Submitted:
27 April 2023
Posted:
28 April 2023
You are already at the latest version
Abstract
The high morbidity and mortality rate of the pulmonary arterial hypertension (PAH) is partially explained by the metabolic deregulation associated with the disease. The present study identified statistically significant increase of the glucose transporter solute carrier family 2 (Slc2a1), beta nerve growth factor (Ngf), and nuclear factor erythroid derived 2, like 2 (Nfe2l2) on three standard PAH rat models and their healthy counterpart. PAH was induced by subjecting the animals to hypoxia (HO group) or by injecting them with monocrotaline in either normal (CM) or hypoxic (HM) atmospheric conditions. The Western blot and double immunofluorescent experiments were complemented with the gene expression profiling of the animal lungs analyzed from the perspective of the Genomic Fabric Paradigm. We found substantial remodeling of the genomic fabrics of the citrate cycle, pyruvate metabolism, glycolysis/gluconeogenesis and fructose and manose pathways. According to the novel transcriptomic distance criterion, the most comprehensive measure of the transcriptomic alteration, glycolysis/gluconeogenesis was the most affected functional pathway in all three PAH models. PAH decoupled the coordinated expression of many metabolic genes and replaced Pmm2 with Pmm1 in the center of the fructose and mannose metabolism. Our data show that metabolic dysregulation is a major pathogenic factor of the PAH.
Keywords:
citrate cycle
; fructose and mannose pathway
; glycolysis/gluconeogenesis pathway
; Il17b
; Ngf
; Nfe2l2
; pmm1
; pmm2
; pyruvate metabolism
; Slc2a1
1. Introduction
Pulmonary arterial hypertension (PAH) is a rare disease with high morbidity and mortality rate. A number of systemic and genetic diseases, lung developmental defects, congenital heart defects and drug toxicity are known to lead to PAH. Survival time in patients with PAH without treatment is reported to be about 2.8 years. Although, modern treatment has slowed the progression of the disease, it fails to halt it [1,2,3].
During the 6th World Symposium on Pulmonary Hypertension (WSPH), PAH was defined as the mean pulmonary artery pressure >20 mmHg and pulmonary vascular resistance >3 Wood units [4]. Irrespective of the underlying malady, endothelial cell (EC) dysfunction plays an important role in the pathophysiology of PAH. EC dysfunction leads to impaired bioavailability of nitric oxide, activation of proliferative pathways, medial hypertrophy, neointima formation, and obstruction in the pulmonary arteries leading to increased pulmonary artery pressure, subsequent right ventricular hypertrophy and heart failure resulting in premature death [5].
The disruption of endothelial caveolin-1 (Cav1), a cell membrane protein, has been shown to play a key role in the initiation and the progression of pulmonary hypertension (PH) in experimental models and also in human PAH. The loss of endothelial Cav1 is followed by an enhanced expression of Cav1 in smooth muscle cells accompanied by a loss of caveolae. This Cav1 is tyrosine phosphorylated which is thought to participate in cell proliferation and migration. It is significant that a reduced number of caveolae has been observed when the smooth muscle cells shift from a contractile to a proliferative (synthetic) phenotype. These changes lead to further cell proliferation, medial hypertrophy and neointima formation. Importantly, neointimal cells in experimental models and human PAH exhibit lack of Cav1 [6,7,8,9].
The proliferative and obstructive vasculopathy of PAH is accompanied by a shift to aerobic glycolysis and mitochondrial fragmentation. Metabolic alterations including glycolysis, increased glutamine utilization and decreased fatty acid oxidation observed in human and experimental models of PAH are reminiscent of cancer. In cancer, hypoxia-inducible factor-1α (HIF-1α) - mediated effects such as metabolic shift, production of reactive oxygen species, inhibition of fatty acid β-oxidation and alteration in the expression of tumor suppressor genes promote tumor progression [10]. In PAH, acquired mitochondrial abnormalities, such as epigenetic silencing of superoxide dismutase (SOD2), disrupt oxygen sensing creating a pseudo-hypoxic environment characterized by normoxic activation of HIF-1α. The resulting metabolic shift to aerobic glycolysis (the Warburg phenomenon) reflects inhibition of pyruvate dehydrogenase by pyruvate dehydrogenase kinases [11]. In addition, Cav1 knockdown in endothelial cells (in-vitro studies), revealed a decrease in glycolytic intermediates and an increase in fatty acids indicative of metabolic switch [12]. Intact plasma membrane is required for compartmentation of glycolysis and gluconeogenesis. Disruption of caveolae results in the inhibition of glycolysis and stimulation of gluconeogenesis [13]. Furthermore, the expression of glucose transporter solute carrier family 2 member 1 (Slc2a1) has been shown to be increased in cancer; and it is thought to regulate glycolysis and proliferation in cancer cells [14].
In hypoxia-induced PAH, endothelial Cav1 dysfunction was shown to be associated with the activation of proliferative pathways, loss of phosphatase and tensin homologue (Pten) and increased expression of Slc2a1 [15]. In thyroid cancer cells, the loss of PTEN expression was found to be accompanied by increased expression of SLC2A1 on the plasma membrane [16]. Caveolin-1 protein expression has been shown to determine the membrane-associated PTEN levels and activity. PTEN via caveolin-1 binding sequence suppresses cell proliferation and thus, maintains homeostasis [17]. Nuclear factor erythroid derived 2, like 2 (Nfe2l2, also known as Nrf2) plays an important role in regulating the cellular redox status and metabolic reprogramming. Cav1 interacts with Nfe2l2 in cytosol as well as in nucleus. Caveolin-1 inhibits cellular antioxidant capacity through direct interaction and suppression of Nfe2l2, and Cav1 knockdown leads to dissociation between Nfe2l2 and its cytoplasmic inhibitor Keap1 (Kelch-like ECH-associated protein 1), thus increasing Nfe2l2 transcriptional activity [18,19]. Under normal conditions, Nfe2l2 protects cells against oxidative stress by activating genes encoding detoxifying and antioxidant proteins. However, Nfe2l2 accumulation has been shown to confer growth and survival in cancer [20,21]. In human PAH and in experimental models of PAH, increased expression of nerve growth factor (NGF) has been reported. NGF promotes cell proliferation, migration and increased activity of pro-inflammatory cytokines. Furthermore, Nfe2l2 was confirmed as the downstream target of NGF to promote the viability, adhesion and migration [22,23,24].
In a previous paper [25], we have reported altered Cav1 expression in the lungs of all the three investigated PAH rat models. In order to test our hypothesis that Cav1 dysfunction has an important role in the pathogenesis of PH, in this study performed on the same rat models of PAH, we examined alterations in the protein levels of Ngf, Nfe2l2, Slc2a1, and the restructuring of several metabolic pathways.
Numerous publications have reported gene expression regulations caused by PAH in lungs from humans (e.g.: [1,26]) and animal models of PAH [27,28]), but they are limited to comparing the expression levels of genes in lungs of PAH and healthy subjects. Our study goes further by adopting the Genomic Fabric Paradigm (GFP) [29] that takes full advantage of the simultaneous profiling of tens of thousands of genes on several biological replicas. Thus, in addition to the average expression level (AVE), GFP attaches to every gene in each condition the independent Relative Expression Variability (REV) among biological replicas and expression correlation (COR) with each other gene. By doing so, GFP approaches the cellular transcriptome as a mathematical object with tens of millions of dimensions, subjected to dynamic sets of transcription controls and expression correlations. GFP increases by four orders of magnitude the amount of workable transcriptomic data obtained from a high throughput platform (RNA-sequencing, microarray) without increasing the experimental costs.
The genomic fabric of a functional pathway is by definition the transcriptome associated to the most inter-coordinated and stably expressed gene network responsible for that functional pathway.
REV shows how sensitive is the transcription of that gene to the slight (not significantly regulating) environmental differences among the biological replicas. As such, REV is in inverse relationship with the strength of the transcription control exerted by the cellular homeostatic mechanisms to limit the expression random fluctuations, indicating the genes whose right expression is critical for the cell life [30]. COR analysis is used to refine the gene networking based on the “Principle of Transcriptomic Stoichiometry” (PTS) [31], a generalization of Dalton’s law from chemistry that imposes the correlated expression of genes whose encoded products are involved in a functional pathway.
2. Materials and Methods
2.1. Animals
Sprague-Dawley rats (weight 150–175 g) were obtained from Charles River Laboratories (Wilmington, MA, USA). Rats were allowed to acclimatize in the animal facility for five days before the start of experimental protocols and had free access to chow and water. Protocol (“Mechanism of neointima formation in pulmonary hypertension”) was approved by the Institutional Animal Care and Use Committee of New York Medical College (approval Letter# 4-1-0113/2014, Banner No: 171020) and respected the guiding principles for the use and care of laboratory animals of the American Physiological Society and the National Institutes of Health.
Rats were divided in 4 groups: Gr.1 Control (CO), Gr.2 Monocrotaline (CM), Gr.3 Hypoxia (HO), and Gr.4 Monocrotaline + Hypoxia (HM). Control rats (Gr.1) were maintained in room air, Gr.2 rats received monocrotaline (40 mg/kg, sc) and were kept in room air, Gr.3 rats were subjected to hypobaric hypoxia (atmospheric pressure 380 mmHg) starting on day 1, and Gr.4 rats received monocrotaline 40 mg/kg and were subjected to hypobaric hypoxia starting on day 1. The hypoxia chamber was opened twice per week for 15 min to weigh the rats, replenish food and water, and to provide clean bedding similar to the other rats in room air. At the end of the 4 weeks, the rats were sacrificed and their tissues were analyzed.
2.2. Hemodynamic Data
As previously described [6,7,8], rats were anesthetized with pentobarbital (60 mg/kg, ip), and ventilated at 70-80 breaths/min through a tracheostomy, cannulated with a PE240 tubing (tidal vol. 0.83 mL/100 g body weight). A thoracotomy was performed; right ventricular systolic pressure (RVSP) measured with a small needle attached to a PE50 tubing, using Grass polygraph (model 7E). After perfusing the lungs with normal saline, heart and lungs were removed. The ratio of the right ventricle (RV) and the left ventricle including septum (LV) was used to assess right ventricular hypertrophy (RVH). Right lung was frozen and stored at -80℃. The heart and the left lung were kept in 10% buffered formaldehyde.
2.3. Estimation of Protein Expression
Western blot analysis was carried out as previously described [6,7,8]. Briefly, the lung tissue was homogenized in a buffer containing 0.1 M PBS (pH 7.4), 0.5% sodium deoxycholate, 1% Igepal, 0.1% SDS, 10 mL/mL phenylmethylsulfonic fluoride (PMSF; 10 mg dissolved in 1 mL of isopropanol), 25 mg/mL aprotinin, and 25 mg/mL leupeptin. PMSF (10 mL/mL) and phosphatase Inhibitor Cocktail 1 (10 mL/mL; Sigma) were added to homogenates placed on ice for 30 min, and then centrifuged at 14,000 rpm for 20 min at 40 C. A total of 50 or100 mg of protein from lung supernatants were loaded and separated on a 10% sodium SDS-polyacrylamide gel (Mini Protean-II, Bio-Rad) and transferred to nitrocellulose membrane (Hybond ECL, Amersham Life Science, MA, USA) using Semi Dry Transfer Cell (Bio-Rad). The membranes were blocked with 5% non-fat milk powder in Tris-Buffered-Saline with Tween buffer (TBST; 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% Tween 20) overnight at 40 C. Membranes were then incubated with antibodies of interest; Slc2a1 (1:400, Abcam) Abcam), Nfe2l2 (1:400 Abcam), or NGF (1:800, Santa Cruz) for 1 hr at room temperature, or overnight at 40 C, according to the manufacturers’ recommendations. The blots were re-probed with β-actin (1:10,000) to assess the protein loading. The protein bands were visualized by chemiluminescence (ECL Western Blotting Analysis System, Amersham International). The relative expression of the proteins was quantified using densitometric scanning and is expressed as the ratio of protein of interest and β-actin.
2.4. Double Immunofluorescence
Double immunofluorescence was carried out as described previously [25]. Briefly, lung sections were mounted on superfrost plus microscope slides (VWR Scientific, PA, USA); and deparaffinized in xylene (5 min x2), rehydrated through a range of aqueous ethyl alcohol solution in H2O, and immersed in PBS for 5 min. Antigen retrieval was performed by incubating the sections in 10 mM citrate buffer, pH 6, in a microwave oven for 5 min. The slides were then incubated in a blocking solution (5% normal donkey serum, 0.5% Triton in PBS) for 1 hr. at room temperature, followed by an overnight incubation at 4o C with the primary antibody Slc2a1 (1:400) in blocking solution. The next day the slides were washed with PBS for 10 min x3 and incubated in Alexa 488 (donkey anti-rabbit, 1:300, green color, Molecular Probes) at room temperature in a dark place for 1 h followed by washing in PBS for 10 min 3. For double staining, the sections were blocked again, as described earlier, and incubated in the smooth muscle α-actin antibody (1:15) overnight; the procedure was repeated using appropriate secondary antibody Alexa 594 (donkey anti-rabbit 1:200, red color, Molecular Probes). The sections were examined with a laser scanning confocal fluorescence microscope (MRC 1000, Bio-Rad). The negative controls were run in the absence of primary antibodies.
2.5. Statistical Analysis
The data are expressed as means ± SEM. For statistical analysis, we used one-way analysis of variance followed by Scheffe’s multiple comparison tests. Differences were considered statistically significant at p < 0.05.
2.6. Gene Expression Data
This study used the gene expression results deposited and publicly available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72707. The data were obtained by profiling the lungs of the three rat models using Agilent 60 mer 4 × 44k whole genome rat V2 arrays (#G2519F) as previously described [25]. Filtered raw data were further processed and the reciprocally independent characteristics AVE (average expression level), REV (relative expression variability) and COR (expression correlation with each other gene) are attached to every single gene in each condition.
Because numerous genes are probed redundantly in the Agilent microarray by more than one spot (non-uniform number of spots), the independent three types of characteristics were computed as:
The expression levels in each condition were normalized to the median expression level of all genes in that condition.
Lower REV values indicate genes whose random fluctuations of the expression level are strongly limited by the cellular homeostatic mechanisms; such genes are most likely critical for the cell survival, phenotypic expression and proliferation. By contrast, higher REVs are associated with less controlled genes that may function as vectors of cell adaptation to the environmental fluctuations [32].
COR analysis is based on the Pearson product-moment correlation coefficient between the (log2) expressions of each gene i across biological replicas with each other gene g in the same group of replicas [30].
COR analysis identifies the (p < 0.05) significantly synergistically (in-phase fluctuations), antagonistically (opposite fluctuations) and independently expressed gene pairs in each condition. One can further determine the coordination score COORD as:
where: SYN, ANT and IND are the percentages of the gene pairs that are p-significantly synergistically, antagonistically or independently expressed within the investigated functional pathway Γ in the given condition.
COR refines the gene wiring in functional pathways built by specialized software like Ingenuity [33], DAVID [34], KEGG [35] etc. that ignore the race/strain, sex, age, and other factors known to influence the incidence of the disease, and the response to a treatment. By combining REV and COR, GFP singles out for each patient, in each condition, the most stably expressed and interconnected gene network responsible for that particular biological process.
2.7. Transcriptomic Analyses
2.7.1. Significant Regulation
Traditional analysis considers a gene as significantly regulated in the disease with respect to the healthy counterpart when the absolute expression ratio (|x|) exceeds an arbitrarily introduced cut-off (e.g. 1.5x or 2.0x) and/or the p-value of the heteroscedastic t-test of the means’ equality is less than 0.05 or 0.01 (another arbitrarily introduced cut-off criterion). While we are kipping the p-value condition, for the expression ratio we determine for each gene, in every pair of conditions to be compared the cut-off “CUT” that takes into account both the biological variability of that gene in both conditions and the technical noise of the probing spot(s) in the two groups of 4 microarrays. Thus, for stably expressed genes (low variability among biological replicas) and accurate probing (low technical noise), CUT is below 1.5 while for other it may exceed 1.5.
2.7.2. Measures of Individual Genes’ Contributions to the Transcriptomic Alteration
In addition to the traditional Percentage of Significantly Regulated (PSR) genes and expression ratio (x), we use the “Weighted Individual (gene) Regulation (WIR) and the 3D vector “Individual (gene) Transcriptomic Trajectory” (ITT), whose module is the “Transcriptomic Distance” (TD). PSR is restricted to the significantly regulated genes (according to arbitrarily introduced criteria) and implicitly considers all genes as uniform +1 or -1 contributors to the overall transcriptomic alteration. In contrast, WIR and TD take into account all genes. While WIR weights the contribution of the AVE change by the normal expression level, the net absolute fold-change and the confidence of the expression regulation, TD goes further by quantifying also the changes in expression variability and expression coordination with all other genes.
3. Results
3.1. Significant Increase of the RVSP and RV/LV Weight Ratio in All Three PAH Models
RVSP and RV/LV weight ratio in the control group (CO) were respectively: 21±0.8 mmHg and 0.23±0.004. RVSP was significantly increased in all PAH groups: 53 ± 3.48*mmHg in HO, 66 ± 8* mmHg in CM, and 90 ± 0.104** mmHg in HM. RV/LV ratio was also significantly increased to: 0.56 ± 0.004* in HO, 0.65 ± 0.004* in CM and 0.65 ± 0.004* in HM. [* = p < 0.05 significant, ** = p < 0.01 significant].
3.2. Monocrotaline Administration Increased the Expression of the Nerve Growth Factor (Ngf)
Figure 1 presents the Western blot analysis of the nerve groth factor (Ngf) in all groups of PAH rats with respect to controls. Of note is the statistically (p < 0.05) significant increase in CM and (p < 0.01) in HM. Although the HO group exhibited an average of 23.5% increase, it was not statistically significant. The data indicates that the administration of the monocrotaline is the main driver of the Ngf increase, with hypoxia enhancing its effect.
3.3. Administration of Monocrotaline to Rats Exposed to Hypoxia Increased the Expression of the Nuclear Factor Erythroid Derived 2, like 2 (Nfe2l2)
Figure 2 presents the Western blot analysis of the abundance of the nuclear factor erythroid 2-related factor 2 (Nfe2l2) in the lungs of the three rat models of pulmonary hypertension with respect to the control rats. Interestingly, only the combination of hypoxia exposure and monocrotaline administration produced statistically significant increase of the Nfe2l2.
3.4. Hypoxia –Induced Pulmonary Hypertension Increased the Expression of Glucose Transporter Solute Carrier Family 2 (Slc2a1)
Figure 3 presents the Western blot analysis of the Slc2a1 in the lungs of the three rat models of pulmonary hypertension and the validation by double immunofluorescence. The increase was statistically (p < 0.05) significant in both groups of hypoxia-expossed animals but not in the CM grup (rats injected with monocrotaline in normal athmospheric conditions).
3.5. Independent Transcriptomic Characteristics
Figure 4 illustrates the independence of the three types of transcriptomic characteristics for 50 genes involved in the KEGG-determined immune response [36]. Correlation with interleukin 17B (Il17b) was selected for this illustration owing to contrasting reports of both pro- and anti-inflammatory function of Il17b in rodents [37]. The impact of PAH on the immune response is important by itself; however any other subset of genes would verify the independence of the three types of characteristics in any condition as we proved in several other genomic studies on human cancers (e.g. [38]) and animal models of neurological diseases (e.g. [39]).
While the independence of th three characteristics is evident by visual inspection, of note are also the substantial differences both among the genes and among the conditions. Thus, within this section, AVE goes from 0.22 median gene expression for interleukin 25 (Il25) in HM (1.12 in CO, 0.82 in HO, and 0.75 in CM) to 302.56 median gene for chemokine (C-X3-C motif) receptor 1 (Cx3cr1) in CM (114.29 in CO, 283.17 in HO, and 45.20 in HM). REV goes from 3.71% for chemokine (C-X-C motif) ligand 12 (Cxcl12) in CO (97.67% in HO, 99.80% in CM, 58.78% in HM) to 159.57% for chemokine (C-C motif) ligand 6 (Ccl6) in HO (38.93% in CO, 115.51% in CM, 57.39% in HM).
The substantial synergistic expression correlation of this subset of genes with Il17b in CO (55.10%) was significantly reduced in the PAH models (22.45% in both HO and CM, and 32.65% in HM). Using the “CORRELATION” software [40], we found that the (p < 0.05) significat coordination score within this 50-gene subset discreased from 31.51% in CO to: 7.02% in HO, 22.04% in CM and 24.37% in HM. Figure 5 shows the remarkable decrease of the coordination score in HO compared to CO for this gene subset.
3.6. Measures of Transcriptomic Alterations
The quantification methods were applied to 50 genes involved in the immune response. Most reports just count how many genes were significantly (according to arbitrarily introduced criteria) up- or down-regulated, listing eventually the genes with the largest positive and negative fold-change. However, we believe that the analysis not only should include all genes but should also go further by showing how much the change of the expression level counts for the expression profile of the entire transcriptome, as by WIR scoring. Given that PAH alters also the homeostatic mechanisms that limit the random expression fluctuations and remodels the gene networks, in addition to WIR, we introduced also the comprehensive quantifier “transcriptomic distance” to the normal state. Figure 6 illustrates our four ways to quantify the contributions of the individual genes to the transcriptomic alterations in the PAH models: uniform, expression ratio “x”, weighted individual (gene) regulation “WIR”, and transcriptomic distance “TD” to the normal/control state (CO).
3.7. Regulation of Glycolysis-Gluconeogenesis
3.8. Remodeling of the Metabolic Genes Network
4. Discussion
In this report, we present the effects of the pulmonary arterial hypertension on the expression of several important genes and proteins, and on the topology of certain metabolic pathways. The transcriptomic analyses were carried on from the perspective of the Genomic Fabric Paradigm [43] that characterizes the expression levels, the control of transcripts’ abundances and the expression inter-coordination of the genes.
Interestingly, the reported in [25] Cav1 overexpression by 39.14x in CM, 13.18x in HO, and 79.06x in HM is qualitatively in line with the overexpression of nerve growth factor (Ngf) in the three models depicted in Figure 1. The results indicate that Cav1 is associated with Ngf. However, an inverse relationship between the protein levels of Cav1 and Ngf was found in the heart of a rat model of induced chronic ischemic heart failure compared to the corresponding control [44]. An increase of Ngf expression associated with Cav1 decrease was reported in the urinary bladder of rats following acute urinary retention after parturition [45]. Our results indicate that expressions of Cav1 and Ngf might be controlled by potentially different upstream factors.
Although none of the hypoxia exposure and monocrotaline treatment applied alone had practically any effect on the abundance of the nuclear factor erythroid derived 2, like 2 (Nfe2l2), together these two PAH inducers triggered a significant increase of Nfe2l2. We interpret the substantial increase of Nfe2l2 in the HM rats as a desperate activation of a molecular mechanism aiming to maintaining the redox balance [46].
Albeit we found that hypoxia is the main trigger of the increase in expression of glucose transporter solute carrier family 2 member 1 (Slc2a1), with monocrotaline administration having also a positive effect, other groups [47] gave more credit to monocrotaline. A significant up-regulation of the SLC2A1 gene was also reported recently in a group of 6 patients with end-stage PAH [48].
While checking the 50 immune-inflammatory response genes to illustrate the independence of the three types of transcriptomic characteristics, we found that the expression inter-coordination of these genes was significantly decreased by PAH in all three models. This result indicates (for the first time to our knowledge) a substantial desynchronization of the expressions of these genes, making the immune-inflammatory response more chaotic which requires use of anti-inflammatory therapeutics [49]. For now, we have no explanation of why hypoxia alone is the most efficient decoupling factor (group HO, Figure 5), albeit monocrotaline administration has also a significant effect in CM while reducing the consequences of hypoxia in HM group.
The traditional percentage of significantly regulated genes has the major handicaps of being based on arbitrarily introduced cut-offs for expression ratio and p-value, and considering all regulated genes as equal +1 or -1 contributors. Therefore, we used instead the weighted individual (gene) regulation (WIR) and the transcriptomic distance (TD). WIR accounts for the total change of the expression level, while TD considers the changes in all three types of the gene expression characteristics: the average expression level, the relative expression variability among biological replicas and the expression coordination with all other genes. As such, WIR and much more TD are far better measures of the contributions of the individual genes to the transcriptome alteration. For instance, the most significantly up-regulated gene in HM, chemokine (C-C motif) ligand 21 (Ccl21), has the expression ratio x = 247.07, WIR = 199.89, and TD = 22.88. Each quantifier (excepting the uniform contribution) can be used to establish the regulation hierarchy of the genes, but the resulted hierarchies are not the same. Thus, the most affected 3 genes in HO as fold-change are: Ccl6 (chemokine (C-C motif) ligand 6, x = 7.53), Ifngr2 (chemokine (C-C motif) ligand 6, x = -6.97), Il34 (interleukin 34, x = 5.71). As WIR, the top 3 affected genes in HO are: Il2rg (interleukin 2 receptor, gamma, WIR = -391), Tnfsf12 (tumor necrosis factor ligand superfamily member 12, WIR = -222), Ifngr2 (WIR = -82.5), while as TD the hierarchy in HO is: Cx3cr1 (chemokine (C-X3-C motif) receptor 1, TD = 15.90), Ccrl2 (chemokine (C-C motif) receptor-like 2, TD = 14.60), Il2rg (TD = 9.29). As expected, the hierarchy of the individual gene contributions to the overall transcriptomic alteration depends also on how PAH was installed. With respect to the TD measure, the most affected genes in CM were: Cx3cr1 (TD = 17.70), Il2rg (TD = 10.80) and Ccl21 (chemokine (C-C motif) ligand 21, TD = 8.17), while in HM they were: Ccl21 (TD = 22.88), Ifitm2 (interferon induced transmembrane protein 2, TD = 13.00) and Il2rg (TD = 9.64).
Ccl21, found by us as up-regulated by 3.26x in HO, 79.95x in CM and 247.09 in HM, has a disputed value as a potential PAH biomarker in systemic sclerosis [51,52]. Nonetheless, our results indicate that the contribution of Ccl21 to the PAH phenotype depends on the disease etiology, hypoxic PAH being the least dependent on (TD = 4.45).
From transcriptomic point of view, the glycolysis/gluconeogenesis was the most affected functional pathway. As presented in Figure 7(d), the average TD was 62.84 for HO rats, 55.13 for CMs and 91.10 for HMs, substantially larger than the average TD of the 50 immune-inflammatory genes from Figure 6(d): 2.42 (HO), 2.98 (CM) and 2.76 (HM). Glycolysis/gluconeogenesis tops the list of altered pathways also by the percentages of the significantly regulated genes in all three PAH models: 42.37% (HO), 55.93% (CM) and 83.05% (HM). Our results confirms that the aberrant glycolysis is a major pathogenic mechanism in the development of PAH [53].
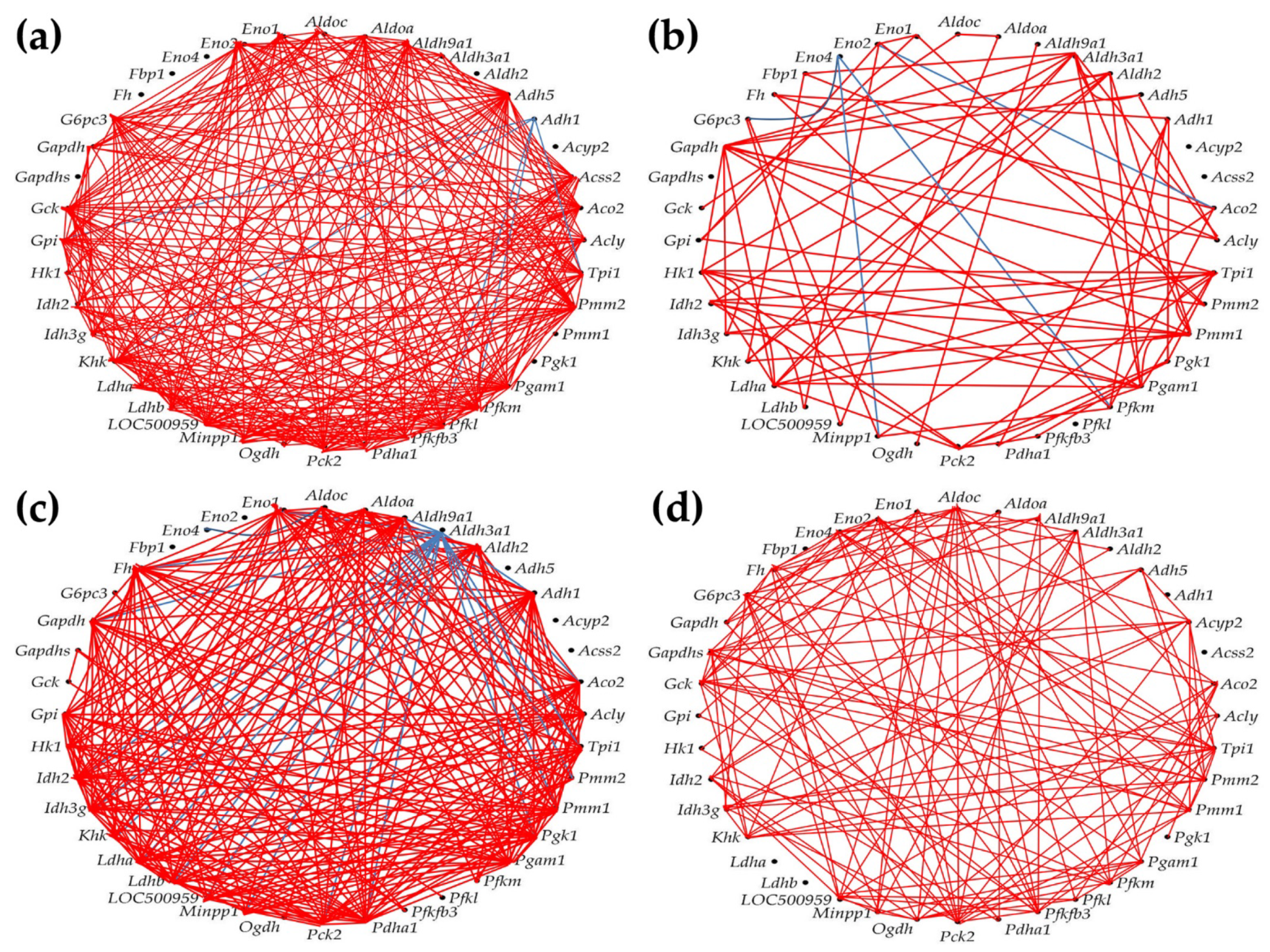
As presented in Figure 8, we found that PAH had a strong impact on the networking of the selected genes. Thus, out of 780 gene pairs, 316 (i.e. 40.5%) were (p < 0.05) significantly synergistically expressed in CO. The number of synergistic pairs was reduced to 79 (10.1%) in HO, 260 (33.3%) in CM and 138 (17.7%) in HM, indicating massive decoupling of the genes in the studied metabolic pathways. There are some remarkable results at the level of individual genes. For instance, the most coupled genes in CO are: Adh5, Pck2 and Pmm2, all with 28 (i.e. 71.8%) synergistically expressed partners within the selection, while: Acyp2, Aldh2, Eno4, Fbp1, Fh, Gapdhs, Pgk1, and Pmm1 have no synergistic partners. Our finding confirms the major roles played by Adh5 [54], Pck2 [55], and Pmm2 [56] for the lung and other organs physiology.
In HO, the most coupled genes are: Aldh3a1 and Gapdh with only 10 (25.5%) synergistic partners, while the uncoupled genes (no partners) are: Acss2, Acyp2, Eno4, Gapdhs, Pfkl. Aldh3a1 is a putative biomarker of lung cancer [57], while Gapdh was reported as critical for stem cell therapy of pulmonary hypertensive females [58].
Very interesting are the results about the phosphomannomutases 1 and 2. Thus, the uncoupled Pmm1 in CO, has 9 (23.1%) synergistic partners in HO, 23 (59.0%) in CM and 10 (25.5%) in HM, while the high interconnection (71.8%) of Pmm2 in CO is reduced to 7.7% in HO, 17.9% in CM, and 25.5% in HM. These results suggest that PAH replaced Pmm2 with Pmm1 in the center of the fructose and mannose metabolism. While the Pmm1 was significantly up-regulated in all three PAH models (by 11.87x in HO, 17.71x in CM and 26.04x in HM), Pmm2 was significantly down-regulated in all three (by -4.65x in HO, -3.50x in CM and -3.14x in HM). Another surprising finding is the switch from 46.2% synergistic partners and 0% antagonistic ones for Aldh3a1 in CO to 0% synergism and 51.3% antagonism in CM, although in the other two PAH models is only synergistically connected, and in all three models it was significantly down-regulated.
5. Conclusions
Altogether, our data indicate the significant association between the metabolism dysregulation and the pulmonary arterial hypertension.
Author Contributions
Conceptualization, R.M. and D.A.I.; methodology, R.M. and D.A.I.; software, D.A.I.; validation, R.M. and D.A.I.; formal analysis, D.A.I.; investigation, S.I. and J.H.; resources, R.M.; data curation, R.M. and D.A.I.; writing—original draft preparation, D.A.I.; writing—review and editing, R.M.; visualization, D.A.I.; supervision, D.A.I.; project administration, R.M.; funding acquisition, R.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and Protocol (“Mechanism of neointima formation in pulmonary hypertension”) was approved by the Institutional Animal Care and Use Committee of New York Medical College (approval Letter# 4-1-0113/2014, Banner No: 171020).
Data Availability Statement
This study used the gene expression results deposited and publicly available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72707.
Conflicts of Interest
The authors declare no conflict of interest.
References
- D’Alonzo, G.E.; Barst, R.J.; Ayers, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991, 115, 343–349. [Google Scholar] [CrossRef]
- Thenappan, T.; Shah, S.J.; Rich, S.; Tian, L.; Archer, S.L.; Gomberg-Maitland, M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010, 35, 1079–1087. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaïci, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Survival in patients with idiopathic, familial and anorexigen-associated pulmonary arterial hypertension in modern management era. Circulation. 2010, 122, 156–163. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Gewitz, M.H. Pulmonary hypertension in infancy and childhood. Heart Dis 2000, 2, 362–368. [Google Scholar]
- Huang, J.; Wolk, J.H.; Gewitz, M.H.; Mathew, R. Caveolin-1 expression during the progression of pulmonary hypertension. Exp Biol Med. 2012, 237, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wolk, J.H.; Gewitz, M.H.; Loyd, J.E.; West, J.; Austin, E.D.; Mathew, R. Enhanced caveolin-1 expression in smooth muscle cells: Possible prelude to neointima formation. World J Cardiol. 2015, 7, 671–684. [Google Scholar] [CrossRef]
- Huang, J.; Mathew, R. Loss of Cavin1 and Expression of p-Caveolin-1 in Pulmonary Hypertension: Possible role in neointima formation. World J Hypertens 2019, 9, 17–29. [Google Scholar] [CrossRef]
- Thyberg, J. Differences in caveolae dynamics in vascular smooth muscle cells of different phenotypes. Lab Invest 2000, 80, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Ajduković, J. HIF-1 — A big chapter in cancer tale. Experimental Oncology 2016, 38, 9–12. [Google Scholar] [CrossRef]
- Ryan, J.; Dasgupta, A.; Huston, J.; Chen, K.H.; Archer, S.L. Mitochondrial dynamics in pulmonary arterial hypertension. J. Mol Med (Berl) 2015, 93, 229–242. [Google Scholar] [CrossRef]
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 Is a Critical Determinant of Autophagy, Metabolic Switching, and Oxidative Stress in Vascular Endothelium. PLoS one. 2014, 9, e87871. [Google Scholar] [CrossRef]
- Vallejo, J.; Hardin, C.D. Metabolic organization in vascular smooth muscle: distribution and localization of caveolin-1 and phosphofructokinase. Am J Physiol Cell Physiol 2004, 286, C43–C54. [Google Scholar] [CrossRef]
- Xiao, H.; Wang, J.; Yan, W.; Cui, Y.; Chen, Z.; Gao, X.; Wen, X.; Chen, J. GLUT1 regulates cell glycolysis and proliferation in prostate cancer. Prostate 2018, 78, 86–94. [Google Scholar] [CrossRef]
- Huang, J.; Frid, M.; Gewitz, M.G.; Fallon, J.T.; Brown, D.; Krafsur, G.; Stenmark, K.; Mathew, R. Hypoxia-induced pulmonary hypertension and chronic lung disease: caveolin-1 dysfunction an important underlying feature. Pulm Circ. 2019, 9, 2045894019837876. [Google Scholar] [CrossRef] [PubMed]
- Morani, F.; Phadngam, S.; Follo, C.; Titone, R.; Aimaretti, G.; Galetto, A.; Alabiso, O.; Isidoro, C. PTEN regulates plasma membrane expression of glucose transporter 1 and glucose uptake in thyroid cancer cells. J Mol Endocrinol. 2014, 53, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Khalil, W.; Kahm, J.; Jessurun, J.; Kleidon, J.; Henke, C.A. Pathologic Caveolin-1 Regulation of PTEN in Idiopathic Pulmonary Fibrosis. Am J Pathol 2010, 176, 2010. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014, 39, 199–2181. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, H.; Zhou, J.S.; Cao, J.F.; Zhou, X.B.; Choi, A.M.K.; Chen, Z.H.; Shen, H.H. Caveolin-1 Inhibits Expression of Antioxidant Enzymes through Direct Interaction with Nuclear Erythroid 2 p45-related Factor-2 (Nrf2). J Biol Chem 2012, 287, 20922–20930. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Panda, H.; Wen, H.; Suzuki, M.; Yamamoto, M. Multifaceted Roles of the KEAP1–NRF2 System in Cancer and Inflammatory Disease Milieu. Antioxidants 2022, 11, 538. [Google Scholar] [CrossRef]
- Rojo, A.I.; Rada, P.; Mendiola, M.; Ortega-Molina, A.; Wojdyla, K.; Rogowska-Wrzesinska, A.; Hardisson, D.; Serrano, M.; Cuadrado, A. The PTEN/NRF2 Axis Promotes Human Carcinogenesis. Antioxidant Redox Signaling 2014, 21, 2498–2541. [Google Scholar] [CrossRef]
- Freund-Michel, V.; Dos Santos, M.C.; Guignabert, C.; Montani, D.; Phan, C.; Fl Coste, F.I.; Tu, L.; et al. Role of nerve growth factor in development and persistence of experimental pulmonary hypertension. Am J Respir Crit Care Med. 2015, 192, 342–355. [Google Scholar] [CrossRef]
- Fang, C.-N.; Tan, H.-Q.; Song, A.-B.; Jiang, N.; Liu, Q.-R.; Song, T. NGF/TrkA promotes the vitality, migration and adhesion of bone marrow stromal cells in hypoxia by regulating the Nrf2 pathway. Metab Brain Dis. 2022. [Google Scholar] [CrossRef]
- Mathew, R.; Huang, J.; Iacobas, S.; Iacobas, D.A. Pulmonary Hypertension Remodels the Genomic Fabrics of Major Functional Pathways. Genes 2020, 11, 126. [Google Scholar] [CrossRef]
- Lago-Docampo, M.; Solarat, C.; Méndez-Martínez, L.; Baloira, A.; Valverde, D. Common Variation in EDN1 Regulatory Regions Highlights the Role of PPARγ as a Key Regulator of Endothelin in vitro. Front Cardiovasc Med. 2022, 25, 823133. [Google Scholar] [CrossRef]
- Castillo-Galán, S.; Riquelme, B.; Iturriaga, R. Crucial Role of Stromal Interaction Molecule-Activated TRPC-ORAI Channels in Vascular Remodeling and Pulmonary Hypertension Induced by Intermittent Hypoxia. Front Physiol. 2022, 13, 841828. [Google Scholar] [CrossRef]
- Thomas, S.; Manivannan, S.; Garg, V.; Lilly, B. Single-Cell RNA Sequencing Reveals Novel Genes Regulated by Hypoxia in the Lung Vasculature. J Vasc Res. 2022, 16, 1–13. [Google Scholar] [CrossRef]
- Iacobas, D.A. Biomarkers, Master Regulators and Genomic Fabric Remodeling in a Case of Papillary Thyroid Carcinoma. Genes 2020, 11, 1030. [Google Scholar] [CrossRef]
- Iacobas, S.; Amuzescu, B.; Iacobas, D.A. Transcriptomic uniqueness and commonality of the ion channels and transporters in the four heart chambers. Sci Rep 2021, 11, 2743. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Lee, P.R.; Cohen, J.E.; Fields, R.D. Coordinated Activity of Transcriptional Networks Responding to the Pattern of Action Potential Firing in Neurons. Genes 2019, 10, 754. [Google Scholar] [CrossRef]
- Victorino, P.H.; Marra, C.; Iacobas, D.A.; Iacobas, S.; Spray, D.C.; Linden, R.; Adesse, D.; Petrs-Silva, H. Retinal Genomic Fabric Remodeling after Optic Nerve Injury. Genes 2021, 12, 403. [Google Scholar] [CrossRef]
- Qiagen Ingenuity Pathway Analysis. Available online: https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/.
- DAVID Functional Annotation Bioinformatics Microarray Analysis. Available online: https://david.ncifcrf.gov.
- Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.kegg.jp/kegg/pathway.html.
- Chemokine signaling pathway. Available online: https://www.genome.jp/kegg-bin/show_pathway?rno04062.
- Ramirez-Carrozzi, V.; Ota, N.; Sambandam, A.; Wong, K.; Hackney, J.; Martinez-Martin, N.; Ouyang, W.; Pappu, R. Cutting Edge: IL-17B Uses IL-17RA and IL-17RB to Induce Type 2 Inflammation from Human Lymphocytes. J Immunol. 2019, 202, 1935–1941. [Google Scholar] [CrossRef]
- Iacobas, S.; Iacobas, D.A. A Personalized Genomics Approach of the Prostate Cancer. Cells 2021, 10, 1644. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Wen, J.; Iacobas, S.; Putterman, C.; Schwartz, N. TWEAKing the Hippocampus: The Effects of TWEAK on the Genomic Fabric of the Hippocampus in a Neuropsychiatric Lupus Mouse Model. Genes 2021, 12, 1172. [Google Scholar] [CrossRef]
- Iacobas, S.; Ede, N.; Iacobas, D.A. The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy. Genes 2019, 10, 560. [Google Scholar] [CrossRef]
- Glycolisis/glucogenesis pathway. Available online: https://www.genome.jp/kegg-bin/show_pathway?rno00010.
- Fructose and manose metabolism. Available online: https://www.genome.jp/kegg-bin/show_pathway?rno00051.
- Pyruvate metabolism. Available online: https://www.genome.jp/kegg-bin/show_pathway?rno00620.
- Iacobas, D.A.; Xi, L. Theory and Applications of the (Cardio) Genomic Fabric Approach to Post-Ischemic and Hypoxia-Induced Heart Failure. J. Pers. Med. 2022, 12, 1246. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, C.; Ye, T.; Chen, X.; Liu, X.; Chen, X.; Sun, Y.; Qu, C.; Liang, J.; Shi, S.; et al. Pinocembrin ameliorates arrhythmias in rats with chronic ischaemic heart failure. Ann Med. 2021, 53, 830–840. [Google Scholar] [CrossRef]
- Chang, S.D.; Lin, Y.H.; Liang, C.C.; Chen, T.C. Effects of sacral nerve stimulation on postpartum urinary retention-related changes in rat bladder. Taiwan J Obstet Gynecol. 2015, 54, 671–677. [Google Scholar] [CrossRef]
- Qin, Y.; Qiao, Y.; Wang, D.; Li, L.; Li, M.; Yan, G.; Tang, C. Target Nuclear Factor Erythroid 2-Related Factor 2 in Pulmonary Hypertension: Molecular Insight into Application. Oxid Med Cell Longev. 2022, 2022, 7845503. [Google Scholar] [CrossRef]
- Li, W.; Chen, W.; Peng, H.; Xiao, Z.; Liu, J.; Zeng, Y.; Huang, T.; Song, Q.; Wang, X.; Xiao, Y. Shikonin improves pulmonary vascular remodeling in monocrotaline-induced pulmonary arterial hypertension via regulation of PKM2. Mol Med Rep. 2023, 27, 60. [Google Scholar] [CrossRef] [PubMed]
- Bekedam, F.T.; Goumans, M.J.; Bogaard, H.J.; de Man, F.S.; Llucià-Valldeperas, A. Molecular mechanisms and targets of right ventricular fibrosis in pulmonary hypertension. Pharmacol Ther. 2023, 244, 108389. [Google Scholar] [CrossRef] [PubMed]
- Smolders, V.F.E.D.; Rodríguez, C.; Blanco, I.; Szulcek, R.; Timens, W.; Piccari, L.; Roger, Y.; Hu, X.; Morén, C.; Bonjoch, C.; et al. Metabolic profile in endothelial cells of chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension. Sci Rep. 2022, 12, 2283. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.M.; Hesselstrand, R.; Fretheim, H.; Ueland, T.; Andreassen, A.K.; Brunborg, C.; Palchevskiy, V.; Midtvedt, Ø.; Garen, T.; Aukrust, P.; et al. CCL21 as a Potential Serum Biomarker for Pulmonary Arterial Hypertension in Systemic Sclerosis. Arthritis Rheumatol. 2018, 70, 1644–1653. [Google Scholar] [CrossRef]
- Zhou, R.P.; Wei, X.; Ma, G.G.; Zhu, F.; Chen, F.H.; Hu, W. Questions Regarding the Value of CCL21 as a Potential Biomarker for Pulmonary Arterial Hypertension in Systemic Sclerosis: Comment on the Article by Hoffmann-Vold et al. Arthritis Rheumatol. 2019, 71, 653–654. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, X.; Wang, L.; Yang, Q.; Ma, Q.; Xu, J.; Wang, J.; Kovacs, L.; Ayonm, R.J.; Liu, Z.; et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc Natl Acad Sci USA 2019, 116, 13394–13403. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.D.; Buxton, I.L.O. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit Rev Biochem Mol Biol. 2017, 52, 340–354. [Google Scholar] [CrossRef]
- Smolle, E.; Leko, P.; Stacher-Priehse, E.; Brcic, L.; El-Heliebi, A.; Hofmann, L.; Quehenberger, F.; Hrzenjak, A.; Popper, H.H.; Olschewski, H.; et al. Distribution and prognostic significance of gluconeogenesis and glycolysis in lung cancer. Mol Oncol. 2020, 14, 2853–2867. [Google Scholar] [CrossRef]
- Pirard, M.; Achouri, Y.; Collet, J.F.; Schollen, E.; Matthijs, G.; Van Schaftingen, E. Kinetic properties and tissular distribution of mammalian phosphomannomutase isozymes. Biochem J. 1999, 339, 201–207. [Google Scholar] [CrossRef]
- Voulgaridou, G.P.; Theologidis, V.; Xanthis, V.; Papagiannaki, E.; Tsochantaridis, I.; Fadouloglou, V.E.; Pappa, A. Identification of a peptide ligand for human ALDH3A1 through peptide phage display: Prediction and characterization of protein interaction sites and inhibition of ALDH3A1 enzymatic activity. Front Mol Biosci. 2023, 10, 1161111. [Google Scholar] [CrossRef]
- Tan, R.; Li, J.; Peng, X.; Zhu, L.; Cai, L.; Wang, T.; Su, Y.; Irani, K.; Hu, Q. GAPDH is critical for superior efficacy of female bone marrow-derived mesenchymal stem cells on pulmonary hypertension. Cardiovasc Res. 2013, 100, 19–27. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Increased expression of Ngf in the PAH models with respect to the control group. The increase was evident in all PAH groups but only in the CM and HM groups was statistically significant.
Figure 1.
Increased expression of Ngf in the PAH models with respect to the control group. The increase was evident in all PAH groups but only in the CM and HM groups was statistically significant.
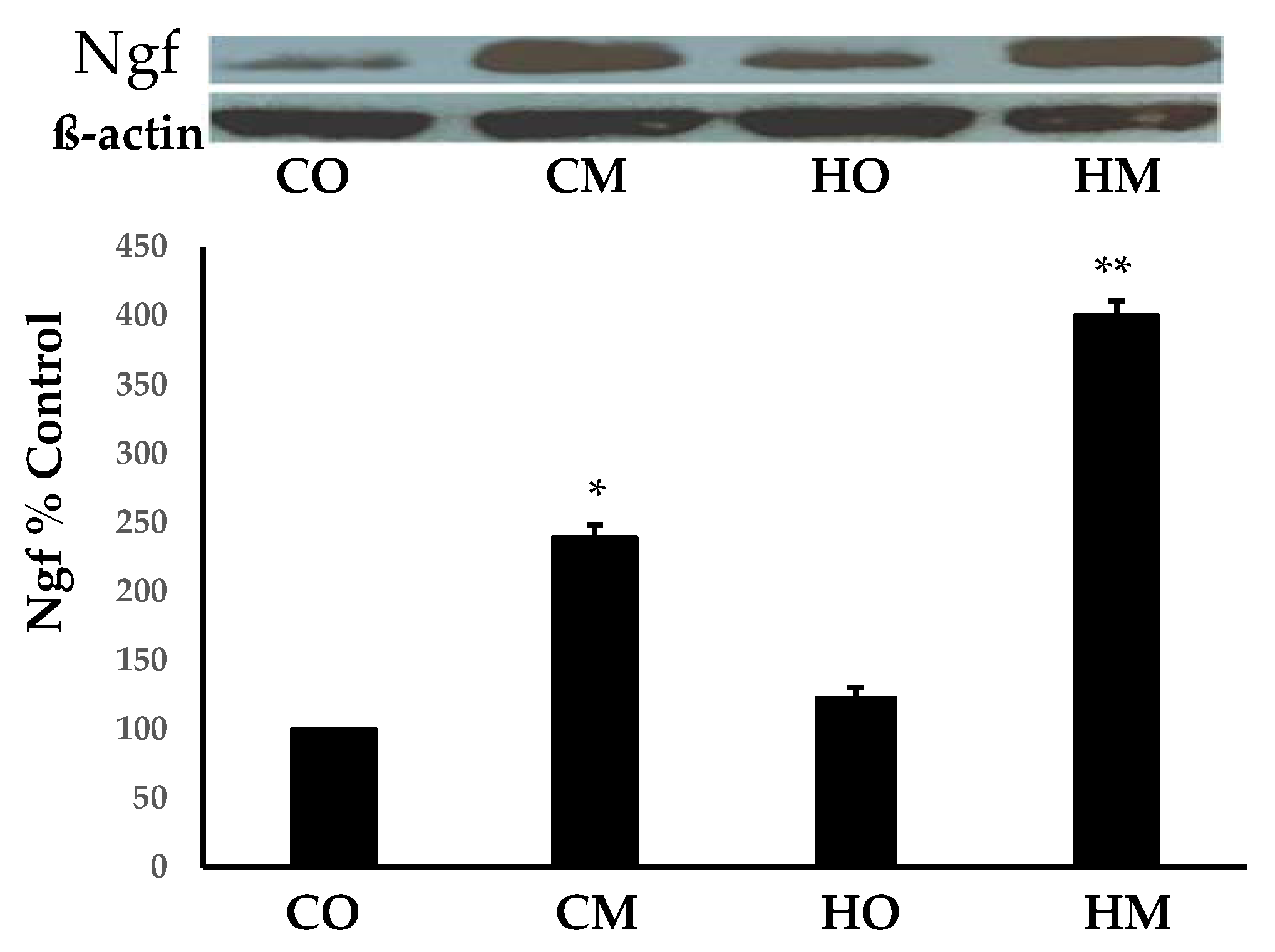
Figure 2.
Alteration of the Nfe2l2 abundance in the lungs of rat PAH models. The increase was statistically (p < 0.05) significant only in the HM group.
Figure 2.
Alteration of the Nfe2l2 abundance in the lungs of rat PAH models. The increase was statistically (p < 0.05) significant only in the HM group.
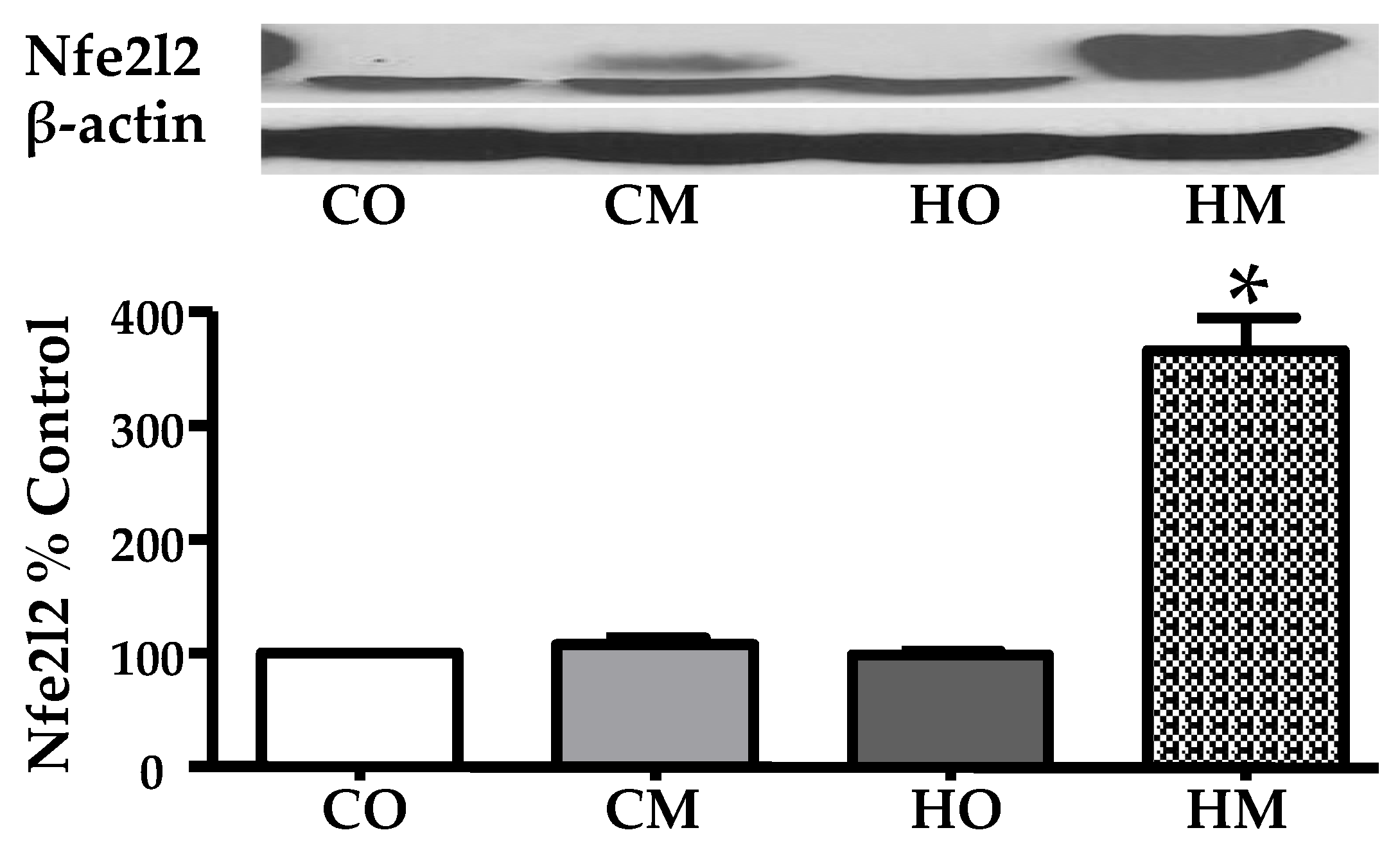
Figure 3.
Western blot and double immunofluorecence showing increased expression of Slc2a1 abundances in the lungs of the three rat PAH models with respect to the control group, although the increase in the CM group was not (p < 0.05) statostically significant.
Figure 3.
Western blot and double immunofluorecence showing increased expression of Slc2a1 abundances in the lungs of the three rat PAH models with respect to the control group, although the increase in the CM group was not (p < 0.05) statostically significant.
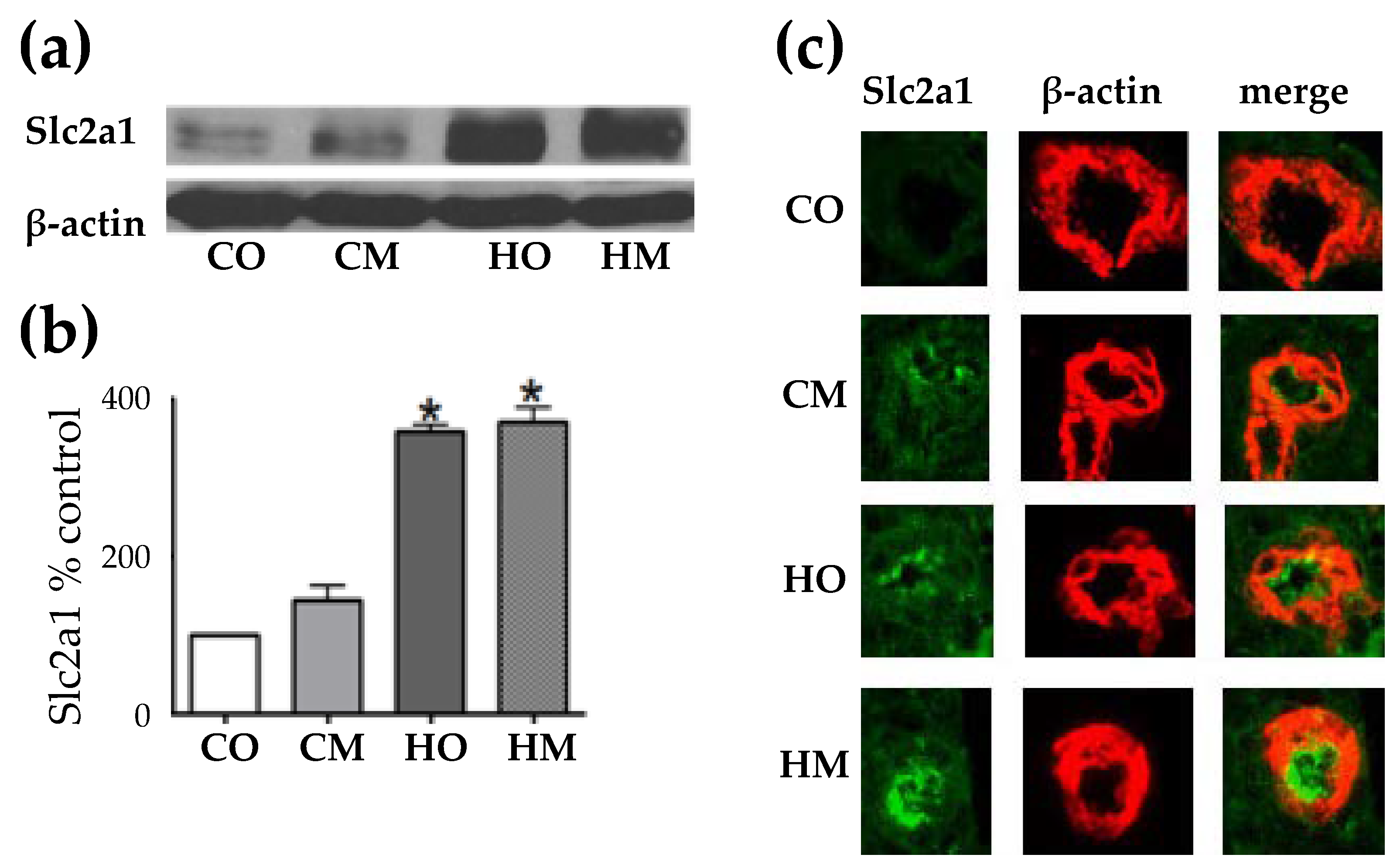
Figure 4.
The independence of the: (a) average expression level (AVE), (b) relative expression variability (REV) and (c) expression correlation (COR, here with Il17b) of 50 genes involved in the immune-inflammatory response in the lungs of the control (CO) and PAH models (HO, CM, HM) rats. The value 1 in panel (c) for Il17b in all conditions is a direct validation of the Pearson correlation coefficient. The correlation was considered as statistically significant if |COR| ≥ 0.95.
Figure 4.
The independence of the: (a) average expression level (AVE), (b) relative expression variability (REV) and (c) expression correlation (COR, here with Il17b) of 50 genes involved in the immune-inflammatory response in the lungs of the control (CO) and PAH models (HO, CM, HM) rats. The value 1 in panel (c) for Il17b in all conditions is a direct validation of the Pearson correlation coefficient. The correlation was considered as statistically significant if |COR| ≥ 0.95.
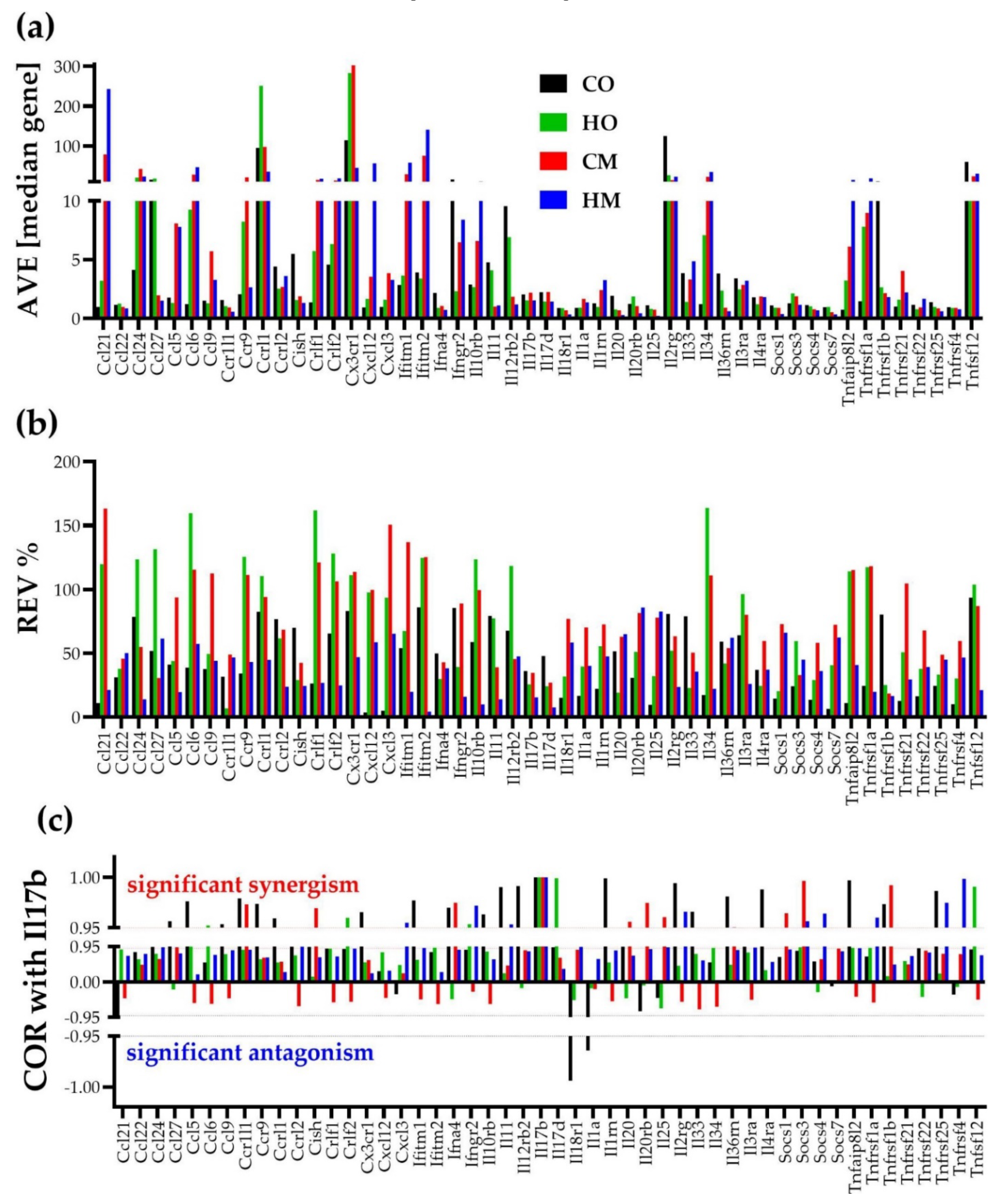
Figure 5.
Significant synergistic, antagonistic and independent pairing of 50 immune-inflammatory response genes in the hypoxia group (HO) compared to the control group (CO). A red/blue/yellow square indicates that the genes labeling the intersecting raw and column are synergistically/antagonistically/independently expressed in that designated condition.
Figure 5.
Significant synergistic, antagonistic and independent pairing of 50 immune-inflammatory response genes in the hypoxia group (HO) compared to the control group (CO). A red/blue/yellow square indicates that the genes labeling the intersecting raw and column are synergistically/antagonistically/independently expressed in that designated condition.
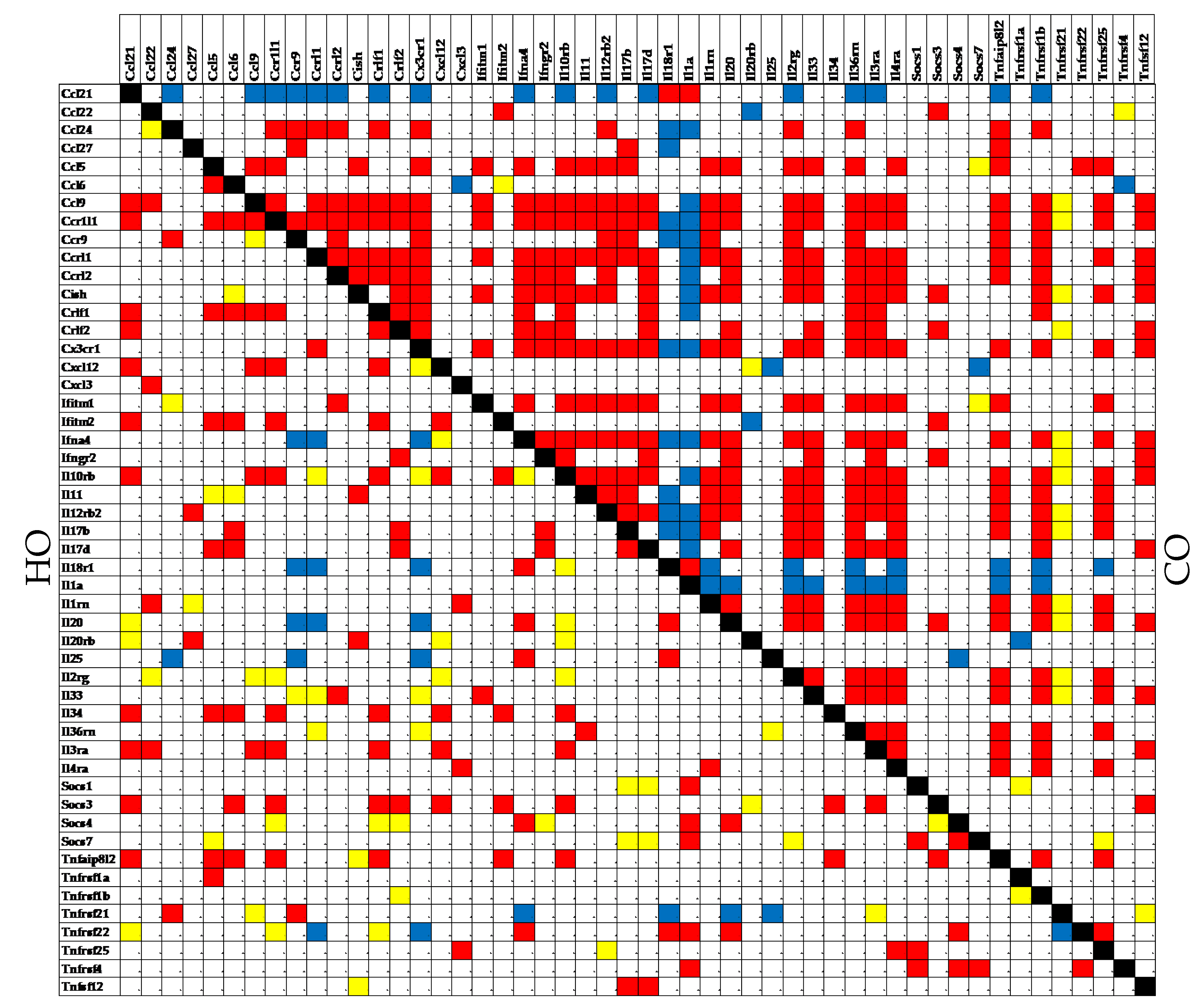
Figure 6.
Four ways to quantify the contributions of the individual genes to the transcriptomic alterations in lungs of the PAH models illustrated by the regulation of 50 immune response genes. (a) Uniform - each significantly regulated gene is a +1 or -1 contributor, all other genes have no contribution). (b) Expression ratio x (negative for down-regulation). (c) Weighted Individual (gene) Regulation (WIR). (d) Transcriptomic distance (TD) to that gene normal transcriptome (CO values for AVE, REV and COR). Note how WIR and TD discriminate the genes according to their contributions the overall alteration of the lung transcriptome.
Figure 6.
Four ways to quantify the contributions of the individual genes to the transcriptomic alterations in lungs of the PAH models illustrated by the regulation of 50 immune response genes. (a) Uniform - each significantly regulated gene is a +1 or -1 contributor, all other genes have no contribution). (b) Expression ratio x (negative for down-regulation). (c) Weighted Individual (gene) Regulation (WIR). (d) Transcriptomic distance (TD) to that gene normal transcriptome (CO values for AVE, REV and COR). Note how WIR and TD discriminate the genes according to their contributions the overall alteration of the lung transcriptome.
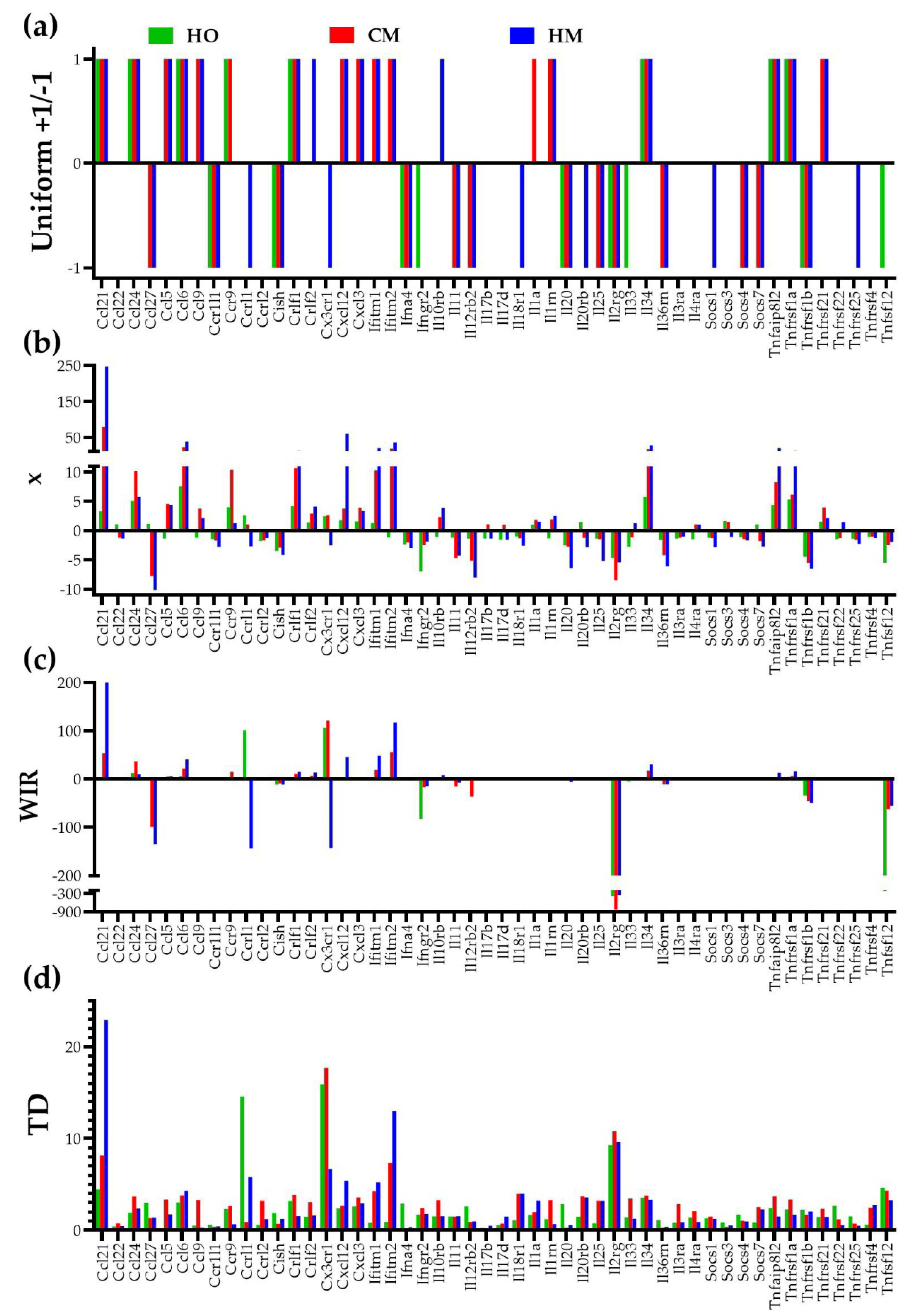
Figure 7.
Regulation of the glycolysis/gluconeogenesis pathway in the PAH rat models with respect to control (CO). (a) Regulated genes in the HO model. (b) Regulated genes in the CM model. (c) Regulated genes in the HM model. (d) Transcriptomic distances separating the transcriptome associated to the glycolysis/gluconeogenesis pathway in the HO, CM and HM PAH models from that in the control (CO) rats. Regulated genes: Acss1/2 (Acyl-CoA synthetase short-chain family member 1/2), Adh1/4/5/6/7 (Alcohol dehydrogenase 1/4/5/6/7), Adpgk (ADP-dependent glucokinase), Aldh1b1 (Aldehyde dehydrogenase 1 family, member B1), Aldh2 (Aldehyde dehydrogenase 2 family), Aldh3a1/2 (Aldehyde dehydrogenase 3 family, member A1/2), Aldh3b1 (Aldehyde dehydrogenase 3 family, member B1), Aldh9a1 (Aldehyde dehydrogenase 9 family, member A1), Aldob/c (Aldolase B/C, fructose-bisphosphate), Bpgm (2,3-bisphosphoglycerate mutase), Dld (Dihydrolipoamide dehydrogenase), Eno1/2/3/4 (Enolase 1/2/3/4), Fbp2 (Fructose-1,6-bisphosphatase 2), G6pc3 (Glucose 6 phosphatase, catalytic, 3), Galm (Galactose mutarotase (aldose 1-epimerase)), Gapdh (Glyceraldehyde-3-phosphate dehydrogenase), Gapdhs (Glyceraldehyde-3-phosphate dehydrogenase, spermatogenic), Gck (glucokinase), Gpi (Glucose-6-phosphate isomerase), Hk2/3 (Hexokinase 2/3), Ldha/b/c (Lactate dehydrogenase A/B/C), Ldha16b (Lactate dehydrogenase A-like 6B), Loc303448 (Similar to glyceraldehyde-3-phosphate dehydrogenase), Minpp1 (Similar to glyceraldehyde-3-phosphate dehydrogenase), Pck1/2 (Phosphoenolpyruvate carboxykinase ½), Pdha1/2 (Pyruvate dehydrogenase (lipoamide) alpha ½), Pdhb (Pyruvate dehydrogenase (lipoamide) beta), Pfkl/m/p (Phosphofructokinase, liver/muscle/platelet), Pgam1/2 (Phosphoglycerate mutase ½), Pgk1/2 (Phosphoglycerate kinase ½), Pklr (Pyruvate kinase, liver and RBC), Tpi1 (Triosephosphate isomerase 1). The tips of the colored cones in panel (d) are the coordinates of the pathway in the 3D pre-Hilbert space of states, while numbers on top of the cones are the average TDs of the composing genes in the indicated model.
Figure 7.
Regulation of the glycolysis/gluconeogenesis pathway in the PAH rat models with respect to control (CO). (a) Regulated genes in the HO model. (b) Regulated genes in the CM model. (c) Regulated genes in the HM model. (d) Transcriptomic distances separating the transcriptome associated to the glycolysis/gluconeogenesis pathway in the HO, CM and HM PAH models from that in the control (CO) rats. Regulated genes: Acss1/2 (Acyl-CoA synthetase short-chain family member 1/2), Adh1/4/5/6/7 (Alcohol dehydrogenase 1/4/5/6/7), Adpgk (ADP-dependent glucokinase), Aldh1b1 (Aldehyde dehydrogenase 1 family, member B1), Aldh2 (Aldehyde dehydrogenase 2 family), Aldh3a1/2 (Aldehyde dehydrogenase 3 family, member A1/2), Aldh3b1 (Aldehyde dehydrogenase 3 family, member B1), Aldh9a1 (Aldehyde dehydrogenase 9 family, member A1), Aldob/c (Aldolase B/C, fructose-bisphosphate), Bpgm (2,3-bisphosphoglycerate mutase), Dld (Dihydrolipoamide dehydrogenase), Eno1/2/3/4 (Enolase 1/2/3/4), Fbp2 (Fructose-1,6-bisphosphatase 2), G6pc3 (Glucose 6 phosphatase, catalytic, 3), Galm (Galactose mutarotase (aldose 1-epimerase)), Gapdh (Glyceraldehyde-3-phosphate dehydrogenase), Gapdhs (Glyceraldehyde-3-phosphate dehydrogenase, spermatogenic), Gck (glucokinase), Gpi (Glucose-6-phosphate isomerase), Hk2/3 (Hexokinase 2/3), Ldha/b/c (Lactate dehydrogenase A/B/C), Ldha16b (Lactate dehydrogenase A-like 6B), Loc303448 (Similar to glyceraldehyde-3-phosphate dehydrogenase), Minpp1 (Similar to glyceraldehyde-3-phosphate dehydrogenase), Pck1/2 (Phosphoenolpyruvate carboxykinase ½), Pdha1/2 (Pyruvate dehydrogenase (lipoamide) alpha ½), Pdhb (Pyruvate dehydrogenase (lipoamide) beta), Pfkl/m/p (Phosphofructokinase, liver/muscle/platelet), Pgam1/2 (Phosphoglycerate mutase ½), Pgk1/2 (Phosphoglycerate kinase ½), Pklr (Pyruvate kinase, liver and RBC), Tpi1 (Triosephosphate isomerase 1). The tips of the colored cones in panel (d) are the coordinates of the pathway in the 3D pre-Hilbert space of states, while numbers on top of the cones are the average TDs of the composing genes in the indicated model.
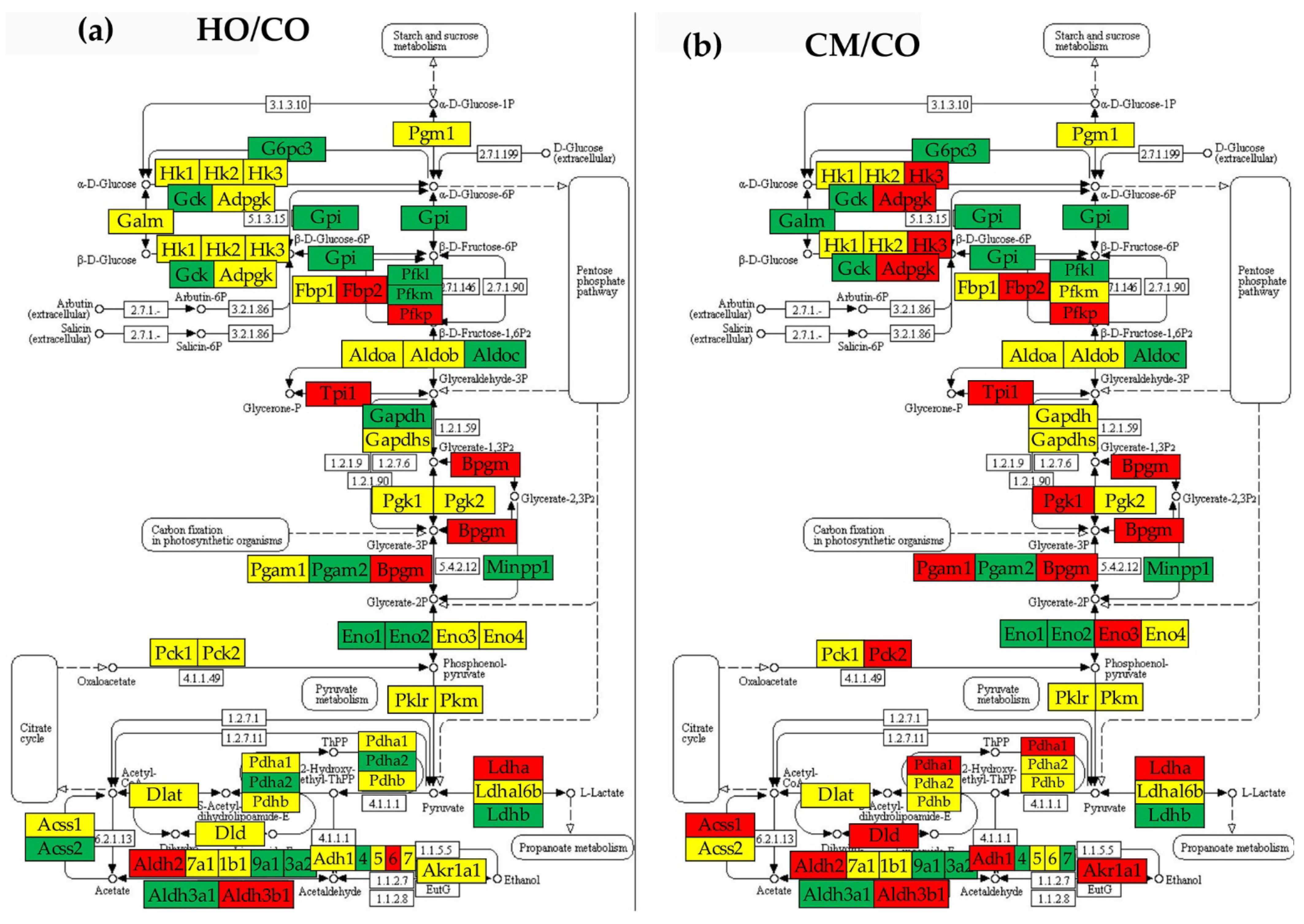
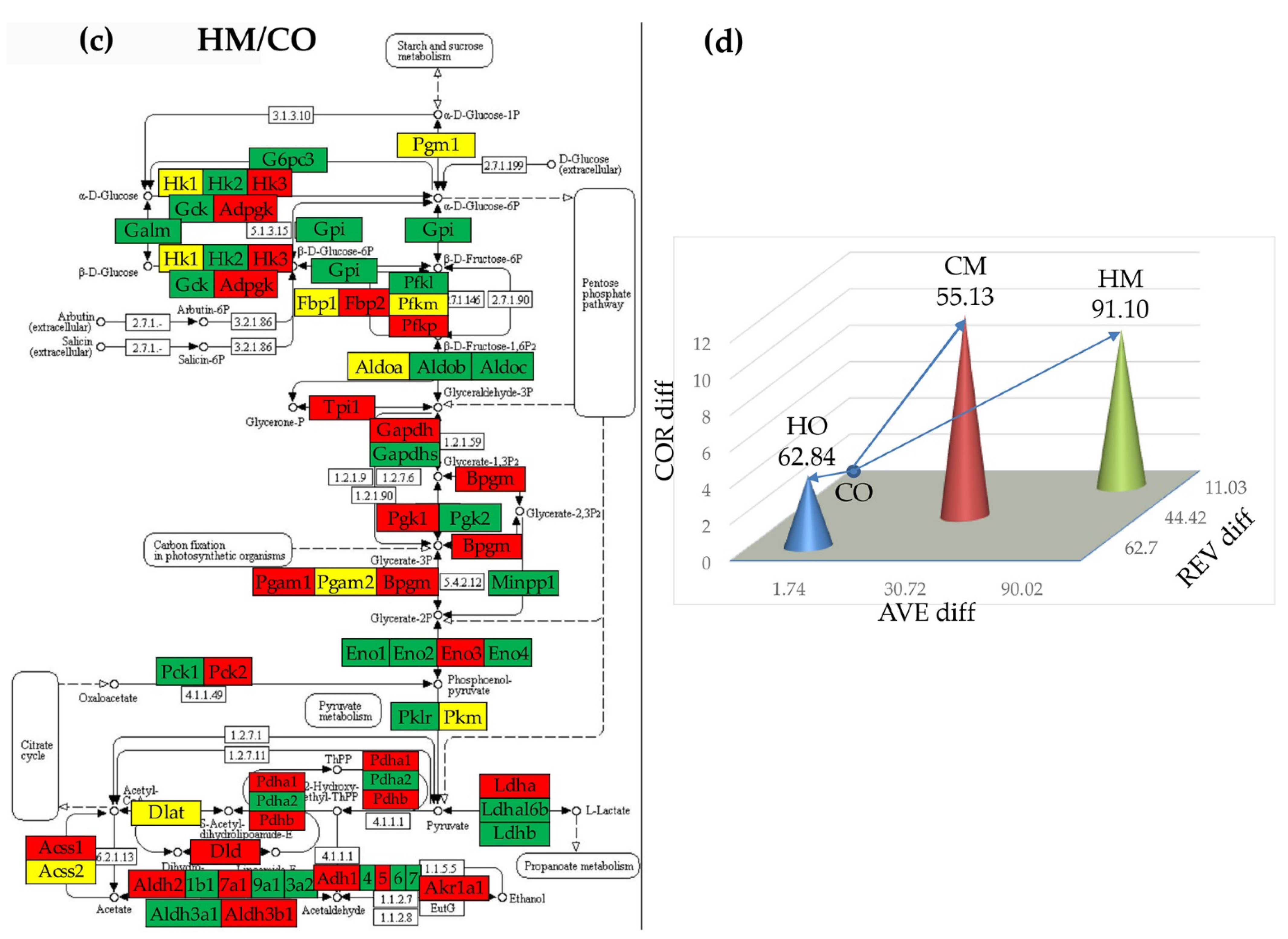
Figure 8.
(p < 0.05) Significant synergistically (red lines) and antagonistically (blue lines) expression coordination of selected 40 metabolic genes in the four rat models. (a) CO. (b) HO. (c) CM. (d) HM. Note that hypoxia, either with or without monocrotaline treatment, significantly decoupled the expression coordination of these genes. Citrate cycle: Acly (ATP citrate lyase), Aco2 (Aconitase 2), Fh (Fumarate hydratase), Idh2 (Isocitrate dehydrogenase 2 (NADP+)), Idh3g (Isocitrate dehydrogenase 3 (NAD), gamma), Ogdh (Oxoglutarate (alpha-ketoglutarate) dehydrogenase), Pck2 (Phosphoenolpyruvate carboxykinase 2), Pdha1 (Pyruvate dehydrogenase (lipoamide) alpha 1). Fructose and mannose metabolism: Aldoa/c (Aldolase A/C, fructose-bisphosphate), Fpbp1 (Fructose-1,6-bisphosphatase 1), Hk1 (Hexokinase 1), Khk (Ketohexokinase), LOC500959 (Triosephosphate isomerase), Pfkfb3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3), Pfkm (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3), Pmm1/2 (Phosphomannomutase 1/2), Tpi (Triosephosphate isomerase 1). Glycolysis/gluconeogenesis pathway: Acss2 (Acyl-CoA synthetase short-chain family member 2), Adh1/5 (Alcohol dehydrogenase 1/5), Aldh2 (Aldehyde dehydrogenase 2 family), Aldh3/9a1 (Aldehyde dehydrogenase 3/9 family, member A1), Aldoa/c, Eno1/2/4 (Enolase 1/2/4), Fbp1, G6pc3 (Glucose 6 phosphatase, catalytic, 3), Gapdh (Glyceraldehyde-3-phosphate dehydrogenase), Gapdhs (Glyceraldehyde-3-phosphate dehydrogenase, spermatogenic), Gck (glucokinase), Gpi (Glucose-6-phosphate isomerase), Hk1, Ldha/b (Lactate dehydrogenase A/B), Minpp1 (Multiple inositol-polyphosphate phosphatase 1), Pck2, Pdha1, Pfkl/m (Phosphofructokinase, liver/muscle), Pgam1 (Phosphoglycerate mutase 1), Pgk1 (Phosphoglycerate kinase 1), Tpi (Triosephosphate isomerase 1). Pyruvate metabolism: Acss2, Acyp2 (Acylphosphatase 2), Adh1/5, Aldh2, Aldh9a1, Fh, Ldha/b, Pck2 (Phosphoenolpyruvate carboxykinase 2), Pdha1.
Figure 8.
(p < 0.05) Significant synergistically (red lines) and antagonistically (blue lines) expression coordination of selected 40 metabolic genes in the four rat models. (a) CO. (b) HO. (c) CM. (d) HM. Note that hypoxia, either with or without monocrotaline treatment, significantly decoupled the expression coordination of these genes. Citrate cycle: Acly (ATP citrate lyase), Aco2 (Aconitase 2), Fh (Fumarate hydratase), Idh2 (Isocitrate dehydrogenase 2 (NADP+)), Idh3g (Isocitrate dehydrogenase 3 (NAD), gamma), Ogdh (Oxoglutarate (alpha-ketoglutarate) dehydrogenase), Pck2 (Phosphoenolpyruvate carboxykinase 2), Pdha1 (Pyruvate dehydrogenase (lipoamide) alpha 1). Fructose and mannose metabolism: Aldoa/c (Aldolase A/C, fructose-bisphosphate), Fpbp1 (Fructose-1,6-bisphosphatase 1), Hk1 (Hexokinase 1), Khk (Ketohexokinase), LOC500959 (Triosephosphate isomerase), Pfkfb3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3), Pfkm (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3), Pmm1/2 (Phosphomannomutase 1/2), Tpi (Triosephosphate isomerase 1). Glycolysis/gluconeogenesis pathway: Acss2 (Acyl-CoA synthetase short-chain family member 2), Adh1/5 (Alcohol dehydrogenase 1/5), Aldh2 (Aldehyde dehydrogenase 2 family), Aldh3/9a1 (Aldehyde dehydrogenase 3/9 family, member A1), Aldoa/c, Eno1/2/4 (Enolase 1/2/4), Fbp1, G6pc3 (Glucose 6 phosphatase, catalytic, 3), Gapdh (Glyceraldehyde-3-phosphate dehydrogenase), Gapdhs (Glyceraldehyde-3-phosphate dehydrogenase, spermatogenic), Gck (glucokinase), Gpi (Glucose-6-phosphate isomerase), Hk1, Ldha/b (Lactate dehydrogenase A/B), Minpp1 (Multiple inositol-polyphosphate phosphatase 1), Pck2, Pdha1, Pfkl/m (Phosphofructokinase, liver/muscle), Pgam1 (Phosphoglycerate mutase 1), Pgk1 (Phosphoglycerate kinase 1), Tpi (Triosephosphate isomerase 1). Pyruvate metabolism: Acss2, Acyp2 (Acylphosphatase 2), Adh1/5, Aldh2, Aldh9a1, Fh, Ldha/b, Pck2 (Phosphoenolpyruvate carboxykinase 2), Pdha1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



