
Submitted:
04 May 2023
Posted:
05 May 2023
You are already at the latest version
Abstract
Recent data suggest that K-Ras4B (hereafter K-Ras) can drive cancer cell stemness via calmodulin (CaM)-dependent, non-canonical Wnt-signalling. Here we examined whether another Ca2+-binding protein, the CaM-related centrin1, binds to K-Ras and could mediate some K-Ras functions that were previously ascribed to CaM.
While CaM and centrin1 appear to distinguish between peptides that were derived from their classical targets, they both bind to K-Ras in cells. Cellular BRET-data suggest that CaM engages more with K-Ras than centrin1 and that the interaction with the C-terminal membrane anchor of K-Ras is sufficient for this. Surprisingly, binding of neither K-Ras nor its membrane anchor alone to CaM or centrin1 is sensitive to inhibition of prenylation. In support of an involvement of the G-domain of K-Ras in cellular complexes with these Ca2+-binding proteins, we find that oncogenic K-RasG12V displays increased engagement with both CaM and centrin1. This is abrogated by addition of the D38A effector-site mutation, suggesting that K-RasG12V is held together with CaM or centrin1 in complexes with effectors.
When treated with CaM-inhibitors the BRET-interaction of K-RasG12V with centrin1 was also disrupted in the low micromolar range, comparable to that with CaM. While CaM predominates in regulating functional membrane anchorage of K-Ras, it has a very similar co-distribution with centrin1 on mitotic organelles. Given these results, a significant overlap of the CaM- and centrin1-dependent functions of K-Ras is suggested.
Keywords:
K-Ras
; centrin
; calmodulin
; mitosis
; centrosome
; BRET
Introduction
KRAS is the most frequently mutated oncogene and in addition mutated in congenital disorders, called RASopathies [1,2]. It is not fully understood why KRAS is more frequently mutated in cancer than the other RAS genes, NRAS and HRAS. Several facets of Ras biology may contribute to the higher exploitation of KRAS, such as its higher expression level, its specific intracellular trafficking and distribution or its distinct nanoscale organization in the plasma membrane that imposes differential effector usage [3,4,5]. Another less characterized difference is the ability of Ras proteins to impose stemness properties in cells [6,7]. Notably, the most common KRAS splice variant, K-Ras4B (hereafter K-Ras), but not H-Ras, mediates stemness properties via calmodulin (CaM)-dependent non-canonical Wnt-signalling [6]. In line with this, CaM-inhibitors block stemness properties of K-RAS-mutant cancer cells [8,9]. However, the exact mechanism of how CaM mediates K-Ras-driven stemness is not resolved.
Previous cellular data showed that K-Ras/ CaM complexes are disrupted by phosphomimetic mutations of Ser181 at the C-terminus of K-Ras. Conversely, CaM binding blocked phosphorylation at that site [10]. Intriguingly, the phosphomimetic mutation of K-RasG12V on Ser181 reduces its ability to drive stemness [6]. Mutations at this site also modulate the interaction with another trafficking chaperone PDE6D [11], which traffics several prenylated proteins to stemness mediating organelles [12]. Hence, CaM may not be alone in mediating the K-Ras-stemness activity.
CaM possesses two Ca2+-binding lobes, which can each encase 15-20-residue long peptide stretches of classical target proteins in their hydrophobic surfaces [13]. Classical target peptides are typically helical, positively charged and contain hydrophobic anchor residues. Very similar biochemical characteristics are found in singly lipidated, polybasic termini of prenylated or myristoylated proteins, which have emerged as non-canonical targets of CaM [14]. CaM facilitates the Ca2+-dependent cytoplasmic solubilization of K-Ras, by sequestering its farnesyl-tail from the aqueous environment [15]. This contrasts to the GTP-Arl2/3 triggered release of PDE6D cargo [16]. PDE6D and CaM share the preference for K-Ras amongst the Ras isoforms, as palmitoylation obstructs access to the hydrophobic pockets, making K-Ras4A, N-Ras and H-Ras clients only in their non-palmitoylated states [17,18]. Both trafficking chaperones are found in the cyto- and nucleoplasm, and on centriolar structures, such as the primary cilium and the centrosomes [16,19,20]. Hence, it is plausible to assume that these two chaperones have overlapping, yet distinct roles to coordinate trafficking of prenylated proteins spatio-temporally.
In cell lysates, CaM engages more with GTP-loaded K-Ras than with its inactive counterpart [17,21]. Furthermore, complexes between K-Ras, CaM and PI3K p110 subunits have been proposed as being relevant for Akt activation during platelet-derived growth factor receptor (PDGFR)-mediated cell migration [22,23]. The fact that the activation state of Ras matters for its interaction with CaM contrasts with in vitro and structural data. Only weak transient contacts of CaM with non-farnesylated K-Ras were observed in NMR-experiments, while the farnesylated poly-lysine stretch of K-Ras comprising residues 180-185 was sufficient for CaM binding [15,24]. In vitro data further suggest that the polybasic and farnesylated C-terminus of K-Ras binds to either of the Ca2+-bound lobes of CaM, but without involvement of the G-domain [25]. Thus, it appears that the farnesylated C-terminus of K-Ras is sufficient for micromolar binding to CaM. However, in cells there may be CaM/ K-Ras complexes that depend on the activation state of K-Ras.
Inhibitors of CaM alter its conformation, thus preventing binding of canonical target peptides and non-canonical targets [9,13,26,27]. The covalent CaM inhibitor ophiobolin A disrupts binding of K-Ras to CaM and K-Ras membrane anchorage by irreversibly modifying Lys75, 77 and 148 of CaM [8,9,28]. We recently developed an alternative, less toxic covalent inhibitor of CaM, called Calmirasone1, which is much more suitable for cell biological applications [9].
Centrin (or caltractin) proteins are highly related to CaM with the same bi-lobal structure, however, only the C-terminal lobe binds and senses Ca2+ with high affinity [29]. This leaves the centrin-specific N-terminus free for mediating self-assembled extended structures of centrins, which are Ca2+ -dependent due to allosteric coupling with the C-terminus [30]. In humans three centrin paralogs (centrin1-3, CETN1-3) are known [31]. While centrin2 and centrin3 are ubiquitously expressed, centrin1 expression is limited to male germ cells, neurons, and ciliated cells [32]. Centrin2 is probably best known for binding and stabilizing XPC (xeroderma pigmentosum group C), which is involved in DNA repair [33]. In addition, centrins have been implicated in nuclear pore functions and proteasomal activities [32]. Like CaM, centrins appear to recognize a hydrophobic motif of 15-20 residues in such classical target proteins [34].
The activity of centrins can be regulated by several phosphorylation and SUMOylation events [34]. Nuclear localization of centrin2 is enhanced by its SUMOylation [35]. Phosphorylation of T118 in the third EF-hand of the centrin2 C-terminal lobe is required for Ca2+-binding and its centrosomal localization [32]. Centrins localize to distal and intermediate regions preferentially of the mother centrioles and are part of a set of 14 ancient and highly conserved centriolar proteins [36,37]. Hence, loss of centrins broadly affects centriolar functions, including organisation of the microtubule network or overall biogenesis of centrioles [32]. Based on the essential roles of centrins in uni-cellular organisms that depend on cilia formation, it is plausible to assume that an important role also exists for centrins in vertebrate/ mammalian ciliogenesis [32]. In line with this, ciliogenesis is reduced upon depletion of centrin2 in hTERT-RPE1 cells [38].
Given the highly similar bi-lobal structure with hydrophobic binding pockets, we hypothesized that centrins also bind to non-classical targets of CaM, such as K-Ras. Here we show that K-Ras binds to centrin1 in cells in a similar manner to CaM. Our results suggest that binding of K-Ras to these Ca2+-binding proteins in cells is largely independent of the prenylation of K-Ras and involves the G-domain. Given that CaM-inhibitors also affect the K-Ras/ centrin1 interaction and the very similar distribution of centrin1 and CaM throughout the cell cycle, the dependence of K-Ras on either protein may be difficult to determine.
Results and Discussion
The Sequence Divergence between CaM and Centrin-1 is Sufficient to Define CaM-Specific Peptides Derived from its Target Proteins
Calmodulin (CaM) and centrin proteins are highly related, both at the sequence level (Figure 1A) with 54 % sequence identity between CaM and centrin1, and structurally (Figure 1B), with the most obvious difference being the N-terminal extension of centrins. While three CaM genes encode proteins with the exact same sequence, the three centrin paralogs are more divergent. Centrin1 and -2 are ~84 % identical in sequence, while centrin3 differs significantly from centrin1 with only 58 % sequence similarity. In several vertebrates, at least two paralog genes from each family of Ca2+-binding proteins are found, supporting their cell biological significance (Figure 1C).
Current evidence suggests that both CaM and centrins have distinct target protein selectivities [34]. We therefore examined if classical CaM target proteins could also bind to centrin1. The plasma membrane calcium transporting ATPase isoform 4b (PMCA) removes intracellular calcium and a 20-residue stretch mediates its regulation by CaM to which it binds with low nanomolar affinity [39]. An even higher, picomolar affinity has been reported for the 19-residues of the CaM-dependent kinase II (CaMKII) [40].
We employed fluorescence polarization experiments to measure the binding of fluorescein-labelled peptides of these target proteins, F-PMCA and F-CaMKII, respectively, to His-tagged CaM and centrin1 [26]. Similar to previous observations with bovine CaM [26], we found that both peptides bound to human CaM with low nanomolar affinity (F-PMCA, KD = 36 ± 5 nM; F-CaMKII, KD = 6.6 ± 0.2 nM) (Figure 1D). By contrast, no binding of either peptide to human centrin1 was observed even at 2 μM centrin1 concentration (Figure 1E). However, when testing a fluorescently labelled 18-residue long centrin1-specific target peptide, F-Sfi1, derived from the mitotic spindle regulator Sfi1, we observed a nanomolar affinity (KD = 30 ± 12 nM) (Figure 1F), which was higher than the reported micromolar affinity [41]. This deviation could be partially explained by the applied methods, as in the latter case isothermal titration calorimetry was used.
Overall, these results suggest that the sequence divergence between CaM and centrin1 is sufficient to define specific binding to their classical targets that contain a distinct peptide recognition sequence.
Cellular BRET Data Suggest that the K-Ras G-Domain Participates in Complexes with Either CaM or Centrin1
Given the high sequence similarity between CaM and centrin1 (Figure 1A), we investigated whether farnesylated K-Ras could bind to centrin1 as a non-canonical target. We therefore established a cellular Bioluminescence Resonance Energy Transfer (BRET)-assay to test binding of wild-type K-Ras or oncogenic K-RasG12V to centrin1 as compared to CaM.
We genetically fused the donor emission enabling Renilla Luciferase derivative Rluc8 to the N-terminus of the K-Ras protein and the acceptor GFP2 to the N-terminus of centrin1 or CaM. If donor- and acceptor-tagged proteins interact, the BRET signal increases with increasing acceptor-to-donor ratio and may reach a saturation value. Commonly the BRETmax value describes an absolute saturation value [42], which is typically not reached in most of the BRET titration experiments and therefore associated with significant extrapolation.
We here introduce the BRETtop value that characterizes the highest BRET value reached within a defined acceptor-to-donor ratio titration range. By keeping the titration range constant, we can compare different BRETtop values with each other. Like BRETmax, BRETtop would also correlate with the relative number of binding-sites and the relative affinities. Therefore, higher BRETtop values indicate a higher interaction probability and strength of examined BRET-pairs in cells.
We previously observed a higher interaction BRET-signal of oncogenic K-Ras as compared to its wild-type (wt) counterpart with CaM [9]. In line with these data, both CaM and centrin1 were significantly more co-immunoprecipitated with oncogenic GFP2-tagged K-RasG12V than with wt K-Ras (Figure 2A,B; Supplementary Materials Figure S1).
Consistent with our previous BRET-data, we also found that K-RasG12V had a significantly higher BRETtop with CaM than wt K-Ras (Figure 2C). Similarly, the BRETtop of K-RasG12V with centrin1 was significantly higher than that of wt K-Ras with centrin1 (Figure 2D). As expected, a control BRET-pair showed significantly lower BRET-values than the weakest BRET-interaction pair studied (Supplementary Materials Figure S2). The higher BRET of K-RasG12V with the Ca2+ -binding proteins was surprising, given the afore mentioned in vitro binding data [15,25]. Consistent with a binding contribution mediated via some effector interaction, addition of the D38A effector-site mutation reduced the BRET to the level of wt K-Ras for both CaM and centrin1 (Figure 2C,D).
CaM-Inhibitors Bind to Centrin
Given that centrin1 possesses lysines on positions 96, 100, 167 and 168 that are homologous to those targeted by covalent CaM inhibitors (Figure 1A), we tested whether covalent CaM inhibitors ophiobolin A and calmirasone1 or the potent non-covalent CaM inhibitor calmidazolium would disrupt binding of K-Ras to centrin1 in cells. Indeed, treatment with any of these CaM inhibitors lowered the BRETtop of K-RasG12V/ centrin1 (Figure 3A). The inhibition of this interaction occurred at IC50 (calmidazolium) = 10.44 ± 0.05 µM and IC50 (calmirasone1) = 41.6 ± 0.3 µM (Figure 3B), the latter of which was comparable to what was previously observed with CaM [9]. For centrin1, fluorescence anisotropy data revealed that CaM-inhibitors can displace fluorescently labelled Sfi1 from it, indicating their direct binding to centrin1 (Figure 3C, Table 1).
The sensitivity of the K-Ras/ centrin1 interaction to CaM-inhibitors suggests conserved inhibitor binding sites and a similar mode of interaction between K-Ras and the Ca2+ -binding proteins. Importantly, treatment with several CaM-inhibitors may therefore also affect centrin1 biology, making it potentially difficult to interpret inhibitor-dependent phenotypic observations.
Inhibition of Prenylation does not Disrupt BRET-Interaction of K-Ras with CaM or Centrin1 in Cells
Agamasu et al., have previously reported that the K-Ras-derived farnesylated and carboxymethylated KSKTKC-peptide is sufficient to bind to CaM in vitro [25]. To test, whether non-prenylated K-Ras can still bind to the Ca2+-binding proteins in cells, we tested the effects of the prenylation inhibitor mevastatin in our BRET-assays. Statins such as mevastatin inhibit the HMG-CoA pathway and thus provision of prenylpyrophosphate substrates for protein prenylation [43]. We therefore expected that treatment of cells with high concentrations of mevastatin would abrogate farnesyl-mediated K-Ras/ CaM interaction. Surprisingly, mevastatin treatment did not significantly affect the BRET-levels of K-Ras with either CaM or centrin1 (Figure 4A,B). The higher BRETtop of K-Ras with CaM (Figure 4A) than with centrin1 (Figure 4B) may relate to the fact that only one Ca2+-binding lobe is found in centrins [32], which may allow for the binding of only one K-Ras per centrin1 protein.
We next examined whether the C-terminal membrane targeting sequence of K-Ras alone (residues 166-188), CTK, was sufficient to mediate binding to CaM as suggested by in vitro data and whether the same would apply for binding to centrin1. In agreement with in vitro data, the BRET between CTK and CaM indicated binding albeit with a lower BRETtop than full length K-Ras (Figure 4C), but still above background (Supplementary Materials Figure S2). The CTK interaction with centrin1 was comparable to that with CaM (Figure 4C,D). As with full-length K-Ras, mevastatin treatment did not decrease the BRET of CTK with either of the Ca2+-binding proteins (Figure 4C,D).
This mevastatin insensitivity was overall unexpected, given the strong contribution of the farnesyl-moiety to CaM-binding in vitro [15,24,25]. Taken together with the activation-state dependent complexation of K-Ras with CaM or centrin1, this may suggest that these proteins exist in cellular complexes that are largely prenylation independent, yet involve the C-terminal poly-lysine stretch of K-Ras and depend on the activation state of K-Ras. Alternatively, similar sized, distinct pools of K-Ras in complex with the Ca2+-binding proteins exist, which require a subset of the aforementioned features.
Membrane Targeting and Anchorage of K-Ras Depends more on CaM than on Centrin1
Prenyl-binding chaperone proteins can effectively facilitate diffusion of their target proteins in cells, as they shield the hydrophobic prenyl-moiety and thus allow for a longer residence in the aqueous cytoplasm [4]. Others suggested that CaM can extract and solubilize K-Ras and act as a trafficking chaperone [15].
We previously showed that inhibition of CaM selectively reduces K-RasG12V- as compared to H-RasG12V-BRET signals that originate from nanoclustering of active Ras on the plasma membrane [9]. This nanoclustering-dependent BRET-signal is sensitive to disruption not only of Ras nanoclustering, but any process upstream that interferes with functional membrane anchorage, such as disrupted trafficking or inhibition of the Ras lipid modification [44]. Similar to CaM inhibition, knockdown of another trafficking chaperone PDE6D, which also binds to prenylated proteins and facilitates K-Ras localization at the plasma membrane, reduces K-Ras membrane anchorage associated FRET [45].
In line with the mevastatin data (Figure 4), binding of K-Ras to CaM or centrin1 was essentially insensitive to inhibition of prenylation by the knockdown of the shared ⍺-subunit of farnesyl- and geranylgeranyl-transferases (FNTA) (Figure 5A–C). Yet, the same treatment significantly abrogated the membrane anchorage-BRET signal of both K-RasG12V or H-RasG12V (Figure 5E,F), consistent with the significance of prenylation for Ras membrane anchorage [46].
As observed previously, knockdown of CaM selectively reduced the membrane anchorage-BRET signal of K-RasG12V (Figure 5E) but not H-RasG12V (Figure 5F). By contrast, knockdown of centrin1 decreased the BRET-signal K-Ras-selectively and to a significantly lesser extent than knockdown of CaM (Figure 5D-F). Immunoblotting confirmed the significant knockdown of FNTA and CETN1 expression in HEK293-ebna cells (Figure 5C,D; Supplementary Materials Figures S3 and S4), while that of CALM1 was previously validated by us using RT-qPCR [9].
These data suggest that CaM is more important to facilitate membrane trafficking of K-Ras in cells than centrin1.
Centrin1 Co-Distributes with CaM during the Cell Cycle
We previously observed that CaM inhibitors decrease stemness properties of KRAS-mutant cancer cell lines [8,9]. The clonogenic growth of cancer cell spheroids is employed as a surrogate measure for cancer cell stemness [47]. We therefore tested the effect of the knockdown of CALM1 and CETN1 on MDA-MB-231 and MCF-7 derived spheroids (Figure 6A,B). Both CALM1 and CETN1 (Supplementary Materials Figures S5 and S6) knockdown decreased the formation of spheroids derived from these cell lines. However, the effect was more pronounced in the KRAS-mutant MDA-MB-231 cell line (Figure 6A). Moreover, the knockdown of CALM1 decreased spheroid growth significantly more in this cell line, which correlated with the overall stronger effect of this knockdown treatment on K-RasG12V membrane anchorage BRET (Figure 5E).
Only limited conclusions in regard to cancer cell stemness can be derived from these experiments, which are essentially assaying the ability of cancer cells to evade anoikis and bear some similarity to culture conditions employed for stem/ progenitor cells.
The fate of stem and progenitor cells is decided during the cell cycle, which can proceed to symmetric or asymmetric cell divisions [48]. Oncogenes are suggested to shift the mode of cell division to more symmetric divisions that produce more stem cells [49]. Stemness can be mediated by centriolar organelles, such as the centrosomes, specifically the mother centrosome [50]. Interestingly, mCherry-tagged CaM localizes during different cell cycle phases to the centrosomes and the midbody in HeLa cells (Figure 6C) [19,27]. The same is essentially seen for endogenous centrin1, which also localizes to these structures (Figure 6C). However, during interphase, CaM has a more pronounced cyto-/nucleoplasmic distribution, while centrin1 discretely localizes to the centrosomes.
Conclusions
Our data show that K-Ras does not only interact with CaM, but also with the highly related protein centrin1. While both Ca2+-binding proteins distribute to similar mitotic structures, notably the centrosomes, CaM appears to have a stronger impact on K-Ras functional membrane organisation at the plasma membrane. Centrin1 may therefore rather function to localize K-Ras to certain structures, such as the centrosomes, while it appears to have only a minor role in K-Ras trafficking. These distinct functions of CaM and centrin proteins are difficult to tell apart using pharmacological inhibitors against CaM, which we found also affect binding of K-Ras to centrin1.
While previous in vitro data demonstrated that the farnesylated C-terminus of K-Ras was sufficient for binding to CaM, our data here suggest that farnesylation is essentially expendable for most of the interaction of K-Ras with either CaM or centrin1 in cells. Given that oncogenic K-Ras engages more with either of these proteins, we propose that most of the K-Ras/ CaM and K-Ras/ centrin1 pairs are found in complexes that can recognize the activation state of K-Ras. This recognition is typically afforded by effectors, hence it is plausible to assume that most of the K-Ras binding to these Ca2+-binding proteins happens in higher order complexes that contain effectors. Others have previously proposed PI3K⍺-containing complexes with K-Ras and CaM [51]. Our data encourage further investigation of these potential complexes and their function inside cells.
Experimental Procedures
Plasmids, siRNAs and Inhibitors
All construct names contain the tag at a position corresponding to its location in the protein sequence, e.g. GFP2-CaM, contains the GFP2-tag at the N-terminus of CaM. All plasmids employed in the study were produced by multi-site gateway cloning [53]. The human CALM1 entry clone with L1 – L2 recombination sites was obtained from the NCI RAS Initiative. The K-Ras4b entry clone was procured from Addgene (RAS mutant clone collection, Kit #1000000089). Custom-synthesised entry clones encoding human CETN1 or the CTK fragment with L1 – L2 recombination sites in pDONR221 vector were commercially obtained from Genecust, France. An LR recombination reaction comprising three entry clones encoding the CMV promoter, a tag (Rluc8 or GFP2) and the protein of interest (CTK, K-Ras wt, CaM and centrin1) and a destination vector, pDest-305 vector was performed to obtain the recombinant plasmids. In a single-site LR recombination reaction, CaM or centrin1 entry clones were combined with the destination vector, pDest-527 to produce bacterial expression plasmids encoding N-terminally His6-tagged CaM and centrin1. The positive clones were selected using ampicillin in E. coli DH10B. The pmCherry-CaM, pEGFP-centrin1 plasmids and plasmids encoding N-terminal Rluc8 or GFP2-tagged K-RasG12V and H-RasG12V were previously described [9,26]. siRNA for CALM1 (Hs_CALM1_6, SI02224222), and FNTA (Hs_FNTA_6, SI02661995) were from Qiagen. The siRNA for CETN1 (ON-TARGETplus SMARTpool siRNA, L-011831-00-0005) and negative control siRNA (ON-TARGETplus Non-targeting pool, D-001810-10-05) were from Dharmacon. Mevastatin (J61357, Alfa Aeser), calmidazolium chloride (sc-201494, Santa Cruz), and ophiobolin A (sc-202266, Santa Cruz) were commercially acquired from the sources given in parenthesis. Calmirasone1 was synthesized as previously described by us [9].
Protein Sequence Analyses
The protein sequences encoded by CALM1-3 and CETN1-3 genes were collected from uniport database (http://unirprot.org/) and multiple sequence alignment was performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). For paralog number analysis, the protein coding genes of calmodulin and centrin were searched for each species in the NCBI protein database (https://www.ncbi.nlm.nih.gov/). The CALM1 or CETN1 genes were given as search query and orthologs were identified from the annotation pipeline. A process flow was then generated using RefSeq to identify a set of comparable proteins including orthologs and similar proteins. Note that only protein encoding genes were considered and pseudogenes were discarded.
Protein Purification
The His6-tagged human CaM and centrin1 proteins were purified as described previously [9]. Briefly, the pDest527-His6-CaM or pDest527-His6-centrin1 plasmid transformed E.coli BL21 (DE3) cells were grown in LB medium supplemented with 100 μg/ml of ampicillin. At 0.4 – 0.6 OD, the culture was induced with 0.5 mM IPTG followed by overnight incubation at 25 °C with shaking. The culture was centrifuged and the obtained cell pellet was suspended in a lysis buffer composed of 20 mM HEPES, pH 7.6, 150 mM NaCl, 5 mM MgCl2, 0.5 mg/ml lysozyme and 700 units DNase I. 20 ml of lysis buffer was used for cell pellet from 1 l of cell culture. The cells were lysed by sonication and the His-tagged proteins were purified using HisTrapTM HP Prepacked Columns (GE Healthcare) using the chromatography system ÄKTAprime plus (GE Healthcare). The columns were equilibrated with a buffer composed of 50 mM Tris HCl, pH 7.5, 150 mM NaCl, and 35 mM imidazole, and the His-tagged proteins were eluted in an elution buffer containing 250 mM of imidazole. The eluted fractions were dialyzed for 16 h at 4 °C in a buffer composed of 50 mM Tris HCl, pH 7.5, 150 mM NaCl, and 2 mM CaCl2. Protein concentration was measured by absorbance using a NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific).
Fluorescence Polarisation Binding Assay
Fluorescence polarisation assays were performed as established previously by us [9,26]. The fluorescein labelled PMCA- and CaMKII-peptides were custom synthesized by Genscript (USA) and Pepmic (China), respectively. The PMCA peptide was derived from 1086-LRRGQ-ILWFR-GLNRI-QTQIK-1105 of human PMCA and fluorescein was attached to the C-terminal native Lys. The CaMKII peptide sequence was derived from 294-NARRK-LKGAI-LTTML-ATRN-312 of human CaMKII and fluorescein was attached to a non-native cysteine added to the N-terminus. The N-terminal His6-tagged CaM or centrin1 proteins were 2-fold diluted in a buffer composed of 20 mM Tris Cl pH 7.5, 50 mM NaCl, 1 mM CaCl2 and 0.005% v/v Tween 20 in a black low volume, round bottom 384-well plate (cat. no. 4514, Corning). Then 10 nM of fluorescein-labelled peptide was added to the protein dilution series. The reaction mix was incubated for 20 min at RT before anisotropy measurements.
The Sfi1 peptide was derived from 670-REVAA-RESQH-NRQLL-RGALR-RWK-692 of human Sfi and the fluorescein was attached to the native C-terminal Lys. Sfi1 peptide titration was performed in a buffer composed of 10 mM HEPES pH 7.4, 100 mM CaCl2 and 0.005 % v/v Tween 20. For binding, to a 2-fold dilution series of centrin1, 100 nM of Sfi1 peptide was added and the reaction mix was incubated for 45 min at RT before anisotropy measurements. For measuring the IC50 of inhibitors to centrin1, to the 3-fold dilution series of inhibitors in the assay buffer, a complex of 100 nM fluorescein labelled Sfi1 peptide and 250 nM His-centrin1 was added in 20 µl volume in a 384-well plate. The fluorescence anisotropy was measured after overnight incubation at RT.
The fluorescence anisotropy was measured on a Clariostar (BMG Labtech) plate reader using the fluorescence intensity signal recorded from vertical (Iv)- and horizontal (Ih)- polarized light using a fluorescence polarisation module (λexcitation 482 ± 8 nm and λemission 530 ± 20 nm). Fluorescence anisotropy was calculated from the measured fluorescence intensities, according to, where r is the fluorescence anisotropy value, Iv and Ih are the fluorescence emission intensities detected with vertical and horizontal polarization respectively. The instrument specific correction factor G(λ) was set to 1, and not determined further. A quadratic equation as described [54,55] by others was defined in Prism (GraphPad) and was used to determine the KD value of the fluorescein tagged peptides to target protein.
Here, Af is the anisotropy value of the free fluorescent probe, Ab is the anisotropy value of the fluorescent probe /protein complex, Lt is the total concentration of the fluorescent probe, KD is the equilibrium dissociation constant, x is total concentration of protein and y is measured anisotropy value. KD is measured in the same unit of x. Note that variations in the active fraction of the home-made proteins and different methods used to determine the protein concentrations the obtained KD values can vary from those reported.
The IC50 value of inhibitors was determined by plotting the log concentration of inhibitor against fluorescence anisotropy values and fitting the data to log inhibitor vs. response – variable slope (four parameters) equation in Prism (GraphPad). The IC50 of the inhibitor was converted into Kd as described earlier using the equation [56],
where [I]50 is the concentration of free inhibitor at 50 % displacement, given as , where [EI]50 is the concentration of centrin1:inhibitor complex in case of 50 % displacement, [P]50 is concentration of free probe, F-Sfi1 at 50 % displacement, [E]0 is concentration of free centrin1 at 0 % displacement, KD,probe is the dissociation constant of the complex of Sfi1 and centrin1.
Co-Immunoprecipitation Experiments
About 800,000 HEK293-ebna cells were seeded in 60 mm dishes and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10 % v/v Foetal Bovine Serum (FBS), 2 mM L-glutamine (cat. no. 25030-024, Gibco, Thermo Fisher Scientific) and 1 % v/v penicillin/ streptomycin (cat. no. 15140122, Gibco, Thermo Fisher Scientific) overnight. The next day cells were transiently transfected with 4 µg plasmids encoding the indicated combinations of constructs using jetPRIME (cat. no. 114-75, Polyplus) according to the manufacturer’s instructions. At 48 h post-transfection, the cells were lysed using 200 μl of Lysis buffer (10 mM Tris Cl pH 7.5, 150 mM NaCl, 2 mM CaCl2, 0.2 % v/v NP40) supplemented with protease inhibitor cocktail (cat. no. A32955, Pierce, Thermo Fisher Scientific). After 30 min incubation on ice, the lysate was cleared by centrifugation for 10 min at 4 °C and 17,000 g. The cleared lysate was transferred to a clean tube and 15 µl sample was withdrawn (as “Input” for Western blot analysis). The lysate was diluted with 300 μl of dilution buffer (10 mM Tris Cl pH 7.5, 150 mM NaCl, 2mM CaCl2) supplemented with protease inhibitor cocktail. Then 25 µl of GFP-trap Beads Slurry (ChromoTek GFP-Trap Agarose, cat. no. gta) were added to the diluted lysate and rotated end-over-end for 1 h at 4 ⁰C. Then the beads were washed 3 times with Wash buffer (10 mM Tris Cl pH 7.5, 150 mM NaCl, 2 mM CaCl2, 0.02 % v/v NP40). Bound proteins were eluted by the addition of 2 × Laemlli buffer and boiling for 10 min at 95 ⁰C. The eluted proteins were subsequently analyzed by SDS-PAGE on 10 % acrylamide gels. Proteins were transferred onto a nitrocellulose membrane 0.2 µm (Bio-Rad) using the Trans-Blot Turbo Transfer system (Bio-Rad) and probed with a primary antibody. The primary antibodies employed were anti-GFP (SAB4301138, Sigma-Aldrich, 1:5000), anti-Renilla Luciferase (ab187338, Abcam, 1:3000), anti β-actin (A5441, Sigma- Aldrich, 1:5000). Anti-rabbit IRDye 680RD or anti-mouse IRDye 800CW secondary antibodies (LI-COR) were used to visualize the proteins on an Odyssey CLx system (LI-COR). The relative expression level of proteins was densitometrically quantified from images of membranes analysed using Image Studio software (LI-COR). For the quantitative analysis of the pull-down proteins, the signal of the Rluc8-tagged prey proteins was normalized with the signal from the GFP-tagged bait protein. Next, the signal intensity of the GFP2-K-RasG12V + Rluc8-CaM transfected sample was used to normalize the other samples.
BRET Donor Saturation Titration Assays
The detailed method of our BRET assay can be found in [9,57]. Briefly, ~200,000 HEK293-ebna cells were seeded per well of a 12-well plate (cat. No. 665180, Greiner Bio-One) and grown in 1 ml of complete DMEM. The next day ~ 1 µg of BRET sensor plasmids was transfected using 2.5 µl of jetPRIME. For titration curves, the concentration of donor plasmid was kept constant at 25 ng and the concentration of acceptor plasmid was varied from 25 ng to 1000 ng. 24 h after transfection, the cells were treated with inhibitors or vehicle control (DMSO at 0.2 % v/v). The next day, cells were collected in PBS and re-plated in white flat bottom 96-well plates (cat. no. 236108, Nunc, Thermo Fisher Scientific). Then the BRET readings were taken on a Clariostar plate reader (BMG Labtech). First the fluorescence intensity (λexcitation 405 ± 10 nm and λemission 515 ± 10 nm) of GFP2 was measured as it is directly proportional to the acceptor concentration (RFU). Next the BRET readings were taken in well-mode by adding coelenterazine 400a (cat. no. C-320, GoldBio) to a final concentration of 10 μM and the luminescence emission intensities were simultaneously recorded at 410 ± 40 nm and at 515 ± 15 nm corresponding to the signal of donor (RLU) and the BRET signal, respectively. The raw BRET ratio was calculated as the ratio of BRET signal /RLU. The BRET ratio was obtained by subtracting the raw BRET ratio from the background raw BRET ratio measured from cells expressing only the donor. The relative expression is calculated as the ratio of RFU /RLU and denoted as [Acceptor] /[Donor]. The BRET ratio vs [Acceptor] /[Donor] ratio data from typically 3 biological repeats were plotted and the data were fitted by a hyperbolic equation in Prism. The BRETtop value represents the top asymptote of the BRET ratio reached within the defined [Acceptor] /[Donor] ratio. The one phase association equation of Prism 9 (GraphPad) was used to predict the top asymptote Ymax-value, which was taken as the BRETtop. Statistical analysis between the BRETtop values was performed using the Extra sum-of-squares F test.
Dose Response Analysis of Inhibitors and siRNA Knockdown in BRET Assays
For dose response analysis of inhibitors, on day one, ~200,000 HEK293-ebna cells were seeded per well of a 12-well plate (cat. No. 665180, Greiner Bio-One) and grown in complete DMEM. On day two ~ 1 µg of BRET sensor plasmids were transfected at an indicated donor /acceptor plasmid ratio using jetPRIME as mentioned in the corresponding figure legends. On day three, the medium was exchanged with fresh medium containing various doses of inhibitors. After 24 h incubation, on day four, the cells were collected in PBS and the BRET assay was performed. The log inhibitor vs BRET ratio was plotted, and the data were fitted by a log (inhibitor) vs. response variable slope (four parameters) equation of Prism and the IC50 values were calculated.
For studying the effect of siRNA-mediated knockdown, on day one the HEK293-ebna cells were seeded in 12-well plates in 1 ml of growth medium. On day two, cells were transfected with 100 nM of siRNA per well using 3.5 µl Lipofectamine RNAiMAX (cat. no. 13778, Thermo Fisher Scientific) and Opti-MEM medium (cat. no. 31985062, Gibco, Thermo Fisher Scientific) as vehicle. On day three, the medium was exchanged and the cells were transfected with ~ 1 µg of BRET sensor plasmids using 3 µl jetPRIME reagent and expressed for 48 h. The transfected donor /acceptor plasmid ratio is indicated in corresponding figure legends. On day five, the BRET assay was performed as indicated above.
siRNA-Mediated Knockdown and Western Blotting
About 300,000 HEK293-ebna cells were seeded per well of a 6-well plate (cat. no. 657160, Cellstar, Greiner Bio-One) and grown in 2 ml of complete DMEM for 24 h. The next day cells were transfected with 100 nM of siRNA using Lipofectamine RNAiMAX, followed by a medium exchange after 4 h. After 48 h, cells were lysed in RIPA buffer (10 mM Tris, 150 mM NaCl, 0.5 mM EDTA, 0.2% v/v NP40) supplemented with protease inhibitor cocktail (cat.no. A32955, Pierce, Thermo Fisher Scientific). The protein amount in cell lysates were quantified using Bio-Rad protein assay kit (cat.no. 5000006). Cell lysate containing 50 µg of protein per lane was resolved in Mini-PROTEAN precast 4 – 20 % acrylamide gels. Proteins were subsequently transferred onto a nitrocellulose membrane 0.2 µm (Bio-Rad) using the Trans-Blot Turbo Transfer system (Bio-Rad) and probed with the mix of primary antibodies against the protein of interest and the loading control. The primary antibodies employed were anti-FNTA (cat. no. ab109738-1001, Abcam, at 1:1000), anti-centrin1 (cat. no. 12794-1-AP, Proteintech, at 1:500) and anti-GAPDH (cat. no. G8796 mouse and G9545 rabbit, Sigma-Aldrich, at 1:10,000). Anti-mouse or anti-rabbit IRDye 800CW or 680RD secondary antibody (LI-COR) were used subsequently to develop the membrane and the proteins were detected using an Odyssey CLx system (LI-COR).
3D Spheroid Assay
MDA-MB-231 and MCF-7 cells were seeded in 12-well plates (cat. No. 665180, Greiner Bio-One) and transfected with either 100 nM negative control siRNA or siRNA targeting CALM1 or CETN1 using Lipofectamine RNAiMAX. The next day, cells were collected by trypsinization and re-plated into low attachment, suspension cell culture 96-well plates (cat. no. 655185, Cellstar, Greiner Bio-One) for 3D spheroid suspension culture. About 1,000 MDA-MB-231 or 2,500 MCF-7 cell were seeded per well of the 96-well plate in 50 µl of RPMI medium (cat. no. 52400-025, Gibco, Thermo Fisher Scientific) or DMEM, respectively, containing 0.5% v/v MethoCult (cat. no. SFH4636, Stemcell technologies), 1x B27 (cat. no. 17504044, Gibco, Thermo Fisher Scientific), 25 ng/ml EGF (cat. no. E9644, Sigma-Aldrich), and 25 ng/ml FGF (cat. no. RP-8628, Thermo Fisher Scientific). Cells were incubated in a cell culture incubator for 6 days and fresh growth medium was supplemented on the third day. After six days of incubation, a 10 % final volume of the alamarBlue reagent (cat. no. DAL1025, Invitrogen, Thermo Fisher Scientific) was added to each well and incubated for 4 h at 37 °C. Then the fluorescence intensity (λexcitation 560 ± 5 nm and λemission 590 ± 5 nm) was measured using the Clariostar plate reader. The obtained fluorescence intensity data were normalized to negative control siRNA corresponding to 100 % sphere formation.
Confocal Microscopy
HeLa cells were seeded on glass coverslips 1.5H (cat. no. LH22.1, Carl Roth) in 6-well plates (cat. no. 657160, Cellstar, Greiner Bio-One) and grown in complete DMEM for 24 h. The next day the cells were transiently co-transfected with pmCherry-CaM and pmGFP-K-RasG12V using jetPRIME. 48 h after transfection, cells were fixed using 4% v/v formaldehyde (cat. no. 43368, Alfa Aesar) in PBS for 10 min at room temperature. The fixation solution was then replaced with PBS-Tween 0.05% v/v (cat. no. 9127.1, CarlRoth). After permeabilization in PBS-Triton X100 0.5% v/v (cat. no. T8787, Merck) for 10 minutes and blocking for 30 min in 2 % v/v solution of BSA (A6588, Applichem) in PBS, the cells were incubated for 1 h at room temperature with primary antibody against centrin1 (rabbit polyclonal, cat no.12794-1-AP, Proteintech). After washing with PBS-Tween 0.05% v/v, the secondary antibody AlexaFluor 667 goat anti-rabbit (cat no. A21244, Life Technologies) was applied for 1 h at room temperature. DNA was stained with a 1 mg/ml solution of DAPI (cat. no. D1306, Thermo Fisher Scientific) in PBS for 10 min. The coverslips were mounted onto glass slides using Vectashield (cat. no. H-1000, Vector Laboratories). Images were captured on a spinning disk confocal microscope (Andor, Oxford Instruments), fitted with a Zyla 5.5 sCMOS camera (Andor, Oxford Instruments), using a plan APO 60×/ 1.40 Ph3 DM oil immersion objective (Nikon) and NIS-Elements Imaging Software (Nikon).
Data and Statistical analysis :Prism 9 (GraphPad) was used for the preparation of plots, data and statistical analysis. The number of independent biological repeats (n) and type of statistical analysis used are indicated in the corresponding figure legends. A p-value < 0.05 is considered statistically significant, and the statistical significance levels are annotated as follows: * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001, or ns = not significant.
Supplementary Materials
The article contains Supplementary Materials.
Data Availability Statement
Any data that support the findings of this study are included within the article and Supplementary Materials.
Acknowledgements
The study was supported by internal funds of the University of Luxembourg and the Luxembourg National Research Fund (FNR) grant C19/BM/13673303-PolaRAS2 to DA.
Conflict of Interests
The authors declare that they have no conflicts of interest with the contents of this article.
References
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Rauen, K.A.; McCormick, F. The duality of human oncoproteins: drivers of cancer and congenital disorders. Nat. Rev. Cancer 2020, 20, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Abankwa, D.; Gorfe, A.A. Mechanisms of Ras Membrane Organization and Signaling: Ras Rocks Again. Biomolecules 2020, 10, 1522. [Google Scholar] [CrossRef] [PubMed]
- Schmick, M.; Kraemer, A.; Bastiaens, P.I. Ras moves to stay in place. Trends Cell Biol. 2015, 25, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Newlaczyl, A.U.; Coulson, J.M.; Prior, I.A. Quantification of spatiotemporal patterns of Ras isoform expression during development. Sci. Rep. 2017, 7, 41297. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, M.P.; Quatela, S.E.; Philips, M.R.; Settleman, J. Activated Kras, but Not Hras or Nras, May Initiate Tumors of Endodermal Origin via Stem Cell Expansion. Mol. Cell. Biol. 2008, 28, 2659–2674. [Google Scholar] [CrossRef] [PubMed]
- Najumudeen, A.K.; Jaiswal, A.; Lectez, B.; Oetken-Lindholm, C.; Guzmán, C.; Siljamäki, E.; Posada, I.M.D.; Lacey, E.; Aittokallio, T.; Abankwa, D. Cancer stem cell drugs target K-ras signaling in a stemness context. Oncogene 2016, 35, 5248–5262. [Google Scholar] [CrossRef]
- Okutachi, S.; Manoharan, G.B.; Kiriazis, A.; Laurini, C.; Catillon, M.; McCormick, F.; Yli-Kauhaluoma, J.; Abankwa, D. A Covalent Calmodulin Inhibitor as a Tool to Study Cellular Mechanisms of K-Ras-Driven Stemness. Front. Cell Dev. Biol. 2021, 9, 665673. [Google Scholar] [CrossRef]
- Alvarez-Moya, B.; López-Alcalá, C.; Drosten, M.; Bachs, O.; Agell, N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene 2010, 29, 5911–5922. [Google Scholar] [CrossRef]
- Dharmaiah, S.; Bindu, L.; Tran, T.H.; Gillette, W.K.; Frank, P.H.; Ghirlando, R.; Nissley, D.V.; Esposito, D.; McCormick, F.; Stephen, A.G.; et al. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEδ. Proc. Natl. Acad. Sci. USA 2016, 113, E6766–E6775. [Google Scholar] [CrossRef] [PubMed]
- Chippalkatti, R.; Abankwa, D. Promotion of cancer cell stemness by Ras. Biochem. Soc. Trans. 2021, 49, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013, 280, 5551–5565. [Google Scholar] [CrossRef]
- Grant, B.M.M.; Enomoto, M.; Ikura, M.; Marshall, C.B. A Non-Canonical Calmodulin Target Motif Comprising a Polybasic Region and Lipidated Terminal Residue Regulates Localization. Int. J. Mol. Sci. 2020, 21, 2751. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.M.M.; Enomoto, M.; Back, S.-I.; Lee, K.-Y.; Gebregiworgis, T.; Ishiyama, N.; Ikura, M.; Marshall, C.B. Calmodulin disrupts plasma membrane localization of farnesylated KRAS4b by sequestering its lipid moiety. Sci. Signal. 2020, 13, eaaz0344. [Google Scholar] [CrossRef] [PubMed]
- A Ismail, S.; Chen, Y.-X.; Rusinova, A.; Chandra, A.; Bierbaum, M.; Gremer, L.; Triola, G.; Waldmann, H.; Bastiaens, P.I.H.; Wittinghofer, A. Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat. Chem. Biol. 2011, 7, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, P.; López-Alcalá, C.; Bosch, M.; Chiloeches, A.; Rocamora, N.; Gil, J.; Marais, R.; Marshall, C.J.; Bachs, O.; Agell, N. Calmodulin Binds to K-Ras, but Not to H- or N-Ras, and Modulates Its Downstream Signaling. Mol. Cell. Biol. 2001, 21, 7345–7354. [Google Scholar] [CrossRef]
- Chandra, A.; Grecco, H.E.; Pisupati, V.; Perera, D.; Cassidy, L.; Skoulidis, F.; Ismail, S.A.; Hedberg, C.; Hanzal-Bayer, M.; Venkitaraman, A.R.; et al. The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nature 2011, 14, 148–158. [Google Scholar] [CrossRef]
- Li, C. J., Heim, R., Lu, P., Pu, Y., Tsien, R. Y., andChang, D. C. (1999) Dynamic redistribution of calmodulin in HeLa cells during cell division as revealed by a GFP-calmodulin fusion protein technique Journal of Cell Science 112, 1567-1577 papers3://publication/uuid/BEF14B51-A6BE-4B81-81D4-72825BA2B6CD.
- Yu, Y.-Y.; Chen, Y.; Dai, G.; Chen, J.; Sun, X.-M.; Wen, C.-J.; Zhao, D.-H.; Chang, D.C.; Li, C.-J. The association of calmodulin with central spindle regulates the initiation of cytokinesis in HeLa cells. Int. J. Biochem. Cell Biol. 2004, 36, 1562–1572. [Google Scholar] [CrossRef]
- Abraham, S.J.; Nolet, R.P.; Calvert, R.J.; Anderson, L.M.; Gaponenko, V. The Hypervariable Region of K-Ras4B Is Responsible for Its Specific Interactions with Calmodulin. Biochemistry 2009, 48, 7575–7583. [Google Scholar] [CrossRef]
- Liao, J.; Planchon, S.M.; Wolfman, J.C.; Wolfman, A. Growth Factor-dependent AKT Activation and Cell Migration Requires the Function of c-K(B)-Ras Versus Other Cellular Ras Isoforms. J. Biol. Chem. 2006, 281, 29730–29738. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Julsgart, J.; Berchtold, M.W. High affinity calmodulin target sequence in the signalling molecule PI 3-kinase. FEBS Lett. 1998, 425, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-J.; Xu, L.-R.; Liao, J.-M.; Chen, J.; Liang, Y. Both the C-Terminal Polylysine Region and the Farnesylation of K-RasB Are Important for Its Specific Interaction with Calmodulin. PLOS ONE 2011, 6, e21929. [Google Scholar] [CrossRef]
- Agamasu, C.; Ghirlando, R.; Taylor, T.; Messing, S.; Tran, T.H.; Bindu, L.; Tonelli, M.; Nissley, D.V.; McCormick, F.; Stephen, A.G. KRAS Prenylation Is Required for Bivalent Binding with Calmodulin in a Nucleotide-Independent Manner. Biophys. J. 2019, 116, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, G.B.; Kopra, K.; Eskonen, V.; Härmä, H.; Abankwa, D. High-throughput amenable fluorescence-assays to screen for calmodulin-inhibitors. Anal. Biochem. 2019, 572, 25–32. [Google Scholar] [CrossRef]
- Berchtold, M.W.; Villalobo, A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2014, 1843, 398–435. [Google Scholar] [CrossRef]
- Faust, F. M., Slisz, M., andJarrett, H. W. (1987) Calmodulin is labeled at lysine 148 by a chemically reactive phenothiazine The Journal of biological chemistry 262, 1938-1941 papers3://publication/uuid/4C146F6A-D207-4E2B-8F14-4E39F6CE9EC0.
- Bhattacharya, S.; Bunick, C.G.; Chazin, W.J. Target selectivity in EF-hand calcium binding proteins. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2004, 1742, 69–79. [Google Scholar] [CrossRef]
- Yang, A.; Miron, S.; Mouawad, L.; Duchambon, P.; Blouquit, Y.; Craescu, C.T. Flexibility and Plasticity of Human Centrin 2 Binding to the Xeroderma Pigmentosum Group C Protein (XPC) from Nuclear Excision Repair, Biochemistry 2006, 45, 3653–3663. [Google Scholar] [CrossRef]
- Friedberg, F. Centrin isoforms in mammals. Relation to calmodulin. Mol. Biol. Rep. 2006, 33, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Dantas, T.J.; Daly, O.M.; Morrison, C.G. Such small hands: the roles of centrins/caltractins in the centriole and in genome maintenance. Cell. Mol. Life Sci. 2012, 69, 2979–2997. [Google Scholar] [CrossRef] [PubMed]
- Puumalainen, M.-R.; Rüthemann, P.; Min, J.-H.; Naegeli, H. Xeroderma pigmentosum group C sensor: unprecedented recognition strategy and tight spatiotemporal regulation. Cell. Mol. Life Sci. 2015, 73, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.M.; Grecu, D.; Assairi, L. Ca2+ signaling and Target Binding Regulations: Calmodulin and Centrin In Vitro and In Vivo. Bioenerg. Open Access 2016, 5, 1–6. [Google Scholar] [CrossRef]
- Klein, U.R.; Nigg, E.A. SUMO-dependent regulation of centrin-2. J. Cell Sci. 2009, 122, 3312–3321. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, A.; Moudjou, M.; Paintrand, M.; Salisbury, J.L.; Bornens, M. Most of centrin in animal cells is not centrosome-associated and centrosomal centrin is confined to the distal lumen of centrioles. J. Cell Sci. 1996, 109, 3089–3102. [Google Scholar] [CrossRef] [PubMed]
- Hodges, M.E.; Scheumann, N.; Wickstead, B.; Langdale, J.A.; Gull, K. Reconstructing the evolutionary history of the centriole from protein components. J. Cell Sci. 2010, 123, 1407–1413. [Google Scholar] [CrossRef]
- Graser, S.; Stierhof, Y.-D.; Lavoie, S.B.; Gassner, O.S.; Lamla, S.; Le Clech, M.; Nigg, E.A. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007, 179, 321–330. [Google Scholar] [CrossRef]
- Liyanage, M.R.; Zaidi, A.; Johnson, C.K. Fluorescence polarization assay for calmodulin binding to plasma membrane Ca2+–ATPase: Dependence on enzyme and Ca2+ concentrations. Anal. Biochem. 2009, 385, 1–6. [Google Scholar] [CrossRef]
- Waxham, M.N.; Tsai, A.-L.; Putkey, J.A. A Mechanism for Calmodulin (CaM) Trapping by CaM-kinase II Defined by a Family of CaM-binding Peptides. J. Biol. Chem. 1998, 273, 17579–17584. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, X.; Yang, B. Calcium-induced human centrin 1 self-assembly and double-regulating the binding with peptide R18-Sfi1p. Int. J. Biol. Macromol. 2019, 128, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, K.D.G.; A Eidne, K. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 2006, 3, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Thurnher, M.; Gruenbacher, G.; Nussbaumer, O. Regulation of mevalonate metabolism in cancer and immune cells. Biochim. et Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 2013, 1831, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Parkkola, H., Siddiqui, F. A., Oetken-Lindholm, C., andAbankwa, D. (2021) FLIM-FRET Analysis of Ras Nanoclustering and Membrane-Anchorage Methods Mol Biol 2262, 233-250. [CrossRef]
- Siddiqui, F.A.; Alam, C.; Rosenqvist, P.; Ora, M.; Sabt, A.; Manoharan, G.B.; Bindu, L.; Okutachi, S.; Catillon, M.; Taylor, T.; et al. PDE6D Inhibitors with a New Design Principle Selectively Block K-Ras Activity. ACS Omega 2019, 5, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Köhnke, M.; Schmitt, S.; Ariotti, N.; Piggott, A.M.; Parton, R.G.; Lacey, E.; Capon, R.J.; Alexandrov, K.; Abankwa, D. Design and Application of In Vivo FRET Biosensors to Identify Protein Prenylation and Nanoclustering Inhibitors. Chem. Biol. 2012, 19, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Minerva Anestesiol. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Vlachou, T.; Carminati, M.; Pelicci, P.G.; Mapelli, M. Molecular mechanisms of asymmetric divisions in mammary stem cells. EMBO Rep. 2016, 17, 1700–1720. [Google Scholar] [CrossRef]
- Cicalese, A.; Bonizzi, G.; Pasi, C.E.; Faretta, M.; Ronzoni, S.; Giulini, B.; Brisken, C.; Minucci, S.; Di Fiore, P.P.; Pelicci, P.G. The Tumor Suppressor p53 Regulates Polarity of Self-Renewing Divisions in Mammary Stem Cells. Cell 2009, 138, 1083–1095. [Google Scholar] [CrossRef]
- Chen, C.; Yamashita, Y.M. Centrosome-centric view of asymmetric stem cell division. Open Biol. 2021, 11, 200314. [Google Scholar] [CrossRef]
- Nussinov, R.; Zhang, M.; Tsai, C.-J.; Jang, H. Calmodulin and IQGAP1 activation of PI3Kα and Akt in KRAS, HRAS and NRAS-driven cancers. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2018, 1864, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Halling, D.B.; Liebeskind, B.J.; Hall, A.W.; Aldrich, R.W. Conserved properties of individual Ca 2+ -binding sites in calmodulin. Proc. Natl. Acad. Sci. USA 2016, 113, 201600385–25. [Google Scholar] [CrossRef] [PubMed]
- Wall, V. E., Garvey, L. A., Mehalko, J. L., Procter, L. V., andEsposito, D. (2014) Combinatorial assembly of clone libraries using site-specific recombination Methods Mol Biol 1116, 193-208. [CrossRef]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.H.; et al. Small molecule inhibition of the KRAS–PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef]
- Vaasa, A.; Viil, I.; Enkvist, E.; Viht, K.; Raidaru, G.; Lavogina, D.; Uri, A. High-affinity bisubstrate probe for fluorescence anisotropy binding/displacement assays with protein kinases PKA and ROCK. Anal. Biochem. 2009, 385, 85–93. [Google Scholar] [CrossRef]
- Sinijarv, H.; Wu, S.; Ivan, T.; Laasfeld, T.; Viht, K.; Uri, A. Binding assay for characterization of protein kinase inhibitors possessing sub-picomolar to sub-millimolar affinity. Anal. Biochem. 2017, 531, 67–77. [Google Scholar] [CrossRef]
- Manoharan, G.B.; Okutachi, S.; Abankwa, D. Potential of phenothiazines to synergistically block calmodulin and reactivate PP2A in cancer cells. PLOS ONE 2022, 17, e0268635. [Google Scholar] [CrossRef]
Figure 1.
Despite its high similarity to CaM, centrin1 does not recognize CaM-target peptides. (A) Multiple sequence alignment of human CaM (CALM1-3) and centrin (CETN1-3) protein paralogs, designated by the encoding gene names. The Ca2+-binding residues are highlighted in cyan. Lysines 75, 77 and 148 of CaM that become covalently modified by CaM inhibitor ophiobolin A, are highlighted in yellow. The same highlight was used for lysine residues at similar positions in centrin1 and centrin2, while no such lysine residues could be identified for centrin3. Note that the CaM protein numbering is started from Ala, as the N-terminal, native Met is removed in most organisms [52]. (B) Structures of human CaM (PDB ID 1CLL) and human centrin1 (PDB ID 2GGM). Calcium ions are marked as green spheres. (C) Analysis of the number of paralog coding genes of CALM1-3 and CETN1-3 in different species. Data were curated from the NCBI protein database. (D-F) Binding of 10 nM fluorescein labelled F-CaMKII and F-PMCA (D, E) or 100 nM F-Sfi1 (F) peptides to His-tagged human CaM or centrin1 was detected using fluorescence anisotropy measurements.
Figure 1.
Despite its high similarity to CaM, centrin1 does not recognize CaM-target peptides. (A) Multiple sequence alignment of human CaM (CALM1-3) and centrin (CETN1-3) protein paralogs, designated by the encoding gene names. The Ca2+-binding residues are highlighted in cyan. Lysines 75, 77 and 148 of CaM that become covalently modified by CaM inhibitor ophiobolin A, are highlighted in yellow. The same highlight was used for lysine residues at similar positions in centrin1 and centrin2, while no such lysine residues could be identified for centrin3. Note that the CaM protein numbering is started from Ala, as the N-terminal, native Met is removed in most organisms [52]. (B) Structures of human CaM (PDB ID 1CLL) and human centrin1 (PDB ID 2GGM). Calcium ions are marked as green spheres. (C) Analysis of the number of paralog coding genes of CALM1-3 and CETN1-3 in different species. Data were curated from the NCBI protein database. (D-F) Binding of 10 nM fluorescein labelled F-CaMKII and F-PMCA (D, E) or 100 nM F-Sfi1 (F) peptides to His-tagged human CaM or centrin1 was detected using fluorescence anisotropy measurements.
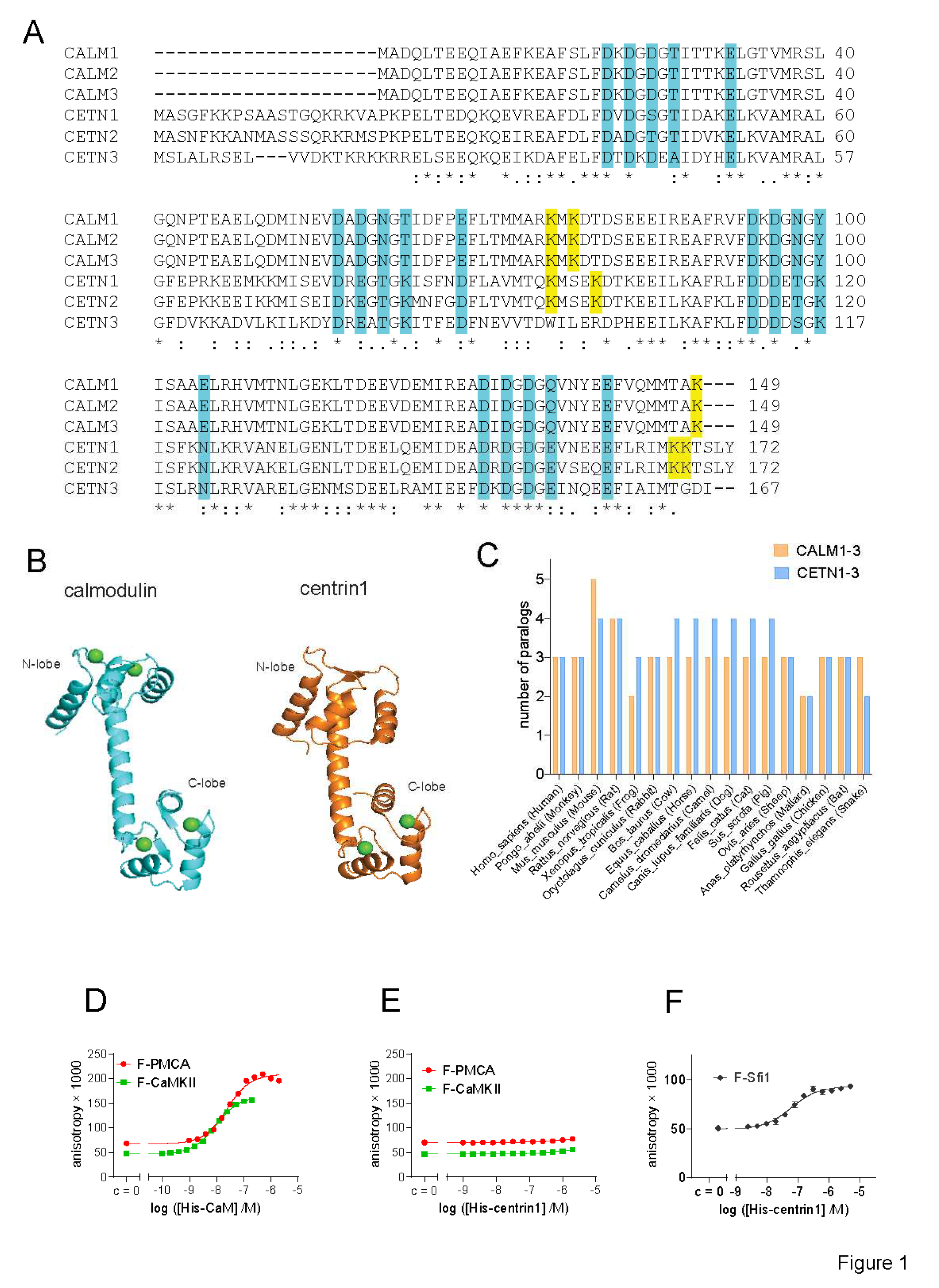
Figure 2.
The interaction of CaM or centrin is increased with active K-Ras. (A) Co-immunoprecipitation of Rluc8-CaM or Rluc8-centrin1 with GFP2-K-RasG12V or GFP2-K-Ras wt. Pull-down was performed using lysates of HEK293-ebna cells transfected with combinations of GFP2-K-RasG12V/ Rluc8-CaM, GFP2-K-RasG12V/ Rluc8-centrin1, GFP2-K-Ras/ Rluc8-CaM, GFP2-K-Ras/ Rluc8-centrin1 and GFP2/ Rluc8 and expressed for 48 h. The GFP2-tagged protein was bound using GFP-beads and the samples were analysed using anti-Rluc8 and anti-GFP antibodies. (B) Immunoprecipitated Rluc8-tagged protein signals were normalized to GFP-tagged protein signals. The signal intensity of the GFP2-K-RasG12V/Rluc8-CaM transfected sample was set to 1 in each experiment and was used to normalize the other samples. The plot shows mean ± SEM and the statistical analysis was performed using one-way ANOVA test. (C, D) Interaction of Rluc8-K-Ras wt, Rluc8-K-RasG12V, and Rluc8-K-RasG12V-D38A with GFP2-CaM (C) or GFP2-centrin1 (D). All samples were treated with 0.2 % v/v DMSO for 24 h, n = 3. Statistics of BRETtop values was analysed using the F-test. BRET donor protein is boxed purple, acceptor protein is boxed green.
Figure 2.
The interaction of CaM or centrin is increased with active K-Ras. (A) Co-immunoprecipitation of Rluc8-CaM or Rluc8-centrin1 with GFP2-K-RasG12V or GFP2-K-Ras wt. Pull-down was performed using lysates of HEK293-ebna cells transfected with combinations of GFP2-K-RasG12V/ Rluc8-CaM, GFP2-K-RasG12V/ Rluc8-centrin1, GFP2-K-Ras/ Rluc8-CaM, GFP2-K-Ras/ Rluc8-centrin1 and GFP2/ Rluc8 and expressed for 48 h. The GFP2-tagged protein was bound using GFP-beads and the samples were analysed using anti-Rluc8 and anti-GFP antibodies. (B) Immunoprecipitated Rluc8-tagged protein signals were normalized to GFP-tagged protein signals. The signal intensity of the GFP2-K-RasG12V/Rluc8-CaM transfected sample was set to 1 in each experiment and was used to normalize the other samples. The plot shows mean ± SEM and the statistical analysis was performed using one-way ANOVA test. (C, D) Interaction of Rluc8-K-Ras wt, Rluc8-K-RasG12V, and Rluc8-K-RasG12V-D38A with GFP2-CaM (C) or GFP2-centrin1 (D). All samples were treated with 0.2 % v/v DMSO for 24 h, n = 3. Statistics of BRETtop values was analysed using the F-test. BRET donor protein is boxed purple, acceptor protein is boxed green.
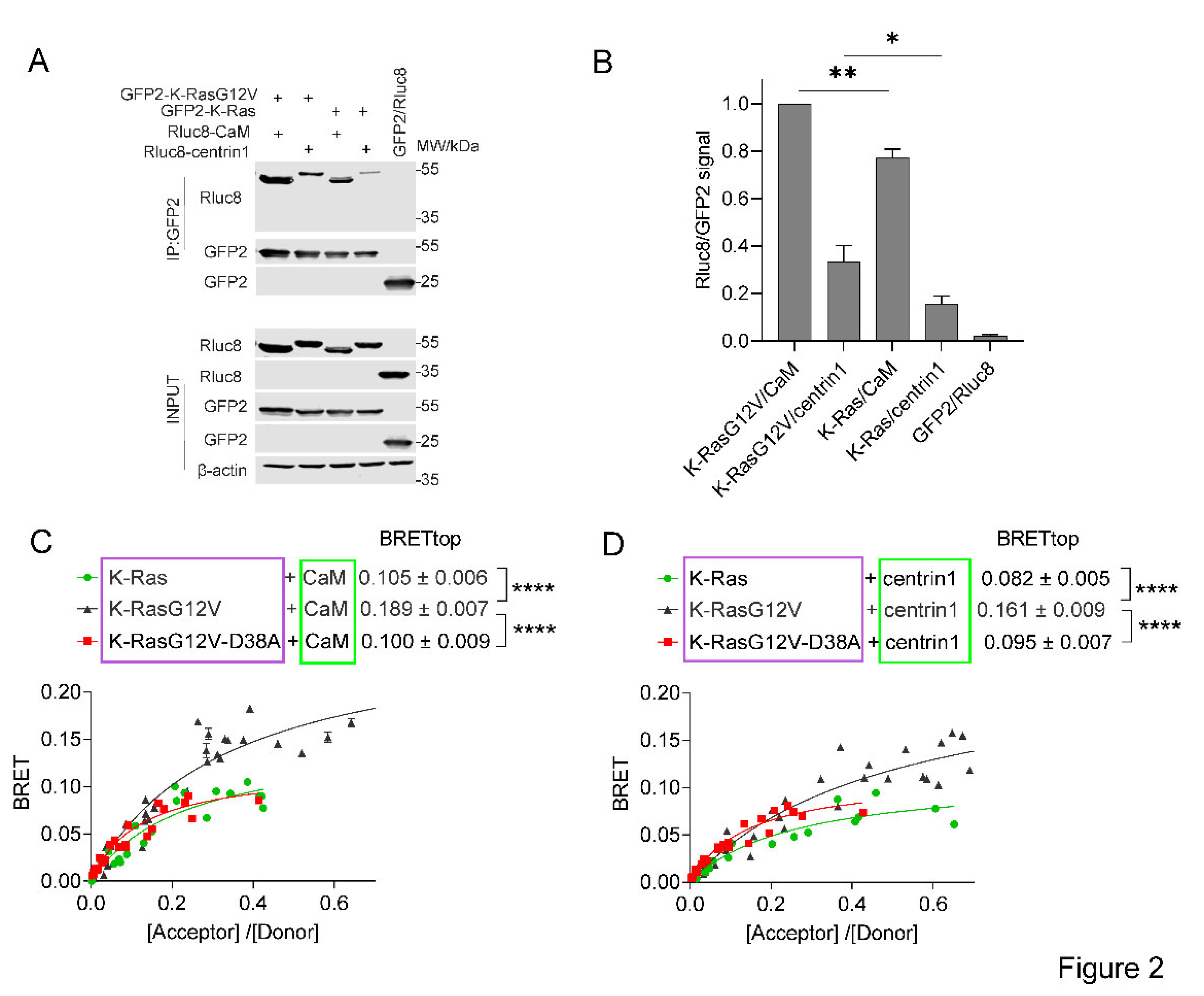
Figure 3.
The interaction of K-Ras with centrin1 is modulated by direct binding of CaM-inhibitors to centrin1. (A) HEK293-ebna cells were transfected with Rluc8-K-RasG12V/ GFP2-centrin1 BRET sensor plasmids for 24 h followed by treatment with ophiobolin A (2.5 µM), calmidazolium (10 µM), calmirsone1 (20 µM) or equal volume of DMSO (0.2% v/v) for another 24 h, n = 3. (B) HEK293-ebna cells were transfected with BRET sensor plasmids Rluc8-K-RasG12V/ GFP2-centrin1 at a ratio of 1/19, respectively, for 24 h followed by a 24 h treatment with 2-fold dilution series of calmidazolium or calmirasone1 ranging from 80 µM to 0.1 µM. Data represent mean ± SEM, n = 2. BRET donor protein is boxed purple, acceptor protein is boxed green. (C) Displacement of fluorescent F-Sfi1 from centrin1 by CaM-inhibitors. The inhibitors were 3-fold diluted in assay buffer, followed by addition of the complex of 100 nM F-Sfi1 and 250 nM His-centrin1. The fluorescence anisotropy was measured after overnight incubation at RT.
Figure 3.
The interaction of K-Ras with centrin1 is modulated by direct binding of CaM-inhibitors to centrin1. (A) HEK293-ebna cells were transfected with Rluc8-K-RasG12V/ GFP2-centrin1 BRET sensor plasmids for 24 h followed by treatment with ophiobolin A (2.5 µM), calmidazolium (10 µM), calmirsone1 (20 µM) or equal volume of DMSO (0.2% v/v) for another 24 h, n = 3. (B) HEK293-ebna cells were transfected with BRET sensor plasmids Rluc8-K-RasG12V/ GFP2-centrin1 at a ratio of 1/19, respectively, for 24 h followed by a 24 h treatment with 2-fold dilution series of calmidazolium or calmirasone1 ranging from 80 µM to 0.1 µM. Data represent mean ± SEM, n = 2. BRET donor protein is boxed purple, acceptor protein is boxed green. (C) Displacement of fluorescent F-Sfi1 from centrin1 by CaM-inhibitors. The inhibitors were 3-fold diluted in assay buffer, followed by addition of the complex of 100 nM F-Sfi1 and 250 nM His-centrin1. The fluorescence anisotropy was measured after overnight incubation at RT.

Figure 4.
Inhibition of prenylation by mevastatin does not disrupt BRET between K-Ras and CaM or centrin1. (A-D) BRET-sensors Rluc8-K-Ras/ GFP2-CaM (A), Rluc8-K-Ras/ GFP2-centrin1 (B), Rluc8-CTK/ GFP2-CaM (C) and Rluc8-CTK/ GFP2-centrin1 (D) were transfected into HEK293-ebna cells, and cells were treated with 10 µM mevastatin or the vehicle control, DMSO 0.2% v/v for 24 h, n = 3. Statistics of BRETtop values was analysed using the F-test. BRET donor protein is boxed purple, acceptor protein is boxed green.
Figure 4.
Inhibition of prenylation by mevastatin does not disrupt BRET between K-Ras and CaM or centrin1. (A-D) BRET-sensors Rluc8-K-Ras/ GFP2-CaM (A), Rluc8-K-Ras/ GFP2-centrin1 (B), Rluc8-CTK/ GFP2-CaM (C) and Rluc8-CTK/ GFP2-centrin1 (D) were transfected into HEK293-ebna cells, and cells were treated with 10 µM mevastatin or the vehicle control, DMSO 0.2% v/v for 24 h, n = 3. Statistics of BRETtop values was analysed using the F-test. BRET donor protein is boxed purple, acceptor protein is boxed green.
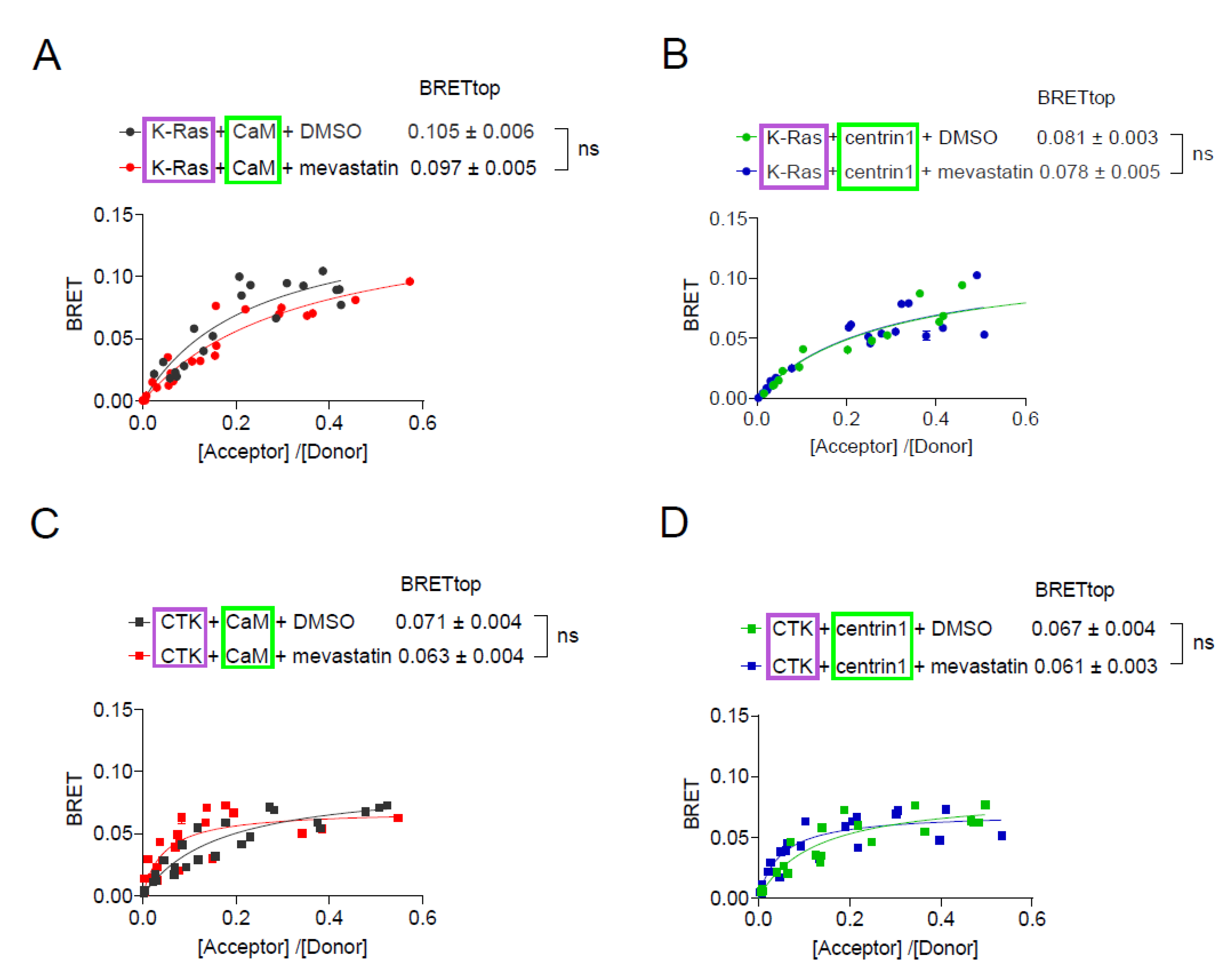
Figure 5.
K-Ras membrane anchorage is selectively affected by CaM-, but less by centrin1-depletion. (A, B) Rluc8-K-Ras was transfected with GFP2-CaM (A) or GFP2-centrin1 (B) plasmids at a donor/acceptor plasmid ratio of 1/5 into HEK293-ebna cells. BRET donor protein is boxed purple, acceptor protein is boxed green. Data represent mean ± SEM, n = 2 to 4. Statistical significance between negative control siRNA and sample siRNA was analysed using Mann-Whitney test. (C, D) HEK293-ebna cells were transfected with 100 nM of negative control siRNA or siFNTA or siCETN1 for 48 h and cell lysates were immunoblotted as indicated. (E, F) HEK293-ebna cells were transfected with 100 nM siRNA for 24 h, followed by BRET sensor transfection. Rluc8-/ GFP2-tagged K-RasG12V (E) or H-RasG12V (F) nanoclustering-BRET sensor plasmids were transfected at a donor/acceptor plasmid ratio of 1/15. BRET donor protein is boxed purple, acceptor protein is boxed green. Data represent mean ± SEM, n = 4. Statistical significance between negative control siRNA and sample siRNA was analysed using Mann-Whitney test.
Figure 5.
K-Ras membrane anchorage is selectively affected by CaM-, but less by centrin1-depletion. (A, B) Rluc8-K-Ras was transfected with GFP2-CaM (A) or GFP2-centrin1 (B) plasmids at a donor/acceptor plasmid ratio of 1/5 into HEK293-ebna cells. BRET donor protein is boxed purple, acceptor protein is boxed green. Data represent mean ± SEM, n = 2 to 4. Statistical significance between negative control siRNA and sample siRNA was analysed using Mann-Whitney test. (C, D) HEK293-ebna cells were transfected with 100 nM of negative control siRNA or siFNTA or siCETN1 for 48 h and cell lysates were immunoblotted as indicated. (E, F) HEK293-ebna cells were transfected with 100 nM siRNA for 24 h, followed by BRET sensor transfection. Rluc8-/ GFP2-tagged K-RasG12V (E) or H-RasG12V (F) nanoclustering-BRET sensor plasmids were transfected at a donor/acceptor plasmid ratio of 1/15. BRET donor protein is boxed purple, acceptor protein is boxed green. Data represent mean ± SEM, n = 4. Statistical significance between negative control siRNA and sample siRNA was analysed using Mann-Whitney test.
Figure 6.
CaM and centrin1 distribute to the centrosomes and their loss more potently affects spheroid formation of KRAS mutant MDA-MB-231 cells. (A, B) Effect of knockdown of CALM1 and CETN1 on spheroids derived from MDA-MB-231 (A) and MCF-7 (B) cells. The knockdown efficiency was compared to spheroids grown from negative control siRNA transfected cells. Data represent mean ± SEM of 4 biological repeats. Statistical analysis was performed using Mann-Whitney test. (C) Representative images of HeLa cells that were co-transfected with mGFP-K-RasG12V (green) and mCherry-CaM (red). Endogenous centrin1 was immunostained (purple) and DNA was stained using DAPI (blue). Cell-cycle stages are indicated on the left. Scale bar is 5 µm.
Figure 6.
CaM and centrin1 distribute to the centrosomes and their loss more potently affects spheroid formation of KRAS mutant MDA-MB-231 cells. (A, B) Effect of knockdown of CALM1 and CETN1 on spheroids derived from MDA-MB-231 (A) and MCF-7 (B) cells. The knockdown efficiency was compared to spheroids grown from negative control siRNA transfected cells. Data represent mean ± SEM of 4 biological repeats. Statistical analysis was performed using Mann-Whitney test. (C) Representative images of HeLa cells that were co-transfected with mGFP-K-RasG12V (green) and mCherry-CaM (red). Endogenous centrin1 was immunostained (purple) and DNA was stained using DAPI (blue). Cell-cycle stages are indicated on the left. Scale bar is 5 µm.
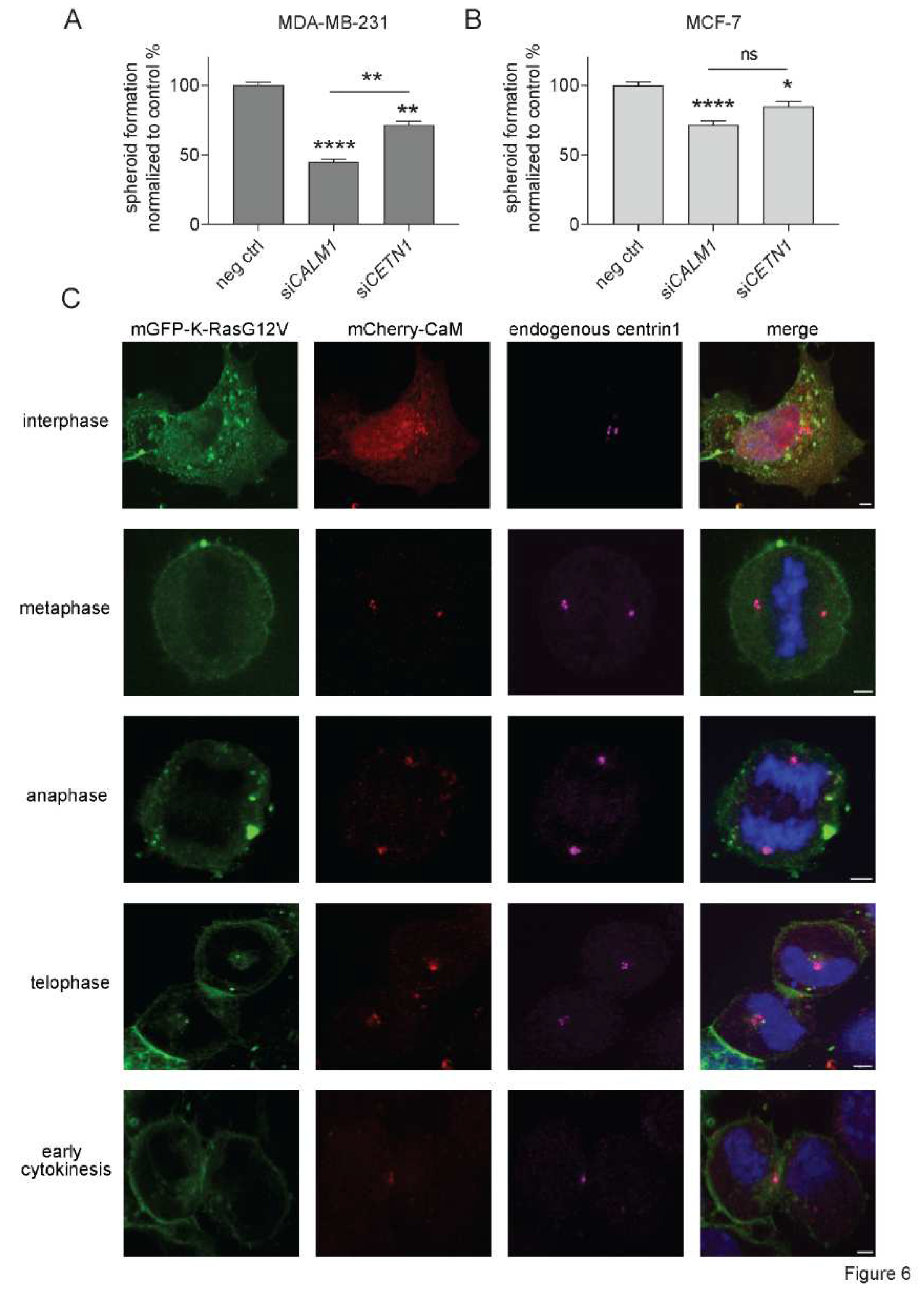

Table 1.
Comparison of Kd values of CaM-inhibitors with centrin1 and CaM determined by fluorescence anisotropy measurements. The competition assay derived Kd values of inhibitors to CaM were previously reported by us using F-PMCA peptide as probe.
Table 1.
Comparison of Kd values of CaM-inhibitors with centrin1 and CaM determined by fluorescence anisotropy measurements. The competition assay derived Kd values of inhibitors to CaM were previously reported by us using F-PMCA peptide as probe.
| inhibitor | centrin1 | CaM |
| Kd (mean ± SEM, n = 2) | Kd (references) | |
| calmidazolium | 1.6 ± 0.3 µM | 13.5 nM [26] |
| W-7 | 18.2 ± 0.3 µM | 1.47 µM[26] |
| ophiobolin A | 49 ± 9 µM | 3.5 µM [9] |
| calmirasone1 | 0.9 ± 0.2 µM | 0.87 µM [9] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



