
Submitted:
27 April 2023
Posted:
27 April 2023
You are already at the latest version
Abstract
Different dopamine receptor (DR) subtypes are involved in pathophysiological conditions such as Parkinson´s Disease (PD), schizophrenia and depression. While many DR-targeting drugs have been approved by the FDA, only a very small number is truly selective for one of the DR subtypes. Additionally, most of them show promiscuous activity at related G-protein coupled receptors, thus suffering from diverse side-effect profiles. Multiple studies have shown that combined in silico / in vitro approaches are a valuable contribution to drug discovery processes. They can also be applied to divulge the mechanisms behind ligand selectivity. In this study, novel DR ligands were investigated in vitro to assess binding affinities at different DR subtypes. Thus, nine D2R/D3R-selective ligands (micro- to nanomolar binding affinities, D3R-selective profile) were successfully identified. The most promising ligand exerted nanomolar D3R activity (KI = 2.3 nM) with 263.7-fold D2R/D3R selectivity. Subsequently, ligand selectivity was rationalized in silico based on ligand interaction with a secondary binding pocket, supporting selectivity data determined in vitro. The developed workflow and identified ligands could aid in the further understanding of structural motifs responsible for DR subtype selectivity, thus, benefitting drug development in D2R/D3R-associated pathologies like PD.
Keywords:
dopamine receptor
; subtype selectivity
; GPCR
; in silico
; molecular docking
; secondary binding pocket
; in vitro
; HTRF
1. Introduction
G-protein coupled receptors (GPCRs) are one of the most prominent protein families targeted in drug research. Currently, they are represented by 475 approved (by the U.S. Food and Drug Administration, FDA) drugs acting on 108 different GPCRs [1]. Sixty-five of them target an essential sub-group of GPCRs, the dopamine receptor (DR) family, consisting of the subtypes 1, 2, 3, 4 and 5 (D1R, D2R, D3R, D4R and D5R, respectively) [2]. The DR family is divided into D1like- (D1R and D5R) and D2like receptors (D2R, D3R and D4R) and plays a crucial role in physiological processes such a motoric function, cognition, sleep and memory [3]. However, it is also involved in many devastating diseases of the central nervous system (CNS) like Parkinson´s Disease (PD), schizophrenia and bipolar disorders. DR-targeting drugs act in different ways acting as; e g. agonists in PD, activating the receptor, antagonists in schizophrenia blocking the receptor or partial agonists used in treating bipolar disorders or addiction [3-5].
While all of the listed diseases are connected to the dopaminergic system, they are also characterized by a distinct dysfunctionality of different dopaminergic projection pathways [6]. In PD, the degeneration of dopaminergic neurons in the substantia nigra leads to reduced dopamine levels, thus, reduced activation of the D2R [4]. In contrast, schizophrenia is defined by hyperproductive, presynaptical dopaminergic neurons in the mesolimbic region, thus overactivating the D2R. At the same time, dopaminergic neurons in the prefrontal cortex are hypofunctional, resulting in insufficient activation of the D1R due to a lack of dopamine [3]. On the one hand, aberrant signalling involving the D3R has been implicated in diseases like PD, restless leg syndrome and depression, where agonists are used to treat motor dysbalances. On the other hand, D3R antagonists have been shown to be useful as antipsychotics [7, 8].
For most of those conditions, DR-targeting drugs have been approved by the FDA, successfully ameliorating major symptoms [2]. However, at the same time, they suffer from major drawbacks due to promiscuous activity at DR subtypes other than the intended ones as well as closely related GPCRs [9]. Levodopa (L-DOPA), the gold standard in treating PD, successfully reduces the major motoric symptoms like bradykinesia and tremor after biotransformation to dopamine, subsequently activating the D2R. However, L-DOPA (and dopamine, respectively) is also known to induce dyskinesia (L-DOPA-induced dyskinesia) due to promiscuous activation of the D1R in long-term treatment conditions [10]. While D2R agonists play an important role in treating PD, antagonists act as potent antipsychotics in different psychiatric disorders associated with the DR family. Those antipsychotics are also tightly connected to serious adverse drug events such as extrapyramidal syndrome and neuroleptic malignant syndrome [11, 12].
D2R-selective drugs are clearly beneficial in treating PD and psychiatric disorders by alleviating the mentioned off-target effects. However, DR-subtype selectivity should not only be seen as a tool to counteract side effects but also to open up novel therapeutic avenues. Selective D3R agonists have been shown to be effective in vivo by mitigating cell death of dopaminergic neurons and improving behavioural performances in mouse models of PD [13, 14]. Interesting results have also been obtained in clinical studies establishing pramipexole (a D3R-preferring ligand) as an effective dopamine substitute in patients not responding to L-DOPA treatment, simultaneously delaying dyskinesia [15]. Another in vivo study indicated the capability of D3R-preferring agonists to reverse motivational deficits related to PD [16]. D3R-selective antagonists present a promising opportunity in the treatment of schizophrenia. They appear to be completely devoid of the D2R-associated side effects described earlier and also treat negative symptoms, which are not covered by conventional antipsychotics [17, 18]. Selective D1R agonists are particularly interesting in treating cognitive deficits affecting patients suffering from schizophrenia targeting the prefrontal cortex. Since the clinical relevance of D1R agonists was recognized early on, several selective compounds with diverse chemical scaffolds have been designed throughout the years [19]. However, many selective ‘successful’ ligands suffer from limited oral bioavailability and poor blood-brain-barrier (BBB) permeability, thus, exposing them to rapid peripheral metabolism. This has mainly been attributed to the presence of catechol functionalities in many of the ligands [20]. Different agonists have shown promising results in improving cognitive impairments and working memory in schizophrenia [21-23]. Unfortunately, other studies provide evidence for D1R agonists being responsible for inducing seizures [24, 25]. While the seizure-inducing mechanisms and the involvement of structure-activity relationships (SAR) is still not fully understood, the development of novel, potentially non-catechol agonists is continuing [19, 26]. All of these findings clearly indicate the benefits of DR-subtype selective drugs. However, they also highlight the necessity to better understand the molecular mechanisms involved in DR-ligand interactions to rationalize the SARs responsible for specific effects.
Drug research in the field of GPCRs has been benefitting from the ‘golden age of GPCR structural biology’ in the discipline of cheminformatics [27]. Different studies have been utilizing computer-assisted drug-design (CADD) methods to investigate GPCRs and also different DR subtypes [28-30]. A particularly interesting study by Bueschbell et al investigated the selectivity of several known DR ligands (e.g. apomorphine and bromocriptine) with homology modelling and molecular docking approaches [31]. The ever-increasing availability of X-ray or cryo-EM structures of the discussed DR subtypes D1R, D2R and D3R aids our ability to comprehend DR ligand selectivity. In total, twelve three-dimensional (3D) protein structures of the D1R, five D2R structures and three D3R structures are accessible in the Protein Data Bank (PDB) database as of March 2023. The advent of cryo-EM technologies enabled the high-resolution depiction of the complex DR subtype structures at ≤ 3 Å, potentially improving molecular docking approaches investigating DR ligand selectivity.
The conserved amino acids creating the orthosteric binding pockets (OBPs) of virtually all DR subtypes are well known and described [31, 32]. Asp3.32 in transmembrane (TM) 3 is responsible for ligand recognition forming a salt bridge with the positively charged amine function of ligands. The serine triade consisting of Ser5.42, Ser5.43 and Ser5.46 positioned in TM5 is important in orienting the respective ligand (especially if a catechol functional group is involved) and considering the ligands binding affinity. An aromatic microdomain in TM6 includes Trp6.48, Phe6.51 and Phe6.52 as well as His/Asn6.55 and is involved in activating the receptor upon interaction with an agonist. Agonist binding induces the so-called ‘rotamer toggle switch’, a domino-like cascade along TM6 reorienting the named amino acids, eventually triggering receptor activation. Less is known about DR sub-domains or structural elements originating from ligands responsible for selectivity. The D1R, although belonging to the D1-like DR family, is phylogenetically closest to the β-adrenergic receptors (βARs) [33]. Consequently, it features distinct motifs, responsible for selectivity. A study by Zhuang et al. suggested the involvement of extracellular loop (ECL) 2, more specifically Ser188, enabling the D1R to accommodate bulkier ligands such as SKF81297 and SKF83959 [34]. In comparison, the same ligands would sterically clash with the corresponding amino acid Ile184 in the ECL2 of the D2R, consequently resulting in D1R-selectivity over the D2-like DR family. Considering selectivity between D2R and D3R, work by Newman et al. revealed a secondary binding pocket (SBP), consisting of multiple amino acids such as Val2.61, Leu2.64, Phe3.28 and conserved Gly and Cys residues located in ECL1 and ECL2, respectively [35]. In more detail, Michino and colleagues suggest the Gly residue in ECL1 to be the critical selectivity determinant [36]. Additionally, studies show that the D3R possesses an intrinsically higher affinity towards ligands such as dopamine and quinpirole. Robinson and colleagues showed that intracellular loop (ICL) 3 might be responsible for this behaviour. Generating D2R hybrids containing the D3R-ICL3 motif could increase ligand affinity 10- to 20-fold compared to the wild-type D2R. A D3R-D2R-ICL3 hybrid showed inverse effects [37]. An overview of the described SBP and the different domains involved in DR subtype selectivity is shown in Figure 1.
A great deal of effort has been invested in CADD-approaches to investigate and discover potential DR subtype-selective ligands, thus, benefitting drug development in e.g. neurodegenerative diseases like PD [28, 30, 31, 34, 38, 39]. However, due to the complexity of DR selectivity, in silico approaches require in vitro validation. In vitro binding affinities at different DR subtypes can be investigated using, e.g. homogenous time-resolved fluorescence (HTRF) assays, which are standardizable, commercially available and also semi-high-throughput compatible [40, 41].
Thus, the aim of this study was to develop a combined in silico / in vitro approach to assess the selectivity of novel DR ligands at different receptor subtypes using a cell-based HTRF assay as well as a molecular docking approach. Discovering DR-selective ligands as well as providing more detailed insights into their binding behaviour would contribute to better pharmacological tools and new starting points in drug development.
2. Materials and Methods
2.1. Materials
Bromocriptine mesylate (50 mg; CAYM14598-50) was acquired from VWR. A68930 hydrochloride (10 mg; A68930 ) was acquired from biotechne tocris. Apomorphine hydrochloride was kindly provided by EVERPharma AT GmbH within the context of a different project. Compounds tested in vitro (shown in Table S5) were acquired either from SPECS (https://www.specs.net/, accessed in April 2021) or Maybrige (https://www.thermofisher.com/at/en/home/industrial/pharma-biopharma/drug-discovery-development/screening-compounds-libraries-hit-identification.html, accessed in April 2021). All tested compounds were dissolved in DMSO (dimethyl sulfoxide, acquired from Sigma-Aldrich) and stored at -80°C until further use.
2.2. Ligand selection for combined in silico / in vitro approach
Compounds selected for combined in silico / in vitro investigations were chosen based on two main criteria. First, compounds identified as active D2R ligands with the previously developed workflow shown in Zell et al [42] were selected for further in vitro investigations. Second, other compounds from this study showing normalized decreased fluorescence (NDF) values ≥ 2-fold increased (during in vitro activity screening, section 2.10) at any of the investigated DR subtypes compared to the other two DR subtypes were included in further investigations.
2.3. Similarity assessment – Tanimoto scoring (TS) matrix
Canonical SMILES codes of all the compounds of interest were imported to Canvas 3.8. In Canvas 3.8, radial fingerprints (ECFP4 [43, 44]) of all the molecules (based on 2D structures) were calculated followed by an automated calculation of a TS [45] for each compared pair. TS matrices were exported to Excel as .csv files and imported to GraphPad Prism 8 to display heatmaps, color-coding the structural similarities. An increasing coefficient indicated an increasing structural similarity. (Dis-)similarities considering chemical scaffolds were further used to assess observed in silico / in vitro phenomena.
2.2. Dataset assembly for molecular docking (ChEMBL validation)
For the validation of the molecular docking approach, DR ligands with known biological activities were extracted from the ChEMBL (https://www.ebi.ac.uk/chembl/) for all three DR subtypes investigated in vitro. Only entries originating from homo sapiens (UniProt accession numbers: D1R, P21728; D2R, P14416 and D3R, P35462) were considered. ChEMBL entries were only selected for further evaluation, if (I) KI values for all three DR subtypes were available for each respective molecule and (II) the in vitro measurements included a valid control. The curated ChEMBL entries were divided into D1R-, D2R-, D2like- and D3R-selective subsets. D1R-, D2R- or D3R-selectivity was assumed for molecules with binding affinities ≤ 1000 nM at the respective subtype and ≥ 1000 nM at the others. Additionally, the Ki values were required to differ at least by a factor of two. D2like-selective compounds showed binding affinities ≤ 500 nM at D2R and D3R. The final datasets consisting of 29 (SC1 to SC29 [46-60]), 25 (SC30 to SC54 [53, 61-72]), 152 (SC55 to SC206 [56, 69, 72-106]) and 78 (SC207 to SC284 [53, 56, 62, 63, 65, 66, 79, 87, 89, 96, 99-101, 103-118]) molecules, respectively, are shown in Tables S1-S4.
2.3. Data set preparation for molecular docking
All compounds were energetically minimized using OMEGA 3.0.1.2 prior to molecular docking. (OpenEye Scientific Software, Santa Fe, NM) [119].
2.4. Molecular docking workflow
Docking was performed using GOLD 5.8.0 [120]. Hydrogens were added to all protein structures. CHEMPLP was used as a scoring function not allowing early termination. For defining the binding site, all atoms within 6 Å of the bound ligand (depending on the cryo-EM structure) were chosen. The number of GA runs was set to 30.
2.4.1. Molecular docking – D1R
2.4.2. Molecular dynamics simulation (MDS) – D2R
The MDS was performed following the workflow shown in Figure 2.
2.4.3. Molecular docking D2R
Molecular docking into the D2R ligand binding site was performed using the MDS-modified (see section 2.4.2) cryo-EM structure of the PDB entry 7jvr [34]. During docking only ASP114R was specified as flexible (1 rotamer (free)).
2.4.4. Molecular docking D3R
2.5. DR subtypes – BLASTP alignment
To identify the analogous amino acids of the SBP of D1R and D2R in respect to D3R (defined in [35, 36]) a BLASTP alignment was performed (https://blast.ncbi.nlm.nih.gov) [122]. The relevant amino acids in regard to the SBP of D3R are shown in Table 3.
The respective amino acids were used during the in silico selectivity assessment during the validation process and the analysis of the novel DR ligands.
2.6. Validation of the molecular docking approach – ChEMBL dataset(s)
Molecular docking results for each ChEMBL dataset (containing 30 poses for each compound) were uploaded to Pipeline Pilot Client 9.1 [123]. Duplicates from each molecular docking output were removed. Only top-ranked poses (based on fitness score) of each docked compound were retained in the datasets used for further evaluation. The subsequent docking analysis (top-ranked poses) was performed using DiscoveryStudio (DS) 2018 Client. The (modified) DR subtype protein structures were loaded into DS. The conserved Gly residues (shown in Table 4) were marked, centroids were calculated and checked as center of mass (COM). Subsequently, the docked DR subtype-selective output files were loaded into the respective DR protein structure. All molecules were marked and COM was calculated with respect to to the Gly residues. Finally, distances between COM (Gly residue) and COM (docked ligands) were calculated in Å.
2.7. Docking analysis – novel DR ligands
The docking analysis of the novel ligands was performed using LigandScout 4.4.4. Docked ligands (sd files) were loaded into the different DR protein structures (D1R into 7jvq [34]; D2R into the MDS-modified D2R 7jvr [34]; and D3R into 7cmv [121], respectively). All 30 poses of each ligand were individually superimposed and the most frequent pose was assessed visually. Subsequently, DR protein structures as well as molecular docking output files were loaded in PyMOL for each DR subtype individually. The protein including the respective most frequent ligand pose (taking the highest-ranking according to fitness score) was extracted as a pdb file. The resulting pdb files were loaded into DS for calculating distances [Å] as shown in section 2.6.
2.8. HTRF-based receptor binding studies
All HTRF assays were performed using a HTRF-compatible plate reader (model Tecan Spark). The respective settings were specifically modified and optimized for the determination of D2R ligand-binding affinities. Binding affinities were determined using the same settings for measurements with D1R and D3R carrier cells. Experiments were performed using two different emission wavelengths at 620 (control) and 665 (D2/3R) / 510 (D1R) nm, respectively. Fluorophores were excited at 320 nm. A dichroic 510 mirror was used, while lag and integration times of 100 and 400 µs were applied, respectively. Flashes were set to 75. Electronic gain was automatically optimized, while the z-position was optimized based on the well with the highest expected signal. Experiments described in 2.9, 2.10, and 2.12 require the use of two 96-well plates. The first plate was used to determine the gain and the z-position. Subsequently, the determined values were set manually for the second plate to enable direct comparison between the different plates.
2.9. Characterization of DR carrier cells (D1R and D3R) – KD determination
The cells used for the subsequent screening and detailed investigation of D1R and D3R ligands were acquired from PerkinElmer/cisbio (Tag-lite Dopamine D1 or D3a-labeled Cells, ready-to-use (transformed and labeled), 200 tests; C1TT1D1 and C1TT1D3A, respectively). The cells were stored in liquid nitrogen until further use. Fluorescent-labelled ligands (Dopamine D2 Receptor red antagonist Fluorescent Ligand (L0002RED), stored at -20°C and Dopamine D1 Receptor green antagonist (L0031GRE), stored at -20°C), assay buffer (Tag-lite Buffer (5x concentrate), 100 mL, stored at +4°C; LABMED), and 96-well plates (HTRF 96-well low-volume white plate; 66PL96005) required for the in vitro assay were also acquired from PerkinElmer/cisbio. The assay was conducted according to the standard operation protocol (SOP) available from PerkinElmer/cisbio. The 96-well plates were incubated at room temperature for 2 h. The 96-well plates were read as described in section 2.8. The respective concentrations of the dilution series were performed in triplicates. In total, KD determination was performed twice.
The characterization of the D2R carrier cells is detailed in [42].
2.10. In vitro screening – assessment of compound activity
Materials described in 2.1 and 2.9 were also used during ligand screening. TLB (1x) was prepared as described earlier. For ligand screening, compounds were prepared at a working solution concentration of 40 µM in 1x TLB. Compound 1, apomorphine, was used as the positive control at the same concentration. The assay was conducted in duplicates and as described in section 2.9.
2.10. Ligand selection for KI determination
Ligand selection was based on NDF values detailed in Table 5. Novel D2R ligands from our previous study (compounds 2, 3, 5, 6, 7 and 9) [42] were selected for selectivity assessment. Additionally, compounds 4, 8 and 10 were investigated due to a NDF fold-difference ≥ 2 of any of the three DR subtypes compared to the other two.
2.12. KI Determination for selected ligands
The materials described in 2.1 and 2.9 were also used for KI determination of the ligands selected after screening. The selected ligands (compounds 1 - 10) were diluted in 1x TLB. Compounds 1, 2 and 4 - 9 were diluted to an initial working solution concentration of 4x10-4 M. Compounds 3 and 10 were diluted to an initial working solution concentration of 1x10-4 M. Different concentrations were chosen due to differences in aqueous solubility of the compounds. The KI was determined in duplicates following the SOP available from PerkinElmer/cisbio as described in section 2.9.
2.13. Data processing, representation and analysis
Saturation binding curves were processed and visualized in GraphPad Prism 8 (Nonlinear regression (curve fit), One site – Fit logIC50 was performed using GraphPad Prism version 8.2.1 for Windows, GraphPad Software, San Diego. California USA, www.graphpad.com). Molecular docking was performed in GOLD 5.8.0 [120]. Docking analysis was performed in LigandScout version 4.4.5 (Inte:Ligand GmbH, Vienna, Austria) [124]. MDS and calculations for distance-based in silico approach were performed in DS Client 2018 (DiscoveryStudio, Accelrys Inc, San Diego, CA, USA). 2D structures of all shown compounds were generated using ChemDraw 19.0 (PerkinElmer, Waltham, MA, USA). SD files used for similarity assessment were generated using PipelinePilot Client 9.1 (Dassault Systems, BIOVIA Discovery Studio, San Diego, CA, USA, 2018). Similarity assessment was performed using Canvas 3.8 (Canvas, Schrödinger, LLC, New York, NY, USA, 2021). Docking alignments and visualization were performed in PyMOL (PyMOL, Schrödinger, LLC, New York, NY, USA, 2021).
3. Results
3.1. Structural summary of the investigated ligands
All compounds investigated in silico and in vitro within this study are shown in Figure 3.
The novel ligands (compounds 2 to 10) investigated with the combined approach within the scope of this study were structurally compared to each other using a Tanimoto scoring (TS) matrix. Thus, observed in silico and / or in vitro phenomena could be potentially correlated to structural (dis-)similarities. The TS matrix is shown in Figure 4, ranging from 0 (green) to 1 (red), corresponding to structurally unrelated and identical compounds, respectively.
Thirty-three out of 36 pairs scored between 0.03 and 0.15, thus, representing a structurally divers compound collection. Only three pairs, i.e. compounds 5 and 6 (TS 0.28), 6 and 10 (TS 0.21) and 9 and 10 (TS 0.21), were characterized by a similarity score of > 0.21, reflecting a higher degree of similarity (considering the use of radial fingerprints).
3.2. In vitro compound screening – an assessment of DR subtype selectivity
The investigated compounds were taken from a previous pharmacophore-based virtual screening study described in Zell et al [42]. All 2D structures (compounds 2 - 10 and SC285 - SC365) and respective NDF values for all DR subtypes are shown in Table S5 and Figure S1 to S9. The activities of all compounds were investigated via competitive binding (in comparison to a fluorescence-labeled ligand) of the respective compounds at D1R / D3R utilizing a HTRF assay using a screening concentration of 10 µM. NDF values of compounds chosen for further evaluation are shown in Table 4.
Based on the resulting NDF values, all compounds but 4 and 8 showed promiscuous receptor activities suggesting diverse selectivity profiles. Only compounds 4 and 8 showed NDF values close to 1 at both D1R and D2R suggesting inactivity at those DR subtypes, respectively selectivity for the D3R.
3.3. KI determination –of the selected compounds at DR subtypes
The selected compounds were investigated in vitro to determine their binding affinities (KI values) at the three different DR subtypes D1R, D2R and D3R. In Figure 5a and b, compounds 2 and 10 are shown as examples. Compound 2 represents a non-selective ligand while compound 10 is characterized by the highest selectivity (for D3R). The remaining binding curves are shown in Figure S10a to h.
The KI values of all investigated ligands as well as the calculated fold-differences for each receptor pair are shown in Table 5.
All compounds investigated in vitro, except compound 1, showed a clear D2like selectivity with preferences for D3R (D1R/D3R fold differences ranging from 3.06 to 1031.4, D2R/D3R fold-differences ranging from 1.66 to 263.7, respectively). While compounds 2, 5, 6, 9 and 10 showed higher affinities for D1R compared to the D2like DR subtypes, the affinities of compounds 3, 4 and 8 were not determinable for D1R, thus considered inactive. Compounds 4 and 8 were also inactive at D2R, thus considered D3R-selective. Only compound 1 was characterized by the lowest binding affinity for D2R. Interestingly, all compounds show the highest affinity at the D3R.
3.4. Dataset assembly – in silico assessment
To validate the molecular docking approach utilized to assess compound selectivity in silico, DR ligands with different selectivities for the DR subtypes D1R, D2R and D3R with known biological activities were extracted from the ChEMBL database. Compounds were only included in the final datasets if (I) their binding affinities were determined in vitro at all three DR subtypes and (II) in vitro measurements included a valid control to assess assay functionality. The curated ChEMBL entries were divided into D1R-, D2R-, D2like- and D3R-selective subsets. D1R-, D2R- or D3R-selectivity was assumed for molecules with binding affinities ≤ 1000 nM at the respective subtype and ≥ 1000 nM at the others. D2like-selective compounds showed binding affinities ≤ 500 nM at D2R and D3R. The final datasets consisting of 29 (SC1 - SC29 [46-60]), 25 (SC30 - SC54 [53, 61-72]), 152 (SC55 - SC206 [56, 69, 72-106]) and 78 (SC207 - SC284 [53, 56, 62, 63, 65, 66, 79, 87, 89, 96, 99-101, 103-118]) molecules, respectively, are shown in Table S1 to Table S4.
3.5. Validation of molecular docking
The utilized molecular docking approach was based on the work of Michino and colleagues [36]. Therefore, different ChEMBL datasets previously defined as DR subtype-selective (see section 3.4) were docked into the 3D protein structures of D1R, D2R and D3R (molecular docking workflow described in section 2.4.1, 2.4.3, and 2.4.4). Due to the high number of investigated ligands (> 300), only the top-ranked poses (considering the fitness score) were considered during further analysis. The COM for all ligands included in each specific DR-selective subset was calculated using DS. Distances of each COM with respect to each DRs conserved Gly residue (shown in Table 3) were calculated in [Å]. The calculated fold-differences for each subset, comparing different DRs with each other, are shown in Figure 6 (absolute distances determined in DS are given in Table S6).
The dashed red line shown in Figure 6 indicates a fold-difference of 1.0, which indicates the same distance between the ligands collective COM and the conserved Gly residue after docking into the respective DR structures. All investigated datasets show a fold-difference close to 1.0 considering the D2R/D3R comparison. This means that they showed an almost identical distance between COM and the Gly residue. In contrast, all datasets showed an increased fold-difference > 1.0, when comparing D1R with D2R or D3R, respectively. Details considering all datasets are shown in Table 6.
Clearly, the approach was incapable of distinguishing D2R- and D3R-selectivity from each other based on the COM-Gly distance. However, the utilized molecular docking approach was capable to identify D2like-selective ligands based on their position within the respective DRs OBP.
3.6. In silico assessment of DR selectivity – interaction with the SBP
For the in silico assessment of the selected compounds 1 – 10, the most frequent poses after docking were used. After calculating the fold-differences based on the distances between each ligands individual COM and the respective Gly residue (within each of the three DR subtypes SBP, shown in Table S7), they were plotted against the fold-differences based on the DR-specific KI values (shown in Table 5) determined in vitro. The resulting scatter plot is shown in Figure 7.
In addition to the individual data points, Figure 7 shows regression curves for all DR pairs. The D1R/D2R curve (dots) was characterized by the steepest slope suggesting the capability of the in silico approach to discriminate D2R-selective ligands. While the slope for the D1R/D3R curve (squares) was less steep, the calculated fold-differences (based on distance, y-axis) was already higher at lower KI-based fold-differences (x-axis) indicating a similar capability to discriminate D3R-selective ligands. The D2R/D3R curve (triangles) was flatter, with individual values scattered around 1.0. Consequently, this reflected the results shown in Figure 6, where D2R/D3R-selectivity couldn´t be discriminated based on the selected approach. In summary, the developed distance-based in silico approach was highly capable to identify D2like-selectivity. This was also indicated by the R2 values (shown in Figure 7) regarding the D1R/D2R and the D1R/D3R comparison showing a positive correlation of increasing binding affinities with increasing selectivity.
3.7. Retrospective analyis of the in silico / in vitro correlation
To get a more detailed insight into the binding mode of each of the investigated ligands at the respective DR subtype, the most frequent docking poses of each compound (Figure 8, Figure 9, Figure 10 and Figures S11 - S16) were visualized in the different binding pockets using PyMOL. Figure 8 and 9 show the different binding poses of the non-selective compound 2 and compound 10, which had the highest D3R-selectivity.
In Figure 8, the tertiary amine functionality (contained in the piperazine motif) of compound 2 is clearly oriented towards the OBP, allowing the formation of the salt-bridge with Asp3.32 (described as the crucial interaction to define a DR ligand). While the position of compound 2 was flipped in D2R in comparison to D1R and D3R (highlighted by the orientation of the chlorine, green), the overall positioning of compound 2 was similar in each DR subtype. Consequently, there was no distinct orientation of any of the poses towards the SBP, resulting in the non-selective binding with KI fold-differences between 1.2 and 2.2 (see Table 5).
In Figure 9, the tertiary amine functionality (contained in the piperazine motif) of compound 10 was again oriented towards the OBP. Thus, the salt-bridge formation with Asp3.32 was possible. In contrast to compound 2, the binding poses of compound 10 were distinctly different in the respective DR subtypes. Comparing the poses in D1R and D3R, the D3R pose (green) was shifted slightly to the right towards the SBP. The D2R binding pose (orange) was clearly different from both D1R and D3R, with, the chloro-substituted ring clearly oriented towards the SBP. While this explained the observed D1R/D2R fold difference of 3.7, it didn’t correlate with the D2R/D3R fold-difference of 331.8. However, the detailed analysis of the binding poses of compound 10 correlated with the observed D2like selectivity determined in vitro. Additionally, it also partially confirmed the retrospective results of the distance-based approach shown Figure 7, highlighting the capability of the developed approach to identify D2like-selectivity.
These findings were also supported by the in-depth analyses of compounds 3, 5, 6, 7 and 9 (PyMOL alignments shown in Figure S11 - S14 and Figure S16), where D2R and D3R poses were distinctly oriented towards the SBP. However, in agreement with the findings considering compound 10, the D2R- and D3R binding poses didn´t correlate with the higher D3R binding affinities found in vitro. Again, the results allowed for the confirmation of D2like-selectivity of the investigated ligands.
Compounds 4 (Figure 10) and 8 (Figure S15) were the only compounds with no determinable binding affinity at D1R and D2R, additionally showing slightly increased distance-based fold-differences (Figure 7 and Table S7), regarding D2R/D3R-selectivity, of 1.10 and 1.04, respectively.
This was also reflected in the binding pose of compound 4, where the D3R pose (green) was oriented closer to the SBP. Similar results were observed in the binding pocket comparison of compound 8.
4. Discussion
The characterized DR ligands showed different selectivity profiles. Interestingly, all ten compounds investigated by the developed in silico / in vitro approach (including the novel compounds 2 to 10) showed either D3R-preferences or clear D3R-selectivity. Compound 2; for example, showed fold-differences of 3.23 and 1.66 for D1R/D3R and D2R/D3R. respectively, thus, exerting D3R-preferences. Compounds 4 and 8 were characterized by no determinable binding affinities at D1R and D2R, consequently they were categorized as D3R-selective. While compound 10 showed low to intermediate binding affinities at D1R (2.38 µM) and D2R (0.61 µM), it also exerted the highest quantifiable selectivity fold-differences with values of 1031.4 and 263.7 for D1R/D3R and D2R/D3R, respectively. Additionally, all investigated compounds but 1 were D2like-selective.
The rather promiscuous behavior of compound 2 is attributed to its structural similarity to clozapine, the prototypical representative of tricyclic antipsychotics (a drug class belonging to the atypical antipsychotics). While clozapine is characterized by its potent antipsychotic effect, it is also known as a so-called ‘dirty drug’ due to its promiscuous activity at a variety of aminergic GPCRs (including dopaminergic, serotonergic and adrenergic receptor families) [125]. Thus, a similar pharmacological profile of compound 2 was expected. This was not only confirmed by the in vitro data but also by the developed in silico approach, correlating the positioning of the ligand within the OBP and SBP with its respective DR subtype selectivity. Even though the binding behavior of compound 2 appeared non-selective, the in silico approach was capable of detecting the slight D2like-preference resulting in distance-based fold-differences of 1.33 and 1.28 for D1R/D2R and D1R/D3R, respectively. Moreover, the compound could be active at other GPCRs which were not investigated within this study.
The comparison of compounds 6 and 10 allowed for very interesting insights into the DR subtype-selectivity profile of structurally similar ligands differing mainly regarding linker lengths. Compounds 6 and 10 are both characterized by two terminal aromatic rings and a linker region consisting of a piperazine motif, an amide functionality and an alkyl chain (see Figure 3). Moreover, they also share binding preferences at the different DR subtypes following D1R > D2R > D3R. Both compounds showed comparable KI-based fold-differences of 5.77 (compound 6) and 3.91 (compound 10) for D1R/D2R. However, the D1R/D3R and D2R/D3R fold-differences increased drastically for compound 10 (1031.4 and 263.7, respectively) compared to compound 6 (20.7 and 3.59, respectively). Michino and colleagues showed similar phenomena in their study investigating the impact of the linker length in analogues of the highly D3R-selective compound R22 ([(R)-N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide]) [36, 79]. The investigated R-22 analogues included C3- to C5-linker regions. C3-linker length results in non-selective binding behavior at D2R and D3R. C5-linker length markedly reduced D2R/D3R selectivity. Only the C4 analogue retained a significant D2R/D3R-selectivity with 45.7 fold-difference. Compound 6, including a C2-linker region showed a comparably reduced D2R/D3R-selectivity of 3.59. In contrast, compound 10, including a C4-linker region exerted a fold-difference of 263.7. While compound 6 and 10 are only partially related (similarities shown in red) to the R22-analogues (see Figure 11), the observed in vitro effects are potentially attributable to the length of the linker region.
Compounds 2, 3, 5, 6, 7 and 9 were already reviewed in our earlier publication investigating their novelty and also the DR-associated effects of the closest structural relatives [42]. While none of the investigated structures yielded exact structural matches, the most similar structures had been associated with different DR-related effects. Structurally similar compounds to 5 and 6 were associated with D4R-selectivity but no defined mode of action (agonism or antagonism) [126]. A compound similar to 2 had been associated with D4R antagonism, while structurally similar ligands regarding compounds 3 and 7 were investigated considering D2R antagonism [127-130]. Only a structurally similar compound to 9 was associated with D3R-selectivity and D2R antagonism [131].The novel ligands included within this study were compared to the literature using SwissTargetPrediction (http://www.swisstargetprediction.ch/) and SwissSimilarity (http://www.swisssimilarity.ch/) [132-134]. Compounds 4 and 8 yielded low scores in SwissTargetPrediction where the identified similar compounds (ChEMBL IDs 59603 and 592377) had been investigated considering D1R- and D2R activity but not D3R selectivity [135, 136]. ChEMBL entry 4081151 was structurally closely related to compound 4 but had only been investigated for kappa opioid receptors [137]. SwissSimilarity match for compound 8 (ChEMBL ID 1094101) was investigated for its binding affinity at serotonergic receptors and aminergic GPCR family members, but not in respect with DRs [138]. Thus, compounds 4 and 8, open up novel insights into D3R-selectivity. Compound 10 resulted in exact structural matches and closely related matches in both SwissTargetPredicition and SwissSimilarity investigating D3R-selectivity. Still, the comparison between compounds 6 and 10 contributes to a better understanding of the role of the linker length on DR subtype selectivity of structurally related but not identical chemical scaffolds.
As mentioned earlier, all novel ligands exerted their highest binding affinities at the D3R. This is partially in accordance with scientific literature, where the D3R shows a high intrinsic binding affinity for agonists like dopamine (420-fold increased affinity) and quinpirole [37, 139]. While this is attributed to intracellular loop 3 in D3R, this characteristic has only been shown for agonists. However, especially the known characteristics of the structurally related compounds of the novel compounds described above suggest a low probability that all investigated ligands are actually agonists. Thus, the increased D3R affinity of compounds 2 to 10 presumably originates from a distinct interaction with the described SBP [36]. The developed in silico approach proposes a workflow to identify D2like-selectivity. However, the static nature of the molecular docking approach doesn’t allow for discrimination of D2R/D3R-selectivity. This limitation can be attributed to the very dynamic nature of the EL structural motifs of the D2like DRs responsible for subtype selectivity. Different studies propose MDS approaches to circumvent the shortcomings of molecular docking approaches and to account for protein flexibility [36, 140].
Thus, the developed in silico / in vitro workflow clearly demonstrated its potential use in preclinical drug research by enabling the identification of D2like-selective ligands independently of chemical scaffolds. This could be especially important in diseases of the CNS, where D1R activation has been associated with induction of seizures. In addition, the D3R is a fast emerging molecular target of interest in treating PD. Thus, the accurate prediction of D2like-selectivity could act as an important starting point in developing truly D3R-selective compounds and also providing pharmacological tools aiding in the understanding of D2like DR subtype selectivity.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1 to Figure S9: Summary of the in vitro screening results of all investigated compounds; Figure S10: Overview of the in vitro determined binding affinities of selected compounds (1 and 3 to 9); Figure S11 to Figure S16: Detailed insights into the docking poses and binding pockets of selected compounds (3, 5, 6, 7, 8 and 9); Table S1: Dataset of D1R-selective compounds (ChEMBL); Table S2: : Dataset of D2R-selective compounds (ChEMBL); Table S3: Dataset of D2like-selective compounds (ChEMBL); Table S4: Dataset of D3R-selective compounds (ChEMBL); Table S5: Overview of 2D structures and NDF values; Table S6: Validation of in silico approach using ChEMBL; Table S7: Summary of the retrospective analysis of investigated compounds.
Author Contributions
Conceptualization. D.S.; methodology. L.Z.; software. Commercially and publicly available software was used; validation. D.S., L.Z. and V.T.; investigation. A.B. and L.Z.; data curation. A.B. and L.Z.; writing—original draft preparation. L.Z.; writing—review and editing. D.S., L.Z. and V.T.; visualization. L.Z.; supervision. D.S. and V.T.; project administration. D.S.. All authors have read and agreed to the published version of the manuscript.
Funding
V.T. is funded by the FWF project T942. L.Z. is funded by PMU-RIF, 2022-PRE-005-Zell.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the Supplementary Material. If further data are required, they are available from the corresponding author upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hauser, A.S.; et al., Trends in GPCR drug discovery: New agents, targets and indications. Nat Rev Drug Discov, 2017. 16(12): P. 829-842. [CrossRef]
- Hauser, A.S.; et al., Pharmacogenomics of GPCR Drug Targets. Cell, 2018. 172(1-2): P. 41-54 e19. [CrossRef]
- Martel, J.C. and S. Gatti McArthur, Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front Pharmacol, 2020. 11: P. 1003. [CrossRef]
- Stocchi, F., M. Torti, and C. Fossati, Advances in dopamine receptor agonists for the treatment of Parkinson's disease. Expert Opin Pharmacother, 2016. 17(14): P. 1889-902. [CrossRef]
- Ashok, A.H.; et al., The dopamine hypothesis of bipolar affective disorder: The state of the art and implications for treatment. Mol Psychiatry, 2017. 22(5): P. 666-679. [CrossRef]
- Beaulieu, J.M. and R.R. Gainetdinov, The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev, 2011. 63(1): P. 182-217. [CrossRef]
- Kiss, B.; et al., Neuronal Dopamine D3 Receptors: Translational Implications for Preclinical Research and CNS Disorders. Biomolecules, 2021. 11(1). [CrossRef]
- Prieto, G.A., Abnormalities of Dopamine D(3) Receptor Signaling in the Diseased Brain. J Cent Nerv Syst Dis, 2017. 9: P. 1179573517726335. [CrossRef]
- Wang, Q.; et al., Subtype selectivity of dopamine receptor ligands: Insights from structure and ligand-based methods. J Chem Inf Model, 2010. 50(11): P. 1970-85. [CrossRef]
- Murer, M.G. and R. Moratalla, Striatal Signaling in L-DOPA-Induced Dyskinesia: Common Mechanisms with Drug Abuse and Long Term Memory Involving D1 Dopamine Receptor Stimulation. Front Neuroanat, 2011. 5: P. 51. [CrossRef]
- Berman, B.D., Neuroleptic malignant syndrome: A review for neurohospitalists. Neurohospitalist, 2011. 1(1): P. 41-7. [CrossRef]
- Sykes, D.A.; et al., Extrapyramidal side effects of antipsychotics are linked to their association kinetics at dopamine D(2) receptors. Nat Commun, 2017. 8(1): P. 763. [CrossRef]
- Lao, C.L.; et al., Intranasal and subcutaneous administration of dopamine D3 receptor agonists functionally restores nigrostriatal dopamine in MPTP-treated mice. Neurotox Res, 2013. 24(4): P. 523-31. [CrossRef]
- Li, C.; et al., Novel D3 dopamine receptor-preferring agonist D-264: Evidence of neuroprotective property in Parkinson's disease animal models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and lactacystin. J Neurosci Res, 2010. 88(11): P. 2513-23. [CrossRef]
- Antonini, A.; et al., Role of pramipexole in the management of Parkinson's disease. CNS Drugs, 2010. 24(10): P. 829-41. [CrossRef]
- Carnicella, S.; et al., Implication of dopamine D3 receptor activation in the reversion of Parkinson's disease-related motivational deficits. Transl Psychiatry, 2014. 4(6): P. e401. [CrossRef]
- Millan, M.J.; et al., S33084, a novel, potent, selective, and competitive antagonist at dopamine D(3)-receptors: I. Receptorial, electrophysiological and neurochemical profile compared with GR218,231 and L741,626. J Pharmacol Exp Ther, 2000. 293(3): P. 1048-62.
- Meltzer, H.Y., Cognitive factors in schizophrenia: Causes, impact, and treatment. CNS Spectr, 2004. 9(10 Suppl 11): P. 15-24. [CrossRef]
- Jones-Tabah, J.; et al., The Signaling and Pharmacology of the Dopamine D1 Receptor. Front Cell Neurosci, 2021. 15: P. 806618. [CrossRef]
- Felsing, D.E., M.K. Jain, and J.A. Allen, Advances in Dopamine D1 Receptor Ligands for Neurotherapeutics. Curr Top Med Chem, 2019. 19(16): P. 1365-1380. [CrossRef]
- Girgis, R.R.; et al., A proof-of-concept, randomized controlled trial of DAR-0100A, a dopamine-1 receptor agonist, for cognitive enhancement in schizophrenia. J Psychopharmacol, 2016. 30(5): P. 428-35. [CrossRef]
- Rosell, D.R.; et al., Effects of the D1 dopamine receptor agonist dihydrexidine (DAR-0100A) on working memory in schizotypal personality disorder. Neuropsychopharmacology, 2015. 40(2): P. 446-53. [CrossRef]
- Abi-Dargham, A.; et al., Dopamine D1R Receptor Stimulation as a Mechanistic Pro-cognitive Target for Schizophrenia. Schizophrenia Bulletin, 2022. 48(1): P. 199-210. [CrossRef]
- O'Sullivan, G.J.; et al., Dopamine D1 vs D5 receptor-dependent induction of seizures in relation to DARPP-32, ERK1/2 and GluR1-AMPA signalling. Neuropharmacology, 2008. 54(7): P. 1051-61. [CrossRef]
- Arnsten, A.F.; et al., Novel Dopamine Therapeutics for Cognitive Deficits in Schizophrenia. Biol Psychiatry, 2017. 81(1): P. 67-77. [CrossRef]
- Kozak, R.; et al., Characterization of PF-6142, a Novel, Non-Catecholamine Dopamine Receptor D1 Agonist, in Murine and Nonhuman Primate Models of Dopaminergic Activation. Front Pharmacol, 2020. 11: P. 1005. [CrossRef]
- Basith, S.; et al., Exploring G Protein-Coupled Receptors (GPCRs) Ligand Space via Cheminformatics Approaches: Impact on Rational Drug Design. Front Pharmacol, 2018. 9: P. 128. [CrossRef]
- Salman, M.M.; et al., Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int J Mol Sci, 2021. 22(9). [CrossRef]
- Lian, P.; et al., A computational perspective on drug discovery and signal transduction mechanism of dopamine and serotonin receptors in the treatment of schizophrenia. Curr Pharm Biotechnol, 2014. 15(10): P. 916-26. [CrossRef]
- Nikolic, K.; et al., Drug Design for CNS Diseases: Polypharmacological Profiling of Compounds Using Cheminformatic, 3D-QSAR and Virtual Screening Methodologies. Front Neurosci, 2016. 10: P. 265. [CrossRef]
- Bueschbell, B.; et al., A Complete Assessment of Dopamine Receptor- Ligand Interactions through Computational Methods. Molecules, 2019. 24(7). [CrossRef]
- Floresca, C.Z. and J.A. Schetz, Dopamine receptor microdomains involved in molecular recognition and the regulation of drug affinity and function. J Recept Signal Transduct Res, 2004. 24(3): P. 207-39. [CrossRef]
- Vass, M.; et al., Aminergic GPCR-Ligand Interactions: A Chemical and Structural Map of Receptor Mutation Data. J Med Chem, 2019. 62(8): P. 3784-3839. [CrossRef]
- Zhuang, Y.; et al., Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell, 2021. 184(4): P. 931-942 e18. [CrossRef]
- Newman, A.H.; et al., Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J Med Chem, 2012. 55(15): P. 6689-99. [CrossRef]
- Michino, M.; et al., A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol Pharmacol, 2013. 84(6): P. 854-64. [CrossRef]
- Robinson, S.W., K.R. Jarvie, and M.G. Caron, High affinity agonist binding to the dopamine D3 receptor: Chimeric receptors delineate a role for intracellular domains. Mol Pharmacol, 1994. 46(2): P. 352-6.
- Ishiki, H.M.; et al., Computer-aided Drug Design Applied to Parkinson Targets. Curr Neuropharmacol, 2018. 16(6): P. 865-880. [CrossRef]
- Elek, M.; et al., Synthesis, in silico, and in vitro studies of novel dopamine D(2) and D(3) receptor ligands. Arch Pharm (Weinheim), 2021. 354(6): P. e2000486. [CrossRef]
- Degorce, F.; et al., HTRF: A technology tailored for drug discovery - a review of theoretical aspects and recent applications. Curr Chem Genomics, 2009. 3: P. 22-32. [CrossRef]
- Yasi, E.A., N.S. Kruyer, and P. Peralta-Yahya, Advances in G protein-coupled receptor high-throughput screening. Curr Opin Biotechnol, 2020. 64: P. 210-217. [CrossRef]
- Zell, L.; et al., Identification of Novel Dopamine D(2) Receptor Ligands-A Combined In Silico/In Vitro Approach. Molecules, 2022. 27(14). [CrossRef]
- Glen, R.C.; et al., Circular fingerprints: Flexible molecular descriptors with applications from physical chemistry to ADME (vol 9, pg 199, 2006). Idrugs, 2006. 9(4): P. 311-311.
- Rogers, D. and M. Hahn, Extended-connectivity fingerprints. J Chem Inf Model, 2010. 50(5): P. 742-54. [CrossRef]
- Tanimoto, T.T., An Elementary Mathematical Theory of Classification and Prediction. 1958: International Business Machines Corporation.
- el Ahmad, Y.; et al., New benzocycloalkylpiperazines, potent and selective 5-HT1A receptor ligands. J Med Chem, 1997. 40(6): P. 952-60. [CrossRef]
- Hubner, H., J. Kraxner, and P. Gmeiner, Cyanoindole derivatives as highly selective dopamine D(4) receptor partial agonists: Solid-phase synthesis, binding assays, and functional experiments. J Med Chem, 2000. 43(23): P. 4563-9. [CrossRef]
- Einsiedel, J., H. Hubner, and P. Gmeiner, Benzamide bioisosteres incorporating dihydroheteroazole substructures: EPC synthesis and SAR leading to a selective dopamine D4 receptor partial agonist (FAUC 179). Bioorg Med Chem Lett, 2001. 11(18): P. 2533-6. [CrossRef]
- Lober, S.; et al., Di- and trisubstituted pyrazolo[1,5-a]pyridine derivatives: Synthesis, dopamine receptor binding and ligand efficacy. Bioorg Med Chem Lett, 2002. 12(4): P. 633-6. [CrossRef]
- Wittig, T.W., M. Decker, and J. Lehmann, Dopamine/serotonin receptor ligands. 9. Oxygen-containing midsized heterocyclic ring systems and nonrigidized analogues. A step toward dopamine D5 receptor selectivity. J Med Chem, 2004. 47(17): P. 4155-8. [CrossRef]
- Seong, C.M.; et al., Discovery of 3-aryl-3-methyl-1H-quinoline-2,4-diones as a new class of selective 5-HT6 receptor antagonists. Bioorg Med Chem Lett, 2008. 18(2): P. 738-43. [CrossRef]
- Enzensperger, C.; et al., Dopamine/serotonin receptor ligands. 16.(1) Expanding dibenz[d,g]azecines to 11- and 12-membered homologues. Interaction with dopamine D(1)-D(5) receptors. J Med Chem, 2007. 50(18): P. 4528-33. [CrossRef]
- Linz, S.; et al., Design, synthesis and dopamine D4 receptor binding activities of new N-heteroaromatic 5/6-ring Mannich bases. Bioorg Med Chem, 2009. 17(13): P. 4448-58. [CrossRef]
- Banister, S.D.; et al., Molecular hybridization of 4-azahexacyclo[5.4.1.0(2,6).0(3,10).0(5,9).0(8,11)]dodecane-3-ol with sigma (sigma) receptor ligands modulates off-target activity and subtype selectivity. Bioorg Med Chem Lett, 2011. 21(12): P. 3622-6. [CrossRef]
- Robaa, D.; et al., Chiral indolo[3,2-f][3]benzazecine-type dopamine receptor antagonists: Synthesis and activity of racemic and enantiopure derivatives. J Med Chem, 2011. 54(20): P. 7422-6. [CrossRef]
- Sampson, D.; et al., Identification of a new selective dopamine D4 receptor ligand. Bioorg Med Chem, 2014. 22(12): P. 3105-14. [CrossRef]
- Zhang, J.; et al., Structural manipulation on the catecholic fragment of dopamine D(1) receptor agonist 1-phenyl-N-methyl-benzazepines. Eur J Med Chem, 2014. 85: P. 16-26. [CrossRef]
- Madapa, S. and W.W. Harding, Semisynthetic Studies on and Biological Evaluation of N-Methyllaurotetanine Analogues as Ligands for 5-HT Receptors. J Nat Prod, 2015. 78(4): P. 722-9. [CrossRef]
- Lee, D.Y.W.; et al., Asymmetric total synthesis of tetrahydroprotoberberine derivatives and evaluation of their binding affinities at dopamine receptors. Bioorg Med Chem Lett, 2017. 27(6): P. 1437-1440. [CrossRef]
- Martini, M.L.; et al., Defining Structure-Functional Selectivity Relationships (SFSR) for a Class of Non-Catechol Dopamine D(1) Receptor Agonists. J Med Chem, 2019. 62(7): P. 3753-3772. [CrossRef]
- Heier, R.F.; et al., Synthesis and biological activities of (R)-5,6-dihydro-N,N-dimethyl-4H-imidazo[4,5,1-ij]quinolin-5-amine and its metabolites. J Med Chem, 1997. 40(5): P. 639-46.
- Thomas, C., H. Hubner, and P. Gmeiner, Enantio- and diastereocontrolled dopamine D1, D2, D3 and D4 receptor binding of N-(3-pyrrolidinylmethyl)benzamides synthesized from aspartic acid. Bioorg Med Chem Lett, 1999. 9(6): P. 841-6. [CrossRef]
- Einsiedel, J.; et al., Phenyloxazoles and phenylthiazoles as benzamide bioisosteres: Synthesis and dopamine receptor binding profiles. Bioorg Med Chem Lett, 2000. 10(17): P. 2041-4. [CrossRef]
- Lehmann, T., H. Hubner, and P. Gmeiner, Dopaminergic 7-aminotetrahydroindolizines: Ex-chiral pool synthesis and preferential D3 receptor binding. Bioorg Med Chem Lett, 2001. 11(21): P. 2863-6. [CrossRef]
- Einsiedel, J.; et al., Stereocontrolled dopamine receptor binding and subtype selectivity of clebopride analogues synthesized from aspartic acid. Bioorg Med Chem Lett, 2003. 13(19): P. 3293-6. [CrossRef]
- Enguehard-Gueiffier, C.; et al., 2-[(4-phenylpiperazin-1-yl)methyl]imidazo(di)azines as selective D4-ligands. Induction of penile erection by 2-[4-(2-methoxyphenyl)piperazin-1-ylmethyl]imidazo[1,2-a]pyridine (PIP3EA), a potent and selective D4 partial agonist. J Med Chem, 2006. 49(13): P. 3938-47. [CrossRef]
- Tietze, R.; et al., Discovery of a dopamine D4 selective PET ligand candidate taking advantage of a click chemistry based REM linker. Bioorg Med Chem Lett, 2008. 18(3): P. 983-8. [CrossRef]
- Balle, T.; et al., Synthesis and structure-affinity relationship investigations of 5-heteroaryl-substituted analogues of the antipsychotic sertindole. A new class of highly selective alpha(1) adrenoceptor antagonists. J Med Chem, 2003. 46(2): P. 265-83. [CrossRef]
- Sromek, A.W.; et al., Synthesis and Evaluation of Fluorinated Aporphines: Potential Positron Emission Tomography Ligands for D2 Receptors. ACS Med Chem Lett, 2011. 2(3): P. 189-194. [CrossRef]
- Banerjee, A.; et al., Click chemistry based synthesis of dopamine D4 selective receptor ligands for the selection of potential PET tracers. Bioorg Med Chem Lett, 2013. 23(22): P. 6079-82. [CrossRef]
- Salama, I.; et al., Synthesis and binding profile of haloperidol-based bivalent ligands targeting dopamine D(2)-like receptors. Bioorg Med Chem Lett, 2014. 24(16): P. 3753-6. [CrossRef]
- Lindsley, C.W. and C.R. Hopkins, Return of D(4) Dopamine Receptor Antagonists in Drug Discovery. J Med Chem, 2017. 60(17): P. 7233-7243. [CrossRef]
- Glase, S.A.; et al., Aryl 1-but-3-ynyl-4-phenyl-1,2,3,6-tetrahydropyridines as potential antipsychotic agents: Synthesis and structure-activity relationships. J Med Chem, 1996. 39(16): P. 3179-87. [CrossRef]
- Yuan, J.; et al., NGB 2904 and NGB 2849: Two highly selective dopamine D3 receptor antagonists. Bioorg Med Chem Lett, 1998. 8(19): P. 2715-8. [CrossRef]
- Birch, A.M.; et al., N-Substituted (2,3-dihydro-1,4-benzodioxin-2-yl)methylamine derivatives as D(2) antagonists/5-HT(1A) partial agonists with potential as atypical antipsychotic agents. J Med Chem, 1999. 42(17): P. 3342-55. [CrossRef]
- Paul, N.M.; et al., Structure-activity relationships for a novel series of dopamine D2-like receptor ligands based on N-substituted 3-aryl-8-azabicyclo[3.2.1]octan-3-ol. J Med Chem, 2008. 51(19): P. 6095-109. [CrossRef]
- Kumar, J.S.; et al., Synthesis and in vivo validation of [O-methyl-11C]2-4-[4-(7-methoxynaphthalen-1-yl)piperazin- 1-yl]butyl-4-methyl-2H-[1,2,4]triazine-3,5-dione: A novel 5-HT1A receptor agonist positron emission tomography ligand. J Med Chem, 2006. 49(1): P. 125-34. [CrossRef]
- Schlotter, K.; et al., Fancy bioisosteres: Novel paracyclophane derivatives as super-affinity dopamine D3 receptor antagonists. J Med Chem, 2006. 49(12): P. 3628-35. [CrossRef]
- Newman, A.H.; et al., N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J Med Chem, 2009. 52(8): P. 2559-70. [CrossRef]
- Ortega, R.; et al., Synthesis, binding affinity and SAR of new benzolactam derivatives as dopamine D3 receptor ligands. Bioorg Med Chem Lett, 2009. 19(6): P. 1773-8. [CrossRef]
- Skultety, M.; et al., Bioisosteric replacement leading to biologically active [2.2]paracyclophanes with altered binding profiles for aminergic G-protein-coupled receptors. J Med Chem, 2010. 53(19): P. 7219-28. [CrossRef]
- Hofling, S.B.; et al., Synthesis, biological evaluation and radiolabelling by 18F-fluoroarylation of a dopamine D3-selective ligand as prospective imaging probe for PET. Bioorg Med Chem Lett, 2010. 20(23): P. 6933-7. [CrossRef]
- Ye, N.; et al., Further SAR study on 11-O-substituted aporphine analogues: Identification of highly potent dopamine D3 receptor ligands. Bioorg Med Chem, 2011. 19(6): P. 1999-2008. [CrossRef]
- Reinart-Okugbeni, R.; et al., Chemoenzymatic synthesis and evaluation of 3-azabicyclo[3.2.0]heptane derivatives as dopaminergic ligands. Eur J Med Chem, 2012. 55: P. 255-61. [CrossRef]
- Spetea, M.; et al., Discovery and pharmacological evaluation of a diphenethylamine derivative (HS665), a highly potent and selective kappa opioid receptor agonist. J Med Chem, 2012. 55(22): P. 10302-6. [CrossRef]
- Majo, V.J.; et al., Synthesis and in vivo evaluation of [(18)F]2-(4-(4-(2-(2-fluoroethoxy)phenyl)piperazin-1-yl)butyl)-4-methyl-1,2,4-triazine-3,5(2H,4H)-dione ([(18)F]FECUMI-101) as an imaging probe for 5-HT1A receptor agonist in nonhuman primates. Bioorg Med Chem, 2013. 21(17): P. 5598-604. [CrossRef]
- Abdelfattah, M.A., J. Lehmann, and A.H. Abadi, Discovery of highly potent and selective D4 ligands by interactive SAR study. Bioorg Med Chem Lett, 2013. 23(18): P. 5077-81. [CrossRef]
- Insua, I.; et al., Synthesis and binding affinity of new 1,4-disubstituted triazoles as potential dopamine D(3) receptor ligands. Bioorg Med Chem Lett, 2013. 23(20): P. 5586-91. [CrossRef]
- van Wieringen, J.P.; et al., Synthesis and characterization of a novel series of agonist compounds as potential radiopharmaceuticals for imaging dopamine D(2)/(3) receptors in their high-affinity state. J Med Chem, 2014. 57(2): P. 391-410. [CrossRef]
- Moller, D.; et al., Functionally selective dopamine D(2), D(3) receptor partial agonists. J Med Chem, 2014. 57(11): P. 4861-75. [CrossRef]
- Sampson, D.; et al., Further evaluation of the tropane analogs of haloperidol. Bioorg Med Chem Lett, 2014. 24(17): P. 4294-7. [CrossRef]
- Weichert, D.; et al., Molecular determinants of biased agonism at the dopamine D(2) receptor. J Med Chem, 2015. 58(6): P. 2703-17. [CrossRef]
- Jorg, M.; et al., Investigation of novel ropinirole analogues: Synthesis, pharmacological evaluation and computational analysis of dopamine D-2 receptor functionalized congeners and homobivalent ligands. Medchemcomm, 2014. 5(7): P. 891-898. [CrossRef]
- Bartuschat, A.L.; et al., Fluoro-substituted phenylazocarboxamides: Dopaminergic behavior and N-arylating properties for irreversible binding. Bioorg Med Chem, 2015. 23(14): P. 3938-47. [CrossRef]
- Moller, D.; et al., 1,4-Disubstituted aromatic piperazines with high 5-HT2A/D2 selectivity: Quantitative structure-selectivity investigations, docking, synthesis and biological evaluation. Bioorg Med Chem, 2015. 23(18): P. 6195-209. [CrossRef]
- Weichert, D.; et al., Structure-guided development of dual beta2 adrenergic/dopamine D2 receptor agonists. Bioorg Med Chem, 2016. 24(12): P. 2641-53. [CrossRef]
- Moller, D.; et al., Discovery of G Protein-Biased Dopaminergics with a Pyrazolo[1,5-a]pyridine Substructure. J Med Chem, 2017. 60(7): P. 2908-2929. [CrossRef]
- Mannel, B.; et al., Hydroxy-Substituted Heteroarylpiperazines: Novel Scaffolds for beta-Arrestin-Biased D(2)R Agonists. J Med Chem, 2017. 60(11): P. 4693-4713. [CrossRef]
- Mannel, B.; et al., beta-Arrestin biased dopamine D2 receptor partial agonists: Synthesis and pharmacological evaluation. Bioorg Med Chem, 2017. 25(20): P. 5613-5628. [CrossRef]
- Stossel, A.; et al., Development of molecular tools based on the dopamine D(3) receptor ligand FAUC 329 showing inhibiting effects on drug and food maintained behavior. Bioorg Med Chem, 2017. 25(13): P. 3491-3499. [CrossRef]
- Omran, A.; et al., Synthesis of 3-(3-hydroxyphenyl)pyrrolidine dopamine D(3) receptor ligands with extended functionality for probing the secondary binding pocket. Bioorg Med Chem Lett, 2018. 28(10): P. 1897-1902. [CrossRef]
- Ashraf-Uz-Zaman, M.; et al., Analogs of penfluridol as chemotherapeutic agents with reduced central nervous system activity. Bioorg Med Chem Lett, 2018. 28(23-24): P. 3652-3657. [CrossRef]
- Chen, P.J.; et al., Design, synthesis, and evaluation of N-(4-(4-phenyl piperazin-1-yl)butyl)-4-(thiophen-3-yl)benzamides as selective dopamine D(3) receptor ligands. Bioorg Med Chem Lett, 2019. 29(18): P. 2690-2694. [CrossRef]
- Tan, L.; et al., Design and Synthesis of Bitopic 2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as Selective Dopamine D3 Receptor Ligands. J Med Chem, 2020. 63(9): P. 4579-4602. [CrossRef]
- Moritz, A.E.; et al., Discovery, Optimization, and Characterization of ML417: A Novel and Highly Selective D(3) Dopamine Receptor Agonist. J Med Chem, 2020. 63(10): P. 5526-5567. [CrossRef]
- Battiti, F.O., A.H. Newman, and A. Bonifazi, Exception That Proves the Rule: Investigation of Privileged Stereochemistry in Designing Dopamine D(3)R Bitopic Agonists. ACS Med Chem Lett, 2020. 11(10): P. 1956-1964. [CrossRef]
- Bergauer, M., H. Hubner, and P. Gmeiner, 2,4-Disubstituted pyrroles: Synthesis, traceless linking and pharmacological investigations leading to the dopamine D4 receptor partial agonist FAUC 356. Bioorg Med Chem Lett, 2002. 12(15): P. 1937-40. [CrossRef]
- Bettinetti, L.; et al., Interactive SAR studies: Rational discovery of super-potent and highly selective dopamine D3 receptor antagonists and partial agonists. J Med Chem, 2002. 45(21): P. 4594-7. [CrossRef]
- Hocke, C.; et al., Synthesis and radioiodination of selective ligands for the dopamine D3 receptor subtype. Bioorg Med Chem Lett, 2004. 14(15): P. 3963-6. [CrossRef]
- Rodriguez Loaiza, P.; et al., Click chemistry based solid phase supported synthesis of dopaminergic phenylacetylenes. Bioorg Med Chem, 2007. 15(23): P. 7248-57. [CrossRef]
- Butini, S.; et al., Discovery of a new class of potential multifunctional atypical antipsychotic agents targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors: Design, synthesis, and effects on behavior. J Med Chem, 2009. 52(1): P. 151-69. [CrossRef]
- Dorfler, M.; et al., Novel D3 selective dopaminergics incorporating enyne units as nonaromatic catechol bioisosteres: Synthesis, bioactivity, and mutagenesis studies. J Med Chem, 2008. 51(21): P. 6829-38. [CrossRef]
- von Coburg, Y.; et al., Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics. Bioorg Med Chem Lett, 2009. 19(2): P. 538-42. [CrossRef]
- Tschammer, N.; et al., Highly potent 5-aminotetrahydropyrazolopyridines: Enantioselective dopamine D3 receptor binding, functional selectivity, and analysis of receptor-ligand interactions. J Med Chem, 2011. 54(7): P. 2477-91.
- Berry, C.B.; et al., Discovery and Characterization of ML398, a Potent and Selective Antagonist of the D4 Receptor with in Vivo Activity. ACS Med Chem Lett, 2014. 5(9): P. 1060-4. [CrossRef]
- Ponnala, S., N. Kapadia, and W.W. Harding, Identification of tris-(phenylalkyl) amines as new selective h5-HT2B receptor antagonists (vol 6, pg 601, 2015). Medchemcomm, 2015. 6(4): P. 732-732. [CrossRef]
- Gadhiya, S.; et al., New Dopamine D3-Selective Receptor Ligands Containing a 6-Methoxy-1,2,3,4-tetrahydroisoquinolin-7-ol Motif. ACS Med Chem Lett, 2018. 9(10): P. 990-995. [CrossRef]
- Karki, A.; et al., Structural manipulation of aporphines via C10 nitrogenation leads to the identification of new 5-HT(7A)R ligands. Bioorg Med Chem, 2020. 28(15): P. 115578. [CrossRef]
- Hawkins, P.C.; et al., Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model, 2010. 50(4): P. 572-84. [CrossRef]
- Jones, G.; et al., Development and validation of a genetic algorithm for flexible docking. J Mol Biol, 1997. 267(3): P. 727-48. [CrossRef]
- Xu, P.; et al., Structures of the human dopamine D3 receptor-G(i) complexes. Mol Cell, 2021. 81(6): P. 1147-1159 e4. [CrossRef]
- Altschul, S.F.; et al., Basic local alignment search tool. J Mol Biol, 1990. 215(3): P. 403-10. [CrossRef]
- BIOVIA, Dassault Systèmes, BIOVIA Discovery Studio, Release 2018. 2018, San Diego: Dassault Systems.
- Wolber, G. and T. Langer, LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model, 2005. 45(1): P. 160-9. [CrossRef]
- Joober, R. and P. Boksa, Clozapine: A distinct, poorly understood and under-used molecule. J Psychiatry Neurosci, 2010. 35(3): P. 147-9. [CrossRef]
- Perrone, R.; et al., A structure-affinity relationship study on derivatives of N-[2-[4-(4-Chlorophenyl)piperazin-1-yl]ethyl]-3-methoxybenzamide, a high-affinity and selective D(4) receptor ligand. J Med Chem, 2000. 43(2): P. 270-7. [CrossRef]
- Smith, J.L.; et al., Inhibition of dengue virus replication by a class of small-molecule compounds that antagonize dopamine receptor d4 and downstream mitogen-activated protein kinase signaling. J Virol, 2014. 88(10): P. 5533-42. [CrossRef]
- Campiani, G.; et al., New antipsychotic agents with serotonin and dopamine antagonist properties based on a pyrrolo[2,1-b][1,3]benzothiazepine structure. J Med Chem, 1998. 41(20): P. 3763-72. [CrossRef]
- Staron, J.; et al., Virtual screening-driven discovery of dual 5-HT(6)/5-HT(2A) receptor ligands with pro-cognitive properties. Eur J Med Chem, 2020. 185: P. 111857. [CrossRef]
- Peprah, K.; et al., Multi-receptor drug design: Haloperidol as a scaffold for the design and synthesis of atypical antipsychotic agents. Bioorg Med Chem, 2012. 20(3): P. 1291-7. [CrossRef]
- Schoemaker, H.; et al., Neurochemical characteristics of amisulpride, an atypical dopamine D2/D3 receptor antagonist with both presynaptic and limbic selectivity. J Pharmacol Exp Ther, 1997. 280(1): P. 83-97.
- Daina, A., O. Michielin, and V. Zoete, SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res, 2019. 47(W1): P. W357-W364. [CrossRef]
- Bragina, M.E.; et al., The SwissSimilarity 2021 Web Tool: Novel Chemical Libraries and Additional Methods for an Enhanced Ligand-Based Virtual Screening Experience. Int J Mol Sci, 2022. 23(2). [CrossRef]
- Zoete, V.; et al., SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J Chem Inf Model, 2016. 56(8): P. 1399-404. [CrossRef]
- Charifson, P.S.; et al., Conformational analysis and molecular modeling of 1-phenyl-, 4-phenyl-, and 1-benzyl-1,2,3,4-tetrahydroisoquinolines as D1 dopamine receptor ligands. J Med Chem, 1989. 32(9): P. 2050-8. [CrossRef]
- Pettersson, F.; et al., Synthesis and evaluation of a set of 4-phenylpiperidines and 4-phenylpiperazines as D2 receptor ligands and the discovery of the dopaminergic stabilizer 4-[3-(methylsulfonyl)phenyl]-1-propylpiperidine (huntexil, pridopidine, ACR16). J Med Chem, 2010. 53(6): P. 2510-20. [CrossRef]
- Zheng, Z.; et al., Structure-Based Discovery of New Antagonist and Biased Agonist Chemotypes for the Kappa Opioid Receptor. J Med Chem, 2017. 60(7): P. 3070-3081. [CrossRef]
- Nievergelt, A.; et al., Identification of serotonin 5-HT1A receptor partial agonists in ginger. Bioorg Med Chem, 2010. 18(9): P. 3345-51. [CrossRef]
- Maramai, S.; et al., Dopamine D3 Receptor Antagonists as Potential Therapeutics for the Treatment of Neurological Diseases. Front Neurosci, 2016. 10: P. 451. [CrossRef]
- Fan, L.; et al., Haloperidol bound D(2) dopamine receptor structure inspired the discovery of subtype selective ligands. Nat Commun, 2020. 11(1): P. 1074. [CrossRef]
Figure 1.
Overview of DR sub-domains relevant in DR subtype selectivity. Red dots highlight the highly conserved amino acids Val2.61, Leu2.64, Phe3.28, GlyECL1 and LeuECL2 in the SBP of D2R and D3R. Zoomed in box of the conserved SBP shows the 3D arrangement. Partial primary sequences (amino acid positions are shown in the index) of ICL3 are shown for both D2R and D3R. (Created with BioRender.com).
Figure 1.
Overview of DR sub-domains relevant in DR subtype selectivity. Red dots highlight the highly conserved amino acids Val2.61, Leu2.64, Phe3.28, GlyECL1 and LeuECL2 in the SBP of D2R and D3R. Zoomed in box of the conserved SBP shows the 3D arrangement. Partial primary sequences (amino acid positions are shown in the index) of ICL3 are shown for both D2R and D3R. (Created with BioRender.com).
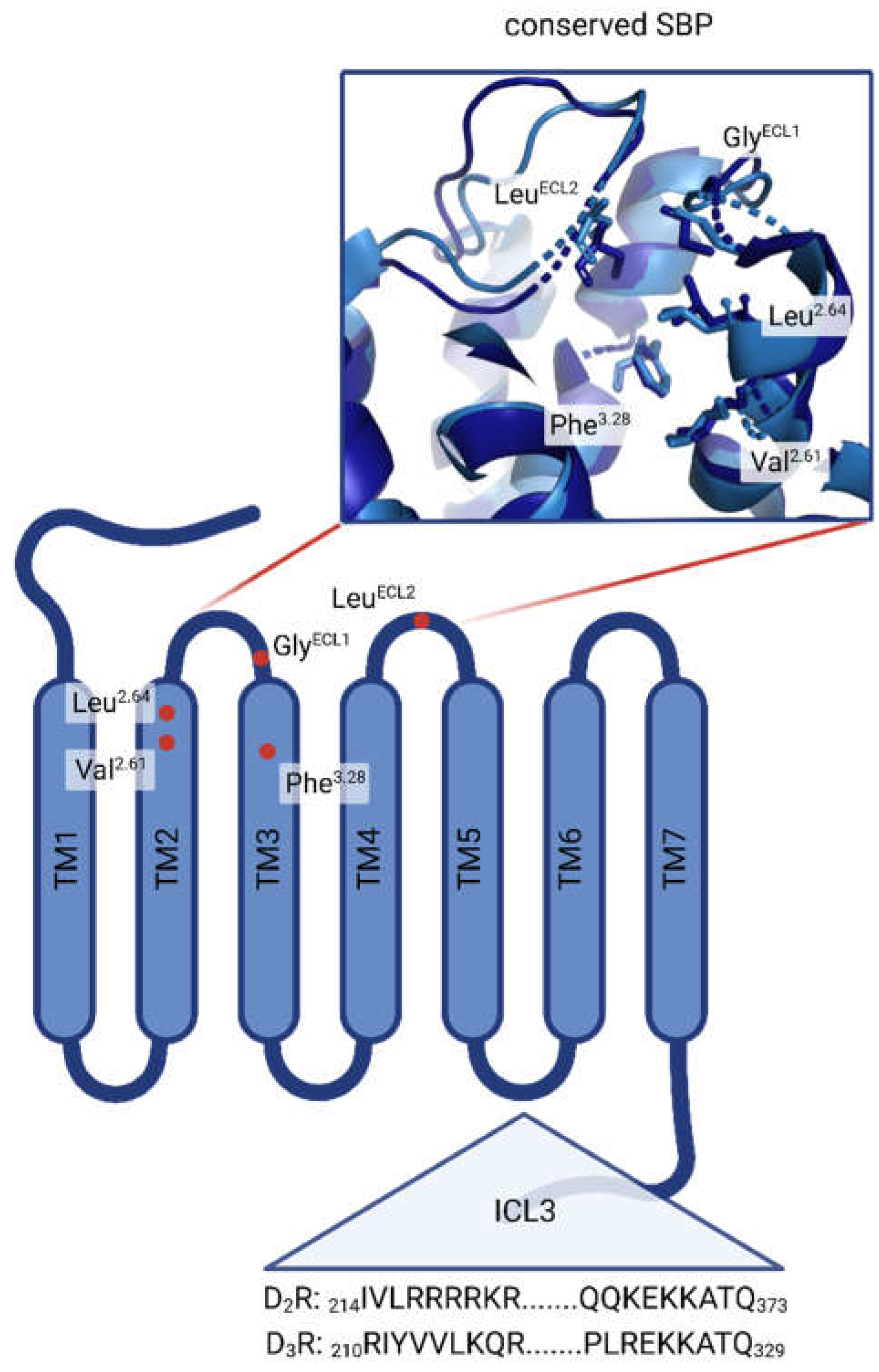
Figure 2.
Summary of the utilized approach to modify PDB entry 7jvr [34] using a MDS approach.a ‘best-fit’ docking pose was assessed based on the correct positioning of apomorphine in the related D1R structure 7jvq [34] with respect to the orientation of the catechol motif towards the serine triade as well as the formation of a salt-bridge with Asp3.32.
Figure 2.
Summary of the utilized approach to modify PDB entry 7jvr [34] using a MDS approach.a ‘best-fit’ docking pose was assessed based on the correct positioning of apomorphine in the related D1R structure 7jvq [34] with respect to the orientation of the catechol motif towards the serine triade as well as the formation of a salt-bridge with Asp3.32.
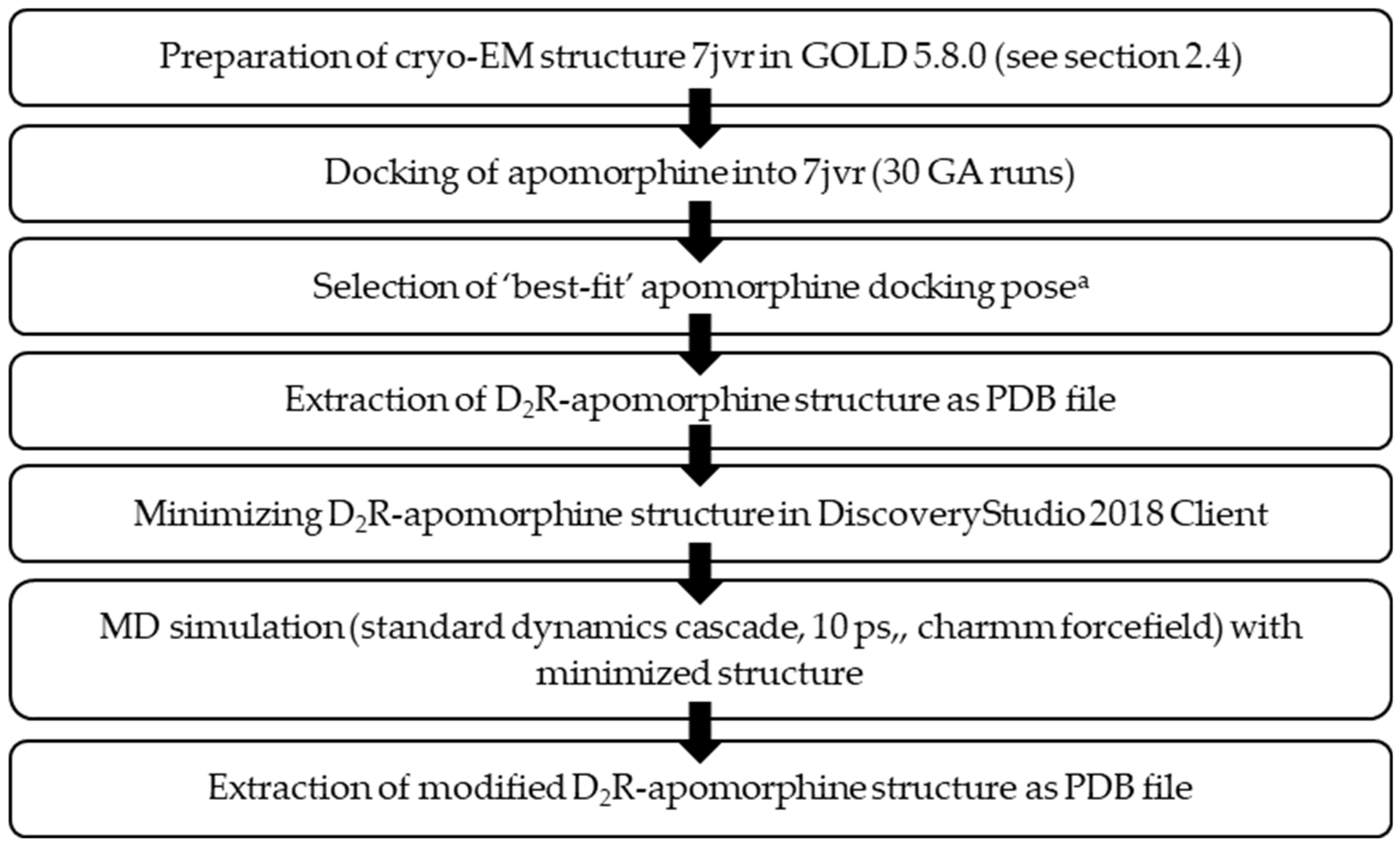
Figure 3.
Overview of the 2D structures of ligands investigated in silico and in vitro.
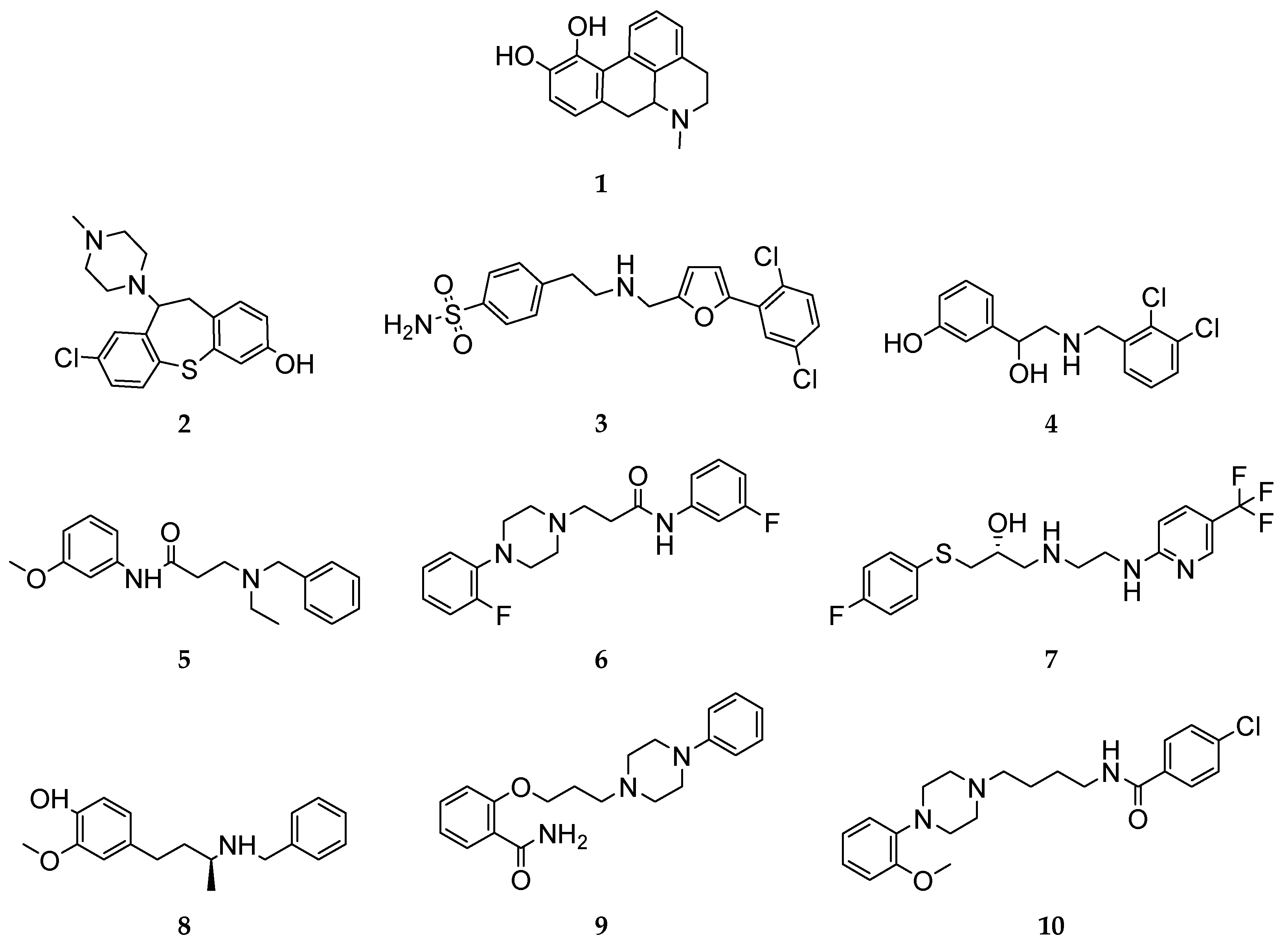
Figure 4.
Overview of the structural (dis-)similarity of the investigated novel ligands (compounds 2 to 10). (Dis-)similarity was assessed utilizing a TS matrix based on radial fingerprints (ECFP4). TS ranging from 0 (green) to 1 (red) showing unrelated and identical structures, respectively.
Figure 4.
Overview of the structural (dis-)similarity of the investigated novel ligands (compounds 2 to 10). (Dis-)similarity was assessed utilizing a TS matrix based on radial fingerprints (ECFP4). TS ranging from 0 (green) to 1 (red) showing unrelated and identical structures, respectively.

Figure 5.
Comparison of the KI values of compounds (a) 2 and (b) 10 determined at the investigated DR subtypes D1R, D2R and D3R. Vertical, dotted lines indicate the respective KI values at the different DR subtypes. KI values [µM] ± SD were determined with n = 6.
Figure 5.
Comparison of the KI values of compounds (a) 2 and (b) 10 determined at the investigated DR subtypes D1R, D2R and D3R. Vertical, dotted lines indicate the respective KI values at the different DR subtypes. KI values [µM] ± SD were determined with n = 6.
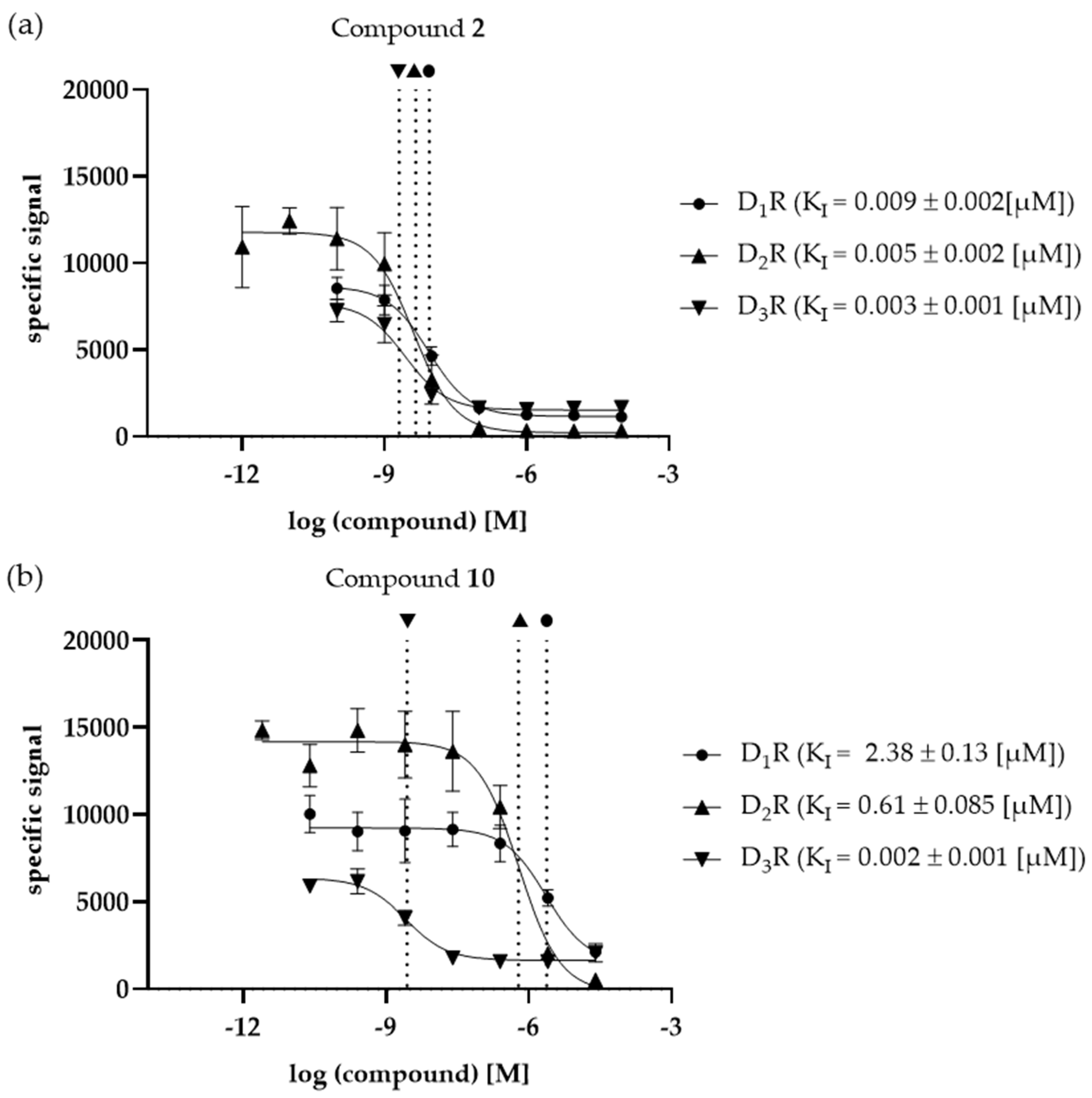
Figure 6.
Comparison of the fold-differences based on the distances [Å] between each DR-selective subsets COM and the respective conserved Gly residue.
Figure 6.
Comparison of the fold-differences based on the distances [Å] between each DR-selective subsets COM and the respective conserved Gly residue.
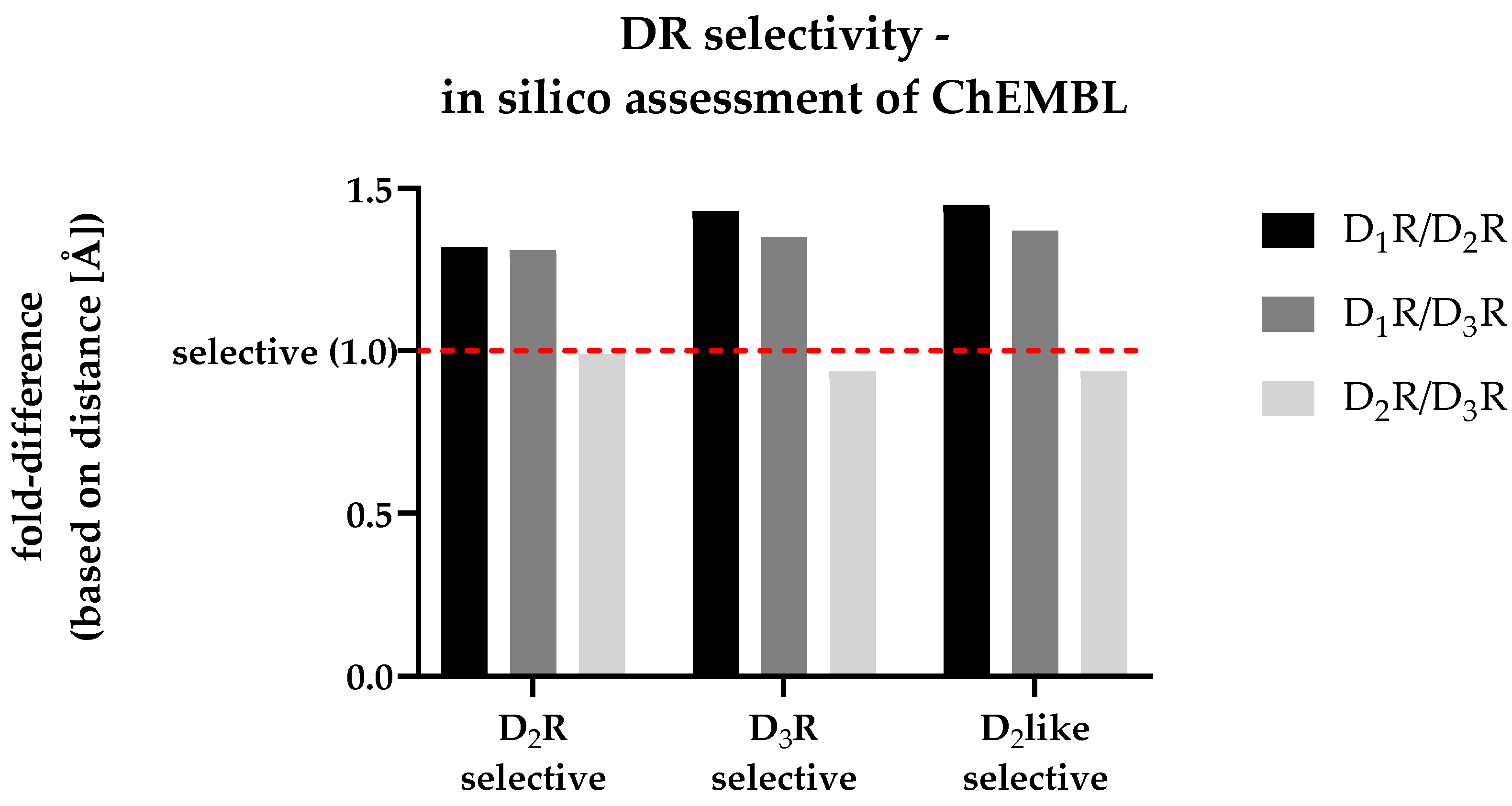
Figure 7.
Correlation of in vitro determined fold-differences (x-axis) and in silico determined distance-based fold-differences (y-axis) to investigate DR subtype selectivity of novel compounds. The dashed red line shows a distance-based fold-difference of 1.0, indicating a non-selective profile of the respective compound based on the in silico analysis.
Figure 7.
Correlation of in vitro determined fold-differences (x-axis) and in silico determined distance-based fold-differences (y-axis) to investigate DR subtype selectivity of novel compounds. The dashed red line shows a distance-based fold-difference of 1.0, indicating a non-selective profile of the respective compound based on the in silico analysis.
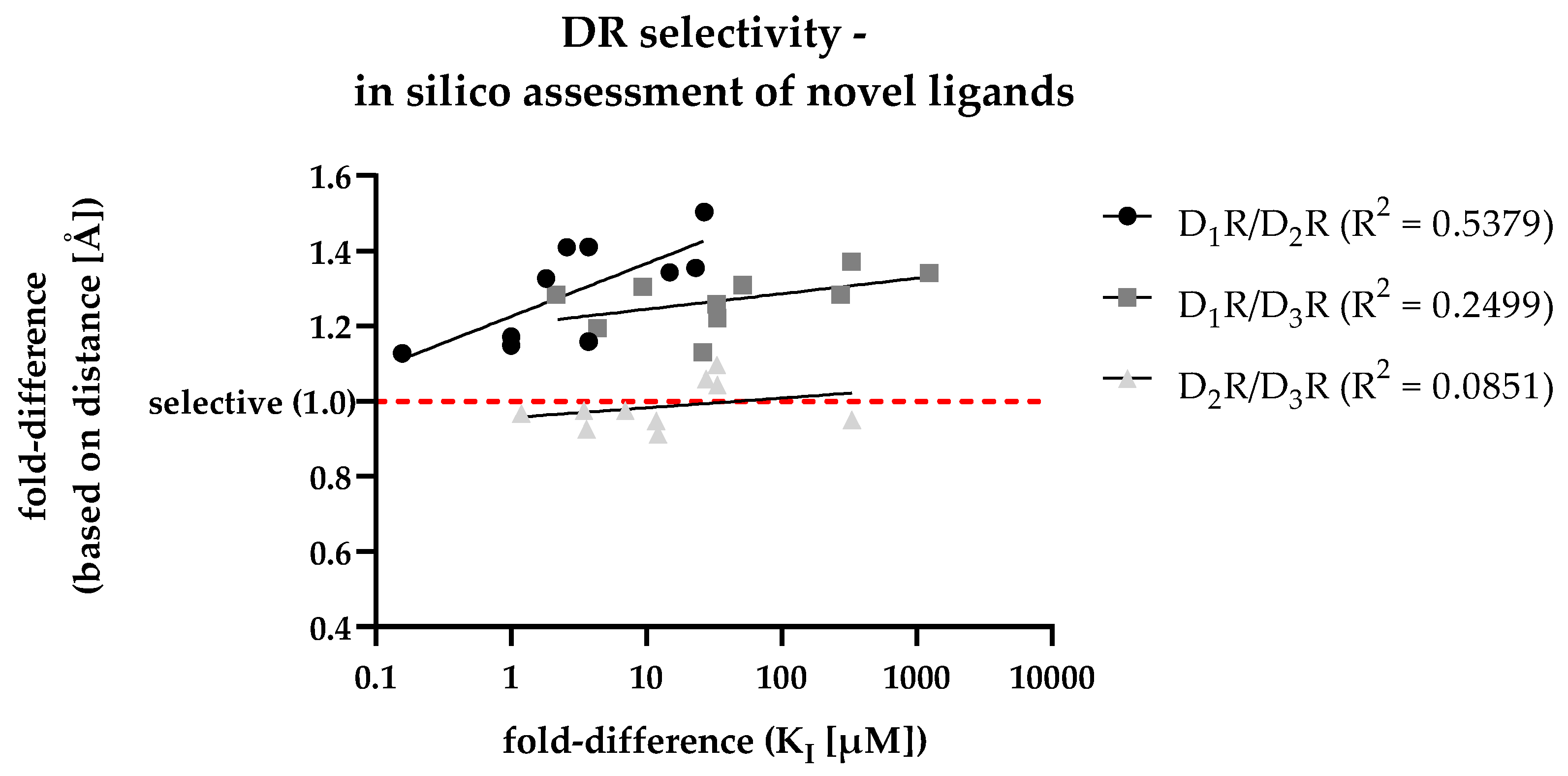
Figure 8.
Alignment of the most frequent poses of compound 2 docked into D1R, D2R and D3R. The surface of the highly conserved OBP is shown in blue (based on D1R) consisting of Asp3.32 (highlighted in cyan) and Ser5.42/5.43/5.46. The conserved SBP-surface is displayed in orange (D2R) and green (D3R) consisting of Val2.61, Leu2.64, GlyEL1, Phe3.28 and CysEL2 (individual amino acid labels shown in Table 3), respectively. KI values determined in vitro are shown for each DR subtype. 2D structure of compound 2 is shown. Amine functional group involved in formation of the salt-bridge is highlighted in red.
Figure 8.
Alignment of the most frequent poses of compound 2 docked into D1R, D2R and D3R. The surface of the highly conserved OBP is shown in blue (based on D1R) consisting of Asp3.32 (highlighted in cyan) and Ser5.42/5.43/5.46. The conserved SBP-surface is displayed in orange (D2R) and green (D3R) consisting of Val2.61, Leu2.64, GlyEL1, Phe3.28 and CysEL2 (individual amino acid labels shown in Table 3), respectively. KI values determined in vitro are shown for each DR subtype. 2D structure of compound 2 is shown. Amine functional group involved in formation of the salt-bridge is highlighted in red.
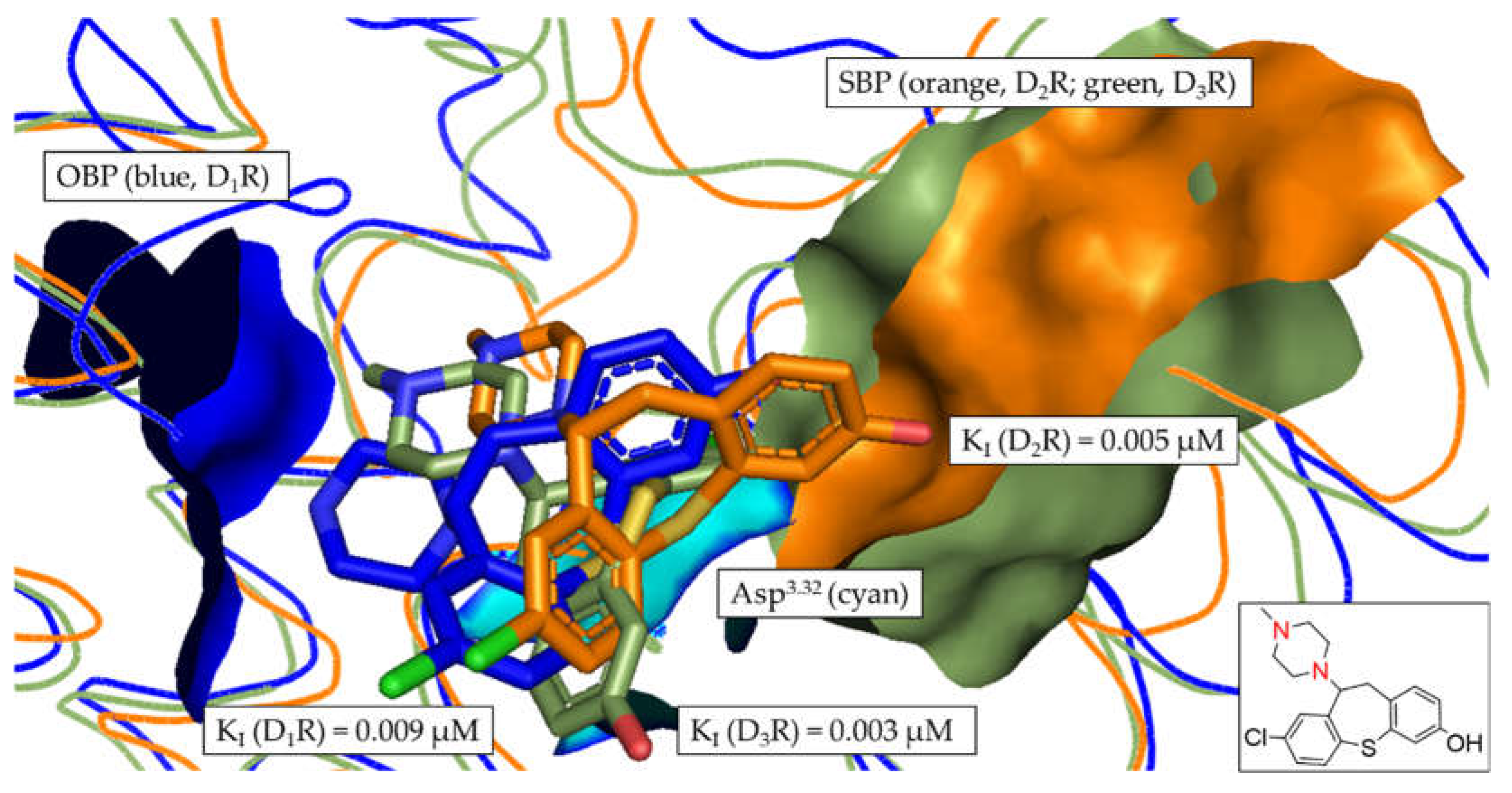
Figure 9.
Alignment of the most frequent poses of compound 10 docked into D1R, D2R and D3R. The surface of the highly conserved OBP is shown in blue (based on D1R) consisting of Asp3.32 (highlighted in cyan) and Ser5.42/5.43/5.46. The conserved SBP-surface is displayed in orange (D2R) and green (D3R) consisting of Val2.61, Leu2.64, GlyEL1, Phe3.28 and CysEL2 (individual amino acid labels shown in Table 3), respectively. KI values determined in vitro are shown for each DR subtype. 2D structure of compound 10 is shown. Amine functional group involved in formation of the salt-bridge is highlighted in red.
Figure 9.
Alignment of the most frequent poses of compound 10 docked into D1R, D2R and D3R. The surface of the highly conserved OBP is shown in blue (based on D1R) consisting of Asp3.32 (highlighted in cyan) and Ser5.42/5.43/5.46. The conserved SBP-surface is displayed in orange (D2R) and green (D3R) consisting of Val2.61, Leu2.64, GlyEL1, Phe3.28 and CysEL2 (individual amino acid labels shown in Table 3), respectively. KI values determined in vitro are shown for each DR subtype. 2D structure of compound 10 is shown. Amine functional group involved in formation of the salt-bridge is highlighted in red.
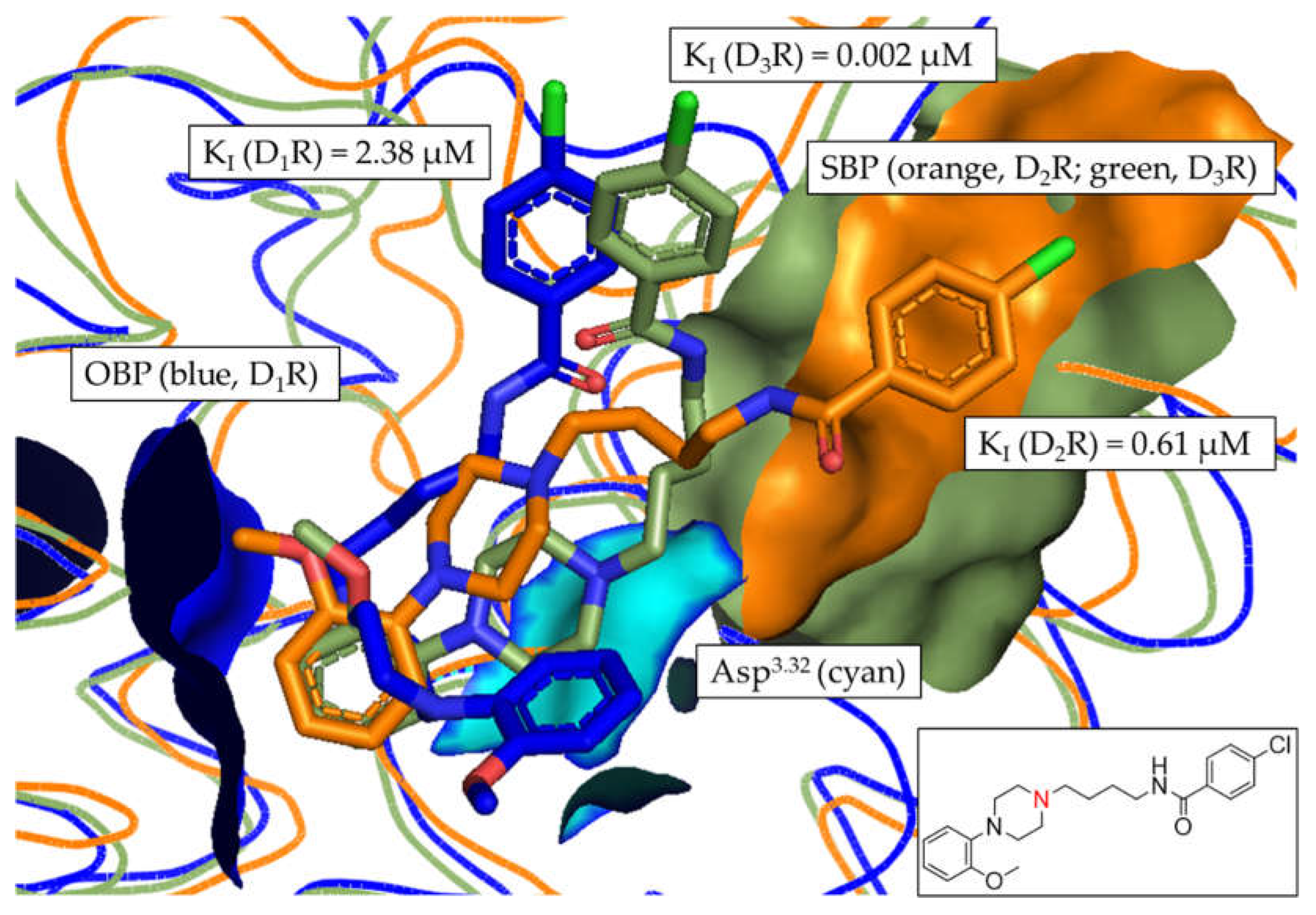
Figure 10.
Alignment of the most frequent poses of compound 4 docked into D1R, D2R and D3R. The surface of the highly conserved OBP is shown in blue (based on D1R) consisting of Asp3.32 (highlighted in cyan) and Ser5.42/5.43/5.46. The conserved SBP-surface is displayed in orange (D2R) and green (D3R) consisting of Val2.61, Leu2.64, GlyEL1, Phe3.28 and CysEL2 (individual amino acid labels shown in Table 3), respectively. KI values determined in vitro are shown for each DR subtype. 2D structure of compound 4 is shown. Amine functional group involved in formation of the salt-bridge is highlighted in red.
Figure 10.
Alignment of the most frequent poses of compound 4 docked into D1R, D2R and D3R. The surface of the highly conserved OBP is shown in blue (based on D1R) consisting of Asp3.32 (highlighted in cyan) and Ser5.42/5.43/5.46. The conserved SBP-surface is displayed in orange (D2R) and green (D3R) consisting of Val2.61, Leu2.64, GlyEL1, Phe3.28 and CysEL2 (individual amino acid labels shown in Table 3), respectively. KI values determined in vitro are shown for each DR subtype. 2D structure of compound 4 is shown. Amine functional group involved in formation of the salt-bridge is highlighted in red.
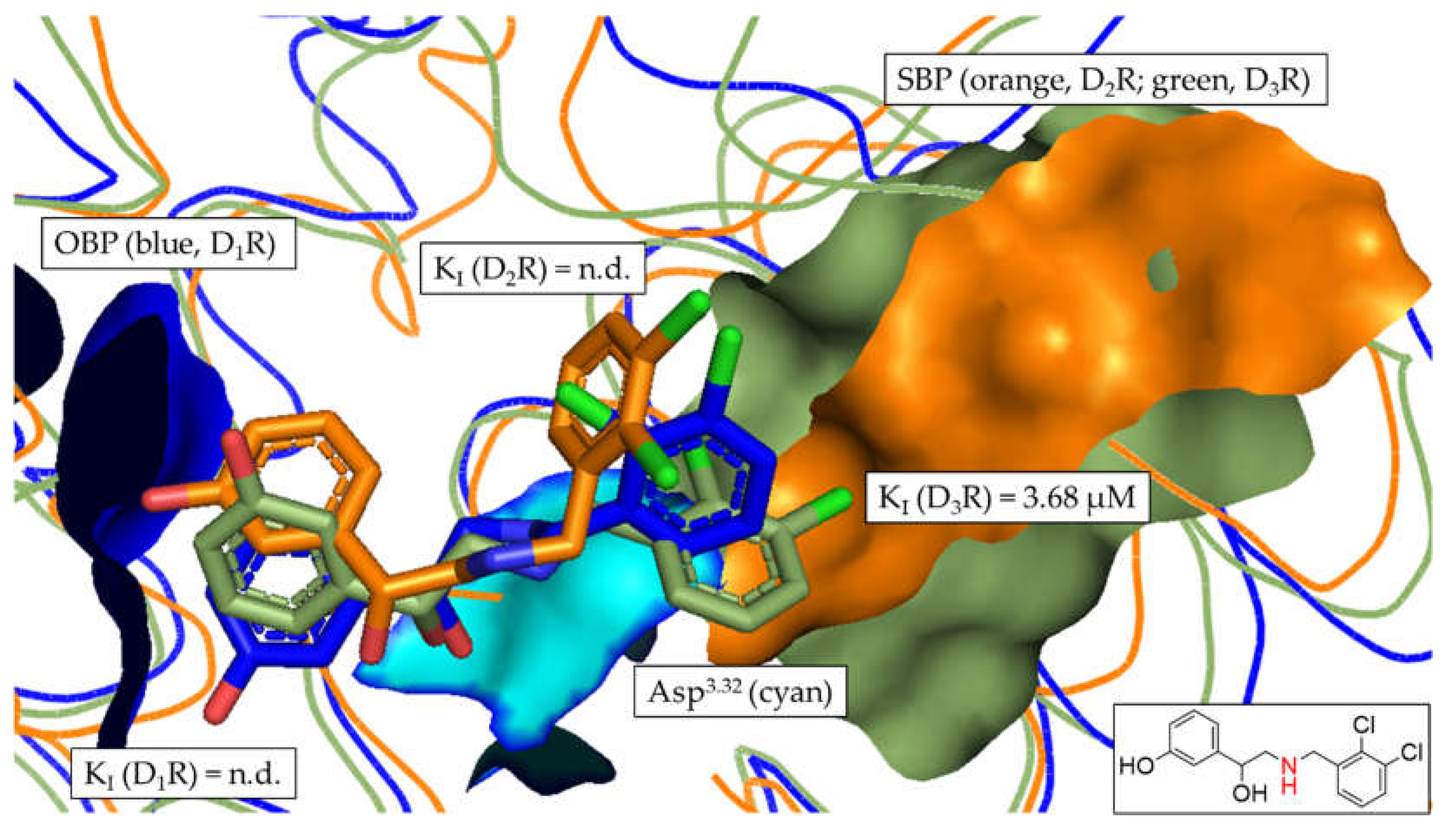
Figure 11.
Comparison of the chemical scaffolds of compounds 6, 10 and the R22-analogue. Structural elements highlighted in red show similarities between the different compounds also indicating the differences in linker length.
Figure 11.
Comparison of the chemical scaffolds of compounds 6, 10 and the R22-analogue. Structural elements highlighted in red show similarities between the different compounds also indicating the differences in linker length.

Table 1.
Summary of the amino acid flexibility settings of the D1R cryo-EM structure used during docking.
Table 1.
Summary of the amino acid flexibility settings of the D1R cryo-EM structure used during docking.
| Setting | Value |
|---|---|
| Flexible Sidechains | ASP103 R, 1 rotamer (free) |
| TRP285 R, 1 rotamer (free) | |
| PHE288 R, 1 rotamer (free) | |
| PHE289, 1 rotamer (free) | |
| ASN292 R, 1 rotamer (free) |
Table 2.
Summary of the amino acid flexibility settings of the D3R cryo-EM structure used during docking.
Table 2.
Summary of the amino acid flexibility settings of the D3R cryo-EM structure used during docking.
| Setting | Value |
|---|---|
| Flexible Sidechains | ASP110 R, 1 rotamer (free) |
| HIS349 R, 8 rotamers (constrained) |
Table 3.
Overview of the amino acids forming the SBP in different DR subtypes. D2like subtypes include D2R and D3R.
Table 3.
Overview of the amino acids forming the SBP in different DR subtypes. D2like subtypes include D2R and D3R.
| DR subtype | |||
|---|---|---|---|
| D3R | D1R | D2R | Status |
| Val86 | Lys81 | Val91 | Conserved in D2like DRs |
| Leu89 | Ala84 | Leu94 | Conserved in D2like DRs |
| Gly94 | Gly88 | Gly98 | Conserved |
| Phe106 | Trp99 | Phe110 | Conserved in D2like DRs |
| Cys181 | Cys186 | Cys182 | Conserved |
Table 4.
Summary of the in vitro screening of known and potential DR ligands considered selective for one of the three investigated subtypes. All measurements were conducted at a concentration of 10 µM (n = 4). Fluorescence decrease was normalized to the control.
Table 4.
Summary of the in vitro screening of known and potential DR ligands considered selective for one of the three investigated subtypes. All measurements were conducted at a concentration of 10 µM (n = 4). Fluorescence decrease was normalized to the control.
| Compound | Normalized Decrease in Fluorescence (NDF) ± SD | ||
|---|---|---|---|
| D1R | D2Ra | D3R | |
| Control | 1 | 1 | 1 |
| 1 | 3.47 ± 1.04 | 9.44 ± 5.97 | 3.82 ± 1.32 |
| 2 | 5.46 ± 1.97 | 40.41 ± 1.39 | 4.16 ± 1.08 |
| 3 | 1.78 ± 1.35 | 3.99 ± 2.58 | 3.96 ± 1.10 |
| 4 | 0.90 ± 0.31 | 0.90 ± 0.39 | 2.75 ± 0.61 |
| 5 | 1.26 ± 0.64 | 15.74 ± 18.15 | 2.65 ± 0.64 |
| 6 | 2.15 ± 1.16 | 8.18 ± 3.62 | 4.21 ± 0.79 |
| 7 | 1.57 ± 0.54 | 10.85 ± 4.93 | 3.79 ± 0.70 |
| 8 | 1.20 ± 0.59 | 1.10 ± 0.52 | 2.63 ± 0.52 |
| 9 | 1.71 ± 0.86 | 22.08 ± 6.62 | 3.97 ± 0.78 |
| 10 | 2.59 ± 1.04 | 22.89 ± 8.41 | 4.09 ± 1.07 |
a Values were extracted from previous experiments detailed in [42].
Table 5.
Summary of the determined KI values of all ligands investigated in vitro. Binding affinities are shown for the three different DR subtypes D1R, D2R and D3R. KI values were determined using n = 6. Apomorphine (1) was used as a control.
Table 5.
Summary of the determined KI values of all ligands investigated in vitro. Binding affinities are shown for the three different DR subtypes D1R, D2R and D3R. KI values were determined using n = 6. Apomorphine (1) was used as a control.
| Cpd. | KI [µM] | selectivity | ||||
|---|---|---|---|---|---|---|
| D1R | D2R | D3R | D1R/D2R | D1R/D3R | D2R/D3R | |
| 1 | 0.36 ± 0.009 | 2.36 ± 0.14 | 0.12 ± 0.048 | 0.15 | 3.06 | 19.8 |
| 2 | 0.009 ± 0.002 | 0.005 ± 0.002a | 0.003 ± 0.001 | 1.95 | 3.23 | 1.66 |
| 3 | n.d.b | 4.66 ± 2.69a | 0.38 ± 0.022 | > 21.4b | 262.2 | 12.2 |
| 4 | n.d.b | n.d.b | 3.68 ± 0.94 | - | > 27.2b | > 27.2b |
| 5 | 46.9 ± 27.4 | 10.95 ± 4.43a | 2.25 ± 0.91 | 4.28 | 20.8 | 4.86 |
| 6 | 7.76 ± 4.41 | 1.35 ± 0.63a | 0.37 ± 0.28 | 5.77 | 20.7 | 3.56 |
| 7 | 8.33 ± 2.17 | 2.78 ± 1.06a | 0.68 ± 0.068 | 3.00 | 12.3 | 4.11 |
| 8 | n.d.b | n.d.b | 2.32 ± 0.92 | - | > 43.1b | > 43.1b |
| 9 | 9.46 ± 1.18 | 0.33 ± 0.093a | 0.024 ± 0.003 | 28.6 | 395.1 | 13.8 |
| 10 | 2.38 ± 0.13 | 0.61 ± 0.085 | 0.002 ± 0.001 | 3.91 | 1031.4 | 263.7 |
a Values were extracted from previous experiments detailed in [42]. b KI values could not be quantitatively determined. To calculate values for selectivity, KI values were assumed to be ≥ 100 µM (highest concentration used during in vitro testing). n.d., not determinable.
Table 6.
Summary of the distance-based docking approach of different DR-subtype selective ChEMBL datasets. Calculated fold-differences were based on distances [Å] between COM and the respective conserved Gly residue.
Table 6.
Summary of the distance-based docking approach of different DR-subtype selective ChEMBL datasets. Calculated fold-differences were based on distances [Å] between COM and the respective conserved Gly residue.
| Dataset | Fold-difference (cons. Gly-COM)) | ||
|---|---|---|---|
| D1R/D2R | D1R/D3R | D2R/D3R | |
| D2R selective | 1.32 | 1.31 | 0.99 |
| D3R selective | 1.43 | 1.35 | 0.94 |
| D2like selective | 1.45 | 1.37 | 0.94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



