Submitted:
28 April 2023
Posted:
29 April 2023
You are already at the latest version
Abstract
The stimulus-secretion coupling of glucose-induced release is generally attributed to the metabolism of the hexose in the β-cells in the glycolytic pathway and the citric acid cycle. Glucose metabolism generates an increased cytosolic concentration of ATP and of the ATP/ADP ratio that closes the ATP-dependent K+-channel at the plasma membrane by the interaction of ATP with the regulatory Kir6.2 channel subunit The resultant depolarization of the β-cells opens voltage-dependent Ca2+-channels at the plasma membrane that allow an increase of the cytosolic cation concentration that triggers the exocytosis of insulin secretory granules. The resultant secretory response evolves in time as a biphasic secretion with a first and transient peak of approximately 10 minutes duration followed by a sustained phase of secretion lasting as long as the stimulus. Whereas the first transient peak can be reproduced by a simple depolarization of β-cells with high extracellular KCl maintaining the KATP-channels open with diazoxide, the sustained phase is agreed to depend on the participation of some metabolic signal that remains to be determined Our group has been investigating for several years the participation of β-cell GABA metabolism, together with that of glucose (the β-cell “specific” nutrient secretagogue) and some other “metabolic” secretagogues (branched-chain alpha-ketoacids and a mixture of L-Leucine + L-glutamine, at supraphysiological concentrations) in their mechanism of stimulation of insulin secretion. All three types of stimuli promote the flux in the GABA shunt of rat islets by different metabolic pathways that end in the production of α-ketoglutarate. This citric acid cycle intermediary is preferentially derived to the GABA shunt instead of its continuous oxidation in the citric acid cycle. Islet content of GABA is significantly suppressed by all the stimuli and blocking the GABA shunt with gabaculine, or ϒ-vinyl-GABA (GABAT inhibitors), diminish the insulin secretory responses as well as total ATP and the ATP/ADP ratio. It is concluded that GABA metabolism is increased in parallel to glucose metabolism and is significantly contributing to the magnitude of the insulin secretory response. Its possible implication in β-cells degradation in type-2 (perhaps also in type 1) diabetes is suggested.
Keywords:
Functional role of GABA in insulin secretion stimulation
; GABA metabolism in pancreatic islets
; GABA and insulin secretion
; interaction of glucose and GABA metabolism in pancreatic islets
; GABA shunt
I. Metabolic role of GABA on the mechanism of stimulation of insulin secretion.
1. Introduction
The stimulus-secretion coupling of
glucose-induced release is generally attributed to the metabolism of the hexose
in the
β
-cells in the
glycolytic pathway and the citric acid cycle. Glucose metabolism generates an
increased cytosolic concentration of ATP and of the ATP/ADP ratio that closes
the ATP-dependent K+-channel at the plasma membrane by the
interaction of ATP with the regulatory Kir6.2 channel subunit (1). The
resultant depolarization of the
β
-cells opens voltage-dependent Ca2+-channels at the
plasma membrane that allow an increase of the cytosolic cation concentration
that triggers the exocytosis of insulin secretory granules (1). The resultant
secretory response evolves in time as a biphasic secretion with a first and
transient peak of approximately 10 minutes duration (in rat perifused islets)
followed by a sustained phase of secretion lasting as long as the stimulus.
Whereas the first transient peak can be reproduced by a simple depolarization
of
β
-cells with high
extracellular KCl maintaining the KATP-channels open with diazoxide
(2), the sustained phase is agreed to depend on the participation of some
metabolic signal that remains to be determined (3,4). Given that extracellular
ATP alone (10 mM) triggers a biphasic insulin secretion in KCl-permeabilized
islets (5), the metabolic signal responsible for the sustained phase may be any
enzymatic pathway capable of leading to ATP synthesis. Our group has been
investigating for several years the participation of
β
-cell GABA metabolism, together with that of glucose (the
β
-cell “specific” nutrient secretagogue) and
some other “metabolic” secretagogues (physiological metabolites at
supraphysiological concentrations) in their mechanism of simulation of insulin
secretion.
ϒ-Aminobutyric acid (GABA) is abundant exclusively in the β-cells of pancreatic islets that are equipped
with all the enzymes required to synthesize and metabolize it in the GABA shunt
(6): 1. Glutamic acid decarboxylase isoenzymes (cytosolic GAD65 and GAD67)
generating GABA by L-glutamate decarboxylation. 2. GABA transaminase (GABAT), a
mitochondrial enzyme exchanging an amino group between GABA and α-ketoglutarate
(αKG or 2-oxoglutarate, 2-OG) to give succinic acid semialdehyde (SSA) and
L-glutamate. 3. NAD+-dependent SSA dehydrogenase oxidizing SSA to
succinic acid that is further metabolized in the Krebs cycle. 4. NAPH-dependent
SSA-reductase reduces SSA to ϒ-hydroxybutyric acid, competes with the previous enzyme for the same
substrate.
Its functional role in
β
-cells has been discussed for many years (7)
but there is not yet any conceptual consensus on its participation on the
mechanism of biphasic insulin secretion triggered by glucose and some other
known metabolic stimuli (fundamentally L-leucine + L-glutamine and some
branched-chain 2-oxoacids (BCKAs)). So far, the most accepted hypothesis seems
to be the autocrine regulation by the released (or co-secreted with insulin?)
of GABA through the stimulation of either GABA-A or -B receptors (8,9). Also, a
possible mechanism of paracrine regulation of glucagon secretion by GABA,
co-secreted with insulin, through the activation of GABAA receptors
of α-cells has been postulated (10). However, we will limit our review to
experimental data dealing with the participation of intracellular GABA in islet
metabolism.
2. Effect of glucose stimulation of insulin secretion on islet GABA content
Since the pioneering work of M.
Erecinska (11) suggesting an energetic role for GABA in
β
-cells contributing to the maintenance of islet ATP/ADP ratio, some
progress has been achieved. A first important finding was the demonstration
that islet GABA synthesis in vitro was dependent on the extracellular
concentration of L-glutamine (12): it reached a maximum between 0.5 to 1 mM,
the physiological value of plasma L-glutamine concentration (13). That means
that rat islet GABA content is regularly maintained at relatively high values,
together with L-glutamate and L-aspartate, in comparison with other L-amino
acids (13). A detailed study of the effect of 20 mM glucose on the spectrum of
islet plasma L-amino acids showed strong, specific, and significant decreases
of islet GABA-content with respect to the values at 0 mM glucose in a range of
L-glutamine concentrations (0.0,0.5 and 10.0 mM). The simultaneous measurements
of GABA release at either 0.5 or 10.0-mM L-glutamine were also strongly reduced
(14). It was concluded that the suppression of islet GABA content did not
depend on an increased rate of release, but it was produced by an increase of
its metabolic rate. This was supported by the finding that inhibition of
glucose metabolism by D-mannoheptulose blocked sugar capacity to suppress islet
GABA content (
Figure S1
in 14 and 15). A significantly positive, linear correlation was
found between islet content and GABA release among all the values obtained at
both 0- and 20-mM glucose: the averaged GABA release at 0 mM glucose was higher
than at 20 mM glucose at 0.5- and 10.0-mM L-glutamine: the lower the GABA
content the lower the rate of release (12,14). The linear correlation obtained
between GABA content and release has been confirmed by other authors that have
supplied experimental evidence of the mediation of GABA release and uptake by
the membrane anion transporters VRAC and TauT, respectively (8). Like the
previous authors, our data speak against the co-secretion of insulin and GABA
under glucose stimulation. By contrast, our data do not predict an increase of
GABA release by high glucose but a decrease (14). Notwithstanding the remaining
release of GABA through the postulated specific anion transporters might
contribute to the pulsatility of the contemporarily stimulated insulin
secretion (8).
3. Effects of branched-chain 2-oxoacids (BCKAs) stimulation of insulin release on islet GABA content
Besides glucose, branched-chain 2-oxo acids (α-ketoisocaproic
acid or KIC, α-keto-β-methyl-valeric acid or KMV), deaminated products of
L-leucine and L-norleucine, respectively; and other branched-chain α-keto
acids) are potent “metabolic” stimuli of islet insulin secretion at
supraphysiological concentrations, in the absence of glucose. They induce a
biphasic secretion of insulin like that of glucose but of variable magnitude
and strongly decreased the islet content of GABA, similarly to glucose (16).
The mentioned branched-chain 2-oxo acids (KIC and KMV) were aminated back to
their corresponding amino acids (L-leu and L-norLeu, respectively) that
diffused and accumulated in the incubation medium without increasing their
islet content (16). These results suggested the hypothesis that the amination
of the branched-chain 2-oxo acids was coupled to the deamination of L-glutamate
to αKG by α-branched-chain amino acid transaminases (BCATs: cytosolic or
mitochondrial). This hypothesis was supported by the suppression of
BCKAs-stimulated insulin secretion with generic or more specific inhibitors of
BCKATs (17). Moreover, a contemporary publication demonstrated that a general
knockout of BCKATm (mitochondrial isoform) in mice led to a block of insulin
secretion stimulation of the majority of regularly used BCKAs without affecting
glucose stimulation (18). These results further support the contention of the
important role of BCKAs-amination in their stimulation of secretion. However,
the authors did not investigate the effects of BCKAs on islet GABA metabolism.
4. Effects of L-leucine plus -L-glutamine stimulation of insulin release on islet GABA content
Another example of a long known,
strong “metabolic” secretagogue mixture is the combination of L-glutamine plus
L-leucine, the latter at supraphysiological concentrations. L-leucine has been
shown to be an allosteric activator of mitochondrial L-glutamate dehydrogenase
(GDH) catalyzing the conversion between L-glutamate and α-KG (19). L-leucine
alone (10 mM) stimulated a predominantly first phase of insulin secretion in
comparison with glucose (20 mM) but in combination with 10 mM-glutamine, it
triggered a stronger biphasic secretion (13,20). L-leucine (10 mM) did not
modify islet GABA content in the absence of extracellular L-glutamine but
significantly stimulated the islet concentrations of L-glutamate and
L-aspartate. At 5 and 10 mM-glutamine, the islet content of all recorded amino
acids increased several folds and that of GABA was significantly decreased by
L-leucine (-38%), 10 mM BCH (GDH activator). (-56%) and 10 mM allylglycine (GAD
inhibitor) (-42%) (13).
5. Conclusions and prospects
In these three main examples of
nutrient and metabolic secretagogues, their metabolism shares a common
metabolic step: the diversion of αKG to the GABA shunt where it would be first
transaminated with GABA by GABAT to render L-glutamate and semialdehyde
succinic acid (SSA). SSA will be oxidized by semialdehyde succinic acid
dehydrogenase (SSAdh) to succinic acid that will in turn enter the Krebs cycle
for its further oxidation. The resulting ATP synthesis would lead to elevated
ATP/ADP ratios and the closure of K+ATP channels that
initiates the stimulation of insulin secretion.
Confirmation of these hypothetical
mechanisms presupposes that interference with GABA shunt may alter Insulin
responses to nutrient and metabolic secretagogues as well as their capacity to
increase their mitochondrial production of ATP. For that purpose, gabaculine
and allylglycine have been used as GABAT- and GAD65-inhibitors, respectively,
to check their effects on insulin secretion, adenine nucleotides concentrations
and GABA metabolism.
6. Postulated metabolic pathway leading to the stimulation of BCKAs-induced insulin secretion (Figure 1)
10mM KIC induced a biphasic
insulin secretion in the absence of glucose and its magnitude was not modified
by either 0.5- or 10.0-mM L-glutamine. Gabaculine (0.25mM) diminished the
height of the first peak of secretion and suppressed significantly by 33% the
second phase of sustained release (16). KIC increased 14CO2-production
from 0.5- and 10.0-mM L-(U-14C) glutamine by 27% and 66%,
respectively (16). Considering that the rate of L-(U-14C)
glutamine-oxidation is stoichiometric with the amount of GABA synthesized
(13), this would facilitate the replenishment of the intracellular pool of
GABA. On the other hand, 0.25 mM gabaculine did not
modify the rate of L-glutamine oxidation in the presence of KIC whereas 40 µM
gabaculine strongly suppressed GABAT activity in rat islet homogenates (16).
Gabaculine (0.25mM) also blocked within 59% the abrupt decreased of intra-islet
oxygen tension caused by 10mM KIC reflecting that the contribution to islet
oxygen consumption promoted by the α-keto acid is partially attributable to an
increased flux in the GABA shunt and part of the Krebs cycle (16).
7. Postulated metabolic pathway leading to the stimulation of insulin secretion by L-Leucine plus L-glutamine (Figure 2)
L-leucine (10 mM) alone does not
stimulate the oxidation of 14CO2-production from 0.5- and
10.0-mM L-(U-14C) glutamine (13).
However, other authors have shown that BCH (GDH activator) and some amino acids
(L-isoleucine and L-norvaline, at 20 mM) stimulate 14CO2-production
from islets pre-labelled with 1 mM L-(U-14C) glutamine (19).
Notwithstanding, 10 mM L-leucine strongly suppressed islet GABA content in the
presence of 0.5- and 10.0-mM L-glutamine (13). L-glutamine
potentiation of L-Leucine stimulated insulin secretion is generally assumed to
be due to the production of αKG secondary to GDH
stimulation. According to our own data, the equilibrium of GDH activity in
islet homogenates favors the amination (A) versus the deamination (D) reaction
(A/D= 8.2) (13). L-leucine (10 mM) increases the ratio more in favor of the
amination (A/D= 10.1; p<0.05 versus the absence of Leu) (13). This condition
would not facilitate an optimal αKG concentration to increase the net flux
trough GABAT. Therefore, possibly GABAT requires a higher supply of GABA
(higher medium L-glutamine concentration) than KIC. In fact, KIC induces a
strong decrease of cellular GABA in the absence of extracellular L-glutamine
whereas L-leucine failed to do it (13). Moreover, the dimethyl ester of
α-ketoglutarate (dmKG), a membrane permeable analogue of αKG, increased the
islet GABA content at 5 mM but decreased it significantly at higher concentrations
in the absence and presence of L-leucine (13).
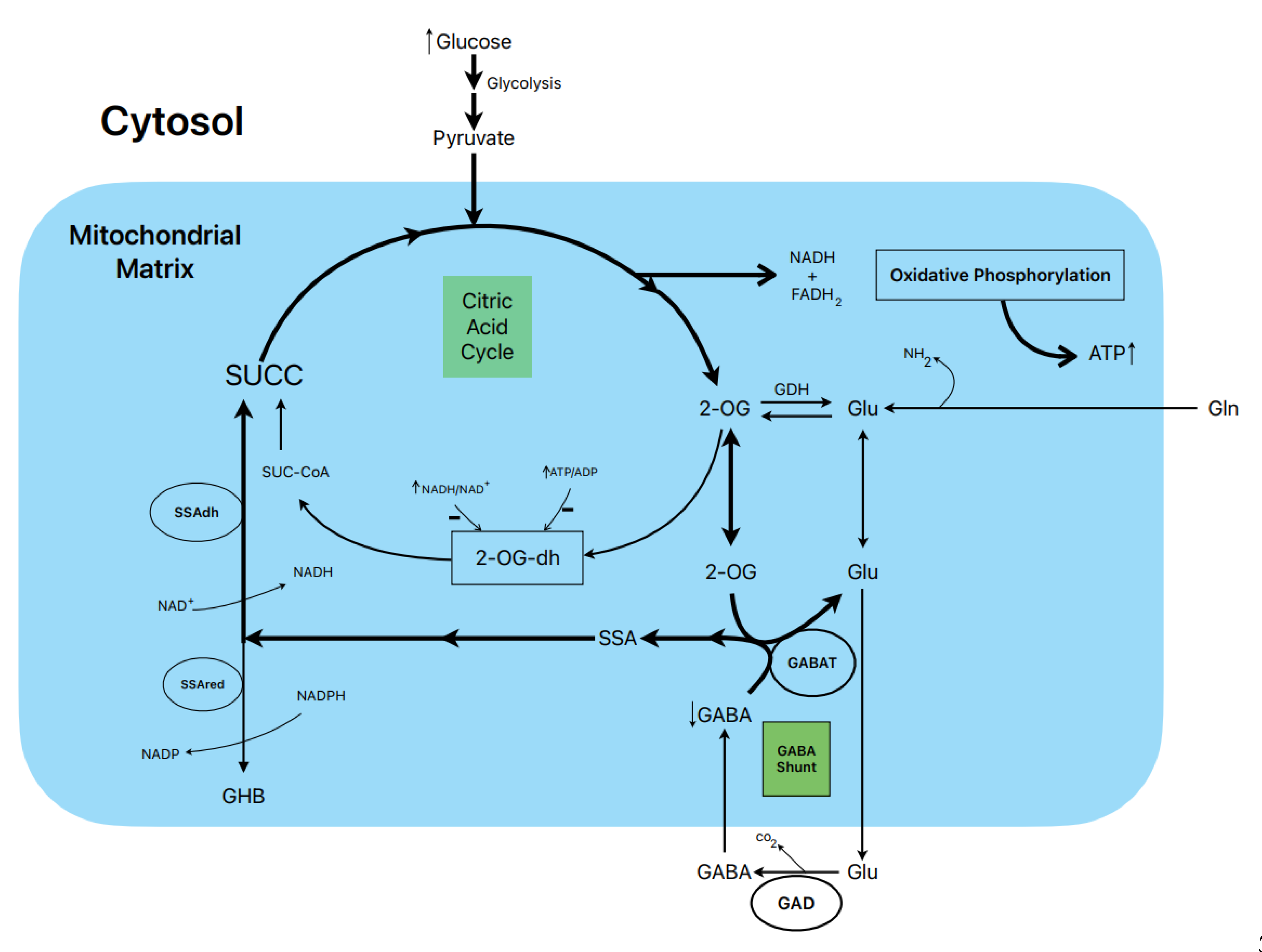
8. Postulated metabolic pathway leading to the stimulation of insulin secretion by glucose (Figure 3)
The biphasic stimulation of
insulin secretion by 20 mM glucose in rat islets was partially suppressed by 1
mM gabaculine in the presence of 10 mM L-glutamine and it was also decreased by
20 mM allylglycine (14,
Figure
3
C and
Figure 2
S, respectively). These
experimental data support that the metabolic flux in the GABA shunt also
contributes to the stimulated secretion. This is also supported by the
reduction of islet ATP contents and the ATP/ADP ratios at 20 mM glucose induced
by 1 mM gabaculine in the absence and presence of 1- and 10-mM L-glutamine
(14).
An important argument to support
what might be named as the “GABA metabolic hypothesis” for the
stimulation of insulin secretion by glucose and other non-physiological
secretagogues is why the GABA shunt might be required to participate in the
metabolic stimulation of insulin secretion. We have proposed that, at least in
islet β-cells, the flux in the Krebs cycle is limited by the lower expression
of the 2-oxo-glutarate (αKG) dehydrogenase gene
compared with the gene of its competitor enzyme in the GABA shunt, GABAT, for
their common substrate 2-oxo-glutarate (αKG) (see supplementary
Figure S4
in 14). In a model of “KCl-permeabilized islets” (5) we have
demonstrated that 5 mM αKG stimulates a sustained phase of insulin secretion
after the peak of release induced by 70 mM KCl (21). It was reversible and
returned to basal levels after withdrawing αKG and was suppressed within 47% by
1 mM gabaculine. In parallel experiments in incubated and permeabilized islets,
the ATP content and ATP/ADP ratio of islets and the amount of ATP diffused and
accumulated in the extracellular medium were measured (21). The islet ATP
content was decreased by 1 mM gabaculine (p<0.05) and the medium ATP
severely reduced within 37% (p<0.0005). Neither the islet ATP content nor
the amount of medium ATP due to metabolism of any other Krebs cycle
intermediary metabolites tested were significantly modified by gabaculine. 10
mM SSA alone, surprisingly permeable trough the plasma membrane, stimulated a
biphasic secretion of insulin of less magnitude than 20 mM glucose and whose
second phase was partially suppressed by 1mM gabaculine (14). It also
significantly increased islet ATP content (+93.5 %) and the ATP/ADP ratio (+84%).
At 1mM glucose, 10 mM SSA depolarized the membrane potential of isolated
β-cells and diminished membrane currents through KATP-channels. The
depolarization capacity of glucose was not altered after pre-incubating the
β-cells for 1-2 h with1 mM gabaculine (results not shown).
9. Conclusion
The GABA shunt in islet β-cells
seems to contribute to the metabolic energy required for a sustained stimulation
of insulin secretion by nutrient and metabolic secretagogues. As judged by the
documented dependence of the metabolic oxidation of αKG and its ATP production
on the functioning of the GABA shunt, its contribution might be significant.
II. Possible implication of the GABA-shunt on β-cell degradation in type 2 diabetes.
1. Introduction
Type 1 diabetes is an autoimmune
disease caused by the activation of CD8+ and CD4+ T
cells targeted to a group of autoantigens released from the β-cells:
(Pre)proinsulin, glutamic acid decarboxylase of 65 KD (GAD65), tyrosine
phosphatase IA2 and the zinc transporter ZnT8. GAD 65 i
s
one of the most prevalent autoantigens found in patients with type 1 diabetes
(22).
2. Intracellular versus extracellular effects of GABA.
GABA is released in a pulsatile
manner by non-diabetic human islets, and it is not modified by a range of
glucose concentrations (1, 3 and 11 mM), as “detected by cytosolic Ca2+ flux in
GABAB receptor-expressing biosensor cells or on total GABA secretion measured
by HPLC” (8). The lack of sensitivity of GABA release to glucose makes it
difficult to understand how it might be synchronized with glucose-induced
insulin secretion. Inhibition of GABA biosynthesis with allylglycine decreased
GABA release and stimulated basal serotonin/insulin secretion that “failed to
display regular secretory pulses” (8). By contrast, blocking islet GABA
metabolism with 10 µ M ϒ-vinyl-GABA
(GABAT inhibitor) increased GABA
release and decreased basal serotonin/insulin secretion at 3 mM glucose (8).
These results are coherent with the assumption that GABA release is governed by
the intracellular content of the ϒ-amino acid. However, these
inhibitors do not mimic the effect of insulin secretagogues that increase GABA
metabolism and consequently decrease islet GABA content and its release as
commented before. Otherwise, as islet GABA content depends on the extracellular
L-glutamine concentration, addition of a physiological (0.5-1.0 mM) or higher
(10 mM) concentration of the α-amino acid would increase islet GABA content,
and therefore its release, resulting in the inhibition of high glucose-induced
insulin secretion against accumulated experimental evidence. In fact, it might
be considered that a physiological L-glutamine concentration should be always
used to study the function of non-cultured islets in in vitro
experiments
. However, it cannot be denied that both
functions of islet GABA, intra- and extra-cellular, might be concordant, mainly
in the inhibitory side of the context.
Importantly, Islets from type 1
and 2 diabetic patients’ donors were shown by immunostainings to be depleted of
GABA in
β
-cells spite the
presence of GAD65 (8). No pulsatile GABA secretion could be observed in type 2
diabetic islets, and it could be rescued by 10
µ
M
ϒ
-vinyl-GABA
(GABAT inhibitor). However, the periodicity of
serotonin/insulin release was diminished (<0.01) by 10 µ M ϒ-vinyl-GABA (8). It might be
interesting to check whether there is any alteration of the gene expression or
the activity of GABA shunt enzymes that might contribute to the derangement of
diabetic islets ‘function.
3. Metabolic changes contributing to Islet degradation in type 2 diabetes
In a recent and exhaustive
publication, the metabolic mechanism(s) that might be implicated in the
degradation of β-cells of type2 diabetic islets due to persistent hyperglycemia
were studied (23). The authors found an elevated expression of most of the
genes codifying glycolytic enzymes and a very strong suppression of
glyceraldehyde phosphate dehydrogenase (GAPDH) activity Hyperglycemia-induced
consequences were prevented by D-mannoheptulose. They checked the hypothesis
that an increase of a glycolytic metabolite between glucokinase and GAPDH
mediates the effects of hyperglycemia trough inhibition of AMPK and activation
of mTORC1 activities, respectively. Most of the consequences of hyperglycemia
could be prevented by an inhibitor of one of the effectors activated by mTORC1,
ribosomal protein S6 kinase (70S6K-Pi), that promotes the translation of mRNAs
exhibiting specific sequences in their 5’-terminus. Besides glycolysis,
hyperglycemia suppressed the expression of some of the genes of the Krebs cycle
enzymes and of the mitochondrial electron chain transporters (23), leading to a
suppression of ATP-linked respiration. However, this down-regulation of
mitochondrial gene expression could not be prevented, like in the case of
glycolysis, by S6K inhibitors. It is unknown whether GAPDH enzymatic activity
may be restored by S6K inhibitors in diabetic islets or INS-1 cells cultured at
25 mM glucose for 48 hours.
Islets and INS-1 cells are
characterized by a “reverse” Pasteur effect when subject to anoxia (under N2):
their glucose utilization rate at high glucose levels is almost completely
depressed instead of being stimulated (24). This “anomalous” feature of β-cells
has been attributed to their low lactic acid dehydrogenase activity that cannot
cope with the re-oxidation of cytosolic NADH at high rates of glycolysis (25).
This function is taken over by the glycerol phosphate shuttle coupled to the
respiratory chain. One might conclude that most of the islet glucose
utilization rate is performed as aerobic glycolysis that depends on the
integrity of mitochondrial respiration. This suggests that a mitochondrial
respiratory defect might be the primary cause of islet degradation in diabetic
islets. This might produce a block of GAPDH and of the reoxidation of glycolytically
produced NADH followed by the upregulation of the gene expression of some
glycolytic genes and the already described consequences (23).
4. Conclusions and perspectives
To our opinion, there is yet no
consensus about the priority of an autoimmune attack against GAD65 versus a
pre-existing metabolic derangement of the β-cells in the development of type-1
diabetes. Studies made in type-1 diabetes patients suggest that “a 2-component
causal model for T1D comprising constitutional metabolic impairments that act
in concert with autoimmunity “might be responsible for the development of the
sickness (26). Moreover, studies in non-inbred BB rats (BB/Hagedorn, a model of
spontaneous autoimmune type 1 diabetes) suggest that “beta cells may have an
inherent sensitivity that possibly makes them susceptible to autoimmune attack”
(27). In contrast, in type-2 diabetic patients, one has identified several risk
factors that might primarily provoke a functional derangement of the β-cells
(26).
In conclusion, many experimental
data support a role for GABA shunt metabolism in the stimulation of insulin
secretion. The fact that GAD65 is one of the most prevalent autoantigens in
type 1 diabetic patients that is immunologically destroyed in the development
of the sickness may indicate that a block of the GABA shunt might be
co-responsible of β-cell degradation. Therefore, any of the enzymes of GABA
shunt might be considered as a risk factor for the triggering of β-cell
malfunction: GAD65, GABAT, SSA-dehydrogenase (NADH-dependent, generating
succinic acid) and, perhaps, SSA-reductase (NADP+-dependent,
producing ϒ-hydroxybutyric acid, GHB). The latter enzyme is, surprisingly,
strongly inhibited (-98%) by 10 mM KIC without affecting its competitor enzyme
(SSA dehydrogenase) for their common substrate SSA (14).
Acknowledgments
The collaboration of all the co-authors of our joint articles was and is greatly appreciated. A particular recognition is due to my grandson, Manny Rodriguez-Tamarit, for incredibly drawing the schemes of the Figures in his mobile.
Abbreviations
Gamma-hydroxybutyric acid (GABA), Semialdehyde succinic acid (SSA), Succinic acid (SUCC), (α-ketoisocaproic acid (KIC), α-keto-β-methyl-valeric acid (KMV), 2-oxoglutarate or α-ketoglutarate (2-OG or α-KG), gamma-hydroxybutiric acid (GHB), 2-OG-dehydogenase (2-OG-dh), SSA-dehydrogenase (SSA-dh), SSA-reductase (SSA-red), GABA-transaminase (GABAT),glutamate dehydrogenase (GDH), 4-methyl valeric acid (MVA).
References
- Ashcroft, F.M.; Rorsman, P. Electrophysiology of the pancreatic β-cell. Prog. Biophys. Mol. Biol. 1989, 54, 87–143. [Google Scholar] [CrossRef] [PubMed]
- Gembal, M.; Gilon, P.; Henquin, J.C. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1992, 89, 1288–1295. [Google Scholar] [CrossRef]
- Gembal, M.; Detimary, P.; Gilon, P.; Gao, Z.Y.; Henquin, J.C. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1993, 91, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Kalwat, M.A. , Cobb M.H., Mechanisms of the amplifying pathway of insulin secretion in the β cell. Pharm. Therap 2017, 179, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Delgado, J.; Deeney, J.T.; Martín-Del-Río, R.; Corkey, B.E.; Tamarit-Rodriguez, J. KCl -Permeabilized Pancreatic Islets: An Experimental Model to Explore the Messenger Role of ATP in the Mechanism of Insulin Secretion. PLOS ONE 2015, 10, e0140096. [Google Scholar] [CrossRef]
- Sörenson, R.L. , Garry D.G., Brelje T.C., Structural and functional considerations of GABA in islets of Langerhans. 1: Diabetes40, 1365. [Google Scholar]
- Hagan, D.W.; Ferreira, S.M.; Santos, G.J.; Phelps, E.A. The role of GABA in islet function. Front. Endocrinol. 2022, 13, 972115. [Google Scholar] [CrossRef]
- Menegaz, D.; Hagan, D.W.; Almaça, J.; Cianciaruso, C.; Rodriguez-Diaz, R.; Molina, J.; Dolan, R.M.; Becker, M.W.; Schwalie, P.C.; Nano, R.; et al. Mechanism and effects of pulsatile GABA secretion from cytosolic pools in the human beta cell. Nat. Metab. 2019, 1, 1110–1126. [Google Scholar] [CrossRef]
- Brice, N.L. , Varadi A., Ashcroft S.J.H., Molnar E., Metabotropic glutamate and GABABreceptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 2002, 45, 242–252. [Google Scholar] [CrossRef]
- Wendt, A.; Birnir, B.; Buschard, K.; Gromada, J.; Salehi, A.; Sewing, S.; Rorsman, P.; Braun, M. Glucose Inhibition of Glucagon Secretion From Rat α-Cells Is Mediated by GABA Released From Neighboring β-Cells. Diabetes 2004, 53, 1038–1045. [Google Scholar] [CrossRef]
- Michalik, M. , Nelson J., Erecinska M., GABA production in rat islets of Langerhans. Diabetes 1993, 42, 1506–1513. [Google Scholar] [CrossRef]
- Smismans, A.; Schuit, F.; Pipeleers, D. Nutrient regulation of gamma-aminobutyric acid release from islet beta cells. Diabetologia 1997, 40, 1411–1415. [Google Scholar] [CrossRef]
- Fernández-Pascual, S. , Mukala-Sengu-Tsshibangu A., Martín-del-Río R., Tamarit-Rodriguez J., Conversion into GABA (ϒ-aminobutyric acid) may reduce the capacity of L-glutamine as an insulin secretagogue. Biochem. J. 2004, 379, 721–729. [Google Scholar] [CrossRef]
- Pizarro-Delgado, J.; Braun, M.; Hernández-Fisac, I.; Martín-Del-Río, R.; Tamarit-Rodriguez, J. Glucose promotion of GABA metabolism contributes to the stimulation of insulin secretion in β-cells. Biochem. J. 2010, 431, 381–390. [Google Scholar] [CrossRef]
- Winnock, F.; Ling, Z.; De Proft, R.; Dejonghe, S.; Schuit, F.; Gorus, F.; Pipeleers, D. Correlation between GABA release from rat islet β-cells and their metabolic state. Am. J. Physiol. Metab. 2002, 282, E937–E942. [Google Scholar] [CrossRef]
- Hernández-Fisac, I.; Fernández-Pascual, S.; Ortsäter, H.; Pizarro-Delgado, J.; Del Río, R.M.; Bergsten, P.; Tamarit-Rodriguez, J. Oxo-4-methylpentanoic acid directs the metabolism of GABA into the Krebs cycle in rat pancreatic islets. Biochem. J. 2006, 400, 81–89. [Google Scholar] [CrossRef]
- Pizarro-Delgado, J.; Hernández-Fisac, I.; Martín-Del-Río, R.; Tamarit-Rodriguez, J. Branched-chain 2-oxoacid transamination increases GABA-shunt metabolism and insulin secretion in isolated islets. Biochem. J. 2009, 419, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jetton, T.L.; Goshorn, S.; Lynch, C.J.; She, P. Transamination Is Required for α-Ketoisocaproate but Not Leucine to Stimulate Insulin Secretion*. J. Biol. Chem. 2010, 285, 33718–33726. [Google Scholar] [CrossRef] [PubMed]
- Sener, A.; Malaisse-Lagae, F.; Malaisse, W.J. Stimulation of pancreatic islet metabolism and insulin release by a nonmetabolizable amino acid. . 1981, 78, 5460–5464. [Google Scholar] [CrossRef]
- Sener, A. , Somers G. , Devis G., Malaisse W.J., The stimulus-secretion coupling of amino acid-induced insulin release. Biosynthetic and secretory responses of rat pancreatic Islet to L-leucine and L-glutamine, Diabetologia 1981, 21, 135–142. [Google Scholar]
- Pizarro-Delgado, J.; Deeney, J.T.; Martín-del-Río, R.; Corkey, B. ; Tamarit-Rodriguez J, Direct stimulation of islet insulin secretion by glycolytic and mitochondrial metabolites in KCl-depolarized islets. Plos One 2014, 11, e0166111. [Google Scholar] [CrossRef]
- Misra, S. Pancreatic autoantibodies: who to test and how to interpret the results. Pr. Diabetes 2017, 34, 221–223a. [Google Scholar] [CrossRef]
- Haythorne, E.; Lloyd, M.; Walsby-Tickle, J.; Tarasov, A.I.; Sandbrink, J.; Portillo, I.; Exposito, R.T.; Sachse, G.; Cyranka, M.; Rohm, M.; et al. Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic β-cells. Nat. Commun. 2022, 13, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hellman, B. , Idahl L.-Å., Sehlin J. Täljedal I.-B., Influence of anoxia on glucose metabolism in pancreatic islets: lack of correlation between fructose-l,6-diphosphate, and apparent glycolytic flux. Diabetologia 1975, 11, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Sekine, N. , Cirulli V., Regazzi R., Brown L.J., Giné E., Tamarit-Rodriguez J., Girotti M., Marie S., MacDonald M.J., Wollheim C.B., Rutter G.A., Low lactate dehydrogenase and mitochondrial glycerol phosphate dehydrogenase in pancreatic β-cells. Potential role in nutrient sensing. J Biol.Chem 1994, 7, 4895–4902. [Google Scholar]
- Evans-Molina, C.; Sims, E.K.; DiMeglio, L.A.; Ismail, H.M.; Steck, A.K.; Palmer, J.P.; Krischer, J.P.; Geyer, S.; Xu, P.; Sosenko, J.M. β Cell dysfunction exists more than 5 years before type 1 diabetes diagnosis. J. Clin. Investig. 2018, 3. [Google Scholar] [CrossRef]
- Medina, A.; Parween, S.; Ullsten, S.; Vishnu, N.; Siu, Y.T.; Quach, M.; Bennet, H.; Balhuizen, A.; Åkesson, L.; Wierup, N.; et al. Early deficits in insulin secretion, beta cell mass and islet blood perfusion precede onset of autoimmune type 1 diabetes in BioBreeding rats. Diabetologia 2017, 61, 896–905. [Google Scholar] [CrossRef]
Figure 1.
Postulated metabolic pathway leading to the stimulation of BCKAs-induced insulin secretion.
Figure 1.
Postulated metabolic pathway leading to the stimulation of BCKAs-induced insulin secretion.
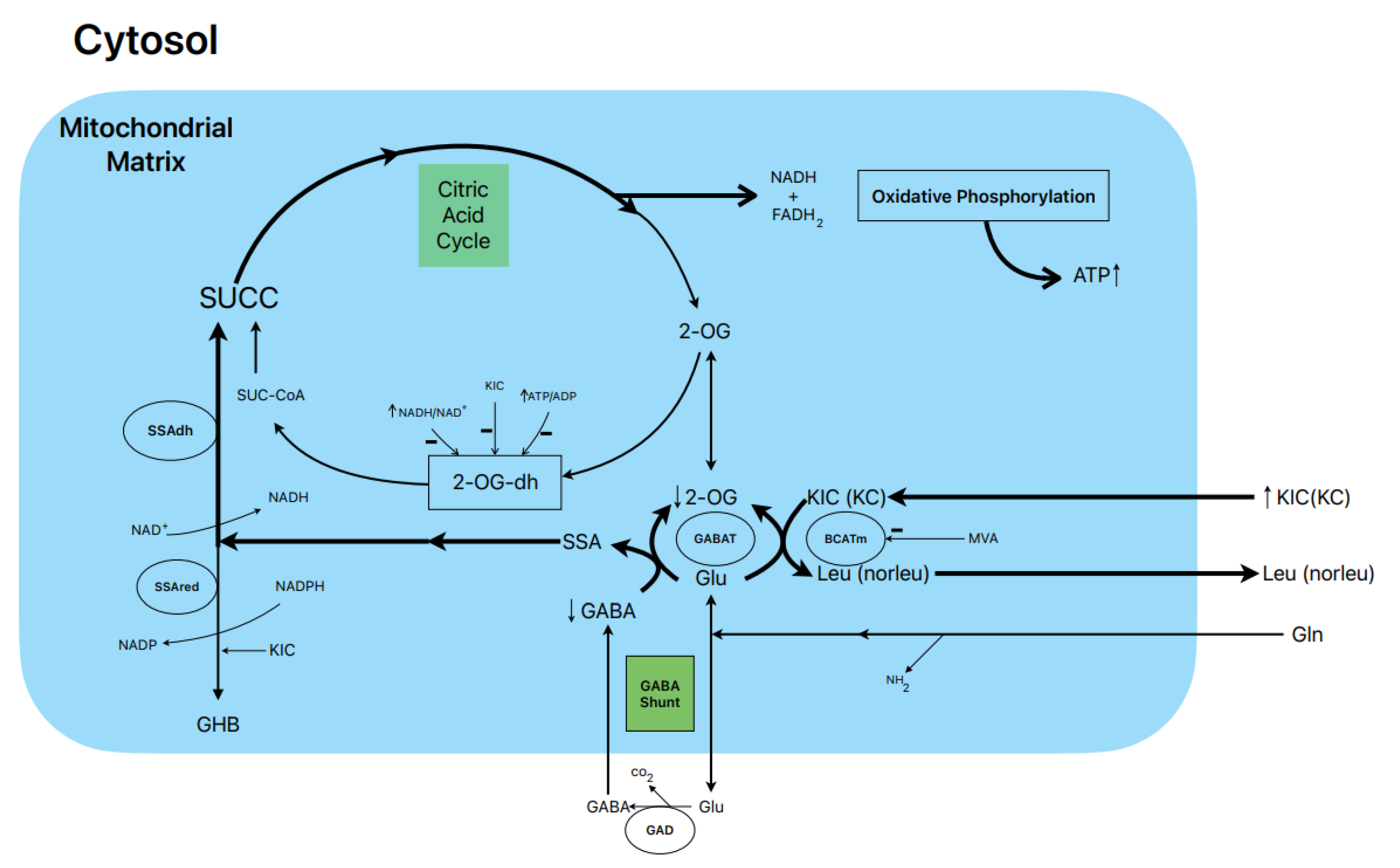
Figure 2.
Postulated metabolic pathway leading to the stimulation of insulin secretion by L-Leucine plus L-glutamine.
Figure 2.
Postulated metabolic pathway leading to the stimulation of insulin secretion by L-Leucine plus L-glutamine.
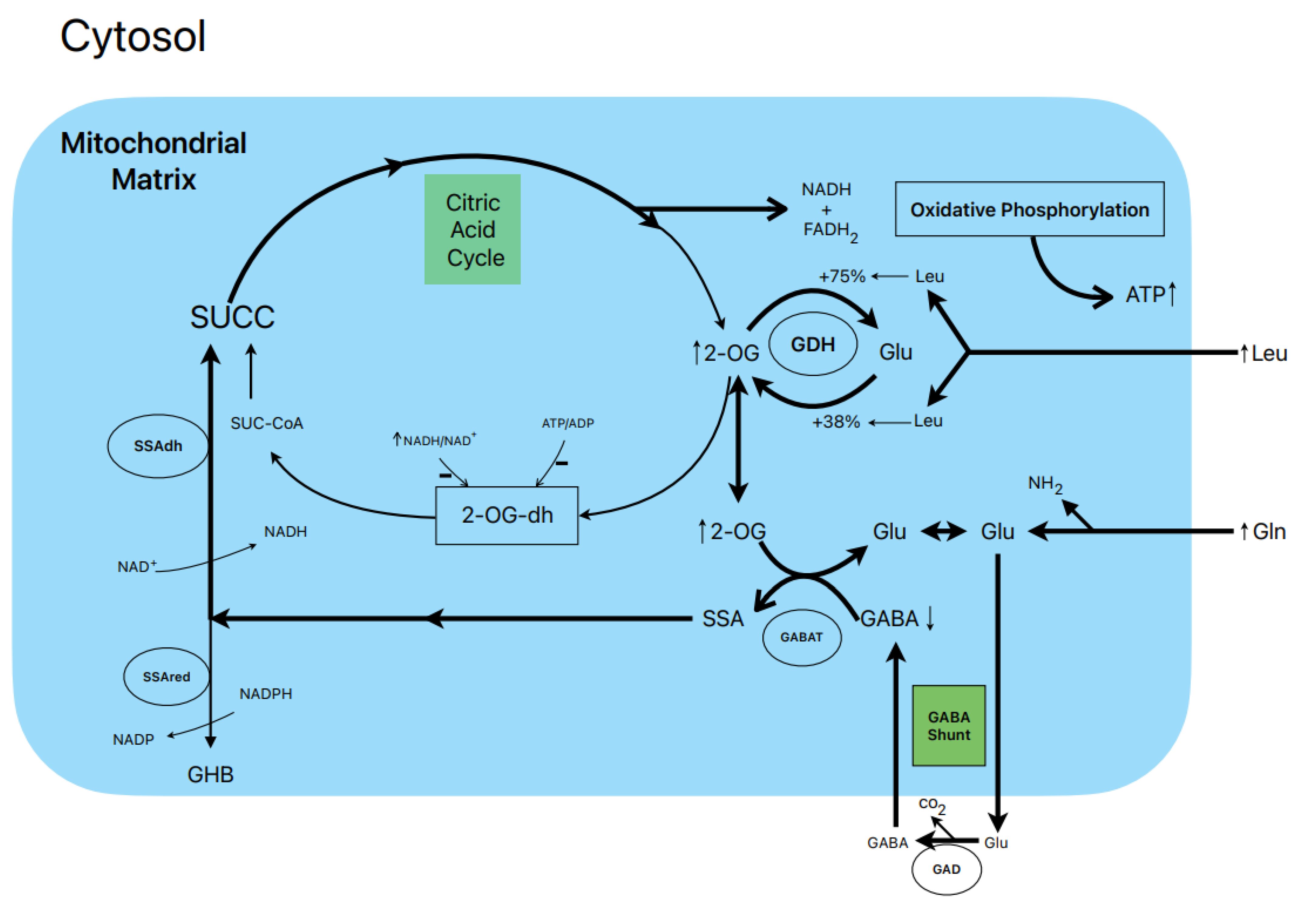
Figure 3.
Postulated metabolic pathway leading to the stimulation of insulin secretion by glucose.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



