
Submitted:
25 April 2023
Posted:
27 April 2023
You are already at the latest version
Abstract
Vascular calcification (VC) is associated with increased cardiovascular risks in patients with chronic kidney disease (CKD). Sodium-glucose cotransporter 2 inhibitors, like empagliflozin, can improve cardiovascular and renal outcomes. We assessed the expression of Runt-related transcription factor 2 (Runx2), interleukin (IL)-1β, IL-6, AMP-activated protein kinase (AMPK), nuclear factor erythroid-2-related factor (Nrf2), and heme oxygenase 1 (HO-1) in inorganic phosphate–induced VC in mouse VSMCs to investigate the mechanisms underlying empagliflozin’s therapeutic effects. We evaluated biochemical parameters, mean artery pressure (MAP), pulse wave velocity (PWV), transcutaneous glomerular filtration rate (GFR), and histology in an in vivo mouse model with VC induced by an oral high-phosphorus diet following a 5/6 nephrectomy in ApoE−/− mice. Compared to the control group, empagliflozin-treated mice showed significant reductions in blood glucose, MAP, PWV, and calcification, as well as increased calcium and GFR levels. Empagliflozin inhibited osteogenic trans-differentiation by decreasing inflammatory cytokine expression and increasing AMPK, Nrf2, and HO-1 levels. Empagliflozin mitigates high phosphate–induced calcification in mouse VSMCs through the Nrf2/HO-1 anti-inflammatory pathway by activating AMPK. Animal experiments suggested that empagliflozin reduces VC in CKD ApoE−/− mice on a high-phosphate diet.
Keywords:
empagliflozin
; vascular calcification
; AMP-activated protein kinase
; nuclear factor erythroid-2-related factor
; heme oxygenase 1
; chronic kidney disease
1. Introduction
Chronic kidney disease (CKD) affects nearly 10% of the global population, amounting to >800 million individuals [1]. Patients with CKD display a significant risk for cardiovascular (CV) incidents; half of all individuals with stage 4–5 CKD have CV disease, and CV-related fatalities account for approximately 40%–50% of all deaths in patients with advanced CKD and end-stage kidney disease. Over 50% of dialysis patients experience vascular calcification, with a 15-30% yearly rise in coronary calcification, and 40-45% of hemodialysis patients display VC in the aortic region, as identified through plain chest radiography[2]. Vascular calcification (VC) is a powerful indicator of CV risk; intimal calcification is related to atherosclerotic plaque formation, whereas medial calcification is characterized by arteriosclerosis [3]. VC is linked to increased CV morbidity and mortality [3-5]. Pulse wave velocity (PWV) is a marker of arterial stiffness and is frequently used to assess the risk of CV disease[6]. Risk factors that drive the progression of CV calcification, including hyperphosphatemia, hypercalcemia, and inflammation, are typically present in individuals with CKD and end-stage kidney disease [7]. High levels of serum phosphorus, particularly inorganic phosphate, are a crucial risk factor for VC [8]. Proinflammatory cytokines, such as interleukin (IL)-1β and IL-6, can also contribute to VC by causing the transformation of vascular smooth muscle cells (VSMCs) into osteoblast-like cells, which then help form calcium phosphate crystals within blood vessel walls [9].
Empagliflozin, a sodium–glucose cotransporter 2 (SGLT2) inhibitor, is mainly used to treat type 2 diabetes by inhibiting renal glucose reabsorption. It also has various positive effects on CV and renal health [10]. It activates AMP-activated protein kinase (AMPK) in lipopolysaccharide treated cardiomyocytes [11]. Empagliflozin improves obesity-related cardiac function by regulating sestrin2-mediated AMPK-mTOR signaling and enhancing the nuclear factor erythroid-2-related factor (Nrf2)–heme oxygenase 1 (HO-1)-mediated oxidative stress response, suggesting antioxidant and anti-inflammatory capabilities [12]. AMPK is an essential cellular energy sensor and a key regulator of metabolic homeostasis. It is also a crucial anti-inflammatory molecule, as it suppresses proinflammatory signaling pathways and decreases reactive oxygen species production [13, 14]. The precise molecular mechanisms of AMPK activation are still being explored. Further research is required to fully understand these effects and determine the optimal use of empagliflozin for vascular protection.
No therapies have successfully reversed VC in patients with CKD, and only a few have successfully retarded its progression [15, 16]. The commonly used five-sixths nephrectomy (5/6 Nx) serves as a standard experimental model for CKD [17, 18]. In this study, we aimed to investigate the effects of empagliflozin on VC model by the surgical approach of a 5/6 Nx mouse model in ApoE-knockout mice (ApoE−/−) fed a high-phosphate diet and identify the possible molecular mechanism of inorganic phosphate–induced VC in VSMCs.
2. Results
2.1. Empagliflozin Enables the AMPK–Nrf2–HO-1 Pathway to Attenuate Phosphorus-Induced Calcium Deposition in Mouse VSMCs
We first examined whether empagliflozin would decrease inorganic phosphate–induced calcium deposition in the mouse VSMCs line (MOVAS cells). Treatment with different empagliflozin concentrations with or without 2.6 mmol/L inorganic phosphate reduced calcium deposition in red nodules stained with Alizarin Red S stain positive for Alizarin Red S stain (Figure 1A, 1B), and intracellular calcium content (Figure 1C).
After cotreatment of MOVAS cells with high levels of phosphate and empagliflozin, quantitative real-time polymerase chain reaction indicated suppressed runt-related transcription factor 2 (Runx2) mRNA expression (Figure 2A), and Western blotting revealed decreased RUNX2, MSX2, IL-1β, and IL-6 protein levels (Figure 2B–E) and significantly restored phosphorylated AMPK, phosphorylated Nrf2, and HO-1 levels (Figure 2F–H).
2.2. Empagliflozin Reduces Mean Arterial Pressure, PWV in The Mouse Model of VC
Arterial blood pressure was recorded in conscious mice at the end of Week 16. Mean arterial pressure in the VC group (141.2 ± 1.9 mmHg) was significantly higher than that in the control group (103.7 ± 1.8 mmHg), and empagliflozin treatment in the VC group (VCE; 119.1 ± 1.7 mmHg) decreased the mean arterial pressure significantly compared with the VC group (Figure 3A). PWV was significantly higher in the VC group (2.18 ± 0.05 m/s) than in the control group (1.23 ± 0.02 m/s) and in the VCE group (1.68 ± 0.06 m/s, p < 0.001, Figure 3B).
2.3. Empagliflozin Improves Renal Function in The Mouse Model of VC
VC group significant increase serum blood urea nitrogen compared with the control group (Figure 4A). The mouse transcutaneous GFR was measured in the conscious state for 90 min; empagliflozin treatment (443.6 ± 32.2 μg/min/100g bw; VCE group) restored the GFR compared with the VC group (279.9 ± 40.4 μg/min/100g bw; Figure 4B).
2.4. Empagliflozin Improves Serum Level of Calcium, Phosphorus and Decrease Glucose in The Mouse Model of VC
After being fed a high-phosphorus diet for 8 weeks, the blood glucose levels in 5/6 Nx ApoE-/- mice treated with empagliflozin (139.3 ± 5.8 mg/dL) were found to be lower than those in the VC group. Moreover, our findings revealed no significant difference between the control and VC mice (Figure 5A). When compared to the control group, the VC group showed increased serum calcium and phosphorus levels (Figure 5B-C). Empagliflozin treatment (VCE) notably raised serum calcium levels (2.18 ± 0.02 mmol/L; Figure 5B) and reduced phosphorus levels (8.73 ± 0.28 mg/dL; Figure 5C) in comparison to the VC group, suggesting its protective effect against vascular calcification induced by a high-phosphorus diet.
2.5. Empagliflozin Decreases Serum Proinflammatory IL-1β, and IL-6 Cytokines in The Mouse Model of VC
We examined the association between inflammatory cytokines and the progression of vascular calcification (VC) in mice. In comparison to the control group, both VC and VCE groups exhibited significantly higher serum concentrations of IL-1β and IL-6. Empagliflozin effectively lowered the IL-1β increase in the VCE group (0.74 ± 0.19 pg/ml) compared to the VC group (1.43 ± 0.26 pg/ml; Figure 6A). Furthermore, our results revealed that the VCE group had substantially reduced IL-6 levels (17.7 ± 5.0 pg/ml) relative to the VC group (39.3 ± 8.2 pg/ml; p < 0.05; Figure 6B).
2.6. Empagliflozin Decreased Aorta Calcium Deposition in Von Kossa Stain
Positive von Kossa staining for calcium phosphate (black) was observed in the VC group and VCE group (Figure 7B-C), whereas no calcium deposition was noted in the control group (Figure 6A). We further performed von Kossa stain analysis by using Image-Pro Plus 6.0, with five data points collected from each section (eight sections per group). The quantitative expression level of positive von Kossa staining was determined using integrated optical density–area analysis (Figure 7D).
2.7. Empagliflozin increased AMPK expression in The Mouse Model of VC
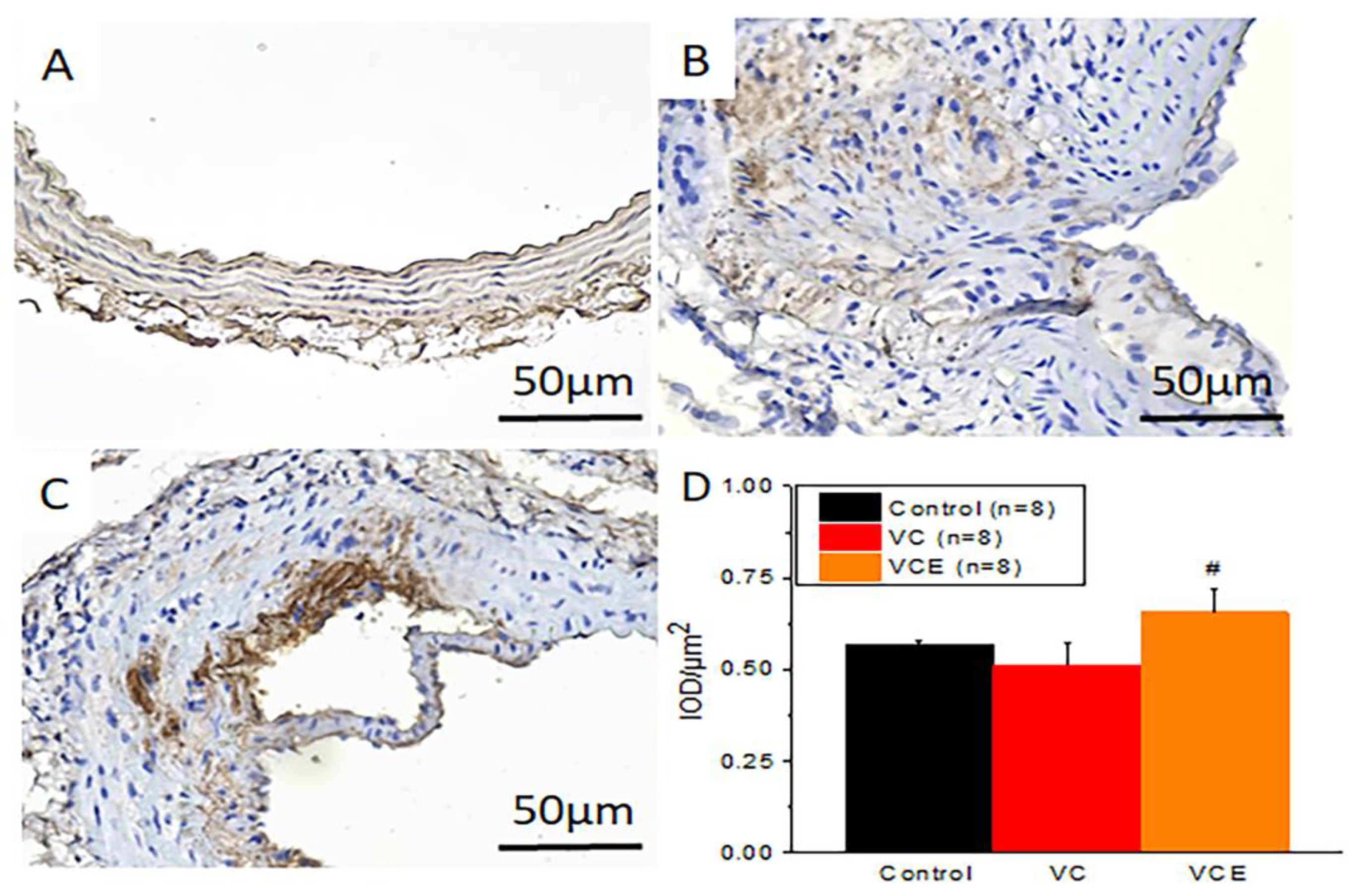
We investigated the potential relationship between empagliflozin treatment and the activation status of AMPK in VC. To do this, immunohistochemistry was conducted on paraffin-embedded aorta samples, and AMPK levels were analyzed. Figure 8 shows representative images of the immunohistochemical staining outcomes. The quantitative expression level of positive AMPK staining was evaluated using integrated optical density-area analysis with Image-Pro Plus 6.0 software (Figure 8D). The findings indicated that AMPK expression was higher in the aorta treated with empagliflozin (VCE group; Figure 8C) compared to the aorta in the VC group (Figure 8B). However, there was no significant difference between the control group and the VCE group.
3. Discussion
This study found that empagliflozin attenuates high phosphate-induced calcification of MOVAS cells by regulating NRF2/HO-1 anti-inflammation pathway through activating AMPK in VSMCs. In addition, animal experiments also confirmed that empagliflozin can decrease VC in 5/6 nephrectomy ApoE -/- mice fed with high-phosphorus diet.
Patients with CKD, diabetes, and hypertension are prone to VC, particularly in the media, where VSMCs transdifferentiate into osteogenic cells or undergo apoptosis [19]. Runx2 is crucial for osteoblast differentiation, and low-to-moderate DNA damage can increase Runx2-mediated osteogenic expression [19, 20]. Our results indicate that empagliflozin decreased calcium deposition by reducing calcium accumulation in VSMCs. AMPK activation downregulated the expression and activity of Runx2, resulting in the inhibition of osteoblastic differentiation of VSMCs [21]. AMPK mediates the antioxidative cascade by activating Nrf2 [22]. Our data indicate that empagliflozin decreased both Runx2 mRNA and protein levels and decreased the expression of proinflammatory IL-6 and IL-1β cytokines by activating AMPK-mediated upregulation of Nrf2-mediated HO-1 expression in VSMCs. In high-fat diet–fed ApoE−/− mice, empagliflozin was reported to activate AMPK, inhibit atherosclerosis progression, and decreased IL-1β and IL-6 levels [23]. In vascular calcification associated with chronic kidney disease (CKD), proinflammatory cytokines such as interleukin-1β (IL-1β) and interleukin-6 (IL-6) may contribute to the transformation of VSMCs into osteoblast-like cells[9]. These inflammatory cytokines can trigger phenotypic alterations in VSMCs, encouraging their conversion into osteoblast-like cells and subsequent mineralization[24]. In our study, we discovered that empagliflozin can enhance AMPK expression (Figure 2F, 6C) and counteract the elevation of serum IL-1β and IL-6 levels triggered by a vascular calcification mouse model (Figure 6). This indicates that empagliflozin may offer anti-inflammatory advantages through AMPK activation, leading to reduced calcium deposition in 5/6 nephrectomy ApoE-/- mice subjected to a high-phosphorus diet-induced VC.
VC can result in arterial stiffening and reduced elasticity, thus increasing blood pressure [25, 26]. It can also lead to endothelial dysfunction, inflammation, oxidative stress, and other pathophysiological changes, which contribute to hypertension [26]. Several studies have demonstrated a significant correlation between PWV and VC, with higher PWV values associated with higher calcification levels [5, 27, 28]. This study indicated that empagliflozin can protect against arterial stiffness, as evidenced by a lower PWV, which can decrease blood pressure.
People with CKD, who typically have a low GFR, are at a higher risk of VC than those with normal kidney function [29-31]. Decreased GFR is associated with an increased risk of VC, likely due to several factors, including alterations in calcium and phosphate metabolism (such as impaired renal excretion), chronic inflammation, oxidative stress [32], and diminished renal blood circulation, thus promoting kidney disease progression [33]. In CKD patients with an estimated GFR below 30 ml/min/1.73 m2, empagliflozin has been shown to decrease the likelihood of kidney disease advancement or cardiovascular-related mortality across a diverse population of individuals at risk for CKD progression [34]. The impaired renal excretion of calcium and phosphate increases their levels in the blood, thus promoting their deposition in the vessel walls. Hyperphosphatemia and hypercalcemia lead to increased calcium and phosphate deposition in blood vessels, triggering RUNX2 expression in vascular smooth muscle cells (VSMCs), which then adopt an osteoblast-like phenotype. Reducing phosphate levels in chronic kidney disease (CKD) patients could potentially enhance cardiovascular outcomes, as hyperphosphatemia contributes to vascular calcification and cardiovascular events [35]. However, the evidence for this strategy remains inconclusive. Our findings suggest that empagliflozin treatment may slow down kidney function decline and reduce serum phosphorus levels in 5/6 nephrectomy ApoE -/- mice with VC induced by a high-phosphorus diet, implying that phosphate reduction in CKD could potentially offer beneficial effects in alleviating VC.
At present, limited research exists examining the connection between empagliflozin and vascular calcification. Medial calcification, which refers to calcification in the smooth muscle layer of the arterial wall, is particularly prevalent among patients with CKD [36]. Our findings from a pathological analysis (Figure 7) reveal a notable decrease in calcium deposits within the tunica media following empagliflozin treatment in CKD ApoE -/- mice subjected to a high-phosphorus diet-induced vascular calcification. As a result, empagliflozin could potentially be used to treat vascular calcification, although further evidence is required to substantiate this hypothesis.
4. Materials and Methods
4.1. Antibodies and Chemicals
Antibodies against the MSX2, AMPKα, Phospho-AMPKα, β-actin, and GAPDH homeobox protein were purchased from Cell Signaling (Cell Signaling Technology, Massachusetts, USA). Antibodies against NRF2, Phospho-NRF2, HO-1, IL-6, and IL-1β were purchased from ABclonal (ABclonal, Massachusetts, USA). Antibodies against Runx2 were purchased from Abcam (Abcam, Cambridge, UK). Methylthymol blue tetrasodium salt was purchased from Thermo Fisher Scientific/ACROS organics (Acros Organicas, Geel, Belgium). Empagliflozin was obtained from AdipoGen Life Sciences (AdipoGen Life Sciences, San Diego, USA). Alizarin Red S was purchased from PanReac Applichem (PanReac Applichem, Monza, Italy). High-glucose Dulbecco modified Eagle’s medium (DMEM), fetal bovine serum, and G418 antibiotics were purchased from Invitrogen (Invitrogen, Carisbad, California, USA). The Bio-Rad protein assay reagent was purchased from (Bio-Rad Laboratories, Hertfordshire, UK). Sodium phosphate monobasic and other reagents were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA). All chemicals were prepared and stored in accordance with the manufacturers’ recommendations.
4.2. MOVAS Cell Culture
The MOVAS cells (ATCC; Manassas, VA, USA; CRL-2797), a mouse VSMCs line, were cultured in high-glucose DMEM supplemented with 10% fetal bovine serum (Life Technologies Inc., Gaithersburg, MD, USA) and 0.2 mg/mL G418 antibiotics (Sigma-Aldrich, St. Louis, MO, USA). Incubation was conducted at 37 ° C in a humidified atmosphere with 5% CO2, and the cells were subcultured every second day in the designed medium.
4.3. Induction of Calcification
VSMCs were cultured in DMEM growth medium and subcultured at 80% confluence. The growth medium was supplemented with (calcification medium) or without (normal medium) 2.6 mM sodium phosphate monobasic for another 10 days to induce calcification. During this time, the cells were subcultured every 2 days. For time course experiments, the first day of culture in the calcification medium was defined as Day 1.
4.4. Alizarin Red S Staining
To determine calcification status, Alizarin Red S staining was performed after 10-day incubation with normal-phosphate or high-phosphate medium and various empagliflozin concentrations. The cells were fixed with 95% ethanol for 30 min at room temperature and then stained with Alizarin Red S (0.1%, pH 4.3) at 37 ° C in a humidified, 5% CO2 atmosphere. The calcified areas were stained red under a light microscope.
4.5. Quantification of Calcium Content
After the indicated incubation period, MOVAS cells were decalcified with 0.6 mol/L HCl at 37 °C overnight, and calcium content in the supernatant was determined colorimetrically by using the methylthymol blue method in accordance with the manufacturer’s protocol. The amount of free calcium was expressed as millimoles per 108 cells.
4.6. Western Blotting
For each of three independent experiments, total protein from MOVAS cells was extracted after treatment with RIPA lysis buffer supplemented with complete protease and a phosphatase inhibitor cocktail. After centrifugation at 12,000 ×g for 30 min, the protein content of the cells was measured using the Bio-Rad protein assay reagent (Bio-Rad Laboratories, Hertfordshire, UK), with BSA as the standard. After protein denaturation, equal amounts of proteins were separated on SDS-PAGE and transferred to PVDF membranes, which were then blocked with 5% nonfat milk. The membranes were incubated overnight at 4 °C with primary antibodies—antibodies against RUNX2, MSX-2, GAPDH, and β-actin, AMPKα, phospho-AMPKα, NRF2, phospho-NRF2, HO-1, IL-6, IL-1β—followed by the addition of appropriate horseradish peroxidase–conjugated secondary antibodies, and detected using ECL (GE). The chemiluminescent signal was detected using a UVP BioSpectrum 810 imaging system (Thermo Fisher Scientific, Waltham, MA, USA). Bands were quantified with ImageJ software.
4.7. Animals
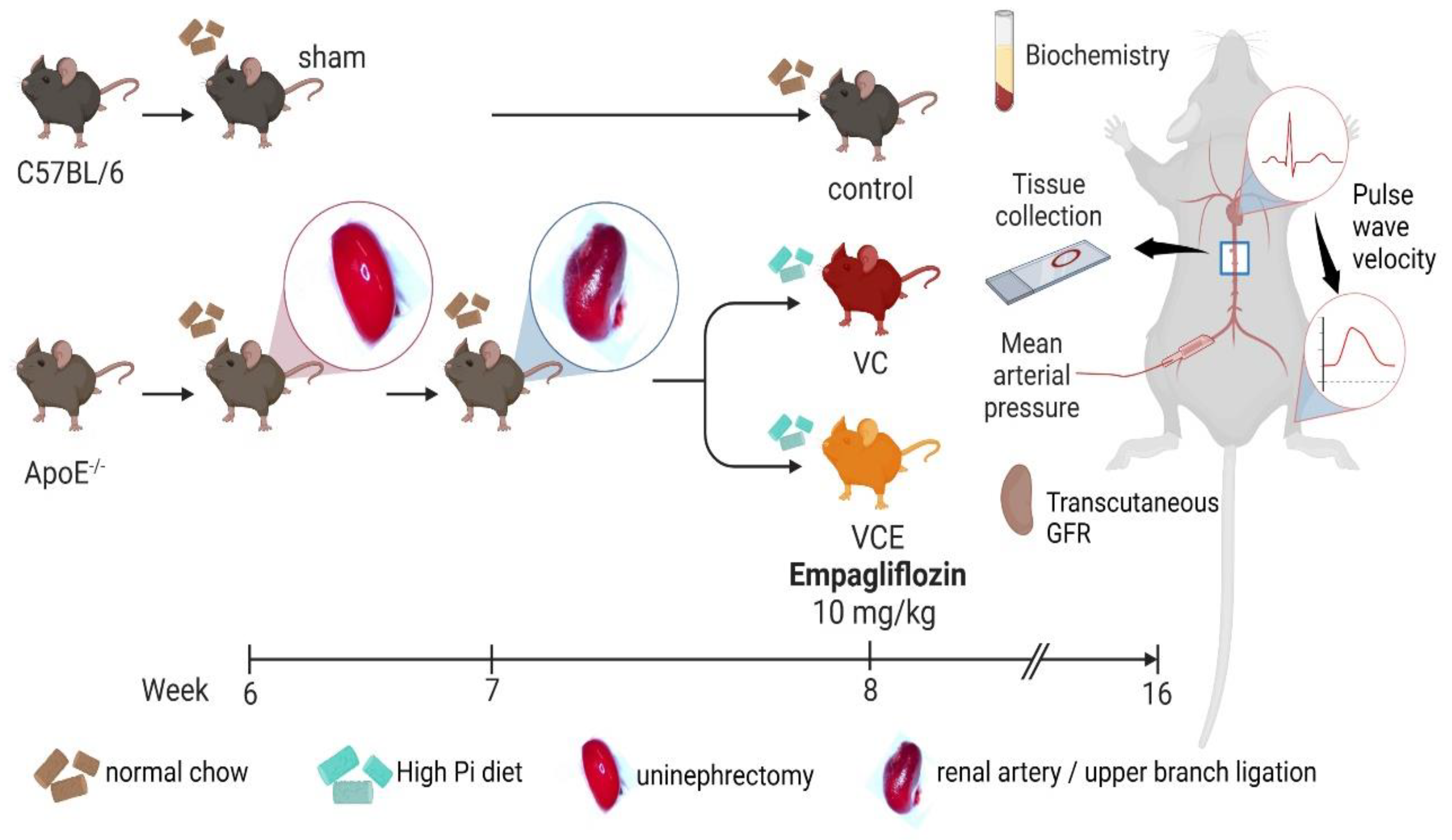
Eight male C57BL6 mice (20–25 g) were purchased from BioLASCO (Taipei, Taiwan), and 16 male Apoetm1Unc/J mice (20–25 g) were obtained from Jackson Laboratory (Bar Harbor, ME, USA). The 24 mice were divided into three equal groups (eight per group): C57BL/6 of sham fed normal chow (the control group), ApoE−/− of 5/6 nephrectomy (5/6 Nx) fed high-phosphorus diet (the VC group), ApoE−/− of 5/6 nephrectomy fed both high-phosphorus diet and empagliflozin (the VCE group; Figure 6). The VCE group received oral empagliflozin 10 mg/kg for 8 weeks after 5/6 nephrectomy. All mice were housed at the Laboratory Animal Center of Tzu Chi University (Hualien, Taiwan). The experimental protocols were approved by the Institutional Animal Care and Use Committee of Hualien Tzu Chi Hospital.
Figure 9.
Schematic of animal experiment. Created with BioRender.com.
4.8. Animal Model of VC
The VC mouse model was established by subjecting them to 5/6 Nx with a high-phosphorus diet as described elsewhere [18]. We modified 5/6 Nx into a two-stage procedure [17, 18]. The mice were anesthetized with isoflurane inhalation (Forane, Baxter, USA) by using a vaporizer (Matrx VIP 3000, Midmark, USA). Under anesthesia, the right kidney was removed in the first stage, and the upper branch of the left renal artery was ligated using a 6-0 catgut absorbable suture 7 days later. Following 5/6 Nx, the VC mice were fed a high-phosphorus (1.5% total phosphorus) diet (Product #D09051102, Research Diets, NJ, USA) for 8 weeks.
4.9. Mean Arterial Pressure
The mice were anesthetized using isoflurane as described earlier. During anesthesia, the femoral artery was cannulated and connected to a pressure transducer to record mean arterial pressure on a polygraph recorder (e-corder 410, eDAQ, Australia). The operation was completed in 15 min, leaving a small section wound (<0.5 cm2). The mice soon awakened, and arterial blood pressure was recorded in the conscious mice [37].
4.10. Pulse Wave Velocity
PWV was acquired from the cardiovascular pulse, distance between locations d, and transit time Δt for the pulse to travel distance d. After isoflurane anesthesia, the mice were placed supine, and distance d between the supraclavicular notch and the ankle of the left hindlimb was measured. Three acupuncture needle electrodes were inserted subcutaneously into the right and left forelimbs and the left hindlimb. The other ends of the electrodes were connected to an amplifier cable for electrocardiography (ECG) of lead Ⅱ (Powerlab/8sp, BIO Amp, ADinstruments, New Zealand). The pulse oximeter (MouseOx, Starr Life Sciences, USA) was placed on the left ankle. The transit time (Δt) was acquired using the pulse wave between the initial peak of the ECG R-wave and arrival peak of pulse oximeter wave (LabChart software v7, Adinstruments, New Zealand). PWV was calculated as follows: PWV = d (meters)/Δt (seconds).
4.11. Transcutaneous GFR
Transdermal GFR in mice was measured using the excretion kinetics of fluorescein-isothiocyanate conjugated sinistrin (FITC-sinistrin) [37]. After hair was removed and the transdermal GFR Monitor (MediBeacon, Louis, MO, USA) was attached, the mice were anesthetized with isoflurane as described earlier and administered FITC-sinistrin (Mannheim Pharma & Diagnostics, Mannheim, Germany) 0.15 mg per gram body weight (g bw) through the retro-orbital venous sinus. Mice recovered from isoflurane anesthesia, and the measurement period was 1.5 h. The GFR was calculated from the measured half-life of FITC-sinistrin clearance using software (MPDStudio, MediBeacon, Louis, MO, USA).
4.12. Tissue Collection, Biochemical Analysis, and Serum Cytokines Analysis
After the animals were sacrificed, blood samples were collected and centrifuged at 10,000 g for 10 min at 4°C to separate the serum. Serum biochemical measurements of glucose, blood urea nitrogen, calcium, and phosphorus were recorded using a biochemistry analyzer (Spotchem SP-4430, Arkray, Minneapolis, MN, USA) [37, 38]. The remaining serum was preserved at −80°C prior to evaluation of IL-1β and IL-6 using an enzyme-linked immunosorbent assay kit and commercial assay kits (R&D Systems, Minneapolis, MN, USA) [38]. The tissue specimens were fixed in 4% buffered formaldehyde and then subjected to von Kossa staining.
4.13. Von Kossa staining
Sections of the aorta were deparaffinized, rehydrated, and incubated in silver nitrate solution (5%) for 60 min with 100-watt incandescent. The sections were incubated in sodium thiosulfate (5%) solution (5%) for 2 min and nuclear fast red solution for 5 min and rinsed in distilled water before and after incubation. The slides were dehydrated, mounted, and observed under light microscopy. Von Kossa stain was evaluated on the basis of the average optical density of positive reactions by using Image-Pro Plus 6.0 software [39].
4.14. Immunohistochamistry staining
Aorta sections, 3 μm thick, were deparaffinized, rehydrated, and underwent microwave-assisted antigen retrieval using Trilogy (Cell Marque, CA, USA). Following this, the sections were treated to block endogenous peroxidase activity with a 3% hydrogen peroxide solution for 5 minutes and a 10% bovine serum albumin-containing phosphate-buffered saline for 30 minutes at room temperature. The primary antibodies used were AMPK (1:150 dilution; Cell Signaling Technology, Massachusetts, USA) for 30 minutes. After washing three times, biotinylated mouse anti-mouse secondary antibodies were applied to the sections for 30 minutes at room temperature. The reaction was visualized using 3,3′-diaminobenzidine, counterstained with Mayer's hematoxylin, and dehydrated with ethanol before being coverslipped for assessment. Light microscopy was used to observe the slides, and the average optical density of positive reactions was determined for immunohistochemical analysis using Image-Pro Plus 6.0 software [37-39].
4.15. Statistical analysis
Data are presented as the mean ± standard error of the mean. For multiple comparisons, significance was assessed using a one-way analysis of variance with Bonferroni’s post hoc test or the Cochran-Armitage test for trend. Statistical Package for the Social Sciences (SPSS) (version 19.0; SPSS Inc., Chicago, IL, USA) was used for statistical analysis. Groups were compared using an unpaired t test. p < 0.05 was considered statistically significant.
5. Conclusions
VC is a complex process driven by numerous factors that lead to calcium phosphate accumulation in arterial walls, and the consequent arteriosclerosis increases blood pressure. Our results indicate that empagliflozin reduces high phosphate–induced calcification in MOVAS cells by activating the AMPK–Nrf2–HO-1 pathway and decreases VC in the aortas of ApoE-/- mice subjected to 5/6 nephrectomy and fed a high-phosphorus diet.
Author Contributions
Conceptualization, C.-J. L. and B.-G.H.; Data curation, C.-W. L., C.-J. L. and Y.-J. H.; Formal analysis, C.-W. L., C.-J. L. and Y.-J. H.; Funding acquisition, B.-G.H.; Methodology, C.-W. L., C.-J. L. and Y.-J. H.; Validation, B.-G.H.; Writing – original draft, C.-W. L. and C.-J. L; Writing – review & editing, B.-G.H.; supervision, B.-G.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the Ministry of Science and Technology, Taiwan (MOST 109-2314-B-303-020).
Institutional Review Board Statement
The experimental protocols were approved by the Institutional Animal Care and Use Committee of Hualien Tzu Chi Hospital (approval number 108-48).
Informed Consent Statement
Not applicable.
Acknowledgments
This manuscript was edited by Wallace Academic Editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kovesdy, C. P. Epidemiology of chronic kidney disease: an update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef]
- Kim, J. S.; Hwang, H. S. Vascular calcification in chronic kidney disease: distinct features of pathogenesis and clinical implication. Korean Circ. J. 2021, 51, 961–982. [Google Scholar] [CrossRef]
- London, G. M.; Guerin, A. P.; Marchais, S. J.; Metivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef]
- Lee, C. J.; Hsieh, Y. J.; Lin, Y. L.; Wang, C. H.; Hsu, B. G.; Tsai, J. P. Correlation between serum 25-hydroxyvitamin D level and peripheral arterial stiffness in chronic kidney disease stage 3-5 patients. Nutrients 2022, 14, 2429. [Google Scholar] [CrossRef]
- Temmar, M.; Liabeuf, S.; Renard, C.; Czernichow, S.; Esper, N. E.; Shahapuni, I.; Presne, C.; Makdassi, R.; Andrejak, M.; Tribouilloy, C.; Galan, P.; Safar, M. E.; Choukroun, G.; Massy, Z. Pulse wave velocity and vascular calcification at different stages of chronic kidney disease. J. Hypertens. 2010, 28, 163–169. [Google Scholar] [CrossRef]
- Miyoshi, T.; Ito, H. Arterial stiffness in health and disease: The role of cardio-ankle vascular index. J. Cardiol. 2021, 78, 493–501. [Google Scholar] [CrossRef]
- Nemcsik, J.; Kiss, I.; Tisler, A. Arterial stiffness, vascular calcification and bone metabolism in chronic kidney disease. World J. Nephrol. 2012, 1, 25–34. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Vascular calcification: Key roles of phosphate and pyrophosphate. Int. J. Mol. Sci. 2021, 22, 13536. [Google Scholar] [CrossRef]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R. V.; Mokas, S.; Lariviere, R.; Richard, D. E. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am. J. Hypertens. 2015, 28, 746–755. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J. M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O. E.; Woerle, H. J.; Broedl, U. C.; Inzucchi, S. E.; Investigators, E.-R. O. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Koyani, C. N.; Plastira, I.; Sourij, H.; Hallstrom, S.; Schmidt, A.; Rainer, P. P.; Bugger, H.; Frank, S.; Malle, E.; von Lewinski, D. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol. Res. 2020, 158, 104870. [Google Scholar] [CrossRef]
- Sun, X.; Han, F.; Lu, Q.; Li, X.; Ren, D.; Zhang, J.; Han, Y.; Xiang, Y. K.; Li, J. , Empagliflozin ameliorates obesity-related cardiac dysfunction by regulating Sestrin2-mediated AMPK-mTOR signaling and redox homeostasis in high-fat diet-induced obese mice. Diabetes 2020, 69, 1292–1305. [Google Scholar] [CrossRef]
- Tong, X.; Ganta, R. R.; Liu, Z. , AMP-activated protein kinase (AMPK) regulates autophagy, inflammation and immunity and contributes to osteoclast differentiation and functionabs. Biol. Cell 2020, 112, 251–264. [Google Scholar] [CrossRef]
- Lei, L.; Chai, Y.; Lin, H.; Chen, C.; Zhao, M.; Xiong, W.; Zhuang, J.; Fan, X. Dihydroquercetin activates AMPK/Nrf2/HO-1 signaling in macrophages and attenuates inflammation in LPS-induced endotoxemic mice. Front. Pharmacol. 2020, 11, 662. [Google Scholar] [CrossRef]
- Ruderman, I.; Holt, S. G.; Hewitson, T. D.; Smith, E. R.; Toussaint, N. D. Current and potential therapeutic strategies for the management of vascular calcification in patients with chronic kidney disease including those on dialysis. Semin. Dial. 2018, 31, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Rementer, C.; Giachelli, C. M. Vascular calcification: an update on mechanisms and challenges in treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, J.; Wang, L.; Cha, B. J.; Jiang, S.; Liu, R. A new low-nephron CKD model with hypertension, progressive decline of renal function, and enhanced inflammation in C57BL/6 mice. Am. J. Physiol. Renal Physiol. 2018, 314, F1008–F1019. [Google Scholar] [CrossRef]
- Shobeiri, N.; Adams, M. A.; Holden, R. M. Vascular calcification in animal models of CKD: A review. Am. J. Nephrol. 2010, 31, 471–481. [Google Scholar] [CrossRef]
- Bostrom, K. I. DNA damage response, Runx2 (runt-related transcription factor 2), and vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1358–1359. [Google Scholar] [CrossRef]
- Liu, T. M.; Lee, E. H. Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng. Part B Rev. 2013, 19, 254–263. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, T.; Min, X.; Yuan, Z.; Cai, Z. AMPK: Potential therapeutic target for vascular calcification. Front. Cardiovasc. Med. 2021, 8, 670222. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhao, T.; Xiao, H. The implication of oxidative stress and AMPK-Nrf2 antioxidative signaling in pneumonia pathogenesis. Front. Endocrinol. (Lausanne) 2020, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Xu, H.; Wu, F.; Tu, Q.; Dong, X.; Xie, H.; Cao, Z. Empagliflozin inhibits macrophage inflammation through AMPK signaling pathway and plays an anti-atherosclerosis role. Int. J. Cardiol. 2022, 367, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Shao, J. S.; Cheng, S. L.; Sadhu, J.; Towler, D. A. Inflammation and the osteogenic regulation of vascular calcification: A review and perspective. Hypertension 2010, 55, 579–592. [Google Scholar] [CrossRef]
- Xiong, Y.; Yu, Y.; Huang, K.; Liao, R.; Wang, L.; Zhang, Z.; Li, J.; Qin, Z.; Sun, S.; Li, Y.; Su, B. Vascular calcification exacerbates abnormal blood pressure variability in chronic kidney disease: a "two-step" study in rats. Cardiorenal Med. 2023, in press. [Google Scholar] [CrossRef]
- Tesauro, M.; Mauriello, A.; Rovella, V.; Annicchiarico-Petruzzelli, M.; Cardillo, C.; Melino, G.; Di Daniele, N. Arterial ageing: from endothelial dysfunction to vascular calcification. J. Intern. Med. 2017, 281, 471–482. [Google Scholar] [CrossRef]
- Wen, W.; Portales-Castillo, I.; Seethapathy, R.; Krinsky, S.; Kroshinsky, D.; Kalim, S.; Goverman, J.; Nazarian, R. M.; Chitalia, V.; Malhotra, R.; Kramann, R.; Malhotra, C. K.; Nigwekar, S. U. Intravenous sodium thiosulphate for vascular calcification of hemodialysis patients-a systematic review and meta-analysis. Nephrol. Dial. Transplant. 2023, 38, 733–745. [Google Scholar] [CrossRef]
- Herrmann, J.; Gummi, M. R.; Xia, M.; van der Giet, M.; Tolle, M.; Schuchardt, M. Vascular calcification in rodent models-keeping track with an extented method assortment. Biology (Basel) 2021, 10, 459. [Google Scholar] [CrossRef]
- Budoff, M. J.; Rader, D. J.; Reilly, M. P.; Mohler, E. R., 3rd; Lash, J.; Yang, W.; Rosen, L.; Glenn, M.; Teal, V.; Feldman, H. I.; Investigators, C. S. Relationship of estimated GFR and coronary artery calcification in the CRIC (Chronic Renal Insufficiency Cohort) Study. Am. J. Kidney Dis. 2011, 58, 519–526. [Google Scholar] [CrossRef]
- Palit, S.; Kendrick, J. Vascular calcification in chronic kidney disease: role of disordered mineral metabolism. Curr. Pharm. Des. 2014, 20, 5829–5833. [Google Scholar] [CrossRef]
- Shi, J.; Yang, Y.; Wang, Y. N.; Li, Q.; Xing, X.; Cheng, A. Y.; Zhan, X. N.; Li, J.; Xu, G.; He, F. Oxidative phosphorylation promotes vascular calcification in chronic kidney disease. Cell Death Dis. 2022, 13, 229. [Google Scholar] [CrossRef]
- Voelkl, J.; Cejka, D.; Alesutan, I. An overview of the mechanisms in vascular calcification during chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2019, 28, 289–296. [Google Scholar] [CrossRef]
- Levey, A. S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- The, E.-K. C. G.; Herrington, W. G.; Staplin, N.; Wanner, C.; Green, J. B.; Hauske, S. J.; Emberson, J. R.; Preiss, D.; Judge, P.; Mayne, K. J.; Ng, S. Y. A.; et al. Empagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar]
- Lioufas, N.; Toussaint, N. D.; Pedagogos, E.; Elder, G.; Badve, S. V.; Pascoe, E.; Valks, A.; Hawley, C.; Committee, I.-C. W. , Can we IMPROVE cardiovascular outcomes through phosphate lowering in CKD? Rationale and protocol for the IMpact of Phosphate Reduction On Vascular End-points in Chronic Kidney Disease (IMPROVE-CKD) study. BMJ Open 2019, 9, e024382. [Google Scholar] [CrossRef] [PubMed]
- Disthabanchong, S. Vascular calcification in chronic kidney disease: Pathogenesis and clinical implication. World J. Nephrol. 2012, 1, 43–53. [Google Scholar] [CrossRef]
- Chi, P. J.; Lee, C. J.; Hsieh, Y. J.; Lu, C. W.; Hsu, B. G. Dapagliflozin ameliorates lipopolysaccharide related acute kidney injury in mice with streptozotocin-induced diabetes mellitus. Int. J. Med. Sci. 2022, 19, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Wu, T. J.; Hsieh, Y. J.; Lu, C. W.; Lee, C. J.; Hsu, B. G. Linagliptin protects against endotoxin-induced acute kidney injury in rats by decreasing inflammatory cytokines and reactive oxygen species. Int. J. Mol. Sci. 2021, 22, 11190. [Google Scholar] [CrossRef]
- Lin, T. C.; Lu, C. W.; Chang, K. F.; Lee, C. J. Juniperus communis extract ameliorates lipopolysaccharide-induced acute kidney injury through the adenosine monophosphate-activated protein kinase pathway. Food Sci. Nutr. 2022, 10, 3405–3414. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Inhibition of inorganic phosphate (Pi)–induced calcification by empagliflozin. (A) Alizarin Red S staining in MOVAS cells. (B) Quantification of Alizarin Red S staining positive in MOVAS cells by using Image-Pro Plus 6.0 software. (C) Intracellular calcium content was determined colorimetrically by using the methylthymol blue method and expressed as millimoles per 108 cells. *p for the high-Pi group compared with the control group. #p for the empagliflozin 1.25 μM group compared with high-Pi group.
Figure 1.
Inhibition of inorganic phosphate (Pi)–induced calcification by empagliflozin. (A) Alizarin Red S staining in MOVAS cells. (B) Quantification of Alizarin Red S staining positive in MOVAS cells by using Image-Pro Plus 6.0 software. (C) Intracellular calcium content was determined colorimetrically by using the methylthymol blue method and expressed as millimoles per 108 cells. *p for the high-Pi group compared with the control group. #p for the empagliflozin 1.25 μM group compared with high-Pi group.
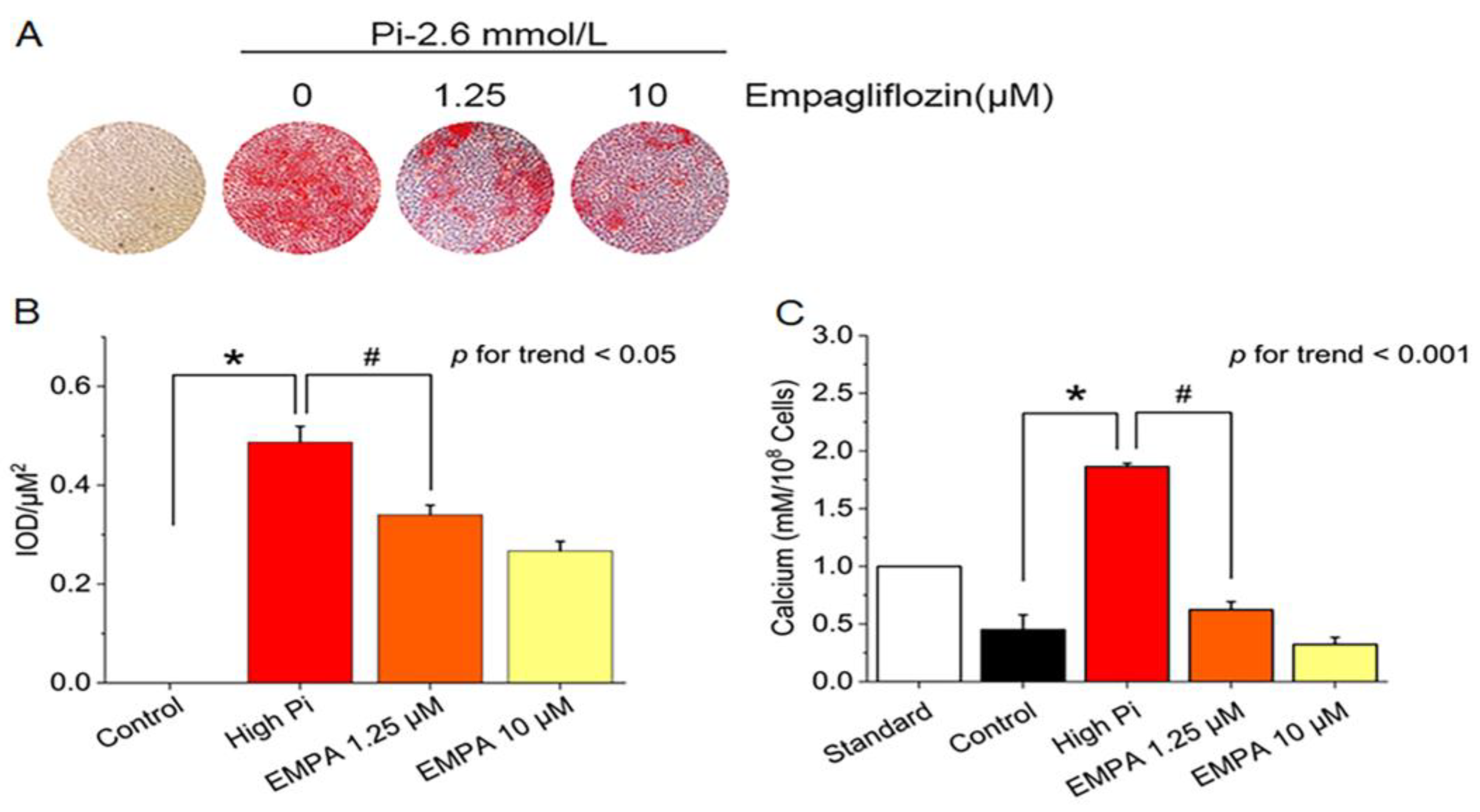
Figure 2.
Empagliflozin attenuated high phosphate–induced VC in MOVAS cells through chondro-osteogenic phenotype switching. (A) Empagliflozin reduced mRNA abundance of RUNX2 in MOVAS cells treated with 2.6 mM phosphate and/or 1.25–10 μM empagliflozin for 10 days. Immunoblot images and protein expression analysis of (B) Runx2, (C) MSX2, (D) IL-1β, (E) IL-6, (F) p-AMPK, (G) p-Nrf2, and (H) HO-1 were represented by empagliflozin in MOVAS cells. Results are presented as mean ± standard error of the mean; each n = 3, and experiments were repeated at least three times independently. *p for the high-Pi group compared with the control group. #p for the empagliflozin 1.25 μM group compared with high-Pi group.
Figure 2.
Empagliflozin attenuated high phosphate–induced VC in MOVAS cells through chondro-osteogenic phenotype switching. (A) Empagliflozin reduced mRNA abundance of RUNX2 in MOVAS cells treated with 2.6 mM phosphate and/or 1.25–10 μM empagliflozin for 10 days. Immunoblot images and protein expression analysis of (B) Runx2, (C) MSX2, (D) IL-1β, (E) IL-6, (F) p-AMPK, (G) p-Nrf2, and (H) HO-1 were represented by empagliflozin in MOVAS cells. Results are presented as mean ± standard error of the mean; each n = 3, and experiments were repeated at least three times independently. *p for the high-Pi group compared with the control group. #p for the empagliflozin 1.25 μM group compared with high-Pi group.
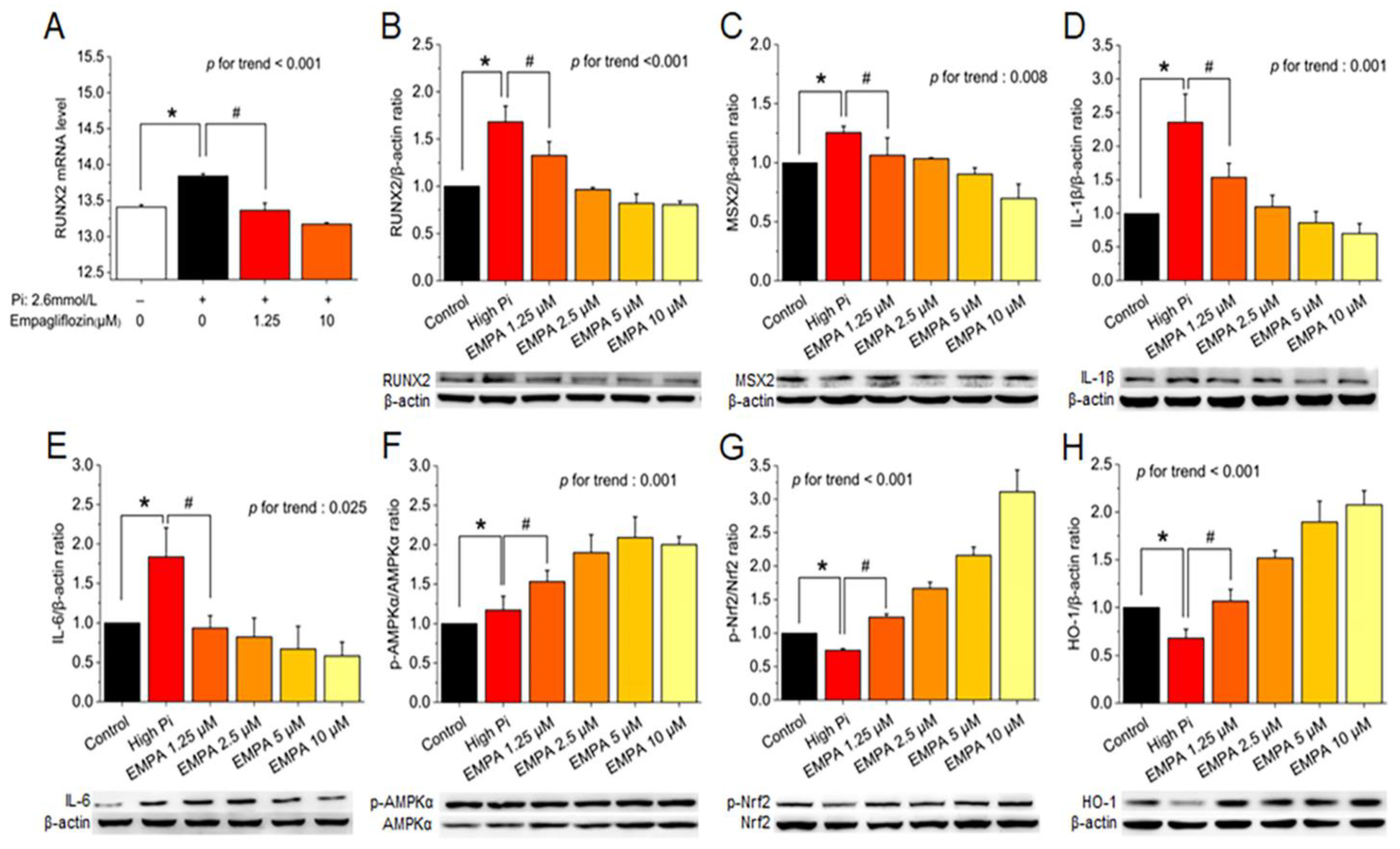
Figure 3.
Physiological characteristics of (A) mean arterial pressure, (B) PWV, and (C) transcutaneous GFR. *p for the VC group compared with the control group. #p for the VCE group compared with VC group.
Figure 3.
Physiological characteristics of (A) mean arterial pressure, (B) PWV, and (C) transcutaneous GFR. *p for the VC group compared with the control group. #p for the VCE group compared with VC group.

Figure 4.
Biomarker of renal function. (A) Serum level of blood urea nitrogen, (B) transcutaneous GFR. *p for the VC group compared with the control group. #p for the VCE group compared with VC group.
Figure 4.
Biomarker of renal function. (A) Serum level of blood urea nitrogen, (B) transcutaneous GFR. *p for the VC group compared with the control group. #p for the VCE group compared with VC group.
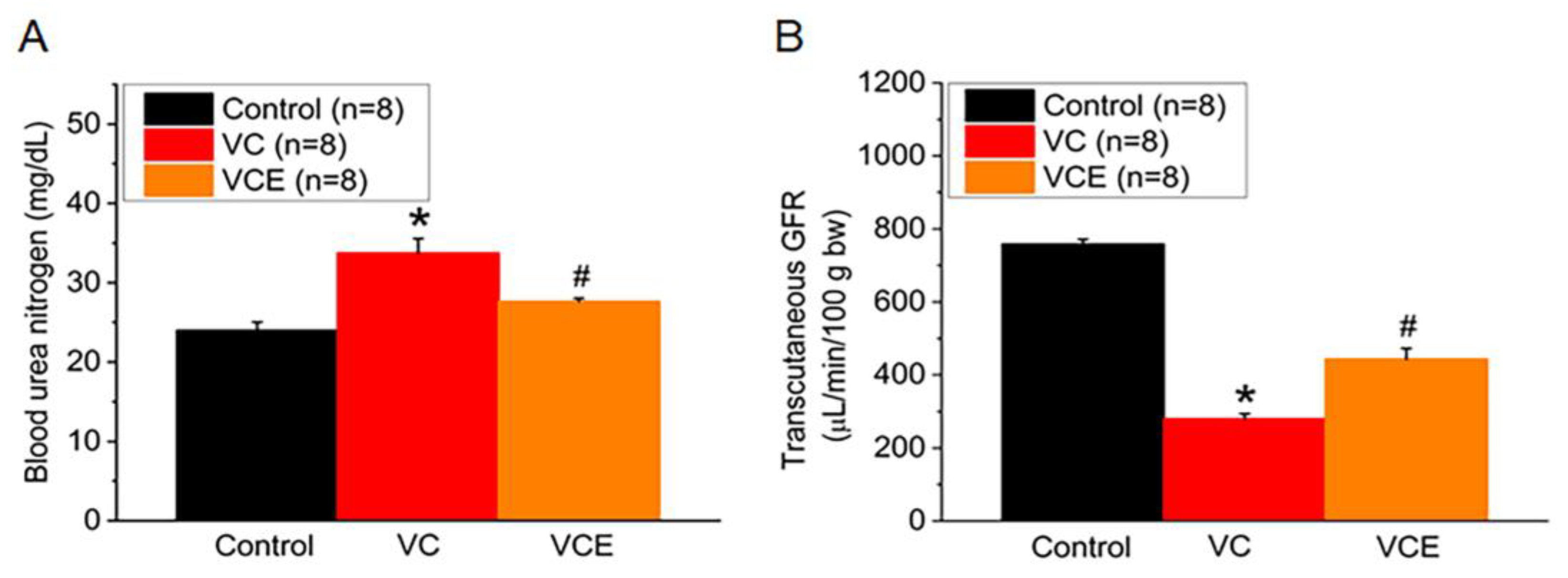
Figure 5.
Serum levels of (A) glucose, (B) calcium, (C) phosphorus. *p for the VC group compared with the control group. # p for the VCE group compared with VC group.
Figure 5.
Serum levels of (A) glucose, (B) calcium, (C) phosphorus. *p for the VC group compared with the control group. # p for the VCE group compared with VC group.
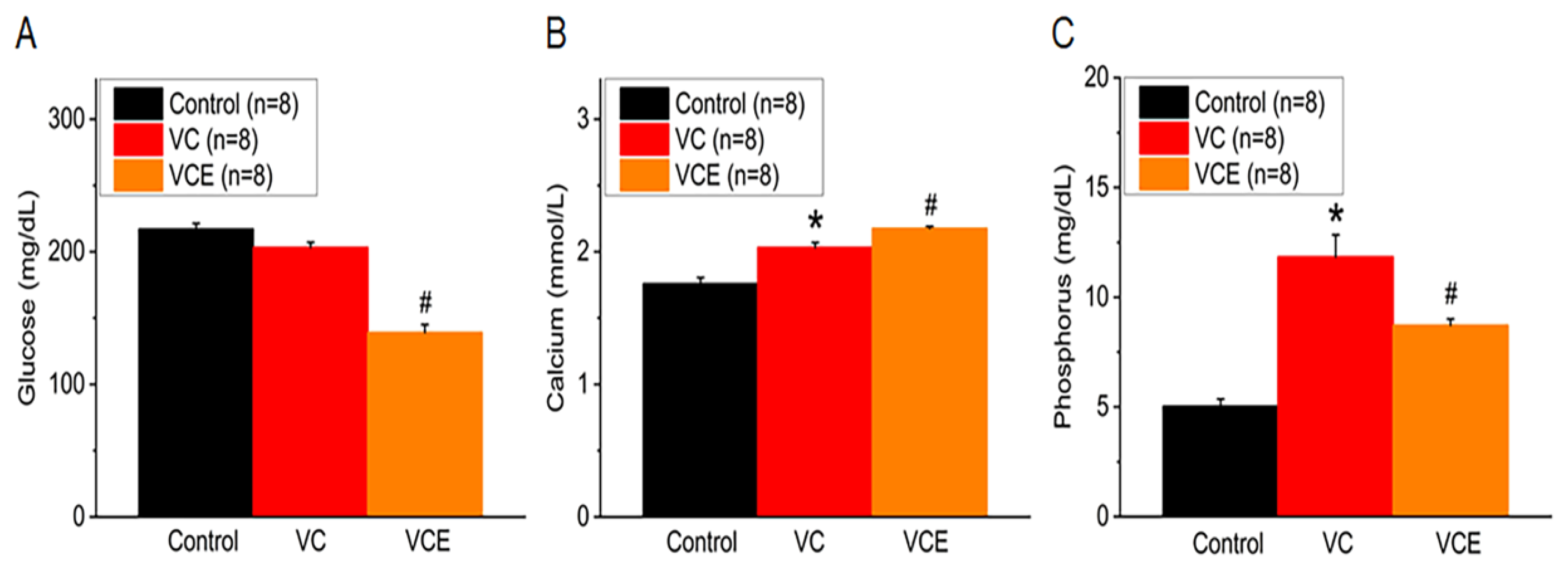
Figure 6.
The quantitative measurement of (A) IL-1β and (B) IL-6 levels in the serum. *p for the VC group compared with the control group. # p for the VCE group compared with VC group.
Figure 6.
The quantitative measurement of (A) IL-1β and (B) IL-6 levels in the serum. *p for the VC group compared with the control group. # p for the VCE group compared with VC group.
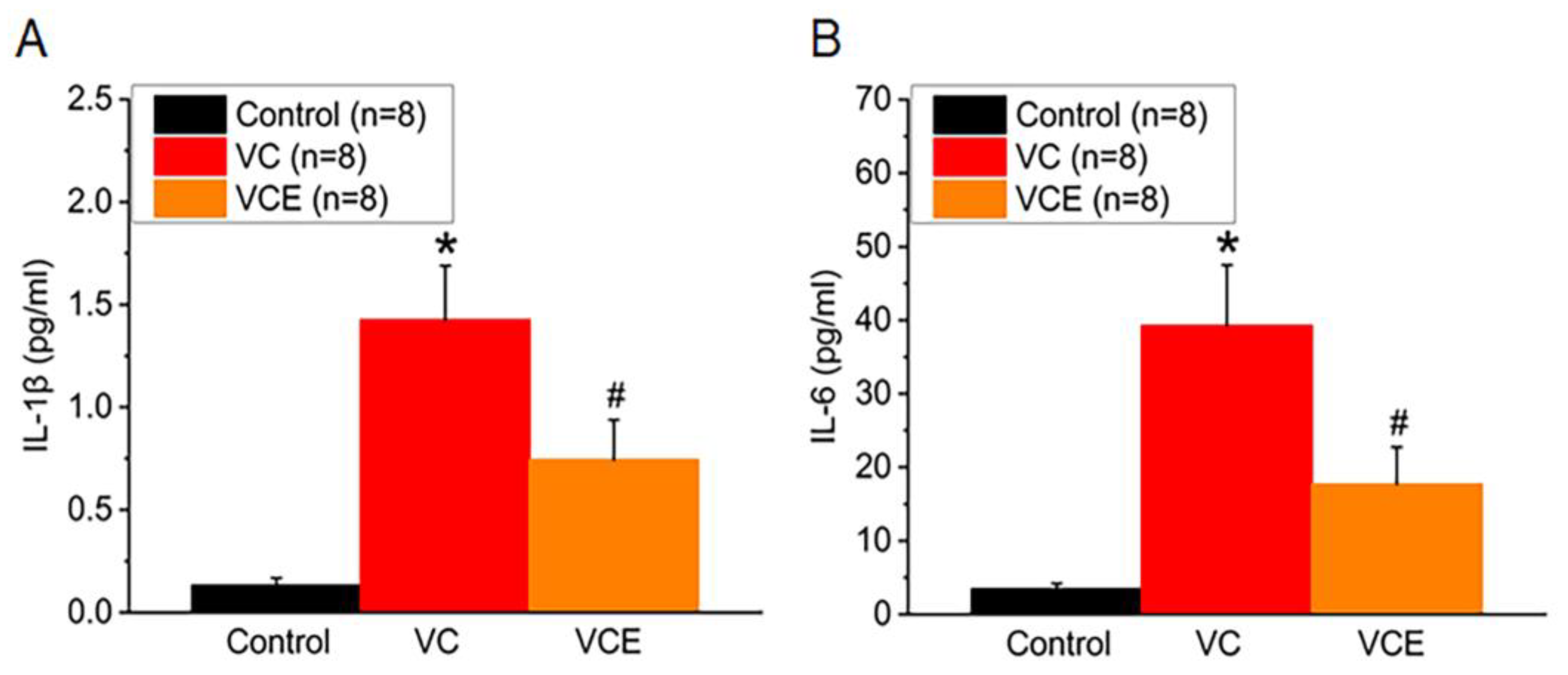
Figure 7.
Calcium deposition represented by von Kossa staining in aorta of the (A) control, (B) VC, and (C) VCE groups. (D) Quantification of von Kossa staining positive in aorta.
Figure 7.
Calcium deposition represented by von Kossa staining in aorta of the (A) control, (B) VC, and (C) VCE groups. (D) Quantification of von Kossa staining positive in aorta.
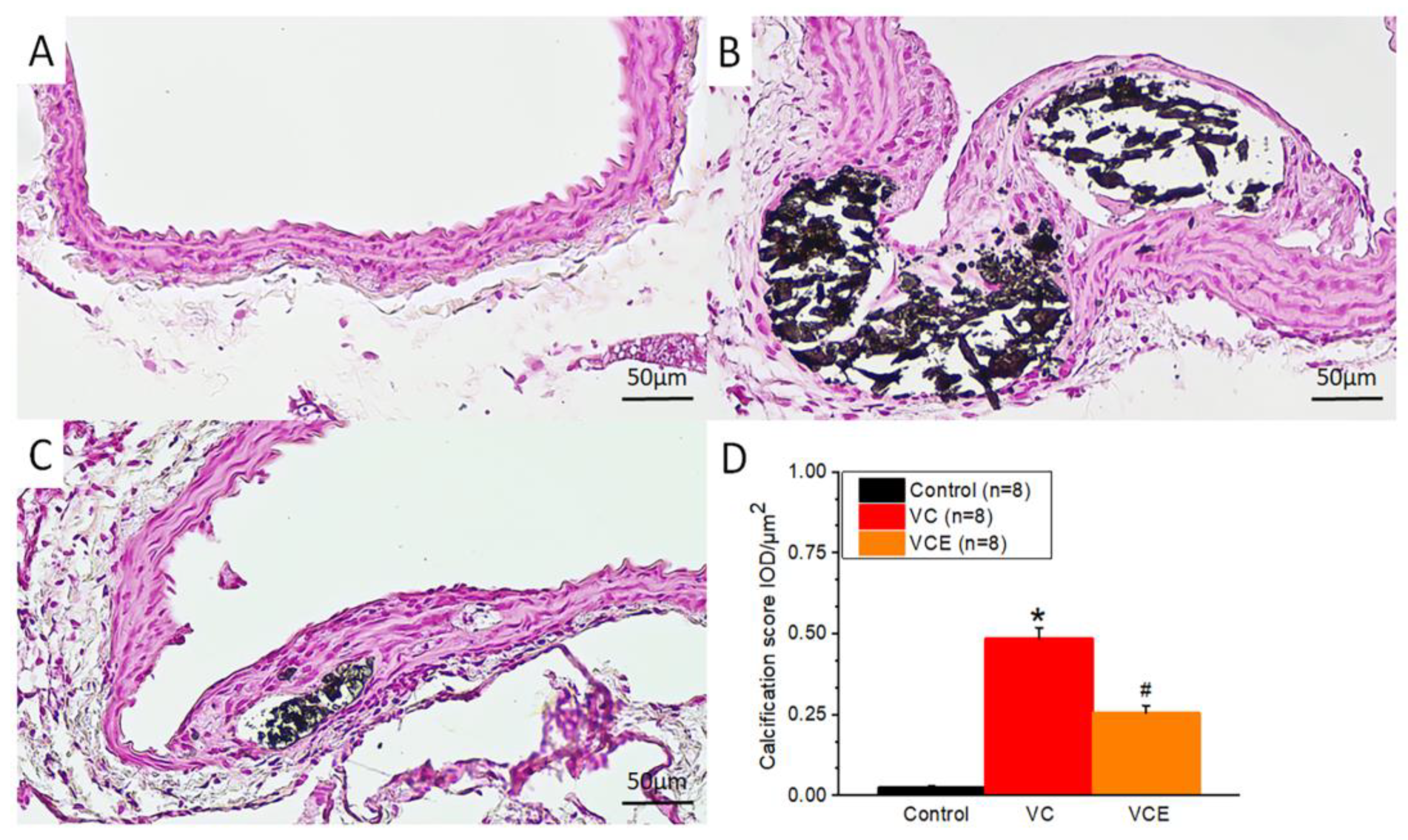
Figure 8.
Immunohistochamistry staining of AMPK in aorta of the (A) control, (B) VC, and (C) VCE groups. (D) Quantification of AMPK positive staining in aorta.
Figure 8.
Immunohistochamistry staining of AMPK in aorta of the (A) control, (B) VC, and (C) VCE groups. (D) Quantification of AMPK positive staining in aorta.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



