
Submitted:
22 April 2023
Posted:
23 April 2023
You are already at the latest version
Abstract
Truffles are ascomycete hypogeous fungi belonging to the Tuberaceae family of the Pezizales order that grow in ectomycorrhizal symbiosis with tree roots and are known for their peculiar aromas and flavors. Axenic culture of truffle mycelium is problematic because it is not possible in many cases, and the growth rate is meager when it is possible. This limitation prompts searching and characterizing new strains that can be handled in laboratory conditions for basic and applied studies. In this work, a new strain of Tuber borchii (strain SP1) has been isolated and cultured, and its transcriptome has been analyzed under different in vitro culture conditions. The results show that the best T. borchii SP1 growth was obtained using maltose-enriched cultures made with soft-agar and in static submerged cultures made at 22ºC. The transcriptome analysis of this strain cultured in different media indicated that most of the gene transcription effort is due to a limited number of genes (20% of genes account for 80% of the transcription), that the transcription profile of the central metabolism genes was similar in the different conditions analyzed with a transcription signal detected for around 80% of the annotated genes. The gene expression profile suggests that T. borchii uses a fermentative rather than respiratory metabolism, even in aerobic conditions. Finally, there is a reduced expression of genes belonging to secondary metabolite clusters, whereas there is a significative transcription of those involved in producing volatile aromatic compounds.
Keywords:
Axenic culture
; Aroma genes
; Central metabolism
; Fermentation
; Morphological characterization
; Secondary metabolite clusters
; Strain isolation
; Transcriptome profile
; Truffle cultivation
; Vegetative mycelium
1. Introduction
Truffles are ascomycete hypogeous fungi belonging to the Tuberaceae family of the Pezizales order [1]. Although different genera of this order produce subterranean ascomata, several authors only consider “true truffles” the species belonging to the genus Tuber [2,3]. Tuber spp grow associated in ectomycorrhizal symbiosis with tree roots and are known for their peculiar aromas and flavors, which are especially appreciated in haute cuisine [4]. There are around 200 species in the genus Tuber [5], the most valuable being: Tuber melanosporum Vittad. (Périgord black truffle), T. magnatum Picco (Italian white truffle), T. aestivum Vittad. (Summer or Burgundy truffle) and T. borchii Vittad. (Bianchetto truffle) [6].
Beyond their use as food, studies show that truffles have potential antioxidant [7], anti-angiogenic, anti-inflammatory [8], and antitumor activity [9], as well as the presence of volatile compounds (VOCs) [1,10,11] responsible for their distinctive aroma. Truffle VOCs are a mixture of 30 to 60 volatile constituents, including alcohols, ketones, aldehydes, and aromatic and sulfur compounds, that produce each specific scent [1].
The Tuber fruiting bodies can be grown naturally and semi-artificially. However, natural truffle production has decreased over time, increasing its price [12,13]. The semi-artificial cultivation requires the controlled mycorrhization of appropriate tree seedlings using Tuber mycelium or spores, which must be planted in suitable soils [14]. Although obtaining fruiting bodies under in vitro conditions without the host plant is impossible, truffle mycelium, including that of T. borchii, can be grown in pure cultures [15]. Different methods have been developed for industrial in vitro cultivation from natural truffle samples [13]. These mycelia usually grow very slowly, and several months are frequently required to obtain enough biomass for mycorrhization or other studies. Because of that, the number of strains characterized for their use as model systems is still scarce. In vitro studies have demonstrated the presence of VOCs in truffle axenic mycelium cultures [16,17,18]. The availability of new strains for genetic and physiological studies in this fungus could facilitate progress in this field of research.
T. borchii is a whitish truffle found in Europe, especially in Italy, during winter and early spring. It grows in cold temperate to Mediterranean climates, in subalkaline, and, less frequently, in slightly acidic soils with trees and shrubs such as oak, poplar, strawberry tree, and pine, among others [19,20]. There are currently 72 pezizomycete genomes available in the Mycocosm database coordinated by the Joint Genome Institute; 10 correspond to Tuber. The main features of the truffle genomes were elucidated after sequencing those of T. melanosporum in 2010 [21]. The other Tuber genomes sequenced are those of T. aestivum, T. borchii, T. brumale Vittad., T. canaliculatum Gilkey., T. gibbosum Harkn., T. indicum Cooke & Massee., T. magnatum, T. melosporum [22], and T. mesentericum Vittad. [23].
The genome sizes of the sequenced truffles are the largest in the Pezizomycetes group (mean genome size of 114.13 Mbp compared with the 71.45 Mbp of the whole group), and eight out of the ten larger genomes in this group correspond to truffles. Moreover, this difference is even more apparent when the genomes of the Tuber species are compared (129.48 Mbp mean genome size of the group). However, the larger genome size of Tuber species is not a consequence of having more gene models (mean numbers 11,621.33 and 11,776.50 for Pezizomycetes, and Tuber, respectively), but is the consequence of an expanded number of transposons and repetitive sequences in truffles as described alter the analysis of the genome of T. melanosporum [21].
The T. borchii reference draft genome (Tbo3840) was sequenced in 2018 [24]. The nuclear genome assembly of this species is 97.18 Mbp in length, and it is relatively small compared to other Tuber and truffle genomes. The genome codes for 12,346 predicted genes fit well with the expected number of genes for truffles and Tuber. The T. borchii genome lacks gene coding for the glycosylhydrolases GH6 and GH7, revealing its mycorrhizal nature.
Beyond gene catalogs, understanding the expression profiles in different metabolic pathways is crucial for a comprehensive knowledge of their life cycle. Moreover, producing complex secondary metabolites and volatile compound mixtures determines Tuber commercial value. The transcriptome profile of the central metabolic pathways can serve as an internal reference for evaluating the activity to produce secondary metabolites, more so when the differences in genetic background, environment, and culture conditions make the comparisons of different isolates difficult.
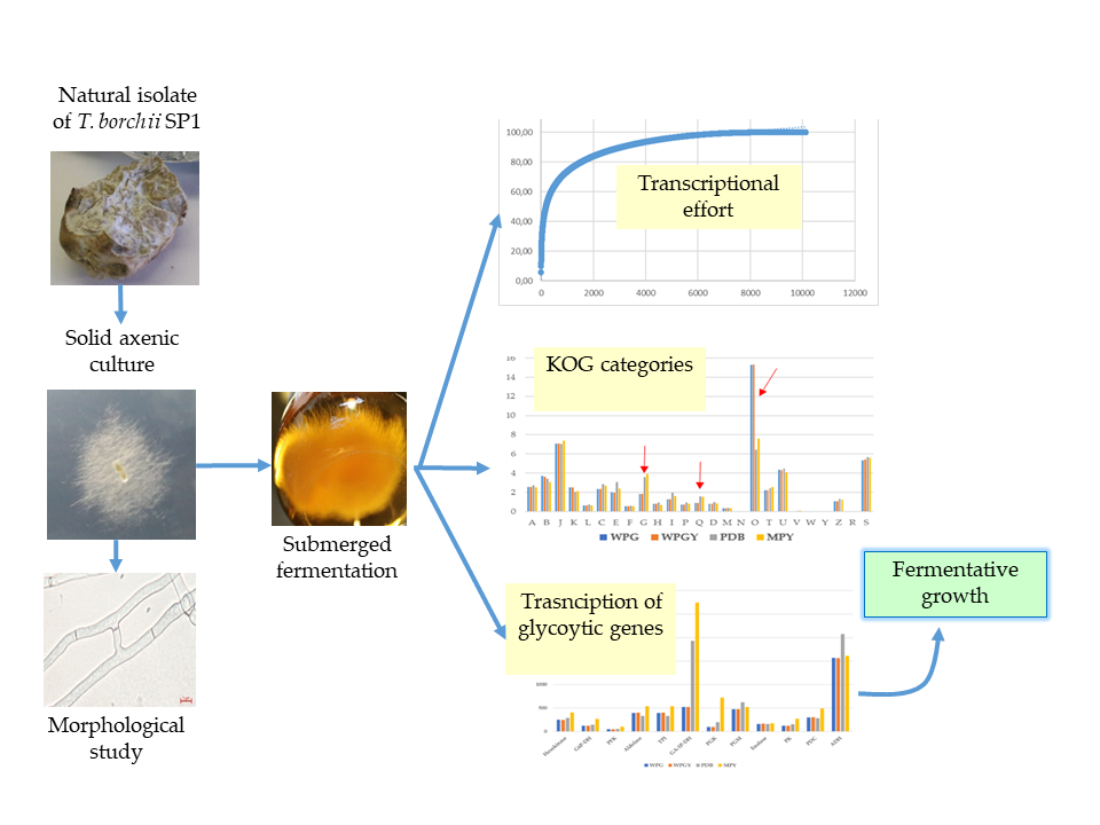
This work aims to characterize a new Spanish T. borchii isolate, grow its mycelium in vitro, initially explore the transcriptome under different culture conditions and study the genes related to the volatile compounds responsible for truffle aroma. The final objective is to increase the available strains for in vitro and in vivo functional studies in this group of organisms.
2. Results
2.1. Culture conditions, growth rate, and biomass production.
Several truffles (T. borchii, T. aestivum, T. magnatum, T. melanosporum ) were obtained from local markets and collectors in different regions of Spain. The truffle surfaces were washed and aseptically broken to transfer gleba fragments to Petri and incubated as described in the Materials and Methods section. There were difficulties isolating the mycelium from the fruiting bodies used in this study (Figure 1A). There was no success in isolating mycelium from T. aestivum, T. magnatum, and T. melanosporum. However, it was possible to isolate mycelium from a T. borchii sample provided by collectors from Castilla y León, Spain, at the end of the winter season using WPS as an isolation medium after eight days of culture.
Once the mycelium was isolated, the strain was molecularly classified by sequencing of the ITS. The top ten blast hits corresponded to sequences of Tuber spp., and the highest similarity corresponded to Tuber borchii Tbo3840 [24](data not shown). From now on, this isolate will be referred to as T. borchii SP1 (Figure 1B).
Different culture media were tested to select the one supporting the highest growth rate for this strain in plate cultures. Figure 1C shows the growth of the colonies after 35 days of incubation at 22ºC in the dark. The cultures made in solid Maltose medium (MPY) performed the best.
When the mycelial was incubated in submerged liquid cultures, growth was detected solely when the culture was made in static mode and not under agitation (200 rpm). To measure the biomass production in liquid cultures of T. borchii, three different culture media were used (WPG, MPY, and PDB), and cultures were continued for 40 days before harvesting the mycelium. It was frequently observed that the mycelium produced in WPG was whiter than that produced in MPY or PBD. Furthermore, when the mycelium grown in MPY or PDB was filtered to remove the broth, it darkened quickly (Figure 2).
The mycelial biomass produced after 40 days of submerged static culture is indicated in Table 1. As in the case of solid cultures, the growth in MPY was higher than in the other media.
2.2. Hyphal morphology
The growth and morphological characteristics of the mycelium of T. borchii SP1 growing in different solid media (MPY, PDB, and WPG) were analyzed. In all the culture media, hyphae were hyaline, branched, septated, and hyphal anastomosis was observed (Figure 3). It was observed that hyphal coarseness and branching angles were similar in the three culture media: the average hyphal thickness varied between 2.7 and 3.7 µm, and the average branching angle was between 47 and 57 degrees. Regarding cell size, the distance between septa was larger in the mycelium grown on MPY medium (43.38 ±16,26) than when grown in PDB or WPG.
2.3. RNA-Seq and transcriptome analysis
The transcriptomes of four different T. borchii SP1 submerged cultures were studied. Table 2 summarizes the results of these analyses. The transcriptome data is presented in extenso in Supplementary Materials Table S1. The total transcriptome counts were initially normalized to RPKM and further to TPM (total count number normalized to 106 in each sample).
The transcriptome reads were mapped to the Tubbor1 reference genome sequence deposited in the JGI database. Gene expression was detected for more than 104 genes in each sample representing between 82.2 (WPG sample) and 86.2 (PDB sample) of the genes annotated for this species.
The functional classification of the expressed genes was made using the KOG database. The expressed genes with functional annotation represented between 80.1 and 81.9 % of the genes with KOG annotation for this species.
Most of the total gene expression in the four samples was due to a reduced number of genes (Figure 4). The cumulative transcriptomes were adjusted to logarithmic curves with a high determination coefficient (R2, 0.984 for WPG, 0.984 for WPGY, 0.977 for PDB, and 0,979 for MPY). Transcription effort was defined as the percentage of the total normalized transcription corresponding to a gene or a group of genes. These results indicate that most of the transcription effort was concentrated on a few genes. Between three (WPG and WPGY samples) and eight (MPY sample) of the 12,300 genes of the truffle genome were responsible for 10% of the total gene expression observed, and 75% of the total expression was due to around 10% of the annotated genes (Table 3), and less than 2,000 genes represented more than 80% of the total gene expression in all cases. In the four studied transcriptomes, the genes that accumulate up to 80% of the total transcription had individual values higher than 100 TPMs, indicating that they are actively transcribed, although to a low level in comparison with the most transcribed genes that had normalized expression levels higher than 59,000 in the WPG and WPGY samples, and around 20,000 in the PDB and MPY samples (see Supplementary Materials – graphics S1, S2, S3, and S4).
Although nearly 80% of the genes for which transcription was detected contribute individually very little to the total transcription effort, their cumulative transcription represented 20%. In summary: 20% of the genes accumulated 80% of the transcription effort, and the remaining 80% accumulated 20%.
Moreover, the proportion of functionally KOG annotated genes among the most expressed ones was lower than for the whole genome. Around 42-44% of the annotated genes in these samples had a presumed KOG function, whereas this percentage was lower among the genes responsible for most of the total gene expression in the analyzed samples. The percentage of T. borchii with a KOG annotation was 46.06%. As shown in Table 3, the genes with a higher expression in our samples were fewer KOG-annotated than the average (Supplementary Materials- Table S2). In conclusion, most of the genes on which most of the transcriptional effort was concentrated coded for genes with unknown functions.
2.4. Comparison of the transcriptomes from the different culture media
It was decided to study the frequency of KOG categories in the expressed genes. As expected, all four samples had the same profile (Figure 5A) because most T. borchii genes were expressed in all four samples. However, considering the transcript counts associated with each category (Figure 5B), category O (Posttranslational modification, protein turnover, chaperones) was more expressed in the wood-plant-based culture media. In contrast, categories G (Carbohydrate transport and metabolism) and Q (Secondary metabolites) were more expressed in the MPY and PDB cultures (see legend of the KOG categories in Supplementary Materials).
2.5. Pathways of central metabolism
The general description of characteristics of T. borchii SP1 can be used as a reference for future studies. Therefore, it was decided to evaluate the genetic expression in the central metabolic routes. Attention was focused on four pathways of central metabolism: glycolysis, TCA, glyoxylate cycle, and oxidative phosphorylation. The transcription of the genes identified in these pathways was monitored, and the main results are presented below.
2.5.1. Central metabolism glycolysis genes
There were 43 gene models annotated in the glycolysis/gluconeogenesis pathway in T. borchii. These genes coded for 11 enzymes of the central pathway plus the alcohol dehydrogenase. Several isozymes were transcribed for some of the genes (three hexokinases, two glyceraldehyde-3P-dehydrogenases, nine phosphoglycerate mutases, two pyruvate kinases, and seven alcohol dehydrogenases) although to different levels (Figure 6). The correlation between the transcription of glycolysis genes in the WPG and WPGY was complete (1,000), and that of PDB with MPY was high (0,885) (Supplementary Materials - Glycolysis). As presented above, the most different transcription profile for these genes was that of the MPY medium.
All the genes, with two exceptions, had transcription values of 500 TPMs or lower. These two exceptions were glyceraldehyde-3P-dehydrogenase (GA3PDH), which showed expression levels in the range of 2000 to 2500 TPMs in the PDB and MPY cultures and that were among the more expressed genes in these two conditions (see above), and the Alcohol dehydrogenase (ADH) gene ID1133725. In the four samples, the ADH gene is among the 125 more expressed genes, suggesting the importance of this fermentation in the biology of the truffle as cultivated in these experiments. Finally, the Phosphoglycerate Kinase (PGK) expression was higher in the MPY culture than in the other transcriptomes.
2.5.2. TCA and glyoxylate cycle
There were 25 genes annotated as coding for enzymes involved in the TCA that covered all the enzymes of the cycle. All the genes, except one, showed expression levels lower than 500 TPMs in the expression range of the glycolytic pathway. This exception was the gene coding for citrate synthase in PDB and MPY, reaching 600 TPMs (Figure 7). The correlation between the expression levels of these genes in the different culture media followed a similar pattern to that observed in the glycolysis genes (data not shown).
The glyoxylate cycle is a shortcut to the citrate cycle involving two enzymes: the isocitrate lyase (IL, two genes) and the malate synthase (MS, one gene). The accumulated transcription of the two IL genes was similar to that of the TCA genes (300-600 TPMs), whereas there was a difference in the transcription of the MS: 36 TPM in WPG and WPGY and 400-500 in the samples grown in rich media (PDB and MPY). The transcription correlation in the minimal media samples was close to 1.0, whereas between the minimal and complex media dropped to 0.5-0.6.
2.5.3. Respiratory chain
There were 11 genes annotated as coding for enzymes involved in the electron transport chain of the oxidative phosphorylation pathway: NADH-dehydrogenase (two genes), succinate dehydrogenase (two genes coding for an enzyme that also participates in the TCA cycle), ubiquinol-cytochrome c reductase (one gene), five genes involved in the coupling of ATP synthesis and H+ transport and one gene coding for an alternative oxidase. The expression level of all the genes was similar in the four conditions analyzed, and all of them (with two exceptions) had expression values ranging from 100 to 700 TPMs, similar to those of the TCA and most of the glycolytic pathway. The exceptions were the two genes coding for enzymes of the NADH-dehydrogenase complex with expression values lower than 100 TPMs in the four conditions (Supplementary Materials - Respiratory chain).
2.6. Genes involved in secondary metabolism pathways – Clusters
It is known that truffles can present different biological activities and, therefore, may present secondary metabolites of interest. It was decided to check the expression of non-ribosomal peptide synthetases (NRPS and NRPS-like) and polyketide-synthases (PKS and PKS-like) gene clusters in SP1 mycelium samples. All the genes identified in these clusters had a null or minimal expression level, representing 0.05% of the transcription effort (Supplementary Materials - Secondary metabolism clusters). The most expressed genes were coding for the same PKS-like clusters in all cases.
2.7. Genes related to the synthesis of volatile compounds
Finally, a search for genes related to the aroma of truffles was carried out, following the studies of Martin et al. [21], who found 92 genes in T. melanosporum species related to truffle flavoring. It was found that 85 of these genes were expressed in the four samples of T. borchii SP1 (Supplementary Materials - aroma genes), and their expression levels ranged from very low (below 10 TPMs) to very high (more than 1000 TPMs). One of the genes (model ID133725, see above the results on the expression of ADH in the central metabolism) coding for a class V alcohol dehydrogenase was the most expressed coding for an enzyme involved in aroma production.
Figure 8 shows the expression of the 10 most expressed aroma genes in the four samples (between 200 and 1900 TPMs). These genes have different functions and are related to different metabolic pathways, such as the synthesis of aldehydes and alcohols, the metabolism of sulfur compounds, and the synthesis of isoprenoids. An enrichment in genes coding for enzymes involved in the production of aldehydes and alcohols group synthesis was detected in this group.
3. Discussion
3.1. SP1 is a new mycelial isolate of Tuber borchii.
Truffles are among the most appreciated edible fungi worldwide; therefore, there is great interest in this research field. In recent decades, research has been conducted on submerged fermentation of fungal mycelia to address the time- and skill-intensive process of truffle cultivation [12,13]. Several research groups have conducted cultivation optimization studies for submerged fermentation of truffle mycelia [25,26]. These studies generally evaluate changes in biomass, exopolysaccharides, enzymes, presence of metabolites, and volatile organic acids for proximity to fruiting bodies.
However, the number of Tuber strains available for laboratory studies is scarce. Tuber spp. mycelia are difficult to cultivate in culture medium; when possible, the growth of the mycelium is extremely slow, and the biomass yield is also minimal [27,28]. For this reason, increasing the number of Tuber strains cultivated in vitro and molecularly characterized is of broad interest.
This paper describes the isolation and characterization of a new Tuber strain called SP1. T. borchii SP1 was collected in the wild from Castilla-León (Spain) soil. During the isolation process, the truffle was cleaned to remove other bacterial or fungal contaminants, and samples were collected from the gleba. The original samples were heavily contaminated with other microorganisms, such as fungi from the genera Cladosporium and Fomitopsis, identified by ITS sequencing as the most frequent (data not shown). This occurs because ectomycorrhizal truffles and fruiting bodies harbor a diverse microbial community, including filamentous fungi, yeasts, and bacteria[29,30,31].
The mycelium recovered from this strain was developed from the glebal tissue (data not shown) and consequently corresponds to the maternal mycelium of this truffle [32,33]. The identification of this T. borchii new strain SP1 isolate was confirmed by sequencing the ITS of the isolated mycelium.
In vitro culturing of T. borchii is difficult because of its low growth rate. Different solid culture media (PDA, MPY, WPGY, and WPG, see Materials and Methods for their composition) were tested to isolate and cultivate the mycelium of the strain SP1. The best growth rate was observed in the cultures performed using MPY or WPG. In MPY, the estimated mycelial growth rate of T. borchii SP1 was 40 cm/yr, considerably faster than the growth achieved for T. borchii by Iotti et al. (2002) [28](Figure 1C). The mycelia grew better when a lower agar concentration was used (data not shown).
The mycelium growing on MPY formed white colonies with straight, septated, sparse hyaline hyphae (Figure 1B). Branches and hyphae anastomoses were infrequently observed (Figure 3). The morphological characteristics of the mycelia growing in the three culture media were similar. However, the cells of the mycelium growing on the maltose culture medium were larger, demonstrating that the nature of the culture medium can influence the morphogenesis of hyphae [34]. No vesicles, which are typical features of Tuber spp. mycelia were observed in T. borchii SP1[28,35].
Submerged cultures of T. borchii SP1 were made using different media (WPG, MPY, and PDB) to select the conditions producing larger biomass. All cultures should be maintained static; growth was not observed under shaking conditions (200 rpm). Corroborating this information, Lacourt et al. 2002 also performed their submerged cultures of T. borchii in static conditions [7].
The culture broth supporting a higher biomass yield was MPY. In this culture medium, the hyphae showed the largest cell sizes under the microscope, and the transcription of some critical genes of the central metabolism also had values higher than in other culture conditions.
Amicucci et al. 2010 [36] found that the carbon source influenced the growth of T. borchii mycelium. They saw that the mycelium grew better in glucose while the hyphae were thinner and less branched in maltose or sucrose [36]. Our results with T. borchii SP1 reflect the opposite, as the mycelium of T. borchii SP1 grew better in the presence of maltose than glucose.
In all cases, the mycelium grew as a single pellet colony that occupied the culture flask. Even when different inoculation points were used to start the culture, the pellets merged into one. The pellet color depended on the culture medium (Figure 3).
In some cases when the mycelia were collected by filtration, they turned brownish rapidly. This color change was more evident when it occurred in the cultures performed in MPY or PDB than in the cultures made using WPG. The brownish of the mycelium in these samples could be correlated with the slightly higher expression of genes in clusters of secondary metabolism in the cultures made in complex media.
In summary, a new strain of T. borchii that can be axenically cultured in maltose-containing plates and static-submerged cultures has been isolated. This strain can be maintained by subculturing indefinitely without signs of strain degeneration.
3.2. RNA-Seq and transcriptome analysis
The transcriptome analyses of the T. borchii SP1 strain described in this paper are preliminary and qualitative and do not pretend to be an exhaustive quantitative study since the difficulties in obtaining enough RNA material prevented making replicas of the different conditions studied. Our objective was to study the expression of different genes and gene families under different culture conditions and to identify common expression patterns that can be used as a reference for future comparative studies.
The transcriptomic profile of T. borchii SP1 growing in static liquid cultures of different media (PDB, MPY, WPGY, and WPG) was studied. The samples were harvested after either 64 (WPGY and WPG) or 85 days of culture (PDB and MPY). In the four media, expression of 10.1x103 to 10.6x103 gene models was detected and identified using the published T. borchii Tbo3840 genome as a reference. These values represent between 86.2 and 88.6% of the annotated genes.
The most expressed gene in the WPG and WPGY samples was ID990338, with an expression level representing 5.97 % of the transcription effort in these conditions. Surprisingly, the transcription level of this gene in the PDB and MPY samples was much lower and represented only 0.19 and 0.28% of the transcription effort, respectively, for these samples. Gene ID990338 codes presumably for an Hsp26/Hsp42 chaperone. This chaperone belongs to the family of the Hsp20 and is a small heat-shock protein that suppresses protein aggregation and protects against cell stress. Hsp20 has been reported to be absent in the T. melanosporum genome and other ascomycetes [37]. However, the search for BlastP hits of this protein reveals its presence in T. brumale, T. aestivum, T. indicum, T. magnatum, and many other ascomycetes. This protein has been used to construct a temperature-tolerant strain of Lentinula edodes [38] and found in tandem repeats in a temperature-adapted Coriolopsis trogii. The model identified in T. borchii corresponds to a secreted protein. Secreted heat shock proteins have been proposed to act as intercellular signals [39]. Their function in Tuber is unknown, but it seems relevant in minimal media compared to complex media.
The most expressed gene in the PDB samples codes for a protein without an assigned function (ID1067414). The transcription effort of this gene in PBD is 2.10% and 1.37% in MPY. The gene coding for this protein is also among the most expressed in WPG and WPGY, representing 0.96 and 0.5 % of the gene expression and being among the 10 most expressed genes in these samples. This gene codes for a small (154 amino acids) presumably secreted protein. Proteins similar to this have been found in other ascomycetes (preferentially other Tuber) and some budding yeasts. It has a peculiar primary structure as it contains 14 Ser and 34 Thr residues.
Finally, the most expressed gene in MPY was ID957843, coding for the Glyceraldehyde 3-phosphate dehydrogenase (GA3PDH). The portion of the total transcription associated with this gene was 1.95, 1.36, 0.35, and 0.35% in the MPY, PDB, WPG, and WPGY samples., respectively. In the two samples derived from complex media (MPY and PDB), this gene was among the ten more expressed, whereas in the samples derived from minimal media (WPG and WPGY), the gene was among the 30 more expressed ones.
3.3. Comparison of the transcriptomes from the different culture media
The lack of technical replicas of the transcriptomes hampers the comparison between transcriptomes in this preliminary analysis. This lack is more relevant when studying the differences between samples but is more tolerable when stressing the similarities between the samples studied. Because of that, some rough qualitative comparisons under different culture conditions can be made. The correlation between the normalized expression values for all genes in all the pairwise combinations of conditions (Supplementary Materials - Correlation coefficient) was studied. The correlation between the gene expression values when WPG and WPGY samples were compared was 99.6%, with R2 being 0.996, thus indicating that both samples were virtually identical. No genes in this comparison fell outside the twofold limit established as a rough comparative criterion. This result suggests that the yeast extract in the WPGY medium did not induce significant gene expression changes in the samples. When all the other comparisons were made, the R2 values were lower (between 0,64 and 0,70), appearing to be the most different sample, the one from the MPY culture. Only a few genes fell out of the twofold expression limits established for these comparisons, most without functional KOG annotation.
The WPG and WPGY samples showed a more similar transcriptome profile than the MPY and PDB samples. There are two possible explanations for this. The wood-plant-based culture media (WPG and WPGY) are synthetic culture media with glucose as the sole carbon source, whereas the PDB and MPY media are undefined culture media with complex carbon sources (maltose and potato dextrose). Furthermore, the WPG and WPGY samples were harvested after 65 days of culture, whereas the MPY and PDB samples were harvested on day 85.
The transcription effort of the genes annotated within the O KOG category (posttranslational modification, protein turnover, and chaperones) in WPG and WPGY media was 15.27 and 15.30%, respectively, whereas in the PDB and MPY media was 6.43 and 7.61%, respectively. The importance of the transcription of gene ID990338 in WPG and WPGY has been described above. This gene codes for a chaperone belonging to KOG Class O, and its contribution to the effort of this class is around 6%. Putting aside the contribution of this gene to the complete transcription of class O, this class is still more transcribed in the minimal media (an effort close to 10%) than in the more complex media. Within the 20 most expressed genes in WPG and WPGY, three code for heat shock proteins (Hsp20 and Hsp70) and one for a Ubiquitin-like protein.
Concerning the genes belonging to the KOG class G (carbohydrate transport and metabolism), the observed enrichment in the expression of the genes involved in the carbon source processing can be a consequence of the complexity of these two media compared with the glucose used as the sole carbon source in WP-based broths.
Finally, the expression of genes classified in the KOG class Q seems higher in the MPY and PDB samples than in the WP-based ones. This category includes the genes involved in secondary metabolite synthesis. This result suggests that the mycelium cultivated in nutritionally richer broths can produce more secondary metabolites than in the basal WP medium.
3.4. Pathways of central metabolism
The NADH-dehydrogenase system is critical for regenerating the NAD+ required for the glycolytic reactions. The combination of the low expression of the NADH-dehydrogenase system combined with the high expression levels of the alcohol dehydrogenase in the four media (see Figure 6) suggests that this fungus displays a metabolism preferentially fermentative in these aerobic conditions. In yeasts, alcoholic fermentation is known as the Crabtree effect in aerobic conditions. This effect occurs by inhibiting the TCA and respiratory chain in conditions of high glucose concentration. The amount of glucose used in our system could be high enough to trigger this effect in T. borchii, preventing the accumulation of biomass in this way.
Besides the high level of ADH expression, the expression of GAPDH, PGK, and PK (responsible for phosphorylation at the substrate level) was also high, especially in the MPY culture medium. The high quantities of PGK and PK produce ATP in more significant amounts, which could justify the better mycelial performance in the culture MPY medium.
3.5. Genes involved in secondary metabolism – Clusters
Secondary metabolites are not produced in the rapid growth phase (trophophase) but rather during the later production phase (idiophase). Generally, the synthesis of these metabolites begins when a nutrient source is depleted, such as carbon or nitrogen [40]. Although the transcriptomic analysis of this study was carried out with samples from days 65 and 85 of growth, it is possible that these periods were insufficient to produce and accumulate secondary metabolites.
Whereas the genes coding for enzymes involved in the central metabolic pathways are dispersed across the genome, those coding for enzymes participating in the secondary metabolism are arranged in biosynthetic clusters [41]. Ascomycete genomes encode an average of ten non-ribosomal protein synthetases (NRPS), 16 polyketide synthetases (PKS), two tryptophan synthetases (TS), and two dimethylallyl tryptophan synthases (DMATS), per genome [42]. The automatic annotation of the T. borchii genome used as a reference for this study, however, identifies seven clusters coding for highly conserved proteins: one non-ribosomal peptide synthetase (NRPS, containing proteins IDs 1077522, 1099828, and 1122331), one type I polyketide synthase (PKS, ID 962914), three NRPS-like (containing proteins IDs 1126844, 966209, 1119995, and 1032817) some of them with PKS domains, and two PKS-like (containing protein ID 1116892, and proteins 1121687, 970716, 1076327, and 970641). This number and distribution are also found in the genome of T. melanosporum. This reduced number of secondary metabolite clusters in the Tuber genomes could result from a miss-annotation of some of them in the highly complex genomes of these species.
The expression of the genes of the secondary metabolism clusters was low (lower than 50 TPMs in all the samples) except for two PKS genes in the PDB cultures that showed around 150 TPMs. Interstingly, the mycelium harvested under these conditions showed a more intense brownish that could correlate with the production of compounds that are easily oxidized upon removing the liquid culture medium.
3.6. Genes related to the synthesis of volatile compounds
Regarding genes related to truffle aroma, it was observed that most of the analyzed genes were present and expressed in the samples. Upon analysis of the ten genes with the most significant expression, it was found that the gene expression was very similar in WPG and WPGY media, suggesting that adding yeast extract did not seem to influence the expression of these genes. It was verified that the PDB sample in some genes presented higher gene expression when compared to the other media. It was also observed that genes were expressed very differently between the different growing conditions. The gene corresponding to phosphoadenosine phosphosulfate reductase (thioredoxin reductase) was expressed almost five times more in PDB when compared to the MPY medium. The thioredoxin reductase was included in this group because it appears in the list of aroma-related genes published by Martin et al. [21]. The thioredoxin reductase also plays a role as an agent protecting the cell against the oxidative stress produced by ROS. As discussed above, the high expression of ADH and the low of the NADH-dehydrogenase genes suggest a fermentative metabolism for T. borchii under the culture conditions used. Fermentative metabolism and hypoxia conditions generate high levels of ROS. The data presented here suggest that the production of Thioredoxin-reductase could help prevent the detrimental effects of these toxic compounds.
4. Materials and Methods
4.1. Strain and culture conditions
Several truffles (T. borchii, T. aestivum, T. magnatum, T. melanosporum ) were obtained from local markets and collectors in different regions of Spain. The truffle surfaces were washed with soapy water (1:200 dilution), brushed to remove excess soil, and dried with absorbent filter paper. Then, the truffles were aseptically broken in two halves, and small pieces of the innermost part of the gleba were transferred to Petri dishes with different culture media supplemented with amoxicillin at 100 mg/mL to avoid bacterial contamination. All cultures were incubated at 22 ±1°C in the dark.
The cultivation process was performed in the following culture media: (i) Potato Dextrose Broth 20 g/L (PDB) (Scharlab) or Potato Dextrose Agar (PDA). (ii) Wood Plant (WP) medium (composition per liter: 0.2g KH2PO4, 0.1g CaCl2•2H2O, 0.3g MgSO4•7H2O, 0.9g K2SO4, 0.1g myoinositol, 2.3 g WoodPlant, 1 mL oligo-elements solution containing, per liter, 22.3 mg MnSO4•H2O, 0.014mg FeSO4•7H2O, 8.6 mg ZnSO4•7H2O, 0.25mg Na2MoO4•2H2O, 0.025mg CuSO4•5H2O) [28] modified. The WP was occasionally supplemented with 20g/L Glucose (WPG), 20g/L glucose plus 5g/L yeast extract (WPGY), 20g/L Dextrose (WPD), 20g/L Dextrose plus 5g/L yeast extract (WPDY) or 20g/L Sucrose (WPS). (iii) Maltose medium (MPY) containing, per liter, 35g Maltose, 5g peptone, 5g yeast extract, 0.5g MgSO4•7H2O, 1g K2SO4, and 0.05 g myoinositol [43] modified. The pH of all culture media was adjusted to 6.3 - 6.4 with NaOH. All liquid cultures were incubated in dark and static conditions at 22±1°C.
When needed, 7g/L Agar was added for solid cultures. Maltose and yeast extract were purchased from Scharlab, peptone from Panreac, WoodPlant from Merck, and Agar from Scharlab.
4.2. Growth rate
The linear growth rate was measured as follows: one plug (0.2 cm2) of actively growing mycelium was placed in the center of the Petri dish, and the linear growth of the colony’s edge was measured in the four perpendicular directions each week until the 35th day of growth. All plates were made in triplicate to calculate the mean growth rate values and standard deviation. ANOVA and Turkey were used to compare the variations between the means of the different groups.
4.3. Biomass
Three plugs (0.2 cm2) of actively growing mycelium from the outermost growth zone of a solid culture were used to inoculate 250 ml flasks containing 100 ml of medium. The flasks were grown at 22°C in dark and static conditions for 40 days. After 40 days, the mycelia were filtered through a micropore filter, weighed and dried at 50°C for 24 hours, and weighed again to obtain fresh and dry weight. The weight of the agar plugs was discounted. There were four replicates for each culture medium.
4.4. Molecular characterization
The mycelium was ground using liquid nitrogen. DNA extraction was performed using the commercial EZNA Fungal DNA Mini Kit (Promega Biotech) following the producer’s protocol. After checking the DNA concentration, a PCR was performed using sequences from the internal transcribed spacer (ITS) region, primers ITS4 5’-TCCTCCCGCTTATTGATA-3’, and ITS1F 5’-CTTGGTCATTTAGAGGAAGTAA-3’1. The PCR conditions were as follows: an initial denaturation step at 95°C for 5 min, followed by 29 cycles consisting of 1 min denaturation at 95ºC, 0.5 min annealing at 53ºC, and 1 min extension at 72ºC. After the last cycle, a 10 min final extension step at 72ºC was added to complete the reaction. The PCR products were sequenced, and the sequences were blasted against the NCBI database to identify the most similar entries. The ITS sequence was deposited in the GenBank NCBI database, with accession number OQ002403.
4.5. Morphology
Morphological data of the mycelium of T. borchii strain SP1 were studied under light microscopy in different culture media. Small pieces (1cm2) of mycelium were analyzed and collected from each Petri dish (three per substrate) after 40 days of mycelial growth. The following parameters were measured: thickness, branching angle, and distance between the septa of the hyphae grown in the different culture media. Photos were taken using a camera (ZEISS Axiocam 208 color/202) attached to the microscope and further processed using the program ZEN 2.3 - blue edition.
4.6. RNA seq
For the transcriptome analysis of the SP1 strain, axenic cultures of the mycelium were performed under different culture conditions. The mycelia were collected after 65 days in WPG and WPGYor 85 days in PDB and MPY of culture.
The mycelial samples were ground using liquid nitrogen and stored at -80°C for transcriptome analysis. A fungal RNA EZNA kit (Omega Bio-Tek, Norcross, GA, USA) was used to extract the total RNA. RNA quality was determined by electrophoresis on 1% (w/v) agarose gels. A Qubit® RNA Assay kit (Invitrogen, Life Technologies Corporation, USA) was used to measure RNA concentration. The mRNA libraries were built using TruSeq® RNA (Illumina, Inc., San Diego, CA, USA), following the manufacturer’s instructions. Sequencing was performed with an Illumina NovaSeq 6000 System (North Caroline genomic lab) using paired-end reads of 150 bp.
4.7. Transcriptome data analysis
The quality of mRNA-seq data was checked using FastQC [44] and trimmed with Trimmomatic [45] to remove reads and sequences containing adapters. The results of reads from all libraries were mapped to the T. borchii Tbo3840 reference genome obtained from the JGI Mycocosm platform [24] using the STAR Galaxy Version 2.7.8 program [46]. The feature Counts [47] program was used to assign the mapped reads generated from RNA sequencing. KOG (EuKaryotic Orthologous Groups) and EggnogMapper [48] were used for protein function annotation. The Kallisto program was used to normalize the read data, which generated values of TPMs that were transformed into RPMKs per gene [49].
4.8. Analysis of metabolic pathways
The analysis of metabolic pathways, glycolysis, citrate cycle (TCA), glyoxylate cycle, and oxidative phosphorylation has been performed based on KEGG annotation of the T. borchii genome available in the mycocosm portal of the Joint Genome Institute. (https://mycocosm.jgi.doe.gov/Tubbor1/Tubbor1.home.html).
5. Conclusions
A new Spanish strain of T. borchii was isolated, the axenic culture conditions described, and the gene expression in different media conditions were analyzed. Significant gene expression was obtained for most of the annotated genes, although most of the transcription effort was concentrated in a few genes (80% of the transcription was due to 20% of the genes). The transcription profiles of the cultures made in minimal medium (WPG and WPGY) were highly similar, whereas there were more differences in the cultures made in complex media (PDB and MPY). Altogether, the analyzed transcriptomes can shed some light on the metabolic processes in T. borchii under the conditions studied. The distribution of gene numbers and expression by KOG categories depends on the culture medium. The transcription of genes involved in the central metabolic pathways was relatively constant in the four conditions studied, giving a reference for other experiments. The high transcription level of ADH genes suggests a fermentative metabolism for t. borchii. This hypothesis is strengthed by the reduced expression level of NADH-dehydrogenase genes (suggesting low respiration), the high expression of genes involved in substrate-level phosphorylation (PGK and PK), and the high expression of ROS-protecting genes (thioredoxin reductase). In vitro culture of truffle mycelium may become a potential alternative resource for secondary metabolite production and biotechnological applications and for studying the factors required for enhancing its vegetative growth.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, E.C.T., L.R. and A.G.P.; methodology, E.C.T., M.A. and G.P.; formal analysis, E.C.T, M.A., A.Z., E.G. and G.P.; funding acquisition, L.R., H.S. and A.G.P.; project administration, H.S and A.G.P.; writing—original draft, E.C.T.; writing—review and editing, E.C.T., A.Z. and A.G.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Research Projects RTI2018-099371-B-I00 (MCIU, AEI, FEDER/UE) and AGL2015-66833-R (MINECO) of the Spanish National Research Programme, H2020 MUSA 727624 (EU), and by funds of the Public University of Navarre (UPNA). E.C.T. was supported by a grant of the Gobierno de Navarra (Ref. 0011-1408-2020-000000) for Industrial Ph.D. students and by mobility grants from Erasmus+ program of Campus Iberus and by a mobility grant of the Gobierno de Navarra.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
References
- Mustafa, A.M.; Angeloni, S.; Nzekoue, F.K.; Abouelenein, D.; Sagratini, G.; Caprioli, G.; Torregiani, E. An Overview on Truffle Aroma and Main Volatile Compounds. Molecules 2020, 25, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Mello, A.; Murat, C.; Bonfante, P. Truffles: Much More than a Prized and Local Fungal Delicacy. FEMS Microbiol Lett 2006, 260, 1–8. [Google Scholar] [CrossRef]
- Zambonelli, A., IM, MC True Truffle (Tuber Spp.) in the World ; 2016; Vol. 47; 333-349. [CrossRef]
- Splivallo, R.; Cullere, L. The Smell of Truffles : From Aroma Biosynthesis to Product Quality. 2016, 393–407. [CrossRef]
- Bonito, G.; Trappe, J.M.; Rawlinson, P.; Vilgalys, R. Improved Resolution of Major Clades within Tuber and Taxonomy of Species within the Tuber Gibbosum Complex. Mycologia 2010, 102, 1042–1057. [Google Scholar] [CrossRef] [PubMed]
- Hall, I.R.; Brown, G.T.; Zambonelli, A. Taming the Truffle. The History Lore and Science of the Ultimate Mushroom; 1st ed.; Portland, OR, USA, 2007, 304.
- Lacourt, I.; Abba, S.; Bonfante, P.; Martin, F. Isolation and Characterization of Differentially Expressed Genes in the Mycelium and Fruit Body of Tuber Borchii. 2002, 68, 4574–4582. 68. [CrossRef]
- Marathe, SJ; Hamzi, W. ; Bashein, A.M.; Deska, J.; Seppänen-Laakso, T.; Singhal, R.S.; Shamekh, S. Anti-Angiogenic and Anti-Inflammatory Activity of the Summer Truffle (Tuber Aestivum Vittad.) Extracts and a Correlation with the Chemical Constituents Identified Therein. Food Research International 2020, 137. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, X.H.; Li, HM; Wang, S. H.; Chen, T.; Yuan, Z.P.; Tang, Y.J. Isolation and Characterization of Polysaccharides with the Antitumor Activity from Tuber Fruiting Bodies and Fermentation System. Appl Microbiol Biotechnol 2014, 98, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Li, YY; Wang, G. ; Li, HM; Zhong, J.J.; Tang, Y.J. Volatile Organic Compounds from a Tuber Melanosporum Fermentation System. Food Chem 2012, 135, 2628–2637. [Google Scholar] [CrossRef] [PubMed]
- Vita, F.; Taiti, C.; Pompeiano, A.; Bazihizina, N.; Lucarotti, V.; Mancuso, S.; Alpi, A. Volatile Organic Compounds in Truffle (Tuber Magnatum Pico): Comparison of Samples from Different Regions of Italy and from Different Seasons. Sci Rep 2015, 5, 12629. [Google Scholar] [CrossRef]
- Shah, N.; Marathe, SJ; Croce, D. ; Ciardi, M.; Longo, V.; Juilus, A.; Shamekh, S. An Investigation of the Antioxidant Potential and Bioaccumulated Minerals in Tuber Borchii and Tuber Maculatum Mycelia Obtained by Submerged Fermentation. Arch Microbiol 2022, 204, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.J.; Liu, R.S.; Li, H.M. Current Progress on Truffle Submerged Fermentation: A Promising Alternative to Its Fruiting Bodies. Appl Microbiol Biotechnol 2015, 99, 2041–2053. [Google Scholar] [CrossRef]
- Zambonelli, A.; Iotti, M.; Hall, I. Current Status of Truffle Cultivation: Recent Results and Future Perspectives. Micol Ital 2015, 44, 31–40. [Google Scholar] [CrossRef]
- Lacourt, I.; Duplessis, S.; Abbà, S.; Bonfante, P.; Martin, F. Isolation and Characterization of Differentially Expressed Genes in the Mycelium and Fruit Body of Tuber Borchii. Appl Environ Microbiol 2002, 68, 4574–4582. [Google Scholar] [CrossRef]
- Splivallo, R.; Bossi, S.; Maffei, M.; Bonfante, P. Discrimination of Truffle Fruiting Body versus Mycelial Aromas by Stir Bar Sorptive Extraction. Phytochemistry 2007, 68, 2584–2598. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, H.; Tang, Y. Comparison of Sterol Composition between Tuber Fermentation Mycelia and Natural Fruiting Bodies. Food Chem 2012, 132, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Tirillini, B.; Verdelli, G.; Paolocci, F.; Ciccioli, P.; Frattoni, M. The Volatile Organic Compounds from the Mycelium of Tuber Borchii Vitt. Phytochemistry 2000, 55, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Murat, C.; Payen, T.; Noel, B.; Kuo, A.; Morin, E.; Chen, J.; Kohler, A.; Krizsán, K.; Balestrini, R.; Da Silva, C.; et al. Pezizomycetes Genomes Reveal the Molecular Basis of Ectomycorrhizal Truffle Lifestyle. Nat Ecol Evol 2018, 2, 1956–1965. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Marcone, M.F. The Biochemistry and Biological Properties of the World’s Most Expensive Underground Edible Mushroom: Truffles. Food Research International 2011, 44, 2567–2581. [Google Scholar] [CrossRef]
- Martin, F.; Kohler, A.; Murat, C.; Balestrini, R.; Coutinho, P.M.; Jaillon, O.; Montanini, B.; Morin, E.; Noel, B.; Percudani, R.; et al. Périgord Black Truffle Genome Uncovers Evolutionary Origins and Mechanisms of Symbiosis. Nature 2010, 464, 1033–1038. [Google Scholar] [CrossRef]
- Moreno, G.; Manjón, J.L. A New Tuber without Spore Ornamentation. Tuber Melosporum Comb. Bol. Soc. Micol. Madrid 2012, 36, 191–196. [Google Scholar]
- Murat, C.; Martin, F. Truffle Genomics: Investigating an Early Diverging Lineage of Pezizomycotina. In True truffle (Tuber spp.) in the world.; Zambonelli A, Iotti M, M.C., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 137–149. [Google Scholar]
- Murat, C.; Kuo, A.; Barry, K.W.; Clum, A.; Dockter, R.B.; Fauchery, L.; Iotti, M.; Kohler, A.; Labutti, K.; Lindquist, E.A.; et al. Draft Genome Sequence of Tuber Borchii Vittad., a Whitish Edible Truffle. American Society for Microbiology 2018, 6, 1–2. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, Z.; Xu, L.; Shi, W.; Liu, X.; Wang, F. Increasing Production of Truffle Polysaccharides in the Solid-State Fermentation of Tuber Melanosporum by Diosgenin Based on Orthogonal Matrix and Nonlinear Regression Analysis. Food Sci Technol Res 2020, 26, 487–494. [Google Scholar] [CrossRef]
- Tang, Y.J.; Wang, G.; Li, YY; Zhong, J. J. Fermentation Condition Outweighed Truffle Species in Affecting Volatile Organic Compounds Analyzed by Chromatographic Fingerprint System. Anal Chim Acta 2009, 647, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A. Atti Del Congresso Internazionale Sul Tartufo. In Proceedings of the MICELI DI FUNGHI IPOGEI IN COLTURA PURA; 1967; pp. 127–133. [Google Scholar]
- Iotti, M.; Amicucci, A.; Stocchi, V.; Zambonelli, A. Morphological and Molecular Characterization of Mycelia of Some Tuber Species in Pure Culture. New Phytologist 2002, 155, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Vahdatzadeh, M.; Deveau, A.; Splivallo, R. The Role of the Microbiome of Truffles in Aroma Formation: A Meta-Analysis Approach. Appl Environ Microbiol 2015, 81, 6946–6952. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Gioacchini, A.M.; Zambonelli, A.; Bertini, L.; Stocchi, V. Determination of Microbial Volatile Organic Compounds from Staphylococcus Pasteuri against Tuber Borchii Using Solid-Phase Microextraction and Gas Chromatography / Ion Trap Mass Spectrometry. RAPID COMMUNICATIONS IN MASS SPECTROMETRY 2015, 19, 3411–3415. [Google Scholar] [CrossRef] [PubMed]
- Perlińska-Lenart, U.; Piłsyk, S.; Gryz, E.; Turło, J.; Hilszczańska, D.; Kruszewska, J.S. Identification of Bacteria and Fungi Inhabiting Fruiting Bodies of Burgundy Truffle (Tuber aestivum Vittad.). Arch Microbiol 2020, 202, 2727–2738. [Google Scholar] [CrossRef]
- Leonardi, P.; Murat, C.; Puliga, F.; Iotti, M.; Zambonelli, A. Ascoma Genotyping and Mating Type Analyses of Mycorrhizas and Soil Mycelia of Tuber Borchii in a Truffle Orchard Established by Mycelial Inoculated Plants. Environ Microbiol 2020, 22, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Murat, C.; Rubini, A.; Riccioni, C.; De la Varga, H.; Akroume, E.; Belfiori, B.; Guaragno, M.; Le Tacon, F.; Robin, C.; Halkett, F.; et al. Fine-Scale Spatial Genetic Structure of the Black Truffle (Tuber Melanosporum) Investigated with Neutral Microsatellites and Functional Mating Type Genes. New Phytologist 2013, 199, 176–187. [Google Scholar] [CrossRef]
- Moore, D. Fungal Morphogenesis. Developmental and Cell Biology Series.; Cambridge, UK.; Cambridge University Press, 1998.
- Graziosi, S.; Hall, I.R.; Zambonelli, A. The Mysteries of the White Truffle: Its Biology, Ecology and Cultivation. Encyclopedia 2022, 2, 1959–1971. [Google Scholar] [CrossRef]
- Amicucci, A.; Zambonelli, A.; Iotti, M.; Polidori, E.; Menotta, M.; Saltarelli, R.; Potenza, L.; Stocchi, V. Morphological and Molecular Modifications Induced by Different Carbohydrate Sources in Tuber Borchii. J Mol Microbiol Biotechnol 2010, 18, 120–128. [Google Scholar] [CrossRef]
- Zampieri, E.; Balestrini, R.; Kohler, A.; Abbà, S.; Martin, F.; Bonfante, P. The Perigord Black Truffle Responds to Cold Temperature with an Extensive Reprogramming of Its Transcriptional Activity. Fungal Genetics and Biology 2011, 48, 585–591. [Google Scholar] [CrossRef]
- Ling, Y.Y.; Ling, Z.L.; Zhao, R.L. Construction of a Heat-Resistant Strain of Lentinus Edodes by Fungal Hsp20 Protein Overexpression and Genetic Transformation. Front Microbiol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A.; Vazquez, D. Extracellular Heat Shock Proteins: A New Location, a New Function. Shock 2013, 40, 239–246. [Google Scholar] [CrossRef]
- Kumar, S.; Sanjeev, C.; Sanjeev, C.; Mohd, S.; Shah, A. Sustainable Management of Potato Pests and Diseases. In; Chandan Maharana, Vinod Kumar Padala, A.B.H., M. Nikhil Raj, Amit Paschapur, Chaitra Bhat, A.K.S., Subbanna, and A.R.N.S., Eds.; 2022, ISBN 9789811676949.
- Keller, N.P. Fungal Secondary Metabolism: Regulation, Function and Drug Discovery. Nat Rev Microbiol 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Pusztahelyi, T.; Holb, I.J.; Pócsi, I. Secondary Metabolites in Fungus-Plant Interactions. Front Plant Sci 2015, 6, 1–23. [Google Scholar] [CrossRef]
- Tang, Y.J.; Zhu, L.L. ; Li, DS; Mi, ZY; Li, HM Significance of Inoculation Density and Carbon Source on the Mycelial Growth and Tuber Polysaccharides Production by Submerged Fermentation of Chinese Truffle Tuber Sinense. Process Biochemistry 2008, 43, 576–586. [Google Scholar] [CrossRef]
- Simon Andrews FastQC. A Quality Control Tool for High Throughput Sequence Data. A Quality Control Tool for High Throughput Sequence Data. 2015.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hern̗andez-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat Biotechnol 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A. Fruiting bodies of the Tuber species used in this work. B, Mycelium growth of T. borchii SP1 after eight days of culture in WPS. The diameter of the colony in the picture was 2 cm. C. Linear growth of T. borchii SP1 in different culture media (*There is a statistically significant difference between MPY and PDA and WPGY p<0.05. ** There is no statistically significant difference between MPY and WPG, p>0.05).
Figure 1.
A. Fruiting bodies of the Tuber species used in this work. B, Mycelium growth of T. borchii SP1 after eight days of culture in WPS. The diameter of the colony in the picture was 2 cm. C. Linear growth of T. borchii SP1 in different culture media (*There is a statistically significant difference between MPY and PDA and WPGY p<0.05. ** There is no statistically significant difference between MPY and WPG, p>0.05).
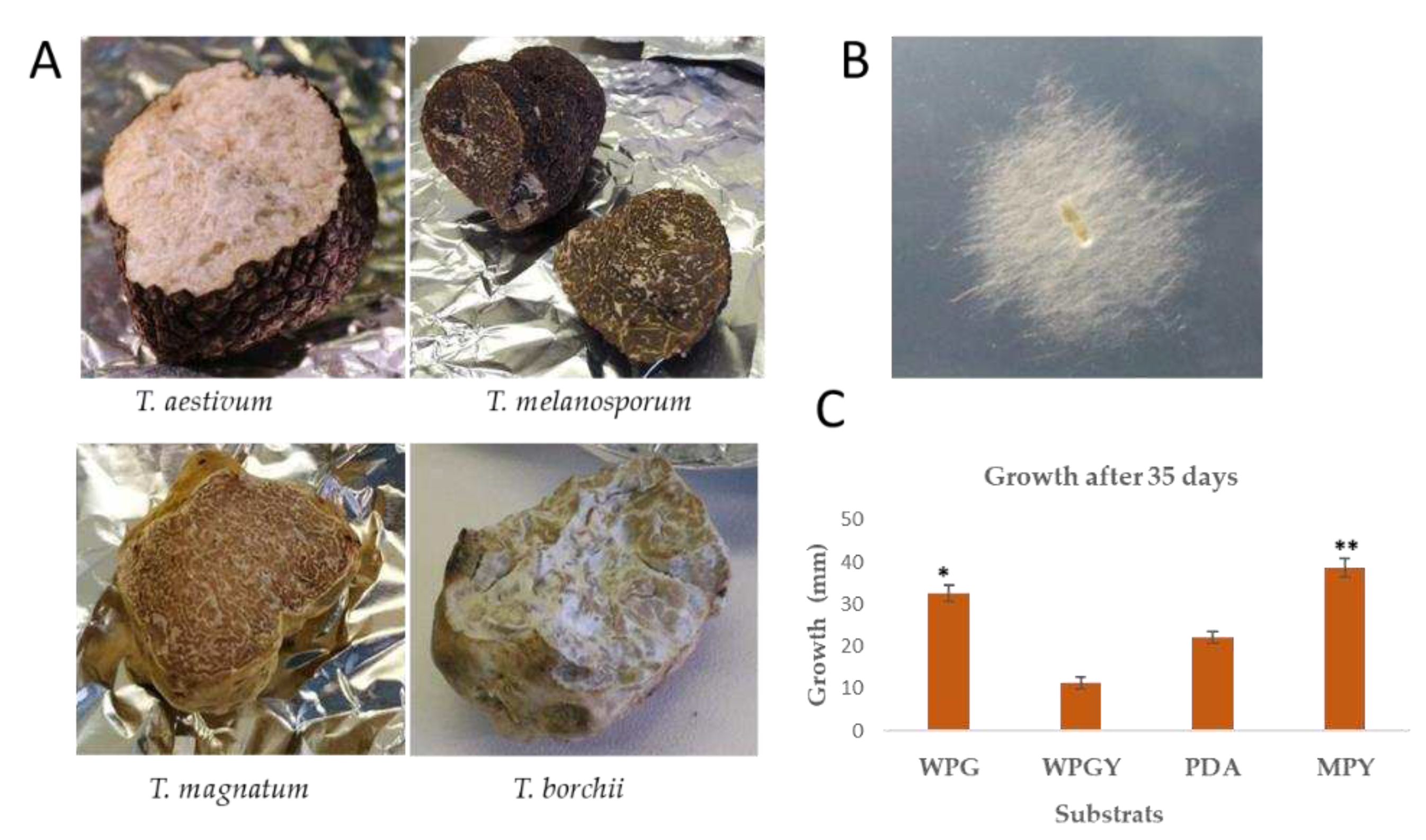
Figure 2.
Vegetative mycelium of T. borchii SP1 was produced in submerged static cultures using PDB, MPY, and WPG (upper row) and the same mycelium after removal of the culture broth by filtration (lower row).
Figure 2.
Vegetative mycelium of T. borchii SP1 was produced in submerged static cultures using PDB, MPY, and WPG (upper row) and the same mycelium after removal of the culture broth by filtration (lower row).

Figure 3.
Representative hyphae of T. borchii after 40 days of growth (40X). Medium MPY (left) and WPG (right).
Figure 3.
Representative hyphae of T. borchii after 40 days of growth (40X). Medium MPY (left) and WPG (right).
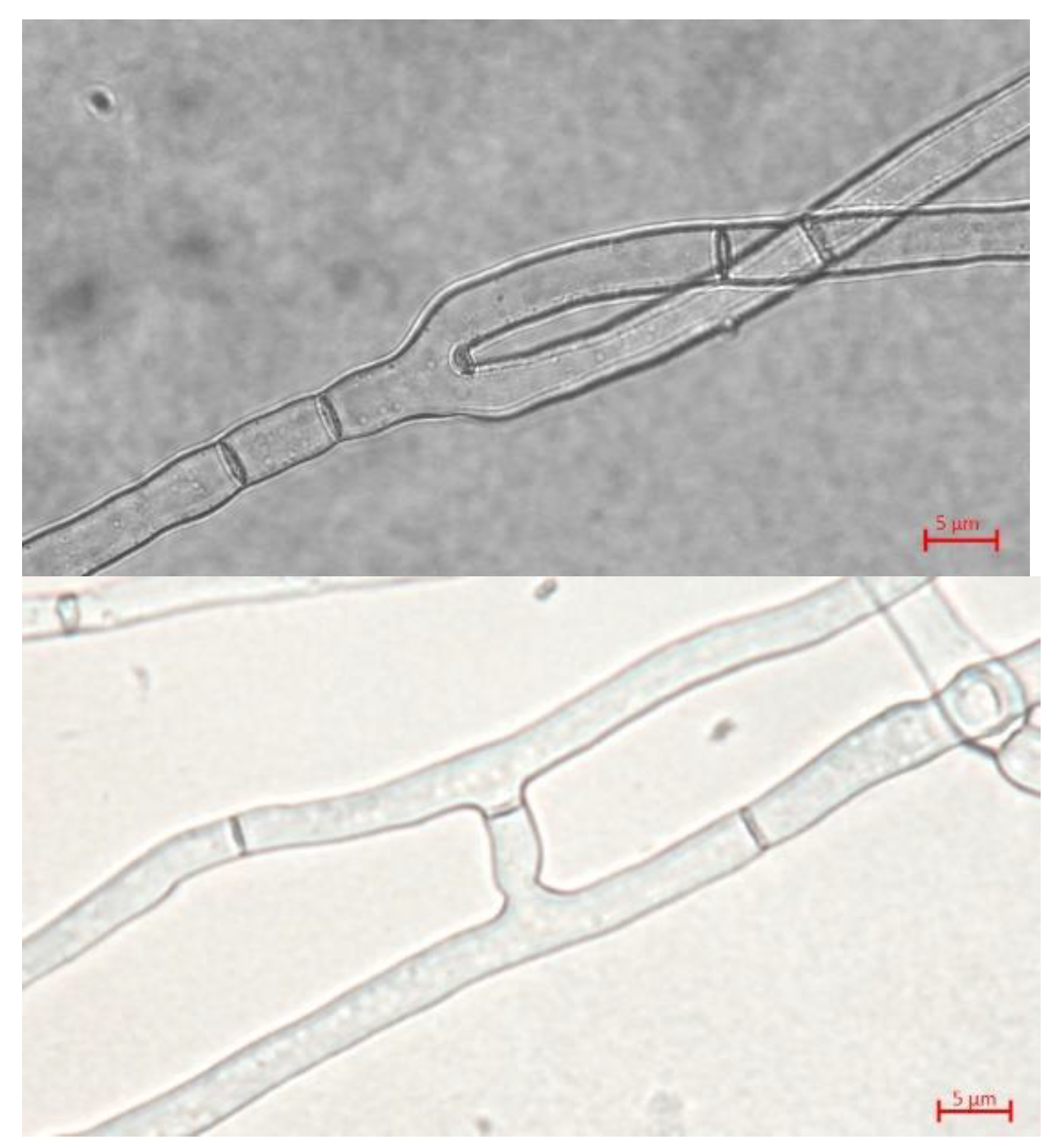
Figure 4.
Cumulative gene expression in T. borchii SP1 cultivated in WPG. The genes were ordered by expression value, and the curve represents the cumulative expression of the transcriptome. Similar curves were obtained for the transcriptomes made in WPGY, PDB, and MPY.
Figure 4.
Cumulative gene expression in T. borchii SP1 cultivated in WPG. The genes were ordered by expression value, and the curve represents the cumulative expression of the transcriptome. Similar curves were obtained for the transcriptomes made in WPGY, PDB, and MPY.
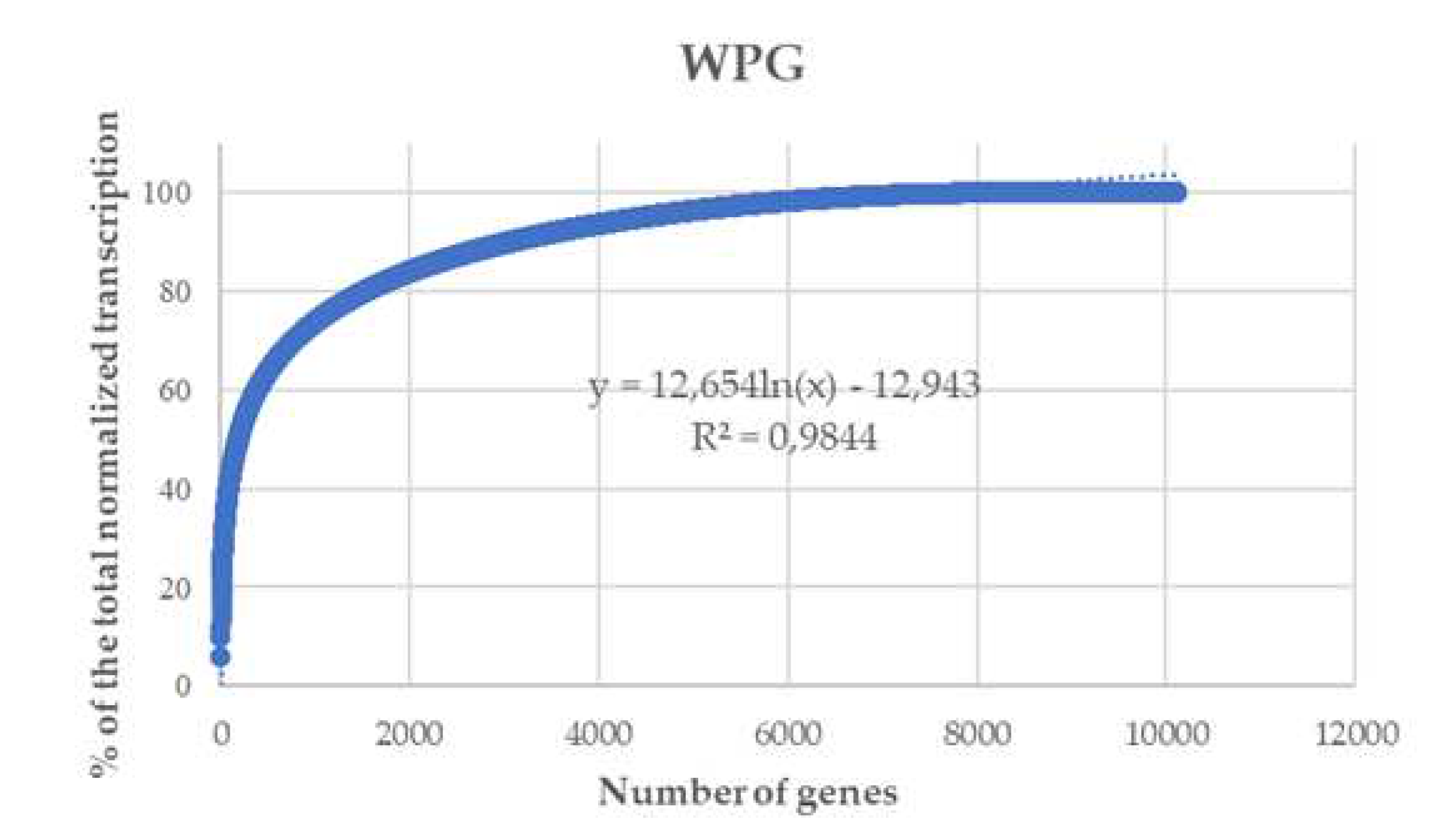
Figure 5.
A. Number of genes in each KOG category in the four samples analyzed. B. Number of TPMs reads associated with each one of the KOG categories in the analyzed samples.
Figure 5.
A. Number of genes in each KOG category in the four samples analyzed. B. Number of TPMs reads associated with each one of the KOG categories in the analyzed samples.
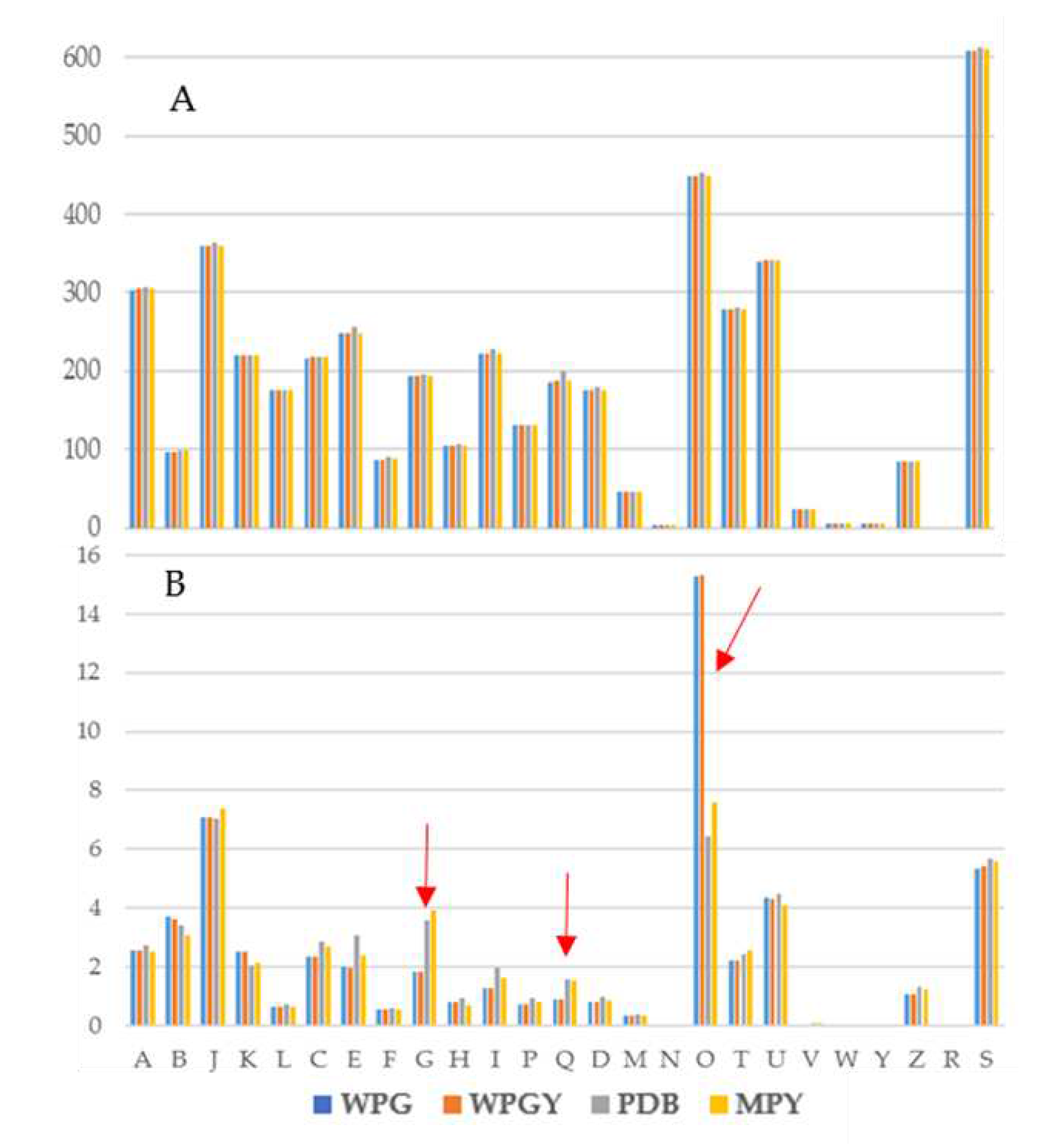
Figure 6.
Transcription of the genes coding for glycolytic enzymes in the four culture media. The columns represent the addition of the transcription of all the genes annotated as coding for the same isozyme.
Figure 6.
Transcription of the genes coding for glycolytic enzymes in the four culture media. The columns represent the addition of the transcription of all the genes annotated as coding for the same isozyme.
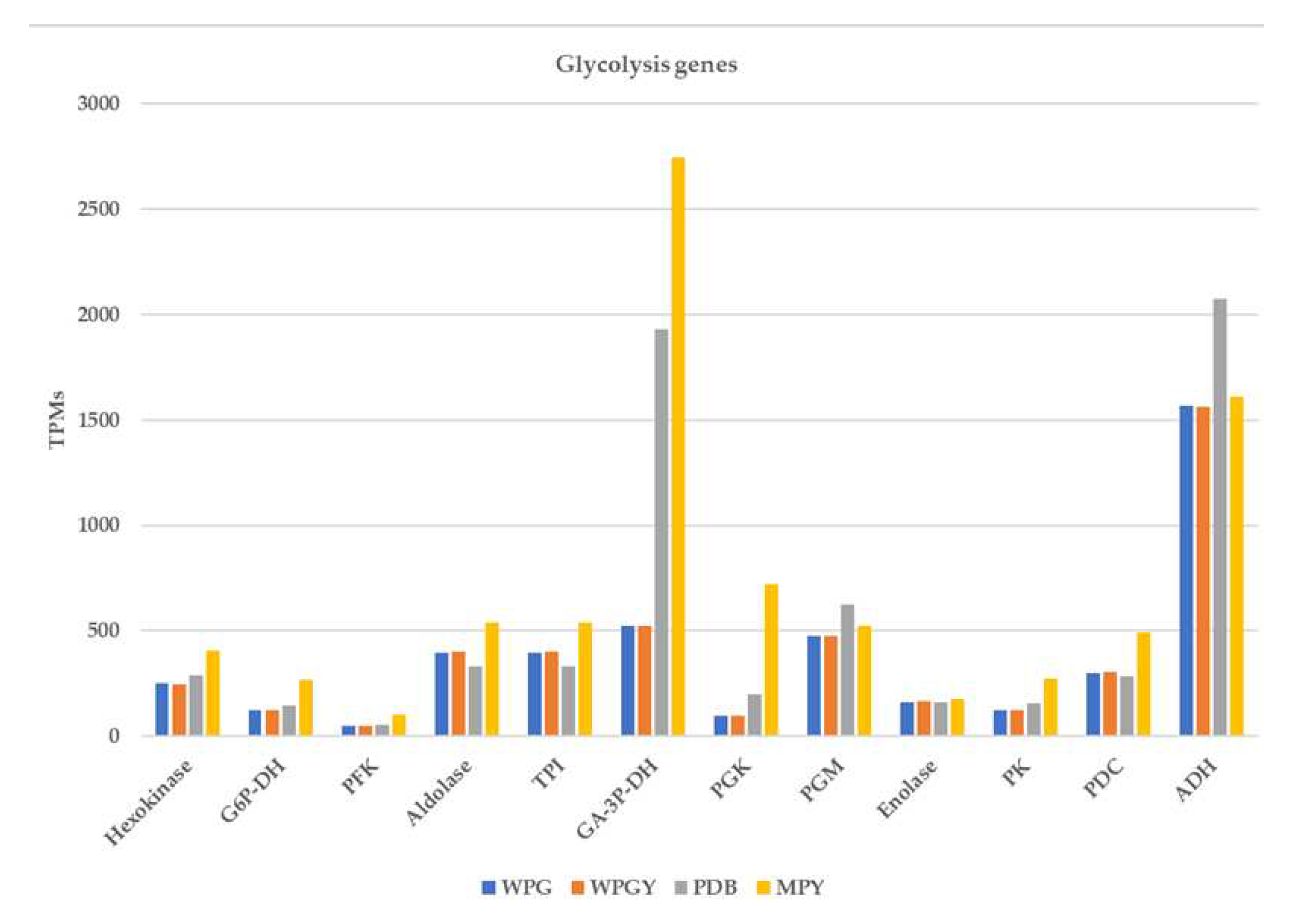
Figure 7.
Transcription of the genes coding for enzymes of the citrate cycle in the four culture media. The columns represent the addition of the transcription of all the genes annotated as coding for the same isozyme.
Figure 7.
Transcription of the genes coding for enzymes of the citrate cycle in the four culture media. The columns represent the addition of the transcription of all the genes annotated as coding for the same isozyme.
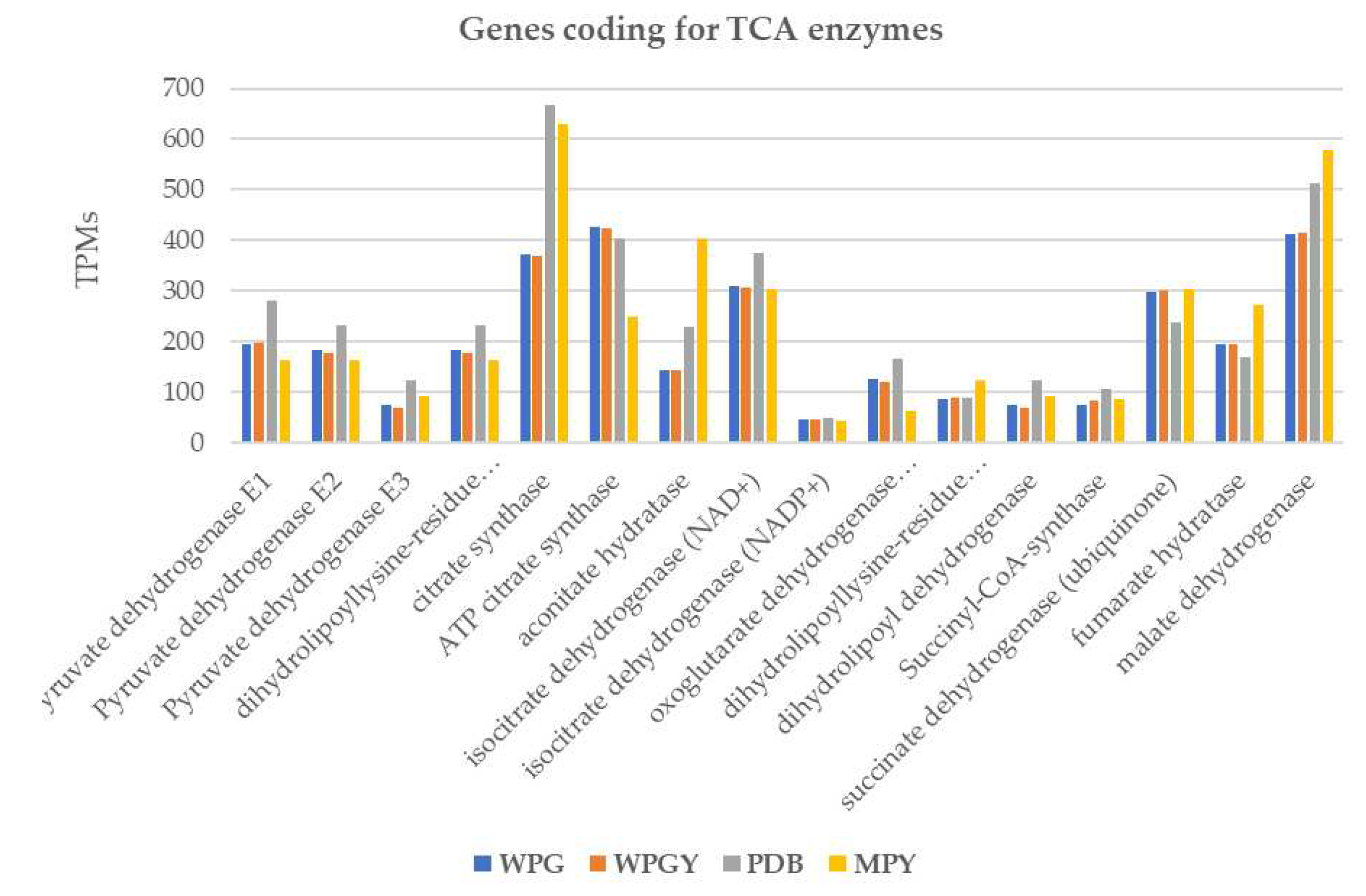
Figure 8.
Genetic expression of the ten most expressed genes related to truffle aroma.
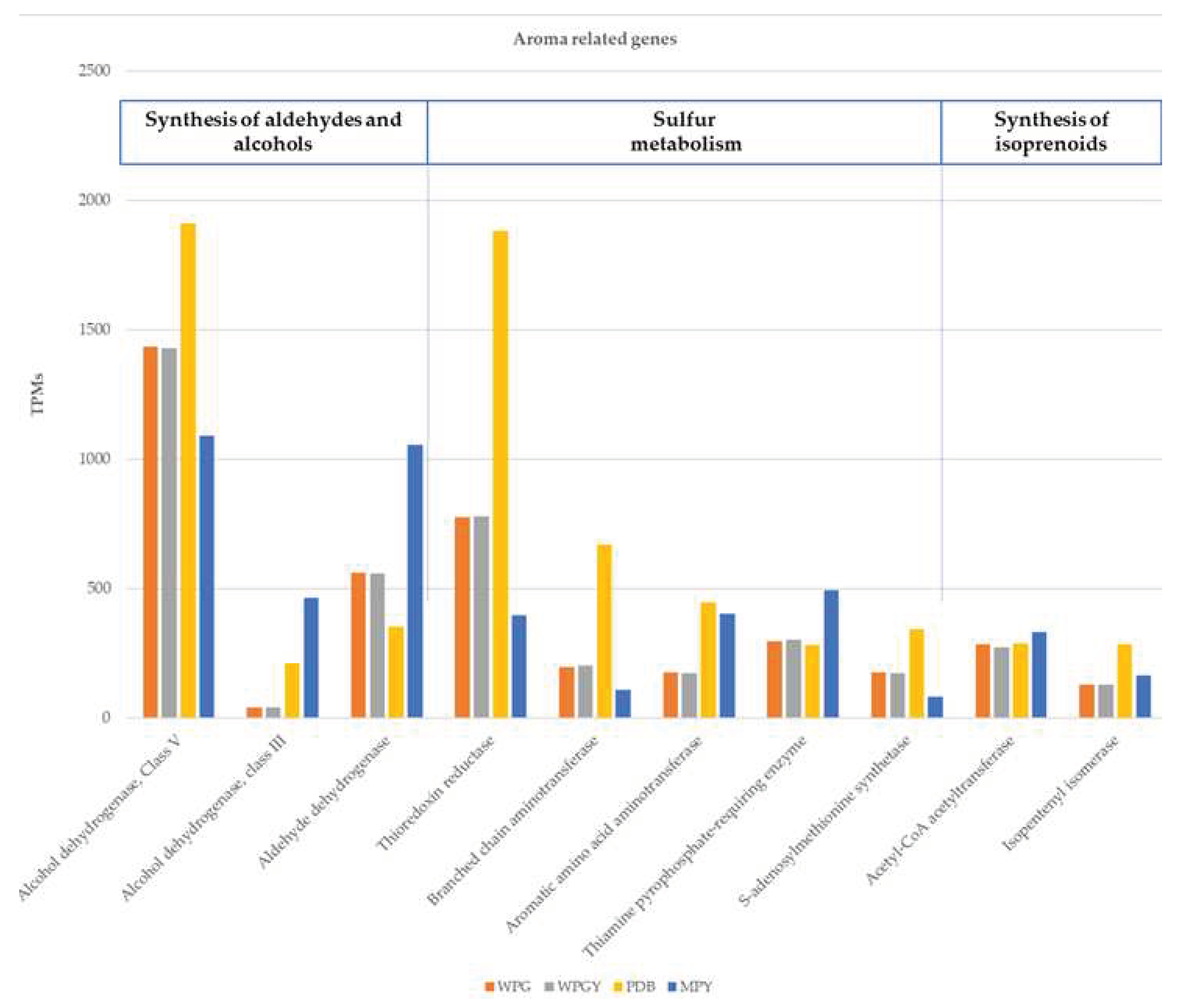

Table 1.
Mean (four repetitions) and standard deviation of the fresh and dry weight of T. borchii SP1 mycelium after 40 days of submerged static growth in different liquid culture media.
Table 1.
Mean (four repetitions) and standard deviation of the fresh and dry weight of T. borchii SP1 mycelium after 40 days of submerged static growth in different liquid culture media.
| Culture medium | Fresh weight (mg) | Dry weight (mg) |
|---|---|---|
| WPG | 535±103 | 41±7 |
| PDB | 470±89 | 38±1 |
| MPY | 1600±270 | 127±22 |
Table 2.
Mean (four repetitions) and standard deviation of the fresh and dry weight of T. borchii SP1 mycelium after 40 days of submerged static growth in different liquid culture medium.
Table 2.
Mean (four repetitions) and standard deviation of the fresh and dry weight of T. borchii SP1 mycelium after 40 days of submerged static growth in different liquid culture medium.
| Sample | Total reads | RPKM | Number of genes expressed (x103) | % of annotated genes | Genes KOG |
|---|---|---|---|---|---|
| WPG | 116,376,579 | 858,673.42 | 10.1 | 82.2 | 4,554 |
| WPGY | 125,457,058 | 848,604.73 | 10.1 | 82.2 | 4,654 |
| MPY | 47,969,567 | 803,655.02 | 10.2 | 82.9 | 4,568 |
| PDB | 157,335,190 | 803,793.44 | 10.6 | 86.2 | 4,620 |
| Genes in the reference genome | 12.3 | 100 | 5,686 | ||
Table 3.
Number of genes accounting for different percentages of total gene expression in the analyzed samples, and the number of them with a KOG annotation.
Table 3.
Number of genes accounting for different percentages of total gene expression in the analyzed samples, and the number of them with a KOG annotation.
| Sample | 10% | 25% | 50% | 75% |
|---|---|---|---|---|
| WPG | 3/1 | 18/9 | 185/108 | 1084/669 |
| WPGY | 3/1 | 19/9 | 186/108 | 1091/669 |
| PDB | 7/2 | 44/21 | 315/171 | 1392/847 |
| MPY | 8/2 | 43/18 | 271/144 | 1250/752 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



