
Submitted:
04 May 2023
Posted:
04 May 2023
You are already at the latest version
Abstract
Noctuidae is known to have high species diversity, although the genomic diversity of Noctuidae species have not been studied extensively. Investigation of transposable elements (TEs) in this family can improve our understanding on the genomic diversity of Noctuidae. In this study, we annotated and characterized genome-wide TEs in ten noctuid species belonging to seven genera. With multiple annotation pipelines, we constructed a consensus sequence library containing 1,038 –2,826 TE consensus. The genome content of TEs showed high variation in the ten Noctuidae genomes, ranging from 11.3% to 45.0%. The relatedness analysis indicated that the TE content, especially the content of LINEs and DNA transposons, are positively correlated with the genome size (r=0.86, p-value=0.001). We identified SINE/B2 as a lineage-specific subfamily in Trichoplusia ni, a species-specific expansion of LTR/Gypsy subfamily in Spodoptera exigua, and a recent expansion of SINE/5S subfamily in Busseola fusca. We further revealed that of the four TE classes, only LINEs showed phylogenetic signal with high confidence. We also examined how the expansion of TEs contributed to the evolution of noctuid genomes. Moreover, we identified a total of 56 horizontal transfer TE (HTT) events among the ten noctuid species and at least three HTT events between the nine Noctuidae species and 11 non-noctuid arthropods. One of HTT events caused by a Gypsy transposon might have caused the recent expansion of Gypsy subfamily in the S.exigua genome. By determining the TE content, dynamics, and HTT events in the Noctuidae genomes, our study emphasized that TE activities and HTT events had substantial impacts on the Noctuidae genome evolution.
Keywords:
Noctuidae
; transposable elements
; genomic diversity
; phylogeny
; horizontal transfer TE (HTT) events
1. Introduction
The family Noctuidae is highly diverse in species with almost 12,000 species, forming the third largest family within the order Lepidoptera [1]. Many members in this family are phytophagous insects and highly harmful to crops or forests. Despite of the large number of previous studies focusing on the morphology, physiology, and biological control of Noctuidae species [2,3], our understanding on the genomic diversity of the Noctuidae species, especially the transposable elements (TEs), is still in its infancy.
TEs are a class of repetitive sequences dispersed throughout the genome. They are essential component of eukaryotic genomes that can jump around the genome within the same chromosome or between different chromosomes, even transfer horizontally between species [4,5]. TEs move through the genomes using either a “cut-and-paste” or a ‘‘copy-and-paste’’ mechanism, and TEs have important impacts on architecture, function, and evolution of the host genome[6,7,8].
In recent decades, many insect genomes have been assembled. Insect genomes vary significantly in size, ranging from as large as 6.5 Gb in Locusta migratoria to as small as less than 0.1 Gb in Tetranychus urticae. However, the number of genes in their genomes are similar, and the difference in genome size is mainly due to variations in TE contents [9]. Previous studies on Arthropoda, Lepidoptera, and the genus Drosophila suggested a positive correlation of genome size to the TE content [7,9,10]. Nevertheless, it is especially challenging to study insect TEs for several reasons. 1) Insect genomes vary greatly in size and in the proportion of TEs. For example, two species in the order Diptera, Aedes aegypti and Belgica Antarctica, have a TE genome content of 55% and 1%, respectively. Even in the same genus, D. simulans and D. ananassae have significant difference in TE content, at 10% and 40% of the genome, respectively [11]. 2) Many lineage-specific TEs exist in different insect genomes, such as the Zisupton subfamily that is specific to coleopterans genomes [11]. 3) The TE composition is also highly variable in insect genomes. For example, DNA transposons the predominant TE class in Heliconius Melpomene, a species in order Lepidoptera, whereas DNA transposons have very low genome proportion in Papilio polytes, another species in order Lepidoptera [7]. 4) TE propagation is significantly different in insect genomes. Wu’s study found that out of 14 arthropod species, only silkworm had a large number of recent expansion TEs, which probably was responsible for the adaptation to domestication in silkworms [9].
In addition to the large TE variation, another challenge in studying insect TEs is the existence of horizontal transfer TE (HTT) events in insect genomes. TEs can transfer from one host to another in two ways. The first is vertical inheritance, where they are passed from parents to offspring. The second way is horizontal transfer, which occurs between organisms that do not mate [12]. The horizontal transfer allows TEs to jump from an old host to a new one. In the old host genome, natural selection and silencing mechanisms can suppress the propagation or delete TEs from the genome. However, when it inserted into a new host genome, it can escape the suppression and extinction[13]. Therefore, HTT plays an important role in the long-term survival of TEs. Since the first HTT event was reported in Drosophila melanogaster[14], a total of 2,836 HTT events have been recorded in HTT-db by 2017 [15]. One of the most recent HTT events occurred after 2010 in D. simulans. A P element horizontal transferred from the D. melanogaster into D. simulans genome and the P element could be found in the populations of D. simulans only after 2010 [16]. Previous study suggested that order Lepidoptera is a hotspot for HTT events [17]. As one of the largest family of Noctuidae in the order Lepidoptera, however, there is no comprehensive study of HTT events among Noctuidae species, as well as between Noctuidae species and non-noctuid arthropods to date.
By 2020, genome assemblies are available for ten species of the Noctuidae. The ten species belong to seven genera. With multiple prediction methods, in this study we annotated and characterized TEs in the genomes of the ten species to reveal the genomic diversity of TEs in Noctuidae and the correlation of TE content to the genome size of noctuids. We also investigated how different TE classes/subfamilies expanded/contracted in the genome of Noctuidae insects. We also estimated HTT events among the genome of Noctuidae species, as well as between Noctuidae species and other arthropods, and elucidated how HTT events affect the evolution of Noctuidae genome.
2. Materials and methods
2.1. Data collection
Ten species of Noctuidae belonging to seven genera with published genomes were selected (Table S1), including Agrotis ipsilon, Busseola fusca, Helicoverpa armigera, Helic. zea, Heliothis virescens, Mamestra configurata, Spodoptera exigua, S. frugiperda, S. litura, and Trichoplusia ni. Two species are in the genus Helicoverpa and three species are in the genus Spodoptera. The genome sequences were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/). Published DNA and protein sequences of repetitive elements were downloaded from Repbase[18]. NR database was downloaded from the NCBI database. The TimeTree[19] was used to derive phylogenetic relationships of the Noctuidae species in conjunction with the literatures, and iTOL[20] was used to generate the phylogenetic tree (Figure 1). Due to the unknown phylogenetic relationship of Helio.virescens[21,22], it was not included in the phylogenetic tree of Figure 1. Genome sequences and TE libraries of 11 non-noctuid arthropods were downloaded from ArTEdb(http://artedb.net/index.html).
2.2. Species-specific TE library construction
We used RepeatModeler 2.0[23] to build consensus sequence libraries for each Noctuidae species. Since version 2.0, RepeatModeleris able to call tools such as LTR_harvest[24] and LTR_retriever[25] to make consensus sequences based on structure, in addition to calling RepeatScout[26] and RECON [27] to create consensus sequences based on repetitive sequence properties(-pa 12 -LTRStruct). Unknown types accounted for the majority of the results output by RepeatModeler2. We therefore attempted to classify the unknown types of consensus sequences by PASTEClassifier[28] and TEclass[29]. Consensus sequences were aligned to TE Repbase proteins using BlastX using default parameters. Only the aligned query sequence is kept. To remove potential protein coding sequences that are not TE-encoded proteins, other sequences were aligned to Swiss-Prot using BlastX(identity > 30%, e-value < 1e-5, percent query coverage > 50%), and the aligned sequences were excluded from the library. To filter non-coding RNAs such as tRNA and rRNA, an ncRNA library was created using tRNA scan-SE [30] and R fam[31]. The consensus sequences were used as queries for Blastn to search against the ncRNA library using default parameters. To obtain the final TE libraries, the aligned query sequences were excluded.
2.3. TE annotation and statistical analysis
Genomes were masked by RepeatMasker version 4.0.9(http://repeatmasker.org/), and the "-lib" parameter was applied to use the custom TE library. We extracted TE copy sequences using the script ONE_CODE_TO_FIND_THEM_ALL.PL [32] with the “—strict” parameter and calculated the number of copies per family for each species via the self-definition function in Python. Pearson correlation analysis was applied via the "scipy.stats.pearsonr" function [33] in Python to analyze the correlation between genome size and TE load.
2.4. TE propagation activity
To estimate TE propagation activity during the evolutionary history of Noctuidae, we performed a copy-divergence analysis of the TE subfamilies based on their Kimura 2-parameter distances. The Kimura 2-Parameter divergence of TEs was calculated using buildSummary.pl and calcDivergenceFromAlign.pl on alignment files. Kimura distances were transformed to the time estimates of variation in TE activity with the following equation: T=K/2r, where r is the neutral mutation rate estimates, and K is the Kimura 2-Parameter divergence of TEs. The neutral mutation rate was set to 2.9×10−9 from H. Melpomene and assumed one generation per year in general [34].
2.5. Calculation of phylogenetic signal
Two tests were applied for phylogenetic signal analysis using the TE load. The first method was Pagel's lambda. The value of lambda ranges from 0 to 1. The higher the value was, the stronger the phylogenetic signal was, indicating that the trait is highly correlated with phylogeny and not random. Another test was Blomberg's K which quantifies the variance of traits relative to what we would expect under Brownian motion (BM). The value of K ranges from 0 to infinity. K=0 means that there was no phylogenetic signal in the continuous trait. K=1 meant that the trait had evolved under BM. K>1 meant that there was more phylogenetic signal than expected under a BM model of trait evolution, which indicated that closely related species shared high similarities in traits. We applied phylogenetic signal analysis via the "phytools.phylosig" function in R [35].According to the phylosig's documentation, lambda value is influenced by the relative height in this function. Therefore, the calculated value may exceed 1.
2.6. Ancestral node reconstruction and predicting the change rate of TEs
The phylogenetic signals of TE estimated by Pagel's lambda method were used to test which standard phylogenetic comparative model was appropriate for ancestral state reconstruction. We used the "Geiger.fitContinuous" function [36]in R to evaluate the value of AICc in each of the following four standard phylogenetic comparative models: Brownian motion (BM), Ornstein-Uhlenbeck (OU), Early-burst (EB), and white noise. We compared the AICc values of the four models, and the results showed that the EB model was the most appropriate (Table S2). The EB model was used for ancestral state reconstruction of TE Loads. Based on the maximum likelihood method, the TE content of the ancestor nodes was reconstructed using the anc.ML function of the R package phytools(models=" EB").
BAMM (Bayesian Analysis of Macroevolutionary Mixtures) is a program for modelling trait evolution in the time-calibrated phylogeny [37,38,39]. We used the program to calculate evolutionary rates for TE loads in a phylogenetic tree. The “BAMMtools.setBAMMpriors” function was applied to estimate the betaInitPrior and betaShiftPrior for BAMM settings. The BAMM outputs were analysed and visualised by BAMMtools[40].
2.7. Identification of TE horizontal transfer events between Noctuidae species
If some TE pairs showed more similarly than their hosts diverge, we suspected that an HTT event occurred [13]. We adopted the dS (synonymous substitutions per synonymous site) to identify HTT events. dS cannot be evaluated on TE class that lacks proteins. Therefore, SINEs were excluded from our analysis. Using ONE_CODE_TO_FIND_THEM_ALL.PL, we extracted the autonomous TE sequences, which were greater than or equal to 80% of the length of the corresponding consensus sequences. TE copies were aligned to the TE protein library extracted from Repbase using tblasn. The best hits were realigned using exonerate, and the proteins of TE copies were extracted. All TE proteins were aligned between every pair of noctuid-selected species using blastp. TE pairs aligned over 300 bp were kept for calculation of dS using KaKs_calculator[41]. Based on the core genes of Lepidoptera, the BUSCO [42] pipeline was used to locate single-copy orthologous genes of the ten species.
Encoded peptide sequences of single-copy genes were aligned between every two noctuids using blastp (10-4). Proteins with reciprocal best hits and the same BUSCO identifier were identified as orthologous sequences. We only kept alignments > 300 amino acids and orthologs for calculation of dS using KaKs_calculator. We define an HTT event as a TE pair with a dS smaller than the top ~2.5% of the total orthologous gene pairs.
2.8. Identification of TE horizontal transfer events between Noctuidae species and other arthropods
The basic principle was to compare TE pairs and host divergence rates. Searching for HTT events only between distant lineages could reduce misclassification of horizontal transfers because the chance of having similar TE pairs that inherited vertically in distant lineage is very low. We divided 9 noctuids and 11 arthropod species into different lineages, and we only considered HTTs between lineages. Due to the unclear evolutionary relationship of Helio. virescens, the analysis was conducted on nine species of nocturnal moth insects instead of ten. Genome and TE libraries of 11 arthropods were downloaded from ArTEdb. We collapsed a clade of species into a lineage if (i) a fraction (>0.3%) [41] of its core orthologous genes showed lower dS than the highest nucleotide divergence of TEs or (ii) these species diverged in the last 40 My[43]. The study here referred to Peccoud's heuristic search approach [43] and the clustering code was written based on reference from Zhang Huahao's vertebrate horizontal transfer TE study [44].
3. Results
3.1. Construction of TE libraries in the ten Noctuidae species
We constructed a repetitive sequence library using RepeatModeler, which contained 1,066-2,904 consensus sequences for each of the 10 different species. Initially, we compared the library to protein sequence databases and removed non-transposable element (TE) encoding proteins, resulting in 1,050-2,835 consensus sequences from the 10 species. We then filtered out RNA and simple repeats, leaving a library with 1,038-2,826 consensus sequences from the 10 species, including 241-603 retrotransposons (RTEs) and 172-350 DNA transposons (DTEs). Most of consensus sequences were unclassified. Next, we used prediction and classification software, such as TEclass, to classify the unclassified sequences and found that the range of retrotransposons in these 10 species was 562-1,617, while that of DNA transposons was 428-1,186(Table 1, Table S3). In general, the bigger the genome size, the more consensus sequences were obtained. Genome sizes of the ten species varied from 299.98 Mb to 559.39 Mb, with the biggest genome in M.configurata and smallest genome in Helic. armigera. Despite the success of the TEclass in classification,3.08%-5.26% consensus sequences failed to be classified and were labeled as unknown repetitive sequences.
3.2. TEcontents
Genome content of TE showed high variations among Noctuidae genomes, accounting for 11.3% - 45.1% of the ten genomes (Table 2). Three species (Helic.armigera, Helic.zea, T.ni) with the smallest genome sizes showed TE contents lower than 20%. B.fusca had the highest TE content (45.1%) and the second largest genome (490 Mb). In general, the bigger the genome was, the higher the TE content.
Four TE classes were annotated while their content varied greatly between ten noctuid genomes (Table 2).LINEs and DNA transposons were dominant accounting for 3.46%-20.16% and4.01%-12.1% of the entire genomes, respectively. SINEs and LTR transposons accounted 0.98%-6.11% and 0.74%-4.22% of the genomes, respectively. Notably, we found large expansion of LINEs (20.16% of the genome) and DNA transposons(12.1% of the genome) in the B. fusca genome compared to other noctuids, which might contribute to the highest TE content (45.1%) in the B. fusca genome. In addition, an expansion of LTR occurred in S. exigua genome with the highest LTR percentage (4.22%) among the ten species while the total TE content of S. exigua was only 30.64%. SINE showed a reduced activity in S. frugiperda compared to other species. S.frugiperda genome had the lowest SINE percentage(0.98%) which was only 16%-47% of that in other noctuid species. Radar plots in Figure S1 showed the differences in TE proportions of noctuid species compared with Helic. armigera (lowest total TE content, 11.33%) and B. fusca(highest total TE content, 45.1%).
3.3. TE subfamilies and copy numbers
A total of 40 subfamilies were identified in the ten Noctuidae species, including 13 LINE subfamilies, 4 SINE subfamilies, 6 LTR subfamilies and 17 DNA subfamilies. A heat map of the 40 subfamilies across the ten species was plotted (Figure 2). The DNA/Helitron subfamily showed high copy numbers consistently in all the ten species, with an average copy number of 141,201 (100,498-236,722). In contrast, copy numbers of other subfamilies varied greatly across species. For example, the SINE/5S subfamily had only130-204 copies in three species of Spodoptera compared with relative high abundance (>10,000 copies) in other species. Meanwhile, LINE/R1 and LINE/CR1 subfamily were abundant in copy number in most noctuids except three species (Helic. armigera, Helic. zea, and T. ni). The three species had much fewer copies of R1 subfamily (13,008 - 14,149 copies) and CR1 subfamily (1,188 - 1,727 copies) than those of other seven species (83,107 -152,596 copies in R1and >100,000 in CR1). We also found several lineage-specific subfamilies. The SINE/B2 subfamily was only found in the T. ni genome with a high copy number (4,784 copies). The DNA/Crypton subfamily was present specifically in Helic. zea (753 copies), A.ipsilon (37 copies) and M.configurata (1,257 copies) genomes.
3.4. Propagation activity of TE subfamilies
Following the methods[7,45], we estimated propagation activity of different TE subfamilies. We converted the 10% divergence rate to 17.24 million years based on estimated neutral mutation rate. Figure 3 showed several subfamilies with distinct propagation activity among the ten species. Given that the SINE/B2 subfamily was specific in T. ni, we found that SINE/B2 began to insert into T. ni genome around 60 Million years ago (Mya) with a propagation peak 20-30 Mya (Figure 3A). According to the phylogenetic tree (Figure 1), T. ni was a basal species diverged from other nine species about 60 Mya. This indicated that SINE/B2 inserted into T. ni after the divergence of the other species and resulted in the lineage-specific subfamily. LINE/CR1 showed continuous propagation in most noctuids except three species (Helic. armigera, Helic. zea and T. ni), which was consistent with the high copy number of the CR1subfamily in many species. However, the timing of CR1propagation varied in different genomes. A burst propagation of CR1 subfamily occurred 30 - 40 Mya in S.exigua, 15-20 Mya in S. frugiperda, in contrast to very recently around 6 Mya in the B. fusca genome (Figure 3B). Figures 3C and 3D showed the distribution of insertion time of LINE/R1 and SINE/5S subfamily in the ten species. Interestingly, both subfamilies had a peak propagation around 6 Mya in the B. fusca leading them to be the two most abundant TE subfamilies among the ten genomes. The DNA/Maverick subfamily was present only in five species with the highest copy numbers in S.frugiperda. Figure 3E showed an obvious peak of propagation around 40 Mya in S.frugiperda.
Next, we compared the activity of TE subfamily among three closely related species in genus Spodoptera. Most TE subfamilies showed similar propagation activity among the three species, except LTR/Gypsy(Figure 3F). Gypsy subfamily showed a high activity in the S. exigua genome recently with a propagation peak about 2 Mya, accumulating 7,393 copies of Gypsy and a genome content of 2.25% in the S. exigua genome which was 4.5 and 6.4 times of that in S.frugiperda and S. litura genomes, respectively. Since S. exigua diverged from other two species about 25 Mya (Figure 1), it was clear that the expansion of Gypsy subfamily was a species-specific event.
3.5. Relatedness of TE content with genome size of noctuid species
The TE content and the genome size of the ten noctuid species showed a strong positive correlation (r=0.86, p-value=0.001). We further investigated which class or superfamily of TEs contributed more to the genome size variation. We listed all subfamilies with r values > 0.2 in Table 3. Both LINEs and DNA transposons showed strong positive correlation with genome size with r > 0.8 and P values<0.005. While LTR and SINEs had moderate or weak correlation with genome size, these correlations are not significant (P values>0.1).
With regard to subfamilies, DNA/TcMar and DNA/Zator subfamilies showed the strongest correlated with genome size (r > 0.7 and P <0.01). Interestingly, the most abundant DNA transposons, the Helitron subfamily, had little correlation with genome size (r = 0.13). Within LINEs, Dong, L2, and R1 subfamilies showed strong correlations with genome size (r > 0.7 and P <0.01), while RTE and CR1had moderate correlations with genome size. All these subfamilies except Dong had high copy numbers in the noctuids genome.
3.6. Phylogenetic signals and changes of TE load
We calculated lambda values and K values to evaluate the phylogenetic signals in the four TE classes (Table 4). Although DNA transposon, LINE, and LTR had lambda values >0.5, only LINE had a p-value <0.05, indicating LINE elements were correlated with phylogeny with high confidence. LINE also had K value> 1, indicating that closely related species shared high similarity in the LINE content. A total of 12 subfamilies had lambda values>0.5, six of which had p-values <0.05and K-values >1 (p<0.05), including DNA/hAT, DNA/TcMar, LINE/CR1, LINE/L2, LINE/RTE and LINE/R1 (Table 4). This result suggests the activity of these subfamilies are highly correlated with the phylogeny.
By predicting TE load and TE change rate on ancestral nodes of phylogenetic tree, we estimated whether the TE expansion activity was associated with the noctuids phylogeny. SINE retrotransposons were excluded for the analysis given that they contained very little phylogenetic signal (Table 4). Four comparative models were evaluated and the EB model was selected (Table S2). The maximum likelihood method was used to infer the TE loads in ancestor nodes (internal nodes). The black and red lines on the phylogenetic tree in Figure 4 represented expanded or reduced TE load in the genomes, respectively. An expansion was found in B. fusca in three class of TE (LTR, LINE and DNA). In contrast, all the three class of TE loads reduced in Helicoverpa lineage. In T. ni lineage, an expansion was found in LTR, and a reduction in both DNA transposons and LINE. In M. configurata/A. ipsilon lineage, an expansion of LINE was observed. In Spodoptera lineage, LTR and LINE expanded, compared to a reduced activity of DNA transposons (Figure 4A, 4B, 4C). The TE expansion activity also varied among species within the same genus. For example, LTR and LINE activity reduced, but DNA transposon expanded in S. frugiperda which was different from other two Spodoptera species.
Next we compared the expansion activity of four LINE subfamilies (CR1, L2, R1 and RTE) across the nine noctuid species (Figure 4D, 4E, 4F, 4G). They were generally similar to each other and consistent with the results of LINE as a whole. However, we noted a reduction of CR1 load (Figure 4D) and R1 load(Figure 4F)in M. configurata, and reduction of CR1 (Figure 4D) and L2 (Figure 4E) in A. ipsilon, while the overall LINE expanded in both species(Figure 4B).RTE subfamily in S. exigua showed a reduced activity(Figure 4G) compared to the expansion of LINE and other LINE subfamilies (CR1, L2, R1). The expansion activity of two DNA subfamilies also was different across the noctuids. There was an expansion in hAT and overall DNA but a reduction in TcMar subfamily in B. fusca(Figure 4H, 4I). In addition, they also showed different expansion activity between the two Helicoverpa species.
We further applied BAMM to predict the change rate of TEs(Table 5). LINE had the fastest change rate, followed by DNA transposons, and LTR had change rate much lower than LINE and DNA.
We next mapped change rates of TE on the phylogenetic tree to demonstrate the expansion activity changed over time(Figure 5). The warmer the color is, the higher change rate was, and the nearer of the color to the species name, it occurred more recently. Evolution of LTR, LINE and DNA transposons(Figure 5A, 5B, 5C) was faster in ancient time than in recent time. While recently, LTR in S. exigua, LINE in B. fusca, and DNA transposons in T.ni showed the fastest change rate among the Noctuidae species. Within LINE subfamilies, change rates varied among different lineages. CR1subfamily in S. exigua, B. fusca and T. ni evolved more rapidly than other species (Figure 5D). While R1 subfamily in M. configurata and T. ni, RTE subfamily in A. ipsilon changed rapidly in recent time (Figure 5E-5G). Within DNA subfamilies, the lineage M. configurata/A. ipsilon showed distinct change rates between the hAT and TcMar subfamily, and the evolution rate of hAT subfamily was relatively fast in M. configurata/A. ipsilon compared with other species (Figure 5H, 5I).
3.7. Transposon horizontal transfer events among Noctuidae species
We identified millions of TE copies based on the constructed TE consensus sequences in the genomes of ten noctuid species. This dataset allowed us to examine the HTT events in these species. To do so, we obtained the single-copy genes in each species and their single-copy homologous genes in other species. dS values in all TEs and the homologous genes were calculated and compared to identify potential HTT events. A strict threshold was adopted in our analysis: a pair of TEs were considered being a HTT event if their dS values are less than the dS value of the 2.5% of the orthologous genes with the lowest dS values in the two species (Table S4).A total of 56 possible HTT events were identified, including 22 DNA transposons, 32 LINEs, and 2 LTR transposons (Figure 6, Table S5). Due to the unclear evolutionary relationship of Helio. virescens, the results related to Helio. virescens are not shown in the figure. However, the analysis in this section does not require a clear evolutionary relationship, so the total number of horizontal gene transfers still includes Helio. virescens. S.exigua was involved in 27 HTT events which was the most among the ten species. We noted that both HTT events involving LTRs occurred in the S. exigua genome. One was LTR/Copia element transferring between the S. exigua and T. ni genome, another one was LTR/Gyspy element transferring between the S. exigua and the M. configurata genome (Table S5). The results were consistent with the above findings that LTR expansion in S. exigua (Figure 4A) and the higher change rate recently in S. exigua(Figure 5A).
TEs from high-copy TE subfamilies were more likely to be identified in HTT events due to misclassification. To evaluate the potential bias, we calculated the number of HTT events per thousand copies in each TE family (Table 6). The DNA/TcMar and LINE/RTE subfamilies were involved in the most number of HTT events, however neither of them had high copy number. In contrast, DNA/Maverick had the highest frequency of HTT per thousand copies (1.72) among the subfamilies but the lowest copy number. While DNA/RC had the lowest frequency of HTT(0.001) per copy with the highest copy number among all subfamilies. The results indicated that subfamilies with high copy numbers were not correlated with more HTT events.
3.8. Transposon horizontal transfer events between 9 Noctuidae species and other arthropods
We further identified HTT events between Noctuidae species and other arthropods. TE libraries of 11 non-Noctuidae arthropods were obtained from the ArTEdb[9] and combined with TEs from nine Noctuidae species for HTT analysis. As stated in the method, this section requires a clear evolutionary relationship, so it does not include Helio. virescens. Therefore, there are nine moth insect species and eleven non-Noctuidae arthropods included in this part. Given that the horizontally transferred TEs could produce multiple copies in the host genome, a HTT event occurred before the species divergence would retain copies of the horizontally transferred TE in all genomes of the diverged species. Therefore, multiple HTT events could be detected in the diverged species, leading to overestimation of the number of horizontally transfer events. To exclude the overestimation, the heuristic methods and clustering algorithms were applied by identification of the minimum number of HTT events for insects[13] and for vertebrates[44]. A total of 37 events were initially identified between the noctuid species and eleven non-noctuid arthropod species. After performing clustering, 37 events can be divided into three minimal events (Table S6, represented using hitGroup), including two horizontal transferred DNA/Helitron occurred between A. pisum and Noctuidae species and one horizontal transferred DNA/Mariner occurred between the M. martensii and the B. fusca (Figure 7). One Helitron HTT event consisted of 12 HTT events involving five noctuid species such as B.fusca, S. frugiperda, S. exigua, S. litura and M. configurata. It estimated the Helitron element inserted into B.fusca genome about 71 Mya. According to the phylogenetic tree, this was before the divergence of the five noctuid species. Therefore the HTT event probably occurred in the common ancestor of these species. Another Helitron HTT event consisted of 19 HTT events involving three species in the genus Spodoptera. The insertion time of the Helitron copy into S.frugiperda was estimated to be 74 Mya, which was before the divergence of the three Spodoptera species, suggesting the HTT event was genus-specific. While the Mariner HTT event contained six HTT events occurred between the M.martensii and B. fusca (Table S6). The estimated insertion time was about 44 Mya. The phylogenetic tree estimated the divergence of B.fusca from common ancestor about 49 Mya, therefore the HTT event was highly likely to be species-specific.
4. Discussion
4.1. TEs shape the genome diversity of Noctuidae species
Unlike protein-coding genes under the selective pressure, TE sequences are usually not subject to selective pressure and thus change rapidly [46]. In addition, TE expansion/contraction occurs in a high frequency in the genome of arthropods[11],leading to enormous variations of TEs in arthropod genomes. To date little has been understood on the TE characteristics and genome-wide diversity in Noctuidae species. This study constructed a consensus sequence library for ten Noctuidae species containing 1,038 – 2,826 TE consensus in each genome, finding TEs showed high variations among Noctuidae species, even among species of the same genus. Genome content of TEs also varied greatly (from 11.33% to 45.1%) among the ten species. The high variation of TE content among close related species was consistent with previous studies on Lepidoptera species, where TEs account for 4.7% - 38.3%of the genomes[7], and on Insecta species, where TEs account for 1% - 55% of the genomes[11]. It was suggested that the increase/decrease of TE content was the most important reason affecting the genome size of arthropods[10].Similarly, in the Noctuidae, we found a strong positive correlation between TE content and genome size (r>0.8, p<0.01). In particular, we revealed that LINE and DNA transposons contributed most to the genome sizes, in contrast to SINEs which had no significant correlation. However, a study based on more than ten arthropods found that LINE, SINE, LTR and DNA transposons were all positively correlated with genome sizes (r>0.6)[9]. The discrepancy might due to the relatively smaller content of LTR and SINE in the noctuid genomes compared to other arthropods.
Noctuid species also exhibited significant differences on the copy numbers and lineage-specific expansion of TE subfamilies. The SINE/5S subfamily was one of examples whose copy number highly varied among closely related species. Copy number of SINE/5Ssubfamily was only 130-204 copies in the three species of genus Spodoptera, but more than 100,000 copies in B. fusca. While among the ten species, B. fusca had the closest phylogenetic relationship to genus Spodoptera. It suggested that an expansion of SINE/5Sin the genome of B. fusca. Activity estimation found propagation peak of SINE/5S about 6 Mya in B. fusca(Figure 4E), suggesting elements in the SINE/5S probably still active recently. Three species in the genus of Spodoptera allow the investigation of lineage-specific TE propagation among closely related species. We found an obvious expansion of LTR/Gypsy subfamily specific in S. exigua but not in S. frugiperda and S. litura. The expansion event occurred very recently with a propagation peak about 2 Mya, long after the divergence of S. exigua from other species (Figure 3F) and represented a species-specific expansion event.
We found the SINE/B2 was a lineage-specific subfamily presented only in T. ni. Since T. ni diverged first from other Noctuidae species in phylogeny, the subfamily might either result from a loss of B2 in other noctuid species, or from an insertion of B2specific into T. ni through horizontal transfer. We further investigated SINE/B2 subfamily in other genomes by comparison ofB2 consensus sequences with the reference genomes of 27 Lepidoptera species (Table S7) using the blastn tool, but did not find the sequence in any genome other than T. ni. The B2 consensus sequence was from RepeatModeler based on the published TE library and was not identified by machine learning, thus the classification was reliable. Therefore, it is highly likely that SINE/B2 inserted into T. ni genome rather than lost in other lineages. The insertion was estimated from 60Mya with a peak propagation around 20-30 Mya. However, where theB2 subfamily came from and how it integrated into T. ni genome requires further study.
4.2. TE expansion activity correlated with phylogeny of Noctuidae species
In addition to be the main contribution factor to genome size of Noctuidae species, we also investigated which class/subfamily of TE was correlated with phylogeny of Noctuidae. Among the four classes of TE, only LINE showed phylogenetic signal with high confidence, indicating the essentially vertical inheritance characteristics of LINE elements in Noctuidae. In particular, four LINE subfamilies, CR1,L2,RTE and R1showed high correlation with Noctuidae phylogeny, all of which were abundant in copy number. In contrast, despite of the high copy number of DNA/Helitron subfamily in noctuid genomes, its correlation with phylogeny was not significant. This was probably because the different integration mechanism of the Helitron. Another potential reason is elements in the Helitron subfamily had involved in horizontal transfer events in the Noctuidae species, which we will discuss below.
We further elucidated whether the expansion of TE class/subfamily contributed to the evolution of noctuid genomes. We noted that the LINE, LTR and DNA transposons all had relatively low activity in the genus Helicoverpa, that was probably the reason that both species in the genus Helicoverpa had the least TE content and the smallest genome sizes. In contrast, the LINE, LTR and DNA transposons all expanded in the genome of B. fusca(Figure 6)accumulating the highest TE content of B. fusca (45.1%) in the ten species. By calculating the change rate of TE, interestingly, we found only LINE and DNA transposons had their expansion occurred very recently (Figure 4) indicating these TEs were highly likely active in B. fusca, especially the LINE/CR1, LINE/R1 and LINE/RTE subfamily who showed recent expansion in B. fusca.
Although LINE and DNA transposons largely impacted the genomes of Noctuidae species, this was not the case in recent evolution of a specific species. For example, the activity of TE class/subfamily in S. exigua was substantially different from other two Spodoptera species. LTR elements expanded in S. exigua genome and the expansion occurred very recently (Figure 3F) accumulating the highest genome content of LTR (4.22%) among the ten species, in contrast to a reduction of LTR in S.frugiperda and S. litura genome. We identified several HTT events related to LTR elements in S. exigua genome (discussed below), which may contribute to its recent expansion. Thus, LTR is the TE class that had the most important impact on the recent evolution of S. exigua.
4.3. HTT events on the genomes of the noctuid moths
Despite of the essential homoplasy free characteristics of TE, horizontally transferred TE (HTT) events have been widely reported in the insect genomes[15]. Peccoud’s study found that as high as 2,248 HTT events occurred among 195 insect species in the last 10 million years, which probably was only a tiny fraction of the actual HTT events between insects [43]. Another study analyzed 460 species of arthropods for horizontal transfer and found significantly more HTT events in Lepidoptera than in other arthropods [17]. Our study identified a total of 56 possible HTT events among the ten noctuid genomes. Previous study indicated that the higher the copy number of a subfamily, the higher the probability its member was misclassified as an HTT event [47]. By calculating frequency of HTT events per thousand TE copies, we did not observe the trend in our results, suggesting our results do not suffer from the copy number bias. A large scale study on the HTT in insect genomes identified a total of 2,248 HTT events, 1,087 of them were associated with DNA/Tc1-Mariner subfamily[43]. In our study, 17 of the 56 HTTs belonged to elements in the Tc1-Mariner subfamily which was the highest among all subfamilies. The Tc1-Mariner is short in length (1 Kb ~ 2 Kb), which may facilitate horizontal transfer through the vector resulting in high frequency of HTT event.
One HTT event involved an LTR/Gypsy element transfer between S.exigua and M. configurata. By calculating divergence of the Gypsy consensus of S.exigua, we estimated the divergence rate of the Gypsy consensus was 1.38%in the S. exigua, converting to an insertion time of 2.37 Mya. It was consistent with the activity estimation of Gypsy with a burst propagation around2 Mya in the S.exigua genome. When TEs inserted into a new genome by horizontal transfer, the new host genome was generally unable to immediately inhibit the replication and translocation of the transposons. If the TE maintained replication capacity in the new genome, they may lead to massive TE replication[13]. Therefore, we inferred that the horizontal transferred Gypsy element could have led to the recently mass replication of Gypsy in the S.exigua genome, accounting for 2.25% of the entire genome, about4.5-6.4 times of that in the closely related species of the same genus.
It is noteworthy that for the HTT events identified between closely related species, no matter how strict the threshold, there is still the possibility that the similarity of vertically inherited TEs exceeds the selection threshold. Previous study suggested that the closer the distribution and the closer the affinity of the species, the higher the frequency of horizontal transfer occurred between species [43]. The study further suggested the method of minimal number of HTT events to identify HTT events between species that diverged more than 40 Mya to avoid exaggerating the number of HTT events. Following the method, we identified three minimal numbers of HTT events between the noctuid moths and eleven non-noctuid arthropods. These HTT events occurred in the genome of five noctuid species including B. fusca, M. configurata, and three Spodoptera species. Previous study suggested that genomes with more HTT events probably had higher TE contents and larger genome sizes [9]. All the five noctuid showed relatively larger genome sizes and higher TE contents compared to other five species, indicating HTT events might have shaped the evolution of Noctuidae genomes by leading to TE expansion.
Among the three minimum number HTT events, one Mariner–related HTT occurred specific in B.fusca. We further searched the Mariner sequence of B.fusca in other arthropod genomes, and found highly similar sequence from Cyphomyrmex costatus in Hymenoptera, supporting the horizontal transfer of this element between different insect genomes. However, whether the Mariner element transferred from M.martensii to the B.fusca genome needs further study with more arthropod genomes.
In conclusion, this study constructed a consensus sequence library for ten Noctuidae species based on multiple methods, significantly improving TE annotation in the Noctuidae genomes. By comparison of the TE genome content, TE composition and propagation activity of TE class/subfamilies among the ten Noctuidae species, this study provided new insights into the essential contributions of TEs to the genome size variation, genomic diversity, and phylogeny of Noctuidae species. We identified lineage-specific TE subfamilies and recent expansion of TE subfamilies in some Noctuidae species, suggesting they were probably still active in the Noctuidae genomes. Moreover, a total of 56 potential HTT events were identified among the noctuid species, and 3 minimum numbers of HTT events between the Noctuidae species and 11 non-noctuid arthropod species. The HTT events could account for the recent expansion of Gypsy subfamily in the S.exigua genome and the species-specific expansion of Mariner subfamily in B. fusca.
Supplementary Materials
Figure S1: Comparison of Helic. armigera, B. fusca and other Noctuids TE loads; Table S1: Species for comparative analysis of TEs in noctuid genomes; Table S2: The value of AICc using 4 comparative models; Table S3: TE consensus libraries in the ten Noctuidae species; Table S4: Cutoffs of dS; Table S5: horizontal transfer of TE; Table S6: Mininum numbers of transfer events of TE; Table S7: Added species for lineage-specific analysis of TEs in noctuid genomes.
Author Contributions
Conceptualization, C.Z., J.X. and J.L.; writing—original draft preparation, methodology and visualization, C.Z.; validation and data curation, L.W.; investigation and formal analysis, L.D.; Writing—review and editing, L.W., J.X. and J.L.; Funding acquisition, B.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Sichuan Key R&D Program (2019YFN0180).
Data Availability Statement
The data supporting the findings of this study are openly available from the NCBI (https://www.ncbi.nlm.nih.gov/) and ArTEdb (http://artedb.net/). Accession numbers are listed in Table S1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Keegan, K.L.;Rota, J.;Zahiri, R.;Zilli, A.;Wahlberg, N.;Schmidt, B.C.;Lafontaine, J.D.;Goldstein, P.Z.;Wagner, D.L. Toward a Stable Global Noctuidae (Lepidoptera) Taxonomy. Insect Systematics and Diversity. 2021, 5, 3. [CrossRef]
- Le Goff, G.;Nauen, R. Recent Advances in the Understanding of Molecular Mechanisms of Resistance in Noctuid Pests. Insects. 2021, 12, 8. [CrossRef]
- Di Lelio, I.;Coppola, M.;Comite, E.;Molisso, D.;Lorito, M.;Woo, S.L.;Pennacchio, F.;Rao, R.;Digilio, M.C. Temperature Differentially Influences the Capacity of Trichoderma Species to Induce Plant Defense Responses in Tomato Against Insect Pests. Front Plant Sci. 2021, 12, 678830. [CrossRef]
- de Almeida, L.M.;Carareto, C.M.A. Multiple events of horizontal transfer of the Minos transposable element between Drosophila species. Molecular Phylogenetics and Evolution. 2005, 35, 3, 583-594.
- Hof, A.E.v.t.;Campagne, P.;Rigden, D.J.;Yung, C.J.;Lingley, J.;Quail, M.A.;Hall, N.;Darby, A.C.;Saccheri, I.J. The industrial melanism mutation in British peppered moths is a transposable element. Nature. 2016, 534, 7605, 102-105. [CrossRef]
- Peng, C.;Niu, L.;Deng, J.;Yu, J.;Zhang, X.;Zhou, C.;Xing, J.;Li, J.J.M.D. Can-SINE dynamics in the giant panda and three other Caniformia genomes. 2018, 9, 1, 1-14. [CrossRef]
- Talla, V.;Suh, A.;Kalsoom, F.;Dincă, V.;Vila, R.;Friberg, M.;Wiklund, C.;Backström, N.J.G.B.;Evolution Rapid increase in genome size as a consequence of transposable element hyperactivity in wood-white (Leptidea) butterflies. 2017, 9, 10, 2491-2505.
- Cordaux, R.;Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009, 10, 10, 691-703. [CrossRef]
- Wu, C.;Lu, J. Diversification of Transposable Elements in Arthropods and Its Impact on Genome Evolution. Genes (Basel). 2019, 10, 5. [CrossRef]
- Sessegolo, C.;Burlet, N.;Haudry, A. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biology Letters. 2016, 12, 8. [CrossRef]
- Petersen, M.;Armisen, D.;Gibbs, R.A.;Hering, L.;Khila, A.;Mayer, G.;Richards, S.;Niehuis, O.;Misof, B. Diversity and evolution of the transposable element repertoire in arthropods with particular reference to insects. BMC Evol Biol. 2019, 19, 1, 11.
- Schaack, S.;Gilbert, C.;Feschotte, C. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol. 2010, 25, 9, 537-46. [CrossRef]
- Bartolome, C.;Bello, X.;Maside, X. Widespread evidence for horizontal transfer of transposable elements across Drosophila genomes. Genome Biol. 2009, 10, 2, R22. [CrossRef]
- Daniels, S.B.;Peterson, K.R.;Strausbaugh, L.D.;Kidwell, M.G.;Chovnick, A.J.G. Evidence for horizontal transmission of the P transposable element between Drosophila species. 1990, 124, 2, 339-355. [CrossRef]
- Dotto, B.R.;Carvalho, E.L.;Silva, A.F.;Duarte Silva, L.F.;Pinto, P.M.;Ortiz, M.F.;Wallau, G.L. HTT-DB: horizontally transferred transposable elements database. Bioinformatics. 2015, 31, 17, 2915-7. [CrossRef]
- Kofler, R.;Hill, T.;Nolte, V.;Betancourt, A.J.;Schlotterer, C. The recent invasion of natural Drosophila simulans populations by the P-element. Proc Natl Acad Sci U S A. 2015, 112, 21, 6659-63. [CrossRef]
- Reiss, D.;Mialdea, G.;Miele, V.;de Vienne, D.M.;Peccoud, J.;Gilbert, C.;Duret, L.;Charlat, S. Global survey of mobile DNA horizontal transfer in arthropods reveals Lepidoptera as a prime hotspot. PLoS genetics. 2019, 15, 2, e1007965-e1007965. [CrossRef]
- Bao, W.;Kojima, K.K.;Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015, 6, 11. [CrossRef]
- Kumar, S.;Stecher, G.;Suleski, M.;Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol Biol Evol. 2017, 34, 7, 1812-1819. [CrossRef]
- Letunic, I.;Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W1, W293-W296. [CrossRef]
- Behere, G.T.;Tay, W.T.;Russell, D.A.;Heckel, D.G.;Appleton, B.R.;Kranthi, K.R.;Batterham, P. Mitochondrial DNA analysis of field populations of Helicoverpa armigera (Lepidoptera: Noctuidae) and of its relationship to H. zea. BMC Evol Biol. 2007, 7, 117. [CrossRef]
- Kergoat, G.J.;Prowell, D.P.;Le Ru, B.P.;Mitchell, A.;Dumas, P.;Clamens, A.-L.;Condamine, F.L.;Silvain, J.-F.J.M.P.;Evolution Disentangling dispersal, vicariance and adaptive radiation patterns: a case study using armyworms in the pest genus Spodoptera (Lepidoptera: Noctuidae). Molecular Phylogenetics and Evolution. 2012, 65, 3, 855-870. [CrossRef]
- Flynn, J.M.;Hubley, R.;Goubert, C.;Rosen, J.;Clark, A.G.;Feschotte, C.;Smit, A.F.J.P.o.t.N.A.o.S. RepeatModeler2 for automated genomic discovery of transposable element families. 2020, 117, 17, 9451-9457. [CrossRef]
- Ellinghaus, D.;Kurtz, S.;Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics. 2008, 9, 18. [CrossRef]
- Ou, S.;Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 2018, 176, 2, 1410-1422.
- Price, A.L.;Jones, N.C.;Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics. 2005, 21, I351-I358. [CrossRef]
- Bao, Z.;Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 8, 1269-76. [CrossRef]
- Hoede, C.;Arnoux, S.;Moisset, M.;Chaumier, T.;Inizan, O.;Jamilloux, V.;Quesneville, H. PASTEC: an automatic transposable element classification tool. PLoS One. 2014, 9, 5, e91929. [CrossRef]
- Abrusan, G.;Grundmann, N.;DeMester, L.;Makalowski, W. TEclass--a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics. 2009, 25, 10, 1329-30. [CrossRef]
- Chan, P.P.;Lin, B.Y.;Mak, A.J.;Lowe, T.M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 16, 9077-9096. [CrossRef]
- Kalvari, I.;Nawrocki, E.P.;Ontiveros-Palacios, N.;Argasinska, J.;Lamkiewicz, K.;Marz, M.;Griffiths-Jones, S.;Toffano-Nioche, C.;Gautheret, D.;Weinberg, Z.;Rivas, E.;Eddy, S.R.;Finn, R.D.;Bateman, A.;Petrov, A.I. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D1, D192-D200. [CrossRef]
- Bailly-Bechet, M.;Haudry, A.;Lerat, E.J.M.D. “One code to find them all”: a perl tool to conveniently parse RepeatMasker output files. 2014, 5, 1, 1-15.
- Virtanen, P.;Gommers, R.;Oliphant, T.E.;Haberland, M.;Reddy, T.;Cournapeau, D.;Burovski, E.;Peterson, P.;Weckesser, W.;Bright, J.;van der Walt, S.J.;Brett, M.;Wilson, J.;Millman, K.J.;Mayorov, N.;Nelson, A.R.J.;Jones, E.;Kern, R.;Larson, E.;Carey, C.J.;Polat, I.;Feng, Y.;Moore, E.W.;VanderPlas, J.;Laxalde, D.;Perktold, J.;Cimrman, R.;Henriksen, I.;Quintero, E.A.;Harris, C.R.;Archibald, A.M.;Ribeiro, A.H.;Pedregosa, F.;van Mulbregt, P.;SciPy, C. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020, 17, 3, 261-272.
- Keightley, P.D.;Pinharanda, A.;Ness, R.W.;Simpson, F.;Dasmahapatra, K.K.;Mallet, J.;Davey, J.W.;Jiggins, C.D. Estimation of the spontaneous mutation rate in Heliconius melpomene. Mol Biol Evol. 2015, 32, 1, 239-43. [CrossRef]
- Revell, L.J.J.M.i.e.;evolution phytools: an R package for phylogenetic comparative biology (and other things). 2012, 3, 2, 217-223.
- Pennell, M.W.;Eastman, J.M.;Slater, G.J.;Brown, J.W.;Uyeda, J.C.;FitzJohn, R.G.;Alfaro, M.E.;Harmon, L.J. geiger v2.0: an expanded suite of methods for fitting macroevolutionary models to phylogenetic trees. Bioinformatics. 2014, 30, 15, 2216-8. [CrossRef]
- Rabosky, D.L.;Donnellan, S.C.;Grundler, M.;Lovette, I.J. Analysis and visualization of complex macroevolutionary dynamics: an example from Australian scincid lizards. Syst Biol. 2014, 63, 4, 610-27. [CrossRef]
- Rabosky, D.L.;Goldberg, E.E. Model inadequacy and mistaken inferences of trait-dependent speciation. Syst Biol. 2015, 64, 2, 340-55. [CrossRef]
- Rabosky, D.L.;Santini, F.;Eastman, J.;Smith, S.A.;Sidlauskas, B.;Chang, J.;Alfaro, M.E. Rates of speciation and morphological evolution are correlated across the largest vertebrate radiation. Nat Commun. 2013, 4, 1958. [CrossRef]
- Rabosky, D.L.;Grundler, M.;Anderson, C.;Title, P.;Shi, J.J.;Brown, J.W.;Huang, H.;Larson, J.G.J.M.i.E.;Evolution BAMM tools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. 2014, 5, 7, 701-707.
- Wang, D.;Zhang, Y.;Zhang, Z.;Zhu, J.;Yu, J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010, 8, 1, 77-80. [CrossRef]
- Manni, M.;Berkeley, M.R.;Seppey, M.;Simao, F.A.;Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol. 2021, 38, 10, 4647-4654. [CrossRef]
- Peccoud, J.;Loiseau, V.;Cordaux, R.;Gilbert, C. Massive horizontal transfer of transposable elements in insects. Proc Natl Acad Sci U S A. 2017, 114, 18, 4721-4726. [CrossRef]
- Zhang, H.H.;Peccoud, J.;Xu, M.R.;Zhang, X.G.;Gilbert, C. Horizontal transfer and evolution of transposable elements in vertebrates. Nat Commun. 2020, 11, 1, 1362. [CrossRef]
- Shao, F.;Han, M.;Peng, Z. Evolution and diversity of transposable elements in fish genomes. Sci Rep. 2019, 9, 1, 15399. [CrossRef]
- Pasyukova, E.G.;Nuzhdin, S.V.;Morozova, T.V.;Mackay, T.F.C. Accumulation of transposable elements in the genome of Drosophila melanogaster is associated with a decrease in fitness. Journal of Heredity. 2004, 95, 4, 284-290.
- Peccoud, J.;Cordaux, R.;Gilbert, C. Analyzing Horizontal Transfer of Transposable Elements on a Large Scale: Challenges and Prospects. Bioessays. 2018, 40, 2. [CrossRef]
Figure 1.
Phylogenetic relationships among the nine noctuid moth species with the silkworm (Bombyx mori) as the outgroup. Helio.virescens is not included in the tree because of its unknown position.
Figure 1.
Phylogenetic relationships among the nine noctuid moth species with the silkworm (Bombyx mori) as the outgroup. Helio.virescens is not included in the tree because of its unknown position.
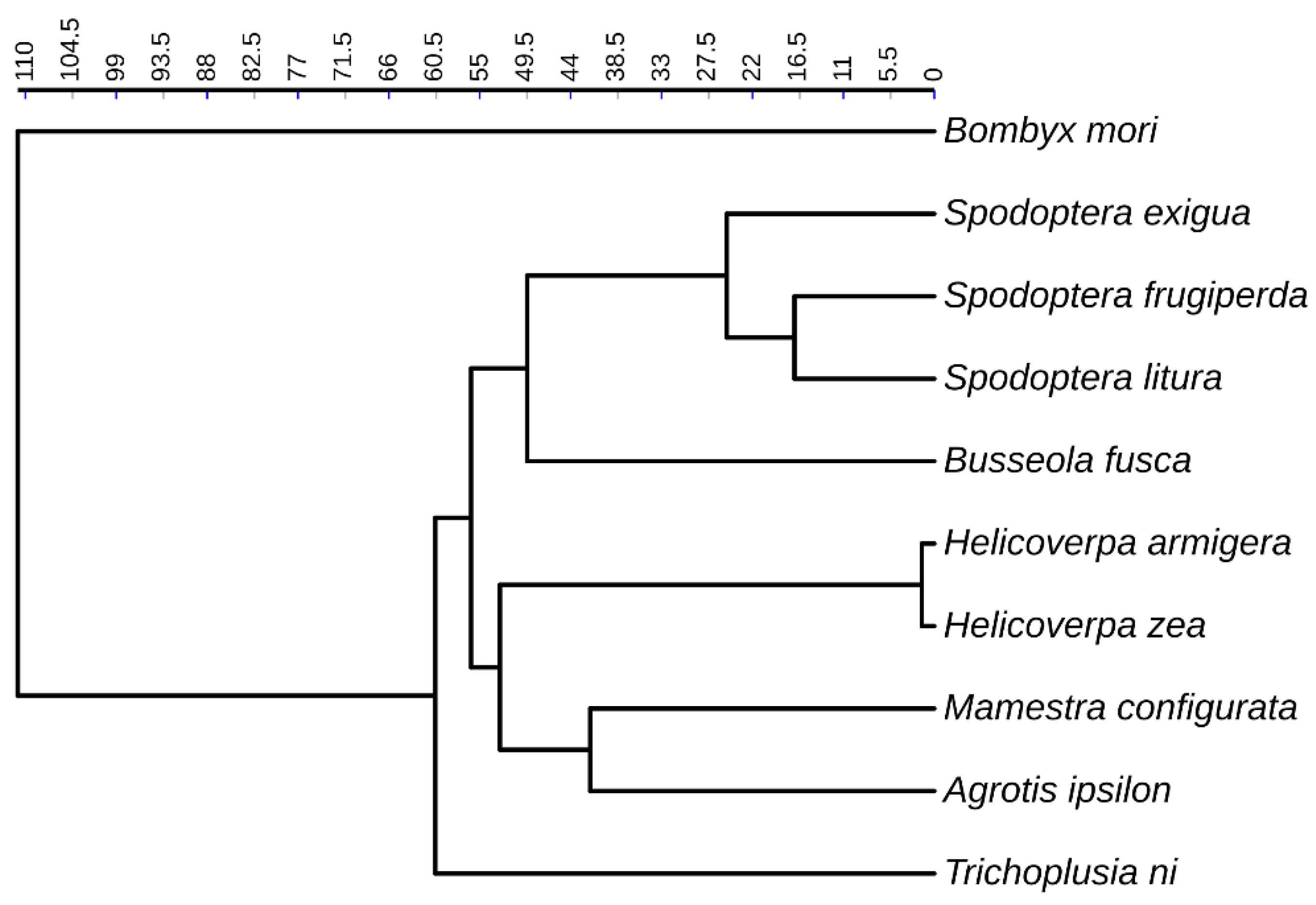
Figure 2.
TE families and their copy numbers of the ten noctuid genomes. Each value was log10 transformed.
Figure 2.
TE families and their copy numbers of the ten noctuid genomes. Each value was log10 transformed.
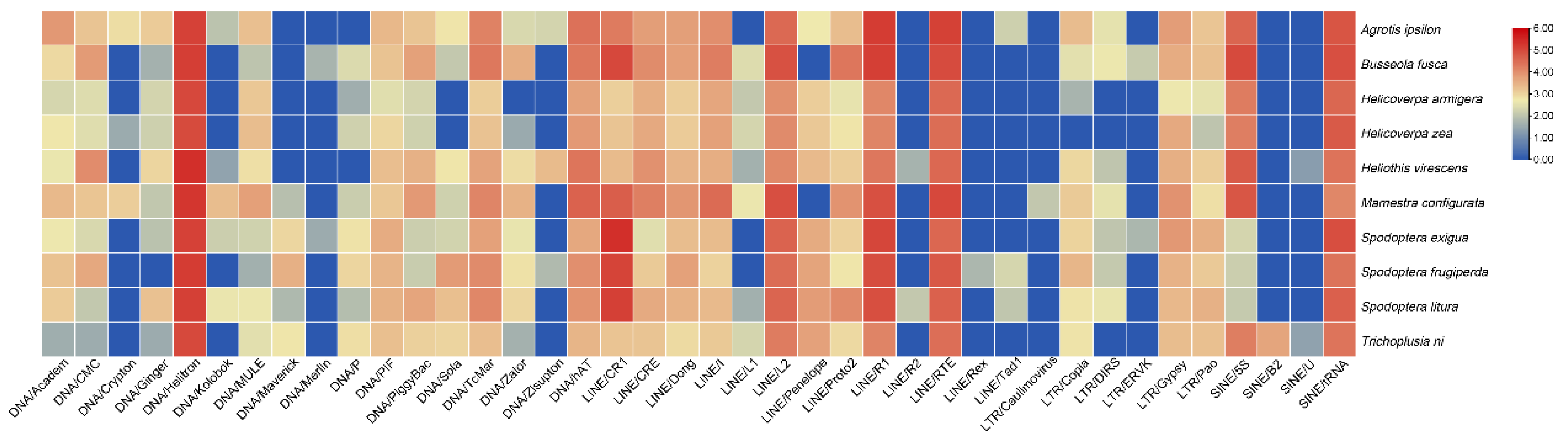
Figure 3.
Illustration of activity of different subfamilies of TEs.
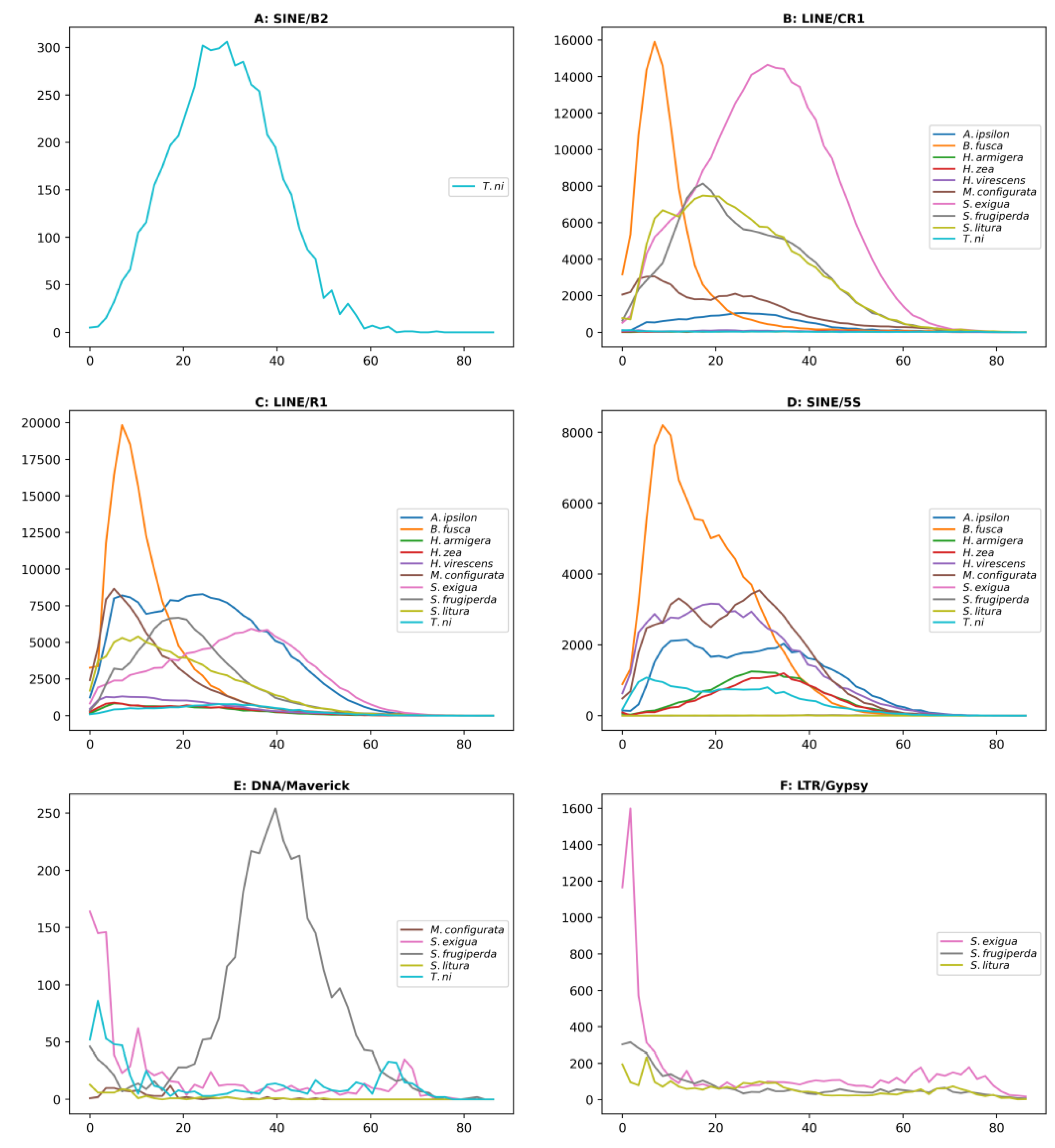
Figure 4.
The expansion activity of TEs in 9 noctuids estimated by TE load. The black and red lines on the phylogenetic tree represented expanded or reduced TE load in the genomes, respectively. A:LTR transposon,B:LINE,C:DNA transposon,D:LINE/CR1,E:LINE/L2,F:LINE/R1,G:LINE/RTE,H:DNA/hAT,I:DNA/TcMar.
Figure 4.
The expansion activity of TEs in 9 noctuids estimated by TE load. The black and red lines on the phylogenetic tree represented expanded or reduced TE load in the genomes, respectively. A:LTR transposon,B:LINE,C:DNA transposon,D:LINE/CR1,E:LINE/L2,F:LINE/R1,G:LINE/RTE,H:DNA/hAT,I:DNA/TcMar.
Figure 5.
The dynamic change rates of TE class/subfamilies in the phylogeny of Noctuidae species.The rate of evolution of the TE load is used to determine the color of the branch, with the rate increasing from cool (blue) to warm (red). A:LTR transposon,B:LINE,C:DNA transposon,D:LINE/CR1,E:LINE/L2,F:LINE/R1,G:LINE/RTE,H:DNA/hAT,I:DNA/TcMar.
Figure 5.
The dynamic change rates of TE class/subfamilies in the phylogeny of Noctuidae species.The rate of evolution of the TE load is used to determine the color of the branch, with the rate increasing from cool (blue) to warm (red). A:LTR transposon,B:LINE,C:DNA transposon,D:LINE/CR1,E:LINE/L2,F:LINE/R1,G:LINE/RTE,H:DNA/hAT,I:DNA/TcMar.
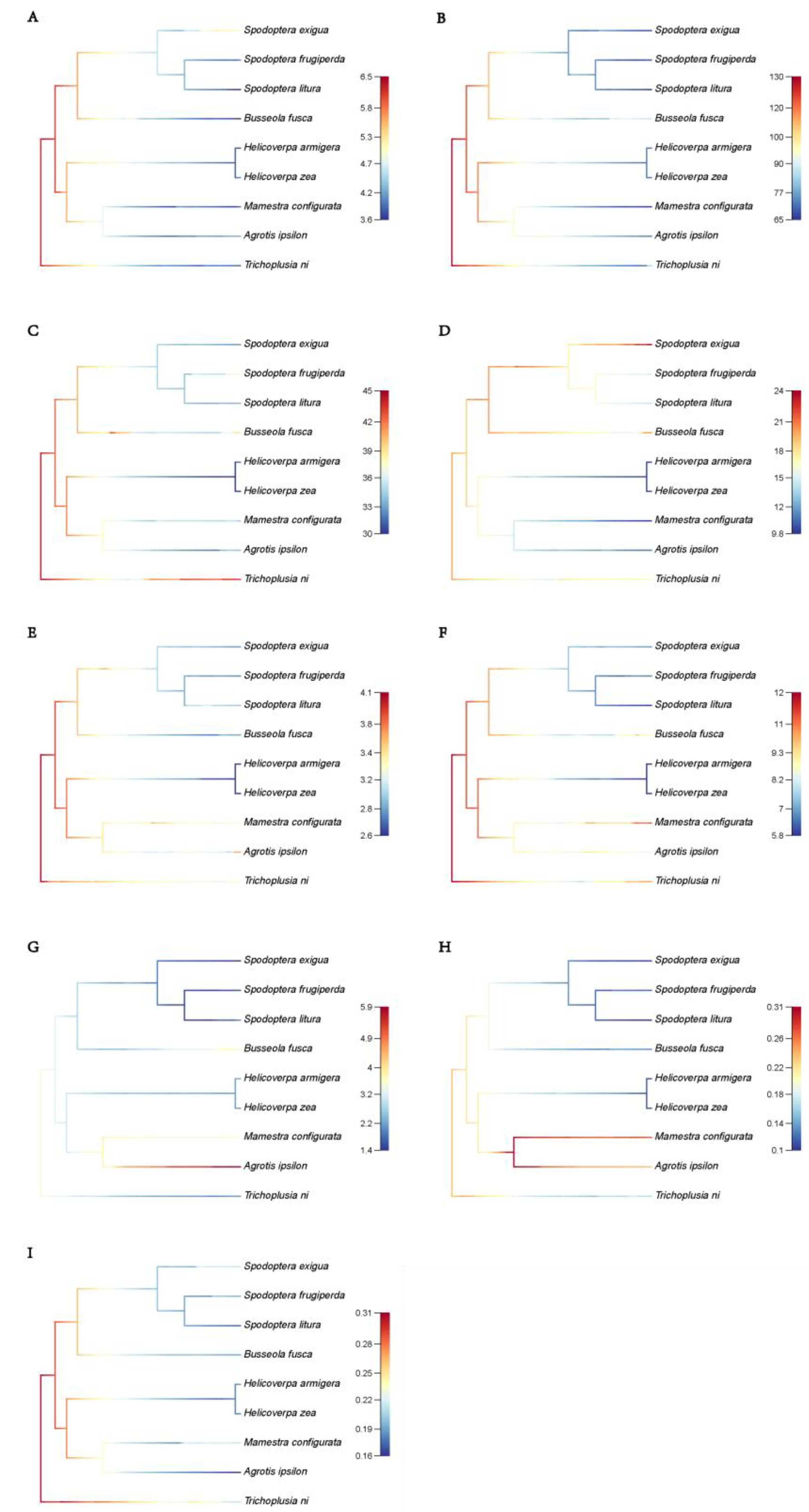
Figure 6.
The potential horizontal transfers events (HTT) among the noctuid species. HTT events from RNA transposons represented by red lines, while from DNA transposons represented by blue lines.
Figure 6.
The potential horizontal transfers events (HTT) among the noctuid species. HTT events from RNA transposons represented by red lines, while from DNA transposons represented by blue lines.
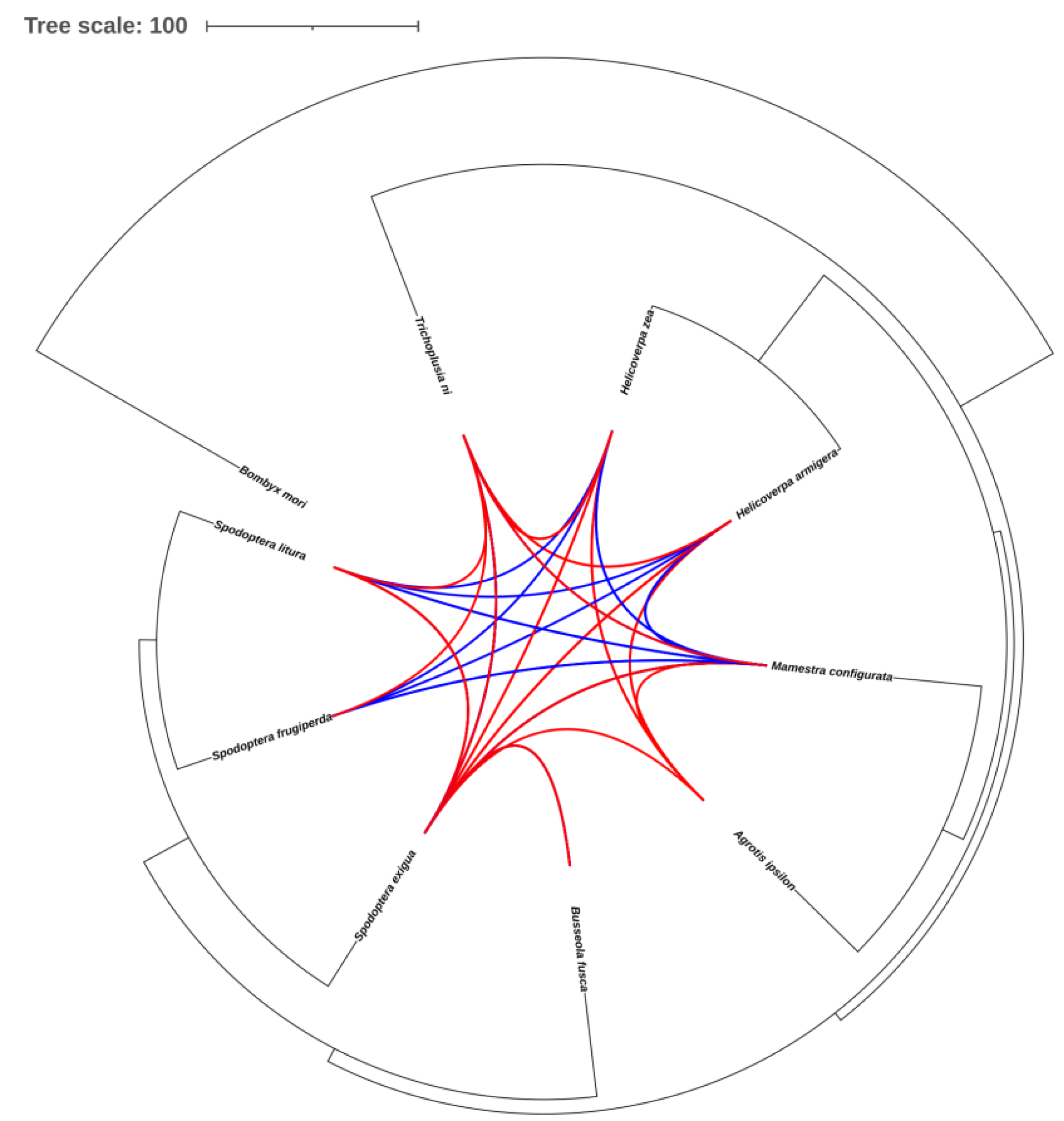
Figure 7.
The minimum number of horizontal transfers events between the 9 noctuid species and eleven non-noctuid arthropod species. HTT events from DNA/Helitron represented by blue lines, while DNA/Mariner represented by red lines.
Figure 7.
The minimum number of horizontal transfers events between the 9 noctuid species and eleven non-noctuid arthropod species. HTT events from DNA/Helitron represented by blue lines, while DNA/Mariner represented by red lines.

Table 1.
Consensus sequences of transposable elements in the ten noctuid species.
| Level | Species name | Genome size(Mb) | RTE | DTE | UnC | Total |
|---|---|---|---|---|---|---|
| Scaffold | Agrotis ipsilon | 486.92 | 1238 | 1120 | 131 | 2489 |
| Scaffold | Busseola fusca | 490.17 | 1617 | 1067 | 136 | 2820 |
| Scaffold | Helicoverpa armigera | 299.98 | 562 | 607 | 52 | 1221 |
| Scaffold | Helicoverpa zea | 306.41 | 638 | 637 | 67 | 1342 |
| Scaffold | Heliothis virescens | 403.15 | 790 | 855 | 85 | 1730 |
| Scaffold | Mamestra configurata | 559.39 | 1509 | 1186 | 131 | 2826 |
| Chromosome | Spodoptera exigua | 446.8 | 831 | 456 | 43 | 1330 |
| Chromosome | Spodoptera frugiperda | 486.23 | 743 | 644 | 63 | 1450 |
| Chromosome | Spodoptera litura | 428.03 | 1006 | 556 | 70 | 1632 |
| Chromosome | Trichoplusia ni | 367.26 | 578 | 428 | 32 | 1038 |
Retrotransposable elements, DNA transposons, and unclassified TEs are represented by RTE, DTE, and UnC, respectively.
Table 2.
Transposable element loads and genome size in the ten noctuid species.
| Species | DTE (%) | LTR (%) | LINE (%) | SINE (%) | UnC(%) | All (%) | Genome size (Mb) |
|---|---|---|---|---|---|---|---|
| Trichoplusia ni | 4.59 | 2.63 | 4.99 | 2.25 | 1.4 | 15.86 | 367.2 |
| Helicoverpa armigera | 4.01 | 0.74 | 3.46 | 2.06 | 1.06 | 11.33 | 299.98 |
| Helicoverpa zea | 4.82 | 1.07 | 4.05 | 2.63 | 1.19 | 13.76 | 306.41 |
| Spodoptera exigua | 4.74 | 4.22 | 17.64 | 2.56 | 1.48 | 30.64 | 446.80 |
| Spodoptera litura | 5.87 | 2.4 | 14.81 | 3.39 | 2.51 | 28.98 | 428.03 |
| Spodoptera frugiperda | 9.12 | 1.94 | 12.54 | 0.98 | 2.17 | 26.75 | 486.23 |
| Agrotis ipsilon | 11.8 | 2.08 | 13.81 | 3.16 | 3.42 | 34.27 | 486.92 |
| Mamestra configurata | 11.24 | 2.12 | 15.59 | 2.37 | 3.4 | 34.72 | 559.39 |
| Busseola fusca | 12.1 | 3.16 | 20.16 | 6.11 | 3.57 | 45.10 | 490.17 |
| Heliothis virescens | 8.39 | 1.3 | 5.64 | 2.92 | 2.29 | 20.54 | 403.15 |
DNA transposons and unclassified TEs are represented by DTE and UnC respectively.
Table 3.
The correlation coefficient between the genome sizes and TE loads.
| Classes of TEs | Subfamily | R value | P value |
|---|---|---|---|
| DNA transposon | 0.81 | 4.21E-03 | |
| DNA/CMC | 0.31 | 0.4 | |
| DNA/TcMar | 0.97 | 2.17E-06 | |
| DNA/Zator | 0.74 | 0.01 | |
| DNA/hAT | 0.46 | 0.18 | |
| DNA/PiggyBac | 0.21 | 0.55 | |
| LINE | 0.83 | 3.2E-03 | |
| LINE/Dong | 0.91 | 2.56E-04 | |
| LINE/L2 | 0.77 | 9.6E-0.3 | |
| LINE/R1 | 0.75 | 0.01 | |
| LINE/RTE | 0.58 | 0.08 | |
| LINE/Jockey | 0.54 | 0.1 | |
| LINE/Proto2 | 0.44 | 0.21 | |
| LINE/CR1 | 0.41 | 0.24 | |
| LINE/CRE | 0.34 | 0.34 | |
| LINE/Rex | 0.25 | 0.49 | |
| LTR | 0.49 | 0.15 | |
| LTR/Copia | 0.4 | 0.26 | |
| SINE | 0.2 | 0.57 | |
| SINE/tRNA | 0.2 | 0.58 |
*The bold line in the table indicated TE classes with significant p-values.
Table 4.
lambda value and K value of the TE load.
| Classes of TEs | Subfamily | Lambda value | P value(lambda) | K value | P value(K) |
|---|---|---|---|---|---|
| DNA | 0.93 | 0.23 | 0.75 | 0.16 | |
| DNA/hAT | 1.02 | 7.1E-03 | 1.13 | 0.01 | |
| DNA/Helitron | 0.89 | 0.72 | 0.6 | 0.22 | |
| DNA/MULE | 0.84 | 0.22 | 0.66 | 0.16 | |
| DNA/TcMar | 0.97 | 0.03 | 1.13 | 0.03 | |
| LINE* | 0.99 | 0.01 | 1.33 | 0.02 | |
| LINE/CR1 | 1.01 | 5.7E-03 | 1.62 | 4E-03 | |
| LINE/Dong | 0.99 | 0.07 | 0.99 | 0.02 | |
| LINE/Jockey | 0.73 | 0.32 | 0.52 | 0.24 | |
| LINE/L2 | 0.99 | 0.03 | 1.19 | 0.03 | |
| LINE/R1 | 1.02 | 6.6E-03 | 1.49 | 0.01 | |
| LINE/RTE | 1.01 | 0.03 | 1.18 | 0.03 | |
| LTR | 0.79 | 0.12 | 0.57 | 0.24 | |
| LTR/Pao | 0.70 | 0.17 | 0.39 | 0.33 | |
| SINE | 6.8E-05 | 1 | 0.42 | 0.43 | |
| SINE/tRNA | 1.01 | 0.21 | 0.82 | 0.1 |
*The bold line in the table indicated TE classes with significant p-values.
Table 5.
Range of change rates of transposons.
| Transposon type | Range of change rates |
|---|---|
| LTR | 3.6 ~ 6.5 |
| LINE | 65 ~ 130 |
| LINE/CR1 | 9.8 ~ 24 |
| LINE/L2 | 2.6 ~ 4.1 |
| LINE/R1 | 5.8 ~ 12 |
| LINE/RTE | 1.4 ~ 5.9 |
| DNA Transposon | 30 ~ 45 |
| DNA/hAT | 0.1 ~ 0.31 |
| DNA/TcMar | 0.16 ~ 0.31 |
Table 6.
The number of HTTs in each TE subfamily.
| Subfamily | Frequency of HTTs in every thousand TE copies | Number of HTTs | Copy number of TEs |
|---|---|---|---|
| LTR/Gypsy | 0.04 | 1 | 22547 |
| LTR/Copia | 0.25 | 1 | 3978 |
| LINE/RTE-RTE | 0.05 | 10 | 183155 |
| LINE/RTE-BovB | 0.03 | 4 | 130583 |
| LINE/R1 | 6.1E-03 | 3 | 485681 |
| LINE/Proto2 | 0.05 | 1 | 21637 |
| LINE/L2 | 0.03 | 6 | 209382 |
| LINE/Dong-R4 | 0.13 | 3 | 22186 |
| LINE/CR1-Zenon | 2.6E-03 | 1 | 380795 |
| LINE/CR1 | 0.25 | 4 | 15874 |
| DNA/Zator | 0.18 | 1 | 5418 |
| DNA/TcMar-Tc1 | 0.44 | 12 | 27452 |
| DNA/TcMar-Mariner | 0.38 | 5 | 13007 |
| DNA/RC | 1.2E-03 | 1 | 774822 |
| DNA/Maverick | 1.72 | 3 | 1741 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



