
Submitted:
23 April 2023
Posted:
30 April 2023
You are already at the latest version
Abstract
The achievement of reproductive competence in male mammals is dependent on the testis. Goat testis’ development and spermatogenesis involve physiological events with high complexity. In the current work, 6 testes were respectively collected from immature, sexually mature and physically mature Qianbei Ma goats (1, 6 and 12 months old, respectively). RNA-Seq was carried out to reveal changes in testis mRNA expression levels in Qianbei Ma goats at various developmental stages, and gene expression profiling at different ages was established. Totally 18 libraries were established for screening genes and pathways associated with testis development and spermatogenic processes. Totally 9,724 upregulated and 4,153 downregulated genes were identified between immature (I) and sexually mature (S) testes; 7 upregulated and 3 downregulated genes were detected between sexually mature (S) and physically mature (P) testes, and approximately 4% of genes were alternately spliced between the I and S groups. Selected genes were verified by qRT-PCR, in agreement with sequencing data indicating their reliability. Those genes have critical functions in various developmental stages of goat testicular development and spermatogenesis. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were carried out to evaluate differentially expressed genes (DEGs). GO analysis suggested DEGs were involved in “reproduction process”, “channel activity” and “cell periphery part” between I and S, and in “ion transport process”, “channel activity” and “transporter complex part” between S and P. KEGG analysis indicated that pathways such as “glycerolipid metabolism”, “steroid hormone biosynthesis” and “MAPK signaling pathway” may be involved in testis development and spermatogenesis. Genes including IGF1, TGFB1, TGFBR1 and EGFR may regulate the development of the testis from immature to sexually mature, which may be key candidate genes for the development of goat testis. These findings provide novel insights into goat testicular development and spermatogenesis.
Keywords:
Qianbei Ma goat
; Testis development
; RNA-Seq
; mRNA expression
1. Introduction
The testis represents a critical organ of the male reproductive system in mammals, producing spermatozoids and androgens. Spermatogenesis constitutes a developmental event, which produces haploid spermatozoa from diploid spermatogonial stem cells during meiosis in the testis [1]. Spermatogenesis comprises three steps, including (1) spermatogonial proliferation and differentiation, (2) meiotic division of spermatocytes and (3) spermatozoid maturation. Spermatogonial stem cell differentiation produces spermatogonia, which after DNA replication produce primary spermatocytes. Next, secondary spermatocytes/spermatids are generated after DNA replication and meiosis from primary spermatocytes. Haploid sperm cells after four or more morphological alterations give rise to sperm cells, including chromatin condensation, acrosome generation, flagella formation and cytoplasmic decrease [2]. Besides spermatogenic cells, spermatogenic mechanisms involve many somatic cells in the testicle, including Sertoli and Leydig cells. In the testis of mammals, spermatogenesis is carried out in seminiferous tubules, where germ cells are associated with Sertoli cells. Associated genes in the latter cells have critical functions in precise steps of spermatogenesis [3].
RNA sequencing (RNA-Seq) allows an expression profiling of genes and may help map and quantify the transcriptome [4,5]. This approach offers many advantages compared to other transcriptomic tools, including high resolution/sensitivity, a broad dynamic range of gene expression, and the identification of new transcript sequences and splice isoforms of previously reported genes [6]. Ramsköld [7] evaluated multiple tissues from mammals by RNA-Seq and reported most genes are specifically expressed in testicular samples. Additionally, considering RNA-Seq-based expression patterns, Djureinovic [8] categorized 20,050 putative human genes, which showed specific expression in the human testicle, whereas 26 additional tissue types were present in 7 people. Their evaluation revealed the testicular tissue had by far the largest quantity of tissue-specific genes. Using microarray analysis, Anand detected differentially expressed genes (DEGs) in testicular samples compared to other tissues, identifying 2868 upregulated transcripts and 2011 downregulated mRNAs [9]. The testicle appears to have a higher degree of metabolic activity relative to other normal tissues. Most current reports assessing the association of testicular development with spermatogenesis have been performed in the human or mouse species, and the goat is scarcely examined.
The Qianbei Ma goat is a fine goat breed in Guizhou Province, which adapts to harsh climatic conditions and prolonged breeding. Normal testicular development and sperm formation is very critical for ensuring high-level semen production and perpetuating species continuity, and genes highly contributing to various steps of testicular development and spermatogenesis likely have functions in fertility. However, gene expression in testicular development and sperm formation in goats is mostly undefined, which deserves further investigation. The present work aimed to perform transcriptome profiling of immature and mature Qianbei Ma goat testis specimens by RNA-Seq and bioinformatic assessment. The findings provide novel insights into the mechanisms regulating goat testicular development and sperm formation.
2. Materials and Methods
2.1. Ethics statement.
This study had approval from the Animal Ethics Committee of Guizhou University (Guiyang, China) (No. EAE-GZU-2021-P024, Guiyang, China; March 30, 2021).
2.2. Animal handling and sample collection.
Permission was granted to geld 18 healthy Qianbei Ma goats in Fuxing Husbandry Co., Ltd., Zunyi, Guizhou, China. Goat ages were obtained from goat farming records. There were 6 immature goats (1 month old, before sexual maturation, i.e., samples I1, I2, I3, I4, I5 and I6), 6 sexually mature goats (6 months old, after sexual maturation but before physical maturation, i.e., samples S1, S2, S3, S4, S5 and S6) and 6 physically mature goats (12 months old, after physical maturation, i.e., samples P1, P2, P3, P4, P5 and P6). We surgically collected the right testes from the 18 goats by castration after anesthesia, followed by storage in RNA/DNA sample protector (Servicebio, Wuhan, China). The testis from each goat was cut longitudinally, and a small amount (3-5 g) of the parenchyma, including seminiferous tubules and Leydig cells, underwent snap freezing in liquid nitrogen and was taken back to the lab for further studies. All castrated goats remained in Fuxing Husbandry Co., Ltd. (Guizhou, China) after our study, for fatten feeding.
2.3. RNA quantitation and quality
Total RNA extraction was carried out from the testicular tissue in groups I, S and P using TRIzol reagent (Servicebio, Wuhan, China) and RNeasy RNA purification kit (Servicebio, Wuhan, China) with DNase as directed by the manufacturer. A NanoDrop™ One spectrophotometer (Thermo Fisher Scientific, USA) was utilized to assess RNA purity and amounts. RNA quality assessment utilized 1% agarose gel electrophoresis. High-quality RNA samples (OD 260/280 of 1.8-2.0, integrity>7.0 and 28S:18S above 1.0) were sequenced on an Illumina NovaSeq 6000 system, generating 150-bp paired end reads.
2.4. Transcriptome sequencing.
Totally 18 libraries were generated. The 6 obtained from immature testis samples were termed I1, I2, I3, I4, I5 and I6; the 6 from sexually mature specimens were S1, S2, S3, S4, S5 and S6, and the 6 from physically mature samples were P1, P2, P3, P4, P5 and P6. Approximately 5 μg RNA/sample constituted the input material for RNA sample preparation. Index-coded samples were clustered with NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® according to a protocol provided by the manufacturer. Upon clustering, the prepared libraries underwent sequencing on an Illumina NovaSeq 6000(Illumina, USA). The image data of the sequences yielded by the high-throughput sequencor underwent conversion into sequence data (reads) by CASA V A base recognition to obtain fastq files. Raw RNA-Seq fastq data next underwent filtration with Fastp v to exclude adapter-containing, N-containing and low-quality (quality score below 20) reads, resulting in clean reads, which were mapped to the goat (Capra hircus) (ARS1.2) reference genome [10] using HISAT2.
2.5. Quantification of gene expression.
2.6. Differential expression analysis.
The DESeq2 software was used to analyze differential expression between treatment and control groups. The Benjamini-Hochberg algorithm was utilized for adjusting p values (p-adj) to control for false discovery rate. |log2 (FoldChange)| ≥ 1 & padj<0.05 was set as the significance threshold for differential expression [13].
2.7. GO and KEGG enrichment analyses of DEGs.
GO and KEGG analyses of DEGs were implemented with ClusterProfiler, correcting for gene length bias. KEGG is an information database based on molecular findings, particularly via genome sequencing and additional high-throughput techniques to produce large-scale molecular data sets, allowing a deep understanding of biological systems (http://www.genome.jp/kegg/) [14]. GO and KEGG terms with |log2 (FoldChange)| ≥1 and padj<0.05 were deemed to be DEGs with significant enrichment [15].
2.8. Prediction of new transcripts and alternative splicing analysis.
StringTie was utilized to build and identify previously reported and new transcripts from HISAT2 alignment data. StringTie utilizes a network-flow algorithm with optional de novo assembly to splice transcripts. Compared with cufflinks, StringTie has the following advantages: it (1) yields more complete transcripts; (2) assembles more accurate transcripts, (3) better estimates the transcript’s expression level and (4) has greater splicing speed [16,17]. rMATS (http://rnaseqmats.sourceforge.net/index.html) was utilized to classify AS events, which were assessed in various samples separately.
2.9. qRT-PCR for RNA-Seq data validation.
TGFBR1, TGFB1, EGFR, IGF1, MAPK3 and SMAD4 were examined by qRT-PCR to validate RNA-Seq findings. Total RNA (1000 ng) was utilized to produce complementary DNA (cDNA) with 2×SYBR Green qPCR Master Mix None ROX (Servicebio, Wuhan, China) at 25°C (5 min), 42°C (30 min) and 85°C (5 s). The primers used for qRT-PCR are shown in Table 4. A CFX96 Real-Time PCR system (Bio-Rad, USA) was utilized for amplification in 15-µL reactions containing 2×qPCR Mix (7.5 µL), forward and reverse primers (10 pmol/µL, 0.75 µL each), cDNA (1000 ng/µL, 2 µL), and Nuclease-Free water (4 µL). The reaction conditions were: 1 cycle at 95°C (30 s), followed by 40 cycles at 95°C (15 s), 60°C (30 s), with fluorescence signals collected every 0.5°C increase from 65°C to 95°C. Melting curves were utilized to assess primer specificity. Assays were carried out in triplicate, and GAPDH was utilized for normalization in data analysis by the 2−∆∆Ct method.
3. Results
3.1. Gene expression profiling during testicular development in Qianbei Ma goats.
To determine genes associated with testicular development and sperm formation, 18 libraries, including 6 each from immature (I1, I2, I3, I4, I5 and I6), sexually mature (S1, S2, S3, S4, S5 and S6) and physically mature (P1, P2, P3, P4, P5, P6) testes, underwent sequencing. Table 1 shows an overview of the information pertaining to raw and clean reads for all libraries. Error rates and GC contents for various libraries were determined for their quality control (Table 2). All 18 libraries had a high quality.
Clean reads underwent alignment to the goat reference genome ARS1.2 that is commonly utilized for goats [10], with HISAT2. Table 3 shows an overview of the information about uniquely mapped clean reads.
Additionally, an overview of the percentages of clean reads mapped to exon borders (junction reads) is shown in Table 4. The RNA-Seq data have been submitted to NCBI (accession number BioProject: PRJNA879963).

Table 4.
Statistics of reads alignment to genomic regions.
| Sample name | Exonic region | Intronic region | Intergenic region |
|---|---|---|---|
| I1 | 4466151533 (79.48%) | 611822545 (10.89%) | 541085383 (9.63%) |
| I2 | 4779622398 (78.65%) | 761383464 (12.53%) | 535946498 (8.82%) |
| I3 | 4432093997 (80.66%) | 570346947 (10.38%) | 492580965 (8.96%) |
| I4 | 4614052978 (78.53%) | 738532433 (12.57%) | 523236881 (8.90%) |
| I5 | 5160159063 (81.96%) | 605739527 (9.62%) | 530252297 (8.42%) |
| I6 | 4298336210 (78.73%) | 599791632 (10.99%) | 561800986 (10.29%) |
| S1 | 5004805293 (79.49%) | 606545683 (9.63%) | 684973145 (10.88%) |
| S2 | 4891821345 (81.09%) | 533206687 (8.84%) | 607543338 (10.07%) |
| S3 | 5173907638 (80.30%) | 600212860 (9.32%) | 668983341 (10.38%) |
| S4 | 4579030919 (79.56%) | 552189052 (9.59%) | 623968138 (10.84%) |
| S5 | 4488498885 (78.27%) | 613561446 (10.70%) | 632649316 (11.03%) |
| S6 | 4595667229 (79.73%) | 543550434 (9.43%) | 624459696 (10.83%) |
| P1 | 5058268703 (79.52%) | 636322977 (10.00%) | 666220762 (10.47%) |
| P2 | 4921081484 (78.42%) | 644761157 (10.27%) | 709616381 (11.31%) |
| P3 | 4871901450 (79.39%) | 593069117 (9.66%) | 671905236 (10.95%) |
| P4 | 4918914052 (80.00%) | 601562419 (9.78%) | 628332353 (10.22%) |
| P5 | 4886718972 (80.63%) | 554456620 (9.15%) | 619529425 (10.22%) |
| P6 | 4658554613 (80.75%) | 537138992 (9.31%) | 573327628 (9.94%) |
Exonic, intronic and intergenic regions: numbers (percentages) of reads with alignment to the exonic, intronic and intergenic regions, respectively.
3.2. Alternative splicing data.
Alternative splicing (AS) represents a commonly encountered phenomenon in eukaryotes, which could lead to the production of various protein forms at different times under different circumstances, increasing species/body fitness. Although AS research is a known subfield of molecular biology, only in recent years has this subfield attracted sufficient attention. AS is critical for the complex proteomes and functions found in higher organisms. In this work, AS events were categorized into 5 types with rMATS (http://rnaseq-mats.sourceforge.net/index.html). By determining the types and amounts of AS events, and analyzing each AS type, a large number of AS events were found in testicular development and sperm formation. In the present work, we detected 13,877 differential genes between immature (I) and sexually mature (S) testes, of which 544 underwent AS events, and 10 differential genes were detected between sexually mature (S) and physically mature (P) testes, with no detected AS events (padj≤0.05). Therefore, approximately 4% of genes showed AS between I and S, and no gene had AS between S and P in this study. The above findings indicated AS was very important in the complexity of gene expression during testis development, especially in the period from immaturity to sexual maturity.
To determine AS types associated with testicular genes, AS events were compared between the S and I groups. The five known types of AS events include retained intron (RI), mutually exclusive exon (MXE), alternative 3′ splice site (A3SS), alternative 5′ splice site (A5SS) and skipped exon (SE). All five types of AS were found in the S vs I group comparison. RI, MXE, A3SS and SE were found in the P vs S group comparison. These findings showed SE as the commonest AS event amounting to 368 in S vs I. Other identified AS events were RI (48), MXE (110), A3SS (66), and A5SS (54) (Figure 1).
3.3. Analysis of DEGs.
DEGs were determined with |log2 (FoldChange)| ≥1 and padj<0.05. As a result, 9,724 upregulated and 4,153 downregulated genes were detected between the I and S groups, and 7 upregulated and 3 downregulated genes were found between the S and P groups (Figure 2A1, Figure 2A2). Venn diagrams showed the I and S libraries had 13451 genes in common, the S and P libraries had 17,448 genes in common (Figure 2B). Figure 2C depicts hierarchical clustering, with DEGs for the 18 libraries grouped into three clusters. The above data suggested the I, S and P libraries had differential expression patterns overall, but similar repetitive expression commonalities across developmental stages.
3.4. GO analysis of DEGs.
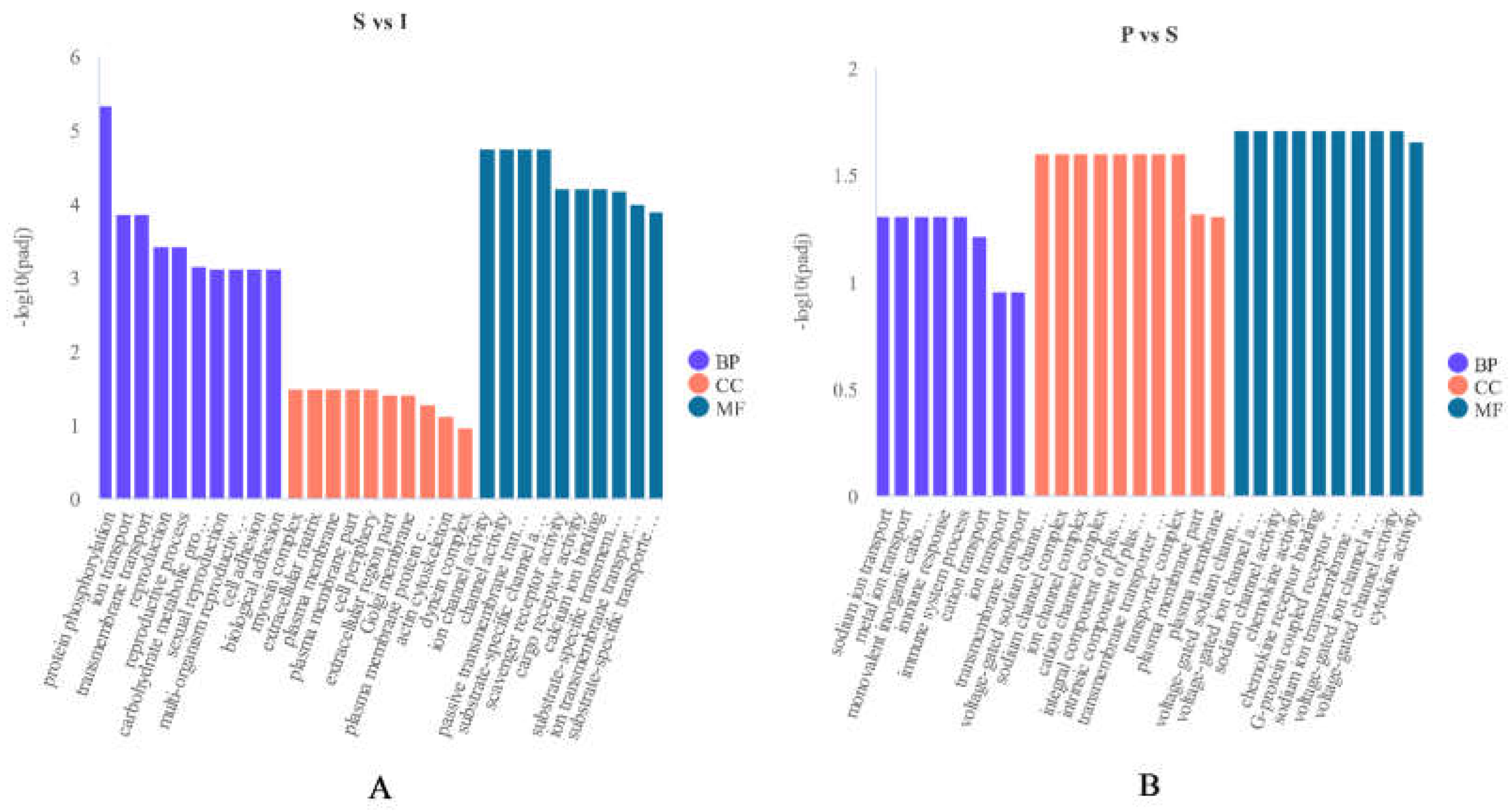
GO analysis was carried out to examine the functions of DEGs in testicular development. Totally 106 GO terms associated with “biological processes”, “molecular functions” and “cellular components” were markedly enriched between immature (I) and sexually mature (S) testes. Approximately 43 GO terms belonged to “biological process”, and mostly involved “protein phosphorylation process”, “transport process” and “reproduction process”. Approximately 56 GO terms belonged to “molecular functions”, and mostly involved “channel activity”, “transporter activity” and “receptor activity”. Approximately 7 GO terms belonged to “cellular components”, and mostly involved “cell periphery part”, “plasma membrane part” and “myosin complex part”. Totally 36 GO terms associated with “biological processes”, “molecular functions” and “cellular components” had significant enrichment in the sexually mature (S) vs physically mature (P) DEG groups. About 5 GO terms belonged to “biological processes”, and mostly involved “ion transport process” and “immune system process”. Approximately 21 GO terms belonged to “molecular functions”, and mostly involved “channel activity”, “chemokine activity” and “cytokine activity”. About 10 GO terms belonged to “cellular components”, and mainly involved “transporter complex part”, “channel complex part” and “component of plasma membrane part”.
Figure 3.
(A): Most enriched GO terms between immature (I) and sexually mature (S) testes. (B): Most enriched GO terms between sexually mature (S) and physically mature (P) testes. Abscissas and ordinates represent enriched GO terms and significance levels of GO enrichment, respectively. Purple, “biological processes”; red, “cellular components”; blue, “molecular functions”.
Figure 3.
(A): Most enriched GO terms between immature (I) and sexually mature (S) testes. (B): Most enriched GO terms between sexually mature (S) and physically mature (P) testes. Abscissas and ordinates represent enriched GO terms and significance levels of GO enrichment, respectively. Purple, “biological processes”; red, “cellular components”; blue, “molecular functions”.
3.5. KEGG pathway analysis of DEGs.
KEGG pathway analysis of DEGs (|log2 (FoldChange)| ≥1 and padj<0.05) was carried out. For I vs S, 8 KEGG pathways were upregulated, including “glycerolipid metabolism”, “protein digestion and absorption” and “steroid hormone biosynthesis”. Totally 90 pathways were downregulated, including “lysosome”, “MAPK signaling pathway” and “chemokine signaling pathway” (Figure 4A). For S vs P, 7 KEGG pathways were upregulated, including “oxidative phosphorylation”, “thermogenesis” and “steroid biosynthesis”. However, no pathways were significantly downregulated (Figure 4B).
3.6. qRT-PCR validation of DEGs.
To verify the DEGs in immature (I), sexually mature (S) and physically mature (P) testes, we selected 6 DEGs, including TGFBR1, TGFB1, EGFR, IGF1, MAPK3 and SMAD4, to validate RNA-Seq data by qRT-PCR. qRT-PCR data corroborated RNA-Seq findings, suggesting the reliability of RNA-Seq data (Figure 5).
4. Discussion
With the current development of detection technology, more and more mRNAs specific to sperm have been reported. RNA-Seq has emerged as a tool for efficiently and inexpensively detecting new transcripts and genes. RNA-Seq methods have been broadly utilized to determine DEGs or gene expression patterns, new transcripts, AS events and SNPS, and have empowered studies examining porcine [18,19], cattle [20,21] and mouse [22,23] testicular development. In goats, the profiles of ovarian [24,25], uterine [26,27] and testicular [28,29] tissues under different conditions were recently compared by RNA-Seq. However, limited data on testicular development in goats are available. Breed and age represent major factors affecting testicular development. Here RNA-Seq was performed to build a complete dataset that details the spatiotemporal transcriptome of the testicular tissue in Qianbei Ma goats. Testicular growth and development constitute the key factors affecting goat reproduction. Therefore, identifying genes regulating testicular growth and development is critical. In this study, 13,887 genes were assessed by RNA-Seq in 6 immature, 6 sexually mature and 6 physically mature testes. Totally 9,724 genes were upregulated and 4,153 were downregulated between immature and sexually mature testes; 7 genes were upregulated and 3 were downregulated between sexually mature and physically mature testes. Using next-generation platforms, we determined most upregulated genes were associated with protein coding and may have functions in testicular development and sperm formation.
AS is an important mechanism in the regulation gene expression and promotes proteome diversity [30]. It is estimated about 95% of human multiple-exon gene expression is associated with AS events [31]. In metazoans, AS is critical for the production of various protein forms with functions in different cell events such as cell growth, differentiation and death [32]. Here, 5 AS events were observed, mostly involving ES. The effects of AS events on the functions of related genes can be predicted by a comprehensive analysis of AS events, and GO and KEGG analysis data [33,34]. In the current study, Sec insertion sequence binding protein 2 (SECISBP2) was the gene with the highest number of SE events, i.e., a total of 10 SE events. Mutation of SECISBP2 alters thyroid hormone metabolism [35,36]. Thyroid hormones can modulate semen quality under physiological conditions by regulating testosterone and changing some semen indexes [37-39].
Combining previous relevant reports and KEGG and GO data in the current study, the genes involved in the regulation of testis development and sperm formation through protein phosphorylation were mainly TGFB1, EGFR and IGF1, which have critical functions in testis growth, hormone secretion, spermatogenesis and Leydig cell differentiation.
Transforming growth factor beta-1 (TGFB1) plays multiple biological roles, including the control of proliferative and differentiation potentials of cells [40]. TGFB1 regulates tight junctions in Sertoli cells and controls spermatogenesis. It modifies the blood-testicular barrier (BTB) by downregulating tight junction proteins [41]. TGFB1 may play an important role in testicular development because of its high expression in the immature testis and markedly reduced expression in sexual maturity, as spermatogenesis begins. A comparable expression pattern was found for TGFB receptor type 1 (TGFBR1) [42]. In addition, loss of TGFB1 resulted in lower testosterone levels in the testis and serum, and decreased the ability to mate with females [43]. Based on previous studies, we speculated that TGFB1 may not only directly regulate goat testicular development and sperm formation, but also ensure the normal development of male external genitalia and affect fertility.
Epidermal growth factor receptor (EGFR) represents a receptor gene for EGF and controls testicular function in the human, mouse, rat and livestock species as well as in alpacas [44]. EGF and EGFR are critical paracrine and/or autocrine modulators of testicular development and sperm formation, and regulate testosterone production by testicular interstitial cells [45,46]. We speculated that EGF and EGFR may also be expressed in various goat testicular cells, and can stimulate testosterone secretion, and regulate testis development and spermatogenesis.
Insulin-like growth factor I (IGF1) contributes to the regulation of testicular function [47]. Pitetti indicated growth factors of the insulin family play essential roles by controlling SC number, testis size and daily sperm production [48,49]. Both IGF1 and its receptor IGF1R are expressed in testicles, and their hormones act directly on male gonads [50,51]. In immature testes, IGF1 promotes the development of sustentacular cells, Leydig cells, and gonocytes. In mature testis, the IGF1 gene induces spermatogenesis and regulates Leydig cell function [52,53]. IGF1 may act as an autocrine/paracrine or endocrine signal to regulate testicular steroid production as well as germ cell and Sertoli cell functions [54]. IGF1 plays different roles in testicular function at different stages of testicular development [47,55]. We speculated that high IGF1 and IGF1R protein amounts in the immature testis may suggest they highly promote the development and differentiation of sustentacular cells, Leydig cells and gonocytes in goat testis during sexual maturity.
Transcriptome data revealed the MAPK pathway is implicated in goat testicular development, while TGFBR1, TGFB1, EGFR and IGF1 were enriched in this pathway and downregulated during sexual maturation, as key genes that regulate testis development and spermatogenesis [56,57]. Multiple reports suggest MAPK signaling is a critical regulator of testis growth and development, testis cell proliferation, differentiation and apoptosis, testosterone secretion, thus affecting male fertility [58-60]. One of the key downstream target genes of MAPK signaling is IGF1, which together with other genes in the pathway, controls testis cell proliferation, testis volume development, hormone secretion and spermatogenesis, and is often reported to be associated with male fertility [48,61,62]. We speculate that MAPK signaling is a critical regulatory pathway in goat testis development and spermatogenesis. As an essential male fecundity-related gene in the MAPK signaling pathway, IGF1 modulates goat testis growth and development and can affect the functions of various cells of goat testis by regulating other downstream genes in the signaling pathway. During male goat sexual maturation, IGF1 regulates the development of testis, spermatogenesis and hormone synthesis through the MAPK signaling pathway and other cooperative genes.
5. Conclusions
This study firstly used RNA-Seq to profile the expression of genes during testicular development in Qianbei Ma goats. We identified 544 genes with AS events between I and S, which suggests that AS of differential genes might be critical in the regulation of testicular development in goats. Totally 8 KEGG pathways were upregulated and 90 were downregulated between I and S. Totally 7 KEGG pathways were upregulated between S and P. Among the 6 screened DEGs (TGFBR1, TGFB1, EGFR, IGF1, MAPK3 and SMAD4) TGFBR1, TGFB1, EGFR, IGF1 and MAPK3 belonged to the "MAPK signaling pathway", corresponding to the GO term "protein phosphorylation". The findings should further our understanding on gene regulation during testicular development and sperm formation.
Author Contributions
Methodology, Y.Z. and X.C.; software, W.T.; investigation, Y.R. and T.J.; resources, X.C.; validation, data curation, X.C.; writing—manuscript drafting, Y.Z.; writing—manuscript revision, X.T., W.G. and K.F.; All authors have read and agreed to the published version of the manuscript.
Funding
The current study was supported by the National Natural Science Foundation of China (No. 32260835), the Science and Technology Project of Guizhou Province (Oian Kehe Foundation-ZK [(2021)] Genera 151), and the Guizhou High Level Innovative Talents Project (Oian Kehe Platform Talents [(2022)] 021-1).
Institutional Review Board Statement
The animals used in our study were consented by their owners. All procedures involving animals were approved and authorized by Guizhou University. The laboratory animal castration protocol for this study was approved by the Laboratory Animal Ethics of Guizhou University (No. EAE-GZU-2021-P024, Guiyang, China; 30 March 2021).
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data from sequencing are available at NCBI under BioProject ID: PRJNA879963.
Acknowledgments
The authors are thankful to Fuxing Husbandry Co., Ltd. (Guizhou, China) for providing the experimental Qianbei Ma goats.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Hecht, N.B. Molecular mechanisms of male germ cell differentiation. BioEssays. 1998, 20, 555–561. [Google Scholar] [CrossRef]
- Eddy, E.M.; O'Brien, D.A. Gene expression during mammalian meiosis. Current Topics in Developmental Biology. 1998, 37, 141–200. [Google Scholar] [PubMed]
- Hess, R.A.; De Franca, L.R. Spermatogenesis and cycle of the seminiferous epithelium. Advances in Experimental Medicine and Biology. 2008, 636, 1–15. [Google Scholar] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Research. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews. Genetics. 2010, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ramsköld, D.; Wang, E.T.; Burge, C.B.; Sandberg, R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Computational Biology. 2009, 5, e1000598. [Google Scholar] [CrossRef] [PubMed]
- Djureinovic, D.; Fagerberg, L.; Hallstrom, B.; Danielsson, A.; Lindskog, C.; Uhlen, M.; Ponten, F. The human testis-specific proteome defined by transcriptomics and antibody-based profiling. Molecular Human Reproduction. 2014, 20, 476–488. [Google Scholar] [CrossRef]
- Anand, M.; Prasad, B.V. The computational analysis of human testis transcriptome reveals closer ties to pluripotency. Journal of Human Reproductive Sciences. 2012, 5, 266–273. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Erratum: Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology. 2016, 34, 888. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M. J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biology. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; Yamanishi, Y. KEGG for linking genomes to life and the environment. Nucleic Acids Research. 2008, 36, D480–484. [Google Scholar] [CrossRef]
- Fu, K.; Chen, X.; Guo, W.; Zhou, Z.; Zhang, Y.; Ji, T.; Yang, P.; Tian, X.; Wang, W.; Zou, Y. Effects of N Acetylcysteine on the Expression of Genes Associated with Reproductive Performance in the Goat Uterus during Early Gestation. Animals. 2022, 12, 2431. [Google Scholar] [CrossRef] [PubMed]
- Shumate, Alaina. Improved Transcriptome Assembly Using a Hybrid of Long and Short Reads with StringTie. PLoS Computational Biology. 2022, 18, 1–19. [Google Scholar]
- Pertea, Mihaela. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-seq Reads. Nature Biotechnology. 2015, 33, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhang, L.; Xu, D.; Ding, H.; Zheng, S.; Liu, M. MeDIP-seq and RNA-seq Analysis during Porcine Testis Development Reveals Functional DMR at the Promoter of LDHC. Genomics. 2022, 114, 110467. [Google Scholar] [CrossRef]
- Zhang, L.; Li, F.; Lei, P.; Guo, M.; Liu, R.; Wang, L.; Yu, T.; Lv, Y.; Zhang, T.; Zeng, W.; Lu, H.; Zheng, Y. Single-cell RNA-sequencing Reveals the Dynamic Process and Novel Markers in Porcine Spermatogenesis. Journal of Animal Science and Biotechnology. 2021, 12, 122. [Google Scholar] [CrossRef]
- Chang, T.; Yang, Y.; Retzel, E.F.; Liu, W. Male-specific Region of the Bovine Y Chromosome Is Gene Rich with a High Transcriptomic Activity in Testis Development. Proceedings of the National Academy of Sciences – PNAS. 2013, 110, 12373–12378. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, M.; Fan, Y.; Li, S.; Lai, Z.; Huang, Y.; Lan, X.; Lei, C.; Chen, H.; Dang, R. Identification and Characterization of Circular RNAs in Qinchuan Cattle Testis. Royal Society Open Science. 2018, 5, 180413. [Google Scholar] [CrossRef]
- Fu, Y.; Wu, P.; Beane, T.; Zamore, P.D.; Weng, Z. Elimination of PCR Duplicates in RNA-seq and Small RNA-seq Using Unique Molecular Identifiers. BMC Genomics. 2018, 19, 531. [Google Scholar] [CrossRef]
- Green, C.D.; Ma, Q.; Manske, G.L.; Shami, A.N.; Zheng, X.; Marini, S.; Moritz, L.; Sultan, C.; Gurczynski, S.J.; Moore, B.B.; Tallquist, M.D.; Li, J.Z.; Hammoud, S.S. A Comprehensive Roadmap of Murine Spermatogenesis Defined by Single-Cell RNA-Seq. Developmental Cell. 2018, 46, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Lu, T.; Zhao, Z.; Liu, G.; Lian, Z.; Guo, Y.; Sun, B.; Liu, D.; Li, Y. Comprehensive Analysis of MRNAs and MiRNAs in the Ovarian Follicles of Uniparous and Multiple Goats at Estrus Phase. BMC Genomics. 2020, 21, 267. [Google Scholar] [CrossRef] [PubMed]
- Zi, X.; Lu, J.; Zhou, H.; Ma, L.; Xia, W.; Xiong, X.; Lan, D.; Wu, X. Comparative Analysis of Ovarian Transcriptomes between Prolific and Non-prolific Goat Breeds via High-throughput Sequencing. Reproduction in Domestic Animals. 2018, 53, 344–351. [Google Scholar] [CrossRef]
- Hong, L.; Hu, Q.; Zang, X.; Xie, Y.; Zhou, C.; Zou, X.; Li, Y.; Deng, M.; Guo, Y.; Liu, G.; Liu, D. Analysis and Screening of Reproductive Long Non-coding RNAs Through Genome-Wide Analyses of Goat Endometrium During the Pre-attachment Phase. Frontiers in Genetics. 2020, 11, 568017. [Google Scholar] [CrossRef]
- Liu, H.; Wang, C.; Li, Z.; Shang, C.; Zhang, X.; Zhang, R.; Wang, A.; Jin, Y.; Lin, P. Transcriptomic Analysis of STAT1/3 in the Goat Endometrium During Embryo Implantation. Frontiers in Veterinary Science. 2021, 8, 757759. [Google Scholar] [CrossRef] [PubMed]
- Bo, D.; Jiang, X.; Liu, G.; Hu, R.; Chong, Y. RNA-Seq Implies Divergent Regulation Patterns of LincRNA on Spermatogenesis and Testis Growth in Goats. Animals. 2021, 11, 625. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, P.; Yang, H.; Wang, Z.; El-Samahy, M.A.; Wang, F.; Zhang, Y. Dietary Supplementation with Metformin Improves Testis Function and Semen Quality and Increases Antioxidants and Autophagy Capacity in Goats. Theriogenology. 2022, 188, 79–89. [Google Scholar] [CrossRef]
- Shen, F.; Geng, Y.; Zhang, L.; Luo, L.; Yan, G.; Hou, R.; Yue, B.; Zhang, X. Transcriptome Analysis Reveals the Alternative Splicing Changes in the Immune-Related Genes of the Giant Panda (Ailuropoda melanoleuca), in Response to the Canine Distemper Vaccine. Zoological Science. 2022, 39, 275. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Hagiwara, M.; Kuroyanagi, H. 2SE01 Regulatory Mechanisms of Alternative Splicing in Metazoans. Seibutsu Butsuri. 2005, 45, S23. [Google Scholar] [CrossRef]
- Liu, X.; He, P.; Zhang, Z.; Gong, P.; Niu, Y.; Bao, Zhen. ; Yang, Y.; Gan, L.; Muhuyati. Candidate Genes and Their Alternative Splicing May Be Potential Biomarkers of Acute Myocardial Infarction: A Study of Mouse Model. BMC Cardiovascular Disorders. 2022, 22, 1–505. [Google Scholar] [CrossRef]
- Zhu, Z.; Ni, S.; Zhang, J.; Yuan, Y.; Bai, Yun. ; Yin, X.; Zhu, Z. Genome-wide Analysis of Dysregulated RNA-binding Proteins and Alternative Splicing Genes in Keloid. Frontiers in Genetics. 2023, 14, 1118999. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L.; Liao, X.; Majed, F.A.; Abdullah, M.S.Y.; Refetoff, S.; Boran, G.; Dumitrescu, A.M.; Lado-Abeal, J.; Moeller, L.C.; Weiss, R.E. Mutations in SECISBP2 Result in Abnormal Thyroid Hormone Metabolism. Nature Genetics. 2005, 37, 1247–1252. [Google Scholar]
- Schoenmakers, E.; Chatterjee, K. Identification of Genetic Disorders Causing Disruption of Selenoprotein Biosynthesis. Methods in Molecular Biology. 2018, 1661, 325–335. [Google Scholar] [PubMed]
- Mohammadabadi, T.; Tabatbaei, S.; Ghezi, Z.; Swelum, A.A. Effect of Dietary Palm Kernel on Semen Quality, Reproductive and Thyroid Hormones and Blood Chemistry Parameters of Arabi Rams. Animal Nutrition and Feed Technology. 2020, 20, 93. [Google Scholar] [CrossRef]
- Mogheiseh, A.; Vara, N.; Ayaseh, M.; Jalali, P. Effects of Cabergoline on Thyroid Hormones and Semen Quality of Dog. Topics in Companion Animal Medicine. 2017, 32, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Quartuccio, M.; Fazio, E.; Medica, P.; Cristarella, S.; Emmanuele, G.; Sinagra, L.; Liotta, L. Correlation between Sperm Parameters and Circulating Thyroid Hormones and Testosterone Concentrations in Labrador Retriever Dog. Italian Journal of Animal Science. 2021, 20, 947–954. [Google Scholar] [CrossRef]
- Ghasemzadeh-Hasankolaei, M.; Sedighi-Gilani, M.A.; Eslaminejad, M.B. Induction of Ram Bone Marrow Mesenchymal Stem Cells into Germ Cell Lineage Using Transforming Growth Factor-β Superfamily Growth Factors. Reproduction in Domestic Animals. 2014, 49, 588–598. [Google Scholar] [CrossRef]
- Rojas-García, P.P.; Recabarren, M.P.; Sir-Petermann, T.; Rey, R.; Palma, S.; Carrasco, A.; Perez-Marin, C.C.; Padmanabhan, V.; Recabarren, S.E. Altered Testicular Development as a Consequence of Increase Number of Sertoli Cell in Male Lambs Exposed Prenatally to Excess Testosterone. Endocrine. 2013, 43, 705–713. [Google Scholar] [CrossRef]
- Ingman, W.V.; Robertson, S.A. The Essential Roles of TGFB1 in Reproduction. Cytokine & Growth Factor Reviews. 2009, 20, 233–239. [Google Scholar]
- McGrath, L.J.; Ingman, W.V.; Robker, R.L.; Robertson, S.A. Exogenous Transforming Growth Factor Beta1 Replacement and Fertility in Male Tgfb1 Null Mutant Mice. Reproduction Fertility and Development. 2009, 21, 561–570. [Google Scholar] [CrossRef]
- He, J.; Dong, C.; You, R.; Zhu, Z.; Lv, L.; Smith, G.W. Localization of Epidermal Growth Factor (EGF) and Its Receptor (EGFR) during Postnatal Testis Development in the Alpaca (Lama Pacos). Animal Reproduction Science. 2009, 116, 155–161. [Google Scholar] [CrossRef]
- Pan, Y.; Cui, Y.; Yu, S.; Zhang, Q.; Fan, J.; Abdul, R. B.; Yang, K. The Expression of Epidermal Growth Factor (EGF) and Its Receptor (EGFR) During Post-Natal Testes Development in the Yak. Reproduction in Domestic Animals. 2014, 49, 970–977. [Google Scholar] [CrossRef]
- Kassab, M.; Abd-Elmaksoud, A.; Ali, M.A. Localization of the Epidermal Growth Factor (EGF) and Epidermal Growth Factor Receptor (EGFR) in the Bovine Testis. Journal of Molecular Histology. 2007, 38, 207–214. [Google Scholar] [CrossRef]
- Cannarella, R.; Condorelli, R.A.; La Vignera, S.; Calogero, A.E. Effects of the Insulin-like Growth Factor System on Testicular Differentiation and Function: A Review of the Literature. Andrology. 2018, 6, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Pitetti, J.; Calvel, P.; Zimmermann, C.; Conne, B.; Papaioannou, M.D.; Aubry, F.; Cederroth, C.R.; Urner, F.; Fumel, B.; Crausaz, M.; Docquier, M.; Herrera, P.L.; Pralong, F.; Germond, M.; Guillou, F.; Jégou, B.; Nef, S. An Essential Role for Insulin and IGF1 Receptors in Regulating Sertoli Cell Proliferation, Testis Size, and FSH Action in Mice. Molecular Endocrinology. 2013, 27, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Cannarella, R.; La Vignera, S.; Condorelli, R.A.; Calogero, A.E. The IGF1/FSH Ratio Correlates with Sperm Count and Testicular Volume. Endocrines. 2022, 3, 624–632. [Google Scholar] [CrossRef]
- Müller, L.; Kowalewski, M.P.; Reichler, I.M.; Kollár, E.; Balogh, O. Different Expression of Leptin and IGF1 in the Adult and Prepubertal Testis in Dogs. Reproduction in Domestic Animals. 2017, 52, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Radovic, S.M.; Starovlah, I.M.; Capo, I.; Miljkovic, D.; Nef, S.; Kostic, T.S.; Andric, S.A. Insulin/IGF1 Signaling Regulates the Mitochondrial Biogenesis Markers in Steroidogenic Cells of Prepubertal Testis, but Not Ovary. Biology of Reproduction. 2019, 100, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Neirijnck, Y.; Calvel, P.; Kilcoyne, K.R.; Kühne, F.; Stévant, I.; Griffeth, R.J.; Pitetti, J.; Andric, S.A.; Hu, M.; Pralong, F.; Smith, L.B.; Nef, S. Insulin and IGF1 Receptors Are Essential for the Development and Steroidogenic Function of Adult Leydig Cells. The FASEB Journal. 2018, 32, 3321–3335. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Conei, D.; Bustos-Obregon, E. Epithelial-Mesenchymal Transitions in the Development of Testis/Interacciones Epitelio-Mesenquimaticas En El Desarrollo Testicular. International Journal of Morphology. 2017, 35, 1444. [Google Scholar] [CrossRef]
- Stratikopoulos, E.; Szabolcs, M.; Dragatsis, I.; Klinakis, A.; Efstratiadis, A. Hormonal Action of IGF1 in Postnatal Mouse Growth. Proceedings of the National Academy of Sciences – PNAS. 2008, 105, 19378–19383. [Google Scholar] [CrossRef]
- Pitetti, J.; Calvel, P.; Romero, Y.; Conne, B.; Vy, T.; Papaioannou, M.D.; Schaad, O.; Docquier, M.; Herrera, P.L.; Wilhelm, D.; Nef, S. Insulin and IGF1 Receptors Are Essential for XX and XY Gonadal Differentiation and Adrenal Development in Mice. PLoS Genetics. 2013, 9, E1003160. [Google Scholar] [CrossRef]
- Warr, N.; Carre, G.; Siggers, P.; Faleato, J.V. , Brixey, R.; Pope, M.; Bogani, D.; Childers, M.; Wells, S.; Scudamore, C.L.; Tedesco, M.; Del Barco, B.I.; Nebreda, A.R.; Trainor, P.A.; Greenfield, A. Gadd45γ and Map3k4 Interactions Regulate Mouse Testis Determination via P38 MAPK-Mediated Control of Sry Expression. Developmental Cell. 2012, 23, 1020–1031. [Google Scholar]
- Liu, J.; Ren, L.; Wei, J.; Zhang, J.; Zhu, Y.; Li, X.; Jing, Li.; Duan, J.; Zhou, X.; Sun, Z. Fine Particle Matter Disrupts the Blood–testis Barrier by Activating TGF-β3/p38 MAPK Pathway and Decreasing Testosterone Secretion in Rat. Environmental Toxicology. 2018, 33, 711–719. [Google Scholar] [CrossRef]
- Shen, L.; Tang, X.; Wei, Y.; Long, C.; Tan, B.; Wu, S.; Sun, M.; Zhou, Y.; Cao, X.; Wei, G. Vitamin E and Vitamin C Attenuate Di-(2-ethylhexyl) Phthalate-induced Blood-testis Barrier Disruption by P38 MAPK in Immature SD Rats. Reproductive Toxicology. 2018, 81, 17–27. [Google Scholar] [CrossRef]
- Luo, D.; He, Z.; Yu, C.; Guan, Q. Role of P38 MAPK Signalling in Testis Development and Male Fertility. Oxidative Medicine and Cellular Longevity. 2022, 2022, 1–12. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Sun, K.; Wang, S.; Gong, D. Polystyrene Microplastics Induce Apoptosis and Necroptosis in Swine Testis Cells via ROS/MAPK/HIF1α Pathway. Environmental Toxicology. 2022, 37, 2483–2492. [Google Scholar] [CrossRef]
- Gonzalez, C.R.; Dorfman, V.B.; Vitullo, A.D. IGF1 Regulation of BOULE and CDC25A Transcripts via a Testosterone-independent Pathway in Spermatogenesis of Adult Mice. Reproductive Biology. 2015, 15, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shan, B.; Duan, Y.; Zhu, J.; Jiang, L.; Liu, Y.; Zhang, Y.; Qi, F.; Niu, S. Effects of Heshouwuyin on Gene Expression of the Insulin/IGF Signalling Pathway in Rat Testis and Spermatogenic Cells. Pharmaceutical Biology. 2020, 58, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
AS events among genes for immature and sexually mature group comparisons.
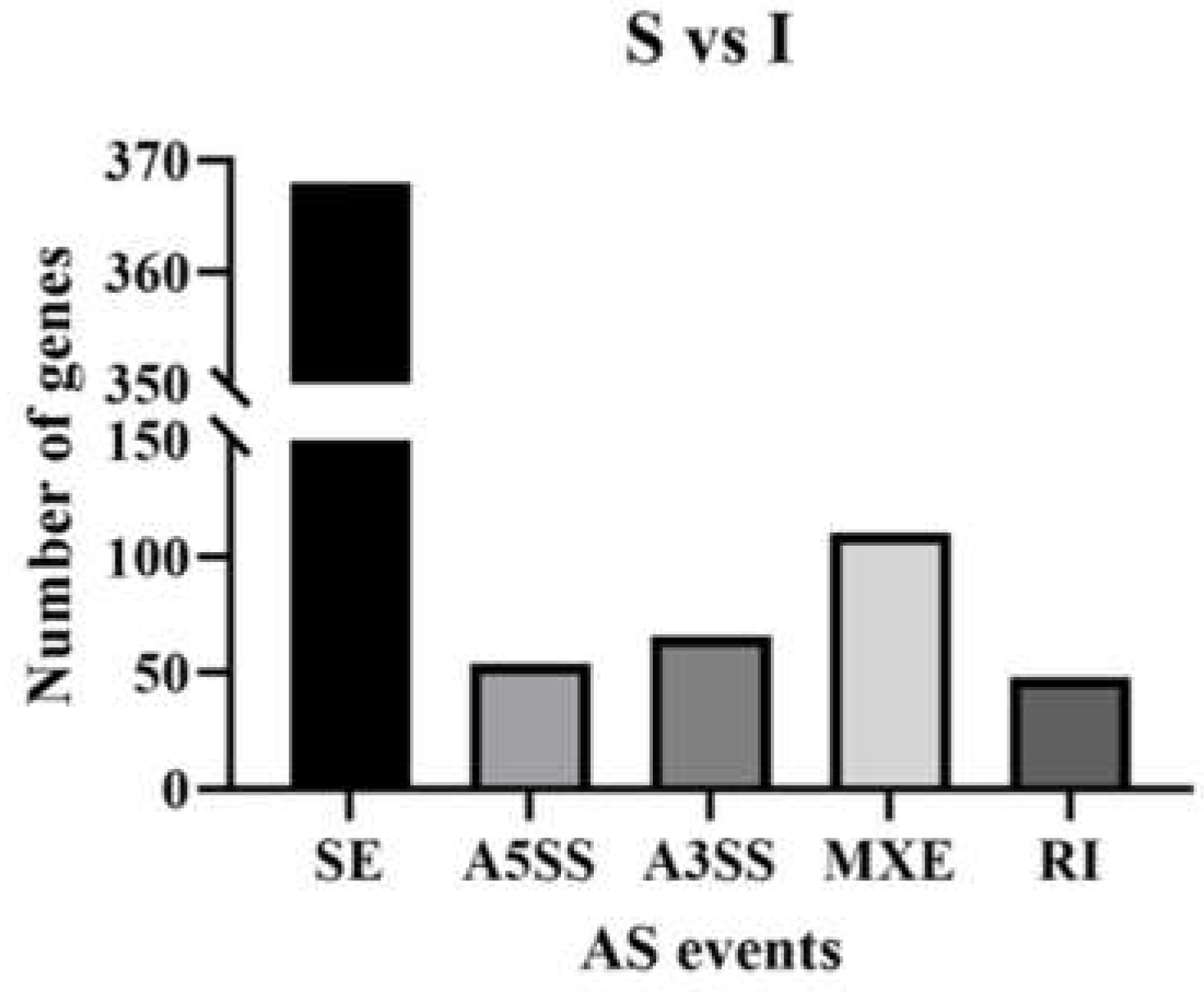
Figure 2.
(A)1: Volcanic plot of differentially expressed genes (DEGs) between the immature and sexually mature groups. (A)2: Volcanic plot of DEGs between the sexually mature and physically mature groups. Highly significant differences in the expression of up- (purple) and down- (red) regulated genes were observed between immature (I) and sexually mature (S) testes, and between sexually mature (S) and physically mature (P) testes. Blue indicates no differential expression. (B): Venn diagram depicting gene expression patterns. The numbers of uniquely and commonly (FPKM>1) expressed genes are shown. (C): Clustering of differentially expressed genes. The overall FPKM hierarchical clustering map was obtained with log10 (FPKM + 1) for normalization. Red and blue represent high and low expression levels, respectively.
Figure 2.
(A)1: Volcanic plot of differentially expressed genes (DEGs) between the immature and sexually mature groups. (A)2: Volcanic plot of DEGs between the sexually mature and physically mature groups. Highly significant differences in the expression of up- (purple) and down- (red) regulated genes were observed between immature (I) and sexually mature (S) testes, and between sexually mature (S) and physically mature (P) testes. Blue indicates no differential expression. (B): Venn diagram depicting gene expression patterns. The numbers of uniquely and commonly (FPKM>1) expressed genes are shown. (C): Clustering of differentially expressed genes. The overall FPKM hierarchical clustering map was obtained with log10 (FPKM + 1) for normalization. Red and blue represent high and low expression levels, respectively.
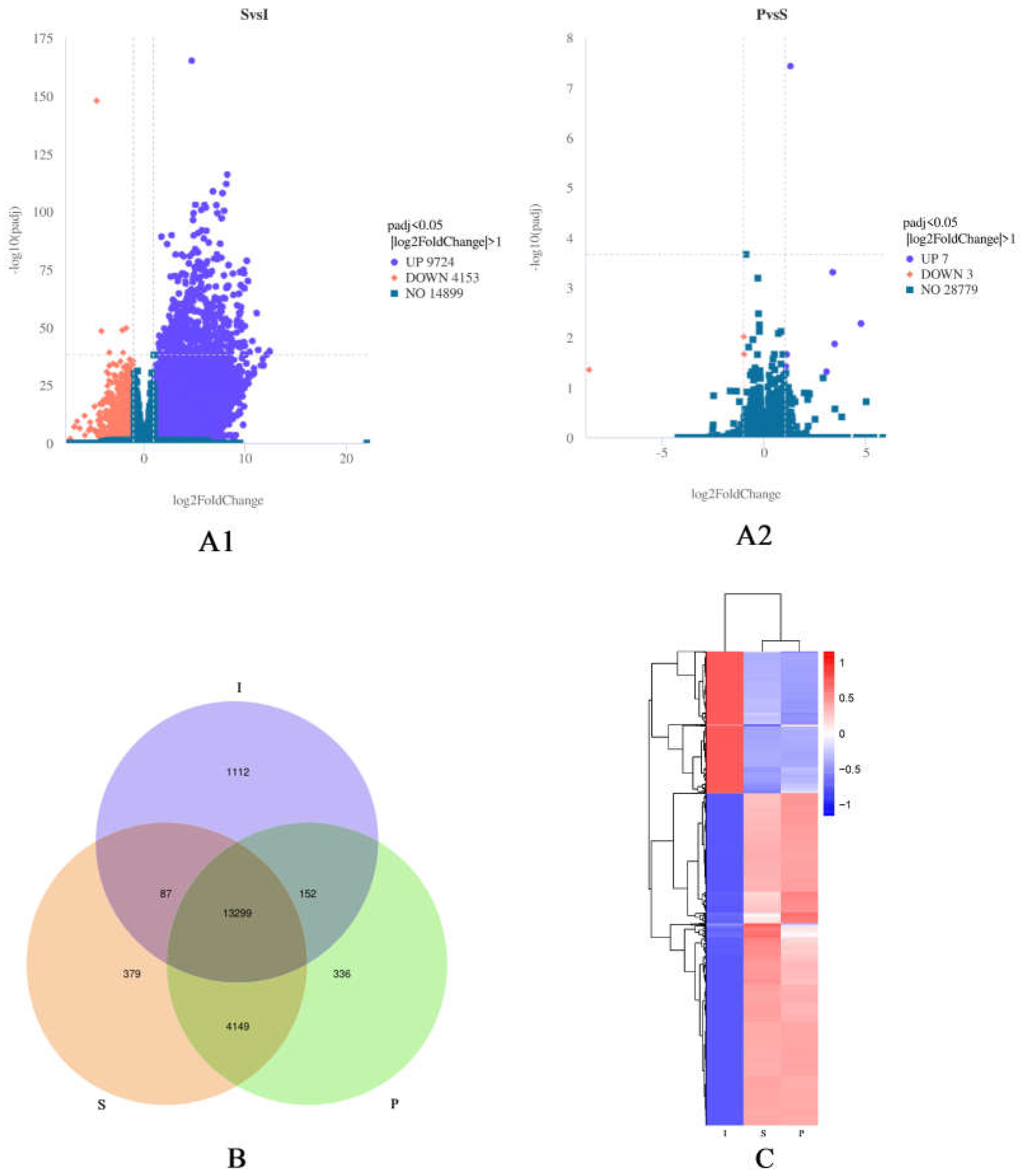
Figure 4.
(A): Scatter plot of differentially expressed KEGG genes in immature (I) and sexually mature (S) testes. (B): Scatter plot of differentially expressed KEGG genes in sexually mature (S) and physically mature (P) testes. Ordinates and abscissas represent the names of KEGG pathways and gene ratios, respectively. Point sizes and colors represent the numbers of DEGs and the ranges of Q values, respectively.
Figure 4.
(A): Scatter plot of differentially expressed KEGG genes in immature (I) and sexually mature (S) testes. (B): Scatter plot of differentially expressed KEGG genes in sexually mature (S) and physically mature (P) testes. Ordinates and abscissas represent the names of KEGG pathways and gene ratios, respectively. Point sizes and colors represent the numbers of DEGs and the ranges of Q values, respectively.
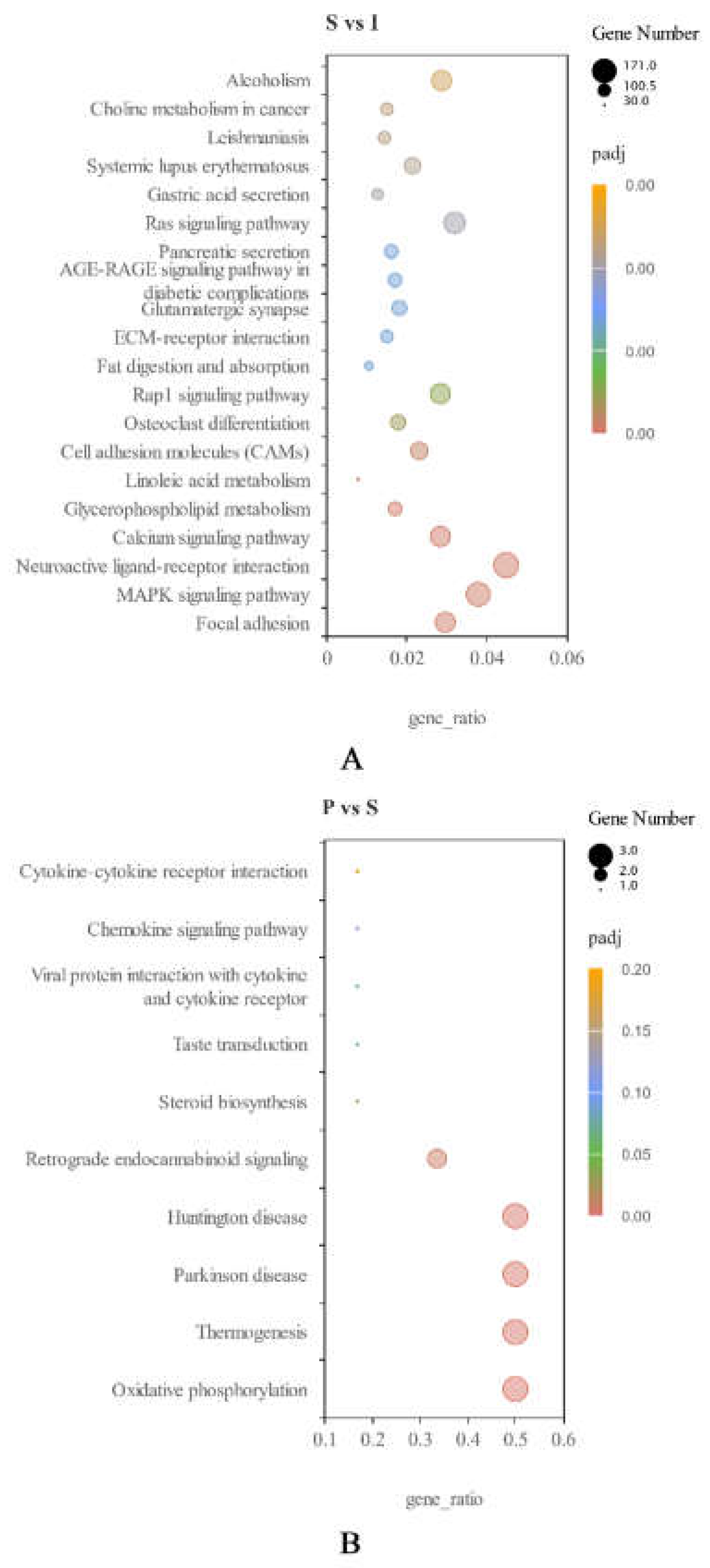
Figure 5.
Relative expression of different genes in sexually mature (S) and physically mature (P) testes compared to immature (I) testes, respectively, using group I as a reference. (A): qPCR relative expression of S vs I and P vs I. (B): RNA-Seq data for S vs I and P vs I.
Figure 5.
Relative expression of different genes in sexually mature (S) and physically mature (P) testes compared to immature (I) testes, respectively, using group I as a reference. (A): qPCR relative expression of S vs I and P vs I. (B): RNA-Seq data for S vs I and P vs I.
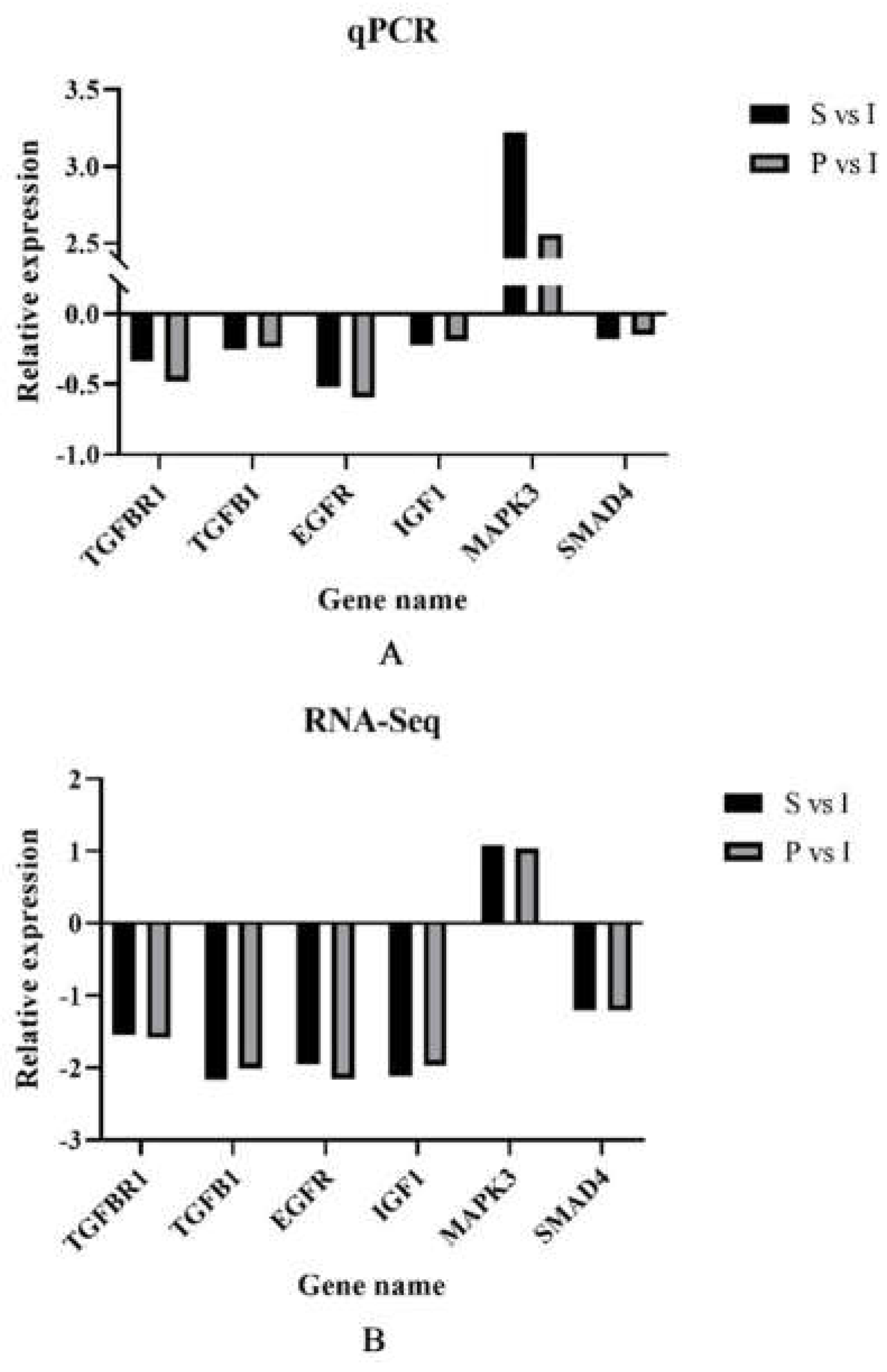
Table 1.
Primers utilized in qRT-PCR.
| Primer name | Gene ID | Primer sequence | Fragment length | annealing temperature |
|---|---|---|---|---|
| GAPDH-F GAPDH-R |
XM_005680968.3 | ATGTTTGTGATGGGCGTGAA GGCGTGGACAGTGGTCATAAGT |
153 bp | 60 ℃ |
| TGFBR1-F TGFBR1-R |
XM_018052233.1 | TTCAAACGTGCTGACATCTATGC ACTGATGGATCGGAAGGTACAAG |
128 bp | 60 ℃ |
| SMAD4-F SMAD4-R |
XM_018039535.1 | CATAACAGCACTACCACCTGGACT GGATGATTAGAAATAGGAGGCTGG |
173 bp | 60 ℃ |
| TGFB1-F TGFB1-R |
NM_001314142.1 | CAACAATTCCTGGCGCTACCT ATGTCCACTTGAAGCGTGTTATCC |
183 bp | 60 ℃ |
| EGFR-F EGFR-R |
XM_018067044.1 | CCGTGCGATTCAGTAACAACC GGTCAATTTCTGGCAGTTCTCCTC |
194 bp | 60 ℃ |
| IGF1-F IGF1-R |
NM_001285697.1 | AATCAGCAGTCTTCCAACCCAA AGCAAGCACAGGGCCAGATA |
114 bp | 60 ℃ |
| MAPK3-F MAPK3-R |
XM_018040780.1 | CTGGACCGGATGTTGACCTTTA CTCCTTCAGTCGTTCCTTGGG |
138 bp | 60 ℃ |
Table 2.
Statistics of RNA-Seq data quality.
| Sample name | Library number | Raw reads (n) | Clean reads (n) | Error rate | Q20 | Q30 | GC pct |
|---|---|---|---|---|---|---|---|
| I1 | 1 | 40405582 | 39128916 | 0.03 | 97.36 | 93.09 | 50.99 |
| I2 | 2 | 43841646 | 42366502 | 0.03 | 97.41 | 93.2 | 51.16 |
| I3 | 3 | 39433208 | 38237518 | 0.03 | 97.42 | 93.25 | 51.42 |
| I4 | 4 | 42396474 | 40926952 | 0.03 | 97.43 | 93.23 | 51.26 |
| I5 | 5 | 45463988 | 43854898 | 0.03 | 97.41 | 93.19 | 51.1 |
| I6 | 6 | 39351212 | 38000364 | 0.03 | 97.48 | 93.36 | 50.91 |
| S1 | 7 | 44934642 | 43739660 | 0.03 | 97.51 | 93.41 | 52.09 |
| S2 | 8 | 43025142 | 41894994 | 0.03 | 97.39 | 93.17 | 52.36 |
| S3 | 9 | 45845898 | 44647422 | 0.03 | 97.59 | 93.55 | 52.15 |
| S4 | 10 | 41237336 | 40020062 | 0.03 | 97.37 | 93.11 | 52.29 |
| S5 | 11 | 41130946 | 39901252 | 0.03 | 97.36 | 93.09 | 51.09 |
| S6 | 12 | 41242240 | 40019490 | 0.03 | 97.51 | 93.4 | 51.9 |
| P1 | 13 | 45377178 | 44131516 | 0.03 | 97.48 | 93.34 | 52.27 |
| P2 | 14 | 44664918 | 43577078 | 0.03 | 97.49 | 93.36 | 52.06 |
| P3 | 15 | 43792502 | 42601198 | 0.03 | 97.47 | 93.32 | 51.98 |
| P4 | 16 | 43921138 | 42702250 | 0.03 | 97.5 | 93.39 | 52.17 |
| P5 | 17 | 43223452 | 42134186 | 0.03 | 97.44 | 93.27 | 52.14 |
| P6 | 18 | 45571906 | 44622618 | 0.03 | 97.3 | 92.91 | 50.37 |
Error rate: overall sequencing error rate for the data; Q20 and Q30: percentages of total bases with Phred values above 20 and 30, respectively; GC pct: percentage of C and G among the 4 bases in clean reads.
Table 3.
Statistics of reads aligned with the reference genome.
| SAMPLE | TOTAL READS | TOTAL MAP | UNIQUE MAP | MULTI MAP | POSITIVE MAP | NEGATIVE MAP |
|---|---|---|---|---|---|---|
| I1 | 39128916 | 37563635(96.00%) | 35626251(91.05%) | 1937384(4.95%) | 17793049(45.47%) | 17833202(45.58%) |
| I2 | 42366502 | 40625554(95.89%) | 38612044(91.14%) | 2013510(4.75%) | 19294015(45.54%) | 19318029(45.60%) |
| I3 | 38237518 | 36735876(96.07%) | 34685761(90.71%) | 2050115(5.36%) | 17323488(45.30%) | 17362273(45.41%) |
| I4 | 40926952 | 39282398(95.98%) | 37389786(91.36%) | 1892612(4.62%) | 18672350(45.62%) | 18717436(45.73%) |
| I5 | 43854898 | 42092675(95.98%) | 40255938(91.79%) | 1836737(4.19%) | 20105549(45.85%) | 20150389(45.95%) |
| I6 | 38000364 | 36500392(96.05%) | 34501227(90.79%) | 1999165(5.26%) | 17230278(45.34%) | 17270949(45.45%) |
| S1 | 43739660 | 42076928(96.20%) | 40551689(92.71%) | 1525239(3.49%) | 20259699(46.32%) | 20291990(46.39%) |
| S2 | 41894994 | 40318684(96.24%) | 39006285(93.10%) | 1312399(3.13%) | 19484346(46.51%) | 19521939(46.60%) |
| S3 | 44647422 | 43061457(96.45%) | 41514183(92.98%) | 1547274(3.47%) | 20739259(46.45%) | 20774924(46.53%) |
| S4 | 40020062 | 38465949(96.12%) | 36829538(92.03%) | 1636411(4.09%) | 18396451(45.97%) | 18433087(46.06%) |
| S5 | 39901252 | 38334062(96.07%) | 36942595(92.59%) | 1391467(3.49%) | 18452256(46.24%) | 18490339(46.34%) |
| S6 | 40019490 | 38522907(96.26%) | 36912252(92.24%) | 1610655(4.02%) | 18439977(46.08%) | 18472275(46.16%) |
| P1 | 44131516 | 42514601(96.34%) | 41073585(93.07%) | 1441016(3.27%) | 20518750(46.49%) | 20554835(46.58%) |
| P2 | 43577078 | 41944011(96.25%) | 40303516(92.49%) | 1640495(3.76%) | 20133389(46.20%) | 20170127(46.29%) |
| P3 | 42601198 | 41016519(96.28%) | 39440892(92.58%) | 1575627(3.70%) | 19701050(46.25%) | 19739842(46.34%) |
| P4 | 42702250 | 41098071(96.24%) | 39528322(92.57%) | 1569749(3.68%) | 19745068(46.24%) | 19783254(46.33%) |
| P5 | 42134186 | 40510371(96.15%) | 38849502(92.20%) | 1660869(3.94%) | 19405731(46.06%) | 19443771(46.15%) |
| P6 | 44622618 | 38550615(86.39%) | 37036723(83.00%) | 1513892(3.39%) | 18501103(41.46%) | 18535620(41.54%) |
Sample: sample name; total reads: number of clean reads upon quality control; total map: number (percentage) of reads aligned to the reference genome; unique map: number (percentage) of reads aligned to a unique region of ARS1.2 (subsequently analyzed for quantitation), multi map: number (percentage) of reads with alignment to many locations of ARS1.2; positive and negative maps: numbers (percentages) of reads with alignment to the positive and negative strands of the reference genome, respectively.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



