
Submitted:
27 April 2023
Posted:
28 April 2023
You are already at the latest version
Abstract
Diabetes mellitus is a global health issue that necessitates the development of novel therapeutic approaches. Today numerous experimental methods have been used and set up to evaluate novel drugs with antidiabetic effects. In addition, several research studies have been done to induce diabetes under in vitro and in vivo conditions, but only a small number of these efforts are currently in a usable stage. This review aims to provide useful guidance for researchers who seek to evaluate the antidiabetic potential of drugs, agents, or new medications using the standard methods published by authorities and academics worldwide organizations and research centers. In this review, we collected 132 relevant articles from prominent databases such as PubMed, ScienceDirect, Scopus and others to summarize in vitro and in vivo experimental validation of antidiabetic treatment from 1943 to present.
Keywords:
Diabetes mellitus
; antidiabetic activity
; in vitro
; in vivo
; assays
1. Introduction
Diabetes mellitus (DM) is a metabolic disorder marked by unnecessarily high blood glucose levels that impose substantial economic costs on society [1], in which impairment or degeneration of the insulin-producing pancreatic beta cells is a crucial aspect that leads to diabetes [2,3]. By 2030, it is estimated that global diabetes prevalence will increase to 578 million (10.2%) from 9.3% in 2019 [4]. Diabetes can be classified into different types, including type 1, type 2, gestational diabetes, steroid use, neonatal diabetes and maturity-onset diabetes of the young MODY. Type 1 diabetes mellitus (T1DM) and Type 2 diabetes mellitus (T2DM) are the two primary subtypes of (DM) which are classified as impaired insulin secretion in T1DM and defective insulin action in T2DM [5,6]. The onset of T2DM is gradual and includes a functional insulin deficiency, an imbalance in insulin levels and insulin sensitivity, which is directly related to aging or obesity [7,8]. Conversely, it is expected that T2DM affects older and middle-aged individuals with chronic hyperglycemia due to poor lifestyle and nutritional choices [9,10]. In contrast, in T1DM, pancreatic beta cells are frequently destroyed due to an autoimmune process, which results in an absent or deficient level of insulin in the body [11,12].
Most cases of T1DM occur in children and teenagers. T1DM and T2DM have separate etiologies and pathogenetic mechanisms that require different courses of treatment [13]. Several antidiabetic medications have been developed to increase insulin sensitivity secretion and reduce the symptoms. However, prolonged usage of these medications may result in harmful and toxic effects [14]. For instance, using metformin as an oral diabetes medication to treat T2DM can result in various side effects. Metformin works at the molecular level to inhibit the liver's mitochondrial respiratory chain, activating AMPK and significantly improving insulin sensitivity [15].
This drug, however, can cause serious side effects such as anorexia, nausea, abdominal pain and diarrhea [16]. The standard test method for the assessment of antidiabetics can be applied to evaluating novel drugs with antidiabetic activities. In the recent decade, several research studies have been published to set up in vitro and in vivo antidiabetic assays. However, only a small number of these efforts are currently in a usable stage. This review offers helpful guidance for researchers who want to evaluate the antidiabetic potential of drugs, agents, or new-developed medications using the standard methods published by authorities and academics worldwide, organizations, and research centers.
2. Antidiabetic assays
This review summarizes the most relevant books and articles from the electronic databases PubMed, Web of Science, Science Direct, Scopus, and Google Scholar. As search criteria, the keywords antidiabetic assays, diabetes induction under in vitro and in vivo conditions, and diabetes were used in various combinations.
Inclusion criteria were determined using two criteria sets, the broad (in vitro and in vivo antidiabetic assays and methods) and detailed (articles with antidiabetic testing). The selected articles and books were published from 1943 onwards (Figure 1).
2.1. Experimental methods
2.1.1. In vitro assays
Double gene knockdown to produce the diabetic cell lines
The development of cell-based screening techniques is required to assess new drugs with potential effects for treating diabetes across the spectrum of dysfunctional genes [17,18,19]. Studies have shown that specific genes are crucial in producing insulin [20,21]. Therefore, the production of genetically modified cells can be used as a first step in evaluating the efficiency of new medications. Various strategies have been considered to assess the antidiabetic activity of new compounds and agents under the in vitro condition. One of the most effective methods is to knock down the expression of specific genes, leading to cellular dysfunction and increasing the risk of diabetes [22]. Researchers have reported that stopping or decreasing the expression (knock down) of Irs-1 and Pdx-1 [23], Shp and Gk, Gk and Irs-1 [24], HNF-1α and Pdx-1 [25], Irs-2, and Kir6.2 [22] can induce diabetes in mice. To produce viable cells for this assay, it is required to identify multiple gene regulations and RNAi sequences and knocking down methods should be considered. Based on research by Saito and coworkers in 2013[22], double knockdown cell lines were produced by stopping or decreasing the expression of Irs-1 and Pdx-1 genes. Pancreatic and duodenal homeobox 1 (Pdx-1) directly activates insulin production genes like insulin I (Ins1) and insulin II (Ins2) to play a significant role in the production of insulin. Moreover, it acts as a transcriptional activator for several genes, including those encoding glucose transporter type 2 insulin, somatostatin, glucokinase, and islet amyloid polypeptide. The nuclear protein that is encoded participates in the early stages of pancreatic development and plays a crucial role in the glucose-dependent control of insulin gene expression. Pancreatic agenesis can be related to gene defects that cause early onset insulin-dependent diabetes mellitus (IDDM) [26,27]. Another gene is insulin receptor substrate 1(Irs-1); defects in this gene lead to insulin resistance or insulin reception deficiency and type 2 diabetes [28,29]. Saito and coworkers in 2013, produced a novel diabetic cell line which can be used as a model for in-vitro evaluation of medical materials. In this regard, a vector was electroporated into a mouse ES cell line (EB3) to decrease the expression of Irs-1 and Pdx-1, and glucose-stimulated insulin secretion was measured using a mouse insulin ELISA kit [22]. A vector was electroporated into mouse ES cells (EB3) to decrease the expression of Irs-1 anusing a mouse Insulin ELISA kit.
α-Glucosidase enzyme inhibition
The enzyme glucosidase catalyses the essential process of digesting carbohydrates and releasing glucose. Hence glucosidase inhibitors are involved in decreasing postprandial blood glucose level by blocking the hydrolysis of glucose through the carbohydrate metabolising enzymes, amylase and glucosidase. Inhibitory activities against these two enzymes can be used to evaluate the potential activity of new antidiabetic agents (Table 1).
Alpha-glucosidase inhibition assay can be perfomed using 5 μl of the sample (drugs or new medications) to 20 μl α-glucosidase enzyme with 50 μg/ml concentration into wells of a 96 well plate and mixed with 60 µL of 67 mM potassium phosphate buffer (pH 6.8). The reactions start with incubation at 37 °C for 5 min. After that, add 10 μl of p-Nitrophenyl-α-glucopyranoside (10 mM pNPG) to each well and incubate the plate at 37 °C for 50 min (Figure 2). In the next step, add 100 μl of sodium carbonate solution (100 mM Na2CO3) to each well, and measure the absorbance with a spectrophotometer at 405 nm [30]. For control, wells use buffer without enzyme and substrate.
Evaluation is then performed using
where A = the absorbance of the blank and B = the absorbance of the sample (drugs or new medications).
α-Amylase enzyme inhibition
α-Amylase is an important enzyme that hydrolysing polysaccharides and glycogen to shorter chains such as maltose and dextrin. Amylase enzyme inhibition could be considered as a valuable strategy for treating carbohydrate absorption diseases like diabetes and obesity [31,32] (Table 1).
To perform this assay, α-amylase solution should be prepared by adding 10 mg of α-amylase in 100 mL of phosphate buffer. In the next step, pipette 5 μl of α-amylase solution into wells of a 96-well plate, then add 15 μl of the sample (drugs or new medications), which was previously mixed in a phosphate buffer and incubate the mixture for 10 min at 37°C. To initiate the reaction, 20 μl of 2% starch solution is added and incubated at 37 °C for exactly 30 min. To stop the reaction, 10 μl of HCl (1M) and 75 μl of iodine reagent are used (Figure 2). Phosphate buffer (pH 6.9) and acarbose (20 mM) are used here in a blank well and positive control, respectively, and the absorbance is measured at 580 nm [33]. To calculate the percentage inhibition, the following equation is used:
where A = the absorbance of the control mixture without any enzyme, B = the absorbance of the reaction mixture containing an enzyme.
Inhibitory effect of Dipeptidyl Peptidase-IV
Dipeptidyl peptidase IV (DPP IV) is an associate and ubiquitous enzyme involved in the incretins metabolism and in the degradation of the glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1). Moreover, it takes part in the glucose-dependent pathway in 50 to 60% of insulin-releasing or secretion. Consequently, DPP-IV inhibition may maintain the insulinotropic activity of GLP-1 and GIP, which improving glucose homeostasis in T2DM (Table 1).
To perform this assay, add 30 μg/Ml of the sample (drugs or new medications) to 0.1 mg/mL of DPP IV enzyme and incubate the mixture in an atmosphere with 5% CO2 at 37◦C for 30 min. In the next step, add 50 μl of prepared Pro p-nitroanilide hydrochloride (Gly-Pro-ρNA) with 10 μM concentration and incubate the solution for 30 min at 37 °C (Figure 2). Use 50 μg/mL of Diprotin A as a standard inhibitor, measure the absorbance by a spectrophotometer at 405 nm and calculate the percentage inhibition of DPP IV activity [34,35]. To calculate the percentage inhibition, use the following equation:
Pancreatic lipase
Pancreatic lipase is necessary for the digestion and absorption of fats from food. The most significant amounts of lipolytic enzymes released through the pancreas are triglyceride lipase [36]. Therefore, inhibiting pancreatic lipase is a potential therapeutic strategy in diabetes treatment [37].
To assess the inhibition of pancreatic lipase, prepare the pancreatic lipase solution by adding 2.5 mg to MOPS-EDTA (10 mM Morpholino propane sulfonic acid and 1 mM ethylene diamine tetra acetic acid, pH 6.8). Then dissolved the mixture to 169 μL of TRIS buffer, which contains (100 mM Tris-HCl, 5 mM CaCl2, pH 7.0) and in the next step add 10 μL of the sample (drugs, agents, or new medications) to 40 μL of the pancreatic lipase enzyme, and add 100 μM tetrahydrolipstatin to 40 μL of the pancreatic lipase enzyme separately in the other wells as a positive control. Incubate the solution for 15 min at 37 °C. Then add 170 μL substrate solution p-nitrophenyl butyrate (p-NPB) 10 mM and incubate the mixtures at 37 °C for 15 min for an enzymatic reaction to proceed (Figure 2). Determine the pancreatic lipase activity (hydrolysis of p-NPB to p-nitrophenol) using the Lipase Assay Kit. Measure the absorbance by ELISA reader at 405 nm [30,38]. To calculate the percentage of lipase inhibition, use the following equation:
PTP-1B inhibition
Upon insulin binding, insulin receptor (IR) autophosphorylation at tyrosine residues is activated, initiating the insulin signaling process [39]. Tyrosine phosphorylation is subsequently used to recruit and activate the immediate effectors' Insulin receptor substrate 1 (IRS-1) and Insulin receptor substrate 2 (IRS-2) 2 to spread intracellular signals. Tyrosine residues in IR and IRS-1 are dephosphorylated by PTP1B, decrease insulin sensitivity [40,41], and inhibit signaling. As a result, insulin signaling is enhanced and T2DM traits are reversed by PTP1B inhibition [42] (Table 1).
To perform PTP-1B inhibition assay, use a non-specific substrate para-Nitrophenyl phosphate (pNPP) containing 1 mM EDTA, 0.5% FCS, 50 mM 3,3-dimethylglutarate and 5 mM glutathione. Then adjust the buffer ionic strength to 0.15 M using sodium chloride (NaCl) at pH 7.4. For the assay, add 50 μM of the sample (drugs, agents, or new medications) to the reaction mixture containing 2.5 mM pNPP and reach the total volume of well to 100 μL. Then add recombinant PTP-1B enzyme to initiate the reaction. The reaction is allowed to proceed for 5-60 min at 37 °C (Figure 2). After that, add 0.5 M sodium hydroxide (NaOH) in 20 μL of ethanol 50% to stop the reaction. Measure the absorbance using an ELISA reader at 405 nm [43].
Glucose uptake assay
The primary energy source for various cell types is glucose. Homeostasis depends on the control of glucose uptake by various tissues. Higher blood glucose levels can lead to diabetes disorder. Thus, it is crucial to investigate the antidiabetic properties of new compounds that enhance glucose absorption and decrease blood glucose levels. This assay is helpful for assessing the antidiabetic effect of substances or crude extracts by increasing glucose absorption. Muscle Adipose Tissue cells and other tissues have the glucose transporter 4 (GLUT4) essential for insulin-dependent glucose absorption for these cells. Various glucose uptake tests are available, categorised into radiolabeled and non-radiolabeled glucose analogues [44,45].
Before starting the assay, to adhere and grow the cells, seed them in the plate (1 × 105 cells/mL), and incubate them in a humidified atmosphere with 5% CO2 at 37◦C for 24 h. To perform this assay, add different sample concentrations (drugs, agents, or new medications) and incubate the cells in a humidified atmosphere with 5% CO2 at 37◦C for 48 h. After that, add 25 μL of the incubation medium and replaced the culture medium (select the medium according to the type of cell line) and incubate for 3 h at 37 °C. In the next step, remove 10 μl of the incubation medium from each well; transfer it into a new 96-well plate. After that, determine the glucose content of the medium by using glucose assay kit. For positive controls add 10 μg/mL berberine and metformin. For negative control add the incubation buffer without any test sample. Finally, measure absorbance by spectrophotometer at 540 nm (Figure 2).
Real-time polymerase chain reaction
Real-time polymerase chain reaction (RT-PCR) can be used to measure the effect of the sample (drugs, agents, or new medications) on the gene expression level that contributes to the production, release, and regulation of insulin (Table 1).
To perform this assay, culture the pancreatic β cells (RIN5F) in the 96-well plates (15 × 103 cells/well) and add different concentrations of a sample (drugs, agents, or new medications) with 5% CO2 at 37◦C for 24 h. Next, extract the total RNA from cells by TRIZOL extraction, and reversly transcribe the cDNA by using cDNA synthesis kit (Figure 2). Perform the RT-PCR using of SYBRGreen Master Mix to detect PCR amplification according to the manufacturer's instructions. Finally, calculate the gene expression levels involved in diabetes (for example, Ins1, Pdx1, Gck, Ptp1b, Slc2a2, and Gapdh as housekeeping gene) in the control and sample groups [46].
Insulin secretion assay
To perform this assay, culture the pancreatic β cells (RIN5F) in a 24-well plate (7 × 105 cells/well) with 5% CO2 at 37◦C. After attachment, remove the medium and replace it with a fresh one which contains different concentrations of the sample (drugs, agents, or new medications) and incubate the plate with 5% CO2 at 37◦C for 24h (Figure 2). Then collect the medium, measure the insulin concentration by ELISA kit, calculate the insulin secretion levels, and compare the treated and normal cells [46,47].

Table 1.
The advantages and disadvantages of in vitro antidiabetic assays.
| Assay | Advantages | Disadvantages |
|---|---|---|
|
α-Glucosidase enzyme inhibition |
|
|
|
α-Amylase enzyme inhibition |
|
|
| Dipeptidyl peptidase IV (DPP IV) |
|
|
|
PTP-1B inhibition |
|
|
|
Glucose uptake assay |
||
| Real-time |
|
|
| Insulin secretion assay |
|
|
|
GLUT4 translocation assay |
|
|
|
GLUT-4 and PPARα expression assay |
|
|
|
Calcium Ca2+ measurement assay |
|
|
| Reporter gene assay |
|
GLUT4 translocation assay
Cells use glucose to produce energy for cell metabolism. Glucose transporters (GLUTs) are transmembrane proteins that move glucose through the cell membranes. Insulin encourages glucose entrance into the adipose and skeletal tissues. There is a direct link between the insulin secretion level and inducing or increasing the GLUT4 translocation. The movement of GLUT4 to the plasma membrane is primarily stimulated by insulin, and it has been shown that insulin stimulates GLUT4 activity in the cell membrane. GLUT4 translocation could be evaluated by assessing the IRAP.
GLUT4 translocation assay can be performed by culturing the L6 cells expressed GLUT4-eGFP and IRAP-mOrange, in α-MEM containing 1% antibiotics (100 μg/mL streptomycin and 100 U/mL penicillin) and 10% fetal bovine serum (FBS) and incubate it at 37◦C with 5% CO2 to express the IRAP-mOrange (L6 IRAP-mOrange). In the next step, add a different concentration of the sample (drugs, agents, or new medications) to the cells. Finally, add 100 nM of insulin for positive control and no sample/insulin for negative control. After that, take microscopic photos of the control and supervise the GLUT4-eGFP and IRAP-mOrange translocation (Figure 2). Take the images after treatment with sample and insulin with excitation laser wavelength of 555 nm every 10 seconds for 30 min [73].
GLUT-4 and PPARα expression assay
One of the essential nuclear receptors is peroxisome proliferator-activated receptor gamma (PPARγ), which increases gene expression and is necessary for the differentiation and growth of adipocytes. It can be activated by natural or synthetic ligands like Thiazolidinediones TZDs and insulin-sensitising drugs frequently employed in managing and treating diabetes disorders [74,75].
To perform this assay, culture the 3T3-L1 fibroblasts cells and differentiate them into the adipocytes by adding 0.5 mM of 3-isobutyl-1-methylxanthine, 0.25 μM of dexamethasone acetate (DA), and 0.8 μM bovine insulin, and incubate the cells with 5% CO2 at 37◦C for 10 days. Then, treat the matured adipocyte phenotype cells with different concentrations of the sample (drugs, agents, or new medications) and incubate the cells with 5% CO2 at 37◦C for 24 h to assess the effect of the sample on PPARγ and GLUT-4 mRNA expression levels (Figure 2). For evaluation of gene expression, isolate the total mRNA from adipocytes and transcribe by using the RT-PCR and amplify the cDNA by using the SYBR Green Master Mix, which contains 0.5 mM of primers for PPARγ, PPAR α and GLUT-4, then measure the expression levels of each gene [76].
Calcium Ca2+ measurement assay
Several biological processes, including bone mineralisation, depend upon calcium. One of the ways to evaluate a new agent or drug is by its impact on G-protein-coupled receptors (GPCRs) through its Ca2+ mobilisation. The GPCR activation by Ca2+ releases inositol phosphate 3 via phospholipase C (PLC IP3). The endoplasmic reticulum is the site of intracellular Ca2+ mobilisation which eventually has an important impact on several cellular functions [67]. For example, indirect and direct Ca2+ release can both result in insulin secretion [68].
In the first step, culture the clonal pancreatic β-cells (BRIN-BD11 cells) as an insulin-releasing cell line and treat them with different sample concentrations (drugs, agents, or new medications). In the next step, measure the changes of Ca2+ ion in cytosolic (intracellular) and membrane potential using a fluorometric imaging plate reader (FLIPR) membrane potential. To perform this assay, seed BRIN-BD11 cells into the wells and incubate the cells with 5% CO2 at 37◦C for 24 h to attach. Then remove the media and pre-incubate the cells with KRB buffer (5·6 mM glucose) at 37°C for 10 min. Subsequently, treat the cells with calcium dye or the FLIPR membrane potential for 60 min and measure the signal intensity changes using a microplate reader. For membrane potential, the excitation, emission, and cut-off use the wavelengths 530, 565, and 550 nm, respectively, and for intracellular calcium, use 485, 525, and 515 nm, respectively [77].
Reporter gene assay
Numerous variable factors play a significant role in diabetes and its related complications. In this regard, different genes, including PPARγ, ABCC8, and KCNJ11 are up- or down-regulated at various stages of diabetes. The reporter gene assay has been used for the assessment of the expression level of specific or desired genes [78].
This assay selects the genes according to their mechanisms, functions, and targets. Various reporter genes are introduced, but the two most common forms of reporter genes are selectable and countable via scoring processes. Countable reporter gene expression can produce a quantifiable phenotype that is easily detected and sensitive, while a selectable reporter gene can be used as a qualitative indicator [49,70]. The primary and common reporter genes are antibiotic resistance genes, luciferase, yellow fluorescent protein, and β-galactosidase, green fluorescent protein. Among them, luciferase is the most often utilised reporter gene with a particular activity and minimal background noise [49].
To perform this assay, culture the Human embryonic kidney 293 cells (HEK293 cells) and transfect the pFR-Luc (UAS-Gal4-luciferase) and pFA-PPARγ into the cells. In the next step, treat the transfected cells with different sample concentrations (drugs, agents, or new medications) and use 2 to 10 μmol/L rosiglitazone or macelignan as a positive control, and incubate the cells with 5% CO2 at 37◦C for 24 h. For PPRE-tk-Luc reporter assay, transfect the PPRE-tk-Luc into the HepG2 cells (human liver cancer cell line) and treat them with sample (drugs, agents, or new medications), rosiglitazone or macelignan and incubate the cells with 5% CO2 at 37◦C for 24 h. then determine the luciferase activities by using a luciferase assay system kit [79].
Collagenase inhibition assay
To perform this assay, add 10 μl of collagenase to 10 μl of the sample (drugs, agents, or new medications with different concentrations), for positive control, use 6 μg/ml EDTA, 2 mg/ml gelatin, and 50 mM (Tris hydrochloride) Tris-HCl buffer at pH 7.4. Then incubate the mixture at 37°C for 1 h. In the next step, add 20 μl of Coomassie brilliant blue (CBB) and centrifuge for 5 min at 500 rcf. After that, remove the supernatant and add 50 μl of washing solution containing 10% acetic acid and 40% methanol, in order to wash the pellet and remove the extra CBB, then dissolve it in the 50 μl of dimethyl sulfoxide. Measure the absorbance by a plate reader at 540 nm [80]. To calculate the percentage of collagenase inhibition, use the following equation:
2.1.2. In vivo assays
Type 1 and type 2 diabetes are endocrine diseases involving numerous physiological systems. Given this, carefully selected animal models should be made depending on the studied condition. Therefore, specific types of 1 and 2 diabetes models are addressed in this review (Figure 3).
Animal models of type 1 diabetes
The majority of diabetes trials are carried out on rodents. These animals may develop diabetes spontaneously, genetically, or virally using chemical agents. The primary feature of T1DM is the autoimmune damage of the pancreatic beta cells, which prevents the pancreas from producing insulin. This lack of insulin production is simulated in animal models using various techniques, such as chemically destroying beta cells or creating rodents with autoimmune diabetes [81].
Chemically induced type 1 diabetes
Due to damage to the endogenous beta cells in chemically produced T1DM models, there is inadequate endogenous insulin production. As a result, hyperglycemia and weight loss occur. In addition to offering a quick and relatively inexpensive way to simulate diabetes in rodents, chemically induced diabetes can also be used to study higher species [82]. Chemicals that cause diabetes can be divided into three categories: 1) those that specifically destroy β cells; 2) those that temporarily restrict insulin generation and/or secretion; and 3) those that reduce insulin's metabolic effectiveness in target tissues. The chemicals of the first category are often attractive because they can cause lesions that resemble T1DM [83]. Diabetes can be induced using either streptozotocin (STZ) or alloxan. Fasting animals are typically more susceptible because alloxan and STZ can compete with glucose due to their structural similarity [84]. Since the relative instability of alloxan and STZ, the solutions should ideally be prepared before injection. In addition, chemicals might be hazardous to other body organs and are a disadvantage of chemically causing diabetes. Additionally, it should be noted that STZ or alloxan administration has been linked to alterations in P450 isozymes in the brain, intestines, liver, lung, kidney, and testis. Thus, this should be taken into consideration when medicines are evaluated in these models [85] (Table 2).
Streptozotocin
The Streptozotocin was reported in 1963 with diabetogenic activity and specifically cytotoxic effects on the pancreatic β cell [86]. A broad-spectrum antibiotic called STZ (2-deoxy-2-(3-methyl-3-nitrosourea) l-D glucopyranose) is synthesised from Streptomyces achromogens [87]. Following intravenous or intraperitoneal dosing, it penetrates the pancreatic β cell by the transmembrane carrier protein GLUT-2 and causes DNA to be alkylated. After PARP is activated, NAD+ is depleted, cellular ATP is decreased, and insulin synthesis is subsequently inhibited. Furthermore, STZ is a power source of free radicals, which can also cause DNA damage and ultimately lead to cell death [81]. Moreover, the DNA alkylating activity of STZ's methyl-nitrosourea moiety determines the toxicity of the compound's mode of action. DNA fragmentation results from the transfer of the methyl group from STZ to the DNA molecule, which damages it through a specific chain of events [88]. STZ is used as a single high dose or several low doses (Table 2).
A single high dose of STZ is between 100 and 200 mg/kg for the mice and 35 and 65 mg/kg for the rats [89]. Male adult Wistar rats (160-240 g) should be kept in carefully controlled lab environments at 25°C, relative humidity of 60°C, and a light/dark cycle of 12 hours. Streptozotocin (60 mg/kg) dissolved in 0.1 M citrate buffer (pH 4.5) is delivered by intravenous injection. Initially, blood glucose increases to 150- 200 mg % within three hours after treatment of STZ. Then, a four-fold increase in serum insulin levels causes a phase of hypoglycemia, which is followed by persistent hyperglycemia [90].
Alloxan
This method was introduced by Frerichs and Creutzfeldt in 1971 [93]. Alloxan (2,4,5,6 tetraoxypyrimidine;5,6-dioxyuracil) is a chemical classified as a pyrimidine derivative. It is a well-known diabetogenic agent used to convince animals to have T1DM. Alloxan causes the death of the pancreatic islet β cells. Animals such as mice, rats, rabbits and dogs are often created with this urea derivative [94]. Alloxan is decreased to dialuric acid and then re-oxidised back to its original state, creating a cycle of superoxide radicals that cause hydrogen peroxide and highly reactive hydroxyl radicals that cause the breakdown of the β cell's DNA [95]. Oxidation of protein SH groups is one of the other mechanisms of beta cell damage by alloxan which affects intracellular calcium homeostasis [96]. There are many animal models of diabetes treatments. In mice, alloxan monohydrate is injected s.c. in the dose of 50 to 200 mg/kg and in Wistar or Sprague Dawley rats weighing 150-200 g, equal to 100-175 mg/kg. In rabbits weighing 2.5-4 kg, during a course of 10 minutes, alloxan monohydrate (5g/100ml, pH 4.5) is infused via the marginal ear vein (150 mg/kg). 70% of the animals become hyperglycemic and uricosuric after these injections. In male dogs weighing 15-20 kg, i.v. injection with 60 mg/kg of alloxan monohydrate is commonly applied. In non-human primates such as monkeys and baboons, alloxan monohydrate is injected i.v. the dose of 65-200 mg/kg to induce diabetes. Subsequently, they are administered glucose solution and regular insulin i.v. for one week along with food ad libitium. [97].
Spontaneous autoimmune
The non-obese diabetic (NOD) mouse and the Biobreeding (BB), KDP, LETL and LEW.1AR1/Ztm-iddm rat are the five most autoimmune models of T1DM [98].
Non-obese diabetic (NOD) mouse
In NOD mice, insulitis occurs between 3 and 4 weeks of age. It was reported that diabetes can develop at any age up to 30 weeks. It usually doesn't become apparent until approximately 90% of the pancreatic insulin has been lost at about 10-14 weeks of age, despite insulitis-inducing β cell damage. These mice lose weight quickly and need insulin therapy when they develop overt diabetes. A structural similarity exists between MHC class 2 in NOD mice and humans, which may make NOD mice and humans more susceptible to or resistant to the disease [99]. Cyclophosphamide can be used to provide NOD mice in a more controlled and rapid manner [100]. Using specific genetic manipulation of NOD mice or developing humanised mouse models incorporating human immune system components are two methods for enhancing the NOD model. Although the NOD mouse has certain limitations, it is widely used because it accurately mimics many elements of the human disease and has been useful in identifying numerous genetic and signaling pathways that might result in type 1 diabetes [101,102].
Biobreeding (BB) rat
Outbred Wistar rats were used to create BB rats, another autoimmune T1DM rat model. After the discovery of spontaneous autoimmune diabetes in a Canadian colony in 1974, two founder colonies-one inbred Biobreeding Diabetes-Prone/Worcester (BBDP/Wor) and one outbred Biobreeding Diabetes-Prone, from which all substrains have descended were established (BBdp) [103]. The phenotype of diabetes in rats is rather severe, and they need insulin therapy to survive. The rats are lymphopenic with a substantial reduction in CD4+ T cells and almost no CD8+ T cells, while having insulitis and having T cells, B cells, macrophages and NK cells present. However, lymphopenia is not a sign of T1DM; it is just considered a disadvantage when the BB is used as a T1DM model. But this method is still preferable for islet transplantation tolerance induction [103], intervention [104] and diabetic neuropathy [105] studies in rats.
Komeda diabetes-prone (KDP) rat
The Komeda diabetes-prone (KDP) rat is one of the most effective spontaneous animal models for autoimmune T1DM research [106]. The genetic investigation of T1DM in KDP rats revealed a genetic predisposition to diabetes, which was explained by two vulnerable loci: MHC on chromosome 20 and IDDM/KDP 1 on chromosome 11. It is also a crucial experimental model in studying autoimmune diseases, specifically autoimmune thyroid disease [107]. Autoimmune destruction of pancreatic cells, quick onset of diabetes regardless of age or gender difference, and no significant T-lymphopenia characterise the KDP rats [106].
Long Evans Tokushima Lean (LETL) rat
The Long Evans Tokushima Lean (LETL) rats, one of the popular spontaneous animal models for IDDM, were found in 1982, and they were descended from a few pairs of outbred Long-Evans rats that were bought from Charles River in Canada [108]. The sudden onset of polyuria, polyphagia, hyperglycemia, weight loss, insulitis, loss of pancreatic β cells, lymphocytes vanish, infiltration of lymphocytes into the lacrimal and salivary glands, pancreatic islet hyperplastic foci, and nodular lesions are the phenotypic characteristics of the LETL rats. The advantages of this method are 1) the disease's severity is the same regardless of an individual's age or gender and 2) no detectable lymphocytopenia [108,109].
LEW.1AR1/-iddm rat
LEW.1AR1/iddm is a T1DM rat model that emerged spontaneously in a colony of congenic Lewis rats with a specific MHC haplotype (LEW.1AR1) bred at Hannover Medical School's Institute of Laboratory Animal Science (Ztm). The LEW-iddm rat, unlike the NOD mouse and BB rat, does not have any other autoimmune disorders. It also lives for a long time beyond the development of overt diabetes and can thus be applied to research diabetic complications [110], mechanisms underlying diabetes development [111] and intervention studies [112].
c-AKITA mouse as a genetically induced insulin-dependent diabetes
The C57BL/6NSlc mouse that led directly to the AKITA mouse in Akita, Japan, had a spontaneous mutation in the insulin 2 gene (Ins2+/C96Y mutation) that prevented proper processing of proinsulin. As a result, too many improperly folded proteins lead to endoplasmic reticulum stress (ERS). Hyperglycemia, hypoinsulinaemia, polyuria, and polydipsia are their notable characteristics. Rarely do untreated homozygotes live for more than 12 weeks. This model is an alternative to STZ-treated mice in transplantation studies because it lacks β cell mass [113]. It has furthermore been used as a model for type 1 diabetic macrovascular disease and neuropathy [114,115]. Additionally, this model frequently mimics some of the pathologies of T2DM and is used to research potential treatments for ERS in the islets [116].
Virus-induced models
Destruction in β cells has started using some viruses in several animal models. Thus, some viruses can be used to induce T1DM. Direct infection of the β cells or the start of an autoimmune reaction against the β cells can both result in destruction. The widely used viruses used to induce diabetes in animal models are Coxsackie B virus, D-variant of encephalomyocarditis virus (EMC-D), reovirus, lymphocytic choriomeningitis virus (LCMV) and Kilham rat virus (KRV) [117,118,119,120,121,122]. Studies on the EMC virus in mice and the KRV in rats provide the most conclusive proof among these viruses that a virus causes T1DM in animals. While KRV is assumed to trigger β cell-specific autoimmunity without infecting β cells, EMC virus is believed to be the primary agent that damages pancreatic β cells [119].
Coxsackie B virus
Diabetes is often induced in mice by Coxsackie viruses, which kill pancreatic acinar cells while conserving the nearby islets of Langerhans. A strong association exists between the coxsackie B4 virus and the development of IDDM in humans. Diabetes driven on by a Coxsackie virus infection causes the release of sequestered islet antigen, which prompts the stimulation of auto-reactive T cells once more [118].
Variant of encephalomyocarditis virus (EMC-D)
In some inbred strains of mice, the EMC-D virus can infect and kill pancreatic β cells, leading to insulin-dependent hyperglycemia. The single-stranded RNA genome of the EMC virus is around 7.8 kB in size. When it infects genetically susceptible strains of mice, the EMC-D virus replicates in pancreatic cells and recruits macrophages to the pancreatic islets. In the islets, these activated macrophages generate soluble mediators, including IL-1b, TNF-α, and nitric oxide, that cause the β cells to undergo apoptosis [119].
Reovirus
Reoviruses can cause encephalitis, hepatitis, myocarditis, adrenalitis, and acinar pancreatitis in neonatal mice. The Langerhans islets, however, had not yet shown signs of infection. Furthermore, although reovirus infection is possible in mouse β cells, reovirus-infected mice did not exhibit overt hyperglycemia. This is because a significant decrease in β cells must happen before hyperglycemia reveals itself [121].
Lymphocytic choriomeningitis virus (LCMV)
RIP-LCMV mice is another experimental approach to induce T1DM. Using this virus, the RIP-LCMV mice produce LCMV antigens transgenically in their cells, which become a target for antiviral T cells after infection. Notably, the viral infection causes immunoregulatory mechanisms in these genetically altered mice identical to those in wild-type mice. However, T1DM still occurs because the initial attack on b cells causes the presentation of autoantigens to autoreactive T cells [122].
Table 2.
The advantages and disadvantages of in vivo antidiabetic assays.
| Chemically induced | Advantages | Disadvantages |
|---|---|---|
|
Streptozotocin (STZ) |
|
|
|
Alloxan |
||
| Spontaneous autoimmune | ||
|
NOD Mouse |
|
|
|
Biobreeding (BB) rat |
||
| KDP rats |
|
|
| LETL rat |
|
|
| LEW.1AR1/-iddm rat |
|
|
| c-AKITA mouse |
|
|
| Virus-induced models | ||
|
Coxsackie B virus, EMC-D, Reovirus, LCMV, KRV |
|
Kilham rat virus (KRV)
In diabetes-resistant BioBreeding (DR-BB) rats, KRV, a tiny DNA parvovirus, induces autoimmune reactions against the B cell rather than directly causing B cell cytolysis. Even though these rats are descended from progenitors that were prone to diabetes, they do not typically get the disease. Unlike cells, KRV mainly affects lymphoid organs such as the spleen, thymus, and lymph nodes. Macrophages seem to be crucial in the development of diabetes in DR-BB rats after KRV infection. In KRV-infected DR-BB rats, the production of proinflammatory cytokines originating from macrophages, such as TNF-a, IL-1b, and IL-12, was strongly linked with an enhanced Th1 immune response [145]. In KRV-infected DR-BB rats, the production of NO by activated macrophages was crucial in the modulation of immunological responses, including the elevation of the Th1 immune response and the activation of b cell-specific cytotoxic T lymphocytes, which led to the development of autoimmune diabetes [120].
Animal models of type 2 diabetes
Significant characteristics of T2DM are insulin resistance and the failure of the β cells to compensate for insulin resistance. Consequently, insulin resistance and/or beta cell failure models are often used as animal models for type 2 diabetes research. Obesity is prevalent in animal models of T2DM, reflecting the human state where obesity is directly associated with the development of T2DM.
Chemically induced T2DM
- Dexamethasone
Wistar rats are injected subcutaneously daily for 10 days with 1 mg/kg of dexamethasone to cause glucocorticoid diabetes. In addition to increasing blood glucose levels and causing insulin resistance, this dose did not affect triglyceride or cholesterol levels. According to this model, the decrease in nitric oxide synthase endothelial activity results in endothelial dysfunction, which may cause insulin resistance. The reduction of nitric oxide and insulin resistance can also be increased by oxidative stress, which can result from dyslipidemia. [146].
Insulin Antibodies-induced Diabetes
The administration of bovine insulin and Freund's adjuvant to guinea pigs induces the formation of anti-insulin antibodies in them. In this regard, bovine insulin is added into a water-oil emulsion based on full Freund's adjuvant or a mixture of paraffin oil and lanolin after being dissolved in acidified water (pH 3.0). Male guinea pigs weighing 300–400 g are subcutaneously given a dose of 1 mg insulin in divided doses. Injections are administered periodically. Two weeks after receiving the second and subsequent doses of antigen, the guinea pigs are bled by heart puncture. Every animal can provide 10 ml of blood once per month. After receiving 0.25–1.0 ml of guinea pig anti-insulin serum intravenously, rats' blood glucose levels rise to 300 mg/dl dose-dependently. Due to the insulin antibodies' ability to neutralise endogenous insulin, guinea pig anti-insulin serum exhibits this particular action. It continues for as long as there is still insulin in circulation, and the antibodies can still react with it. When using a slow intravenous infusion or intramuscular injection, its effects last longer than a few hours. Toxic to animals, ketonemia, ketonurea, glycosuria, and acidosis can result from high doses and prolonged administration [147].
Dithizone
Dithizone (8-(p- toluene- sulfonylamino)– quinolone) is an organosulfur compound that acts as a zinc-chelating agent used to generate diabetes in experimental animal models. When dithizone is administered to animals, the blood's typical levels of zinc, iron, and potassium rise above average. After passing across membranes, dithizone forms a compound with zinc that triggers the release of protons and so provides diabetogenicity. T2DM can be induced in cats, rabbits, golden hamsters, and mice by injecting different chelators such as dithizone, 8-(p-toluenesulfonylamino)-quinoline (8-TSQ), and 8-(benzenesulfanylamino)-quinoline (8-BSQ) at a single dose of 40–100 mg/kg. Injection of dithizone in rabbits results in a triphasic glycemic response. A normoglycemic phase is observed after 2 hours, followed by an initial phase of hyperglycemia after 8 hours, and a persistent secondary phase of hyperglycemia after 24 to 72 hours [148].
Monosodium glutamate
Monosodium glutamate can be used to trigger insulin secretion by elevating plasma glutamate content. When monosodium glutamate is administered to mice, obesity and increased insulin levels are the effects. Additionally, it raises blood levels of triglycerides, total cholesterol, and glucose. A commercially available 99%-pure food-grade monosodium glutamate is supplemented with a daily dose of 2 mg/g body weight and put to drinking water for rats with monosodium glutamate treatment. Every one or two weeks, food intake and body weight are recorded, and after a 12-hour fast, rats are intraperitoneally injected with nembutal and slaughtered at 1, 3, or 9 months. Pancreatic tissue and blood are taken for morphological and functional research [149].
Gold thioglucose
T2DM can also be brought on by the chemical gold thioglucose, which induces obesity. Over time, experimental animals given gold thioglucose intraperitoneally develop obesity, hyperinsulinemia, and hyperglycemia. The gold thioglucose is transported to a cell that causes necrotic lesions responsible for obesity and hyperphagia. Gold thioglucose also enhances body lipid content, hepatic lipogenesis, and triglyceride production. To get six to eight obese mice, 12-20 mice must be assigned to gold thioglucose groups as the expected rates of gold thioglucose-induced obesity varied from 40 to 80%, depending on the mouse strain. Saline or gold thioglucose are administered intraperitoneally to the mice at the recommended doses of 0.6, 0.8, 0.4, and 0.8 g/kg for B6, DBA, BKs, and BDF strain of mice, respectively. Mice that developed obesity after 4 or 6 weeks (BKs mice) are assigned for additional investigations if they gained 8 g or more weight gain compared to the average weight gain recorded in the control mice [150].
Surgically induced diabetes
- Non-obese diabetic animals with partial pancreatectomy
A model of non-obese diabetic animals with partial pancreatectomy is frequently used to explore the capacity of cells or their progenitors for regeneration. This model entails partial or complete removal of the pancreas through surgery within three months of the removal of 95% of the pancreas. Rats, dogs and pigs can be also used in this model. In this model, a 60% partial pancreatectomy only results in a mild increase in cell mass and does not result in higher blood glucose concentrations. Mild hyperglycemia is caused after pancreatectomy in around 90% of cases, and this is then followed by pancreatic regeneration. This model's primary disadvantage is that it is invasive, especially regarding healthy tissues as opposed to the pancreas [151].
The ventromedial hypothalamus -dietary obese rat
Adult rats given high fat, high sucrose diets and bilateral electrolytic lesions of the ventromedial hypothalamus (VMH) are used to create a model of T2DM. Severe obesity, hyperinsulinaemia, hypertriglyceridaemia, insulin resistance, decreased glucose tolerance, and mild to severe fasting hyperglycemia are some of its characteristics. In addition, in these VMH-lesioned rats, significant hyperphagia and poor modulation of insulin secretory response are reported despite elevated leptin levels and extremely high insulin secretory capacity, respectively [152].
Monogenic models of obesity
Monogenic models of obesity are frequently used in T2DM studies, even though monogenic mutations are infrequently the cause of obesity in humans. The most popular monogenic models of obesity include leptin signaling defects. Leptin promotes satiety, animals lacking functioning leptin experience hyperphagia and ensuing obesity. These models include the leptin-deficient Lepob/ob mouse, the leptin receptor-deficient Leprdb/db mouse, and the Zucker diabetic fatty (ZDF) rats. These models are frequently employed to evaluate novel treatments for T2DM [153,154,155].
Lep mouse ob/ob
A spontaneous mutation that was found in an outbred colony at Jackson Laboratory in 1949 gave rise to the Lep ob/ob mouse, a model of extreme obesity. Although the trait was bred into C57BL/6 mice, the mutant protein wasn't discovered to be leptin until 1994. At two weeks of age, the mice begin to gain weight and develop hyperinsulinemia. Blood glucose concentrations begin to rise at 4 weeks and continue to grow until they reach their peak at 3 to 5 months, after which they start to decline as the mouse ages. Hyperlipidemia, infertility, a problem with temperature control, and decreased physical activity are other metabolic abnormalities reported in this model [153].
Diabetes mouse (db/db)
A leptin receptor autosomal recessive mutation on chromosome 4 is performed to develop the diabetes mouse (db/db). The leptin receptor gene carries a mutation that changes Gly to Thr. These mice are plainly overweight, have high blood sugar levels, and have excessive appetite. As a result, these mice develop a severe form of diabetes, which is indicated by the onset of hyperglycemia and hypoinsulinemia [154].
Zucker diabetic fatty (ZDF) rat
Another experimental animal model for T2DM is ZDF rats. These rats were produced in Indianapolis' Walter Shaw's Laboratory from an outbred zucker rat colony (USA) in 1991. In this animal, a basic autosomal recessive leptin receptor gene (fa) on chromosome 5 spontaneously mutates, leading to insulin resistance, hyperphagia, and obesity. For the research of T2DM and the development of the condition from prediabetic to diabetic state, ZDF male rats are most frequently used [155].
Polygenic models of obesity
Obesity polygenic models might offer a more accurate representation of the human condition. Polygenic mice models for diabetes, glucose intolerance and obesity are available, allowing a range of genotypes and susceptibilities to be investigated. However, no wild-type controls exist, in contrast to the monogenic models. Additionally, these models have a more male sex bias (Leiter, 2009).
KK mice
A mildly obese and hyperleptinemic strain called KK mice was developed from wild-derived ddY mice in Japan in 1957 by Kondo. They exhibit insulin resistance in both muscle and adipose tissue and develop severe hyperinsulinemia. In addition, the pancreatic islets are degranulated and hypertrophic. This strain of mice also exhibits symptoms of diabetic nephropathy. The KK-AY mice are a descendant of this strain and were produced by adding the yellow obese AY gene to the KK strain [156].
New Zealand Obese (NZO) mice
The New Zealand Obese (NZO) mice were developed as a polygenic model of obesity through selective breeding. Due to leptin resistance, it is hyperphagic and obese, and by 9–12 weeks of age, these mice are hyperleptinaemic. They are sensitive to centrally administered leptin but resistant to peripherally administered leptin, indicating a deficiency in leptin transport across the blood-brain barrier. Additionally, these mice exhibit hyperinsulinaemia due to hepatic insulin resistance that develops at a young age and appears to be caused by a problem controlling hepatic fructose-1,6-bisphosphatase. Blood glucose levels are high, and decreased glucose tolerance is evident. About 50% of males eventually get diabetes, which worsens with age [81].
The TALLYHO/Jng (TH) mice
As a naturally occurring model of obesity and T2DM, the TALLYHO/Jng (TH) mice spontaneously acquire hyperglycemia and hyperinsulinaemia. This model was developed through selective breeding in a colony of Theiler Original mice. TALLYHO/Jng (TH) mice had higher amounts of plasma triglycerides, cholesterol, and free fatty acids, contributing to increased adiposity. Only male mice get hyperglycemia, which appears as early as 10 to 14 weeks of age. Hyperinsulinaemia is visible, and the pancreatic islets are hypertrophied and degranulated [157].
NONcNZO10/LtJ mice
Combining quantitative trait loci from New Zealand Obese (NZO/HlLt) and Non-obese Nondiabetic (NON/LtJ) mice, a new mouse model of obesity-induced diabetes of NONcNZO10/LtJ mice was created. T2DM in NONcNZO10/LtJ (RCS10) male mice are characterised by maturity-onset obesity, hyperglycemia, and insulin resistance. Insulin resistance in the liver and skeletal muscle first appeared in 8-week-old RCS10 mice due to marked reductions in insulin-stimulated glucose uptake and GLUT4 expression in muscle, which were followed by the development of obesity and corresponding increases in hepatic lipid content. By 13 weeks of age, the development of obesity and hyperglycemia increased insulin resistance in skeletal muscle, the liver, and the heart, which was accompanied by increases in the lipid content of each organ. This polygenic mouse model of T2DM should therefore offer new insights into the etiology of the disease.This polygenic mouse model of T2DM offers new insights into the etiology of the disease as obesity develops with maturity but, plasma insulin levels are only moderately elevated. In this model, insulin action and glucose metabolism in skeletal muscle and liver are also reduced at an early prediabetic age [158].
The Otsuka Long-Evans Tokushima Fat (OLETF) rat
The spontaneously diabetic rat that was found in an outbred colony of Long Evans Rats in 1984 served as the ancestor of the Otsuka Long-Evans Tokushima Fat rat (OLETF). The Tokushima Research Institute used selective breeding to create the OLETF strain, which exhibits late-onset hyperglycemia and mild obesity after 18 weeks, and males inherit diabetes from their mothers. There are three stages of histological alteration in the pancreatic islets. First, cellular infiltration and degradation are visible at 6–20 weeks old. Between 20 and 40 weeks later, there is a stage of hyperplasia. The ultimate stage is distinguished by the fibrosis and replacement of the islets with connective tissue [159].
Obese spontaneously hypertensive rat strain (SHROB or Koletsky rat)
Obese spontaneously hypertensive rat strain (SHROB or Koletsky rat) was created in 1970 as a genetic mutation that caused obesity in the progeny of a cross between a female SHR rat of the Kyoto-Wistar strain and a normotensive Sprague-Dawley male rat. The obese Koletsky rat is a unique strain with traits similar to human obese hypertension and Syndrome X, including hyperlipidemia, hyperinsulinemia, normoglycemia, glomerulopathy with proteinuria, and spontaneous genetic hypertension [160]. This model is frequently used to study how endocrine and metabolic abnormalities relate to obesity. It is considered as a crucial animal model for the investigation of the roles played by hyperlipidemia and high blood pressure in the pathogenesis of arthrosclerosis [161].
Non-obese models of type 2 diabetes
Lean animal models of T2DM should also be researched because not all type 2 diabetes patients are obese. These include models with insufficient β cells, which ultimately cause overt T2DM in people. These models include an islet amyloid polypeptide (IAPP) mouse and Goto-Kakizaki (GK) rat.
Islet amyloid polypeptide (IAPP) mouse
The primary component of the islet amyloid found at autopsy in patients with T2DM is human islet amyloid polypeptide (hIAPP), a pancreatic islet protein with 37 amino acids. Severe glucose intolerance in hIAPP transgenic females was correlated with a downregulation of GLUT-2 mRNA expression [162]. In numerous hIAPP models, it has been demonstrated that upregulating hIAPP expression increases cell toxicity. In addition, reproducing β cells are more vulnerable to hIAPP toxicity, which limits their ability to respond to an increase in insulin demand [163]
Goto-Kakizaki (GK) rat
The insulin-resistant Goto-Kakizaki (GK) rats are a non-fat, highly developed strain of Wistar rodents that rapidly develop T2DM. It has reduced pancreatic β cells and their activities. Because the disease development of this rat was associated with chronic inflammation, it was used in T2DM pathophysiology and therapeutic research [164].
3. Conclusion
Globally, diabetes is a leading cause of death and disability. Millions of people suffer from diabetes, and healthcare costs are escalating. Some in vivo and in vitro models of diabetes have provided valuable insight into the pathogenesis of human diabetes. Various model systems have been used to assess the antidiabetic activity of new treatments, each with its pros and cons. Results obtained from both methods should be considered together to understand the compound's antidiabetic activity comprehensively. By choosing the appropriate model system, researchers can reveal, assess and evaluate the antidiabetic activity of various medications or newly-developed agents.
Author Contributions
Collecting data and writing - original draft; S. G.and A. B., Writing - review & editing. E. M. O., R.A.and E.B.
Conflicts of Interest
The authors declare that there is no conflict of interest
References
- Sattar, N. Advances in the clinical management of type 2 diabetes: a brief history of the past 15 years and challenges for the future. BMC Med 2019, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front Endocrinol (Lausanne) 2013, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- De Tata, V. Age-related impairment of pancreatic Beta-cell function: pathophysiological and cellular mechanisms. Front Endocrinol (Lausanne) 2014, 5, 138. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P., et al., Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract 2019, 157, 107843. [CrossRef] [PubMed]
- Grammatiki, M., S. Karras, and K. Kotsa, The role of vitamin D in the pathogenesis and treatment of diabetes mellitus: a narrative review. Hormones (Athens) 2019, 18, 37–48. [CrossRef] [PubMed]
- Lo-Ciganic, W., et al., Identifying type 1 and type 2 diabetic cases using administrative data: a tree-structured model. J Diabetes Sci Technol 2011, 5, 486–493. [CrossRef] [PubMed]
- Daryabor, G., et al., The Effects of Type 2 Diabetes Mellitus on Organ Metabolism and the Immune System. Front Immunol 2020, 11, 1582. [CrossRef]
- Wu, Y., et al., Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int J Med Sci 2014, 11, 1185–1200. [CrossRef]
- Galaviz, K.I., et al., Lifestyle and the Prevention of Type 2 Diabetes: A Status Report. Am J Lifestyle Med 2018, 12, 4–20. [CrossRef]
- Ley, S.H., et al., Prevention and management of type 2 diabetes: dietary components and nutritional strategies. Lancet 2014, 383, 1999–2007. [CrossRef]
- Roep, B.O., et al., Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat Rev Endocrinol 2021, 17, 150–161. [CrossRef] [PubMed]
- Soleimanpour, S.A. and D.A. Stoffers, The pancreatic β cell and type 1 diabetes: innocent bystander or active participant? Trends Endocrinol Metab 2013, 24, 324–331. [CrossRef] [PubMed]
- Chiang, J.L., et al., Type 1 Diabetes in Children and Adolescents: A Position Statement by the American Diabetes Association. Diabetes Care 2018, 41, 2026–2044. [CrossRef] [PubMed]
- Gavanji, S., et al., Cytotoxic Activity of Herbal Medicines as Assessed in Vitro: A review. Chem Biodivers 2023, 20, e202201098.
- Rena, G., D.G. Hardie, and E.R. Pearson, The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [CrossRef] [PubMed]
- Marín-Peñalver, J.J., et al., Update on the treatment of type 2 diabetes mellitus. World J Diabetes 2016, 7, 354–395. [CrossRef] [PubMed]
- Melloul, D., S. Marshak, and E. Cerasi, Regulation of insulin gene transcription. Diabetologia 2002, 45, 309–326. [CrossRef]
- Dedoussis, G.V., A.C. Kaliora, and D.B. Panagiotakos, Genes, diet and type 2 diabetes mellitus: a review. Rev Diabet Stud 2007, 4, 13–24. [CrossRef]
- Mambiya, M., et al., The Play of Genes and Non-genetic Factors on Type 2 Diabetes. Front Public Health 2019, 7, 349. [CrossRef]
- Kubota, N., et al., Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab 2008, 8, 49–64. [CrossRef]
- Guo, S., et al., The Irs1 branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis. Mol Cell Biol 2009, 29, 5070–5083. [CrossRef] [PubMed]
- Saito, M., et al., Development of novel cell lines of diabetic dysfunction model fit for cell-based screening tests of medicinal materials. Cytotechnology 2013, 65, 105–118. [CrossRef]
- Kulkarni, R.N., et al., PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest 2004, 114, 828–836. [CrossRef] [PubMed]
- Terauchi, Y., et al., Development of non-insulin-dependent diabetes mellitus in the double knockout mice with disruption of insulin receptor substrate-1 and beta cell glucokinase genes. Genetic reconstitution of diabetes as a polygenic disease. J Clin Invest 1997, 99, 861–866. [CrossRef] [PubMed]
- Shih, D.Q., et al., Profound defects in pancreatic beta-cell function in mice with combined heterozygous mutations in Pdx-1, Hnf-1alpha, and Hnf-3beta. Proc Natl Acad Sci U S A 2002, 99, 3818–3823. [CrossRef] [PubMed]
- Pedica, F., et al., PDX-1 (pancreatic/duodenal homeobox-1 protein 1). Pathologica 2014, 106, 315–321.
- Al-Quobaili, F. and M. Montenarh, Pancreatic duodenal homeobox factor-1 and diabetes mellitus type 2 (review). Int J Mol Med 2008, 21, 399–404.
- Shaw, L.M. The insulin receptor substrate (IRS) proteins: at the intersection of metabolism and cancer. Cell Cycle 2011, 10, 1750–1756. [Google Scholar] [CrossRef]
- Kovacs, P., et al., The role of insulin receptor substrate-1 gene (IRS1) in type 2 diabetes in Pima Indians. Diabetes 2003, 52, 3005–3009. [CrossRef]
- Odeyemi, S. and J. Dewar, In Vitro Antidiabetic Activity Affecting Glucose Uptake in HepG2 Cells Following Their Exposure to Extracts of Lauridia tetragona (L.f.) RH Archer. Processes 2019, 8, 33. [Google Scholar] [CrossRef]
- Sales, P.M., et al., α-Amylase inhibitors: a review of raw material and isolated compounds from plant source. J Pharm Pharm Sci 2012, 15, 141–183. [CrossRef] [PubMed]
- Brayer, G.D., Y. Luo, and S.G. Withers, The structure of human pancreatic alpha-amylase at 1.8 A resolution and comparisons with related enzymes. Protein Sci 1995, 4, 1730–1742. [CrossRef] [PubMed]
- Odeyemi, S.W. and A.J. Afolayan, Identification of Antidiabetic Compounds from Polyphenolic-rich Fractions of Bulbine abyssinica A. Rich Leaves. Pharmacognosy Res 2018, 10, 72–80. [Google Scholar] [PubMed]
- Proença, C., et al., The dipeptidyl peptidase-4 inhibitory effect of flavonoids is hindered in protein rich environments. Food Funct 2019, 10, 5718–5731. [CrossRef] [PubMed]
- Purnomo, Y., et al.
- Zhu, G., et al., Structure and Function of Pancreatic Lipase-Related Protein 2 and Its Relationship With Pathological States. Front Genet 2021, 12, 693538. [CrossRef]
- Sellami, M., et al., Inhibition of pancreatic lipase and amylase by extracts of different spices and plants. Int J Food Sci Nutr 2017, 68, 313–320. [CrossRef]
- Kim, Y.S., et al., Anti-obesity effect of Morus bombycis root extract: anti-lipase activity and lipolytic effect. J Ethnopharmacol 2010, 130, 621–624. [CrossRef]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol 2014, 220, T1–t23. [Google Scholar] [CrossRef]
- Goldstein, J.C., et al., The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nature cell biology 2000, 2, 156–162. [CrossRef]
- Bakke, J. and F.G. Haj, Protein-tyrosine phosphatase 1B substrates and metabolic regulation. Semin Cell Dev Biol 2015, 37, 58–65. [Google Scholar] [CrossRef]
- Teimouri, M., et al., The role of protein tyrosine phosphatase 1B (PTP1B) in the pathogenesis of type 2 diabetes mellitus and its complications. J Physiol Biochem 2022, 78, 307–322. [CrossRef] [PubMed]
- Tiong, S.H., et al., Antidiabetic and antioxidant properties of alkaloids from Catharanthus roseus (L.) G. Don. Molecules 2013, 18, 9770–9784. [CrossRef] [PubMed]
- Yamamoto, N., et al., Measurement of Glucose Uptake in Cultured Cells. Curr Protoc Pharmacol 2015, 71, 12–14.
- Yamamoto, N. and H. Ashida, Evaluation Methods for Facilitative Glucose Transport in Cells and Their Applications. 2012.
- Bahrami, G., et al., Molecular mechanism of the anti-diabetic activity of an identified oligosaccharide from Rosa canina. Res Pharm Sci 2020, 15, 36–47. [CrossRef] [PubMed]
- Khazaei, M. and M. Pazhouhi, Protective effect of hydroalcoholic extracts of Trifolium pratense L. on pancreatic β cell line (RIN-5F) against cytotoxicty of streptozotocin. Res Pharm Sci 2018, 13, 324–331. [Google Scholar]
- Zhang, Y., et al. Inhibition on α-Glucosidase Activity and Non-Enzymatic Glycation by an Anti-Oxidative Proteoglycan from Ganoderma lucidum. Molecules 2022, 27, 1457. [CrossRef]
- Vhora, N., et al., Recent Advances in In-Vitro Assays for Type 2 Diabetes Mellitus: An Overview. Rev Diabet Stud 2020, 16, 13–23. [CrossRef]
- Feingold, K.R. Oral and Injectable (Non-Insulin) Pharmacological Agents for the Treatment of Type 2 Diabetes; MDText.com, Inc. Copyright © 2000-2023; Endotext, Feingold, K.R., et al., Eds.; MDText.com, Inc.: South Dartmouth (MA), 2000. [Google Scholar]
- Rao, P.S. and G.K. Mohan, In vitro alpha-amylase inhibition and in vivo antioxidant potential of Momordica dioica seeds in streptozotocin-induced oxidative stress in diabetic rats. Saudi J Biol Sci 2017, 24, 1262–1267. [CrossRef]
- Visvanathan, R., et al., Critical review on conventional spectroscopic α-amylase activity detection methods: merits, demerits, and future prospects. J Sci Food Agric 2020, 100, 2836–2847. [CrossRef]
- Fleury, L., et al., In Vivo and In Vitro Comparison of the DPP-IV Inhibitory Potential of Food Proteins from Different Origins after Gastrointestinal Digestion. Int J Mol Sci 2022, 23, 8365. [CrossRef]
- Yang, S.J., et al., In vitro protein tyrosine phosphatase 1B inhibition and antioxidant property of different onion peel cultivars: A comparative study. Food Sci Nutr 2019, 7, 205–215. [CrossRef] [PubMed]
- Saidu, Y., et al., In vitro screening for protein tyrosine phosphatase 1B and dipeptidyl peptidase IV inhibitors from selected Nigerian medicinal plants. J Intercult Ethnopharmacol 2017, 6, 154–157.
- Deora, G.S., et al., Design, synthesis and biological evaluation of novel arylidine-malononitrile derivatives as non-carboxylic inhibitors of protein tyrosine phosphatase 1B. Medicinal Chemistry Research 2013, 22, 5344–5348. [CrossRef]
- Seong, S.H., et al., Protein tyrosine phosphatase 1B and α-glucosidase inhibitory activities of Pueraria lobata root and its constituents. J Ethnopharmacol 2016, 194, 706–716. [CrossRef] [PubMed]
- Paudel, P., et al., Protein Tyrosine Phosphatase 1B Inhibition and Glucose Uptake Potentials of Mulberrofuran G, Albanol B, and Kuwanon G from Root Bark of Morus alba L. in Insulin-Resistant HepG2 Cells: An In Vitro and In Silico Study. Int J Mol Sci 2018, 19, 1542. [CrossRef]
- Hsu, B.-Y., et al., Hypoglycemic activity of Chenopodium formosanum Koidz. components using a glucose uptake assay with 3T3-L1 adipocytes. Food Bioscience 2018, 24, 9–16. [CrossRef]
- Blass, B.E. Chapter 4 - In vitro Screening Systems. In Basic Principles of Drug Discovery and Development; Blass, B.E., Ed.; Academic Press: Boston, 2015; pp. 143–202. [Google Scholar]
- Jesus, A.R., et al., Targeting Type 2 Diabetes with C-Glucosyl Dihydrochalcones as Selective Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: Synthesis and Biological Evaluation. J Med Chem 2017, 60, 568–579. [CrossRef]
- Della Mea, V., et al., Image selection in static telepathology through the Internet. J Telemed Telecare 1998, 4 (Suppl 1), 20–22. [CrossRef]
- Della Mea, V., et al., Image sampling in static telepathology for frozen section diagnosis. J Clin Pathol 1999, 52, 761–765. [CrossRef]
- Huang, Y.T., et al., Clinical Application of a Real-Time Telepathology System for Frozen Section Diagnosis in Comparison With Optical Microscope. Front Med (Lausanne) 2019, 6, 215.
- Bleve, G., et al., Development of reverse transcription (RT)-PCR and real-time RT-PCR assays for rapid detection and quantification of viable yeasts and molds contaminating yogurts and pasteurized food products. Appl Environ Microbiol 2003, 69, 4116–4122. [CrossRef] [PubMed]
- Diaz-Vegas, A., et al., A high-content endogenous GLUT4 trafficking assay reveals new aspects of adipocyte biology. Life Sci Alliance 2023, 6. [CrossRef]
- Levy, J. Abnormal cell calcium homeostasis in type 2 diabetes mellitus: a new look on old disease. Endocrine, 1999, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gomes Castro, A.J., et al., The potent insulin secretagogue effect of betulinic acid is mediated by potassium and chloride channels. Arch Biochem Biophys 2018, 648, 20–26. [CrossRef]
- Schmidt, S., et al., Extracts from Leonurus sibiricus L. increase insulin secretion and proliferation of rat INS-1E insulinoma cells. J Ethnopharmacol 2013, 150, 85–94. [CrossRef] [PubMed]
- Saji, H., et al., Monitoring the escape of transgenic oilseed rape around Japanese ports and roadsides. Environ Biosafety Res 2005, 4, 217–222. [CrossRef]
- Hill, S.J., J.G. Baker, and S. Rees, Reporter-gene systems for the study of G-protein-coupled receptors. Curr Opin Pharmacol 2001, 1, 526–532. [CrossRef]
- Kotarsky, K., et al., Progress in methodology. Improved reporter gene assays used to identify ligands acting on orphan seven-transmembrane receptors. Pharmacol Toxicol 2003, 93, 249–258. [CrossRef]
- Lv, Y., et al., Antidiabetic effects of a lipophilic extract obtained from flowers of Wisteria sinensis by activating Akt/GLUT4 and Akt/GSK3β. Food Nutr Res 2020, 64. [CrossRef]
- Hu, Y., et al., Pathogenic role of diabetes-induced PPAR-α down-regulation in microvascular dysfunction. Proc Natl Acad Sci U S A 2013, 110, 15401–15406. [CrossRef]
- Celi, F.S. and A.R. Shuldiner, The role of peroxisome proliferator-activated receptor gamma in diabetes and obesity. Curr Diab Rep 2002, 2, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Colin-Lozano, B., et al., Synthesis, In Vitro, In Vivo and In Silico Antidiabetic Bioassays of 4-Nitro(thio)phenoxyisobutyric Acids Acting as Unexpected PPARγ Modulators: An In Combo Study. Pharmaceuticals (Basel) 2022, 15, 102. [CrossRef] [PubMed]
- Ansari, P., et al., Insulin secretory and antidiabetic actions of Heritiera fomes bark together with isolation of active phytomolecules. PLoS One 2022, 17, e0264632.
- Nakajima, Y. and Y. Ohmiya, Bioluminescence assays: multicolor luciferase assay, secreted luciferase assay and imaging luciferase assay. Expert Opin Drug Discov 2010, 5, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J., et al., Effects of a multi-herbal extract on type 2 diabetes. Chin Med 2011, 6, 10. [CrossRef] [PubMed]
- Sagbo, I.J., et al., In Vitro Antidiabetic Activity and Mechanism of Action of Brachylaena elliptica (Thunb.) DC. Evid Based Complement Alternat Med 2018, 2018, 4170372.
- King, A.J. The use of animal models in diabetes research. British journal of pharmacology 2012, 166, 877–894. [Google Scholar] [CrossRef]
- Dufrane, D., et al., Streptozotocin-induced diabetes in large animals (pigs/primates): role of GLUT2 transporter and β-cell plasticity. Transplantation 2006, 81, 36–45. [CrossRef]
- Baily, C. and O. Baily, Production of diabetes mellitus inrabbits with alloxan. A preliminary report. J Am Med 1943, 122, 1165–1166. [Google Scholar]
- Bansal, R., N. Ahmad, and J.R. Kidwai, Alloxan-glucose interaction: Effect on incorporation of14C-leucine into pancreatic islets of rat. Acta diabetologia latina 1980, 17, 135–143. [CrossRef]
- Lee, J.H., et al., Pharmacokinetics of drugs in rats with diabetes mellitus induced by alloxan or streptozocin: comparison with those in patients with type I diabetes mellitus. Journal of Pharmacy and Pharmacology 2010, 62, 1–23. [CrossRef] [PubMed]
- Rakieten, N., M.L. Rakieten, and M.R. Nadkarni, Studies on the diabetogenic action of streptozotocin (NSC-37917). Cancer Chemother Rep 1963, 29, 91–98.
- Wu, J. and L.-J. Yan, Streptozotocin-induced type 1 diabetes in rodents as a model for studying mitochondrial mechanisms of diabetic β cell glucotoxicity. Diabetes, metabolic syndrome and obesity: targets and therapy 2015, 8, 181.
- Lenzen, S. The mechanisms of alloxan-and streptozotocin-induced diabetes. Diabetologia 2008, 51, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Furman, B.L. Streptozotocin-induced diabetic models in mice and rats. Current protocols in pharmacology 2015, 70, 5.47.1–5.47.20. [Google Scholar] [CrossRef] [PubMed]
- Krisanapun, C., et al., Aqueous extract of Abutilon indicum Sweet inhibits glucose absorption and stimulates insulin secretion in rodents. Nutrition research 2009, 29, 579–587. [CrossRef]
- Thayer, T.C., S.B. Wilson, and C.E. Mathews, Use of nonobese diabetic mice to understand human type 1 diabetes. Endocrinology and Metabolism Clinics 2010, 39, 541–561. [CrossRef]
- Qi, W., et al., Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nature medicine 2017, 23, 753–762. [CrossRef]
- Frerichs H, C.W. Der experimentelle chemische; Diabetes; Springer: Berlin Heidelberg; New York, 1971. [Google Scholar]
- Ravichandra, V. and P. Paarakh, Evaluation of anti-diabetic potentials of methanolic extract of Ficus microcarpa leaves in alloxan induced diabetic rats. Int J Pharm Pharm Sci 2013, 5, 369–373. [Google Scholar]
- Szkudelski, T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiological research 2001, 50, 537–546. [Google Scholar]
- im Walde, S.S., et al., Molecular target structures in alloxan-induced diabetes in mice. Life sciences 2002, 71, 1681–1694. [CrossRef] [PubMed]
- Vogel, H.G.; et al. Drug discovery and evaluation: pharmacological assays; Springer, 1997; Volume 2. [Google Scholar]
- Chatzigeorgiou, A., et al., The use of animal models in the study of diabetes mellitus. In Vivo 2009, 23, 245–258.
- Wicker, L.S., et al., Type 1 diabetes genes and pathways shared by humans and NOD mice. Journal of autoimmunity 2005, 25, 29–33. [CrossRef] [PubMed]
- Caquard, M., et al., Diabetes acceleration by cyclophosphamide in the non-obese diabetic mouse is associated with differentiation of immunosuppressive monocytes into immunostimulatory cells. Immunology letters 2010, 129, 85–93. [CrossRef] [PubMed]
- Yang, Y. and P. Santamaria, Dissecting autoimmune diabetes through genetic manipulation of non-obese diabetic mice. Diabetologia 2003, 46, 1447–1464. [Google Scholar] [CrossRef] [PubMed]
- Niens, M., et al., Prevention of “Humanized” diabetogenic CD8 T-cell responses in HLA-transgenic NOD mice by a multipeptide coupled-cell approach. Diabetes 2011, 60, 1229–1236. [CrossRef]
- Mordes, J.P., et al., Rat models of type 1 diabetes: genetics, environment, and autoimmunity. Ilar Journal 2004, 45, 278–291. [CrossRef]
- Holmberg, R., et al., Lowering apolipoprotein CIII delays onset of type 1 diabetes. Proceedings of the National Academy of Sciences 2011, 108, 10685–10689. [CrossRef]
- Zhang, W., et al., C-peptide improves neuropathy in type 1 diabetic BB/Wor-rats. Diabetes/metabolism research and reviews 2007, 23, 63–70. [CrossRef]
- Yokoi, N., et al., A non-MHC locus essential for autoimmune type I diabetes in the Komeda Diabetes-Prone rat. The Journal of clinical investigation 1997, 100, 2015–2021. [CrossRef]
- Yokoi, N., et al., Genetic reconstitution of autoimmune type 1 diabetes with two major susceptibility genes in the rat. Diabetes 2007, 56, 506–512. [CrossRef]
- Ishida, K., et al., Which is the primary etiologic event in Otsuka Long-Evans Tokushima Fatty rats, a model of spontaneous non—insulin-dependent diabetes mellitus, insulin resistance, or impaired insulin secretion? Metabolism 1995, 44, 940–945. [CrossRef] [PubMed]
- Kumar, S., et al., Acute and chronic animal models for the evaluation of anti-diabetic agents. Cardiovascular diabetology 2012, 11, 1–13.
- Mathews, C.E. Utility of murine models for the study of spontaneous autoimmune type 1 diabetes. Pediatric diabetes 2005, 6, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Peschke, E., et al., The insulin–melatonin antagonism: studies in the LEW.1AR1-iddm rat (an animal model of human type 1 diabetes mellitus). Diabetologia 2011, 54, 1831–1840. [CrossRef] [PubMed]
- Jörns, A., et al., Pathology of the pancreas and other organs in the diabetic LEW. 1AR1/Ztm-iddm rat, a new model of spontaneous insulin-dependent diabetes mellitus. Virchows Archiv 2004, 444, 183–189. [CrossRef] [PubMed]
- Mathews, C.E., S.H. Langley, and E.H. Leiter, New mouse model to study islet transplantation in insulin-dependent diabetes mellitus. Transplantation 2002, 73, 1333–1336. [CrossRef]
- Zhou, C., et al., Hyperglycemic Ins2AkitaLdlr−/− mice show severely elevated lipid levels and increased atherosclerosis: a model of type 1 diabetic macrovascular disease [S]. Journal of lipid research 2011, 52, 1483–1493. [CrossRef]
- Drel, V.R., et al., Poly (ADP-ribose) polymerase inhibition counteracts renal hypertrophy and multiple manifestations of peripheral neuropathy in diabetic Akita mice. International journal of molecular medicine 2011, 28, 629–635.
- Chen, H., et al., Apelin alleviates diabetes-associated endoplasmic reticulum stress in the pancreas of Akita mice. Peptides 2011, 32, 1634–1639. [CrossRef]
- Szopa, T.M., et al., Diabetes mellitus due to viruses — some recent developments. Diabetologia 1993, 36, 687–695. [CrossRef] [PubMed]
- Jaïdane, H., et al., Coxsackievirus B4 and type 1 diabetes pathogenesis: contribution of animal models. Diabetes/Metabolism Research and Reviews 2009, 25, 591–603. [CrossRef] [PubMed]
- Yoon, J.-W. and H.-S. Jun, Viruses Cause Type 1 Diabetes in Animals. Annals of the New York Academy of Sciences 2006, 1079, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Mendez, I.I., et al., Immunoregulatory Role of Nitric Oxide in Kilham Rat Virus-Induced Autoimmune Diabetes in DR-BB Rats1. The Journal of Immunology 2004, 173, 1327–1335. [CrossRef] [PubMed]
- Onodera, T., et al., Virus-Induced Diabetes Mellitus: Reovirus Infection of Pancreatic β Cells in Mice. Science 1978, 201, 529–531.
- Filippi, C.M. and M.G.v. Herrath, Good and bad sides of viruses in Type 1 diabetes. Future Virology 2009, 4, 407–410. [Google Scholar] [CrossRef]
- Masiello, P., Animal models of type 2 diabetes with reduced pancreatic beta-cell mass. Int J Biochem Cell Biol 2006, 38, 873–893. [CrossRef]
- Kumar, S., et al., Acute and chronic animal models for the evaluation of anti-diabetic agents. Cardiovasc Diabetol 2012, 11, 9. [CrossRef]
- Botolin, S. and L.R. McCabe, Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology 2007, 148, 198–205. [Google Scholar] [CrossRef]
- Nyman, J.S., et al., Increasing duration of type 1 diabetes perturbs the strength-structure relationship and increases brittleness of bone. Bone 2011, 48, 733–740. [CrossRef]
- Silva, M.J., et al., Type 1 diabetes in young rats leads to progressive trabecular bone loss, cessation of cortical bone growth, and diminished whole bone strength and fatigue life. J Bone Miner Res 2009, 24, 1618–1627. [CrossRef] [PubMed]
- King, A.J. The use of animal models in diabetes research. Br J Pharmacol 2012, 166, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.E., et al., Non-obese diabetic mice rapidly develop dramatic sympathetic neuritic dystrophy: a new experimental model of diabetic autonomic neuropathy. Am J Pathol 2003, 163, 2077–2091. [CrossRef] [PubMed]
- von Herrath, M. and G.T. Nepom, Animal models of human type 1 diabetes. Nat Immunol 2009, 10, 129–132. [Google Scholar] [CrossRef]
- von Herrath, M.G. and G.T. Nepom, Lost in translation: barriers to implementing clinical immunotherapeutics for autoimmunity. J Exp Med 2005, 202, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.A. and J.C. Alcolado, Animal models of diabetes mellitus. Diabet Med 2005, 22, 359–370. [Google Scholar] [CrossRef]
- Mordes, J.P., et al., Rat models of type 1 diabetes: genetics, environment, and autoimmunity. Ilar j 2004, 45, 278–291. [CrossRef]
- Anderson, M.S. and J.A. Bluestone, The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 2005, 23, 447–485. [Google Scholar] [CrossRef]
- Yokoi, N., et al., Establishment and characterization of the Komeda diabetes-prone rat as a segregating inbred strain. Exp Anim 2003, 52, 295–301. [CrossRef]
- Kawano, K., et al., New inbred strain of Long-Evans Tokushima lean rats with IDDM without lymphopenia. Diabetes 1991, 40, 1375–1381. [CrossRef]
- Eisenbarth, G.S. Type 1 diabetes: molecular, cellular and clinical immunology. Adv Exp Med Biol 2004, 552, 306–310. [Google Scholar] [PubMed]
- Komeda, K., et al., Establishment of two substrains, diabetes-prone and non-diabetic, from Long-Evans Tokushima Lean (LETL) rats. Endocr J 1998, 45, 737–744. [CrossRef] [PubMed]
- Lenzen, S. Animal models of human type 1 diabetes for evaluating combination therapies and successful translation to the patient with type 1 diabetes. Diabetes Metab Res Rev 2017, 33. [Google Scholar] [CrossRef] [PubMed]
- Al-Awar, A., et al., Experimental Diabetes Mellitus in Different Animal Models. J Diabetes Res 2016, 2016, 9051426.
- Sin, J., et al., Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [CrossRef] [PubMed]
- Jaïdane, H. and D. Hober, Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab 2008, 34, 537–548. [Google Scholar] [CrossRef]
- Kemball, C.C., M. Alirezaei, and J.L. Whitton, Type B coxsackieviruses and their interactions with the innate and adaptive immune systems. Future Microbiol 2010, 5, 1329–1347. [CrossRef] [PubMed]
- Garmaroudi, F.S., et al., Coxsackievirus B3 replication and pathogenesis. Future Microbiol 2015, 10, 629–653. [CrossRef]
- Chung, Y.H., et al., Role of macrophages and macrophage-derived cytokines in the pathogenesis of Kilham rat virus-induced autoimmune diabetes in diabetes-resistant BioBreeding rats. The Journal of Immunology 1997, 159, 466–471. [CrossRef]
- Talebianpoor, M.S., et al., Antidiabetic Activity of Hydroalcoholic Extract of Myrtus communis (Myrtle) Fruits in Streptozotocin-Induced and Dexamethasone-Induced Diabetic Rats. Pharmacognosy Research 2019, 11.
- Moloney, P. and M. Coval, Antigenicity of insulin: diabetes induced by specific antibodies. Biochemical journal 1955, 59, 179.
- Dewangan, H., et al., Past and Future of in-vitro and in-vivo Animal Models for Diabetes: A Review. Indian Journal of Pharmaceutical Education and Research 2017, 51, s522–s530. [CrossRef]
- Nagata, M., et al., Type 2 diabetes mellitus in obese mouse model induced by monosodium glutamate. Experimental Animals 2006, 55, 109–115. [CrossRef] [PubMed]
- Karasawa, H., K. Takaishi, and Y. Kumagae, Obesity-Induced Diabetes in Mouse Strains Treated With Gold Thioglucose: A Novel Animal Model for Studying β-Cell Dysfunction. Obesity 2011, 19, 514–521. [CrossRef] [PubMed]
- Kottaisamy, C.P.D., et al., Experimental animal models for diabetes and its related complications—a review. Laboratory Animal Research 2021, 37, 1–14.
- Tripathy, B., et al., Various models used to induce Diabetes: A comprehensive review.
- Lindström, P., The physiology of obese-hyperglycemic mice [ob/ob mice]. TheScientificWorldJournal 2007, 7, 666–685. [CrossRef] [PubMed]
- Chen, W., et al., P633H, a novel dual agonist at peroxisome proliferator-activated receptors α and γ, with different anti-diabetic effects in db/db and KK-Ay mice. British journal of pharmacology 2009, 157, 724–735. [CrossRef] [PubMed]
- Srinivasan, K. and P. Ramarao, Animal model in type 2 diabetes research: An overview. Indian journal of medical research 2007, 125, 451. [Google Scholar]
- Chakraborty, G., et al., Age dependence of glucose tolerance in adult KK-Ay mice, a model of non–insulin dependent diabetes mellitus. Lab animal 2009, 38, 364–368. [CrossRef]
- Kim, J.H., et al., Type 2 diabetes mouse model TallyHo carries an obesity gene on chromosome 6 that exaggerates dietary obesity. Physiological genomics 2005, 22, 171–181. [CrossRef]
- Cho, Y.-R., et al., Hyperglycemia, maturity-onset obesity, and insulin resistance in NONcNZO10/LtJ males, a new mouse model of type 2 diabetes. American Journal of Physiology-Endocrinology and Metabolism 2007, 293, E327–E336. [CrossRef] [PubMed]
- Kawano, K., et al., OLETF (Otsuka Long-Evans Tokushima Fatty) rat: a new NIDDM rat strain. Diabetes research and clinical practice 1994, 24, S317–S320. [CrossRef] [PubMed]
- Friedman, J.E., et al., Metabolic consequences of a nonsense mutation in the leptin receptor gene (fak) in the obeses spontaneously hypertensive Koletsky rat (SHROB). Experimental and Clinical Endocrinology & Diabetes 1997, 105, 82–84.
- Tarrés, M., et al., The eSS rat. A model of non-insulin-dependent human diabetes. The American journal of pathology 1992, 141, 761.
- Höppener, J.W.M., et al., Human Islet Amyloid Polypeptide Transgenic Mice: In Vivo and Ex Vivo Models for the Role of hIAPP in Type 2 Diabetes Mellitus. Experimental Diabetes Research 2008, 2008, 697035.
- Matveyenko, A.V., et al., Successful versus failed adaptation to high-fat diet–induced insulin resistance: The role of IAPP-induced β-cell endoplasmic reticulum stress. Diabetes 2009, 58, 906–916. [CrossRef]
- Xue, B., et al., Adipose tissue deficiency and chronic inflammation in diabetic Goto-Kakizaki rats. PLoS one 2011, 6, e17386.
Figure 1.
Methods for antidiabetic activity evaluation.
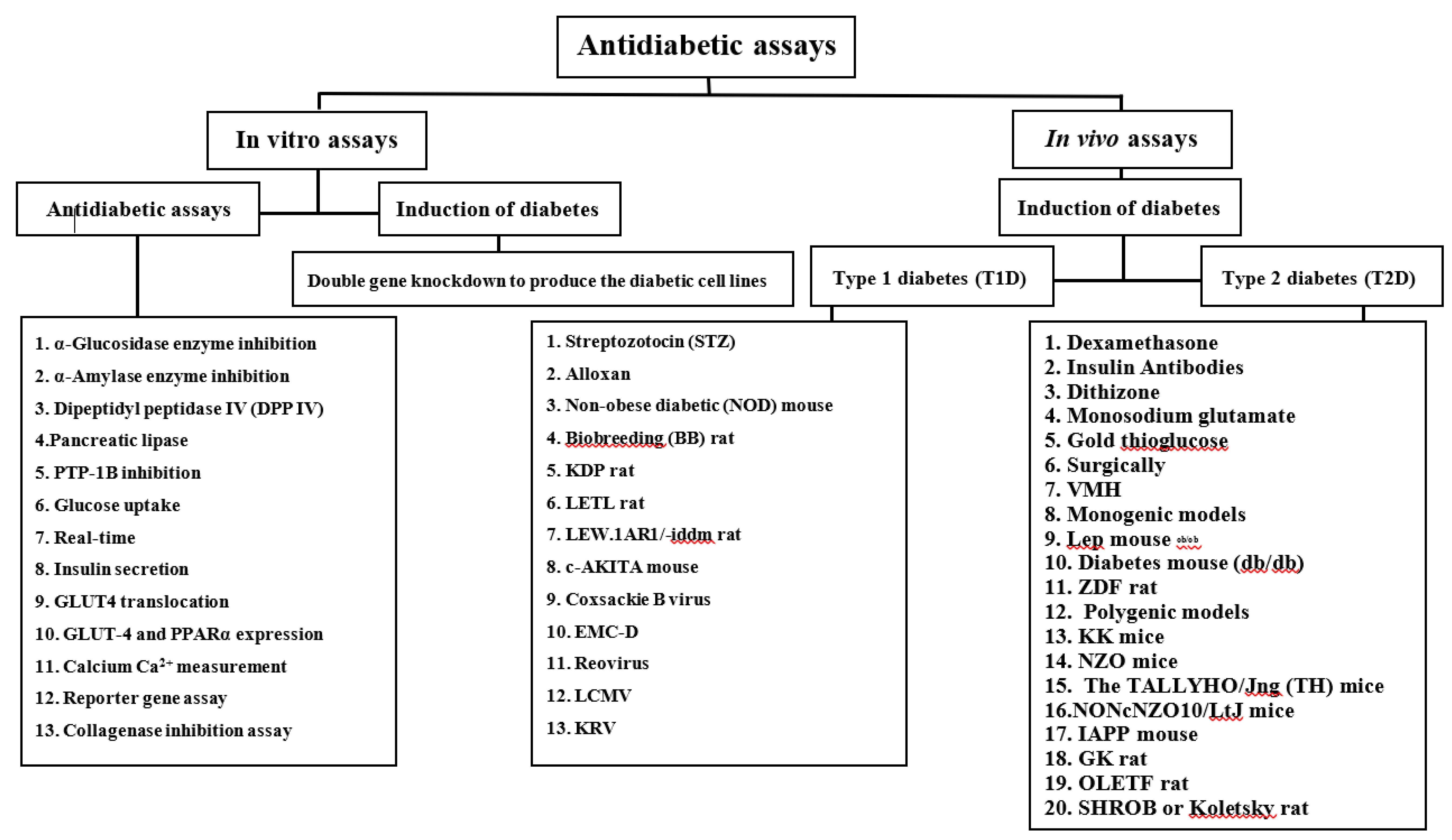
Figure 2.
In vitro antidiabetic assays (https://biorender.io).
Figure 2.
In vitro antidiabetic assays (https://biorender.io).
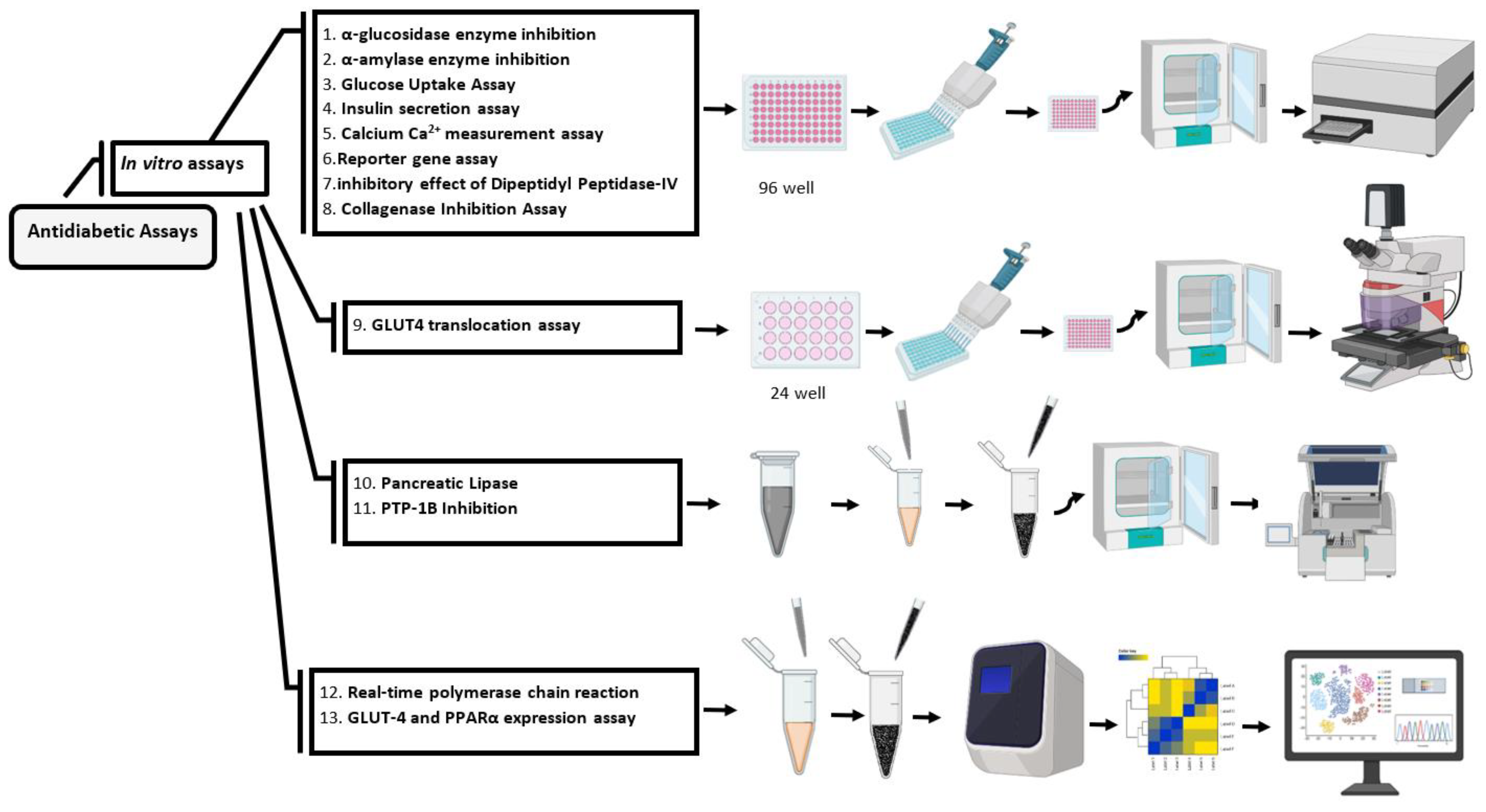
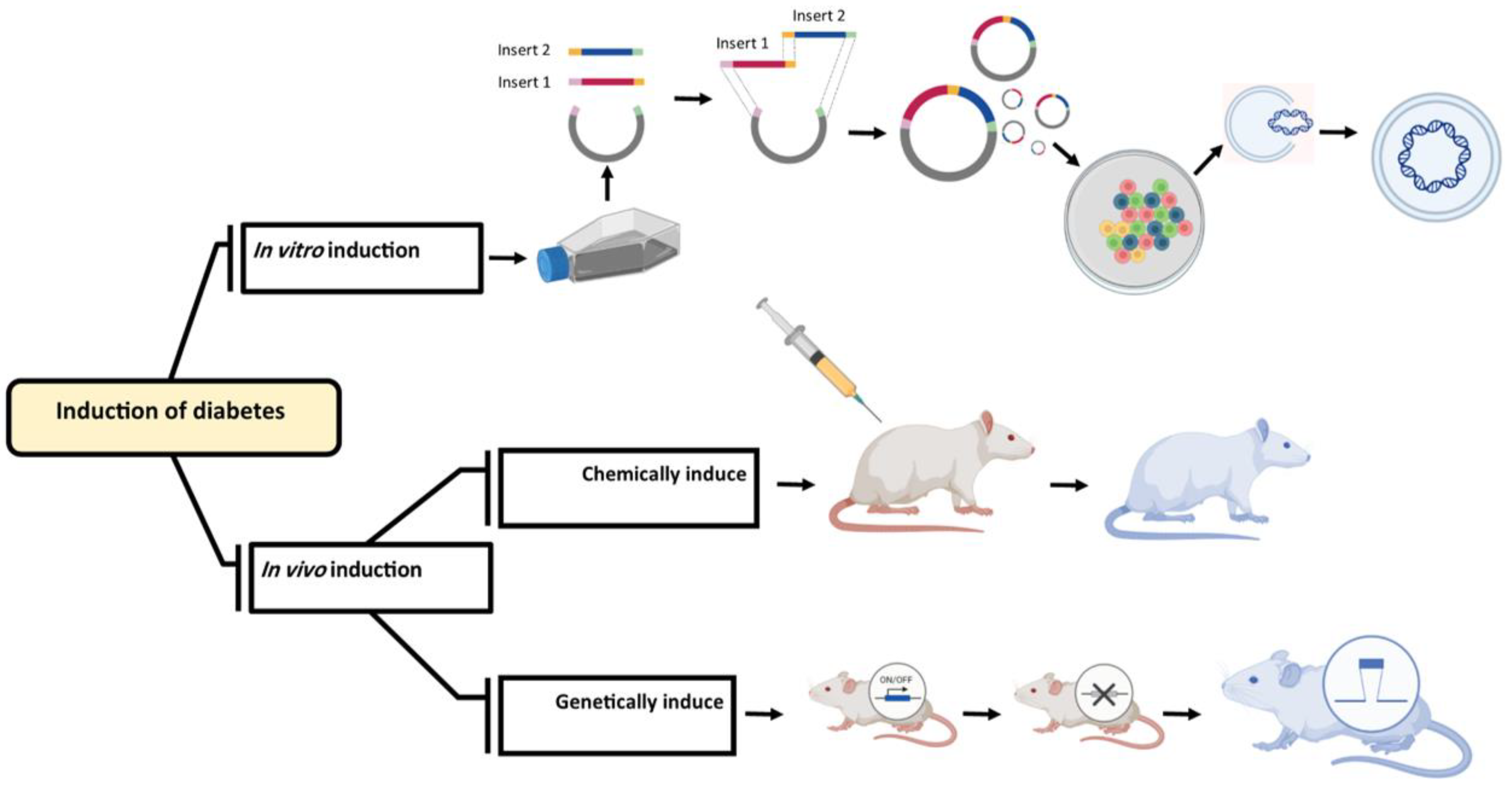
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



