Submitted:
28 April 2023
Posted:
28 April 2023
You are already at the latest version
Abstract
The FDA has concluded that a biosimilar candidate capable of demonstrating pharmacodynamic biomarkers in a healthy subject need not be tested for clinical efficacy in patients, regardless of if the biomarker correlates with clinical response. Since monoclonal antibodies (mAbs) do not trig-ger pharmacodynamic response, they can be substituted with robust functional disqualifying them for this waiver that can be overcome by allowing comparison of functional properties that eventually result in clinical response. This suggestion is based on the FDA's preference for more sensitive testing methods. However, clinical efficacy testing in patients is the least sensitive method, as confirmed by statistical modeling, a fact that regulatory agencies need to admit. In this paper, I present a logical and rational argument to establish the biosimilarity of products that do not have pharmacodynamic biomarkers based on their orthogonally proven functional bio-similarity. This understanding will significantly lower the development cost of biosimilars, a goal that the FDA outlined in all its guidance.
Keywords: biosimilarity 1; biosimilars 2; pharmacodynamic biomarkers 3; monoclonal antibodies 4; FDA 5; clinical efficacy testing in healthy subjects 6; clinical efficacy testing in patients 7; functional assays 8; receptor binding 9.
Keywords:
biosimilarity
; biosimilars
; pharmacodynamic biomarkers
; monoclonal antibodies
; FDA
; clinical efficacy testing in healthy subjects
; clinical efficacy testing in patients
; functional assays
; receptor binding
1. Introduction
The legislation introducing biosimilars arrived in 2015 in the EU and in 2019 in the US. As of April 2023, 47 biosimilars were approved in the US, including the molecules that are approved as chemical drugs, and 74 in the EU (Table 1), representing 19 molecules, including what are not considered proteins in the US, out of more than 200 available recombinant therapeutic protein molecules1 available as a choice.
Biosimilars have “no clinically meaningful differences between the biological product and the reference (originator) product in terms of safety, purity, and potency,” degines the FDA.2 This status is granted based on a stepwise comparative testing plan, starting with structural and functional assessment, followed by clinical pharmacology profiling. In January 2023, the US law, BPCIA, was amended to remove the terms “animal toxicology,” and replaced with “nonclinical.” Further, if there remains any “residual uncertainty,” “additional clinical studies” that may include additional clinical pharmacology profiling or testing in patients. However, almost all biosimilars approved by the FDA conducted extensive patient testing, adding substantial costs.
Of the total cost of developing biosimilars ranging between $100-$300 million,3 on average, the clinical efficacy testing in patients, enrolling a median of 538 participants (interquartile range, 372–644 patients) has a median cost of each is $27.6 million ($18.0 million–$36.7 million) giving an average cost per enrollee at about $55,000; also these trials last a median of 55 weeks (range 46–78 weeks).4 Oncology drugs present the highest cost category. More complex trial protocols take longer to design, obtain approvals (institutional and FDA), recruit patients from contracted providers, analyze the resulting data, and submit the results.
As of April 2023, there were 94,910 subjects enrolled in 170 active or completed phase 3 biosimilar trials with study sizes ranging from 3 to 4,994 subjects; 100 studies were marked for cancer (26 as lymphoma, 34 for breast cancer, 25 metastatic, 21 for HER2, 16 for adenocarcinoma), 18 for macular degeneration, 31 for rheumatoid arthritis, 24 for psoriasis, and 17 for osteoporosis. All completed trials met the equivalence criteria.5 Based on the average cost per enrollees, the cost of current studies amounts to over $5 Billion.
When the EMA and the FDA issued their first guidelines for biosimilar approvals, it was considered essential to demonstrate efficacy in the patients. As a result, these guidelines have undergone many revisions, lowering the barrier to clinical testing. Significant changes include the removal of “animal toxicology” and replacing it with “nonclinical” testing,6 waiver of immunogenicity testing if immunogenicity does not impact pharmacokinetics,7 waiver of clinical efficacy testing in patients where pharmacodynamic biomarkers are available,8 9 and suggestions to apply AI and in silico approaches to demonstrate biosimilarity.10 11
The FDA’s decision to waive efficacy testing in patients is most significant, but many therapeutic proteins, such as antibodies, do not demonstrate a pharmacodynamic response, excluding them from this concession. However, the need to remove clinical efficacy remains urgent since these studies are least sensitive of all other testing as demonstrated in this paper, leaving them merely as a conservative “checklist” item. For products that do not display a pharmacodynamic response, a comparison of functional properties with the reference product, comprising biological assays and receptor-binding tests, should be sufficient, since it is these functionalities that lead to pharmacodynamic responses or clinical responses in patients.
2. Clinical Efficacy Testing
Clinical efficacy testing in patients is the established standard for evaluating the safety and efficacy of new drugs. Clinical trial designing starts with formulating the expected outcome12 that explicitly encompasses the population, intervention, comparator, and outcomes (PICO) to be measured.13 However, this standard is now subject to change based on the new analysis by the FDA questioning14 the validity of the traditional clinical trial systems, labeling them as “broken,”15 suggesting an adaptive approach to clinical trial designs16 that ‘‘allows for prospectively planned modifications to one or more aspects of the design based on accumulating data from subjects in the trial.”17
Adaptive trials may improve efficiency and statistical power compared to more conventional designs, thereby allowing the resolution of clinical inquiries in a shorter time or with fewer participants than non-adaptive designs. However, adaptive trials have potential sources of bias that must be avoided by careful design.18 The most common adaptive approaches include sequential group design, flexible sample size re-estimation, adaptive randomization, biomarker adaptive design, and multi-arm platform design. One adaptive design request unique to testing biosimilars involves requesting a narrow healthy subject or patient inclusion criteria to enhance the homogeneity of the study population and thereby reduce variability in participants, improving the precision of treatment that will allow significant reaction in the study size.
A justification for lowering the study size comes from the fact that the testing of biosimilars is intended to compare the product attributes, not characterize them in a target population.19 These considerations are essential since most trials are conducted as parallel designs requiring a larger population and typically comparing one treatment against no treatment, placebo, or standard of care. While a crossover design eliminates variation in baseline factors between the study arms, increasing the power to detect a significant treatment effect,20 but this design can be used only when the treatment effect is predictably time-limited and cannot be used if the treatment is curative or long-lasting.
The clinical efficacy testing of biosimilars in patients brings unique challenges as described below suggesting that a more rational evaluation of the safety and efficacy of biosimilar candidates can be made without engaging the patients.
Extrapolation of Indication
A unique regulatory allowance for biosimilars is the extrapolation of all indications based on testing in only one indication if testing in patients is considered necessary. This decision is supported by the regulatory argument that the biosimilar candidate has already demonstrated high similarity of analytical assessment and clinical pharmacology. However, this also means that where a molecule demonstrates multiple modes of action, this difference is not relevant.21 If this is the case, then the similarity of the biosimilar candidate demonstrated before conducting the efficacy testing should be sufficient, removing this circular argument for requiring any efficacy testing.22 This suggestion also applies to the argument of testing patient immunogenicity since this is already tested in healthy subjects, and if immunogenicity is considered dependent on the nature of the disease and the condition of the patients, then this should be tested in all indications.
As an example, adalimumab (Humira) is a recombinant human mAb against TNFα (tumor necrosis factor-alpha), with at least 12 indications (autoimmune diseases) approved worldwide, including, among others, rheumatoid arthritis (RA), Crohn’s disease (CD), ulcerative colitis (UC), ankylosing spondylitis (AS), plaque psoriasis (PPs), psoriatic arthritis (PsA), polyarticular juvenile idiopathic arthritis (PJIA), and more. In addition, other TNFα-targeted biological drugs include infliximab (Remicade, a mAb against TNFα) and Enbrel (etanercept, a fusion protein against TNFα). This indicated that TNFα (an important inflammatory cytokine) is one of the shared therapeutic targets of various autoimmune-associated inflammatory diseases and that these TNFα-targeted protein drugs are effective in treating these diseases associated with autoimmune disorders and inflammation through inhibition of TNFα activity based on the class effect. Furthermore, different biologicals can also have the same indication, such as rheumatoid arthritis can be treated either with TNFα-targeted biological drugs, such as adalimumab, infliximab, and Enbrel or with rituximab (Rituxan), which is a mAb against antigen CD20 on B lymphocytes. Thus, a disease may have more than one therapeutic target, such as TNFα and CD20 for rheumatoid arthritis, and concomitant use of biological drugs acting on different targets is expected to exert synergistic or additional efficacy for a certain disease.
Based on their pharmacological activity, therapeutic proteins replace a deficient protein, augment an existing pathway, create a novel function or activity, interfere with an organism or a molecule, and deliver other effector proteins, cytotoxic drugs, radionuclides, or nucleotides. On a molecular type, they are antibodies, c fusion proteins, anticoagulants, blood factors, bone morphogenetic proteins, engineered protein scaffolds, enzymes, growth factors, hormones, interferons, interleukins, and thrombolytics. Their molecular mechanism of activity includes binding non-covalently to target, e.g., mAbs, affecting covalent bonds, e.g., enzymes, or making nonspecific interactions, e.g., serum albumin. Proteins can bind to multiple ligands, and the binding mechanisms can differ.23
Testing biosimilars in one clinical indication is no more than a checklist that is further questioned based on the statistical modeling weaknesses, as shown below.
Statistical Modeling
Statistical analyses used for clinical assessments of biosimilar candidates differ from those used for the regulatory approval of the reference product. Understanding the different analyses, comparisons, endpoints, populations used, and calculations of margins and sample size are the key variables that make study designs highly complex and as demonstrated below, of lesser value since the methods of equivalence testing used to evaluate biosimilarity are much more robust than the efficacy testing.
A new biological drug is subjected to a superiority trial compared to placebo control.24 In contrast, biosimilar products must demonstrate similar efficacy and safety to the originator, mainly as equivalence trials,25 26 where the objective is to show that differences in response are not clinically significant,27 meaning that clinical responses are close enough so neither the biosimilar nor the comparator (originator) is superior or inferior to the other. This is usually shown via demonstration that the treatment difference will likely lie within a specific range of clinically acceptable differences. The equivalence trial design requires establishing an acceptable range of clinically acceptable differences. Equivalence is shown when the confidence interval (CI) falls within the lower and upper equivalence margins set before the experiment or study.28 A noninferiority trial design can sometimes be used if adequate scientific justification is provided. A noninferiority design uses only one margin (the lower or upper limit, depending on what is appropriate for the specific study or endpoint). It tends to require a smaller sample size than an equivalence trial design.29 The FDA and EMA discourage noninferiority trials to avoid the possibility of higher efficacy of the biosimilar that may translate into higher adverse events.
The margins for demonstrating equivalence (and noninferiority) are generally based on the effect size. They should be justified on clinical and statistical grounds and use robust statistical rationale and clinical criteria to determine a margin value.30 To demonstrate biosimilarity, the margin is the largest difference between the potential biosimilar and originator that is judged as clinically acceptable but should be smaller than the minimum difference reported between the originator and placebo for clinically relevant results.31 While this consideration applies to a new entity trial against a placebo, the comparison of a biosimilar candidate with its reference product is akin to comparing the same product in two groups of subjects. In this case, the actual mean difference between treatment and reference will be zero. The variability of an acceptable effect size of the reference in placebo-controlled trials is used to calculate margins.
Determining the equivalence margin, δ is the most critical step in equivalence/noninferiority testing. A small value of δ determines a narrower equivalence region, making it more difficult to establish equivalence/noninferiority. The equivalence margin determines the test result and gives scientific credibility to a study. The value and impact of a study depend on how well the equivalence margin can be justified in terms of relevant evidence and sound clinical considerations. Frequently, regulatory issues also must be considered5.
Power is the probability of correctly establishing the research hypothesis. Power analysis, also called sample size determination, calculates the number of observations needed to achieve a desired power. Table 2 shows the sample size calculated for testing equivalence between a biosimilar candidate and its reference product, assuming that the true efficacies are 33% and 28% for the reference product and the biosimilar candidate, respectively.
Since a small difference in the equivalence margin significantly changes the sample size, the arbitrary selection of equivalence margins makes a comparison of clinical efficacy in patients for biosimilars highly unreliable.
Non-inferiority trials show that a biosimilar is no worse than the reference product and are justified only on the grounds of lower cost of treatment. Like the equivalence studies, investigators must set the maximum acceptable difference between the treatments.33 The treatment effect in these trials must be confidently above this margin. Still, unlike the equivalence design, it is unnecessary to be confident that the treatment effect is below some acceptable superiority margin. With non-inferiority designs, one-sided hypothesis testing is used. Such studies should not be used for biosimilar products since it is the response that leads to toxicity.
A recent report examined the published studies wherein the 38 efficacy testing in patients showed only two clinical safety failures, one for somatropin immunogenicity attributed to higher amounts of host cell protein (HCP) impurities in the biosimilar that were missed out in testing; the second example was erythropoietin where two patients developed neutralizing antibodies that were associated with the residual tungsten in the syringe that catalyzed the formation of insoluble aggregates; a repeat study with a tungsten-free syringe was successful. Based on current guidelines, these differences will not be identified as failed studies; thus, all 38 studies showed no clinical efficacy differences.34
Another statistical challenge comes in establishing the probability of concluding that a failed study is a study failure. The Bayesian probability that a failure is a failed study is based on the alpha value of 0.05, meaning that 95% of the time, if a study fails, it will be determined as a failure [thus, the probability is 95%] and beta value of 0.2 [that a study that was not a failed study shows up failed study], and a prior probability (Figure 1).
P(A) = P(A|B) * P(B) + P(A|¬B) * (1 - P(B))
P(A) = posterior probability that a failed study is a failed; P(A|B) = 0.95 (probability of a failed study given that it is a failed study); P(A|¬B) = 0.20 (probability of a failed study not being a failed study); P(B) = 0.01 to 0.10 (prior probability of a study failing), which is almost zero, but for a practical calculation Figure 1 shows a range of 1 to 10%.
This calculation is more convincing since it is based on real-world data; if a study has never failed, then future studies are also not likely to fail, and if it does, then the confidence that the study is failed is very low.
3. Pharmacodynamic Biomarkers
The limitations of efficacy testing in patients are well-recognized by regulatory agencies. To overcome these concerns, the FDA’s Division of Applied Regulatory Science (DARS)35 has recently published its recommendations to remove this testing for biosimilars36 based on comparing pharmacodynamic (PD) properties between a biosimilar candidate and its reference product. It is now labeled as clinical efficacy testing in healthy subjects. A PD biomarker is not required to be a surrogate endpoint or have an established relationship with clinical efficacy outcomes.37 38 Examples include the absolute neutrophil count area under the effect time curve as a more reliable endpoint than the clinical efficacy endpoint of the duration of severe neutropenia.39 DARS made these conclusions based on its investigations40 and clinical studies it has conducted41 42 43 to define the best practices for characterizing the PD biomarkers for various drug classes. These studies evaluated the use of human plasma proteomic and transcriptomic analysis to find novel biomarkers for the approval of biosimilars.44 A joint FDA/Duke Margolis Workshop45 has covered the study findings that encourage a broader debate on using PD biomarkers to develop biosimilars.
The FDA has also validated that PD biomarker identification can be made using large-scale proteomic approaches and other technologies46 where PD biomarkers are not readily available. The FDA has also confirmed that the PD biomarkers need not correlate with a clinical response to allow their use to support the claim of biosimilarity. A biosimilar development plan aims to demonstrate similarity to the reference product, not the focus of the reference product, where the safety and effectiveness are established independently. Therefore, the correlation between the PD biomarker and clinical outcomes, while beneficial, is not required.47 48
Additionally, evaluating PK and PD similarity to detect differences between a proposed biosimilar and its reference product may be more sensitive than evaluating clinical efficacy endpoint (s), should differences exist. For example, quantitative analysis showed that the PD biomarker, the area under the effect-time curve of an absolute neutrophil count, is a more sensitive endpoint than the clinical efficacy endpoint of the duration of severe neutropenia.49
Although PK similarity has been evaluated for every FDA-approved biosimilar with systemic exposure to date, as of June 2022, only 11 of 36 approved biosimilars had included PD similarity data to support biosimilarity (specifically, 3 filgrastim biosimilars, 4 pegfilgrastim biosimilars, 2 insulin glargine biosimilars, and 1 epoetin biosimilar). Notably, these products all had well-characterized PD biomarkers (absolute neutrophil count for filgrastim products and pegfilgrastim products; CD34+ cells for filgrastim products; reticulocyte count and hemoglobin level for epoetin alfa products; and glucose infusion rate for insulin glargine products).
The standards for surrogate biomarkers used to support the approval of novel drugs are fundamentally different from the standards for PD biomarkers meant to assist a demonstration of biosimilarity.50 This provides opportunities for biomarkers used as secondary and exploratory endpoints in new drug development programs to support biosimilar testing. In addition, many opportunities are available to identify new PD biomarkers or fill information gaps on existing biomarkers to facilitate using PD biomarker data in clinical pharmacology studies instead of comparative clinical efficacy studies.
The FDA guidance describes five characteristics of PD biomarkers (Table 3) to assist sponsors planning to use PD biomarkers as components of a biosimilar development. PD similarity data to demonstrate no clinically meaningful differences in a biosimilar development program can be based on a single scientifically appropriate PD biomarker or more than one PD biomarker. Data to demonstrate biomarker suitability may be obtained from regulatory documents for the reference product and other approved biosimilar products (for example, product labels and review documents).51 Data to demonstrate PD biomarker suitability may also be obtained from peer-reviewed publications, including systematic reviews, research articles, clinical case studies, and reports on real-world data, and could include data for products with similar mechanism(s) of action as the reference product.
Modeling and simulation using dose- or exposure-response data may provide information on dose–response relationships, sensitive dose ranges, variability in PD biomarker responses, and sensitivity of study populations concerning PD biomarker responses.52 53 54 55 56 57
4. Taking a Step Back from PD Markers
For products that do not display PD biomarkers, such as monoclonal antibodies, other “omic” technologies like transcriptomics and metabolomics may offer a chance to find new, sensitive, and robust candidate biomarkers for further exploration as PD biomarkers.58 However, a more rational approach will be to take a step back in the testing cycle of biosimilars and examine if ex vivo testing can provide evidence of biosimilarity that is more sensitive and reliable in identifying any “clinically meaningful difference” in the language of the FDA guidelines.
Since the pharmacodynamic response is triggered by receptor binding, cell-based bioassays, or potency assays, such as ELISA, binding assays, competitive assays, cell signaling, ligand binding, proliferation, and proliferation suppression, should provide a sufficient functional comparison of a biosimilar candidate with its reference product. Furthermore, functional tests for the mode of action (MOA), such as testing for apoptosis, complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis, and antibody-dependent cellular cytotoxicity, are generally not required, can be added to provide a higher degree of confidence of safety and efficacy.
Monoclonal antibodies (mAbs) bind to specific protein epitope targets on target cells resulting in a therapeutic response. Characterizing the mAb’s affinity for binding include target antigen and affinity for binding to specific Fc receptors (Fc(RI, Ia, IIa, IIb, IIIa, IIIb; Fc(RN, Effector functions like ADCC and CDC, molecular properties like charge, pI, hydrophobicity, and glycosylation, and off-target binding employing in-silico or in vitro techniques like baculovirus ELISA tools are all robust and objective to establish functional similarity.59 60 Additional tests can be added based on specific applications such as for TNFα blockers: C1q; CDC; Induction of regulatory macrophage; inhibition of T-Cell proliferation (MLR); LTα; MLR; mTNFα; Off-target cytokines; Reverse signaling; sTNFα; Suppression of cytokine secretion; tmTNF-α. The functional assays form more robust markers to establish efficacy comparisons than the testing in patients, without the necessity to demonstrate any PD response for mABs.61 62 However, the functional tests (ADCC, ADCP, and CDC) are of little value when the drug targets a soluble antigen. 63 64
A collection of functional assays pertinent to a range of biological activities can be employed for a product having multiple biological activities. For instance, some proteins have a variety of functional domains that express enzymatic and receptor-binding functions. The metric for biological activity is potency. Analytical studies to evaluate these features are easily accessible when immunochemical properties are made part of the activity assigned to the product (for instance, antibodies or antibody-based products). The functional assays form more robust markers to establish efficacy comparisons than the testing in patients, without the necessity to demonstrate any PD response for mABs.65 66
It is, therefore, a logical argument to utilize these more objective tests to establish proof of biosimilarity than clinical efficacy testing for a myriad of reasons, as presented above.
5. In Silico Modeling
DARS promotes evaluating safety issues, mostly what could be anticipated by accumulating molecular target information. Knowing a drug’s molecular targets enables early detection of its effects and potential safety issues for new molecules. Still, the exact modeling can also be applied in a comparator mode to study biosimilar candidates. For instance, DARS has created several computational techniques comprising MLs to predict negative responses to drugs driven by contact with biological receptors that target multiple drugs with similar structures,67 68 leading to many useful methods to predict adverse events. However, the criteria for surrogate biomarkers that support the approval of new drugs differ from the pharmacodynamic markers likely to support a claim of biosimilarity.69
Furthermore, the DARS has also experimented with several advanced modeling plans to forecast a syndrome called cytokine release that is fatal for many biological products70 71 and showed that non-clinical models could effectively demonstrate this adverse event, particularly for the checkpoint inhibitor cancer drugs where computational, in vitro, or conventional non-clinical methodologies are not most suitable.72
The in silico models used to predict the activity and immunogenicity of new molecules can also be used to compare biosimilars with their reference products. For instance, the primary amino acid sequence determines how proteins are arranged in three dimensions. So, suppose the primary sequence of a biosimilar candidate is identical or does not have any known structural elements to interfere with the mode of action of the reference product. In that case, it will be reasonable to conclude that the biosimilar candidate will have the same 3D structure displaying the exact domains that lead to receptor binding and immunogenicity. These attributes are now studied using the modeling tools like the AI-based AlphaFold2 and ESM-fold; the thermodynamic instability of pre-translation modifications is riskier for comparison is also readily concluded.73
The key steps involved in protein receptor binding predictions using AI-based technology:
- Data collection and preprocessing: Large datasets of protein-ligand complexes, including their structures, binding affinities, and functional annotations, are collected from various sources. The data may be preprocessed to remove redundant or noisy information and ensure consistency.
- Feature extraction: Relevant features, such as protein and ligand descriptors, physicochemical properties, and structural motifs, are extracted from the protein-ligand complex data. These features are used to represent the complex in a machine-readable format.
- Model training: Machine learning algorithms, such as deep learning methods like convolutional neural networks (CNNs) or recurrent neural networks (RNNs), or other techniques like support vector machines (SVMs) or random forests, are trained using the preprocessed data and extracted features. The models learn patterns and relationships from the data to make predictions.
- Model validation: The performance and generalizability of the trained models are evaluated using a variety of assessment metrics, including accuracy, precision, recall, and F1 score, as well as cross-validation approaches. It is possible to tweak model parameters to raise prediction precision.
- Prediction and interpretation: The trained models are then used to predict receptor binding for new, unseen protein-ligand complexes. These predictions may include identifying the binding sites, estimating binding affinities, and predicting the binding modes or conformations. The predictions may be further analyzed and interpreted to understand the key features or residues involved in the protein-ligand interactions and to gain insights into the underlying molecular mechanisms.
- Validation and experimental verification: Predicted protein-ligand interactions may be validated experimentally using molecular docking, molecular dynamics simulations, or biochemical assays to confirm their accuracy and reliability. This step helps to refine and improve the predictive models.
- Applications: Applications for the predicted protein receptor binding data include the search for new drugs. It can guide the design and optimization of novel drug candidates with improved binding affinities and selectivity. It can also be used in personalized medicine to understand the interactions between drugs and individual patients’ proteins, leading to more targeted and effective treatments.
Protein structure prediction techniques based on deep learning have attained previously unheard-of levels of accuracy. A modeling algorithm like Automated Pairwise Peptide-Receptor Analysis for Screening Engineered Proteins (APPRAISE) fills in the gaps between the ability to predict the structures of candidate proteins and the ability to determine which of those proteins are most likely to bind to a target receptor. It works by first creating models of proteins competing for binding to a target using a well-known structure-prediction tool like AlphaFold2-multimer or ESMFold. Then, the APPRAISE performs a scoring analysis that considers biophysical and geometrical constraints.74
In summary, using AI-based technology, protein receptor binding predictions can significantly accelerate the drug discovery process, improve personalized medicine, and deepen our understanding of protein-ligand interactions that can be valuable in establishing silico biosimilarity.
7. Conclusion
Given the higher sensitivity of analytical assessment, nonclinical functional testing, and clinical pharmacology evaluation compared with clinical efficacy testing in patients, it is now recognized by regulatory agencies that the studies with higher sensitivity should supersede the least sensitive studies since these studies can never fail, and a failed study cannot be confirmed as a failure for statistical reasons. Since clinical efficacy testing in patients is the last step, it can be readily waived since a biosimilar candidate has already demonstrated biosimilarity based on more robust and objective testing methods. The FDA has already agreed to waive this testing for drugs with pharmacodynamic biomarkers.75 This waiver should extend to all other biosimilar products based on their comparison of functional properties, such as receptor binding studies, proteomic comparisons, and applying AI-based and in silico approaches to establish biosimilarity. Returning to functional biosimilarity will be a giant scientific leap in making biosimilars affordable.
Funding
No funding was received for this work.
Conflicts of Interest
Author declares no conflicts of interest.
References
- Inxight Drug Database. ttps://drugs.ncats.io/substances?facet=Development%20Status%2FUS%20Approved%20Rx&facet=Substance%20Class%2Fprotein&facet=Substance%20Form%2FPrincipal%20Form&page=1. (accessed on 25 April 2023).
- Isakov L, Jin B, Jacobs IA. Statistical Primer on Biosimilar Clinical Development. Am J Ther. 2016 Nov/Dec;23(6):e1903-e1910. [CrossRef]
- Fontanillo, M. , et al. (2022) Three imperatives for R&D in biosimilars https://www.mckinsey.com/industries/life-sciences/our-insights/three-imperatives-for-r-and-d-in-biosimilars. (accessed on 25 April 2023).
- Moore TJ, Mouslim MC, Blunt JL, Alexander GC, Shermock KM. Assessment of Availability, Clinical Testing, and US Food and Drug Administration Review of Biosimilar Biologic Products. JAMA Intern Med. 2021;181(1):52–60. [CrossRef]
- Clinical Trials Database. https://clinicaltrials.gov/ct2/results?cond=&term=biosimilar&cntry=&state=&city=&dist=&recrs=e. (accessed on 25 April 2023).
- Han, JJ. FDA Modernization Act 2.0 allows for alternatives to animal testing. Artif Organs. 2023 Mar;47(3):449-450. [CrossRef]
- U.S. Food and Drug Administration: Clinical Immunogenicity Considerations for Biosimilar and Interchangeable Insulin Products https://www.fda. (accessed on 25 April 2023)1330.
- U.S. Food and Drug Administration: Role of pharmacodynamic biomarkers in biosimilar development. https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/role-pharmacodynamic-biomarkers-biosimilar-drug-development. (accessed on 25 April 2023).
- U.S. Food and Drug Administration: BIOSIMILAR BIOLOGICAL PRODUCT REAUTHORIZATION PERFORMANCE GOALS AND PROCEDURES FISCAL YEARS 2023 THROUGH 2027 https://www.fda.gov/media/152279/download. 1522; (accessed on 25 April 2023).
- U.S. Food and Drug Administration: Clinical trial guidelines. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trials-guidance-documents. (accessed on 25 April 2023).
- U.S. Food and Drug Administration Biosimilars Action Plan. https://www.fda. (accessed on 25 April 2023)1145.
- Hulley SB, Cummings SR, Browner WS, Grady DG, Newman TN. Designing clinical research. 3rd ed. Philadelphia: Wolters Kluwer. Lippincott Williams & Wilkins; 2007.
- Sackett DL, Strauss SE, Richardson WS. Evidence-based medicine: how to practice and teach EBM. London: Churchill-Livingstone; 2000.
- Jussi Paananen, Vittorio Fortino, An omics perspective on drug target discovery platforms, Briefings in Bioinformatics, Volume 21, Issue 6, November 2020, Pages 1937–1953. 20 November. [CrossRef]
- Cohen, D. FDA official: “clinical trial system is broken”. BMJ. 2013 Dec 5;347:f6980. [CrossRef] [PubMed]
- Adaptive Platform Trials Coalition. Adaptive platform trials: definition, design, conduct and reporting considerations. Nat Rev Drug Discov. 2019 Oct;18(10):797-807. [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA modernizes clinical trial designs and approaches for drug development, proposing new guidance on the use of adaptive designs and master protocols https://www.fda.gov/news-events/fda-brief/fda-brief-fda-modernizes-clinical-trial-designs-and-approaches-drug-development-proposing-new. (accessed on 25 April 2023).
- Mauer M, Collette L, Bogaerts J, European Organisation for R, Treatment of Cancer Statistics D.. Adaptive designs at European Organisation for Research estimation based on interim-effect size. Eur J Cancer 2012;48:1386–91.
- Piantadosi, S. Clinical trials: a methodologic perspective. 3rd ed. Hoboken, HJ, USA: John Wiley & Sons Inc; 2017.
- Senn, S. Cross-over trials in clinical research. Crichester: Wiley; 2002.
- Talevi, A. Multi-target pharmacology: possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front Pharmacol. 2015 Sep 22;6:205. [CrossRef]
- Tesser JR, Furst DE, Jacobs I. Biosimilars and the extrapolation of indications for inflammatory conditions. Biologics. 2017 Feb 17;11:5-11. [CrossRef]
- Ma B, Shatsky M, Wolfson HJ, Nussinov R. Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 2002 Feb;11(2):184-97. [CrossRef]
- U.S. Food and Drug Administration. Guidance for Industry E9 Statistical Principles for Clinical Trials. U.S. Department of Health and Human Services Food and Drug Administration; 1998. Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073137.pdf. (accessed on 25 April 2023)2015; 1.
- U.S. Food and Drug Administration. Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Draft Guidelines. U.S. Department of Health and Human Services Food and Drug Administration; 2015. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. (accessed on 25 April 2023).
- World Health Organization. Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs). Available at: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed April 7, 2015. (accessed on 25 April 2023).
- U.S. Food and Drug Administration. Guidance for Industry E9 Statistical Principles for Clinical Trials. U.S. Department of Health and Human Services Food and Drug Administration; 1998. Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073137.pdf. /; (accessed on 25 April 2023).
- Walker E, Nowacki AS. Understanding equivalence and noninferiority testing. J Gen Intern Med. 2011;26:192–196.
- World Health Organization. Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs). Available at: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed , 2015. 7 April.
- Dasgupta A, Lawson KA, Wilson JP. Evaluating equivalence and noninferiority trials. Am J Health Syst Pharm. 2010;67:1337–1343.
- Njue, C. Statistical considerations for confirmatory clinical trials for similar biotherapeutic products. Biologicals. 2011;39:266–269.
- Walker E, Nowacki AS. Understanding equivalence and noninferiority testing. J Gen Intern Med. 2011 Feb;26(2):192-6. [CrossRef]
- D’Agostino Sr RB, Massaro JM, Sullivan LM. Non-inferiority trials: design concepts and issues – the encounters of academic consultants in statistics. Stat Med 2003;22:169–86. 1.
- Schiestl M, Ranganna G, Watson K, Jung B, Roth K, Capsius B, Trieb M, Bias P, Maréchal-Jamil J. The Path Towards a Tailored Clinical Biosimilar Development. BioDrugs. 2020 Jun;34(3):297-306.
- https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/division-applied-regulatory-science.
- Chiu, K. , et al., (2023). New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Frontiers in Medicine, 9. [CrossRef]
- US Food and Drug Administration. FDA Guidance: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product <https://www.fda.gov/media/88622/ download> (2016).
- Chiu K, et al., New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Front Med (Lausanne). 2023 Jan 19;9:1109541. [CrossRef]
- Li, L. et al. Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for GCSF in breast cancer patients. Clin Pharmacol Ther 104, 742–748 (2018). 2018.
- Li J, Florian J, Campbell E, Schrieber S, Bai J, Weaver J, et al. Advancing biosimilar development using pharmacodynamic biomarkers in clinical pharmacology studies. Clin Pharmacol Ther. (2020) 107:40–2. [CrossRef]
- Sheikhy M, Schrieber S, Sun Q, Gershuny V, Matta M, Bai J, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development- (I) a randomized trial with PCSK9 inhibitors. Clin Pharmacol Ther. (2022). [CrossRef]
- Gershuny V, Sun Q, Schrieber S, Matta M, Weaver J, Ji P, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development - (II) a randomized trial with IL-5 antagonists. Clin Pharmacol Ther. (2022). [CrossRef]
- Florian JG, Sun Q, Schrieber S, Matta M, Hazel A, Sheikhy M, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development – (III) a randomized trial with interferon beta-1a products. Clin Pharmacol Ther. (2022). [CrossRef]
- Hyland P, Chekka L, Samarth D, Rosenzweig B, Decker E, Mohamed E, et al. Evaluating the utility of proteomics for the identification of circulating pharmacodynamic biomarkers of IFNbeta-1a biologics. Clin Pharmacol Ther. (2022). [CrossRef]
- Florian JS, et al., Pharmacodynamic biomarkers for biosimilar development and approval: a workshop summary. Clin Pharmacol Ther. (2022). [CrossRef]
- Wang YM, Strauss DG. Advancing Innovations in Biosimilars. Clin Pharmacol Ther. 2023 Jan;113(1):11-15. [CrossRef] [PubMed]
- US Food and Drug Administration . FDA Guidance: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product <https://www.fda.gov/media/82647/download> (2015). (accessed on 25 April 2023). (accessed on 25 April 2023).
- US Food and Drug Administration . FDA Guidance: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product <https://www.fda.gov/media/88622/download> (2016). (accessed on 25 April 2023). (accessed on 25 April 2023).
- Li, L. et al. Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for G-CSF in breast cancer patients. Clin Pharmacol Ther 104, 742–748 (2018).
- US Food and Drug Administration . FDA draft guidance: Biomarker Qualification: Evidentiary Framework <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM628118.pdf> (2018). (accessed on 25 April 2023). (accessed on 25 April 2023).
- US Food and Drug Administration . Drugs@FDA. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm. (accessed on 25 April 2023).
- Li, L. et al. Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for G-CSF in breast cancer patients. Clin Pharmacol Ther 104, 742–748 (2018).
- Wang, Y.C. et al. Use of pharmacodynamic/response biomarkers for therapeutic biologics regulatory submissions. Biomark Med 13(10), 805–809 (2019).
- Wang, Y.C. et al. Role of modeling and simulation in the development of novel and biosimilar therapeutic proteins. J Pharm Sci 108(1), 73–77 (2019).
- Wang, Y. & Huang, S.M. Commentary on fit-for-purpose models for regulatory applications. J Pharm Sci 108(1), 18–20 (2019).
- Zhu, P. , Hsu, C.H. , Liao, J. , Xu, S. , Zhang, L. & Zhou, H. Trial design and statistical considerations on the assessment of pharmacodynamic similarity. AAPS J 21(3), 47 (2019).
- Zhu, P. , Ji, P. & Wang, Y. Using clinical PK/PD studies to support No clinically meaningful differences between a proposed biosimilar and the reference product. AAPS J 20(5), 89 (2018).
- Strauss DG, Wang YM, Florian J, Zineh I. Pharmacodynamic Biomarkers Evidentiary Considerations for Biosimilar Development and Approval. Clin Pharmacol Ther. 2023 Jan;113(1):55-61. [CrossRef]
- Hotzel, I. Hotzel, I., et al., A strategy for risk mitigation of antibodies with fast clearance mAbs, 4 (2012), pp. 753-760.
- Sharma, TW., et al., In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci, 111 (2014), pp. 18601-18606.
- Cymera F, Becka H, Rohde A, Reusch D. Therapeutic monoclonal antibody N-glycosylation—structure, function and therapeutic potential. Biologicals. 2018;52:1–11.
- Prior S, et al., International standards for monoclonal antibodies to support pre- and post-marketing product consistency: evaluation of a candidate international standard for the bioactivities of rituximab. MAbs. 2018;10(1):129–42.
- Ryding J, Stahl M, Ullmann M. Demonstrating biosimilar and originator antidrug antibody binding comparability in antidrug antibody assays: a practical approach. Bioanalysis. 2017 Sep;9(18):1395-1406. [CrossRef] [PubMed]
- Wang X, An Z, Luo W, Xia N, Zhao Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell. 2018 Jan;9(1):74-85. [CrossRef]
- Cymera F, Becka H, Rohde A, Reusch D. Therapeutic monoclonal antibody N-glycosylation—structure, function and therapeutic potential. Biologicals. 2018;52:1–11.
- Prior S, et al., International standards for monoclonal antibodies to support pre- and post-marketing product consistency: evaluation of a candidate international standard for the bioactivities of rituximab. MAbs. 2018;10(1):129–42.
- Daluwatte C, Schotland P, Strauss D, Burkhart K, Racz R. Predicting potential adverse events using safety data from marketed drugs. BMC Bioinformatics. (2020) 21:163. [CrossRef]
- Schotland P, Racz R, Jackson D, Soldatos T, Levin R, Strauss D, et al. Target adverse event profiles for predictive safety in the postmarket setting. Clin Pharmacol Ther. (2021) 109:1232–43. [CrossRef]
- Wang YC, Strauss DG, Huang SM. Use of pharmacodynamic/response biomarkers for therapeutic biologics regulatory submissions. Biomark Med. 2019 Jul;13(10):805-809. [CrossRef] [PubMed]
- Yan H, Bhagwat B, Sanden D, Willingham A, Tan A, Knapton A, et al. Evaluation of a TGN1412 analogue using in vitro assays and two immune humanized mouse models. Toxicol Appl Pharmacol. (2019) 372:57–69. [CrossRef]
- Yan H, Semple K, Gonzalez C, Howard K. Bone marrow-liver-thymus (BLT) immune humanized mice as a model to predict cytokine release syndrome. Transl Res. (2019) 210:43–56. [CrossRef]
- Weaver J, Zadrozny L, Gabrielson K, Semple K, Shea K, Howard KEBLT-. Immune humanized mice as a model for nivolumab-induced immune-mediated adverse events: comparison of the NOG and NOG-EXL strains. Toxicol Sci. (2019) 169:194–208. [CrossRef]
- Niazi SK. Molecular Biosimilarity—An AI-Driven Paradigm Shift. International Journal of Molecular Sciences. 2022; 23(18):10690. [CrossRef]
- https://github.com/AppraiseDev/Appraise.
- Chiu K, Racz R, Burkhart K, et al.(2023). New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Front Med.2022;9:1109541.
Figure 1.
The Bayesian probability of a failed trial is an actual failure.
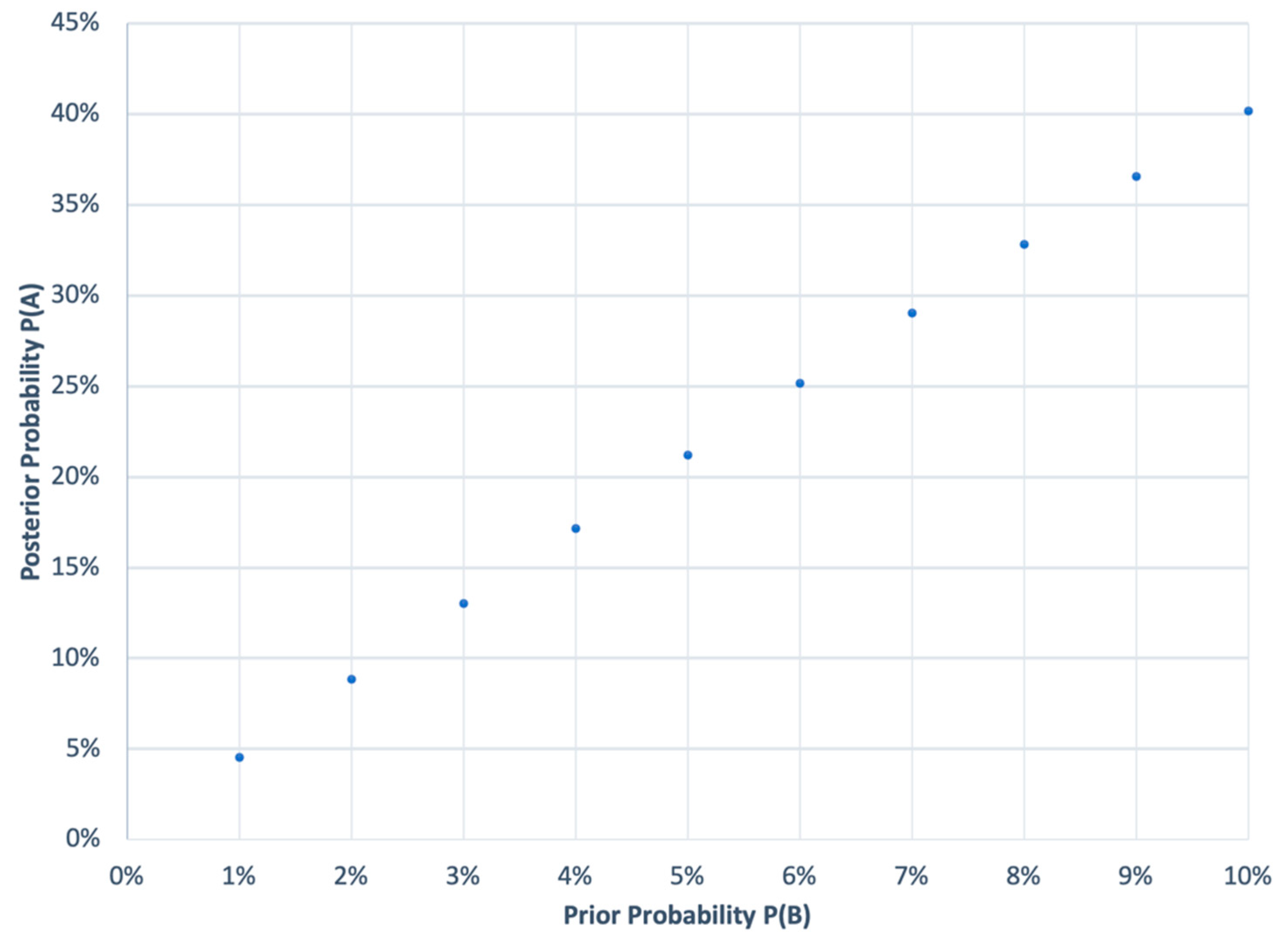

Table 1.
Biologicals approved in EU and US; (products marked with an asterisk were not approved under the 351k legislation in the US).
Table 1.
Biologicals approved in EU and US; (products marked with an asterisk were not approved under the 351k legislation in the US).
| Molecule | Product | EU | US |
|---|---|---|---|
|
Adalimumab | 10 | 8 |
|
Bevacizumab | 8 | 4 |
|
Enoxaparin sodium* | 2 | 2 |
|
Epoetin alfa/zeta | 5 | 1 |
|
Etanercept | 3 | 2 |
|
Filgrastim | 7 | 2 |
|
Follitropin alfa* | 2 | 1 |
|
Insulin glargine | 2 | 2 |
|
Insulin regular | 1 | 0 |
|
Infliximab | 4 | 4 |
|
Insulin glargine | 1 | 2 |
|
Insulin aspart | 2 | 0 |
|
Insulin lispro | 1 | 1 |
|
Pegfilgrastim | 8 | 5 |
|
Ranibizumab | 1 | 2 |
|
Rituximab | 5 | 3 |
|
Somatropin* | 1 | 2 |
|
Teriparatide* | 4 | 1 |
|
Trastuzumab | 6 | 5 |
|
Total | 73 | 47 |
Table 2.
Sample Size to Achieve 0.80 Power to Test Equivalency at the α = 0.05 Significance Level for True Proportions of 0.28 and 0.33.32.
Table 2.
Sample Size to Achieve 0.80 Power to Test Equivalency at the α = 0.05 Significance Level for True Proportions of 0.28 and 0.33.32.
| Equivalence margin | Sample size (per group) |
| 0.06 | 26,185 |
| 0.07 | 6,547 |
| 0.08 | 2,910 |
| 0.09 | 1,637 |
| 0.10 | 1,048 |
| 0.11 | 728 |
| 0.12 | 535 |
Table 3.
Potential evidence to address the five characteristics of a PD biomarker to be used in PD similarity studies.
Table 3.
Potential evidence to address the five characteristics of a PD biomarker to be used in PD similarity studies.
| Characteristics | Evidence |
|---|---|
| The relation of the PD biomarker to the drug’s mechanism of action (to the degree that the reference product’s mechanism of action is known) | Data demonstrating the relevance of a PD biomarker (and the pharmacological effect it illustrates) to all or some of a product’s known mechanisms of action |
| The period at which the PD biomarker first changed concerning dose and when it returned to baseline once dosing was stopped | Data demonstrating the complete PD biomarker response profile includes the onset time, response duration, and return to baseline. |
| The dynamic range of the PD biomarker over the exposure range of the biological product | PD response data obtained from a range of doses that characterize the range of PD biomarker responses to demonstrate dynamic range, dose dependence, and magnitude of response. The PD responses associated with the range of concentrations in the observed PK profile are appropriately captured. |
| The PD biomarker’s susceptibility to variations between the suggested biosimilar product and the reference product | Dose-response relationship data to determine the sensitive dose range and estimate the variability in PD biomarker response |
| The analytical validity of the PD biomarker assay | Data demonstrating the accuracy, precision, specificity, sensitivity, and reproducibility of the PD biomarker assay |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



