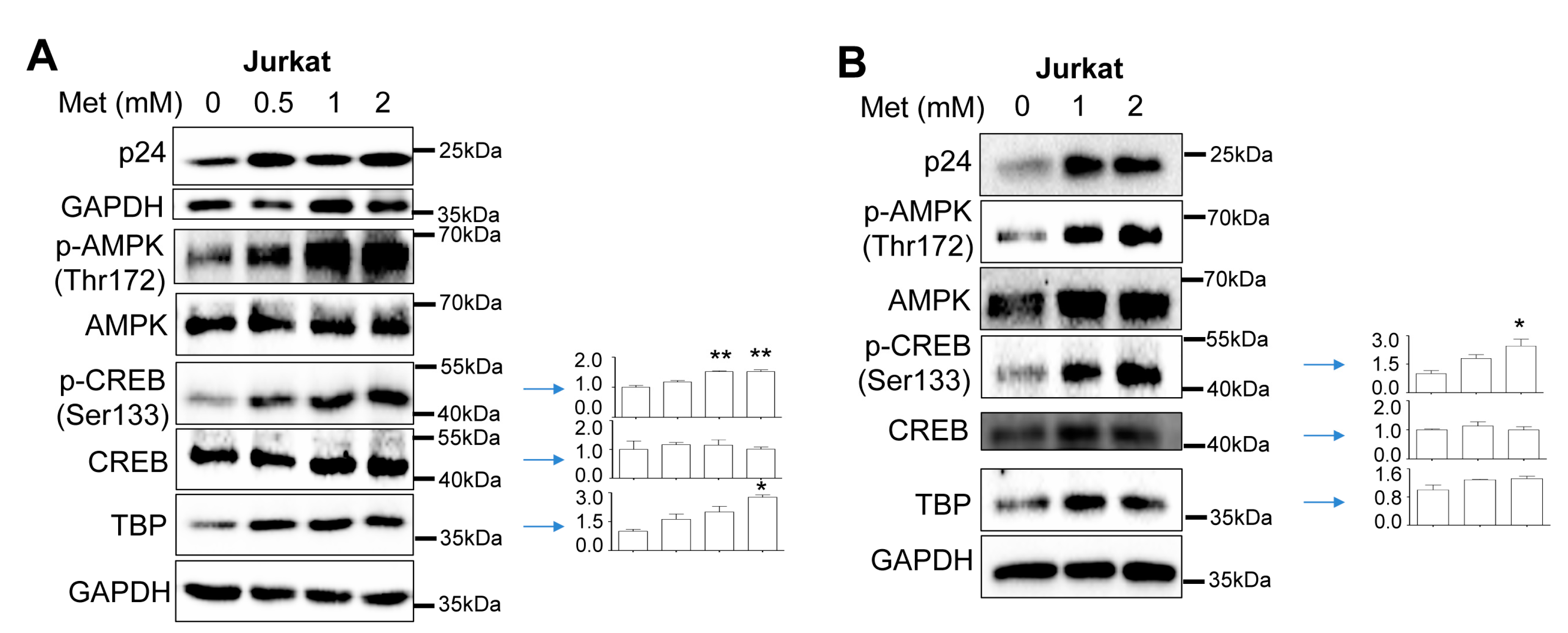
Figure 6.
Effects of Metformin on recruitment of transcription factors CREB and TBP onto HIV LTR promoter. 293T were plated in a 10 cm tissue culture dish at a density of 2x 106 per plate, transfected with 20 μg HIV pNL4-3, cultured for 16 h, replaced with fresh medium, added PBS or Metformin (2 mM), and continued to culture for 48 h. The cells were washed twice with ice-cold PBS and processed for cross-linking and the chromatin immunoprecipitation assay using an anti-CREB, anti-p-CREB, anti-TBP, anti-CBP, anti-p300, or rabbit IgG. The DNA associated with the immunoprecipitates was purified, subjected to qPCR with specific primers spanning the DNA binding sites of each transcription factor, and normalized to corresponding input DNA which was determined using U6 primers. The data were Mean ± SD of 6 replicates. .
Figure 6.
Effects of Metformin on recruitment of transcription factors CREB and TBP onto HIV LTR promoter. 293T were plated in a 10 cm tissue culture dish at a density of 2x 106 per plate, transfected with 20 μg HIV pNL4-3, cultured for 16 h, replaced with fresh medium, added PBS or Metformin (2 mM), and continued to culture for 48 h. The cells were washed twice with ice-cold PBS and processed for cross-linking and the chromatin immunoprecipitation assay using an anti-CREB, anti-p-CREB, anti-TBP, anti-CBP, anti-p300, or rabbit IgG. The DNA associated with the immunoprecipitates was purified, subjected to qPCR with specific primers spanning the DNA binding sites of each transcription factor, and normalized to corresponding input DNA which was determined using U6 primers. The data were Mean ± SD of 6 replicates. .
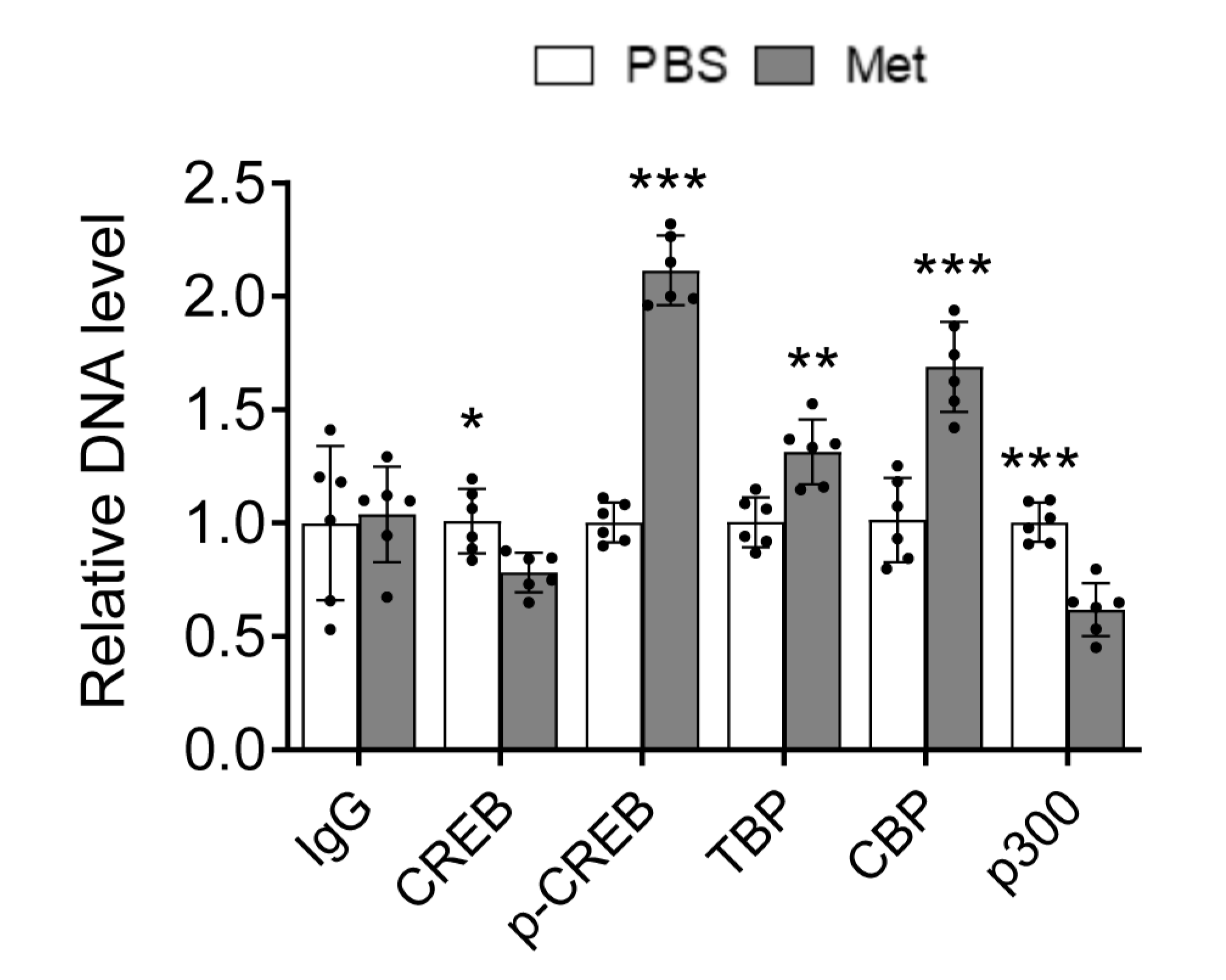
Figure 7.
Effects of Metformin on HIV replication in human PBMC. A-D. Freshly isolated human PBMC were cultured in the presence of anti-human CD3 antibody (1 μg/ml) and CD28 (2 μg/ml) for 72 h, infected with HIV NL4-3 (MOI: 0.5) by spinoculation as above, washed with fresh medium, added Metformin, and cultured in the human IL-2 (100 IU/ml) for 72 h. The cells were harvested for Western blotting against an anti-p24 or GAPDH antibody, followed by densitometry quantitation for p24 (A), and for total RNA isolation, followed by qRT-PCR for gag-pol RNA (B) or total viral RNA (C). The culture supernatant was collected for the RTase activity assay (D). E-H. Similar experiments were performed except for that human PBMC were infected with HIV NL4-3 (MOI=0.1) in the presence of Metformin and human IL-2 (100 IU/ml). The data were representative of six independent experiments (A & E) or Mean ± SD of multiple replicates (B-D & F-H).
Figure 7.
Effects of Metformin on HIV replication in human PBMC. A-D. Freshly isolated human PBMC were cultured in the presence of anti-human CD3 antibody (1 μg/ml) and CD28 (2 μg/ml) for 72 h, infected with HIV NL4-3 (MOI: 0.5) by spinoculation as above, washed with fresh medium, added Metformin, and cultured in the human IL-2 (100 IU/ml) for 72 h. The cells were harvested for Western blotting against an anti-p24 or GAPDH antibody, followed by densitometry quantitation for p24 (A), and for total RNA isolation, followed by qRT-PCR for gag-pol RNA (B) or total viral RNA (C). The culture supernatant was collected for the RTase activity assay (D). E-H. Similar experiments were performed except for that human PBMC were infected with HIV NL4-3 (MOI=0.1) in the presence of Metformin and human IL-2 (100 IU/ml). The data were representative of six independent experiments (A & E) or Mean ± SD of multiple replicates (B-D & F-H).
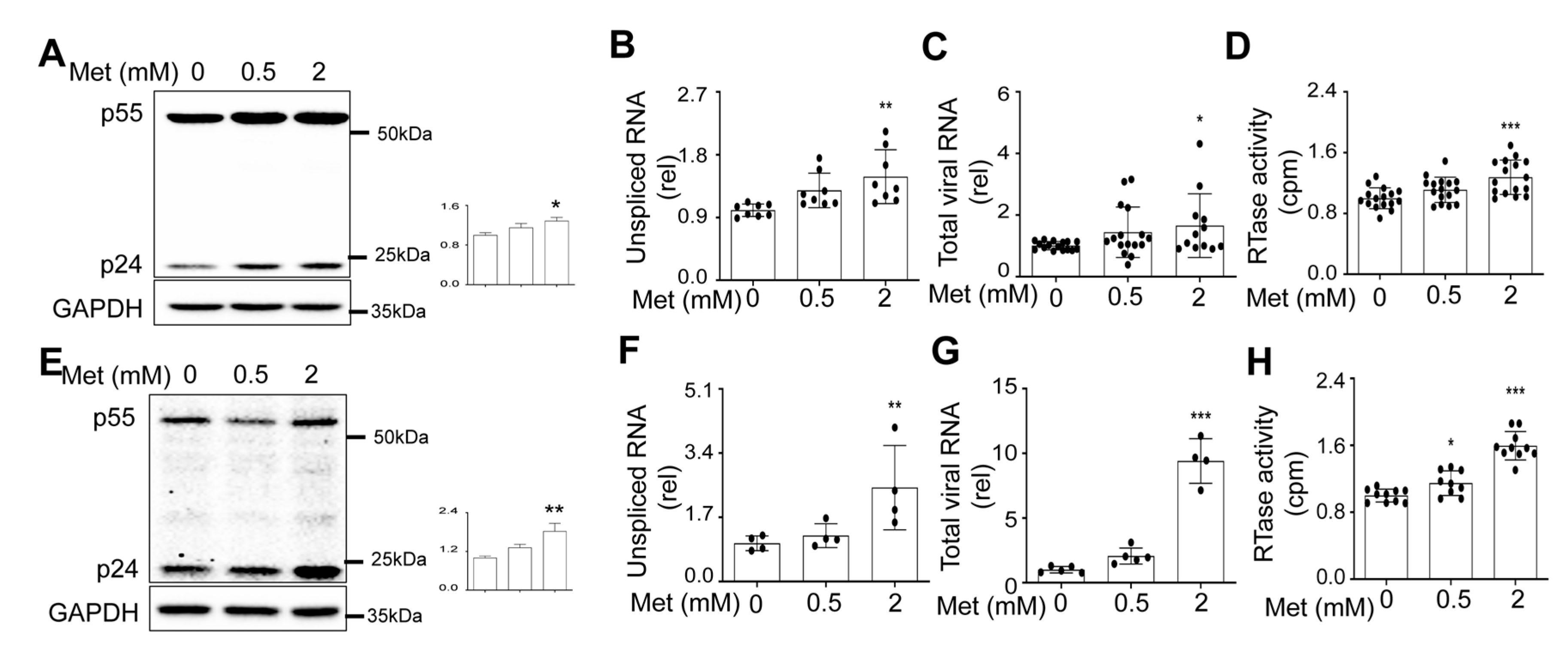
Figure 8.
Effects of Metformin on CREB and TBP expression and their recruitment to the HIV LTR promoter in human PBMC. Human PBMC from Figure 8A-D were harvested for Western blotting (A) or ChIP assay (B); and human PBMC from Figure 8E-H were harvested for Western blotting (C) or ChIP assay (D). The graphed data were representative and Mean ± SD of 8 replicates for p-CREB, 3 replicates for CREB, and 4 replicates for TBP in figure A, and 4 replicates for p-CREB, 4 replicates for CREB, and 6 replicates for TBP in figure C. p24 and AMPK/p-AMPK represent six and two independent experiments, respectively (A & C). The data in figures B & D were Mean ± SD of 8 replicates for p-CREB or 4-6 replicates for TBP.
Figure 8.
Effects of Metformin on CREB and TBP expression and their recruitment to the HIV LTR promoter in human PBMC. Human PBMC from Figure 8A-D were harvested for Western blotting (A) or ChIP assay (B); and human PBMC from Figure 8E-H were harvested for Western blotting (C) or ChIP assay (D). The graphed data were representative and Mean ± SD of 8 replicates for p-CREB, 3 replicates for CREB, and 4 replicates for TBP in figure A, and 4 replicates for p-CREB, 4 replicates for CREB, and 6 replicates for TBP in figure C. p24 and AMPK/p-AMPK represent six and two independent experiments, respectively (A & C). The data in figures B & D were Mean ± SD of 8 replicates for p-CREB or 4-6 replicates for TBP.
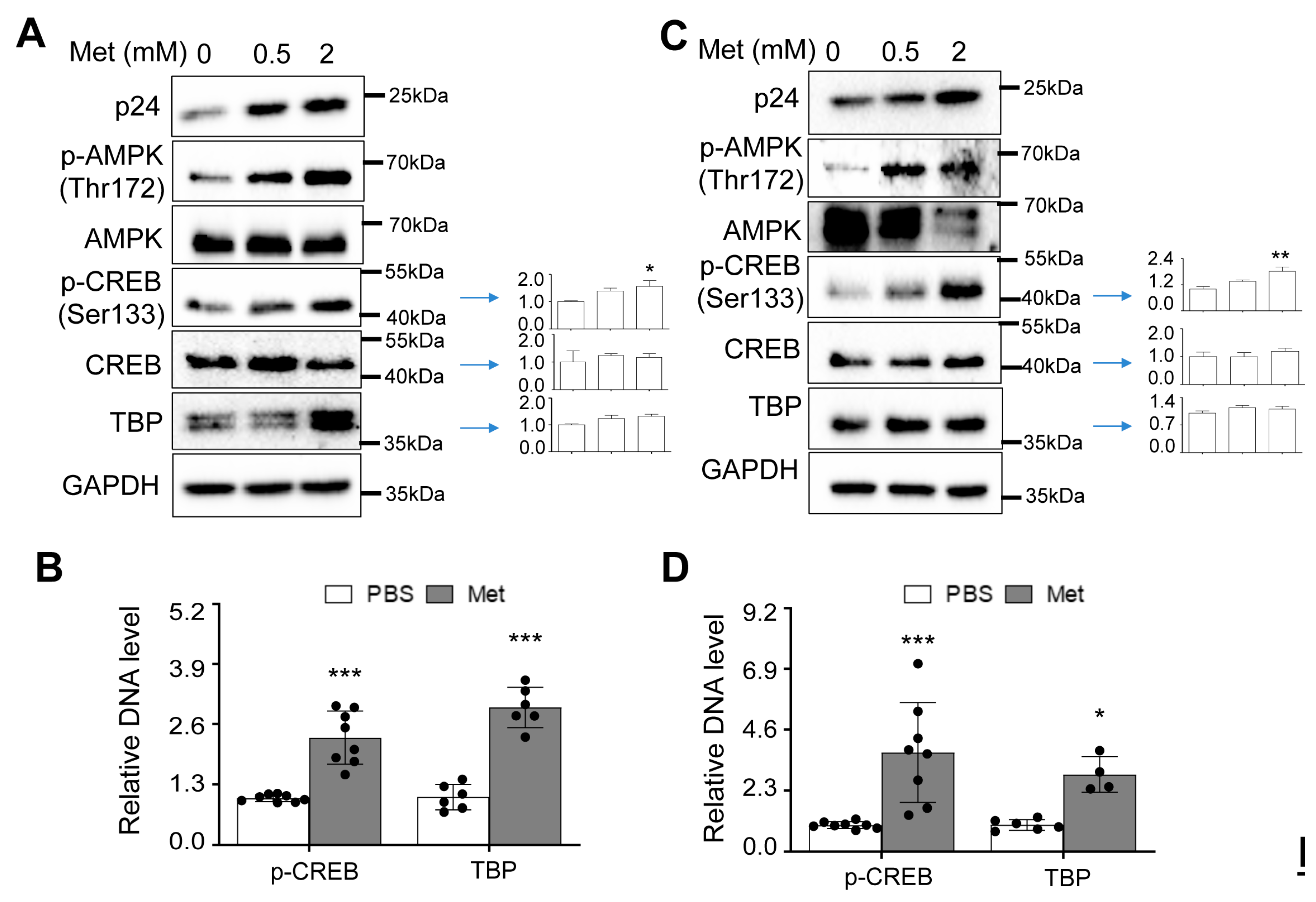
Figure 9.
Effects of 666-15 on HIV gene expression in the presence of Metformin. 293T were plated at a 6-well cell culture plate at a density of 2 x 105 per well, transfected with 3.5 μg pcDNA3, or pNL4-3, cultured for 16 h, changed medium, treated with Metformin (0.5 mM) or Metformin (0.5 mM) plus 666-15 (0.5 µM) for 48 h. PBS (Metformin solvent) was added as a treatment control for Metformin. DMSO (666-15 solvent) was added as a treatment control for 666-15. The cells were harvested for Western blotting (A), followed by densitometry quantitation for p24 (B) and p-CREB (C), and normalized to CREB. The data were representative of thee independent experiments (A) and Mean ± SD of 3 replicates (B) or 2 replicates (C). The results were analyzed by one-way ANOVA followed by a post-hoc Tukey test.
Figure 9.
Effects of 666-15 on HIV gene expression in the presence of Metformin. 293T were plated at a 6-well cell culture plate at a density of 2 x 105 per well, transfected with 3.5 μg pcDNA3, or pNL4-3, cultured for 16 h, changed medium, treated with Metformin (0.5 mM) or Metformin (0.5 mM) plus 666-15 (0.5 µM) for 48 h. PBS (Metformin solvent) was added as a treatment control for Metformin. DMSO (666-15 solvent) was added as a treatment control for 666-15. The cells were harvested for Western blotting (A), followed by densitometry quantitation for p24 (B) and p-CREB (C), and normalized to CREB. The data were representative of thee independent experiments (A) and Mean ± SD of 3 replicates (B) or 2 replicates (C). The results were analyzed by one-way ANOVA followed by a post-hoc Tukey test.
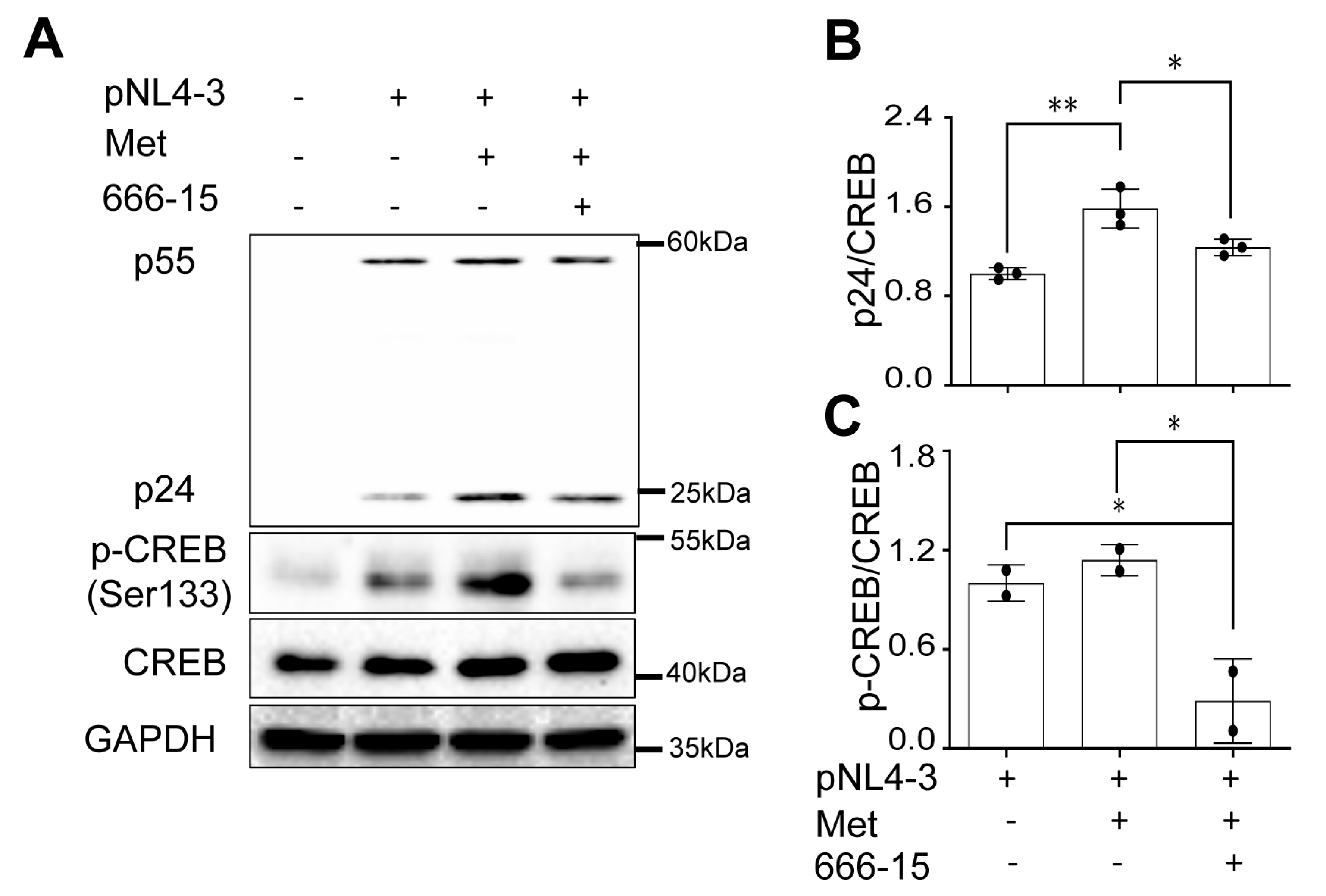
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



