
Submitted:
04 May 2023
Posted:
05 May 2023
You are already at the latest version
Abstract
The metal chelator PBT2 (5,7-dichloro-2-[(dimethylamino)methyl]-8-hydroxyquinoline) acts as a terdentate ligand capable of forming binary and ternary Cu2+ complexes. It was clinically trialed as an Alzheimer’s disease (AD) therapeutic but failed to progress beyond phase II. The β-amyloid (Aβ) peptide associated with AD was recently concluded to form a unique Cu(Aβ) complex that is inaccessible to PBT2. Herein, it is shown that the species ascribed to this binary Cu(Aβ) complex in fact corresponds to ternary Cu(PBT2)NIm[Aβ] complexes formed by anchoring of Cu(PBT2) on imine nitrogen (NIm) donors of His side chains. The primary site of ternary complex formation is His6, having a conditional stepwise formation constant at pH 7.4 (K [M−1] ) of log K = 6.4 ± 0.1, and a second site is supplied by His13 or His14 (log K = 4.4 ± 0.1). The stability of Cu(PBT2)NIm[H13/14] is comparable with that of the simplest ternary complexes involving free imidazole (log K = 4.22 ± 0.09) and histamine (log K = 4.00 ± 0.05). The 100-fold larger formation constant for Cu(PBT2)NIm[H6] indicates that outer-sphere ligand–peptide interactions strongly stabilize its structure. Despite the relatively high stability of Cu(PBT2)NImH6, PBT2 is a promiscuous Cu2+-binding ligand capable of forming a ternary Cu(PBT2)NIm complex with any ligand containing NIm donor. These ligands include histamine, L-His, and ubiquitous His side chains of peptides and proteins in the extracellular milieu, whose combined effect should outweigh that of a single Cu(PBT2)NIm[H6] complex regardless of its stability. We therefore conclude that PBT2 is capable of accessing Cu(Aβ) complexes with high stability but not specificity. The results have implications for future AD therapeutic strategies and understanding the role of PBT2 in the bulk transport of transition metal ions. Given the repurposing of PBT2 as a drug for breaking antibiotic resistance, ternary Cu(PBT2)NIm and analogous Zn(PBT2)NIm complexes may be relevant to its antimicrobial properties.
Keywords:
8-hydroxyquinoline
; PBT2
; amyloid
; copper
; terdentate
; ternary
; antimicrobial
1. Introduction
The compound 5,7-dichloro-2-[(dimethylamino)methyl]-8-hydroxyquinoline (PBT2) is a terdentate Cu2+ and Zn2+ chelator that was previously trialled as a therapeutic to treat Alzheimer’s disease (AD). Its anticipated mechanism of action was based on the controversial “metals hypothesis”, which proposed that AD results from aberrant interactions of the β-amyloid (Aβ) peptide with endogenous transition metal ions, notably Cu2+, causing Aβ to misfold and generate reactive oxygen species (ROS) [1,2]. PBT2 was proposed to prevent these interactions, sequester transition metal ions thought to be trapped within extracellular β-amyloid aggregates, and then enable cellular uptake of these ions by ionophore action [3]. However, repeated phase II clinical trials ultimately provided no evidence for cognitive efficacy of PBT2 in patients with prodromal or mild AD [4,5].
Using electron paramagnetic resonance (EPR) spectroscopy, the Cu2+ coordination of this class of ligand (L) was first characterized using the non-chlorinated homologue of PBT2 (Figure 1), which was shown to form a terdentate CuL complex and a five-coordinate CuL2 complex [6]. The proposed structure of CuL2 has been replicated in the crystal structure of PBT2 [7], and the UV-vis and EPR spectroscopic properties of Cu(PBT2) and Cu(PBT2)2 have been shown to mirror those of the non-chlorinated homologue [6,7,8,9] (Table S1). Early EPR studies also showed that both ligands form ternary CuLNImX complexes (Figure 1) in which the labile Cl− ligand of CuL is replaced with an imine (NIm) donor ligand provided by X = imidazole, histamine, L-His, or proteins such as α-synuclein and Aβ (see, in particular, Figure S33 of ref. [6]). However, despite recently identifying a similar EPR spectroscopic signature using a Cu/PBT2/Aβ1–42 mixture, George and coworkers ascribed the signal to a unique PBT2-inaccessible Cu(Aβ) species and concluded that there was no evidence to support formation of a ternary Cu2+ complex [10]. Since this could be interpreted by some as a reason for the failure of PBT2 in AD clinical trials, it is important to resolve this contradiction.
To address the above issue, we used EPR spectroscopy to analyze the species distributions in Cu/PBT2/Aβ mixtures in unprecedented detail. We identified ternary Cu(PBT2)NImX complex formation with X = His6 and His13/14 of Aβ1–40 and derived the corresponding stepwise conditional formation constants at pH 7.4. For comparison, we also determined the conditional formation constants for X = imidazole and histamine using PBT2 and its non-chlorinated homologue. The stepwise conditional formation constant for Cu(PBT2)NImH6 was found to be 3.5-fold larger than that for Cu(PBT2)2 at pH 7.4 and 100-fold larger than the stepwise formation constants for Cu(PBT2)NImH13/14 and Cu(PBT2)NImX (X = imidazole, histamine).
2. Results
To characterize the Cu2+ complexes formed by PBT2 in the presence of Aβ, we titrated Cu2+ into an equimolar mixture of PBT2 and Aβ1–40 in PBS pH 7.4 and acquired the corresponding EPR spectra (Figure 2a). To determine the species distribution, each spectrum was decomposed into a linear superposition of basis spectra (Figure 2b) corresponding to Cu(PBT2), Cu(PBT2)2, Cu(Aβ1–40), Cu2(Aβ1–40), and the putative Cu(PBT2)NImAβ complex. The spectrum of Cu(PBT2) was obtained at equimolar Cu/PBT2 stoichiometry and that of Cu(PBT2)2 at sub-stoichiometric ratios (Figure S1). The spectra of Cu(Aβ) and Cu 2(Aβ) were acquired at Cu/Aβ ratios of 1:1 and 2.5:1 (Figure S2). Attempts to use linear combinations of normalized Cu(PBT2), Cu(PBT2)2, Cu(Aβ), and Cu 2(Aβ) spectra failed to reproduce the EPR spectra of Cu/PBT2/Aβ1–40 n:1:1 (n = 0–2.67), indicating that additional species must be formed. Indeed, the dominant spectral features did not resemble any of those of the above four species (Figure 2b; Figure S3). Rather, they corresponded closely to those of previously characterized CuLNIm complexes (Table 1) [6,8,11], indicating that Cu(PBT2) can anchor on the imine nitrogen (NIm) of the His side chains of Aβ1–40.
To quantify the number and stabilities of ternary Cu(PBT2)NImAβ interactions, it is important to consider how the His residues participate in binary Cu(Aβ) complexes. At pH 7.4, Cu(Aβ) is dominated by {NH2D1, C=OD1, NImH6, NImH13} and {NH2D1, C=OD1, NImH6, NImH14} coordination spheres with indistinguishable EPR spectra [12,13,14]. Thus, His6 is absolutely required to form Cu(Aβ), whereas only one of His13 or His14 is needed. Thus, anchoring of Cu(PBT2) on His6 to form Cu(PBT2)NImH6 will occur at the expense of Cu(Aβ), whereas anchoring on one of His13 or His14 to form Cu(PBT2)NImH13/14 will not. Sequential binding of a second Cu2+ ion by Cu(Aβ) generates Cu2(Aβ). The coordination in Cu2(Aβ) remains poorly defined, with one suggestion that sequential binding of a second Cu2+ ion changes the coordination of the first [15]. However, it will be shown below that a satisfactory explanation of the species distributions requires that His6 remains Cu2+-bound and His13 or His14 non-coordinated in Cu2(Aβ).
The conditional constants () for formation of Cu(PBT2), Cu(PBT2)2, Cu(Aβ), and Cu2(Aβ) at pH 7.4 have all been previously determined (Table S2), which greatly simplified the task of determining the species distribution of the Cu/PBT2/Aβ1–40 n:1:1 system. Using these values and the EPR basis spectra (Figure 2b), we fitted the series of EPR spectra in Figure 2a as a function of the unknown constants c and c (see Section 4.3 for detail). The best agreement between the experimental and theoretical species distributions (Figure 2c) was obtained for log c = 6.4 ± 0.1 and log c = 4.4 ± 0.1 (Table 2). Allowing the conditional formation constants c and c to vary beyond their generally accepted ranges worsened the fit. Although we did not refine the value of c, we found that large changes from its published value also worsened the fit unless c was set to a value beyond its accepted range.
The general appearance of the species distributions for Cu/PBT2/Aβ1–40 n:1:1 (Figure 2c, Figure S5) can be understood as follows. For small n, the 1000-fold greater stability of Cu(PBT2) compared with Cu(Aβ) (Table 2) ensures that Cu2+ will first bind to PBT2, with the identity of the fourth in-plane ligand then being determined by the relative magnitudes of the stepwise constants c c. Thus, for n < 1, Cu2+ is predominantly bound in a Cu(PBT2)NImH6 complex with a minor quantity of Cu(PBT2)2. For n = 1, Cu(PBT2)2 is almost entirely replaced by Cu(PBT2)NImH6 and Cu(PBT2)NImH13/14, which only require one PBT2 molecule per Cu2+ ion. For n > 1, with no free PBT2 molecules available, the additional Cu2+ is coordinated in the next most stable binary complex, which is Cu(Aβ) (log c = 10.0 ± 0.1). However, because Cu(Aβ) coordination requires His6, some Cu(PBT2) detaches from His6 and anchors instead on a His13 or His14 side chain, albeit with lower stability, to form Cu(PBT2)NImH13/14. As the available sites at His6 are gradually filled by Cu(Aβ), stepwise addition of a second Cu2+ ion to the peptide occurs (log c = 8.0 ± 0.1) to form Cu2(Aβ). Maximum occupancy of the Cu/PBT2/Aβ1–40 n:1:1 system is reached at n = 3, with two Cu2+ ions are bound to the “first” and “second” sites of Aβ, and a third Cu2+ ion bound either to Cu(PBT2) that is free (minor) or anchored to His13/14 of Cu2(Aβ) in a Cu(PBT2)NImH13/14 complex (major).
More than three Cu2+ ions cannot be accommodated by an equimolar PBT2/Aβ mixture, with the excess Cu2+ ions existing as aqueous copper that will precipitate as [Cu(OH)2]n at pH 7.4. Inclusion of Cu(PBT2)NImH13/14, whose formation does not depend on the Cu2+ loading state of Aβ, was essential to fit the experimental data. Alternative explanations of the physical origin of the lower-affinity ternary complex, such as a change in coordination of the first-bound Cu2+ ion in Cu2(Aβ), can be ruled out because the concentration of Cu2(Aβ) relative to that of the ternary complex is too low at n < 2 (Figure S5).
The fact that c is 100-fold larger than c indicates that either Cu(PBT2)NImH6 is stabilized by favorable outer-sphere ligand–peptide interactions and/or that Cu(PBT2)NImH13/14 is destabilized by such interactions. To distinguish between these possibilities, we repeated the EPR analyses using Cu/PBT2/X 1:1:n systems with relatively unstructured NIm donor ligands from X = imidazole (Figures S6–S8) and histamine (Figures S9–S11). The EPR spectra of the isolated ternary Cu(PBT2)NImX complexes were characterized by the same parameters as Cu(PBT2)NImAβ spectra (Table 1), confirming that each of these ternary Cu2+ complexes involves monodentate NIm coordination of the co-ligand (Figure 1). The difference between c and c was within experimental error, whereas c was slightly lower than these constants (Table 2). Thus, we may conclude that the stability of Cu(PBT2)NImH13/14 is not strongly influenced by outer-sphere ligand–peptide interactions, whereas such interactions greatly enhance the stability of Cu(PBT2)NImH6.
To independently verify the EPR method for deriving the conditional formation constants, we performed the same analyses for ternary complexes of the non-chlorinated homologue of PBT2 (L′) with imidazole (Figures S12–S14) and compared the value with that previously determined using potentiometric titrations [11]. After pH correction of the absolute stability constants (Table S3), c was not significantly different from that determined here using EPR (Table S2) and, similar to PBT2, slightly higher than c (Figures S15–S17).
3. Discussion
EPR spectroscopy isolated a common Cu(PBT2)NImAβ spectrum for (Figure S3) for both Cu(PBT2)N ImH6 and Cu(PBT2)N ImH13/14 because they have very similar first coordination spheres. Nevertheless, as has been shown for other terdentate Cu2+ chelators [27,28], ternary complexes with different NIm donor ligands can be distinguished based on their distinct formation constants, which are determined by outer-sphere interactions to which continuous-wave EPR is typically insensitive. Importantly, the spectroscopic signature of Cu(PBT2)NImAβ isolated here for Cu/PBT2/Aβ1–40 n:1:1 closely matches that reported for the species isolated in Cu/PBT2/Aβ1–42 1:2:1 [10]. In the latter study, the authors ascribed this to a unique PBT2-inaccessible Cu(Aβ) complex and concluded that there was no evidence for ternary complex formation. However, we have shown that CuLNImX complexes with this spectroscopic signature are formed by PBT2 with a number of NIm donor ligands X (Table 1, Table S1). Moreover, we demonstrated the requirement for two such complexes — Cu(PBT2)NImH6 and Cu(PBT2)NImH13/14 — with distinct stabilities (Table 2) to explain the species distributions of Cu/PBT2/Aβ1–40 mixtures. The relatively high stability of Cu(PBT2)NImH6 compared with complexes formed with other NIm donors might result from stabilizing pi–pi stacking of the aromatic rings of PBT2 and Phe4 or Tyr10, although a combination of electrostatic, steric, and hydrogen-bonding effects may contribute.
Despite the large ternary formation constant for Cu(PBT2)NImH6, PBT2 remains a promiscuous Cu2+ chelator because it is capable of forming a ternary Cu(PBT2)NIm complex with all NIm donor ligands, including ubiquitous His side chains of peptides and proteins in the biological milieu, whose combined effect should outweigh that of a single Cu(PBT2)NImAβ complex regardless of its stability. We therefore conclude that PBT2 is capable of accessing Cu(Aβ) complexes with high stability but not specificity. Potential functional implications of Cu(PBT2)NIm complexes have been discussed in our previous studies of the non-chlorinated PBT2 homologue. First, as an alternative to acting as a mobile ion carrier (ionophore) in a lipid membrane, endocytosis of extracellular proteins on which Cu(PBT2) is anchored, followed by release of Cu2+ in low-pH and/or reducing intracellular compartments, may contribute to bulk transport of these ions [6]. Second, their production of ROS in the presence of ascorbate [11], in addition to the modulation of cellular ROS signaling following exposure to this class of ligand [16], contrasts with the originally intended ROS-silencing of function of PBT2 [17].
Recently, PBT2 has found renewed interest as an antimicrobial compound. Notably, a number of gram-positive bacteria became re-sensitized to previously resistant classes of antibiotic when these antibiotics were supplemented with PBT2 and Zn2+ in mouse models of wound healing [18] and pneumonia [19]. These results have been attributed multiple bactericidal mechanisms associated with intracellular Zn2+ accumulation, including impairment of Mn-dependent superoxide dismutase and production of ROS [20]. Although ligands generally have a greater affinity for Cu2+ compared with Zn2+ [21], the above effects were observed in response to co-administration of PBT2 (~1 μM) with an excess of Zn2+ (~100 μM). Therefore, remains unclear whether ternary Cu(PBT2)NIm complexes may be formed under these conditions. However, given the ability of PBT2 to also form terdentate Zn2+ chelates [7], the role of similar Zn(PBT2)NIm complexes in the antimicrobial activity of Zn/PBT2 may be speculated.
4. Materials and Methods
4.1. Sample preparation
Aβ1–40 (purity >95%) was synthesized in the Peptide Technology Facility of the Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne. The lyophilised peptide was dissolved at a nominal concentration of 1 mg/mL in 1,1,1,3,3,3-hexafluoroisopropanol and portioned, then the solvent was allowed to evaporate. The resulting peptide film was resuspended at 4°C in 20% v/v 20 mM NaOH, 70% v/v ultrapure water (MilliQ, Merck), and 10% v/v 10× phosphate buffered saline (PBS; 10 mM phosphate buffer, 2.7 mM KCl, 137 mM NaCl; Sigma). After centrifugation at 14000×g for 15 min at 4°C, the supernatant was retained and the peptide concentration immediately determined using ε214 = 74925 M−1cm−1 [22]. A concentrated stock of 65CuCl2 was prepared by stirring 65CuO (65Cu, > 99%; Cambridge Isotope Laboratories) in concentrated HCl, evaporating excess HCl under heat, then adding ultrapure water. PBT2 was synthesized as previously described [23], and a 1mM stock solution was prepared by solubilising the hydrochloride salt directly in PBS.
From the above stock solutions, PBT2 and then 65CuCl2 were added to portions of Aβ1–40 to achieve final molar Cu/PBT2/ ratios of n:1:1 (n = 0.33–2.67) with [Aβ1–40] = 200 μM. Control samples containing Cu/PBT2 0.5:1 and 1:1 and Cu/Aβ1–40 1:1 and 2.5:1 were also prepared. Glycerol (10% v/v) was added to the Cu/PBT2 control samples to prevent formation of a concentrated solute phase upon freezing. Immediately after Cu addition, the final solution pH was measured using a micro-probe (Hanna Instruments, Italy) and adjusted using concentrated NaOH or HCl as required. Samples were transferred to quartz EPR tubes (Wilmad, SQ-707) and snap frozen in liquid nitrogen within two minutes of Cu addition.
4.2. EPR spectroscopy
X-band continuous-wave EPR spectra were acquired using a Bruker ESP380E spectrometer fitted with a rectangular TE102 microwave cavity and a quartz cold finger insert. Microwave frequencies were measured using an EIP Microwave 548A frequency counter and g factors calibrated against the F+ line in CaO (g = 2.0001 ± 0.0002) [24]. Experimental conditions are indicated in the figure captions. Background correction was performed by subtraction of the buffer-only spectrum.
The “pepper” function in EasySpin v.5.2.33 [25,26] was used to simulate basis spectra using the static Hamiltonian
where S and I are the electron and 65Cu nuclear vector spin operators, g and A are the 3×3 electron Zeeman and 65Cu electron-nuclear hyperfine coupling matrices, βe is the Bohr magneton, βn is the nuclear magneton and B is the applied magnetic field vector. Rhombic symmetry or higher was assumed for g and A, with principal values of gx, gy, and gz and Ax, Ay, and Az, respectively. the Hamiltonian in equation 3. Assuming at least rhombic symmetry, the principal values of g and A and the lineshape parameters (g–A strain model) were varied iteratively using the “esfit” module in EasySpin to achieve a least squares minimization of the difference between the experimental and simulated spectra.
4.3. Determination of the ternary formation constants for Cu/PBT2/Aβ1–40
The Cu(PBT2)NImAβ species was assumed to arise from the superposition of two ternary complexes. The first complex involved anchoring of Cu(PBT2) on the His6 side chain (NImH6), which is also required for Cu(Aβ) formation; the second involved anchoring on a side chain of either His13 or His14 (NImH13/14), which is not required for Cu(Aβ) or Cu2(Aβ) formation. Thus, Cu(Aβ) and Cu2(Aβ) can simultaneously accommodate an Cu(PBT2)NImH13/14 complex but not a Cu(PBT2)NImH6 complex. Under these assumptions, Cu/PBT2/Aβ1–40 n:1:1 can be modeled as Cu/L/A/B n:1:1:1, where ligand L is PBT2, ligand A acts like Aβ, and ligand B is treated an isolated NImH13/14 donor. The relevant formation constants are
where the cumulative conditional (pH-dependent) constants () are defined by
and the stepwise conditional formation constants (ck) are defined by
To determine the ternary formation constants c and c, we made use of the previously publishes values for c, c, c and c at pH 7.4 (Table 2, Table S2), then initial guesses for c and c were made and their values systematically varied as follows:
1) For each value of c and c, the theoretical distributions of CuL, CuL2, CuA, Cu2A, CuLA, and CuLB were calculated for the condition Cu/PBT2/Aβ1–40 1:1:1 ≡ Cu/L/A/B 1:1:1:1 under which spectral features attributable to the ternary species were maximal (Figure 2c);
2) The theoretical speciation in step 1 provided weighting factors that were used to algebraically subtract the normalized spectra of CuL, CuL2, and CuA (Figure 2b) from the experimental spectrum of Cu/PBT2/Aβ1–40 1:1:1 ≡ Cu/L/A/B 1:1:1:1, thus yielding a weighted summation of indistinguishable CuLA and CuLB spectra;
3) Linear combinations of the normalized CuL, CuL2, CuA, Cu2A, CuLA, CuLB basis spectra were used to reconstruct the experimental EPR spectra at all intermediate stoichiometries Cu/PBT2/Aβ1–40 n:1:1 ≡ Cu/L/A/B n:1:1:1 (0.33 ≤ n ≤ 2.67), the weightings being iteratively varied using a generalized reduced gradient nonlinear solver (Frontline Systems, Inc., www.solver.com) to minimize the root-mean-squared deviation between the reconstructions and the experimental spectra;
4) The deviation between the fitted and experimental values of [CuL], [CuL2], [CuA], [Cu2A], and [CuLNImAβ] = [CuLA] + [CuLB] for all values of n was calculated;
5). New values of c and c were chosen and steps 1–4 repeated until the root-mean-squared deviation was minimised.
The above method assumed that the frozen-solution EPR spectra of Cu(PBT2)NImH6 and Cu(PBT2)NImH13/14 are indistinguishable, which is justified by the identification of a common set of spin Hamiltonian parameters for numerous Cu(PBT2)NIm complexes with different NIm donor ligands (Table 1, Tale S1). This assumption was also shown to be true for ternary Cu2+ complexes of different NIm donors with other terdentate ligands such as the non-chlorinated homologue of PBT2 (Table S1), the GHK tripeptide [27,28], and α-synuclein [29].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figures S1–S17; Tables S1–S3; Equations S1–S5.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The support of the Brain–Immune Communication laboratory, Institute Pasteur is gratefully acknowledged. PBT2 was kindly supplied by Dr Vijaya Kenche, Florey Institute of Neuroscience and Mental Health, Australia.
Conflicts of Interest
The author declares no conflict of interest.
References
- Bush, A.I. Metals and neuroscience. Curr. Opin. Chem. Biol. 2000, 4, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I. Drug development based on the metals hypothesis of Alzheimer’s disease. J. Alzheimers Dis. 2008, 15, 223–240. [Google Scholar] [CrossRef]
- Adlard, P.A.; Bica, L.; White, A.R.; Nurjono, M.; Filiz, G.; Crouch, P.J.; Donnelly, P.S.; Cappai, R.; Finkelstein, D.I.; Bush, A.I. Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer’s disease. PLoS ONE 2011, 6, e17669. [Google Scholar] [CrossRef] [PubMed]
- Sampson, E.L.; Jenagaratnam, L.; McShane, R. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst. Rev. 2014, CD005380. [Google Scholar] [CrossRef] [PubMed]
- www.alzforum.org/news/research-news/pbt2-takes-dive-phase-2-alzheimers-trial . 1 April 2014.
- Kenche, V.B.; Zawisza, I.; Masters, C.L.; Bal, W.; Barnham, K.J.; Drew, S.C. Mixed ligand Cu2+ complexes of a model therapeutic with Alzheimer's amyloid-β peptide and monoamine neurotransmitters. Inorg. Chem. 2013, 52, 4303–4318. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Vendier, L. ; Stigliani, J-L., Meunier, B.; Robert, A. Structures of the Copper and Zinc Complexes of PBT2, a Chelating Agent Evaluated as Potential Drug for Neurodegenerative Diseases. Eur. J. Inorg. Chem. 2017; 600–608. [Google Scholar] [CrossRef]
- Sgarlata, C.; Arena, G.; Bonomo, R.P.; Giuffrida, A.; Tabbì, G. Simple and mixed complexes of copper(II) with 8-hydroxyquinoline derivatives and amino acids: Characterization in solution and potential biological implications. J. Inorg. Biochem. 2018, 180, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Summers, K.L.; Roseman, G.P.; Sopasis, G.J.; Millhauser, G.L.; Harris, H.H.; Pickering, I.J.; George, G.N. Copper(II) Binding to PBT2 Differs from That of Other 8-Hydroxyquinoline Chelators: Implications for the Treatment of Neurodegenerative Protein Misfolding Diseases. Inorg. Chem. 2020, 59, 17519–17534. [Google Scholar] [CrossRef]
- Summers, K.L.; Roseman, G.; Schilling, K.M.; Dolgova, N.V.; Pushie, M.J.; Sokaras, D.; Kroll, T.; Harris, H.H.; Millhauser, G.L.; Pickering, I.J.; George, G.N. Alzheimer’s Drug PBT2 Interacts with the Amyloid β 1–42 Peptide Differently than Other 8-Hydroxyquinoline Chelating Drugs. Inorg. Chem. 2022, 61, 14626–14640. [Google Scholar] [CrossRef]
- Mital, M.; Zawisza, I.A.; Wiloch, M.Z.; Wawrzyniak, U.E.; Kenche, V.; Wróblewski, W.; Bal, W.; Drew, S.C. Copper Exchange and Redox Activity of a Prototypical 8-Hydroxyquinoline: Implications for Therapeutic Chelation. Inorg Chem. 2016, 55, 7317–7319. [Google Scholar] [CrossRef]
- Drew, S. C.; Barnham, K. J. The Heterogeneous Nature of Cu2+ Interactions with Alzheimer’s Amyloid-β Peptide. Acc. Chem. Res. 2011, 44, 1146–1155. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; La Penna, G. Metal ions and intrinsically disordered proteins and peptides: from Cu/Zn amyloid-β to general principles. Acc. Chem. Res. 47, 2252–2259. [CrossRef]
- Drew, S.C.; Noble, C.J.; Masters, C.L.; Hanson, G.R.; Barnham, K.J. Pleomorphic copper coordination by Alzheimer’s amyloid-β peptide. J. Am. Chem. Soc. 2009, 131, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Alies, B.; Renaglia, E.; Rózga, M.; Bal, W.; Faller, P.; Hureau, C. Cu(II) affinity for the Alzheimer's peptide: tyrosine fluorescence studies revisited. Anal. Chem. 2013, 85, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.L.; Tumpach, C.; Collins, S.J.; Drew, S.C. A 2-substituted 8-hydroxyquinoline stimulates neural stem cell proliferation by modulating ROS signalling. Cell Biochem. Biophys. 2016, 74, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Drew, S.C. The case for abandoning therapeutic chelation of copper ions in Alzheimer’s disease. Front. Neurosci. 2017, 11, 317. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, L.; De Oliveira, D.M.P.; El-Deeb, I.M.; Brazel, E.B.; Harbison-Price, N.; Ong, C.Y.; Rivera-Hernandez, T.; Ferguson, S.A.; Cork, A.J.; Phan, M.D.; Soderholm, A.T.; Davies, M.R.; Nimmo, G.R.; Dougan, G.; Schembri, M.A.; Cook, G.M.; McEwan, A.G.; von Itzstein, M.; McDevitt, C.A.; Walker, M.J. Chemical Synergy between Ionophore PBT2 and Zinc Reverses Antibiotic Resistance. mBio. 2018, 9, e02391–18. [Google Scholar] [CrossRef] [PubMed]
- Brazel, E.B.; Tan, A.; Neville, S.L.; Iverson, A.R.; Udagedara, S.R.; Cunningham, B.A.; Sikanyika, M.; De Oliveira, D.M.P.; Keller, B.; Bohlmann, L.; El-Deeb, I.M.; Ganio, K.; Eijkelkamp, B.A.; McEwan, A.G.; von Itzstein, M.; Maher, M.J.; Walker, M.J.; Rosch, J.W.; McDevitt, C.A. Dysregulation of Streptococcus pneumoniae zinc homeostasis breaks ampicillin resistance in a pneumonia infection model. Cell Rep. 2022, 38, 110202. [Google Scholar] [CrossRef] [PubMed]
- Harbison-Price, N.; Ferguson, S.A.; Heikal, A.; Taiaroa, G.; Hards, K.; Nakatani, Y.; Rennison, D.; Brimble, M.A.; El-Deeb, I.M.; Bohlmann, L.; McDevitt, C.A.; von Itzstein, M.; Walker, M.J.; Cook, G.M. Multiple Bactericidal Mechanisms of the Zinc Ionophore PBT2. mSphere. 2020, 5, e00157–20. [Google Scholar] [CrossRef]
- Irving, H.; Williams, R.J.P. The stability of transition-metal complexes. J. Chem. Soc. 1953, 3192–3210. [Google Scholar] [CrossRef]
- .Kuipers, B.J.H.; Gruppen, H. Prediction of Molar Extinction Coefficients of Proteins and Peptides Using UV Absorption of the Constituent Amino Acids at 214 nm To Enable Quantitative Reverse Phase High-Performance Liquid Chromatography-Mass Spectrometry Analysis. J. Agric. Food Chem. 2007, 55, 5445–5451. [Google Scholar] [CrossRef]
- Barnham, K.J.; Gautier, E.C.L.; Kok, G.B.; Krippner, G. Preparation of 8-hydroxyquinolines for treatment of neurological conditions. 2008. U.S. Pat. Appl. Publ. Patent version number 20080161353 A1; United States,
- Wertz, J.E.; Orton, J.W.; Auzins, P. Electron spin resonance studies of radiation effects in inorganic solids. Discuss. Faraday Soc. 1961, 31, 140–150. [Google Scholar] [CrossRef]
- Stoll, S.; Britt, R.D. General and efficient simulation of pulse EPR spectra. Phys. Chem. Chem. Phys. 2009, 11, 6614–6625. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Bossak-Ahmad, K.; Wiśniewska, M.; Bal, W.; Drew, S.C.; Frączyk, T. Ternary Cu(II) complex with GHK peptide and cis-urocanic acid as a potential physiologically functional copper chelate. Int. J. Mol. Sci. 2020, 21, 6190. [Google Scholar] [CrossRef] [PubMed]
- Bossak-Ahmad, K.; Bal, W.; Frączyk, T.; Drew, S.C. Ternary Cu2+ Complexes of Human Serum Albumin and Glycyl-L-histidyl-L-lysine. Inorg. Chem. 2021, 60, 16927–16931. [Google Scholar] [CrossRef]
- Drew, S.C. α-synuclein and β-amyloid form a bridged copper complex. Appl. Magn. Reson. 2015, 46, 1041–1052. [Google Scholar] [CrossRef]
Figure 1.
Structure of (a) binary and (b) ternary Cu2+ complexes of PBT2 (R1 = Cl) and its non-chlorinated homologue (R1 = H). The imine nitrogen (NIm) is supplied by ligands including imidazole (R2 = H), histamine (R2 = CH2CH2NH3+), L-His (R2 = CH2(COO−)CHNH3+), and His side chains (R2 = protein).
Figure 1.
Structure of (a) binary and (b) ternary Cu2+ complexes of PBT2 (R1 = Cl) and its non-chlorinated homologue (R1 = H). The imine nitrogen (NIm) is supplied by ligands including imidazole (R2 = H), histamine (R2 = CH2CH2NH3+), L-His (R2 = CH2(COO−)CHNH3+), and His side chains (R2 = protein).
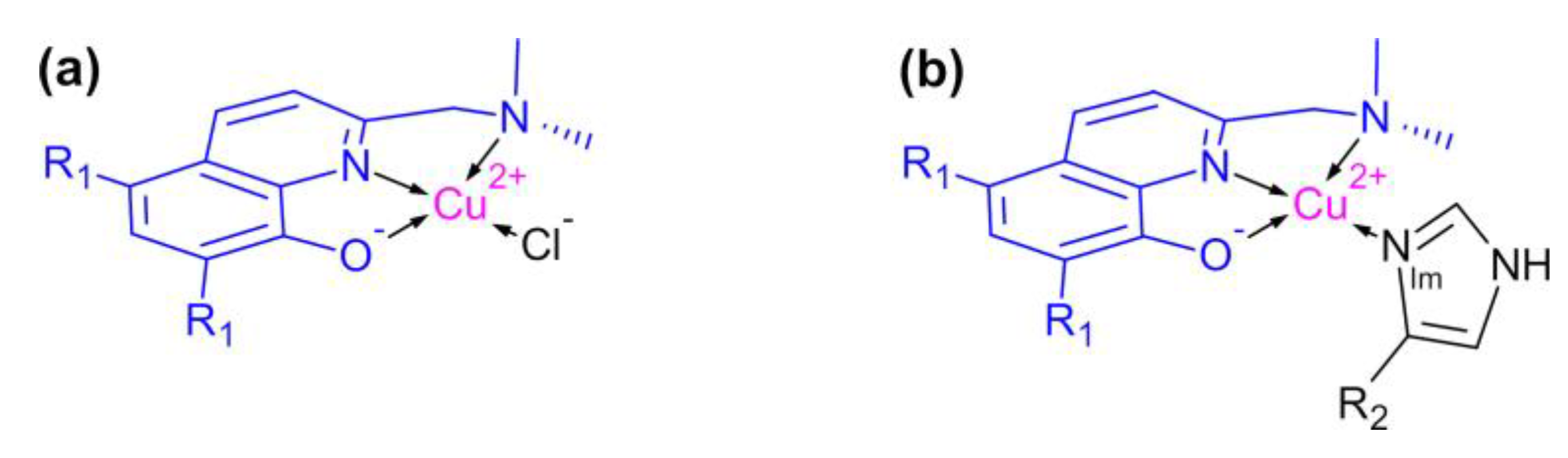
Figure 2.
Determination of the number and stabilities of ternary Cu(PBT2)(Aβ1–40) interactions. (a) EPR spectra of 65Cu/PBT2/Aβ1–40 n:1:1 (n = 0.1–2.67) in PBS pH 7.4 (0.2 mM PBT2/Aβ1–40). (b) Normalized basis set used for the decomposition of the spectra in panel a (see Figures S1–S3 for details). Dotted lines show spectra simulated using the parameters in Table 1. The Cu2Aβ1–40 simulation comprised spectra using “first site” parameters (50%) and “second site” parameters (50%). Vertical scales in panels a and b are different. (c) Experimental species distributions (points) resulting from normalisation and decomposition of the spectra in panel a (Figure S4), and theoretical distributions (lines) calculated using the relevant formation constants in Table 2 (see also Figure S5 for a comparison of relative and absolute Cu2+ speciation). The solid blue line in panel c shows the sum of the Cu(PBT2)NImH6 (dotted line) and Cu(PBT2)NImH13/14 (dashed line). Cu(aq) denotes aqueous Cu2+ (pink). Experimental conditions: temperature, 77 K; microwave power, 10 mW; microwave frequency, 9.425 GHz; modulation amplitude, 8 G; sweep time, 84 s; time constant, 82 ms; averages, 4.
Figure 2.
Determination of the number and stabilities of ternary Cu(PBT2)(Aβ1–40) interactions. (a) EPR spectra of 65Cu/PBT2/Aβ1–40 n:1:1 (n = 0.1–2.67) in PBS pH 7.4 (0.2 mM PBT2/Aβ1–40). (b) Normalized basis set used for the decomposition of the spectra in panel a (see Figures S1–S3 for details). Dotted lines show spectra simulated using the parameters in Table 1. The Cu2Aβ1–40 simulation comprised spectra using “first site” parameters (50%) and “second site” parameters (50%). Vertical scales in panels a and b are different. (c) Experimental species distributions (points) resulting from normalisation and decomposition of the spectra in panel a (Figure S4), and theoretical distributions (lines) calculated using the relevant formation constants in Table 2 (see also Figure S5 for a comparison of relative and absolute Cu2+ speciation). The solid blue line in panel c shows the sum of the Cu(PBT2)NImH6 (dotted line) and Cu(PBT2)NImH13/14 (dashed line). Cu(aq) denotes aqueous Cu2+ (pink). Experimental conditions: temperature, 77 K; microwave power, 10 mW; microwave frequency, 9.425 GHz; modulation amplitude, 8 G; sweep time, 84 s; time constant, 82 ms; averages, 4.
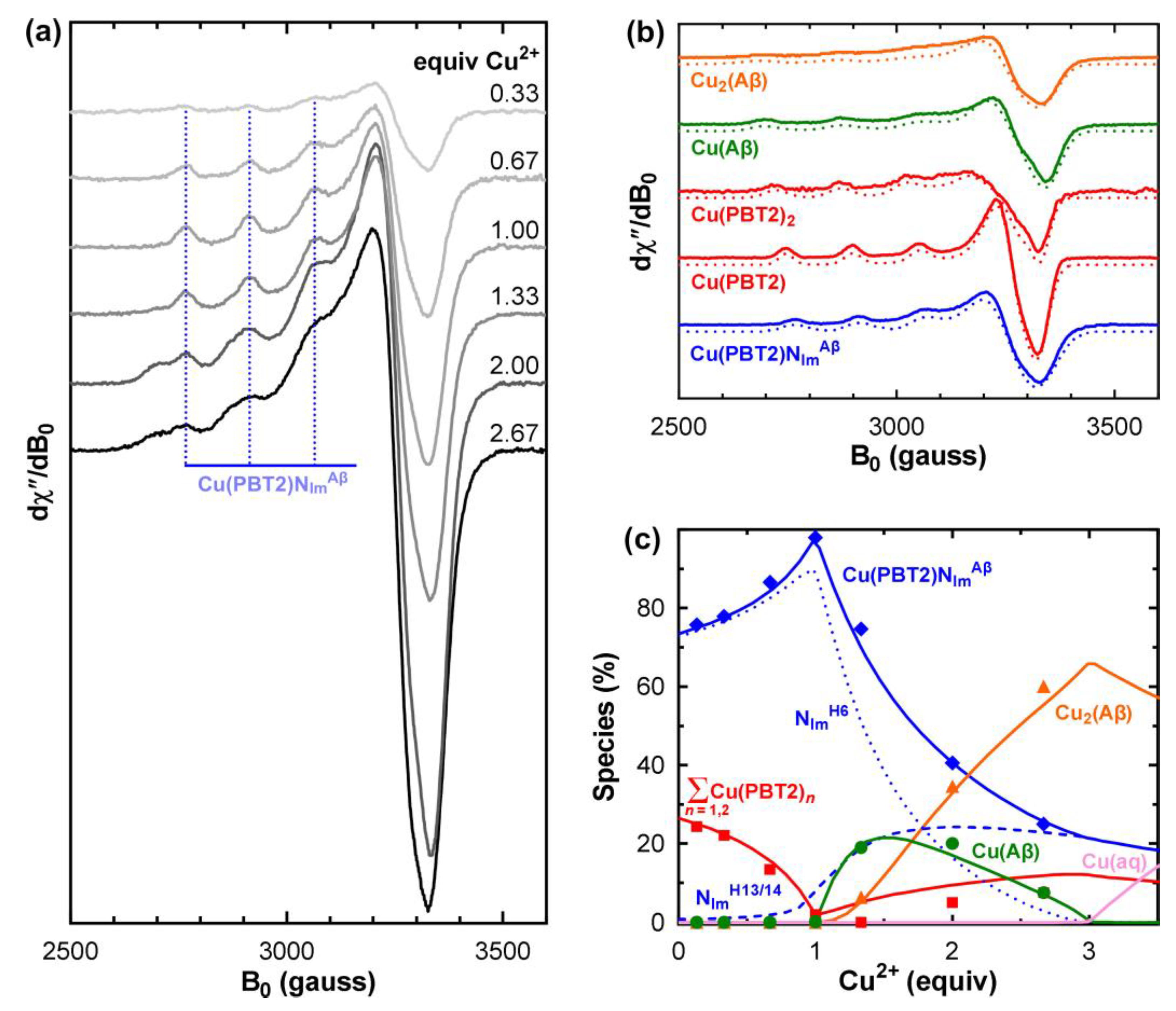

Table 1.
Principal electron Zeeman (gz) and nuclear hyperfine (Az) parameters of binary and ternary Cu2+ complexes of PBT2 and its non-chlorinated homologue with Aβ1–40 and other imidazole-bearing (NIm) co-ligands. A detailed comparison with previously obtained parameters is shown in Table S1.
Table 1.
Principal electron Zeeman (gz) and nuclear hyperfine (Az) parameters of binary and ternary Cu2+ complexes of PBT2 and its non-chlorinated homologue with Aβ1–40 and other imidazole-bearing (NIm) co-ligands. A detailed comparison with previously obtained parameters is shown in Table S1.
| Complex | gz | Az (63Cu) a | Reference | ||
|---|---|---|---|---|---|
| L = PBT2 | |||||
| CuL | 2.259 ± 0.002 | 151 ± 1 | This work | ||
| CuL2 | 2.283 ± 0.002 | 148 ± 3 | This work | ||
| CuLNImX | |||||
| X = Aβ1–40 | 2.249 ± 0.002 | 147 ± 2 | This work | ||
| X = imidazole | 2.248 ± 0.001 | 143 ± 1 | This work | ||
| X = histamine | 2.248 ± 0.001 | 143 ± 1 | This work | ||
| X = Aβ1–42 | 2.242 ± 0.002 | 142 ± 3 | 10b | ||
| L = non-chlorinated PBT2 homologue | |||||
| CuL | 2.255 ± 0.001 | 153 ± 1 | 6 | ||
| CuL2 | 2.267 ± 0.001 | 149 ± 1 | 6 | ||
| CuLNImX | |||||
| X = imidazole | 2.245 ± 0.001 | 144 ± 1 | This work, 6, 11 | ||
| X = histamine | 2.245 ± 0.001 | 145 ± 1 | This work, 6, 11 | ||
| Aβ | |||||
| Cu(Aβ1–40) | 2.268 ± 0.002 | 174 ± 2 | This workc | ||
| Cu2(Aβ1–40) | |||||
| first site | 2.268 ± 0.002 | 174 ± 2 | This workd | ||
| second site | 2.309 ± 0.005 | 168 ± 5 | This workd | ||
a Hyperfine parameters are expressed in units of 10−4 cm−1 (1 × 10−4 cm−1 = 2.9979 MHz). To aid comparison with other studies, hyperfine couplings have been converted from those obtained using 65Cu to those expected for 63Cu using the scaling factor |gn(65Cu) / gn(63Cu)| = 1.07. b Species was not assigned to a ternary complex and spectral isolation was approximate. cParameters are those for the dominant species at pH 7.4. dParameters assume that the coordination of the first-bound ion (“first site”) are the same as those for Cu(Aβ).
Table 2.
Stepwise conditional formation constants (pH 7.4) used to simulate the species distributions of Cu/L/NImX mixtures for X = Aβ1–40 (Figure 2c), imidazole (Figure S6c), and histamine (Figure S9c). A detailed comparison of Cu2+ formation constants, including those for the non-chlorinated homologue of PBT2 and various NIm donors, is provided in Table S2.
Table 2.
Stepwise conditional formation constants (pH 7.4) used to simulate the species distributions of Cu/L/NImX mixtures for X = Aβ1–40 (Figure 2c), imidazole (Figure S6c), and histamine (Figure S9c). A detailed comparison of Cu2+ formation constants, including those for the non-chlorinated homologue of PBT2 and various NIm donors, is provided in Table S2.
| Complex | Formation constanta | c/ (1 M−1)] at pH 7.4 | Reference |
|---|---|---|---|
| CuL | c | 13.61 ± 0.05 | 8 |
| CuL2 | c | 5.95 ± 0.07 | 8 |
| CuLNImAβ (His6) | c | 6.4 ± 0.1 | This work |
| CuLNImAβ (His13/14) | c | 4.4 ± 0.1 | This work |
| CuLNImimidazole | c | 4.22 ± 0.09 | This work |
| CuLNImhistamine | c | 4.00 ± 0.05 | This work |
| Cu(Aβ1–40) | c | 10.0 ± 0.1 | This work |
| Cu2(Aβ1–40) | c | 8.0 ± 0.1 | This work |
a Formation constants are defined in Equations 2–4 (Section 4.3).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



