
Submitted:
03 May 2023
Posted:
04 May 2023
You are already at the latest version
Abstract
Platinum-resistant high-grade serous ovarian cancer (HGSOC) is invariably a fatal disease, so a central goal in ovarian cancer research is to develop novel strategies to overcome platinum resistance. Treatment is thus moving towards personalized therapy. However, validated molecular biomarkers to predict patients’ risk of developing platinum resistance are still lacking. Extracellular vesicles (EVs) are promising candidates for biomarkers. EpCAM-specific EVs are largely unexplored biomarkers in predicting chemoresistance. Using transmission electron microscopy, nanoparticle tracking analysis and flow cytometry, we compared the characteristics of EVs released from a cell line derived from a clinically confirmed cisplatin-resistant patient (OAW28) and EVs released from two cell lines from tumors sensitive to platinum-based chemotherapy (PEO1, OAW42). We showed that characteristics of EVs released from the HGSOC cell line of chemoresistant patients had higher heterogeneity in size, a larger proportion of medium/large (>200nm) EVs and a higher number of released EpCAM-positive EVs of different sizes, although expression of EpCAM predominated, especially on those larger than 400 nm. We also determined a strong positive correlation between the concentration of EpCAM-positive EVs and expression of cellular EpCAM. These results may contribute to prediction of platinum resistance in the future, although they should first be validated in clinical samples.
Keywords:
cell lines
; chemoresistance
; EpCAM
; extracellular vesicles
; HGSOC 1
1. Introduction
Ovarian cancer (OC), despite its infrequent incidence, is the most lethal malignancy of the female reproductive system, with most patients presenting with advance stage tumors (FIGO stage III, IV) [1]. Although there has been an overall decreasing trend of incidence and mortality of OC over the past decade, a substantial increase in incidence has been observed in younger females [2]. In addition to the late detection of the disease, the main reason for the poor prognosis is resistance to pharmacotherapy. Despite novel targeted drugs such as poly (ADP-ribose) polymerase inhibitors (PARPis), carboplatin with added paclitaxel remains the standard first-line treatment for advanced OC. Moreover, PARPis are indicated for maintenance treatment in the first line and relapsed settings of patients who have responded to platinum-based medicine [3,4]. Since platinum-resistant OC is invariably a fatal disease, a central goal in OC research is the development of novel strategies to overcome platinum resistance. Treatment for OC is therefore moving towards personalized therapy. However, selection criteria are not entirely clear, since at present there are no validated molecular predictive biomarkers for platinum resistance [4].
One of the major challenges in the search for clinical biomarkers for OC is that OC is not a single disease, but rather a myriad of different malignancies with different targets that share a common anatomical location at presentation. In our article, therefore, we have focused on the high-grade serous subtype of ovarian cancer (HGSOC), as the dominant and most lethal subtype of OC [5].
In recent years, tumor-derived extracellular vesicles (EVs) have received significant attention as a promising source of minimally invasive biomarkers, including in OC [6,7,8,9]. According to the International Society of Extracellular Vesicles (ISEV), the term EVs is appropriate terminology for heterogeneous populations of cell-derived nanoscale vesicles released under physiological and pathological conditions, which can be isolated from cell culture supernatant or body fluids [10]. Due to their important role in communication between cells [11], it is not surprising that EVs’ cargo, concentration and size have been shown to be unique in tumor-states. EVs are therefore ideal candidates for aiding prediction of chemoresistance to platinum-based medicines. Although among published studies, those showing that the total concentration of OC-derived EVs has the potential for providing information on diagnosis predominate [12,13,14], the results of some studies do not confirm this [7,15], so the question arises as to whether determining the EV concentration alone is sufficient for OC diagnosis. Apart from one preliminary study, the conclusion of which was that determining the EVs’ protein level could be useful in evaluating patients’ response to therapy, we did not find any published study on the importance of determining the total EV concentration as a predictive biomarker in OC [16].
Among specific EV cargos, particular attention has been paid to the epithelial cellular adhesion molecule (EpCAM) [6,7,14,17,18]. Its expression is upregulated in solid epithelial cancer and stem cells. EpCAM can be found in disseminated tumor cells, circulating tumor cells and EVs [19]. However, (to our surprise), the relevance of analysis of EVs in predicting chemoresistance has been largely unexplored. Research on EpCAM specific EVs might also shed light on the contradictory results of immunohistochemical evaluation of EpCAM overexpression in ovarian tumors in relation to its prognostic value, in terms of overall survival and response to chemotherapy [20,21,22,23]. Moreover, the determination of EpCAM specific EVs might be helpful in evaluating the potential of therapeutic strategies targeting EpCAM and could explain the disparate results sometimes obtained concerning the effectiveness of EpCAM-based medicine [24,25].
Patients’ body fluids, especially blood, as a source of EVs for the study of biomarkers, have several limitations, principally the extremely high heterogeneity of vesicles and low proportion of EVs of tumor origin [26,27]. EVs isolated from conditioned media of relevant cultured cells can therefore elucidate these shortcomings. Moreover, the use of well-characterized cancer cell lines to model HGSOC in vitro can be particularly valuable in the age of personalized therapy. They at least increase the likelihood that the conclusions reached in an in vitro setting will be transferable to a clinical one. Unfortunately, many of the top cell lines in terms of publication figures poorly recapitulate the genetic features of HGSOC [28,29,30]. In contrast, most suitable cell lines for an HGSOC model (e.g. KURAMOCHI; COV362, OAW28) are rarely used in laboratories [28]. An additional problem in cell-line models is a lack of patient-derived cell lines of chemoresistant HGSOC. The majority of cell lines used to represent platinum-resistant disease have been generated in vitro by exposing cells to platinum-based medicines. The type of drug exposure used to elicit the resistant phenotype is therefore unrealistic and, as a consequence, the mechanism is likely to be different from those acting within a patient's body [5].
In order to identify the characteristics of EVs that have the potential to predict resistance to platinum-based medicine in HGSOC, we compared EVs released from a cell line derived from a clinically confirmed chemoresistant patient and EVs released from two cell lines from tumors sensitive to platinum-based chemotherapy. We showed that the EV concentration did not differ significantly, although a significantly higher concentration of EpCAM specific EVs and a higher proportion of medium/large particles was typical of the chemoresistant HGSOC cell line. A strong correlation between EpCAM specific EV concentration and cell membrane EpCAM expression supports the use of EpCAM specific EVs as cellular surrogates for EpCAM as a biomarker. Although the results of the current study should be validated in clinical samples, they reveal novel characteristics of EVs that may have the potential to aid the prediction of platinum resistant HGSOC and thereby enable the selection of personalized therapy.
2. Results
EVs were isolated from conditioned media of three serous ovarian cancer cell lines by the commercial reagent. One cell line was derived from a clinically confirmed cisplatin-chemoresistant patient (OAW28) and the other two were derived from patients with tumors sensitive to platinum-based chemotherapy (PEO1, OAW42). A graphic presentation of the methods is shown in Figure 1. We first confirmed the presence of EVs in isolated samples by transmission electron microscopy (TEM). The micrographs showed that the EVs were successfully isolated from conditioned media of all three cell lines (Figure 2A-C). We then determined the EVs' characteristics by different complementary methodologies to identify possible differences between EVs released from cells derived from chemoresistant and those released from cells derived from sensitive tumors. We used the following methods: 1) TEM, 2) nanoparticle tracking analysis in scatter mode (S-NTA) and 3) fluorescence-triggered flow cytometry (FT-FCM). The results of the EVs' concentration were normalized by the number of viable cells. Enumeration of EVs by FT-FCM was performed using volumetric measurement (events/mL). EpCAM-positive vesicles were also specifically isolated using anti-EpCAM magnetic beads and analysed by bead-based flow cytometry (BB-FCM). Additionally, the expression of EpCAM on cell membranes was analyzed using FCM.
2.1. Size distribution of EVs: OC cells derived from the clinically confirmed chemoresistant patient released more medium/large EVs than OC cells from patients with tumours sensitive to platinum-based chemotherapy
TEM analysis showed that isolates from conditioned media of OAV28, PEO1 and OAW42 cells consisted of EVs with a spherical shape morphology (Figure 2A-C). Variations in the size of EVs between cell types were observed.
In order to define further the size distribution of EVs, we used S-NTA. S-NTA is the appropriate method for determining the distribution of sizes because it is based on the tracking of individual particles. Since S-NTA is not specific for EVs and detects any structure that scatters light, we refer to ''particles'' rather than ''vesicles'' in this context. The size distribution of particles from all three cell lines appeared non-symmetrical. with the highest peak ranging between approximately 100 to 140 nm in diameter and several peaks at multiple diameters greater than 200 nm (Figure 3A-C). The modal diameter of OAW28-derived EVs (134.8 ± 6.3 nm) was significantly larger than that from PEO1-derived EVs (107.3 ± 3.4 nm; p = 0.007), although no significant difference in modal diameter was found in comparison to OAW42-derived EVs (137.3 ± 4.2 nm; Figure 3D). On the other hand, NTA revealed a significantly larger mean diameter of EVs from OAW28 cells (234.4 ± 1.6 nm) than those originating from the other two cell lines (p < 0.001) (Figure 3D), which measured 162.5 ± 5.9 nm and 17.3 ± 7.3 nm in PEO1 cell and OAW42 cells, respectively. We therefore took a closer look at the distribution of sizes represented by so-called percentile values. The D10/D50/D90 values were as follows: OAW28-derived EVs: 123.8/206.7/379.7 nm; PEO1-derived EVs: 101.6/139.9/259 nm; OAW42-derived EVs: 79.5/133.3/251 nm. All percentile values of EVs derived from OAW28 cells were significantly (p < 0.001) higher than those of PEO1-EVs and OAW42-EVs (Figure 3D). The D50 value (206.7 ± 7.2 nm) of OAW28-derived EVs showed that approximately 50 % of released EVs were medium/large in size (> 200 nm) (Figure 3D), confirmed also by TEM (Figure 2A). PEO1 and OAW42 released fewer medium/large in size (> 200 nm) EVs, 23 % and 15 %, respectively (Figure 3B-C). On the other hand, PEO1-EVs and OAW42-EVs did not significantly differ in D50 and D90 values. However, a higher proportion of EVs below 100 nm was revealed by the D10 value of OAW42-derived EVs (79.5 ± 8.4 nm) which was statistically significantly smaller compared to OAW28-derived EVs (139.9 ± 3.9 nm; p < 0.001) and to PEO1-derived EVs (133.3 ± 6.4 nm; p < 0.02) (Figure 3D).
2.2. EV concentration: the determination of the level of EVs released from OC cells from the clinically confirmed chemoresistant patient is possibly not enough to predict the response to platinum-based therapy
To determine the EV concentrations, the same samples were analyzed by two methods, S-NTA and FT-FCM (Figure 4A-B). As already noted, we refer to ''particles'' rather than ''vesicles'' in the context of S-NTA analysis. The mean concentration of particles detected by S-NTA for OAW28 cells was 6.12 ± 0.36 × 108/ml per 106 cells and the particle concentrations of PEO1 and OAW cells were 5.43 ± 0.08 × 108/ml and 2.48 ± 0.15 × 108/ml per 106 cells, respectively (Figure 4A). For the FT-FCM analysis, we labelled the EVs with calcein. Calcein is activated by an enzyme within an EV, so its advantage is the differentiation of intact EVs from membrane fragments [33,34]. The results of FT-FCM showed that OAW28 cells produced 2.11 ± 0.07 × 108/ml calcein-positive EVs per 106 cells whereas PEO1 and OAW42 cells produced 2.27 ± 0.03 × 108/ml and 0.7 ± 0.02 × 108/ml calcein-positive EVs per 106 cells, respectively, in the same time period (Figure 4B). According to the results of both methods, the number of OAW28-derived calcein-positive EVs detected by FT-FCM and OAW28-derived particles detected by S-NTA did not significantly differ from those of PEO1 cells. However, the number of calcein-positive EVs/particles released from OAW42 cells was significantly lower (p < 0.001) than the number of calcein-positive EVs/particles released from OAW28 and PEO1 cells (Figure 4A-B). Despite the lower particle number detected by FT-FCM, the two methods, FT-FCM and S-NTA, produced results with high concordance (r = 0.95, p < 0.001).
2.3. EpCAM specific EVs: a promising biomarker to predict chemoresistance to platinum-based therapy
EpCAM-positive vesicles were detected by FT-FCM (Figure 5A). The mean concentration of EpCAM-positive EVs for OAW28 cells was 9.19 ± 0.32 × 107/ml per 106 cells, while for PEO1 and OAW cells the concentrations were 1.78 ± 0.16 × 107/ml and 0.25 ± 0.005 × 107/ml per 106 cells, respectively, released in the same time period (Figure 5A left axis). The mean concentration of EpCAM-positive EVs released from OAW28 cells was significantly higher (p < 0.001) than the mean concentration released from PEO1 cells and from OAW42 cells. In addition, the number of EpCAM-positive EVs released from PEO1 was significantly higher (p = 0.002) than the number of EVs released by OAW42.
To confirm the results of FT-FCM analysis, EVs were also specifically isolated using anti-EpCAM magnetic beads and analyzed by BB-FCM (Figure 5A right axis). The results obtained by this method confirmed that the number of EpCAM-positive EVs released from OAW28 (80.23 ± 2.72 % EpCAM-positive beads) was significantly higher (p < 0.001) than the number of EpCAM-positive EVs released from PEO1 (37.34 ± 1.2 % EpCAM-positive beads) or OAW42 (17.78 ± 1.45 % EpCAM-positive beads) cells. Even so, the concentration of PEO1-EVs was significantly higher (p < 0.001) than those released from OAW42 cells.
The presence of EpCAM on the membrane of EVs was also confirmed by TEM. TEM showed the presence of EpCAM in the different size ranges of the larger and smaller EVs (Figure 5B). However, immunogold labelling predominated in EVs that were ≥ 400 nm in size. A higher number of EpCAM molecules was found on larger EVs (Figure 5B).
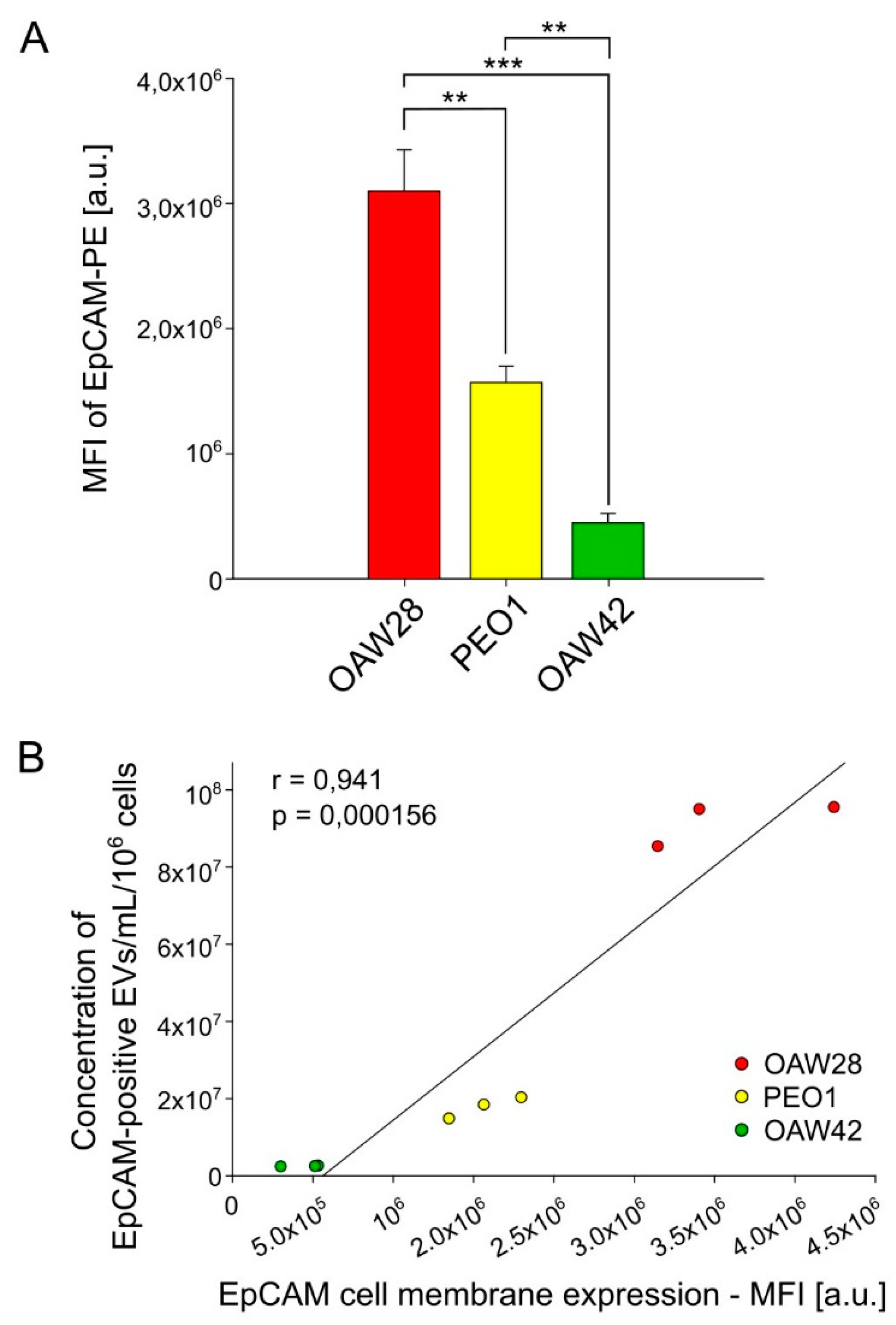
If EpCAM-positive EVs are generated by outward budding from the plasma membrane, then a higher expression of EpCAM on the plasma membrane should generate a higher level of EpCAM-positive EVs. To ascertain whether this positive correlation exists, we determined the EpCAM expression on the plasma membrane by FCM (Figure 6A-B). The expression of EpCAM on the cell membrane was significantly higher on the OAW28 cell line than on the PEO1 (p = 0.004) and OAW42 (p < 0.001) cell lines (Figure 6A). EpCAM expression was also significantly higher on the PEO1 cell line than on the OAW42 cell line (p = 0.009). We next compared the expression of EpCAM on the cell membrane with the concentration of EpCAM-positive EVs. The cellular protein and EpCAM-positive EV concentration showed a strong positive correlation (r = 0.950, p < 0.001, Figure 6B).
3. Discussion
Despite novel targeted drugs, platinum-resistant ovarian cancer remains a fatal disease. Moreover, there are currently no validated molecular predictive biomarkers for platinum resistance. Identification of novel predictive biomarkers to circumvent platinum resistance is thus highly desirable [4]. Tumour-derived EVs are one promising source of such biomarkers, especially since their cargo, concentration and size have been shown to be unique in tumour-states [35,36,37,38,39]. EV publications in the field of OC chemoresistance are primarily dedicated to research of EV-mediated drug-resistance mechanisms [39,40]. The main proposed roles of EVs in this field are drug-export, antigen sink, transfer of resistant phenotype and change in the tumour microenvironment [37,39]. However, the potential of EVs for predicting response to platinum-based therapy in OC has been largely unexplored.
In this study, three OC cell lines, OAW28, OAW24 and PEO1, were used as the EV source, in order to examine the characteristics of EVs from a patient who did not respond to cisplatin (OAW28) compared to two patients with a complete response (PEO1, OAW42) [31,32]. Since EVs are a promising tool for liquid biopsy, we chose cell lines that had been isolated from patients’ ascites [31,32]. We ensured that all three cell lines were well characterized. The OAW28 cell line is among the twelve best candidates for in vitro studies of HGSOC. Evaluation was made on the basis of correlation of genomic characteristics between tumour samples and cell lines. For this purpose, Domcke et.al. made a data comparison between The Cancer Genome Atlas, (TCGA) and The Broad-Novartis Cancer Cell Line Encyclopaedia (CCLE) [28]. Moreover, among 39 OC cell lines tested, the highest concentration of carboplatin causing 50 % growth inhibition was determined for the OAW28 cell line [29]. In view of novel strategies in the therapy of HGSOC, it is also important that OAW28 is without BRCA1/2 mutations [28,29]. For direct comparison of HGSOC EV characteristics, we chose another HGSOC cell line, PEO1 [29,30]. Interestingly, the PEO1 cell line has a reversion of the deleterious BRCA2 mutation as evidence of selective pressure against BRCA1/2 mutations. Additionally, loss of heterozygosity (LOH) was detected at both BRCA1 and BRCA2 loci [41]. The third cell line, OAW42, is probably not of the HGSOC subtype, although the original annotation was serous [28,29,32]. However, during culturing, OAW42 cells show ‘epithelial’ morphological characteristics [Beaufort 2014]. We chose this cell line in order to determine whether differences in EV characteristics could be the result of different tumour subtypes. OAW42 shows no TP53 mutation but a PIK3CA mutation [28,29]. Examination of the BRCA1 status gave contradictory results; a BRCA1 mutation was detected in one study but this result was not confirmed in two other studies [28,29,41]. In addition, OAW42 did not show LOH at the BRCA1/2 locus [41].
Cells release a heterogeneous population of EVs with different sizes (30–10,000 nm) [42]. EVs are a mixture of exosomes and ectosomes (microparticles/microvesicles), which differ in size and biogenesis. Exosomes are released on the exocytosis of multivesicular bodies (MVBs). Ectosomes, on the other hand, bud directly outwards from the plasma membrane and tend to be larger in size. Due to overlapping size ranges, current EV isolation methods are not able to differentiate precisely between subtypes of EVs and so assigning an EV to a particular biogenesis pathway remains difficult. We therefore refer to “small EVs” (sEVs) and “medium/large EVs” (m/lEVs) based on the size range: < 200 nm [small] and > 200 nm [large and medium], respectively, as recommended by the International Society for Extracellular Vesicles [34]. TEM analysis revealed that all three cell lines released spherical EVs with different sizes, although a more heterogeneous size distribution and higher proportion of m/lEVs was observed among EVs released from OAW28 cells, which was confirmed by the results of S-NTA. On the other hand, the EVs from all three cell lines were most commonly found in the size range from 100 to 140 nm, which falls within the size range of sEVs. This result is comparable with a previously published study obtained with an OVCAR3 cell line, as another cell line of the HGSOC subtype, and analysed with S-NTA [17,29]. However, the most commonly found size of EVs released from OAW28 cells was larger than that from PEO1-derived EVs, while OAW42-derived and OAW28-derived EVs did not differ. This observation indicates that the size of the major peak of EVs depends on other factors, such as different histological subtype [17]. On the other hand, a higher proportion of m/lEVs released from OAW28 was observed compared to PEO1-derived and OAW42-derived EVs. Therefore, this result is probably not due to differences between EVs released from different histological subtypes but may indicate an association of a higher number of m/lEVs with resistance to chemotherapy. The difference in the size distribution of EVs is probably a very early sign of a changed state in the cell, which is reflected in the biogenesis of EVs. It was observed that the proportion of EVs of different sizes released from HeLa cells was modified on silencing of some cell components involved in EV formation [35]. One problem with determining the significance of the modified size distribution is that its analysis also depends on the method of EV isolation. Although the size distribution determined by S-NTA showed a major EV peak independently of the isolation method, we still do not know which subpopulations are lost during the isolation process [42]. This problem is even greater when EVs are isolated from a patient’s body fluid. Although studies on the association of a larger size of EVs with a worse prognosis already exist for EVs from the blood of patients with pancreatic ductal adenocarcinoma [43,44], such research in the field of HGOSC is still lacking. Despite problems with EVs analysis, tumour-derived large EVs have progressively received increasing interest due to their peculiar content and function [45].
Since quantification of EVs is difficult, we used two methods to confirm our results: S-NTA and FT-FCM. Particle counting by S-NTA may result in overestimation of EV counts since the technique is not specific to EVs and also registers co-isolated particles. On the other hand, FT-FCM, due to the detection limit, registers larger particles than S-NTA. We used calcein as a generic fluorescent marker for detecting EVs by FT-FCM. The advantage of calcein is differentiation of intact EVs from membrane fragments, while the disadvantage is that it requires the presence of enzyme in the EVs. For both reasons, particle counting by FT-FCM may result in underestimation of EV counts. However, the results of S-NTA and FT-FCM were in excellent correlation (r = 0.95, p < 0.001), despite the lower particle number detected by FT-FCM, which is a logical consequence of the above explanation.
In the present study, in which the results were obtained using the two methods mentioned above, we showed that the EV concentration between OAW28-derived EVs and PEO1-derived EVs did not differ significantly, which indicates that HGSOC cells from patient resistant to platinum-based therapy do not release more EVs than cells from tumours sensitive to platinum-based therapy. The detected concentration of EVs from OAW28 and PEO1 cell lines analysed by S-NTA in our study were in accordance with previous reports on EV concentration released from another HGSOC cell line (OVCAR3) in the same period of time and analysed with the same method [17,29]. Intriguingly, the concentration of EVs produced by OAW42 cells was lower than the concentration of EVs produced by OAW28 and PEO1 cells. This difference in the production of EVs between two cell lines from tumours sensitive to platinum-based therapy might be attributable to different histological OC subtypes. However, detected EVs released from OAW42 were much smaller when compared with the results of a previous study. Zhang et al. measured the concentration of EVs released from cell lines with HGSOC, endometrioid and clear cell histology, but the numbers of EVs produced by cells from different histological OC subtypes did not differ [17]. The lower EV concentration produced by the OAW42 cell line might therefore be a consequence of its special genomic profile or some other unknown factor [28,29]. Szajnik et al. investigated the association between plasma EV level (measured as protein level) and response to primary therapy with carboplatin/paclitaxel in 12 OC patients with different types of histology. The levels of EV proteins varied greatly among patients but the response to therapy was not associated with the level of plasma EV proteins before treatment started, which is in agreement with the results of our study. However, the clinical response to therapy in the majority of patients was associated with a decrease in EV protein levels. These results, while preliminary and performed with only few patients, should be further evaluated [16].
It is likely that specific EVs might be more informative in predicting the response to therapy than total EV levels. Among specific cargoes of EVs, we chose EpCAM as a marker of epithelium-derived cells. EpCAM-positive vesicles were detected in plasma, ascites and pleural effusion of OC patients, as well as in conditioned media of OC cell lines [6,7,14,17,18]. However, the relevance of analysis of EpCAM-positive EVs in predicting chemoresistance has been largely unexplored. The results of our study, obtained by two methods, FT-FCM and BB-FCM, showed that cells derived from the patient resistant to platinum-based therapy (OAW28) released a much larger number of EpCAM-positive EVs than cells from tumours sensitive to platinum-based therapy, regardless of the histological subtype. EpCAM-positive vesicles released from OAW28 cells accounted for 43 % of total EVs as detected by FT-FCM. A previous study reported a lower percentage of EpCAM-positive EVs released from another HGSOC cell line (~24 %, OVCAR-3), an endometrioid cell line (~18 %, ES-2) and a clear cell carcinoma cell line (~15 %, IGROV) detected by fluorescent-NTA [17,29].
EpCAM-positive vesicles showed the clinical usefulness of monitoring the clinical response to platinum-based therapy. The EVs' EpCAM levels decreased among responding patients, whereas increased levels were associated with non-responding patients as detected by nano-plasmonic sensor. However, the difference was not significant since the study was performed in 8 OC patients with different types of histology. The levels of the EVs' EpCAM before therapy varied greatly among patients, but the relation of the response to therapy with these levels was not assessed [7]. EpCAM was also included in the EV signature (a weight sum of eight EV protein markers), which provides a complementary approach for monitoring treatment response in metastatic breast cancer [39].
An interesting question that arises is what is the content of EpCAM-positive EVs? Taylor et.al. isolated EpCAM-positive EVs from the sera of 50 patients with HGSOC and detected 8 microRNA (miR-21, miR-141, miR-200a, miR-200b, mir200c, miR-203, miR-205 and miR-214) in EVs whose levels were increased compared to those with benign disease [6]. Although the purpose of this study was to discover diagnostic biomarkers, there are other published studies that have examined the presence of noncoding RNA (microRNA and circular RNA) in EVs and their utility as biomarkers of response to chemotherapy [39,46,47,48]. Moreover, in prostate cancer, miR-200c and miR-205 were shown to induce expression of EpCAM mRNA and protein [49].
We discovered that cells from the chemoresistant patient released EpCAM-positive EVs of different sizes (sEVs and m/lEVs). However, a higher proportion of m/lEVs was observed compared to those from cells from the tumours sensitive to chemotherapy. Additionally, immunogold labelling predominated in EVs that were ≥ 400 nm in size and a higher number of EpCAM molecules was found on larger EVs. Gercel-Taylor et al. also observed the presence of EpCAM in various size ranges of vesicles derived from sera of advanced HGSOC patients [13].
We hypothesised that larger EpCAM-positive EVs with a higher number of EpCAM molecules can bind more effectively to tumour-reactive autoantibodies and to therapeutic EpCAM antibodies and thereby reduce the binding of antibodies to tumour cells. It has been discovered that ectopic overexpression of EpCAM and HER2 on cancer cells induces humoral immune responses in patients. Both antigens are also targets for a number of therapeutic monoclonal antibodies. In the first situation, this will impair antibody-dependent cellular cytotoxicity (ADDC), which is a major immune effector function toward tumour cells [50]. EpCAM specific autoantibodies have been found in sera of OC patients [51]. This phenomenon may be an additional explanation for the contradictory results of EpCAM overexpression in ovarian tumours in relation to its prognostic value [20,21,22,23]. In the second situation, this may contribute to therapeutic failure of EpCAM monoclonal antibodies. This has been suggested in the case of the HER-2 specific therapeutic antibody trastuzumab [50]. A higher level of HER2-specific autoantibodies was found in sera from patients with HER2-expressing mammary carcinoma [52]. It has been proposed that the antibody has a higher binding affinity to full-sized HER2 than to soluble HER2 extracellular domains generated by proteolytic cleavage [53]. It would therefore be worth exploring the role of this indirect mechanism of EpCAM on EVs in therapeutic failure.
The results of the present study showed a strong positive correlation between the concentration EpCAM-positive EVs and expression of EpCAM on the cell membrane. This finding supports the use of EpCAM specific EVs as cellular surrogates for EpCAM and could therefore be included in liquid biopsy to shed light on the contradictory results of immunohistochemical evaluation of EpCAM overexpression in ovarian tumours as a biomarker for predicting response to chemotherapy. Moreover, it would be interesting to examine in a future study the clinical usefulness of EpCAM-specific EVs in combination with other promising molecular biomarkers or other innovative approaches for the prediction of platinum resistant HGSOC.
In conclusion, we showed for the first time that a more heterogeneous size distribution, with a higher proportion of larger EVs, is characteristic of EVs released from the HGSOC cell line of a clinically confirmed resistant patient, which may reflect the changed state of the cell, reflected in the biogenesis of EVs. In addition, these cells released a higher number of EpCAM specific EVs of different sizes, although expression of EpCAM predominated on larger EVs, especially on those larger than 400 nm. The association of a higher concentration of EpCAM specific EVs with a higher expression of EpCAM on cell membranes demonstrates the feasibility of applying EpCAM specific EVs as a biomarker for cellular EpCAM. Although the results of the current study should be validated in clinical samples, they reveal novel characteristics of EVs that may have the potential to aid the prediction of platinum resistant HGSOC and thereby enable the selection of personalized therapy.
4. Materials and Methods
4.1. Cell culture
We included in this study three well characterized ovarian cancer cell lines purchased from the European collection of cell culture (ECACC via Sigma): OAW28 (ECACC 85101601), PEO1 (ECACC 10032308) and OAW42 (ECACC 85073102). In vitro cultured OAW28, OAW24 and PEO1 cells were used as the EV source for this study, in order to examine the characteristics of EVs from a patient who did not respond to cisplatin (OAW28) compared to EVs from two patients with a complete response (PEO1, OAW42) [31,32]. OAW28 and PEO1 are representatives of HGSOC, while OAW42 cells were described to be of serous origin without mutation in the TP53 gene [28,29]. Cell lines were cultured according to the manufacturer’s instructions. OAW28 and OAW42 cells were grown in DMEM medium (Sigma-Aldrich, #D6546) and supplemented with 2 mM glutamine, 20 UI/l bovine insulin purchased from Sigma-Aldrich, 1 mM sodium pyruvate, 10 % FBS [Gibco/Thermo Fischer Scientific Inc.; #10500-037, Lot 08G1094K], 100 U/ml penicillin, 100 µg/ml streptomycin and 50 µg/ml gentamycin purchased from Gibco/Invitrogene. PEO1 cells were grown in RPMI 1640 medium (Gibco, #A10491-01), supplemented with 1 mM sodium pyruvate, 10 % FBS [#10500-037, Lot 08G1094K], 100 U/ml penicillin, 100 µg/ml streptomycin and 50 µg/ml gentamycin purchased from Gibco/Invitrogene. The cell lines were maintained at 37 °C in an incubator with humidified air with 5 % CO2. For EV isolation, cells were seeded at a density of 1.5 × 106 and grown in a 175 cm2 flask until ~80 % confluence, rinsed three times with Dulbecco's phosphate-buffered saline (DPBS, without Ca2+ and Mg2+; Gibco/Thermo Fischer Scientific Inc., #14190-94) and grown in 30 ml serum-free medium. After 24 h of incubation, the conditioned medium was collected and centrifuged for 30 min at 2000 × g, RT to eliminate cell and cell debris contamination. Supernatant 10 mm above the pellet was transferred into a plastic tube and mixed gently before proceeding with isolation of EVs. For analysis of expression of EpCAM on the cell membrane and for cell/viability counts, attached cells were fully disaggregated by TripLETM select enzyme (Gibco/ Thermo Fischer Scientific Inc.; #12563-029). Cell lines were used from passages 4–7.
4.2. Cell counts and viability measurement
To normalize the amount of released EVs, we chose a number of live cells [34]. For viability measurement, the cell suspension was mixed with an equal volume of 0.4 % trypan blue solution (Invitrogen/Thermo Fischer Scientific Inc.; #T102882) to count live and dead cells using an image-based automated cell counter (Countess II FL; Thermo Fisher Scientific Inc., Waltham, USA) The required viability was > 90 %.
4.3. EV enrichment (isolation)
The cell culture conditioned medium sample (7 ml) was transferred to a fresh tube and 3.5 ml of Exosome Isolation Reagent (Invitrogen/Thermo Fischer Scientific Inc., #4478359) was added and mixed thoroughly by pipetting up and down. The solution was incubated overnight at 4 °C. The following day, the samples were centrifuged at 10,000 x g for 60 min. The pellet was then resuspended in 200 µl DPBS and stored at 2–8 °C for a maximum of up to seven days before downstream analysis or further purification by the immunomagnetic bead method.
4.4. Transmission electron microscopy (TEM)
A 10 µl drop of EVs was loaded onto a glow-discharged formvar/carbon-coated grid, adsorbed vesicles were fixed with 2 % paraformaldehyde (in PBS), washed in distilled water, stained by negative contrast with 1 % aqueous uranyl acetate, briefly washed in distilled water and air-dried. Samples were examined with a Philips CM100 transmission electron microscope operated at 80 kV. Images were captured with an AMT camera (Advanced Microscopy Techniques Corp., Woburn, USA).
For EpCAM immunolabelling, a 10 µl drop of extracellular vesicles was loaded onto a glow-discharged formvar/carbon-coated grid. Adsorbed vesicles were fixed with 2 % paraformaldehyde (in PBS), blocked in 0.5 % BSA and incubated with mouse monoclonal anti-EpCAM (CD326) primary antibodies, clone 1B7 (Invitrogen/ Thermo Fisher Scientific, Waltham, USA; #12-9326-42), diluted to 1:100 (1 % BSA in PBS), for 30 min at room temperature. Vesicles were washed in PBS and incubated with 18 nm colloidal gold goat anti-mouse IgG (Jackson ImmunoResearch, Cambridgeshire, CB7 4EX United Kingdom; #115-215-146) secondary antibodies, washed in distilled water and stained with 1 % aqueous uranyl acetate. Air-dried samples were examined with a Philips CM100 transmission electron microscope operated at 80 kV and equipped with an AMT camera.
4.5. Nanoparticle tracking analysis in scatter mode (S-NTA)
Isolated EVs from conditioned media were diluted in filtered DPBS (without Ca2+ and Mg2+) and analysed for particle size distribution and concentration by S-NTA using a NanoSight NS300 instrument (488 nm laser) connected to an automated sample assistant (both Malvern Panalytical, Worcestershire, UK). The samples were diluted to a concentration allowing accurate particle tracking (according to the manufacturer, app. 20–100 particles in the field of view). Each sample was measured (recorded) 4 times with 80 s acquisitions at 25 °C and processed using NTA software (version 3.3). The camera level was adjusted according to the sample. Analysis settings were maintained for all readings: detection threshold 5, water viscosity, automatic blur size and automatic (10.2–15.7 pix) maximum jump distance. Data obtained were particle concentration (number of particles/ml of cell cultured conditioned media), mode (nm), mean (nm) and percentile values: D10, D50, D90. Particle size values indicating that 10, 50 and 90 %, respectively, of the distribution was below this value. To calculate the number of particles for each sample, the dilution factor during sample preparation was taken into account. Reference nanospheres (100 nm polystyrene, Malvern Pananalytical, #LT3100A) were analysed on the same day to compensate for day-to-day variation in instrument performance. Raw data were analysed by NanoSight NTA 3.3 software (Malvern Panalytical, Worcestershire, UK).
4.6. Fluorescence-triggered flow cytometry (FT-FCM) of EVs
A flow cytometer (CytoFLEX, Beckman Coulter, Brea, USA) was used to determine the concentration of EpCAM-positive and calcein-positive EVs in the conditioned media of OC cell lines. Fluorescence-based detection of EVs using FCM was employed to better resolve EVs from the background noise and because it is independent of size and refractive index [54]. Calcein-acetoxymethyl ester (AM) green (referred to as calcein) (Thermo Fischer Scientific; #C3100MP) was used as the generic fluorescent marker for detection of EVs. Calcein AM is non-fluorescent until it passively enters EVs, after which the lipophilic blocking groups are cleaved by non-specific esterase. As a result, calcein is trapped inside the EVs and emits a green fluorescent signal (emission max. 516 nm) following excitation with a blue (488 nm) laser [33,34,55]. For sample preparation, antibodies were centrifuged at 20,000 × g for 30 minutes at RT to remove any fluorescent antibody aggregates, as described elsewhere [56]. To stain, 20 µl of sample was incubated with 1.25 µl PE-conjugated anti-EpCAM (CD326) primary antibodies, clone 1B7 (Invitrogen/ Thermo Fisher Scientific, Waltham, ZDA; #12-9326-42) or PE-conjugated isotype antibody, mouse IgG1 kappa (Invitrogen/ Thermo Fisher Scientific, Waltham, ZDA; #12-1714-42) and kept in the dark for 2 h at room temperature. Due to some overlap between the emission spectra of PE and calcein, which could lead to a false positive signal, a separate sample was stained with calcein [57]. One hundred µl of calcein (10 µM) was added to 20 µl of sample and incubated for 30 minutes in the dark on ice. After incubation, we diluted samples in DPBS to a count rate below 2000 events/s to prevent swarm detection [58].
The stained samples were measured by flow cytometry within 1.5 hour, using fluorescence triggering, timed at a slow flow rate for 120 s. Enumeration of EVs was performed using volumetric measurement (events/mL). Calibrating the sample flow rate was conducted following the CytoFLEX instructions by water weight difference during 18-min acquisition with a slow flow rate. To set up VSSC detection, we replaced the 450 nm filter with a 405 nm filter, and appropriately labelled the detector as VSSC. For daily verification of the flow cytometer's optical alignment and fluidics system, we used CytoFLEX Daily QC Fluorospheres (Beckman Coulter, #B53230) with settings optimized for EV detection. The upper size limit for the EV gate was set by detecting 1000 nm fluorescent-green silica beads (Kisker-biotech, #PSI-G1.0] using VSSC triggering [56]. The threshold for fluorescence-triggering of calcein-positive events was set by the plain cell culture medium samples, which were treated with calcein in the same way as the EVs in the culture conditioned medium samples. The fluorescence signal from PE was used to trigger detection of EVs labelled with anti PE-conjugated antibodies. The gate for PE was derived by measuring corresponding isotype control bodies. Another negative control was performed using anti-EpCAM-PE in DPB and unstained EV samples. The addition of 0.2% Triton 100 (Sigma, #T8787) to EV samples for 20 minutes at room temperature was accompanied by a near total disappearance of the EVs. Analysis of the acquired data was performed using CytExpert 2.3 software (Beckman Coulter, Brea, USA). To calculate the number of events for each sample, the dilution factor during sample preparation and staining was taken into account. Due to the detection limit of our flow cytometer, the EVs measured in this study predominantly correspond to ectosomes (microparticles), not to exosomes.
4.7. Anti-human EpCAM antibody-coated magnetic bead-based EV isolation
EpCAM-positive EV subsets from a pre-enriched EV sample were isolated using Exosome - Human EpCAM Flow Detection (Invitrogen/ Thermo Fischer Scientific Inc., #10624D) following the manufacturer’s instructions. DynabeadsTM are magnetic polystyrene beads (2.7 µm diameter) coated with primary monoclonal antibody specific for the EpCAM membrane antigen expressed on the EVs. The assay buffer was prepared from DPBS with added 0.1 % BSA as the blocking agent and 0.2 µm filtered. Dynabeads were resuspended by vortexing for 30 seconds, 20 µl was withdrawn and transferred to a tube with 1 ml of assay buffer. The tube was placed in a DynaMag™-2 magnetic separator (Thermo Fischer Scientific Inc., #12321D) for 2 minutes. After the buffer had been carefully removed, 90 µl of fresh assay buffer and 10 µl of pre-enriched EV sample were added and incubated at 2–8 °C overnight on a HulaMixer (Thermo Fischer Scientific Inc., #15920D). The next day, the assay buffer was added and the tube placed in a magnetic separator for 1–2 min before removing all supernatants. The procedure with the magnetic separator was repeated 3-times to wash the bead-bound EVs. For analysis with bead-based flow cytometry, the sample was finally diluted in 300 µl of assay buffer.
4.8. Bead-based flow cytometry (BB-FCM) of EpCAM-positve EVs
EVs were also analysed by bead-based flow cytometry (BB-FCM) as a semi quantitative method for assessing approximate EpCAM-positive concentrations [59]. Because of the size of the Dynabeads™ magnetic beads, EVs are easily detected by flow cytometry while bound to the surface of the beads, enabling the detection of EpCAM on the EV membrane. A total of 100 µl of bead-bound EV sample was stained using 20 µl (0.06 µg) of RPE-conjugated anti-EpCAM (CD326) primary antibodies, clone EBA (Biosciences/Becton Dickinson, #347198). After 45 min of incubation in the dark on an orbital shaker (RT, 1000 rpm), 300 µl of assay buffer was added to each tube and the tubes placed in a magnetic separator for 1–2 min before removing the buffer. We repeated the procedure with the magnetic separator once more. The final EV sample for flow cytometry was diluted in 300 µl DPBS. Flow cytometry was performed using a CytoFLEX flow cytometer (Beckman Coulter, Brea, USA). Isotype control antibodies (Mouse IgG1-RPE isotype control, Biosciences/Becton Dickinson; #559320) and an unstained EV sample were used as negative control. Gating of bead-bound EVs was performed based on FSC/SSC parameters, so that non-bead events and bead doublets were excluded from the analysis, and beads were selected using the corresponding channels of fluorescence. Results were expressed as EpCAM-positive beads. Analysis of the acquired data was performed using CytExpert 2.3 software (Beckman Coulter, Brea, USA).
4.9. Flow cytometry of EpCAM cell membrane expression
Expression of EpCAM on the cell membrane was analysed using a CytoFLEX flow cytometer (Beckman Coulter, Brea, USA). For analysis with the flow cytometer, 2 × 105 cells were needed for each sample, which were resuspended in 100 µl of DPBS. 1.25 µl PE-conjugated anti-EpCAM (CD326) primary antibodies, clone 1B7 (Invitrogen/ Thermo Fisher Scientific, Waltham, ZDA; #12-9326-42) were added to the first sample. The second sample was an isotype control, so 1.25 µl of PE-conjugated isotype antibody, mouse IgG1 kappa (Invitrogen/ Thermo Fisher Scientific, Waltham, ZDA; #12-1714-42) was added to the cells. The third sample consisted of unstained cells. The samples were incubated in the dark for 45 minutes at 4 °C. They were then washed three times by centrifugation (3 minutes, 200 × g, at room temperature) and resuspended in between in 500 µl DPBS. Final cell samples for analysis were resuspended in 200 µl DPBS. Fluorescence gating parameters were established using antibody isotype controls, and values above a 99 % negative staining threshold were considered positive. Using isotype antibodies, we confirmed that the binding of primary antibodies was specific. Unstained cells were used to control cellular autofluorescence. The concentration of the isotype antibodies should be the same as the concentration of the primary antibodies. We set up the instrument appropriately with the help of this control, which also allowed us to set up the gate appropriately to capture the events we wanted to analyse. A total of 5000 cells was measured. Mean fluorescent intensity (MFI) was used to compare expression of EpCAM between different cell lines. Analysis of the acquired data was performed using CytExpert 2.3 software (Beckman Coulter, Brea, USA).
5.0. Statistical Analysis
Statistical analysis was performed using SigmaPlot v.12.5 (Systat Software, Palo Alto, USA). All quantitative measurements were conducted as three independent measurements, which were made in duplicate. One-way ANOVA was used as an appropriate method to test for significant differences between more than two normally distributed samples, followed by Holm-Sidak's multiple comparison test. Person’s correlation coefficient was used to calculate the direction and strength of the relationship between variables. A p value of < 0.05 was considered significant. All data are presented as mean ± standard error of mean (SEM).
Author Contributions
Conceptualization, K.Č., M.H.; methodology, K.Č., N.R.; performance of experiments, K.Č., N.K., N.R., L.V..; data curation, K.Č., N.K.; writing—original draft preparation, K.Č., N.K., N.R.; writing—review and editing, K.Č., N.K., N.R., M.H., P.V., B.K.; visualization, N.K., N.R.; resources and funding acquisition, K.Č., P.V., B.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Slovenian Research Agency through the research programs P3-067 and P3-0108 and by the University Medical Centre Ljubljana (Project Number: 20180033).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
The authors thank Andreas Spittler (Medical University Vienna) for valuable advices and technical support in performing flow cytometry, Nevenka Dolžan for technical assistance and Martin Cregeen for assistance with the English translation of the text.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dalmartello, M.; La Vecchia, C.; Bertuccio, P.; Boffetta, P.; Levi, F.; Negri, E.; Malvezzi, M. European cancer mortality predictions for the year 2022 with focus on ovarian cancer. Ann Oncol 2022, 33, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chung Chan, W.; Ho Ngai, C.; Lok, V.; Zhang, L.; Lucero-Prisno 3rd, D.E.; Xu, W.; Zheng, Z.J.; Elcarte, E.; Withers, M.; Wong, M.C.S.; On Behalf of Ncd Global Health Research Group of Association of Pacific Rim Universities Apru. Worldwide Burden, Risk Factors, and Temporal Trends of Ovarian Cancer: A Global Study. Cancers 2022, 14, 2230. [Google Scholar] [CrossRef] [PubMed]
- Valabrega, G.; Scotto, G.; Tuninetti, V.; Pani, A.; Scaglione, F. Differences in PARP Inhibitors for the Treatment of Ovarian Cancer: Mechanisms of Action, Pharmacology, Safety, and Efficacy. Int J Mol Sci 2021, 22, 4203. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Sessa, C.; du Bois, A.; Ledermann, J.; McCluggage, W. G.; McNeish, I.; Morice, P. , Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO–ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent disease. Ann Oncol 2019, 30, 672–705. [Google Scholar] [CrossRef] [PubMed]
- Lisio, M. A.; Fu, L.; Goyeneche, A.; Gao, Z. H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int J Mol Sci 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Gercel-Taylor, C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol 2008, 110, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Im, H.; Shao, H.; Park, Y.I.; Peterson, V.M.; Castro, C.M.; Weissleder, R.; Lee, H. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat Biotechnol 2014, 32, 490–495. [Google Scholar] [CrossRef]
- Reiner, A. T.; Tan, S.; Agreiter, C.; Auer, K.; Bachmayr-Heyda, A.; Aust, S.; Pecha, N.; Mandorfer, M.; Pils, D.; Brisson, A. R.; et al. EV-Associated MMP9 in High-Grade Serous Ovarian Cancer Is Preferentially Localized to Annexin V-Binding EVs. Dis Markers 2017, 9653194. [Google Scholar] [CrossRef]
- Lucidi, A.; Buca, D.; Ronsini, C.; Tinari, S.; Bologna, G.; Buca, D.; Leombroni, M.; Liberati, M.; D'Antonio, F.; Scambia, G.; et al. Role of Extracellular Vesicles in Epithelial Ovarian Cancer: A Systematic Review. Int J Mol Sci 2020, 19, 8762. [Google Scholar] [CrossRef]
- Witwer, K. W.; Buzás, E. I.; Bemis, L. T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-'t Hoen, E. N.; Piper, M. G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles 2013, 27, 2. [Google Scholar] [CrossRef]
- McMahon, H. T.; Missler, M.; Li, C.; Südhof, T. C. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell 1995, 83, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Dolo, V.; D'Ascenzo, S.; Violini, S.; Pompucci, L.; Festuccia, C.; Ginestra, A.; Vittorelli, M. L.; Canevari, S.; Pavan, A. Matrix-degrading proteinases are shed in membrane vesicles by ovarian cancer cells in vivo and in vitro. Clin Exp Metastasis 1999, 17, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Gercel-Taylor, C.; Atay, S.; Tullis, R. H.; Kesimer, M.; Taylor, D. D. Nanoparticle analysis of circulating cell-derived vesicles in ovarian cancer patients. Anal Biochem 2012, 428, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Roca, E.; Lacroix, R.; Judicone, C.; Laroumagne, S.; Robert, S.; Cointe, S.; Muller, A.; Kaspi, E.; Roll, P.; Brisson, A. R.; et al. Detection of EpCAM-positive microparticles in pleural fluid: A new approach to mini-invasively identify patients with malignant pleural effusions. Oncotarget 2016, 7, 3357–3366. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, C. M.; Oakes, M. L.; Murakami, T.; Muto, M. G.; Berkowitz, R. S.; Ng, S. W. Comparison of benign peritoneal fluid- and ovarian cancer ascites-derived extracellular vesicle RNA biomarkers. J Ovarian Res 2018, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Szajnik, M.; Derbis, M.; Lach, M.; Patalas, P.; Michalak, M.; Drzewiecka, H.; Szpurek, D.; Nowakowski, A.; Spaczynski, M.; Baranowski, W.; Whiteside, T. L. Exosomes in Plasma of Patients with Ovarian Carcinoma: Potential Biomarkers of Tumor Progression and Response to Therapy. Gynecol Obstet 2013, 4, 3. [Google Scholar]
- Zhang, W.; Peng, P.; Kuang, Y.; Yang, J.; Cao, D.; You, Y.; Shen, K. Characterization of exosomes derived from ovarian cancer cells and normal ovarian epithelial cells by nanoparticle tracking analysis. Tumour Biol 2016, 37, 4213–4221. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, Y.; Zeng, Y.; He, M. A microfluidic ExoSearch chip for multiplexed exosome detection towards blood-based ovarian cancer diagnosis. Lab Chip 2016, 16, 489–496. [Google Scholar] [CrossRef]
- Mohtar, M. A.; Syafruddin, S. E.; Nasir, S. N.; Low, T. Y. Revisiting the Roles of Pro-Metastatic EpCAM in Cancer. Biomolecules. 2020, 10, 255. [Google Scholar] [CrossRef]
- Tayama, S.; Motohara, T.; Narantuya, D.; Li, C.; Fujimoto, K.; Sakaguchi, I.; Tashiro, H.; Saya, H.; Nagano, O.; Katabuchi, H. The impact of EpCAM expression on response to chemotherapy and clinical outcomes in patients with epithelial ovarian cancer. Oncotarget 2017, 8, 44312–44325. [Google Scholar] [CrossRef]
- Spizzo, G.; Went, P.; Dirnhofer, S.; Obrist, P.; Moch, H.; Baeuerle, P. A.; Mueller-Holzner, E.; Marth, C.; Gastl, G.; Zeimet, A. G. Overexpression of epithelial cell adhesion molecule (Ep-CAM) is an independent prognostic marker for reduced survival of patients with epithelial ovarian cancer. Gynecol Oncol 2006, 103, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Woopen, H.; Pietzner, K.; Richter, R.; Fotopoulou, C.; Joens, T.; Braicu, E. I.; Mellstedt, H.; Mahner, S.; Lindhofer, H.; Darb-Esfahani, S.; et al. Overexpression of the epithelial cell adhesion molecule is associated with a more favorable prognosis and response to platinum-based chemotherapy in ovarian cancer. J Gynecol Oncol 2014, 25, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Battista, M. J.; Cotarelo, C.; Jakobi, S.; Steetskamp, J.; Makris, G.; Sicking, I.; Weyer, V.; Schmidt, M. Overexpression of epithelial cell adhesion molecule protein is associated with favorable prognosis in an unselected cohort of ovarian cancer patients. J Cancer Res Clin Oncol 2014, 140, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Herreros-Pomares, A.; Aguilar-Gallardo, C.; Calabuig-Fariñas, S.; Sirera, R.; Jantus-Lewintre, E.; Camps, C. EpCAM duality becomes this molecule in a new Dr. Jekyll and Mr. Hyde tale. Crit Rev Oncol Hematol 2018, 126, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Pan, M.; Schinke, H.; Canis, M.; Baeuerle, P. A. Expression and function of epithelial cell adhesion molecule EpCAM: where are we after 40 years? Cancer Metastasis Rev 2020, 39, 969–987. [Google Scholar] [CrossRef] [PubMed]
- Alberro, A.; Iparraguirre, L.; Fernandes, A.; Otaegui, D. Extracellular Vesicles in Blood: Sources, Effects, and Applications. Int J Mol Sci. 2021, 22, 8163. [Google Scholar] [CrossRef] [PubMed]
- Rikkert, L. G.; Beekman, P.; Caro, J.; Coumans, F. A. W.; Enciso-Martinez, A.; Jenster, G.; Le Gac, S.; Lee, W.; van Leeuwen, T. G.; Loozen, G. B.; et al. Cancer-ID: Toward Identification of Cancer by Tumor-Derived Extracellular Vesicles in Blood. Front Oncol 2020, 10, 608. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D. A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 2013, 4, 2126. [Google Scholar] [CrossRef]
- Beaufort, C. M.; Helmijr, J. C.; Piskorz, A. M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W. F.; Heine, A. A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K. M.; Curtis, M.; Eckert, M. A.; Chiang, C. Y.; Tyanova, S.; Montag, A.; Lastra, R. R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat Commun 2016, 7, 12645. [Google Scholar] [CrossRef]
- Wolf, C. R.; Hayward, I. P.; Lawrie, S. S.; Buckton, K.; McIntyre, M. A.; Adams, D. J.; Lewis, A. D.; Scott, A. R.; Smyth, J. F. Cellular heterogeneity and drug resistance in two ovarian adenocarcinoma cell lines derived from a single patient. Int J Cancer 1987, 39, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A. P.; Dent, M.; Pejovic, T.; Hubbold, L.; Radford, H. Characterisation of seven human ovarian tumour cell lines. Br J Cancer 1996, 74, 722–727. [Google Scholar] [CrossRef]
- Gray, W. D.; Mitchell, A. J.; Searles, C. D. An accurate, precise method for general labeling of extracellular vesicles. MethodsX 2015, 2, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K. W.; Aikawa, E.; Alcaraz, M. J.; Anderson, J. D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G. K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L. F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J Cell Sci 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Kosaka, N.; Kogure, A.; Yamamoto, T.; Urabe, F.; Usuba, W.; Prieto-Vila, M.; Ochiya, T. Exploiting the message from cancer: the diagnostic value of extracellular vesicles for clinical applications. Exp Mol Med 2019, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Carollo, E.; Melling, G. E.; Carter, D. R. F. Extracellular Vesicles: Emerging Modulators of Cancer Drug Resistance. Cancers 2021, 3, 749. [Google Scholar] [CrossRef] [PubMed]
- Croft, P. K.; Sharma, S.; Godbole, N.; Rice, G. E.; Salomon, C. Ovarian-Cancer-Associated Extracellular Vesicles: Microenvironmental Regulation and Potential Clinical Applications. Cells 2021, 10, 2272. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, S.; Liu, C.; Han, Z.; Liu, Y.; Deng, J.; Li, Y.; Wu, X.; Cai, L.; Qin, L.; et al. Protein analysis of extracellular vesicles to monitor and predict therapeutic response in metastatic breast cancer. Nat Commun 2021, 12, 2536. [Google Scholar] [CrossRef]
- Vera, N.; Acuña-Gallardo, S.; Grünenwald, F.; Caceres-Verschae, A.; Realini, O.; Acuña, R.; Lladser, A.; Illanes, S. E.; Varas-Godoy, M. Small Extracellular Vesicles Released from Ovarian Cancer Spheroids in Response to Cisplatin Promote the Pro-Tumorigenic Activity of Mesenchymal Stem Cells. Int J Mol Sci 2019, 20, 4972. [Google Scholar] [CrossRef]
- Stordal, B.; Timms, K.; Farrelly, A.; Gallagher, D.; Busschots, S.; Renaud, M.; Thery, J.; Williams, D.; Potter, J.; Tran, T. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol Oncol 2013, 7, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Allelein, S.; Medina-Perez, P.; Lopes, A. L. H.; Rau, S.; Hause, G.; Kölsch, A.; Kuhlmeier, D. Potential and challenges of specifically isolating extracellular vesicles from heterogeneous populations. Sci Rep 2021, 11, 11585. [Google Scholar] [CrossRef] [PubMed]
- Badovinac, D.; Goričar, K.; Zavrtanik, H.; Petrič, M.; Lavrin, T.; Mavec, N.; Dolžan, V.; Tomažič, A.; Lenassi, M. Plasma Extracellular Vesicle Characteristics Correlate with Tumor Differentiation and Predict Overall Survival in Patients with Pancreatic Ductal Adenocarcinoma Undergoing Surgery with Curative Intent. J Pers Med 2021, 11, 77. [Google Scholar] [CrossRef]
- Allenson, K.; Castillo, J.; San Lucas, F. A.; Scelo, G.; Kim, D. U.; Bernard, V.; Davis, G.; Kumar, T.; Katz, M.; Overman, M. J.; et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann Oncol 2017, 28, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Pezzicoli, G.; Tucci, M.; Lovero, D.; Silvestris, F.; Porta, C.; Mannavola, F. Large Extracellular Vesicles-A New Frontier of Liquid Biopsy in Oncology. Int J Mol Sci 2020, 21, 6543. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kim, H. S.; Park, S. J.; Lee, E. J.; Kim, S. I.; Song, G.; Lim, W. Inhibition of miR-214-3p Aids in Preventing Epithelial Ovarian Cancer Malignancy by Increasing the Expression of LHX6. Cancers (Basel) 2019, 11, 1917. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, M.; Sharma, S.; Guanzon, D.; Ali, A.; Zuñiga, F.; Shiddiky, M. J. A.; Yamauchi, Y.; Salas-Burgos, A.; He, Y.; Pejovic, T.; et al. miRNa signature in small extracellular vesicles and their association with platinum resistance and cancer recurrence in ovarian cancer. Nanomedicine 2020, 28, 102207. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Gui, R. Circulating exosomal circFoxp1 confers cisplatin resistance in epithelial ovarian cancer cells. J Gynecol Oncol 2020, 31, e75. [Google Scholar] [CrossRef]
- Massoner, P.; Thomm, T.; Mack, B.; Untergasser, G.; Martowicz, A.; Bobowski, K.; Klocker, H.; Gires, O.; Puhr, M. EpCAM is overexpressed in local and metastatic prostate cancer, suppressed by chemotherapy and modulated by MET-associated miRNA-200c/205. Br J Cancer 2014, 111, 955–964. [Google Scholar] [CrossRef]
- Battke, C.; Ruiss, R.; Welsch, U.; Wimberger, P.; Lang, S.; Jochum, S.; Zeidler, R. Tumour exosomes inhibit binding of tumour-reactive antibodies to tumour cells and reduce ADCC. Cancer Immunol Immunother 2011, 60, 639–648. [Google Scholar] [CrossRef]
- Kim, J. H.; Herlyn, D.; Wong, K. K.; Park, D. C.; Schorge, J. O.; Lu, K. H.; Skates, S. J.; Cramer, D. W.; Berkowitz, R. S.; Mok, S. C. Identification of epithelial cell adhesion molecule autoantibody in patients with ovarian cancer. Clin Cancer Res 2003, 9, 4782–4791. [Google Scholar] [PubMed]
- Disis, M. L.; Pupa, S. M.; Gralow, J. R.; Dittadi, R.; Menard, S.; Cheever, M. A. High-titer HER-2/neu protein-specific antibody can be detected in patients with early-stage breast cancer. J Clin Oncol 1997, 15, 3363–3367. [Google Scholar] [CrossRef] [PubMed]
- Maple, L.; Lathrop, R.; Bozich, S.; Harman, W.; Tacey, R.; Kelley, M.; Danilkovitch-Miagkova, A. Development and validation of ELISA for herceptin detection in human serum. J Immunol Methods 2004, 295, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Arraud Nicolas Arraud, N.; Gounou, C.; Turpin, D.; Alain R Brisson, A. R. Fluorescence triggering: A general strategy for enumerating and phenotyping extracellular vesicles by flow cytometry. Cytometry A 2016, 89, 184–95. [Google Scholar] [CrossRef] [PubMed]
- Kutschera, M.; Pischlaeger, A.; Sztulman, L.; Kietaibl, S.; Spittler, A. Extracellular vesicles in autologous cell salvaged blood in orthopedic surgery. Surgeries 2021, 2, 84–91. [Google Scholar] [CrossRef]
- Aass, H.C.D.; Øvstebø, R.; Trøseid, A.S.; Kierulf, P.; Berg, J.P.; Henriksson, C.E. Fluorescent particles in the antibody solution result in false TF- and CD14-positive microparticles in flow cytometric analysis. Cytometry A 2011, 79, 990–999. [Google Scholar] [CrossRef]
- Kuiper, M.; van de Nes, A.; Nieuwland, R.; Varga, Z.; van der Pol, E. Reliable measurements of extracellular vesicles by clinical flow cytometry. Am J Reprod Immunol 2021, 85, e13350. [Google Scholar] [CrossRef]
- Wisgrill, L.; Lamm, C.; Hartmann, J.; Preißing, F.; Dragosits, K.; Bee, A.; Hell, L.; Thaler, J.; Ay, C.; Pabinger, I.; Berger, A.; Spittler, A. Peripheral blood microvesicles secretion is influenced by storage time, temperature, and anticoagulants. Cytometry A 2016, 89, 663–672. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, Y.; Cao, Y.; Yu, P.; Zhang, L.; Liu, Z.; Ping, Y.; Wang, D.; Zhang, G.; Sang, Y.; Wang, X.; Tao, Z. A Multivariate Diagnostic Model Based on Urinary EpCAM-CD9-Positive Extracellular Vesicles for Prostate Cancer Diagnosis. Front Oncol 2021, 11, 777684. [Google Scholar] [CrossRef]
Figure 1.
Graphic presentation of the methods.
Figure 2.
Transmission electron microscopy analysis of EVs released from OAW28 (A), PEO1 (B), and OAW42 (C) cells. Negative contrast staining shows the typical vesicular morphology of EVs. In the isolate of OAW28, the EVs are larger (A, arrowhead) than the EVs of PEO1 (B, arrowhead) and OAW42 (C, arrowhead) cells.
Figure 2.
Transmission electron microscopy analysis of EVs released from OAW28 (A), PEO1 (B), and OAW42 (C) cells. Negative contrast staining shows the typical vesicular morphology of EVs. In the isolate of OAW28, the EVs are larger (A, arrowhead) than the EVs of PEO1 (B, arrowhead) and OAW42 (C, arrowhead) cells.
Figure 3.
Nanoparticle tracking analysis with representative live stream views of EVs released from OAW28 (A), PEO1 (B) and OAW42 (C) cells. OAW28 cells released more medium/large in size (> 200 nm) EVs (A) than PEO1 (B), and OAW42 (C) cells. The bar graph shows a comparison of mode, mean, D10, D50, D90 diameter among all three cell lines. Particle size distribution D10, D50, and D90 corresponding to the percentages 10 %, 50 %, and 90 % of particles under the reported particle size (D). Data are presented as mean ± SEM of three independent measurements made in duplicate. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 3.
Nanoparticle tracking analysis with representative live stream views of EVs released from OAW28 (A), PEO1 (B) and OAW42 (C) cells. OAW28 cells released more medium/large in size (> 200 nm) EVs (A) than PEO1 (B), and OAW42 (C) cells. The bar graph shows a comparison of mode, mean, D10, D50, D90 diameter among all three cell lines. Particle size distribution D10, D50, and D90 corresponding to the percentages 10 %, 50 %, and 90 % of particles under the reported particle size (D). Data are presented as mean ± SEM of three independent measurements made in duplicate. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 4.
Concentration of total EVs (detected as particles/calcein-positive EVs) released in 24 hours from OAW28 (red bar), PEO1 (yellow bar) and OAW42 (green bar) cells analysed by nanoparticle tracking analysis (A) and fluorescence-triggered flow cytometry (B). Data are presented as mean ± SEM of three independent measurements made in duplicate. ***p < 0.001.
Figure 4.
Concentration of total EVs (detected as particles/calcein-positive EVs) released in 24 hours from OAW28 (red bar), PEO1 (yellow bar) and OAW42 (green bar) cells analysed by nanoparticle tracking analysis (A) and fluorescence-triggered flow cytometry (B). Data are presented as mean ± SEM of three independent measurements made in duplicate. ***p < 0.001.
Figure 5.
Concentration of EpCAM-positive EVs released in 24 hours from OAW28 (red bar), PEO1 (yellow bar) and OAW42 (green bar) cells analysed by fluorescent-triggered flow cytometry (left axis). Percentage of EpCAM-positive beads detected in isolated samples of EVs released in 24 hours from OAW28 (red bar), PEO1 (yellow bar) and OAW42 (green bar) cells analysed by bead-based flow cytometry (right axis) (A). Data are presented as mean ± SEM of three independent measurements made in duplicate. **p < 0.01, ***p < 0.001. TEM micrograph showing the presence of EpCAM on the surface of large EVs (arrowheads) and small EVs (arrows) (B).
Figure 5.
Concentration of EpCAM-positive EVs released in 24 hours from OAW28 (red bar), PEO1 (yellow bar) and OAW42 (green bar) cells analysed by fluorescent-triggered flow cytometry (left axis). Percentage of EpCAM-positive beads detected in isolated samples of EVs released in 24 hours from OAW28 (red bar), PEO1 (yellow bar) and OAW42 (green bar) cells analysed by bead-based flow cytometry (right axis) (A). Data are presented as mean ± SEM of three independent measurements made in duplicate. **p < 0.01, ***p < 0.001. TEM micrograph showing the presence of EpCAM on the surface of large EVs (arrowheads) and small EVs (arrows) (B).
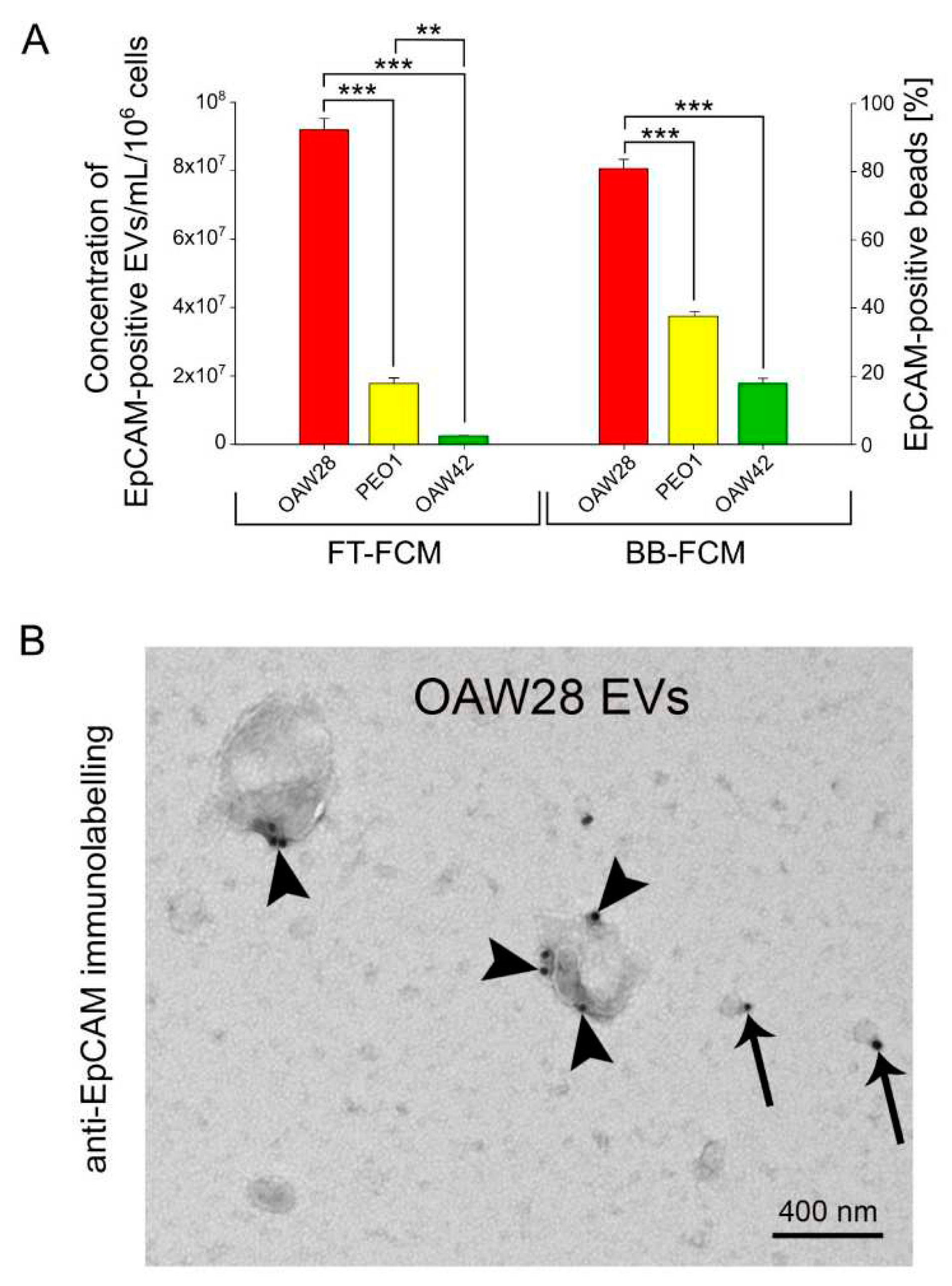
Figure 6.
Expression of extracellular EpCAM domain on OAW28 (red bar), PEO1 (yellow bar), and OAW42 (green bar) cells quantified by flow cytometry. MFI – mean fluorescent intensity. PE – phycoerythrin. Data are presented as mean ± SEM of three independent measurements made in duplicate. **p < 0.01, ***p < 0.001. (A). Positive correlation between EpCAM expression on cell membrane and concentration of EpCAM-positive EVs released from OAW28, PEO1 and OAW42 cells (B). Data are presented as three independent measurements made in duplicate for each cell line.
Figure 6.
Expression of extracellular EpCAM domain on OAW28 (red bar), PEO1 (yellow bar), and OAW42 (green bar) cells quantified by flow cytometry. MFI – mean fluorescent intensity. PE – phycoerythrin. Data are presented as mean ± SEM of three independent measurements made in duplicate. **p < 0.01, ***p < 0.001. (A). Positive correlation between EpCAM expression on cell membrane and concentration of EpCAM-positive EVs released from OAW28, PEO1 and OAW42 cells (B). Data are presented as three independent measurements made in duplicate for each cell line.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



