
Submitted:
30 April 2023
Posted:
01 May 2023
You are already at the latest version
Abstract
Streptococcus pneumoniae is the major cause of invasive pneumococcal disease (IPD). Since 1998, multilocus sequence typing (MLST) has been used for identifying the genotypes of strains of S. pneumoniae and helped reveal a diversity of local and regional epidemiological patterns for IPD, resulting in an archived MLST dataset of over 74,000 isolates. However, the global patterns of MLST sequence type (ST) and allele type (AT) distributions remain largely unexplored. In this study, we investigated the spatial and temporal patterns of AT and ST distributions of S. pneumoniae. We extracted S. pneumoniae MLST data from PubMLST.org and conducted various population genetic and phylogenetic analyses. Our analyses demonstrated both shared and unique distributions of STs and ATs among continental and national/regional populations. Among the 17915 STs in the dataset, 36 STs representing 15263 isolates were broadly shared among all six continents, consistent with recent gene flow and clonal dispersal of this pathogen. The analysis of molecular variance revealed that >96% genetic variations were found within individual continental and national/regional populations. However, though low (<4%), statistically significant genetic differentiation among continental and national populations were observed. Comparisons between non-clone-corrected and clone-corrected datasets showed that localized clonal expansion contributed significantly to the observed genetic differentiations among continents and countries/regions. Temporal analyses of the isolates showed that implementation of pneumococcal conjugate vaccine impacted the distributions of STs. Linkage disequilibrium analyses identified evidence for non-random recombination in all continental populations of this species. We discussed the implications of our analyses to the global epidemiology and future vaccine developments for S. pneumoniae.
Keywords:
pneumococcal disease
; serotype
; geographic differentiation
; vaccination
; temporal variation
; recombination
1. Introduction
Streptococcus pneumoniae is a Gram-positive, lancet-shaped bacterium that contributes significantly to microbial diseases worldwide. S. pneumoniae is commonly found in the nasopharynx of humans where 40-50% of healthy children and 20%-30% of healthy adults are carriers [1]. However, S. pneumoniae can cause a diversity of pneumococcal diseases (PD) such as Otitis Media (OM), pneumonia, sinusitis, septicemia, and meningitis [2,3,4]. For example, children ≤ 4 years old have a high incidence rate of OM [5], with certain local populations in Sub-Saharan Africa and Asia showing 100% incidence rates for children ages 1-4 [6]. S. pneumoniae can spread from the nasopharyngeal into the lower airways and cause pneumonia. The estimated worldwide incidence rate of community-acquired pneumonia ranges between 1.5-14 cases per 1000/year [7]. Pneumonia can be caused by multiple viral, bacterial, and fungal pathogens and is responsible for approximately 2.5 million deaths worldwide/year [8,9]. Among bacterial pneumonia, about 90% is caused by S. pneumoniae [10]. In addition, S. pneumoniae can pass the blood-brain barrier (BBB) to cause pneumococcal meningitis. Meningitis is estimated at over 1.2 million cases worldwide each year, and if untreated, fatality rate can reach 70% and leaving 1 in 5 survivors with permanent brain damage [11].
Among the >100 known serotypes in S. pneumoniae, 16 cause approximately 90% of PDs worldwide [12]. For example, Hausdorff et al. [13] found that serotypes 4, 6, 9, 14, 18, 19, and 23 cause approximately 70-88% of PDs in young children in North America, Europe, Africa, and several regions in other continents. While much is known about the association between serotypes and PD, the relatively low number of serotypes in this species offered limited discriminating power to distinguish strains and was insufficient for many types of epidemiological investigations about this pathogen. In 1998, a multilocus sequence typing (MLST) scheme was established for genotyping strains of S. pneumoniae. In this scheme, DNA sequences at the following seven housekeeping genes were recommended to identify the genotypes for individual isolates: d-alanine-d-alanine ligase (ddl), signal peptidase I (spi), transketolase (recP), shikimate dehydrogenase (aroE), glucose-6-phosphate dehydrogenase (gdh), glucose kinase (gki), and xanthine phosphoribosyltransferase (xpt) [12]. At each locus, a unique sequence was assigned a distinct allele and combinations of allele types (ATs) at the seven housekeeping loci were used to provide a sequence type (ST) for each isolate [12]. The MLST scheme showed much greater discriminatory power over serotyping and provided a consistent method for many researchers around the world to expand on the MLST database of S. pneumoniae over the following 25 years.
As of November 2022, there were 74346 S. pneumoniae isolates with MLST data at the seven loci deposited at PubMLST.org. These isolates came from diverse geographical populations and spanned over nine decades. Using MLST data, previous studies have described the associations between STs, serotypes, antibiotic resistance, and/or PD prevalence among various demographic groups [e.g., 14-22]. However, most of the studies have focused on local level, with a few included isolates and data from multiple countries. Interestingly, several studies identified the emergence of non- pneumococcal conjugate vaccine (PCV) serotypes following the introduction of PCVs. As of 2020, the WHO documented that 149 countries have introduced PCV [10]. However, the effects of PCV introduction on ST distributions at the global level have not been investigated.
In this study, we aimed to use the archived global MLST data on S. pneumoniae to address several population genetic and epidemiological questions. First, how are STs distributed at the continental and national/regional levels? Second, are geographic populations genetically differentiated at the continental and national/regional levels? Third, what is the effect of PCV implementation on the distributions of STs? Because PCVs were designed to target the dominant serotypes, we expect that certain STs will be more impacted by PCV implementation than other STs. And lastly, is there evidence for recombination within individual continental populations and the global population of S. pneumoniae?
2. Materials and Methods
2.1. Collection and Organization of Archived S. pneumoniae MLST data
Allelic profiles, geographical, temporal, and ecological information, and nucleotide sequences of all STs were retrieved from the S. pneumoniae PubMLST database (pubMLST.org). The selection of gene loci for MLST, the primers, and the criteria for identifying ATs and STs were described in the original MLST scheme by Enright and Spratt [12]. Meta data of the isolates associated with each ST, including their geographical location, ecological niche, and time of isolation were all organized into Microsoft Excel. In total, information for 74346 isolates were retrieved in November 2022 for this study [16].
2.2. Phylogenetic Analysis of STs and ATs
Allele sequences at each of the 7 housekeeping loci and allelic profiles for each ST were retrieved from PubMLST [16]. To determine the relationships among ATs at each housekeeping locus, DNA sequences of all ATs were imported into Molecular Evolutionary Genetics Analysis (MEGA) software and were aligned using the Multiple Sequence Comparison by Log-Expectation (MUSCLE) tool as described in the MEGA X-Help manual [23,24]. The aligned AT sequences for each locus were then utilized to construct a neighbour-joining (NJ) tree using a Kimura 2-parameter (K2P) model [23]. The newly constructed NJ trees for each locus were imported into the Interactive Tree of Life (iTOL) online tool for better visualization [25].
To infer the relationships among STs, their allelic sequences at the seven loci were concatenated using MobaXterm based on their AT numbers for each ST [26]. The newly concatenated sequences were then aligned through Multiple Alignment using Fast Fourier Transform (MAFFT) [27]. Similar to the AT tree construction, the aligned ST sequences were utilized to construct a NJ tree using a K2P model [23]. The newly constructed ST tree was imported into iTOL along with the continental, ecological, and temporal data for each ST [25].
2.3. Population Genetics Analysis
To determine genetic differences among populations of S. pneumoniae, three sample types were analyzed: (i) the entire S. pneumoniae population (as of November 2022), (ii) the STs associated with serotypes used in the PCV-13 vaccination program (which contained STs associated with serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F), and (iii) non-PCV-13 STs (which contained STs associated with serotypes 6C, 8, 9N, 11A, 12F, 15BC, 22F, 23B, 35B, and 38) that are part of the top 50 most frequent STs. Our focus on the top 50 most frequent STs was to identify how some of the STs might have changed due to PCV implementation.
To investigate the population genetic patterns, the retrieved S. pneumoniae allelic profiles were imported into Microsoft Excel for each of the three sample types and three datasets were created for each type: (i) ecological niche-based dataset, (ii) country and continent-based dataset, and (iii) temporal and continent-based dataset. For the country and continent-based datasets, countries with <10 isolates were combined with geographically the closest country within the same continent as one sample. For the temporal/continental datasets, isolates prior to the year 1999 were omitted due to small sample sizes. Furthermore, each dataset consisted of a non-clone corrected (NCC) type and a clone-corrected (CC) type. The NCC datasets contain all isolates pertaining to that set. The CC datasets contain only one isolate of each ST as a representative within each sub-population.
An analysis of molecular variance (AMOVA) test was performed to estimate genetic variation within each population and to quantify genetic differences among sub-populations within each dataset, as described in the GenALeX 6.5 manual [28]. Because GenALeX 6.5 as well as all other known population genetic analyses programs only allowed a maximum of 999 ATs for each locus, rare alleles at two loci with > 999 ATs each (xpt and ddl) were combined to fit the program requirement. Specifically, rare alleles that were closely related to each other based on their phylogenetic relationships were combined. In our analyses, only STs with represented isolates and required metadata as reported in the pubMLST website were included in our analyses. Furthermore, allelic profiles of all STs were inputted into Multilocus V1.3. Genotypic diversity and linkage disequilibrium analysis were determined using formulas as described in the Multilocus V1.3 manual [29]. Datasets were divided by continent and a PCV dataset was created for PCV-13 STs and non-PCV-13 (top 50) STs.
3. Results
3.1. Global Distribution of Isolates and Sequence Types
As of November 2022, 74346 isolates belonging to 17915 STs of S. pneumoniae were successfully extracted from PubMLST and imported into Microsoft Excel for analysis. The geographic distributions of these isolates and STs are shown in Table 1. The extracted information included isolates from 6 continents and 115 countries/regions. Based on continental origins, Europe contributed the highest proportion of isolates (32.403%), followed by Africa (25.68%), Asia (23.274%), North America (12.731%), South America (3.891%), and Oceania (1.246%). Among the 74346 isolates, 576 (0.775%) had no associated geographic information. At the country level, the highest number of isolates came from the USA (11.679%), followed closely by South Africa (11.55%) and the UK (8.569%). The USA, South Africa, and the UK also had the highest number of uniquely represented STs among the countries with 2005, 2013, and 2323 unique STs respectively. The lowest number of isolates were found in Sudan, Cuba, Greenland, Monaco, Azerbaijan, and Wallis and Futuna Islands, with one isolate from each country/region (Table 1).
Table 2 shows the breakdown of the top 50 globally most frequent STs. ST180 was the most frequently observed and made up 1.98% of the entire dataset. Overall, the top 50 most frequent STs made up 30.914% of isolates in the entire dataset. Among the top 50 STs, 39 were represented by those in PCV-13, including ST180, while 11 were non-PCV-13 STs. Similar to the skewed ST frequencies, the frequencies of ATs at each locus were also highly skewed (Table 2). For aroE, AT7 was the most frequent (21.80%, represented in 22% of STs). For gdh, AT5 was the most frequent (21.56%, in 24% of STs). For gki, AT2 was most frequent (21.39%, in 18% STs). For recP, AT1 was most frequent (27.57%, in 26% STs). For spi, AT6 was most frequent (37.40%, in 34% STs). For xpt, AT1 was most frequent (19.55%, in 14% STs). Finally, for ddl, AT14 was most frequent (33.90%, in 36% STs). We note that while for six of the seven loci, the most frequent AT at the whole population level was also found in the highest number of STs. However, for gki, AT4 was represented by more STs (n(ST)=11), but fewer isolates (n=4 568) than AT2.
Table 3 shows the geographical, ecological, and temporal distributions of 17915 STs of S. pneumoniae. At the geographical level, 36 STs (n=15263 isolates) were found in all six continents, 27 STs (n=4970 isolates) were found in five of six continents, 55 STs (n=5069 isolates) were found in four of six continents, 184 STs (n=6517 isolates) were found in three of six continents, 737 STs (n=7796 isolates) were found in two of six continents, and 15929 STs (n=33607 isolates) were found in one continent only. The remaining STs were represented by isolates with no associated geographical information. Europe had the highest number of unique STs (n=7986, n(ST)=4 813). At the ecological level, 21 STs (n=4665) were found in both ecological niches (clinical and veterinarian) and 16998 STs (n=69119) were found in one ecological niche only. The ecological niche information was unknown for the remaining 896 STs. Most isolates and the majority of STs were from clinical sources (n(ST)=16987, n=69098 isolates). Based on temporal information, the isolates were grouped into the following five time intervals: Int 1: <1949; Int 2: 1950-1969; Int 3: 1970-1989; Int 4: 1990-2009; Int 5: 2010-2022. At the temporal level, only 1 ST (n=717) was found in all five-time intervals, 12 STs (n=1390) were found in four of five intervals, 72 STs (n=10690) were found in three of five intervals, 1584 STs (n=31349) were found in two of five intervals, and 13627 STs (n=20660) were found in one interval only. Temporal information was unknown for the remaining 2619 STs. Majority of STs in the dataset were collected between 1990-2009 only (n(ST)= 6727). Majority of isolates were collected between 1990-2022 (n=31132).
Geographical and temporal distributions of top five STs in representative countries and time intervals were further highlighted (Table 4). Geographical distributions were shown at the country level where countries with the most collected isolate data on each continent were shown. Interestingly, we see diverse ST distributions among countries, with different countries often having different most frequent STs. However, there were some overlaps among countries. For example, ST199 was among the most frequent in the USA, UK, and Australia. Together, the top 5 most frequent STs in each country make up approximately 12-22% of their respective populations, with the highest proportion found in the USA (22.19%). We note that among the common STs in Table 4, ST4133 was associated with a nontypeable serotype and PCV-13 STs were represented in 17 of the 30 most frequent country-ST combinations. At the temporal level, ST199 and ST180 were among the most frequent, found in 4 of the 6 time intervals, and PCV-13 STs were found 18 of the 30 time interval-ST combinations.
Table 5 shows the global frequencies of ATs. The length of housekeeping loci ranges from 405-486 bp. ddl has the greatest number of ATs with 1157. The most frequent ATs for each housekeeping loci were found in both PCV-13 and non-PCV-13 (top 50) STs. We found AT6 of the spi gene to be represented the most among the dataset (35.31%).
3.2. Phylogenetic Analysis
In total, 17915 STs of S. pneumoniae were concatenated at seven housekeeping loci and aligned for phylogenetic analysis. Figure 1 shows a NJ tree of 17915 STs of S. pneumoniae using a K2P model. Geographical, ecological, and temporal data were respectively added through iTOL to demonstrate their distributions. The NJ tree shows both divergent and close relationships among STs. The most divergent STs were ST13466 and ST9066. Since this tree (Figure 1) contains too many STs for clear visualization of each ST, further phylogenetic analysis was performed for subsets of STs. Specifically, we analyzed the top 50 globally most frequent STs (Supplementary Figure 1). The most divergent STs among the top 50 most frequent were ST558 and ST416. Geographically, though highly abundant, ST4414 was found only from Asia. As the fifth most abundant, ST191 was the only ST found among all five time intervals. Among the top 50 STs, we found no clear phylogenetic clustering between those covered by PCV-13 and those belonging to non-PCV-13 STs. Phylogenetic analysis was also performed for PCV-13 STs and those not covered by PCV-13 (Supplementary Figure 2 and Figure 3). With only a few exceptions, in general, STs that were associated with the same serotypes were found to be phylogenetically closely related to each other among both PCV-13 ST and non-PCV-13 (top 50) ST trees. Phylogenetic trees showing the relationship between AT sequences among seven housekeeping loci can be seen in Supplementary Figures S4–S10.
3.3. AMOVA
Due to the large sample size of the non-clone-corrected data in the total sample, AMOVA test of the non-clone-corrected data among geographic populations were conducted separately for among countries within individual continents. For clone-corrected data, a two-level hierarchical AMOVA was conducted to assess population genetic differentiation at both the continental and country/region levels. Table 6 shows the results of AMOVA test among geographical populations. For clone-corrected data, genetic variation within countries contributed 98.515% to the total observed genetic variation. Genetic variation among countries and among continents contributed 1.074% and 0.411% respectively to the total genetic variations. For the non-clone-corrected data, genetic variation within countries contributed 97.914%, 96.975%, 98.086%, 97.322%, 97.731%, and 96.080% of the observed genetic variation in Africa, Asia, Europe, North America, South America, and Oceania, respectively. The remaining variances were attributed to among countries within individual continents. The among continents, among countries, and within countries contributions were all statistically significant with p<0.01.
Further, two-level hierarchical AMOVA testing was conducted for both PCV-13 and non-PCV-13 (top 50) STs at the geographical level (Supplementary Tables S1 and S2). Only non-clone-corrected data was used for both datasets due to lack of sample size for clone-corrected data. For PCV-13 STs, genetic variation within countries contributed 90.172% to the total observed genetic variation. Genetic variation among countries and among continents contributed 7.436% and 2.392% respectively (Supplementary Table 1). For non-PCV-13 (top 50) STs, genetic variation within countries contributed 90.568% of the total observed genetic variation. Genetic variation among countries and among continents contributed to 7.539% and 1.893% respectively (Supplementary Table 2). The among continents, among countries, and within countries’ contributions were all statistically significant with p<0.01.
Table 7 shows the results of AMOVA test at the temporal level. In this analysis, populations were defined at intervals beginning in 1999, then every three years until 2022. For clone-corrected data, genetic variation within time intervals contributed 99.180% to the total observed genetic variation. Genetic variation among time intervals and among continents contributed 0.189% and 0.631% respectively. For non-clone-corrected data, genetic variation within time intervals contributed 99.556%, 99.393%, 99.546%, 97.947%, 98.891%, and 98.519% to the observed genetic variation in Africa, Asia, Europe, North America, South America, and Oceania, respectively. The remaining amounts were attributed to among time intervals. The among continents, among time intervals, and within time intervals’ contributions were all statistically significant with p<0.01.
The two-level hierarchical AMOVA testing was also conducted for PCV-13 and non-PCV-13 (top 50) populations at the temporal level (Supplementary Table S3). Due to sample size issues, only six countries were used for analysis as described in Supplementary Table S3. In addition, only the non-clone-corrected data was used due to the small sample sizes for clone-corrected data. Here, genetic variation within time intervals contributed 94.784% to the total observed genetic variation for the six countries. Genetic variation among time intervals and among continents contributed 4.173% and 1.043% respectively. All values were statistically significant with p<0.01.
Table 8 shows the results of AMOVA test between the two ecological niches at the global level. For clone-corrected data, genetic variation within ecological niches contributed 98.478% to the total observed genetic variation, with between ecological niches contributing 1.522%. For non-clone-corrected data, only European samples were used due to small sample size for veterinary populations in other continents. Genetic variation within ecological niches and between ecological niches contributed 87.922% and 12.078% to the total observed genetic variation respectively in the European samples. All values were statistically significant with p<0.01.
3.4. Effects of PCV on Population Structure
Next, we analyzed the effects of PCV implementation on relative frequencies of the top 50 most common STs associated with PCV-13 and non-PCV-13 (Figure 2). The associations between STs and PCV-targeted serotypes are shown in Supplementary Table S4. We tested whether there was a rise of non-PCV-13 STs after PCV implementation. A line histogram was produced and as seen in Figure 2, A noticeable trend was observed following 1981-1985, where there was a decrease in relative frequency of PCV-13 STs, followed by a steady frequency following global PCV-13 implementation, and a notable decrease following global PCV-13 implementation. The opposite was observed with non-PCV-13 (top 50) STs where there was an increase after 1981-1985, followed by a steady frequency upon global PCV implementation, and a significant increase in relative frequency after global PCV-13 implementation. However, we note that the relative isolate frequencies taken from 1936-1975 and 2021-2022 did not follow the trends as expected based on PCV-13. This was likely due to small sample sizes collected during these time intervals (Figure 2, Supplementary Table S5).
Aside from the global analyses, changes in ST frequencies related to PCV-13 implementation was also conducted in countries where sample sizes were large (>600 isolates; Supplementary Figure S11). Since PCV-13 implementation differed among some of the countries and continents, we hypothesized that the changes in relative frequencies of STs over time may differ among geographic regions. Indeed, the frequencies of PCV-13 associated and not associated STs differed among geographic regions, with the implementation of PCV-13 showing different but notable effects among the US, South Africa, and UK (Supplementary Figure 11 and Supplementary Table 6 and Table 7). However, the effects of PCV-13 implementation on ST changes were not observed in Brazil and Australia (Supplementary Figure 11). At the continental level, the relative frequencies of PCV-13 associated STs increased from ~2.3% during the 2001-2005 interval to about 9% from 2006-2020 in Asia. However, in Europe, there was a large decrease from ~21% during 2001-2005 to ~10% during 2006-2020. In North America where PCV-13 was implemented the earliest, the decreases in PCV-13 associated STs happened earlier, from ~15% during 1996-2000 to 5.5% or less after 2000 (Supplementary Table 6).
3.5. Recombination & Linkage Disequilibrium
Evidence for potential recombination within individual geographic populations of S. pneumoniae was investigated using two indicators: phylogenetic incompatibility and index of association. In these two analyses, only clone-corrected data were used and due to the large sample size at the global level crushing the program Multilocus, only samples at individual continents were analyzed. An additional dataset was analyzed to test for recombination among STs associated with PCV-13 (Table 9). All datasets, with the exception of the PCV dataset, showed 0% phylogenetic compatibility, indicative of recombination between pairs of loci. However, all index of association values were statistically significant with p<0.01, rejecting the hypothesis of random recombination.
4. Discussion
In this study, we extracted and analyzed the genetic, geographical, ecological, and temporal data of the global S. pneumoniae isolates deposited within PubMLST up to November 2022. The extracted global population included 74346 isolates representing 17915 STs. The isolates were distributed among 6 continents and 115 countries. Among the six continents, Europe contributed the most isolates (32.403%). Among the 115 countries, USA had the most isolates (11.679%). The patterns of distribution of isolates among continents and countries likely reflected sampling and research efforts by scientists working on this pathogen as well as the differences in relative importance of S. pneumoniae to public health and in available resource for research among countries and continents.
Our analyses revealed a diversity of STs within most countries and continents. Indeed, most STs (15929 of 17915 STs) were found in only one continent. However, although only 36 STs were shared among all six continents, they represented over 20.53% of all the isolates in the database, consistent with recent gene flow and that a relatively small number of STs dominated the global S. pneumoniae population. Our results are similar to previous research results that showed a few serotypes causing most pneumococcal diseases [12]. A similar pattern was seen across temporal scales where a relatively small number of STs persisted across decades. Among these, ST191 was collected across all time intervals (Figure S1). ST191 is among the top 50 most frequent STs and is associated with serotype 7F, a PCV-13 target [30,31]. ST191 was a frequent ST prior to PCV-13 implementation. However, introduction of PCV-13 has resulted in a reduction of ST191 frequency over time. While incomplete sampling could have contributed to the low number of shared STs across the analyzed time frames, the persistence of ST191 suggests its significant adaptability in human populations. Interestingly, between 1990-2022, only 1532 STs were collected, representing about 10% of all STs in the database but these STs account for 41.87% of all the isolates in the dataset. The overrepresented STs indicate that a relatively small number of STs are highly transmissible and/or pathogenic to humans. Further genetic and genomic studies of isolates of these frequent STs (e.g., ST199, ST180, and ST81) could help reveal the potential mechanisms for their broad distribution and/or persistence in humans.
Among geographical regions, we observed differences in the prevalence of PCV-13 and non-PCV-13 (top 50) STs. In Thailand, 4/5 of the most frequently represented STs belonged to serotypes targeted by PCV-13, including ST4414 (of serotype 19F), a top 50 PCV-13 ST, found so far only in Asia, with 99.53% of the strains of this ST reported from Thailand (Figure S1). The high frequency of STs associated with PCV-13 is consistent with the lack of PCV immunization program in Thailand [32]. In South Korea, the introduction of PCV-13 resulted in the emergence of novel STs due to serotype replacement [33]. A similar pattern was found in other countries such as South Africa, the UK, and the USA where the implementation of PCV into their immunization programs [32] caused STs not associated with PCV-13 to increase in their relative prevalence in these countries.
Phylogenetic analyses of all STs revealed both divergent STs and closely related STs (Figure 1). Interestingly, our phylogenetic analyses revealed that two STs, ST14613 and ST17858, in the current MLST database for S. pneumoniae were very divergent from most other STs in the database. Further analyses through BLAST revealed that these two STs belonged to Streptococcus mitis, a close relative of S. pneumoniae. Significantly, S. mitis has been found to be a source for capsular polysaccharide variation for S. pneumoniae through horizontal gene transfer and can contribute to vaccine escape in S. pneumoniae [34].
Similar to the overall phylogenetic relationships among all STs, the top 50 most frequent STs in the database also showed both significant divergence and close relatedness among each other. Though small clusters of PCV-13 associated STs were found, STs associated with PCV-13 were overall inter-mixed with those not associated with PCV-13 (Supplementary Figures S2 and S3). Interestingly, 7 STs (ST156, ST81, ST63, ST193, ST320, ST695, and ST172) were each associated with more than one serotype covered by PCV-13. Three STs (ST199, ST162, and ST156) contained strains that were initially associated with PCV-13 prior to PCV implementation but changed their serotypes following PCV implementation. Previous literature has also identified ST199 switched from serotype 19A to 15B following PCV-7 implementation [35]. These findings suggest evidence of capsular switching, a process by which a new capsule operon is acquired through horizontal gene transfer (HGT) [36]. Due to flexibility and ease of DNA uptake in pneumococci, high rates of recombination via frequent HGT within the cps operon could have occurred, causing increases in non-PCV-13 serotypes [37]. Further genomic comparisons should help identify the relationship between STs and cps operon allele variation and how mutation and/or HGT might have impacted capsular switching and serotype changes in the global population.
Analysis of molecular variance revealed statistically significant genetic differentiations among geographical, temporal, and ecological populations of S. pneumoniae, rejecting the null hypothesis of no genetic differentiations among sub-populations in each the three types of analyses. However, in all cases, both the non-clone-corrected datasets and the clone-corrected datasets revealed that most genetic variations were found within subpopulations rather than between subpopulations. Among these, the highest inter-subpopulation differentiation (~12%) was observed for the non-clone corrected data between clinical and veterinary samples. Further investigation revealed that 46/112 veterinary isolates represented STs that were unique to veterinary niches. Among those 46 isolates, 26 belonged to ST6937 and 10 isolates belonged to ST6934. Localized selection and clonal expansion have likely contributed to the observed patterns. Differences between clinical and veterinary/environmental samples have also been reported in other microbial pathogens such as Campylobacter jejuni and Streptococcus agalactiae [38,39].
Population genetic analyses between pre- and post- PCV implementation revealed a decrease in relative frequency of PCV-13 associated STs and an increase in frequency of non-PCV-13 (top 50) STs as well as significant genetic differentiations between them based on AMOVA (Figure 2). These global patterns are consistent with previous literature in England and Wales which found that PCV implementation led to a rise in non-PCV serotype infections [40]. Interestingly in our analysis, we noticed a significant rise in some non-PCV-13 (top 50) STs that were not associated with new serotypes targeted by the upcoming PCV-20 (serotypes 35B, 23B, 9N, 15BC, and 6C). On the other hand, STs associated with serotypes 10A, 15B, and 33F, which are targeted by the new PCV-20, were not among the top 50 most represented STs. Thus, our results suggest that PCV-20 is not as optimized as it could be. We believe that further vaccine development and optimizations should take the global ST distributions into account to produce higher-valent PCVs. At the national level, we observed unique trends for countries that have implemented PCV and countries that have not (Figure S11). In Thailand, which has not implemented PCV into their national immunization program (NIP), the relative frequency of PCV-13 associated STs increased over time, reaching almost 100%. In countries that have implemented PCV into their NIP, a reduction in PCV-13 associated STs is typically seen after PCV-13/PCV-10 introduction together with an increase in non-PCV-13 (top 50) STs. This observation is consistent with the previous finding that ST4414, a unique PCV-13 ST only found in Asia, has contributed to increased PCV-13 ST frequency in Thailand (Figure S1).
S. pneumoniae has shown to be capable of importing DNA through transformation and homologous recombination, generating recombinant genotypes [41]. In this study, we found that strains of the same ST or closely related STs were often associated the same serotypes. However, differences were also found, consistent with recombination and horizontal gene transfer in natural populations of this species. In our analyses, all continent populations of S. pneumoniae showed evidence of recombination among the seven loci used for MLST. However, none of the analyzed populations showed evidence of random recombination, even in the clone-corrected samples.
In conclusion, global analyses of published MLST data for S. pneumoniae revealed great diversity and distribution of STs spatially, temporally, and phylogenetically. Analysis of molecular variance quantified the genetic variations within and among S. pneumoniae subpopulations. Implementation of PCV revealed an impact on both PCV-13 STs and non-PCV-13 (top 50) STs globally. We also demonstrated non-random association among alleles at the seven MLST loci. It is important to note that MLST data for S. pneumoniae only reflects data collected and deposited by researchers. The distributions observed in this study may underrepresent the actual distributions of S. pneumoniae. As new PCVs are being produced to reduce the incidence of pneumococcal diseases among global populations, the rise of non-PCV serotypes due to serotype replacement and capsular switching caused by recombination and horizontal gene transfer remains a major concern. At present, two new higher-valent PCVs, PCV-15 and PCV-20, are being implemented across the world to cover a wider range of serotypes causing pneumococcal diseases. However, information on the coverage and effectiveness of these new vaccines at the global level is scarce. At present, PCV-13 is still recommended by the US Centers for Disease Control and Prevention (CDC) for infants and younger children. However, as shown in our analyses, after PCV-13 implementation, new serotypes and new STs not associated with PCV-13 emerged and spread, reduced the effectiveness of PCV-13 [42]. New PCVs should be designed to target the most prevalent STs and serotypes that are not covered by previous PCVs in order to maximize the efficacy of the new vaccines. Indeed, genotype information from global isolates should be continuously deposited into the pubmlst database for monitoring the potential spatial and temporal patterns related to S. pneumoniae and pneumococcal diseases. Such data, in combination with those on host and environmental factors related to S. pneumoniae and pneumococcal diseases could help develop effective public health policies against this important human pathogen (Xu 2022).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Phylogenetic tree showing relationship among the top 50 globally most frequent sequence types (ST) observed in S. pneumoniae isolates deposited in PubMLST.org as of November 2022.; Figure S2: Phylogenetic tree showing the relationship among 146 STs associated with PCV-13 serotypes; Figure S3: Phylogenetic tree showing the relationship among 107 STs associated with non-PCV-13 serotypes but were present in the top 50 STs from each of the six continents; Figures S4-S10: Phylogenic tree relationships among allele types (AT) at seven housekeeping loci of S. pneumoniae; Figure S11: Line graphs with markers showing the relative isolate frequency (%) of PCV-13 associated STs (blue) and non-PCV-13 (top 50) associated STs (orange) over time (years) in six countries with the highest isolate counts in each respective continent. Table S1: Analysis of molecular variance of PCV-13 STs at the geographical level; Table S2: Analysis of molecular variance of non-PCV-13 (top 50) STs at the geographical level; Table S3: Analysis of molecular variance of PCV-13 and non-PCV-13 (top 50) STs at the country level over time; Table S4: Distribution of serotypes among different PCVs; Table S5: Temporal distribution of PCV-13 STs and non-PCV-13 (top 50) STs as of November 2022; Table S6: Geographical distribution of PCV-13 STs as of November 2022; Table S7: Geographical distribution of non-PCV-13 (top 50) STs as of November 2022.
Author Contributions
Conceptualization, J.X.; methodology, J.D., M.H.; software, J.D., M.H., J.X.; validation, J.D., M.H., J.X.; formal analysis, J.D.; investigation, J.D.; resources, M.H., J.X.; data curation, J.D.; writing—original draft preparation, J.D.; writing—review and editing, J.X.; visualization, J.D., M.H.; supervision, J.X.; project administration, J.X.; funding acquisition, J.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research and the APC were funded by the Global Science Initiative, grant number GSI2020-03.
Institutional Review Board Statement
Not applicable. The study analyzed publicly available data deposited in NCBI.
Informed Consent Statement
Not applicable.
Data Availability Statement
This study did not report any new raw data. All analytical results supporting our conclusions are reported in the main manuscript file and in supplementary files.
Acknowledgments
We thank all researchers who contributed to the S. pneumoniae pubMLST data analyzed here.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Ghaffar, F.; Friedland, I.R.; McCracken, G.H. Dynamics of nasopharyngeal colonization by Streptococcus pneumoniae. Pediatr. Infect. Dis. J. 1999, 18, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Middle ear infection (otitis media). Available online: https://www.nhsinform.scot/illnesses-and-conditions/ears-nose-and-throat/middle-ear-infection-otitis-media. (Accessed on 29 March 2023).
- Meningitis. Available online: https://www.mayoclinic.org/diseases-conditions/meningitis/symptoms-causes.(Accessed on 29 March 2023).
- Pneumonia. Available online: https://www.mayoclinic.org/diseases-conditions/pneumonia/symptoms-causes.(Accessed on 29 March 2023).
- Knott, L. Acute Otitis Media in Adults. Patient. 2022. [Google Scholar]
- Monasta, L.; Ronfani, L.; Marchetti, F.; Montico, M.; Brumatti, L.V.; Bavcar, A.; Grasso, D.; Barbiero, C.; Tamburlini, G. Burden of Disease Caused by Otitis Media: Systematic Review and Global Estimates. PLoS One. 2012, 7, e36226. [Google Scholar] [CrossRef] [PubMed]
- Regunath, H.; & Oba, Y. Community-Acquired Pneumonia. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430749/.(Accessed on 14 April 2023).
- Cilloniz, C. Pneumonia causes 2.5 million deaths around the world each year. Available online: https://www.clinicbarcelona.org/en/news/pneumonia-causes-2-5-million-deaths-around-the-world-each-year.(Accessed on 26 March 2023).
- World Health Organization. Pneumonia. Available online: https://www.who.int/health-topics/pneumonia.(Accessed on 26 March 2023).
- Centers for Disease Control and Prevention. Global Pneumococcal Disease & Vaccine. Available online: https://www.cdc.gov/pneumococcal/global.html.(Accessed on 21 March 2023).
- Ceyhan, M.; Gürler, N.; Ozsurekci, Y.; Keser, M.; Aycan, A.E.; Gurbuz, V.; Salman, N.; Camcioglu, Y.; Dinleyici, E.C.; Ozkan, S.; et al. Meningitis caused by Neisseria Meningitidis, Hemophilus Influenzae Type B and Streptococcus Pneumoniae during 2005–2012 in Turkey. Hum. Vaccin. Immunother. 2014, 10, 2706–2712. [Google Scholar] [CrossRef] [PubMed]
- Enright, M.C. & Spratt, B.G. A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiol. 1998, 144, 3049–3060. [Google Scholar]
- Hausdorff, W.P.; Bryant, J.; Paradiso, P.R.; Siber, G.R. Which pneumococcal serogroups cause the most invasive disease: implications for conjugate vaccine formulation and use, part I. Clin. Infect. Dis. 2000, 30, 100–121. [Google Scholar] [CrossRef]
- Moore, M.R.; Gertz Jr, R.E.; Woodbury, R.L.; Barkocy-Gallagher, G.A.; Schaffner, W.; Lexau, C.; Gershman, K.; Reingold, A.; Farley, M.; Harrison, L.H.; et al. Population Snapshot of Emergent Streptococcus pneumoniae Serotype 19A in the United States, 2005. J. Infect. Dis. 2008, 197, 1016–1027. [Google Scholar] [CrossRef]
- Ko, K.S. & Song, J. Evolution of erythromycin-resistant streptococcus pneumoniae from Asian countries that contains erm(B) and mef(A) genes. J. Infect. Dis. 2004, 190, 739–747. [Google Scholar]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome. Open. Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Seidl, K.; Leimer, N.; Marques, M.P.; Furrer, A.; Holzmann-Bürgel, A.; Senn, G.; Zbinden, R.; Zinkernagel, A.S. Clonality and antimicrobial susceptibility of methicillin-resistant Staphylococcus aureus at the University Hospital Zurich, Switzerland between 2012 and 2014. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 1–7. [Google Scholar] [CrossRef]
- Chongtrakool, P.; Puangprasart, U.; Phongsamart, W.; Tribuddharat, C.; Pummangura, C.; Srifuengfung, S. Invasive streptococcus pneumoniae serotype 19a in Thailand (2008-2018). Southeast. Asian. J. Trop. Med. 2022, 53, 73–90. [Google Scholar]
- Duarte, C.; Sanabria, O.; Moreno, J. Molecular characterization of Streptococcus pneumoniae serotype 1 invasive isolates in Colombia. Rev. Panam. Salud. Publica. 2013, 33, 422–426. [Google Scholar] [PubMed]
- Cassiolato, A.P.; Almeida, S.C.G.; Andrade, A.L.; Minamisava, R.; Brandileone, M.C. Expansion of the multidrug-resistant clonal complex 320 among invasive Streptococcus pneumoniae serotype 19A after the introduction of a ten-valent pneumococcal conjugate vaccine in Brazil. PLoS One. 2018, 13, e0208211. [Google Scholar] [CrossRef] [PubMed]
- Reinert, R.R.; Reinert, S.; van der Linden, M.; Cil, M.Y.; Al-Lahham, A.; Appelbaum, P. Antimicrobial Susceptibility of Streptococcus pneumoniae in Eight European Countries from 2001 to 2003. Antimicrob. Agents. Chemother. 2005, 49, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Golden, A.R.; Rosenthal, M.; Fultz, B.; Nichol, K.A.; Adam, H.J.; Gilmour, M.W.; Baxter, M.R.; Hoban, D.J.; Karlowsky, J.A.; Zhanel, G.G. Characterization of MDR and XDR Streptococcus pneumoniae in Canada, 2007–2013. J. Antimicrob. Chemother. 2015, 70, 2199–2202. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic. Acids. Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic. Acids. Res. 2021, 49, 293–296. [Google Scholar]
- MobaXterm documentation. Available online: https://mobaxterm.mobatek.net/documentation.html.(Accessed on 14 April 2023).
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Peakall, R. & Smouse, P. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinform. 2012, 28, 2537–2539. [Google Scholar]
- Agapow, P-M. & Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes. 2001, 1, 101–102. [Google Scholar]
- Gertz Jr., R. E.; McEllistrem, M.C.; Boxrud, D.J.; Li, Z.; Sakota, V.; Thompson, T.A.; Facklam, R.R.; Besser, J.M.; Harrison, L.H.; Whitney, C.G.; et al. Clonal distribution of invasive pneumococcal isolates from children and selected adults in the United States prior to 7-valent conjugate vaccine introduction. J. Clin. Microbiol. 2003, 41, 4194–4216. [Google Scholar]
- Serrano, I.; Melo-Cristino, J.; Carrico, J.A.; Ramirez, M. Characterization of the Genetic Lineages Responsible for Pneumococcal Invasive Disease in Portugal. J. Clin. Microbiol. 2005, 43, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- International Vaccine Access Center (IVAC). Available online: https://view-hub.org/vaccine/pcv.(Accessed on 3 April 2023).
- Kim, G.R.; Kim, E.; Kim, S.H.; Lee, H.K.; Lee, J.; Shin, J.H.; Kim, Y.R.; Song, S.A.; Jeong, J.; Uh, Y.; et al. Serotype Distribution and Antimicrobial Resistance of Streptococcus pneumoniae Causing Invasive Pneumococcal Disease in Korea Between 2017 and 2019 After Introduction of the 13-Valent Pneumococcal Conjugate Vaccine. Ann. Lab. Med. 2022, 43, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, G.; Junges, R.; Morrison, D.A.; Petersen, F.C. Competence in Streptococcus pneumoniae and Close Commensal Relatives: Mechanisms and Implications. Front. Cell. Infect. Microbiol. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Makarewicz, O.; Lucas, M.; Brandt, C.; Herrmann, L.; Albersmeier, A.; Ruckert, C.; Blom, J.; Goesmann, A.; van der Linden, M.; Kalinowski, J.; et al. Whole Genome Sequencing of 39 Invasive Streptococcus pneumoniae Sequence Type 199 Isolates Revealed Switches from Serotype 19A to 15B. PLoS. One. 2017, 12, e0169370. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, V.; Stevenson, A.; Figueira, M.; Orthopoulos, G.; Trzcinski, K.; Pelton, S.I. Capsular switching as a strategy to increase pneumococcal virulence in experimental otitis media model. Microbes. Infect. 2014, 16, 292–299. [Google Scholar] [CrossRef]
- Hanage, W.P.; Finkelstein, J.A.; Huang, S.S.; Pelton, S.I.; Stevenson, A.E.; Kleinman, K.; Hinrichsen, L.; Fraser, C. Evidence that pneumococcal serotype replacement in Massachusetts following conjugate vaccination is now complete. Epidemics. 2010, 2, 80–84. [Google Scholar] [CrossRef]
- Dingle, K.E.; Colles, F.M.; Wareing, D.R.; Ure, R.; Fox, A.J.; Bolton, F.E.; Bootsma, H.J.; Willems, R.J.; Urwin, R.; Maiden, M.C. Multilocus sequence typing system for Campylobacter jejuni. J. Clin. Microbiol. 2001, 39, 14–23. [Google Scholar] [CrossRef]
- Jones, N.; Bohnsack, J.F.; Takahashi, S.; Oliver, K.A.; Chan, M.; Kunst, F.; Glaser, P.; Rusniok, C.; Crook, D.W.M.; Harding, R.M.; et al. Multilocus sequence typing system for group B streptococcus. J. Clin. Microbiol. 2003, 41, 2530–2536. [Google Scholar] [CrossRef]
- Ladhani, S.N.; Collins, S.; Djennad, A.; Sheppard, C.L.; Borrow, R.; Fry, N.K.; Andrews, N.J.; Miller, E.; Ramsay, M.E. Rapid increase in non-vaccine serotypes causing invasive pneumococcal disease in England and Wales, 2000–2017: a prospective national observational cohort study. Lancet. Infect. Dis. 2018, 18, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Chaguza, C.; Andam, C.P.; Harris, S.R.; Cornick, J.E.; Yang, M.; Bricio-Moreno, L.; Kamng’ona, A.W.; Parkhill, J.; French, N.; Heyderman,; et al. Recombination in Streptococcus pneumoniae Lineages Increase with Carriage Duration and Size of the Polysaccharide Capsule. mBio. 2016, 7, 1053–16. [Google Scholar] [CrossRef] [PubMed]
- Pneumococcal Vaccines. Available online: https://www.immunize.org/askexperts/experts_pneumococcal_vaccines.asp.(Accessed on 19 April 2023).
- Xu, J. Assessing global fungal threats to humans. mLife, 2022, 1, 223–240. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic tree showing the relationships among 17915 sequence types (ST) of S. pneumoniae deposited in PubMLST as of November 2022. Innermost ring indicates ecological niche information. Middle ring indicates temporal information. Outermost ring indicates geographical information. Square symbols for each category represent the presence of that ST once in that population subset. Nodes with 200-500 leaves were collapsed for better visualization.
Figure 1.
Phylogenetic tree showing the relationships among 17915 sequence types (ST) of S. pneumoniae deposited in PubMLST as of November 2022. Innermost ring indicates ecological niche information. Middle ring indicates temporal information. Outermost ring indicates geographical information. Square symbols for each category represent the presence of that ST once in that population subset. Nodes with 200-500 leaves were collapsed for better visualization.
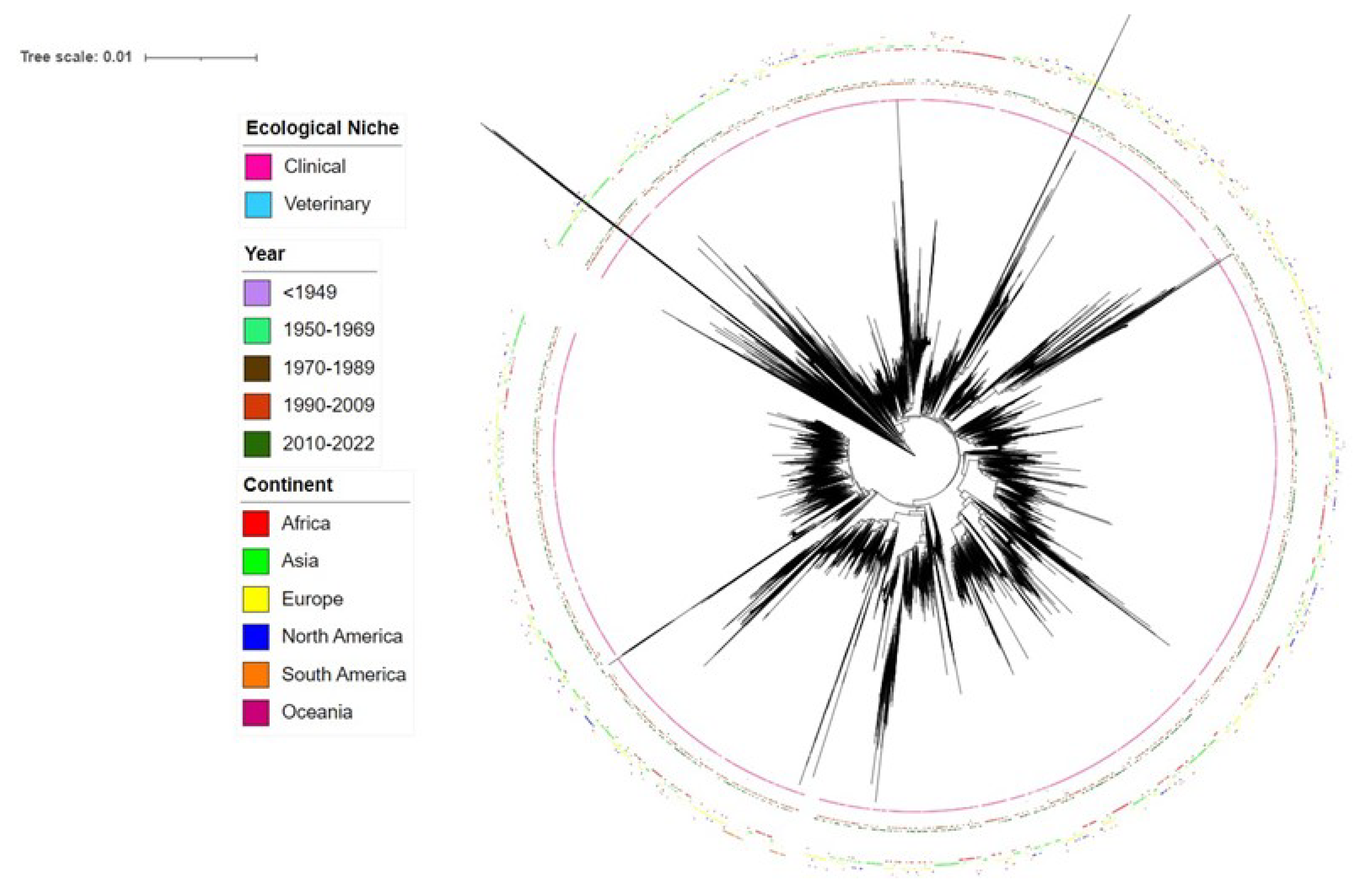
Figure 2.
Line graph with markers showing the relative isolate frequency (%) of PCV-13 associated STs (blue) and non-PCV-13 (top 50) associated STs (orange) over time (years). Purple line indicates initial implementation of PCV-13, in North America. Black line indicates implementation of PCV-13 globally. Y-axis values are standardized and are relative to the total population of isolates globally as of November 2022. Arrows indicate time intervals in which there were no isolates representing PCV-13 or non-PCV-13 STs collected.
Figure 2.
Line graph with markers showing the relative isolate frequency (%) of PCV-13 associated STs (blue) and non-PCV-13 (top 50) associated STs (orange) over time (years). Purple line indicates initial implementation of PCV-13, in North America. Black line indicates implementation of PCV-13 globally. Y-axis values are standardized and are relative to the total population of isolates globally as of November 2022. Arrows indicate time intervals in which there were no isolates representing PCV-13 or non-PCV-13 STs collected.
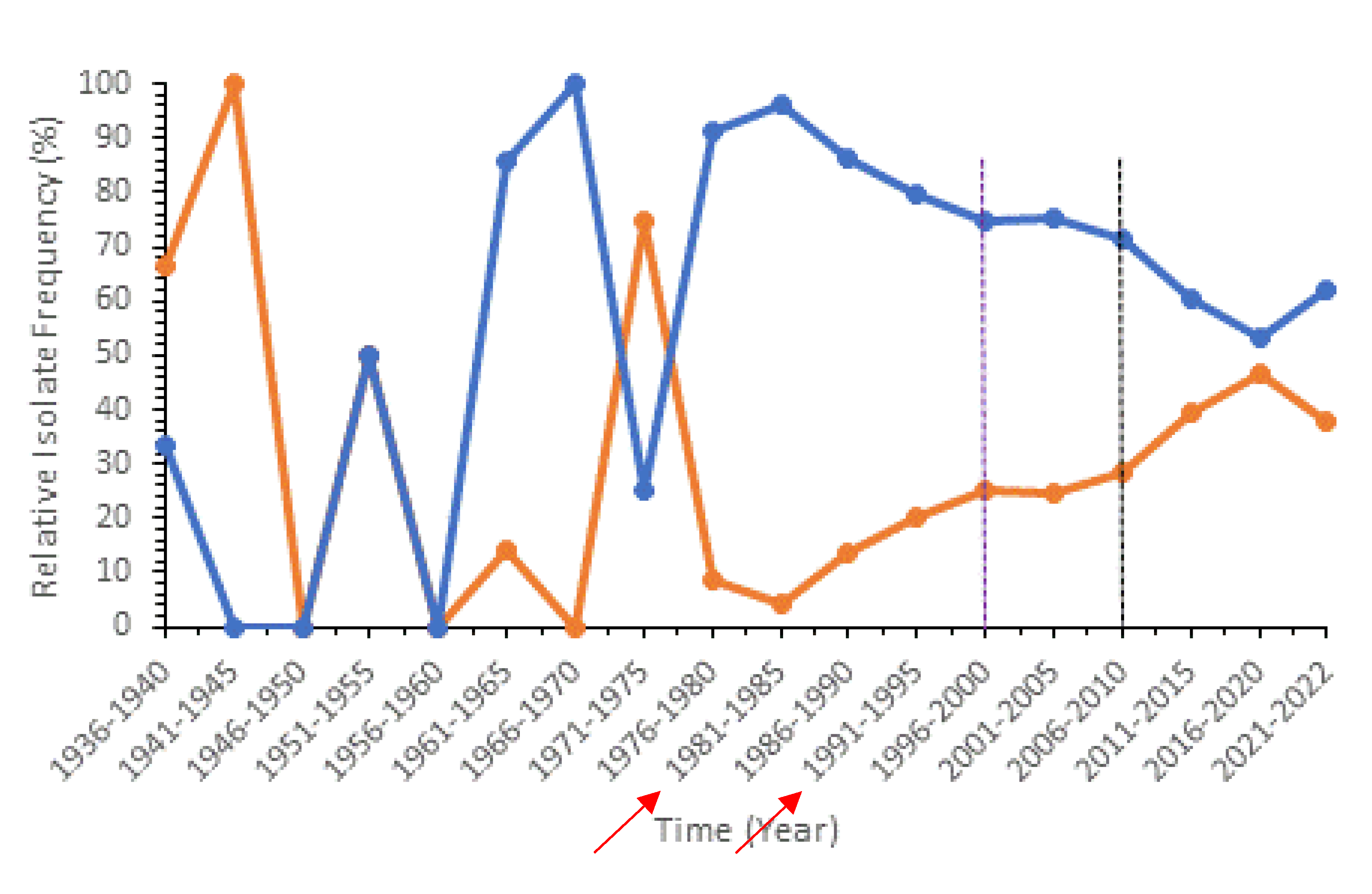

Table 1.
Geographic distributions of isolates of S. pneumoniae deposited within the PubMLST database.
Table 1.
Geographic distributions of isolates of S. pneumoniae deposited within the PubMLST database.
| Region | n(nST(s)) | % | Region | n(nST(s)) | % | Region | n(nST(s)) | % |
|---|---|---|---|---|---|---|---|---|
| Asia | 17303 | 23.274% | Africa | 19092 | 25.680% | Europe | 24090 | 32.403% |
| Thailand | 4412(521) | 5.934% | South Africa | 8587(2013) | 11.55% | UK | 6371(2323) | 8.569% |
| Japan | 3910(706) | 5.259% | The Gambia | 4307(696) | 5.793% | The Netherlands | 3962(610) | 5.329% |
| China | 1862(921) | 2.505% | Malawi | 2097(335) | 2.821% | Germany | 2625(551) | 3.531% |
| Israel | 1385(294) | 1.863 | Kenya | 1735(816) | 2.334% | Iceland | 2135(206) | 2.872% |
| Nepal | 1080(530) | 1.453 | Mozambique | 608(153) | 0.818% | Spain | 1771(696) | 2.382% |
| India | 1033(588) | 1.389 | Ethiopia | 326(181) | 0.438% | Czech Republic | 1386(362) | 1.864% |
| Cambodia | 847(224) | 1.139% | Ghana | 323(121) | 0.434% | Poland | 846(362) | 1.138% |
| South Korea | 552(262) | 0.742% | Nigeria | 261(141) | 0.351% | Russia | 806(376) | 1.084% |
| Taiwan | 371(270) | 0.499% | Togo | 162(52) | 0.218% | France | 742(247) | 0.998% |
| Bangladesh | 326(239) | 0.438% | Egypt | 154(84) | 0.207% | Portugal | 619(444) | 0.833% |
| Saudi Arabia | 325(234) | 0.437% | Niger | 149(44) | 0.2% | Norway | 403(394) | 0.542% |
| Qatar | 240(158) | 0.323% | Senegal | 79(40) | 0.106% | Finland | 309(196) | 0.416% |
| Singapore | 182(172) | 0.245% | Morocco | 63(36) | 0.085% | Italy | 290(136) | 0.39% |
| Malaysia | 161(93) | 0.217% | Tunisia | 50(50) | 0.067% | Belarus | 254(72) | 0.342% |
| Vietnam | 158(69) | 0.213% | Cameroon | 41(25) | 0.055% | Turkey | 231(160) | 0.311% |
| Pakistan | 147(108) | 0.198% | Burkina Faso | 38(36) | 0.051% | Denmark | 225(149) | 0.303% |
| Iraq | 89(42) | 0.12% | Tanzania | 33(18) | 0.044% | Greece | 220(59) | 0.296% |
| Myanmar | 58(39) | 0.078% | Gabon | 17(11) | 0.023% | Sweden | 216(114) | 0.291% |
| Kuwait | 37(32) | 0.05% | Botswana | 16(11) | 0.022% | Hungary | 172(91) | 0.231% |
| Sri Lanka | 26(18) | 0.035% | Uganda | 12(11) | 0.016% | Belgium | 158(25) | 0.213% |
| Mongolia | 25(7) | 0.034% | Congo [DRC] | 10(9) | 0.013% | Slovenia | 117(51) | 0.157% |
| Oman | 20(18) | 0.027% | C.A.R | 7(5) | 0.009% | Switzerland | 83(44) | 0.112% |
| Lebanon | 17(12) | 0.023% | Benin | 7(7) | 0.009% | Ireland | 36(32) | 0.048% |
| Philippines | 14(11) | 0.019% | Ivory Coast | 4(3) | 0.005% | Austria | 31(22) | 0.042% |
| Iran | 10(10) | 0.013% | Algeria | 3(2) | 0.004% | Bulgaria | 25(18) | 0.034% |
| Indonesia | 7(5) | 0.009% | Zambia | 2(2) | 0.003% | Lithuania | 14(9) | 0.019% |
| Syria | 5(4) | 0.007% | Sudan | 1(1) | 0.001% | Latvia | 11(9) | 0.015% |
| Jordan | 4(4) | 0.005% | Slovakia | 9(6) | 0.012% | |||
| Croatia | 8(6) | 0.011% | ||||||
| South America | 2893 | 3.891% | North America | 9465 | 12.731% | Romania | 8(8) | 0.011% |
| Brazil | 1331(640) | 1.79% | USA | 8683(2005) | 11.679% | Armenia | 5(4) | 0.007% |
| Peru | 1155(240) | 1.554% | Canada | 732(246) | 0.985% | Monaco | 1(1) | 0.001% |
| Trinidad and Tobago | 110(56) | 0.148% | Mexico | 42(33) | 0.056% | Azerbaijan | 1(1) | 0.001% |
| Argentina | 79(71) | 0.106% | Costa Rica | 4(1) | 0.005% | |||
| Venezuela | 49(25) | 0.066% | Guatemala | 2(2) | 0.003% | Oceania | 926 | 1.246% |
| Colombia | 46(39) | 0.062% | Cuba | 1(1) | 0.001% | Australia | 632(296) | 0.85% |
| Chile | 43(35) | 0.058% | Greenland | 1(1) | 0.001% | Papua New Guinea | 140(88) | 0.188% |
| Uruguay | 42(31) | 0.056% | New Zealand | 107(57) | 0.144% | |||
| Bolivia | 36(31) | 0.048% | Fiji | 32(15) | 0.043% | |||
| Ecuador | 2(2) | 0.003% | New Caledonia | 14(6) | 0.019% | |||
| Wallis and Futuna Islands | 1(1) | 0.001% |
n: total isolate(s), (nST(s)): total number of unique sequence type(s), %: percent of isolates from the continent/country out of the total S. pneumoniae isolates deposited in the MLST database.
Table 2.
Allelic profiles of the top 50 most frequent sequence types in the global population of S. pneumoniae.
Table 2.
Allelic profiles of the top 50 most frequent sequence types in the global population of S. pneumoniae.
| Sequence Type | n | aroE | gdh | gki | recP | spi | xpt | ddl |
|---|---|---|---|---|---|---|---|---|
| ST180* | 1461 | 7 | 15 | 2 | 10 | 6 | 1 | 22 |
| ST199* | 1363 | 8 | 13 | 14 | 4 | 17 | 4 | 14 |
| ST217* | 1099 | 10 | 18 | 4 | 1 | 7 | 19 | 9 |
| ST63* | 1060 | 2 | 5 | 36 | 12 | 17 | 21 | 14 |
| ST191* | 932 | 8 | 9 | 2 | 1 | 6 | 1 | 17 |
| ST433** | 825 | 1 | 1 | 4 | 1 | 18 | 58 | 17 |
| ST320* | 807 | 4 | 16 | 19 | 15 | 6 | 20 | 1 |
| ST81* | 761 | 4 | 4 | 2 | 4 | 4 | 1 | 1 |
| ST62** | 733 | 2 | 5 | 29 | 12 | 16 | 3 | 14 |
| ST558** | 703 | 18 | 12 | 4 | 44 | 14 | 77 | 97 |
| ST156* | 676 | 7 | 11 | 10 | 1 | 6 | 8 | 1 |
| ST53** | 508 | 2 | 5 | 1 | 11 | 16 | 3 | 14 |
| ST289* | 500 | 16 | 12 | 9 | 1 | 41 | 33 | 33 |
| ST236* | 493 | 15 | 16 | 19 | 15 | 6 | 20 | 26 |
| ST162* | 479 | 7 | 11 | 10 | 1 | 6 | 8 | 14 |
| ST338* | 470 | 7 | 13 | 8 | 6 | 1 | 6 | 8 |
| ST90* | 467 | 5 | 6 | 1 | 2 | 6 | 3 | 4 |
| ST306* | 460 | 12 | 8 | 13 | 5 | 16 | 4 | 20 |
| ST4414* | 426 | 15 | 16 | 19 | 15 | 6 | 20 | 4 |
| ST439** | 419 | 1 | 8 | 9 | 2 | 6 | 4 | 6 |
| ST9* | 410 | 1 | 5 | 4 | 5 | 5 | 1 | 8 |
| ST2062* | 407 | 1 | 5 | 53 | 32 | 14 | 20 | 199 |
| ST989** | 369 | 12 | 5 | 89 | 8 | 6 | 112 | 14 |
| ST124* | 368 | 7 | 5 | 1 | 8 | 14 | 11 | 14 |
| ST176* | 330 | 7 | 13 | 8 | 6 | 10 | 6 | 14 |
| ST230* | 329 | 12 | 19 | 2 | 17 | 6 | 22 | 14 |
| ST66** | 322 | 2 | 8 | 2 | 4 | 6 | 1 | 1 |
| ST218* | 318 | 10 | 20 | 14 | 1 | 6 | 1 | 29 |
| ST802* | 308 | 10 | 13 | 53 | 1 | 72 | 38 | 31 |
| ST193* | 304 | 8 | 10 | 2 | 16 | 1 | 26 | 1 |
| ST276* | 296 | 2 | 19 | 2 | 17 | 6 | 22 | 14 |
| ST172* | 292 | 7 | 13 | 8 | 6 | 25 | 6 | 8 |
| ST271* | 282 | 4 | 16 | 19 | 15 | 6 | 20 | 26 |
| ST393** | 267 | 10 | 43 | 41 | 18 | 13 | 49 | 6 |
| ST315* | 262 | 20 | 28 | 1 | 1 | 15 | 14 | 14 |
| ST1262** | 258 | 7 | 41 | 2 | 6 | 10 | 26 | 1 |
| ST242* | 256 | 15 | 29 | 4 | 21 | 30 | 1 | 14 |
| ST1692** | 252 | 1 | 5 | 7 | 12 | 17 | 158 | 14 |
| ST847* | 248 | 7 | 11 | 4 | 1 | 6 | 112 | 14 |
| ST618* | 243 | 13 | 8 | 4 | 1 | 7 | 19 | 14 |
| ST416* | 223 | 1 | 13 | 14 | 4 | 17 | 51 | 14 |
| ST3111* | 219 | 61 | 60 | 67 | 16 | 10 | 104 | 14 |
| ST448** | 217 | 8 | 5 | 2 | 27 | 2 | 11 | 71 |
| ST458* | 208 | 2 | 32 | 9 | 47 | 6 | 21 | 17 |
| ST1201* | 203 | 1 | 5 | 1 | 12 | 17 | 3 | 8 |
| ST3081* | 200 | 10 | 18 | 4 | 1 | 7 | 232 | 9 |
| ST36* | 199 | 1 | 8 | 4 | 1 | 1 | 4 | 6 |
| ST138* | 196 | 7 | 5 | 8 | 5 | 10 | 6 | 14 |
| ST473* | 196 | 7 | 25 | 4 | 4 | 15 | 20 | 28 |
| ST205* | 189 | 10 | 5 | 4 | 5 | 13 | 10 | 18 |
n: number of isolates; *, STs covered in PCV-13 vaccine; **, STs not covered in PCV-13.
Table 3.
Distribution of 17915 sequence types of S. pneumoniae at the geographical, ecological, and temporal levels.
Table 3.
Distribution of 17915 sequence types of S. pneumoniae at the geographical, ecological, and temporal levels.
| Distribution Patterns | Continent(s)/ecological niche(s)/year interval(s) | Number of sequence types | Number of isolates |
|---|---|---|---|
| Geographical | |||
| In all six continents | 36 | 15263 | |
| In five continents only | |||
| As+Eu+NA+SA+O | 7 | 1039 | |
| Af+Eu+NA+SA+O | 1 | 30 | |
| Af+As+NA+SA+O | 0 | 0 | |
| Af+As+Eu+SA+O | 0 | 0 | |
| Af+As+Eu+NA+O | 9 | 1419 | |
| Af+As+Eu+NA+SA | 10 | 2482 | |
| In four continents only | |||
| Eu+NA+SA+O | 3 | 245 | |
| Af+NA+SA+O | 0 | 0 | |
| Af+As+SA+O | 0 | 0 | |
| Af+As+Eu+O | 8 | 710 | |
| Af+As+Eu+NA | 18 | 1881 | |
| As+Eu+NA+SA | 7 | 905 | |
| As+NA+SA+O | 0 | 0 | |
| Af+As+NA+O | 0 | 0 | |
| Af+As+NA+SA | 1 | 18 | |
| Af+As+Eu+SA | 5 | 461 | |
| Af+Eu+NA+SA | 1 | 79 | |
| As+Eu+SA+O | 0 | 0 | |
| As+Eu+NA+O | 10 | 666 | |
| Af+Eu+NA+O | 2 | 104 | |
| Af+Eu+SA+O | 0 | 0 | |
| In three continents only | |||
| Af+As+Eu | 61 | 3049 | |
| As+Eu+NA | 48 | 1425 | |
| Eu+NA+SA | 15 | 369 | |
| NA+SA+O | 0 | 0 | |
| Af+SA+O | 0 | 0 | |
| Af+As+O | 1 | 15 | |
| Af+Eu+NA | 16 | 303 | |
| Af+Eu+SA | 7 | 85 | |
| Af+Eu+O | 3 | 116 | |
| Af+NA+O | 0 | 0 | |
| Af+NA+SA | 2 | 34 | |
| Af+As+NA | 5 | 95 | |
| Af+As+SA | 0 | 0 | |
| As+Eu+SA | 10 | 221 | |
| As+Eu+O | 0 | 0 | |
| Eu+NA+O | 14 | 766 | |
| Eu+SA+O | 1 | 4 | |
| As+SA+O | 0 | 0 | |
| As+NA+SA | 1 | 35 | |
| As+NA+O | 0 | 0 | |
| In two continents only | |||
| Af+As | 95 | 1728 | |
| Af+Eu | 117 | 1403 | |
| Af+NA | 21 | 162 | |
| Af+SA | 5 | 32 | |
| Af+O | 1 | 55 | |
| As+Eu | 179 | 1799 | |
| As+NA | 44 | 280 | |
| As+SA | 13 | 115 | |
| As+O | 8 | 105 | |
| Eu+NA | 150 | 1503 | |
| Eu+SA | 43 | 251 | |
| Eu+O | 36 | 226 | |
| NA+SA | 20 | 111 | |
| NA+O | 3 | 21 | |
| SA+O | 2 | 5 | |
| In one continent only | |||
| Af | 3968 | 11646 | |
| As | 4205 | 9164 | |
| Eu | 4813 | 7986 | |
| NA | 1770 | 2700 | |
| SA | 888 | 1725 | |
| O | 285 | 386 | |
| Ecological | |||
| In both niches | 21 | 4665 | |
| In one niche only | |||
| Clinical | 16987 | 69098 | |
| Veterinary | 11 | 21 | |
| Temporal | |||
| In all five intervals | 1 | 717 | |
| In four intervals only | |||
| Int 1+Int 2+Int 3+Int 4 | 0 | 0 | |
| Int 2+Int 3+Int 4+Int 5 | 7 | 942 | |
| Int 1+Int 3+Int 4+Int 5 | 2 | 403 | |
| Int 1+Int 2+Int 4+Int 5 | 2 | 41 | |
| Int 1+Int 2+Int 3+Int 5 | 1 | 4 | |
| In three intervals only | |||
| Int 1+Int 2+Int 3 | 0 | 0 | |
| Int 2+Int 3+Int 4 | 0 | 0 | |
| Int 3+Int 4+Int 5 | 53 | 9729 | |
| Int 1+Int 4+Int 5 | 9 | 418 | |
| Int 1+Int 2+Int 5 | 0 | 0 | |
| Int 1+Int 2+Int 4 | 1 | 3 | |
| Int 1+Int 3+Int 4 | 0 | 0 | |
| Int 1+Int 3+Int 5 | 0 | 0 | |
| Int 2+Int 4+Int 5 | 9 | 540 | |
| Int 2+Int 3+Int 5 | 0 | 0 | |
| In two intervals only | |||
| Int 1+Int 2 | 3 | 7 | |
| Int 2+Int 3 | 0 | 0 | |
| Int 3+Int 4 | 36 | 180 | |
| Int 4+Int 5 | 1532 | 31132 | |
| Int 1+Int 5 | 3 | 6 | |
| Int 1+Int 4 | 4 | 9 | |
| Int 1+Int 3 | 0 | 0 | |
| Int 2+Int 4 | 4 | 10 | |
| Int 2+Int 5 | 0 | 0 | |
| Int 3+Int 5 | 2 | 5 | |
| in one interval only | |||
| Int 1 | 23 | 25 | |
| Int 2 | 43 | 63 | |
| Int 3 | 128 | 185 | |
| Int 4 | 6727 | 10505 | |
| Int 5 | 6706 | 9882 |
As: Asia; Af: Africa; Eu: Europe; NA: North America; SA: South America; O: Oceania; Int 1: <1949; Int 2: 1950-1969; Int 3: 1970-1989; Int 4: 1990-2009; Int 5: 2010-2022.
Table 4.
Geographical and temporal distribution of top five STs collected per region or time interval.
Table 4.
Geographical and temporal distribution of top five STs collected per region or time interval.
| Geographical | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Thailand (n=4412) | South Africa (n=8587) | UK (n=6371) | USA (n=8683) | Brazil (n=1331) | Australia (n=632) | ||||||
| Sequence Type | % (n) | Sequence Type | % (n) | Sequence Type | % (n) | Sequence Type | % (n) | Sequence Type | % (n) | Sequence Type | % (n) |
| ST4414* | 9.61% (424) | ST217* | 5.38% (462) | ST199*** | 3.61% (230) | ST199*** | 6.25% (543) | ST156*** | 5.48% (73) | ST63* | 5.22% (33) |
| ST802* | 3.81% (168) | ST2062* | 4.41% (379) | ST62** | 2.94% (187) | ST558** | 6.22% (540) | ST66** | 2.33% (31) | ST191* | 5.06% (32) |
| ST315* | 3.22% (142) | ST230* | 1.89% (162) | ST439** | 2.21% (141) | ST320* | 3.66% (318) | ST1118* | 2.03% (27) | ST306* | 4.43% (28) |
| ST4413* | 2.72% (120) | ST53** | 1.75% (150) | ST162*** | 2.01% (128) | ST338* | 3.54% (307) | ST180* | 1.95% (26) | ST199*** | 3.96% (25) |
| ST4133 | 2.65% (117) | ST1094* | 1.68% (144) | ST433** | 1.70% (108) | ST63* | 2.52% (219) | ST733* | 1.73% (23) | ST66** | 2.69% (17) |
| Temporal | |||||||||||
| ≤ 2000 (n= 6499) | 2001-2005 (n= 7211) | 2006-2010 (n= 25497) | 2011-2015 (n=20227) | 2016-2020 (n=5459) | 2021-2022 (n=395) | ||||||
| Sequence Type | % (n) | Sequence Type | % (n) | Sequence Type | % (n) | Sequence Type | % (n) | Sequence Type | % (n) | Sequence Type | % (n) |
| ST81* | 4.66% (303) | ST199*** | 2.34% (169) | ST199*** | 1.86% (475) | ST180* | 2.61% (527) | ST180* | 3.5% (191) | unclassified | 49.62% (196) |
| ST90* | 2.92% (190) | ST217* | 1.98% (143) | ST4414* | 1.64% (418) | ST63* | 2.47% (499) | ST433** | 2.02% (110) | ST180* | 2.03% (8) |
| ST199*** | 2.75% (179) | ST81* | 1.82% (131) | ST217* | 1.59% (406) | ST558** | 1.99% (403) | ST53** | 1.61% (88) | ST1262** | 0.76% (3) |
| ST124* | 1.89% (123) | ST9* | 1.69% (122) | ST320* | 1.59% (406) | ST433** | 1.78% (361) | ST416* | 1.39% (76) | ST81* | 0.51% (2) |
| ST180* | 1.65% (107) | ST156*** | 1.53% (110) | ST63* | 1.49% (381) | ST199*** | 1.76% (355) | ST320* | 1.34% (73) | ST17503 | 0.51% (2) |
n: number of isolates; %: percent of isolates relative to respective geographical/temporal population; *, PCV-13 ST; **, non-PCV-13 (top 50) ST; ***, PCV-13 and non-PCV-13 (top 50) ST.
Table 5.
The number of allele types and the most frequent alleles in the global population of S. pneumoniae.
Table 5.
The number of allele types and the most frequent alleles in the global population of S. pneumoniae.
| Gene | Gene name | Length (bp) | Total number of ATs | Most frequent AT | Most frequent AT % (n) |
|---|---|---|---|---|---|
| aroE | shikimate dehydrogenase | 405 | 614 | 7* | 19.58% (14 556) |
| gdh | phosphate dehydrogenase | 460 | 790 | 5* | 22.93% (17 047) |
| gki | glucose kinase | 483 | 821 | 4* | 22.89% (17 010) |
| recP | transketolase | 450 | 557 | 1* | 23.46% (17 443) |
| spi | signal peptidase I | 474 | 771 | 6* | 35.31% (26 252) |
| xpt | xanthine phosphoribosyltransferase | 486 | 1078 | 1* | 22.96% (17 069) |
| ddl | d-alanine-d-alanine ligase | 441 | 1157 | 14* | 25.78% (19 169) |
Bp: base pair; %: percent of relative to total S. pneumoniae population; n: number of isolates; *, represented by PCV-13 STs and non-PCV-13 STs.
Table 6.
Results of analysis of molecular variance among geographical populations.
| Clone-Corrected | ||||
| df | MS | Est. Var | % | |
| Total Population | ||||
| Among continents | 5 | 72.252 | 0.013 | 0.411%** |
| Among countries | 78 | 10.452 | 0.034 | 1.074%** |
| Within countries | 21804 | 3.119 | 3.119 | 98.515%** |
| Total | 21887 | 3.165 | 100% | |
| Non-Clone-Corrected | ||||
| df | MS | Est. Var | % | |
| Africa | ||||
| Among countries | 19 | 51.117 | 0.066 | 2.086%** |
| Within countries | 19031 | 3.098 | 3.098 | 97.914%** |
| Total | 19050 | 3.164 | 100% | |
| Asia | ||||
| Among countries | 23 | 63.727 | 0.095 | 3.025%** |
| Within countries | 17105 | 3.044 | 3.044 | 96.975%** |
| Total | 17128 | 3.140 | 100% | |
| Europe | ||||
| Among countries | 26 | 49.909 | 0.059 | 1.914%** |
| Within countries | 23809 | 3.024 | 3.024 | 98.086%** |
| Total | 23835 | 3.083 | 100% | |
| North America | ||||
| Among countries | 2 | 64.164 | 0.085 | 2.678%** |
| Within countries | 9430 | 3.089 | 3.089 | 97.322%** |
| Total | 9432 | 3.174 | 100% | |
| South America | ||||
| Among countries | 8 | 18.667 | 0.070 | 2.269%** |
| Within countries | 2844 | 3.015 | 3.015 | 97.731%** |
| Total | 2852 | 3.085 | 100% | |
| Oceania | ||||
| Among countries | 4 | 17.249 | 0.124 | 3.920%** |
| Within countries | 915 | 3.039 | 3.039 | 96.080%** |
| Total | 919 | 3.163 | 100% |
df: degrees of freedom; MS: mean square; Est. Var: estimated variance; %: percentage of variance; **, p<0.01.
Table 7.
Analysis of molecular variance at the temporal level.
| Clone-Corrected | ||||
| df | MS | Est. Var | % | |
| Total Population | ||||
| Among continents | 5 | 66.615 | 0.020 | 0.631%** |
| Among time intervals | 29 | 6.383 | 0.006 | 0.189%** |
| Within time intervals | 19775 | 3.143 | 3.143 | 99.180%** |
| Total | 19809 | 3.169 | 100% | |
| Non-Clone-Corrected | ||||
| df | MS | Est. Var | % | |
| Africa | ||||
| Among time intervals | 5 | 35.897 | 0.014 | 0.444%** |
| Within time intervals | 16413 | 3.142 | 3.142 | 99.556%** |
| Total | 16418 | 3.156 | 100% | |
| Asia | ||||
| Among time intervals | 5 | 46.212 | 0.019 | 0.607%** |
| Within time intervals | 16002 | 3.112 | 3.112 | 99.393%** |
| Total | 16007 | 3.131 | 100% | |
| Europe | ||||
| Among time intervals | 5 | 42.549 | 0.014 | 0.454%** |
| Within time intervals | 17537 | 3.068 | 3.068 | 99.546%** |
| Total | 17542 | 3.083 | 100% | |
| North America | ||||
| Among time intervals | 5 | 83.128 | 0.064 | 2.053%** |
| Within time intervals | 8188 | 3.054 | 3.054 | 97.947%** |
| Total | 8193 | 3.118 | 100% | |
| South America | ||||
| Among time intervals | 4 | 15.304 | 0.034 | 1.109%** |
| Within time intervals | 2426 | 3.034 | 3.034 | 98.891%** |
| Total | 2430 | 3.067 | 100% | |
| Oceania | ||||
| Among time intervals | 5 | 8.545 | 0.046 | 1.481%** |
| Within time intervals | 776 | 3.059 | 3.059 | 98.519%** |
| Total | 781 | 3.105 | 100% |
df: degrees of freedom; MS: mean square; Est. Var: estimated variance; %: percentage of variance; **, p<0.01.
Table 8.
Analysis of molecular variance at the ecological level.
| Non-Clone-Corrected (Europe) | Clone-Corrected (global) | |||||||
|---|---|---|---|---|---|---|---|---|
| df | MS | Est. Var | % | df | MS | Est. Var | % | |
| Total Population | ||||||||
| Among ecological niches | 1 | 97.238 | 0.422 | 12.078%** | 1 | 6.312 | 0.049 | 1.522%** |
| Within ecological niches | 23 827 | 3.072 | 3.072 | 87.922%** | 17 038 | 3.171 | 3.171 | 98.478%** |
| Total | 23 828 | 3.494 | 100% | 17 039 | 3.220 | 100% |
df: degrees of freedom; MS: mean square; Est. Var: estimated variance; %: percentage of variance; **, p<0.01.
Table 9.
Summary of genotypic diversity and phylogenetic incompatibility based on the sequence type and serotype information assigned by the MLST database.
Table 9.
Summary of genotypic diversity and phylogenetic incompatibility based on the sequence type and serotype information assigned by the MLST database.
| Population | Number | Phylogenetic Compatibility (% of 21 Pairs) | Index of Association |
|---|---|---|---|
| Africa | 4393 | 0% | 0.89** |
| Asia | 4781 | 0% | 0.89** |
| Europe | 5630 | 0% | 0.87** |
| North America | 2214 | 0% | 0.86** |
| South America | 1078 | 0% | 0.85** |
| Oceania | 430 | 0% | 0.89** |
| PCV* | 246 | 4.76% | 0.55** |
*, PCV-13 and non-PCV STs were used; **, p<0.01.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



