Submitted:
07 May 2023
Posted:
08 May 2023
You are already at the latest version
Abstract
Energy is needed by cancer cells to stay alive and communicate with their surroundings. The primary organelles for cellular metabolism and energy synthesis are mitochondria. Researchers recently proved that cancer cells can steal immune cells' mitochondria using nanoscale tubes. This finding demonstrates the dependence of cancer cells on normal cells for their living and function. It also denotes the importance of mitochondria in cancer cells’ biology. Emerging evidence has demonstrated how mitochondria are essential for cancer cells to survive in the harsh tumor microenvironment, evade the immune system, obtain more aggressive features, and resist treatments. For instance, functional mitochondria can improve cancer resistance against radiotherapy by scavenging the released reactive oxygen species. Therefore, targeting mitochondria can potentially enhance oncological outcomes, according to this notion. The patients' reactions to radiation are varied, ranging from a complete response to even cancer progression during treatment. This concept illustrates how different levels of mitochondrial metabolism might contribute to this heterogeneity. Considering this notion can help to improve personalized oncological treatments. This article outlines the importance of mitochondrial metabolism in cancer biology and personalized treatments.
Keywords:
Mitochondria
; Personalized Oncology
; cancer stem cell
; T Cell
1. Introduction
Cancer is a heterogeneous illness made up of various biological entities that require various therapies. Due to this problem, the world is moving away from one-size-fits-all cancer treatment regimens toward ones that are risk-adapted [1]. Recent researches aim to identify the predictive factors influencing outcomes to personalized therapies and enhance quality of life while preserving efficacy. Predictive indicators for therapy response and toxicity are as important to illness prognostic factors.
It has been demonstrated that normal cells are necessary for the survival and functionality of cancer cells. Saha et al noteworthy demonstrated that cancer cells can steal mitochondria from immune cells (CD8+ T cells and natural killer [NK] cells) via nanoscale tube-like structures[2]. In addition to supplying energy, mitochondria are crucial organelles for the survival and development of cancer cells. Furthermore, mitochondria play a crucial role in the biology of cancer stem cells (CSCs), supporting their chemo- and radioresistance [3].
Here we give a thorough understanding of the crucial function of mitochondria in cancer metabolism and how it can serve as a basis to improve personalized treatments, with a special stress on radiotherapy. The radiotherapy schedule is based on the total dose, per fraction dose, overall treatment time, the interval between fractions, and dose rate. Personalized Radiotherapy aims to modulate the RT schedule to improve the therapeutic ratio. More than half of cancer patients receive radiation therapy (RT), which is a crucial component of their regimen [4]. Delivering the highest dose to the target while protecting normal tissues as much as possible is the fundamental tenet of RT. Most of the RT recommendations in use are based on population averages. This mindset has two problems: tumors vary in terms of their genetic and epigenetic signature, and people with tumors vary in terms of their racial, ethnic, and genetic features [5]. Enhancing evidence has demonstrated the effects of patient variables, including as age, gender, ethnicity, comorbidities, and even biological and lifestyle factors, on radiation response and toxicities [6]. This strategy has become a discipline in Oncology called Personalized Cancer Treatment including Radiotherapy. The varied function of mitochondria in cancer metabolism is discussed in the following section, along with how essential healthy mitochondria are for the survival and development of cancer.
2. The Pivotal Role of Mitochondria in Cancer Cells’ Metabolism
Influencing cancer cells' survival in the tumor microenvironment, immune evasion, progression, and resistance to treatment are a few areas of mitochondria's advantages for cancer cells (Figure 1). We previously demonstrated how mitochondria play a crucial role in cancer development, immune evasion, survival, and therapeutic resistance [7]. The benefits of mitochondria for cancer cells are categorized as follows:
(A) Surviving in the tumor microenvironment's (TME): via promoting/meditating glycolysis, reactive oxygen species (ROS) clearance, cell cycle arrest, maintain pH homeostasis, autophagy, mitochondrial hijacking, and angiogenesis.
(B) Immune evasion via facilitating TME acidification, glucose influx, PD-1 upregulation on T cells (by mitochondrial hijacking) [8], recruiting myeloid-derived suppressor cells (MDSCs), PD-L1 overexpression on cancer cells, MHC-1 downregulation, and the secretion of immunosuppressants, mitochondria help cancer cells avoid being recognized by immune cells. Additionally, T cells' mitochondrial hijacking depletes their energy and prevents long-term cancer-fighting action.
(C) Aggressiveness: mitochondria are crucial for cancer progression via mediating genomic instability, quiescence evasion, and epithelial-to-mesenchymal transition (EMT). Production of reactive oxygen species (ROS) mediates these activities.
(D) Treatment resistance: mitochondria can protect cancer cells from chemotherapy and RT by eliminating the released ROS. Additionally, they increase chemotherapy resistance by encouraging the function of efflux pumps (by providing ATP) and inducing cell cycle arrest. Additionally, mitochondrial hijacking from T cells impairs the long-term effects of anti-PD-1 treatment.
3. Mitochondria Individualized Role in Cancer Metastasis
Metastasis happens in a very diverse and individualized pattern. The players in the molecular pathway of metastasis and the therapeutic response to metastasis should also be considered in a personalized and idealized context. EMT is the prerequisite for metastasis by inhibiting cell adhesion and promoting local migration, vascular invasion, and resistance to apoptotic stimuli [9]. EMT and cancer-cell stemness are correlated phenomena regulated by common mediators, including HIFs, SNAIL, and SLUG/SOX9 [10,11]. Mitochondrial ROS (mtROS) can promote EMT through different pathways, such as MAPK, PI3K/Akt/mTOR, and VEGFA–SOX2–SNAI2 pathways [11-13].In addition, mitochondria are involved in cancer proliferation, invasion, and metastasis by providing the crosslink between β1 integrin and the extracellular matrix is [14]. This process is mediated by lysyl oxidase (LOX), which is enhanced by mitochondria via stabilizing and securing HIF-1α function [15,16]. Therefore, targeted anti-mitochondrial therapy has the potential to disrupt EMT and metastasis. Precisional targeting of cancer-specific mitochondria can reduce their ability to de-differentiate, proliferate, and metastasize, and help to improve the treatment results and overall prognosis.
4. Targeting Mitochondria: A Practical Strategy for Personalized Cancer Treatment
As a result of developments in medical genetics and molecular biology, the function of mitochondria in several cellular functions, including apoptosis, redox balance, macromolecule production, and calcium homeostasis, has been demonstrated [17,18]. In contrast to the ancient Warburg theory, the mitochondria of cancer cells are functional, supporting their survival and function[7]. As noted earlier, mitochondria can contribute the development, progression, and metastasis of cancer. In addition, it has a crucial role in treatment resistance. As noted in section 2, functional mitochondria can help cancer cells to overcome chemotherapy effects by scavenging released ROS and activating multidrug resistance pumps [7]. Also, they can improve the resistance against immunotherapy, by inhibiting the immune cells entry to the TME by depleting the glucose content of TME, acidifying the TME, and mediating the mitochondria hijacking from immune cells[7,19]. Next, we outline how mitochondria can improve the cancer cells resistance against radiotherapy. In a recent study, Taghizadeh-Hesary et al. demonstrated that mitochondria have a contributing role in tumor response to radiotherapy. They demonstrated that mitochondria are involved in so-called 6Rs of radiobiology [20](Figure 2). The details of this link were presented as follows:
(a) Repair: DNA damage is the primary cause of RT's cytotoxic effects. Cancer cells with improved DNA repair mechanisms can counteract this effect. Mitochondrial can support ATP-dependent proteins responsible for DNA integrity-related, including PARP-1 [21], XRCC1 [22], ATM[23], and DNA ligases [24], by providing enough ATP molecules..
(b) Repopulation: Mitochondria can support cancer cells proliferation by supplying the building materials, including nucleic acids, amino acids, and lipids through stabilizing HIF-1 and metabolic switching to glycolysis.[25] .
(c) Reoxygenation: HIF-1 can mediate tissue reoxygenation by promoting the expression of different angiogenic factors and shielding endothelial cells from radiation effects [26]. HIF-1 needs functional mitochondria to function properly [16]. Consequently, healthy mitochondria can aid in the reoxygenation of tumor tissue.
(d) Redistribution: Cyclin-Cdk complexes carefully control the cancer cells' cell cycle [27]. The radiosensitive phases of cell cycle are G2 and M and the radioresistant phases are G1 and S. [28]. Cell cycle progression depends on dynamic responses of mitochondria in order that during the G1 and S phases, mitochondria fuse to form a hyperactive network; after that, they undergo fission to ensure proper partitioning between the two daughter cells [27]. In addition, functional mitochondria can help the cell cycle progression by supplying enough energy [29].
e) Reactivation: Emerging evidence has demonstrated that cancer cells can evade the activated immune cells using immune inhibitory molecules, such as programmed death-ligand 1 (PD-L1) [30]. In the hypoxic TME, HIF-1α increases PD-L1 expression on cancer cells [31]. Functional mitochondria can facilitate this process by stabilizing HIF-1α [16]. On the other hand, Akbari et al. found that programmed death protein-1 (PD-1) expression of T cells is inversely related to their mitochondrial capacity; so that a decrease in T cells’ mitochondrial capacity can give rise to PD-1 overexpression on T cells and their inactivation [8]. A recent study by Saha et al. demonstrated that cancer cells can hijack mitochondria from T cells via nanotubes [2]. With this strategy, cancer cells can attain both goals (increasing PD-L1 and PD-1 expression on cancer cells and T cells, respectively), thereby enhancing the immunoescape.
f) Radiosensitivity: Functional mitochondria can reduce the radiosensitivity of cancer cells by scavenging the released ROS and mediating the removal of damaged mitochondria, a process called mitophagy. Hitherto, numerous biological factors have been linked to the intrinsic radiosensitivity of cancer cells, including p53, transforming growth factor beta (TGF-β), and isocitrate dehydrogenase 1 (IDH1) among others. For instance, p53 can improve radioresistance by enhancing the mitochondrial DNA integrity and PGC-1α (peroxisome proliferator-activated receptor γ coactivator-1α) overexpression. For the detailed mechanisms of other corresponding factors, the readers are referred to the study by Taghizadeh-Hesary et al [20]. (Table 1)
This section illustrated how functional mitochondria can improve the tumor resistance against the various treatments. Therefore, inhibiting the cancer cells’ mitochondria can potentially improve the treatment results.
5. Enhancing the Normal Cells’ Mitochondria Reduces the Radiotherapy Toxicity
Normal tissue response to irradiation is primarily determined by Repair, Repopulation, and Radiosensitivity, among others [78]. As alluded to above, functional mitochondria are essential for DNA repair, cellular proliferation, and scavenging radiation-induced ROSs. Radiation-induced inflammation is an important phase in the development of normal tissue toxicity, happening upon ROS release from damaged cells [79]. Functional mitochondrial can alleviate the resultant inflammation (and thus the ultimate tissue damage) by scavenging ROSs [80]. With this information in mind, enhanced mitochondrial content of normal cells can reduce radiation-induced toxicities. In the following, it is outlined how mitochondrial metabolism is linked to the recognized predictive factors of radiosensitivity of normal tissue.
Recruiting genotypic and proteomic data of patients with breast or head and neck cancer, a series of proteins are recognized as determinant for normal tissue toxicity to radiation; including CHIT1, PDGFB, STIM1, and THPO proteins as improving radiosensitivity, and SERPINC1 and SLC4A as enhancing radioresistance [81]. Mitochondrial metabolism interprets the mechanism of action of STIM1, SERPINC1, and SLC4A. STIM1 (stromal interaction molecule 1) regulates intracellular calcium level [82] and downregulates mitochondrial metabolism as its knock-out leads to more metabolically active mitochondria [83]. STIM1 exacerbates radiation toxicity by preventing mitochondrial function from neutralizing the radiation-induced ROSs. Apoptosis and mitochondrial dysfunction are instead encouraged by SERPINC1 knockout because it activates the Bax pathway [84]. In the mitochondrial anti-oxidative system, SLC4 (solute carrier 4) scavenges ROS to improve radioresistance [85]. Hence, SERPINC1 and SLC4 may enhance radioresistance by enhancing mitochondrial metabolism and their capacity to scavenge ROS molecules.
TGF-β overexpression increases the susceptibility of radiation-induced pulmonary fibrosis [86] and its activation affects mitochondrial respiration via impairing the mitochondrial complex IV in lung epithelial cells [87].
The JAK/STAT signaling in human cells has been considered as a radioprotective factor. STAT3 activation improves the radioresistance of normal cells by increasing the generation of NADPH (for redox homeostasis) and ATP (for DNA stability); hence, enhances the mitochondrial electron transport chain in normal cells [88].
Radiation toxicities are more likely to affect older people. Higher ROS production and decreased antioxidant capability in older people have been blamed for this impact [89]. As people get older, there is mounting evidence that their ability to produce ATP and NADPH is reduced because of an accumulation of mtDNA mutations and ROS damage to the mitochondrial substructures [90]. The cellular redox processes (such as glutathione) and the ATP-dependent enzymes responsible for repairing DNA damage are each impaired, necessitating NAPDH to function [91]. As a result, its relationship with radiation damage may be influenced by aging's impact on mitochondrial metabolism.
Several mechanisms have been proposed to explain how smoking during RT may increase the frequency and severity of radiation-induced acute and delayed toxicities [92]. Through endothelial damage and coagulation, it impairs tissue repair and triggers an inflammatory cascade, which increases the rate and severity of acute radiation toxicities and causes late toxicities [93]. Both acute and late radiation toxicities from tobacco smoke affect mitochondria negatively. Smoke exposure alters the mitochondrial membrane potential, which causes the release of ROS from the mitochondria and ultimately results in cellular death DAMPs are then released into the extracellular matrix, where they connect to toll-like receptors (TLRs) on tissue macrophages and trigger the NF-kB pathway. Inflammatory cytokines are released as a result, which damages healthy tissue and exacerbates acute radiation-induced inflammation [93]. The main cause of delayed radiation toxicities, which manifest at least three months after RT, is the replacement of normal tissues by fibrotic tissues with inadequate blood flow [94]. In order for tissue regeneration and angiogenesis to be mediated by wound macrophages—the key players in wound healing—proper mitochondrial function is a crucial precondition and determining factor in the early stages of wound repair [95]. Therefore, increased radiation toxicity in smokers is justified by mitochondrial damage.
Alcohol intake can also enhance the incidence and severity of tissue fibrosis after radiation exposure, which can aggravate radiation-induced toxicities [96]. In order for macrophages to effectively repair the damaged tissue, as mentioned above, functional mitochondria are necessary [95]. Since ethanol can harm normal cells' mitochondria by inducing oxidative stress, its detrimental effects on mitochondrial metabolism may contribute to the radiotherapy's delayed toxicities [97]. As a result, continued use of cigarettes or alcohol during RT may each cause certain radiation-induced toxicities.
6. Immune Cells’ Mitochondria: A Chance to Improve Treatment Results
In addition to immunotherapy, powerful immune system can improve the treatment results of radiotherapy and chemotherapy []. To improve the normal cells’ mitochondrial content and activity several strategies can be employed y. The mitochondria quality can increase by two strategies; (1) improving the lifestyle by regular exercise [98], specific diets (low-specific dynamic action diet [99], branched-chain amino acid-rich diet [100], and Mediterranean diet [101]); good sleep [102], healthy weight [103], alcohol abstinence [104], and smoking cessation [105]; and (2) mitochondria boosting agents (e.g., coenzyme Q10, activators of adenosine AMPK, acetyl-L-Carnitine; mammalian target of rapamycin [mTOR], PGC-1α, etc.) [106,107]. In addition, the human gut microbiota is another modulator of mitochondrial fitness. It has been demonstrated that microbiota-derived metabolites are necessary for the proper action of mitochondrial metabolisms, including glycolysis, tricarboxylic acid (TCA) cycle, oxidative phosphorylation, as well as amino acid and fatty acid metabolism. The mitochondrial boosting strategies are diverse. Detailed information is presented in the following sources [107,108].
7. Heteroplasmy Provides Unique Profiles in Cancer
Heteroplasmy is the presence of more than one type of organellar genome (mitochondrial DNA or plastid DNA) within a cell. The amount of heteroplasmy is determined during oogenesis and is inherited from the mother. There are variations in the percentage of mutant alleles between oocytes and then between children. Heteroplasmy or the presence of at least two mtDNA variants within the single cell, and its level (the proportion of mutated mtDNA) are frequently seen with and in accompanying with tumor heterogeneity. One of the major challenges to understanding and elucidating the role of the variations in tumor growth is the heteroplasmy levels of the mtDNA variants. In turn, intratumor genetic heterogeneity affects personalized medicine strategies in a significant way since it can reduce the effectiveness of treatments and result in treatment resistance. It is interesting to note that numerous studies have linked heteroplasmic levels to both cancer risk and survival [109-113]. It would be essential to advance knowledge of the biological mechanisms at play, including proliferation, metastasis, and intratumoral heterogeneity, as well as the clinical implications of heteroplasmy, via recognizing the crucial role of heteroplasmy in cancer. The high mutation rate found in mtDNA, which is between 10 and 17 times higher than that of the nuclear genome, is explained by the lack of histones, effective DNA repair mechanisms, and closeness to reactive oxygen species (ROS) produced by the OXPHOS system (mostly from Complex I and III) (nDNA) [114-117]. In humans, mtDNA is only inherited via the maternal line as a single unit called a haplotype, which may be shared by populations with similar ancestries. Factually, a set of haplotypes or a haplogroup can be used to distinguish across populations or ethnic groupings while certain haplogroups have advantages for environmental adaptation but are also linked to cancer [118-125].
The degree of heteroplasmy varies greatly between different cancer kinds and individuals. It has been demonstrated that when tumors progressed, heteroplasmy varied amongst tissues. Based on the idea that some heteroplasmic variations are finally able to become dominant or lost in cancer cells based on their tumor-promoting impact, a likely bottleneck process was proposed. The G1576C and G12009A mutations are the most prevalent in tumor cells compared to normal cells (7.8% versus 0.35 % and 68.8% versus 0.35 %, respectively) [126].
Although very limited number of studies have been done on the mechanisms of heteroplasmy shifting in cancer, there is proof that cell niche and the nucleus-mitochondrial environment regulate the OXPHOS system's energy performance, choosing particular mutant alleles [127] . For instance, it has been demonstrated that fumarate accumulation in renal cancer alters the mitochondrial content by inactivating core components necessary for mtDNA replication [128]. Alterations in DNA polymerase gamma (POLG) and mitochondrial transcription factor A (TFAM) expression, mutations in nDNA-encoded genes involved in mitochondrial biogenesis, nuclear and mitochondrial epigenetic modifications, as well as intrinsic and extrinsic stimuli, may all result in anomalies in mtCNVs [129-131]. Examples include the dysregulated expression of nuclear genes such as dynamin 1 (DRP1), mitofusin 1 (MFN1) and 2 (MFN2) mitochondrial fusion and fission proteins, BCL2 inter-acting protein 3 (BNIP3), PTEN-induced kinase 1 (PINK1), and hypoxia inducible factor 1 (HIF1), observed in lung, bladder, and breast cancers [132,133]. The role of the tumor microenvironment in altering the allelic frequencies of mtDNA mutations was also hypothesized based on an investigation of primary tumors and their distant metastasis [134]. Additionally, NOX2-derived redox signaling has been shown to be used by bone marrow stromal cells to transfer functioning mitochondria to acute myeloid leukemia blasts [135,136]. Together, these pathways may be crucial for the emergence of a tumorigenic environment-adaptive-unique response that is represented in the alteration of the allelic frequencies of mtDNA mutations. The nuclear insertions of mitochondrial origin (NumtS), which have been linked to cancer, should be considered in next investigations on heteroplasmy. NumtS, or mtDNA segments integrated into the nucleus during evolution, are thought to occur at a rate of ~5 x 10e-6 per germ cell every generation [137].
Currently, methods based on mitochondrial gene editing have been proposed as a treatment choice for reestablishing the OXPHOS system in conditions brought on by mtDNA mutations. A possible therapeutic target for cancer has been suggested to include components involved in mitochondrial biogenesis and metabolism [138-140].
8. Conclusions
This article illustrated how mitochondria is involved the tumor response to different treatments as well as the normal tissue toxicity. With this concept in mind, future works can design more personalized treatments to improve the treatment results with fewer toxicities. Although there are clear links between heteroplasmy and cancer-related phenotypes, it is still unclear whether heteroplasmy or the variation in mtDNA copy number in cancer is a cause or an effect.
References
- Trapani, D.; Franzoi, M.A.; Burstein, H.J.; Carey, L.A.; Delaloge, S.; Harbeck, N.; Hayes, D.F.; Kalinsky, K.; Pusztai, L.; Regan, M.M.; et al. Risk-adapted modulation through de-intensification of cancer treatments: an ESMO classification. Ann Oncol 2022, 33, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol 2022, 17, 98–106. [Google Scholar] [CrossRef]
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J.; Aznar, M.C.; Bacchus, C.; Coppes, R.P.; Deutsch, E.; Georg, D.; Haustermans, K.; Hoskin, P.; Krause, M.; Lartigau, E.F.; et al. Personalised radiation therapy taking both the tumour and patient into consideration. Radiother Oncol 2022, 166, A1–a5. [Google Scholar] [CrossRef] [PubMed]
- Abdelkarem, O.A.I.; Choudhury, A.; Burnet, N.G.; Summersgill, H.R.; West, C.M.L. Effect of Race and Ethnicity on Risk of Radiotherapy Toxicity and Implications for Radiogenomics. Clin Oncol (R Coll Radiol) 2022, 34, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh-Hesary, F.; Akbari, H.; Bahadori, M.; Behnam, B. Targeted Anti-Mitochondrial Therapy: The Future of Oncology. Genes 2022, 13, 1728. [Google Scholar] [CrossRef]
- Akbari, H.; Taghizadeh-Hesary, F.; Bahadori, M. Mitochondria determine response to anti-programmed cell death protein-1 (anti-PD-1) immunotherapy: An evidence-based hypothesis. Mitochondrion 2022, 62, 151–158. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Fazilaty, H.; Gardaneh, M.; Akbari, P.; Zekri, A.; Behnam, B. SLUG and SOX9 Cooperatively Regulate Tumor Initiating Niche Factors in Breast Cancer. Cancer Microenviron 2016, 9, 71–74. [Google Scholar] [CrossRef]
- Fazilaty, H.; Gardaneh, M.; Bahrami, T.; Salmaninejad, A.; Behnam, B. Crosstalk between breast cancer stem cells and metastatic niche: emerging molecular metastasis pathway? Tumour Biol 2013, 34, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jang, K.; Miller, P.; Picon-Ruiz, M.; Yeasky, T.M.; El-Ashry, D.; Slingerland, J.M. VEGFA links self-renewal and metastasis by inducing Sox2 to repress miR-452, driving Slug. Oncogene 2017, 36, 5199–5211. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Amendola, P.G.; Reuten, R.; Erler, J.T. Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- van Gisbergen, M.W.; Offermans, K.; Voets, A.M.; Lieuwes, N.G.; Biemans, R.; Hoffmann, R.F.; Dubois, L.J.; Lambin, P. Mitochondrial Dysfunction Inhibits Hypoxia-Induced HIF-1α Stabilization and Expression of Its Downstream Targets. Front Oncol 2020, 10, 770. [Google Scholar] [CrossRef] [PubMed]
- Paupe, V.; Prudent, J. New insights into the role of mitochondrial calcium homeostasis in cell migration. Biochem Biophys Res Commun 2018, 500, 75–86. [Google Scholar] [CrossRef]
- McCann, E.; O'Sullivan, J.; Marcone, S. Targeting cancer-cell mitochondria and metabolism to improve radiotherapy response. Transl Oncol 2021, 14, 100905. [Google Scholar] [CrossRef]
- Houshyari, M.; Taghizadeh-Hesary, F. Is Mitochondrial Metabolism a New Predictive Biomarker for Antiprogrammed Cell Death Protein-1 Immunotherapy? JCO Oncol Pract 2022, Op2200733. [Google Scholar] [CrossRef]
- Taghizadeh-Hesary, F.; Houshyari, M.; Farhadi, M. Mitochondrial metabolism: a predictive biomarker of radiotherapy efficacy and toxicity. J Cancer Res Clin Oncol 2023. [Google Scholar] [CrossRef]
- Bentle, M.S.; Reinicke, K.E.; Bey, E.A.; Spitz, D.R.; Boothman, D.A. Calcium-dependent modulation of poly(ADP-ribose) polymerase-1 alters cellular metabolism and DNA repair. J Biol Chem 2006, 281, 33684–33696. [Google Scholar] [CrossRef] [PubMed]
- Lévy, N.; Martz, A.; Bresson, A.; Spenlehauer, C.; de Murcia, G.; Ménissier-de Murcia, J. XRCC1 is phosphorylated by DNA-dependent protein kinase in response to DNA damage. Nucleic Acids Res 2006, 34, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.; Gueven, N.; Keating, K.; Ramsay, J.; Lavin, M.F. ATP activates ataxia-telangiectasia mutated (ATM) in vitro. Importance of autophosphorylation. J Biol Chem 2003, 278, 9309–9317. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem 2008, 77, 313–338. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: historical dogma versus current understanding. J Physiol 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Rakotomalala, A.; Escande, A.; Furlan, A.; Meignan, S.; Lartigau, E. Hypoxia in Solid Tumors: How Low Oxygenation Impacts the "Six Rs" of Radiotherapy. Front Endocrinol (Lausanne) 2021, 12, 742215. [Google Scholar] [CrossRef]
- Leal-Esteban, L.C.; Fajas, L. Cell cycle regulators in cancer cell metabolism. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2020, 1866, 165715. [Google Scholar] [CrossRef]
- Syljuåsen, R.G. Cell cycle effects in radiation oncology. Radiation Oncology; Wentz, F., Ed.; Springer: Berlin/Heidelberg, Germany 2019. [Google Scholar]
- Salazar-Roa, M.; Malumbres, M. Fueling the Cell Division Cycle. Trends Cell Biol 2017, 27, 69–81. [Google Scholar] [CrossRef]
- Suwa, T.; Kobayashi, M.; Nam, J.-M.; Harada, H. Tumor microenvironment and radioresistance. Experimental & Molecular Medicine 2021, 53, 1029–1035. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res 2014, 74, 665–674. [Google Scholar] [CrossRef]
- Kong, X.; Yu, D.; Wang, Z.; Li, S. Relationship between p53 status and the bioeffect of ionizing radiation. Oncol Lett 2021, 22, 661. [Google Scholar] [CrossRef] [PubMed]
- Ericson, N.G.; Kulawiec, M.; Vermulst, M.; Sheahan, K.; O'Sullivan, J.; Salk, J.J.; Bielas, J.H. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet 2012, 8, e1002689. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Chen, L.; Yu, B.; Xue, Y.; Guo, R.; Su, J.; Liu, Y.; Sun, L. p53/PGC-1α-mediated mitochondrial dysfunction promotes PC3 prostate cancer cell apoptosis. Mol Med Rep 2020, 22, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Jarvis, I.W.H.; Bottai, M.; Dreij, K.; Stenius, U. TGF beta promotes repair of bulky DNA damage through increased ERCC1/XPF and ERCC1/XPA interaction. Carcinogenesis 2018, 40, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. Journal of Hematology & Oncology 2022, 15, 135. [Google Scholar] [CrossRef]
- Hu, J.W.; Sun, P.; Zhang, D.X.; Xiong, W.J.; Mi, J. Hexokinase 2 regulates G1/S checkpoint through CDK2 in cancer-associated fibroblasts. Cell Signal 2014, 26, 2210–2216. [Google Scholar] [CrossRef]
- Rydström, J. Mitochondrial NADPH, transhydrogenase and disease. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2006, 1757, 721–726. [Google Scholar] [CrossRef]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduction and Targeted Therapy 2020, 5, 231. [Google Scholar] [CrossRef]
- Tran, A.N.; Lai, A.; Li, S.; Pope, W.B.; Teixeira, S.; Harris, R.J.; Woodworth, D.C.; Nghiemphu, P.L.; Cloughesy, T.F.; Ellingson, B.M. Increased sensitivity to radiochemotherapy in IDH1 mutant glioblastoma as demonstrated by serial quantitative MR volumetry. Neuro Oncol 2014, 16, 414–420. [Google Scholar] [CrossRef]
- Stuani, L.; Sabatier, M.; Saland, E.; Cognet, G.; Poupin, N.; Bosc, C.; Castelli, F.A.; Gales, L.; Turtoi, E.; Montersino, C.; et al. Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J Exp Med 2021, 218. [Google Scholar] [CrossRef]
- Li, S.; Sun, C.; Gu, Y.; Gao, X.; Zhao, Y.; Yuan, Y.; Zhang, F.; Hu, P.; Liang, W.; Cao, K.; et al. Mutation of IDH1 aggravates the fatty acid-induced oxidative stress in HCT116 cells by affecting the mitochondrial respiratory chain. Mol Med Rep 2019, 19, 2509–2518. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Calissano, P.; Bobba, A.; Azzariti, A.; Marra, E.; Passarella, S. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J Biol Chem 2000, 275, 37159–37166. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Frontiers in Cell and Developmental Biology 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Guillot, C.; Favaudon, V.; Herceg, Z.; Sagne, C.; Sauvaigo, S.; Merle, P.; Hall, J.; Chemin, I. PARP inhibition and the radiosensitizing effects of the PARP inhibitor ABT-888 in in vitrohepatocellular carcinoma models. BMC Cancer 2014, 14, 603. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.D.; Parveen, A.; Yadav, D.K. Role of PARP in TNBC: Mechanism of Inhibition, Clinical Applications, and Resistance. Biomedicines 2021, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Arbini, A.A.; Guerra, F.; Greco, M.; Marra, E.; Gandee, L.; Xiao, G.; Lotan, Y.; Gasparre, G.; Hsieh, J.T.; Moro, L. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis 2013, 2, e82. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis 2014, 5, e1437. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; García Carrancá, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Frontiers in Oncology 2019, 9. [Google Scholar] [CrossRef]
- Zhao, Y.; Yi, J.; Tao, L.; Huang, G.; Chu, X.; Song, H.; Chen, L. Wnt signaling induces radioresistance through upregulating HMGB1 in esophageal squamous cell carcinoma. Cell Death Dis 2018, 9, 433. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Livesey, K.M.; Kroemer, G.; Billiar, T.R.; Van Houten, B.; Zeh, H.J., 3rd; Lotze, M.T. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab 2011, 13, 701–711. [Google Scholar] [CrossRef]
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm (2020) 2021, 2, 315–340. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-κB) Doing in and to the Mitochondrion? Frontiers in Cell and Developmental Biology 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Pour Khavari, A.; Liu, Y.; He, E.; Skog, S.; Haghdoost, S. Serum 8-Oxo-dG as a Predictor of Sensitivity and Outcome of Radiotherapy and Chemotherapy of Upper Gastrointestinal Tumours. Oxid Med Cell Longev 2018, 2018, 4153574. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Sun, M.; Li, G.H.; Wu, Y.Z.; Wang, Y.; Jin, F.; Zhang, Y.Y.; Yang, L.; Wang, D.L. Activation of the phosphorylation of ATM contributes to radioresistance of glioma stem cells. Oncol Rep 2013, 30, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.S.; Lin, Z.P.; Sartorelli, A.C.; Bonawitz, N.D.; Shadel, G.S. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest 2007, 117, 2723–2734. [Google Scholar] [CrossRef]
- Vaezi, A.; Feldman, C.H.; Niedernhofer, L.J. ERCC1 and XRCC1 as biomarkers for lung and head and neck cancer. Pharmgenomics Pers Med 2011, 4, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xu, J.; Ji, G.; Liu, Q.; Shao, W.; Chen, Y.; Gu, J.; Weng, Z.; Zhang, X.; Wang, Y. Deficiency of X-ray repair cross-complementing group 1 in primordial germ cells contributes to male infertility. The FASEB Journal 2019, 33, 7427–7436. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhang, S.; Pang, J.; Yin, J.; Zhang, J.; Mu, D.; Tang, S.; Li, L.; Bao, H.; Wu, X. Genomic Profiling Reveals Novel Predictive Biomarkers for Chemo-Radiotherapy Toxicity and Efficacy in Non-Small-Cell Lung Cancer. International Journal of Radiation Oncology, Biology, Physics 2021, 111, e437. [Google Scholar] [CrossRef]
- Fan, F.; Zhuang, J.; Zhou, P.; Liu, X.; Luo, Y. MicroRNA-34a promotes mitochondrial dysfunction-induced apoptosis in human lens epithelial cells by targeting Notch2. Oncotarget 2017, 8, 110209–110220. [Google Scholar] [CrossRef]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J Clin Biochem Nutr 2015, 56, 91–97. [Google Scholar] [CrossRef]
- Zeb, A.; Choubey, V.; Gupta, R.; Kuum, M.; Safiulina, D.; Vaarmann, A.; Gogichaishvili, N.; Liiv, M.; Ilves, I.; Tämm, K.; et al. A novel role of KEAP1/PGAM5 complex: ROS sensor for inducing mitophagy. Redox Biology 2021, 48, 102186. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Fan, J.; Chung, T.-W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.-L.; Polakiewicz, Roberto D. ; Roesel, Johannes L.; et al. Tyrosine Phosphorylation of Mitochondrial Pyruvate Dehydrogenase Kinase 1 Is Important for Cancer Metabolism. Molecular Cell 2011, 44, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, B.; Xiao, H.; Dong, J.; Li, Y.; Zhu, C.; Jin, Y.; Li, H.; Cui, M.; Fan, S. LncRNA HOTAIR enhances breast cancer radioresistance through facilitating HSPA1A expression via sequestering miR-449b-5p. Thoracic Cancer 2020, 11, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Xiong, Q.; Wu, Y.; Chen, Y.; Chen, Z.; Fleming, J.; Gao, D.; Bi, L.; Ge, F. Quantitative Proteomics Analysis Reveals Novel Insights into Mechanisms of Action of Long Noncoding RNA Hox Transcript Antisense Intergenic RNA (HOTAIR) in HeLa Cells. Mol Cell Proteomics 2015, 14, 1447–1463. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhou, X.; Wu, Y.; Wang, Y.; Chen, L.; Li, P.; Liu, S.; Sun, S.; Ren, Y.; Mei, M. Targeting HOTAIR induces mitochondria related apoptosis and inhibits tumor growth in head and neck squamous cell carcinoma in vitro and in vivo. Current Molecular Medicine 2015, 15, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Urushihara, Y.; Murata, Y.; Fujishima, Y.; Hosoi, Y. AMPK increases expression of ATM through transcriptional factor Sp1 and induces radioresistance under severe hypoxia in glioblastoma cell lines. Biochem Biophys Res Commun 2022, 590, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Pedersen, H.; Anne Adanma Obara, E.; Elbæk, K.J.; Vitting-Serup, K.; Hamerlik, P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. International Journal of Molecular Sciences 2020, 21, 1588. [Google Scholar] [CrossRef]
- Liu, X.; Shan, G. Mitochondria Encoded Non-coding RNAs in Cell Physiology. Frontiers in Cell and Developmental Biology 2021, 9. [Google Scholar] [CrossRef]
- Ma, J.; Lu, Y.; Zhang, S.; Li, Y.; Huang, J.; Yin, Z.; Ren, J.; Huang, K.; Liu, L.; Yang, K.; et al. β-Trcp ubiquitin ligase and RSK2 kinase-mediated degradation of FOXN2 promotes tumorigenesis and radioresistance in lung cancer. Cell Death Differ 2018, 25, 1473–1485. [Google Scholar] [CrossRef]
- Mrozowski, RM. Targeting the Ser/Thr protein kinase RSK to reduce breast cancer metastasis, : Vanderbilt University; 2015.
- Chu, C.; Niu, X.; Ou, X.; Hu, C. LAPTM4B knockdown increases the radiosensitivity of EGFR-overexpressing radioresistant nasopharyngeal cancer cells by inhibiting autophagy. Onco Targets Ther 2019, 12, 5661–5677. [Google Scholar] [CrossRef] [PubMed]
- Milkereit, R.; Persaud, A.; Vanoaica, L.; Guetg, A.; Verrey, F.; Rotin, D. LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nature Communications 2015, 6, 7250. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Yadav, P.; Sainis, K.B.; Shankar, B.S. TNF-α and IGF-1 differentially modulate ionizing radiation responses of lung cancer cell lines. Cytokine 2018, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.; Jung, H.; Lee, H.; Singh, K.; Roy, M.; Gohel, D.; Kim, H.B.; Mane, M.; Vasiyani, H.; Currim, F.; et al. TNF-α differentially modulates subunit levels of respiratory electron transport complexes of ER/PR +ve/−ve breast cancer cells to regulate mitochondrial complex activity and tumorigenic potential. Cancer & Metabolism 2021, 9, 19. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondrial complex III: an essential component of universal oxygen sensing machinery? Respir Physiol Neurobiol 2010, 174, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Dörr, W. Radiobiology of tissue reactions. Ann ICRP 2015, 44, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Nicolatou-Galitis, O.; Bossi, P.; Orlandi, E.; René-Jean, B. The role of benzydamine in prevention and treatment of chemoradiotherapy-induced mucositis. Support Care Cancer 2021, 29, 5701–5709. [Google Scholar] [CrossRef] [PubMed]
- Holley, A.K.; Miao, L.; St Clair, D.K.; St Clair, W.H. Redox-modulated phenomena and radiation therapy: the central role of superoxide dismutases. Antioxid Redox Signal 2014, 20, 1567–1589. [Google Scholar] [CrossRef]
- Drobin, K.; Marczyk, M.; Halle, M.; Danielsson, D.; Papiez, A.; Sangsuwan, T.; Bendes, A.; Hong, M.G.; Qundos, U.; Harms-Ringdahl, M.; et al. Molecular Profiling for Predictors of Radiosensitivity in Patients with Breast or Head-and-Neck Cancer. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Dai, N.; Groenendyk, J.; Michalak, M. Binding Proteins | Ca2+ Binding/Buffering Proteins: ER Luminal Proteins☆. In Encyclopedia of Biological Chemistry III (Third Edition), Jez, J., Ed.; Elsevier: Oxford, 2021; pp. 534–546. [Google Scholar]
- Henke, N.; Albrecht, P.; Pfeiffer, A.; Toutzaris, D.; Zanger, K.; Methner, A. Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J Biol Chem 2012, 287, 42042–42052. [Google Scholar] [CrossRef]
- Dewson, G. Bax to the wall: Bax-and Bak-induced mitochondrial dysfunction in apoptosis. Trends in Biochemical Sciences 2001, 26, 353. [Google Scholar] [CrossRef]
- Bonanno, J.A.; Shyam, R.; Choi, M.; Ogando, D.G. The H(+) Transporter SLC4A11: Roles in Metabolism, Oxidative Stress and Mitochondrial Uncoupling. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Sprung, C.N.; Forrester, H.B.; Siva, S.; Martin, O.A. Immunological markers that predict radiation toxicity. Cancer Lett 2015, 368, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Lee, J.H.; Hwang, S.C.; Choi, K.S.; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef]
- Meier, J.A.; Larner, A.C. Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol 2014, 26, 20–28. [Google Scholar] [CrossRef]
- Hernández, L.; Terradas, M.; Camps, J.; Martín, M.; Tusell, L.; Genescà, A. Aging and radiation: bad companions. Aging Cell 2015, 14, 153–161. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes (Basel) 2017, 8. [Google Scholar] [CrossRef]
- Lawler, J.M.; Demaree, S.R. Relationship between NADP-specific isocitrate dehydrogenase and glutathione peroxidase in aging rat skeletal muscle. Mech Ageing Dev 2001, 122, 291–304. [Google Scholar] [CrossRef]
- Wang, K.; Tepper, J.E. Radiation therapy-associated toxicity: Etiology, management, and prevention. CA Cancer J Clin 2021, 71, 437–454. [Google Scholar] [CrossRef]
- Madani, A.; Alack, K.; Richter, M.J.; Krüger, K. Immune-regulating effects of exercise on cigarette smoke-induced inflammation. J Inflamm Res 2018, 11, 155–167. [Google Scholar] [CrossRef]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: mechanisms and implications for therapy. J Cancer Res Clin Oncol 2015, 141, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Willenborg, S.; Sanin, D.E.; Jais, A.; Ding, X.; Ulas, T.; Nüchel, J.; Popović, M.; MacVicar, T.; Langer, T.; Schultze, J.L.; et al. Mitochondrial metabolism coordinates stage-specific repair processes in macrophages during wound healing. Cell Metabolism 2021, 33, 2398–2414. [Google Scholar] [CrossRef] [PubMed]
- Pratson, C.L.; Larkins, M.C.; Karimian, B.H.; Curtis, C.M.; Lepera, P.A.; Brodish, B.N.; Ju, A.W. The Impact of Smoking, Alcohol Use, Recurrent Disease, and Age on the Development of Neck Fibrosis in Head and Neck Cancer Patients Following Radiation Therapy. Frontiers in Oncology 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Memme, J.M.; Erlich, A.T.; Phukan, G.; Hood, D.A. Exercise and mitochondrial health. J Physiol 2021, 599, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Luoma, R.L.; Butler, M.W.; Stahlschmidt, Z.R. Plasticity of immunity in response to eating. J Exp Biol 2016, 219, 1965–1968. [Google Scholar] [CrossRef]
- D'Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-Chain Amino Acid Supplementation Promotes Survival and Supports Cardiac and Skeletal Muscle Mitochondrial Biogenesis in Middle-Aged Mice. Cell Metabolism 2010, 12, 362–372. [Google Scholar] [CrossRef]
- Khalil, M.; Shanmugam, H.; Abdallah, H.; John Britto, J.S.; Galerati, I.; Gómez-Ambrosi, J.; Frühbeck, G.; Portincasa, P. The Potential of the Mediterranean Diet to Improve Mitochondrial Function in Experimental Models of Obesity and Metabolic Syndrome. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Rodrigues, N.R.; Macedo, G.E.; Martins, I.K.; Gomes, K.K.; de Carvalho, N.R.; Posser, T.; Franco, J.L. Short-term sleep deprivation with exposure to nocturnal light alters mitochondrial bioenergetics in Drosophila. Free Radic Biol Med 2018, 120, 395–406. [Google Scholar] [CrossRef]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci 2018, 192, 26–32. [Google Scholar] [CrossRef]
- Abdallah, M.A.; Singal, A.K. Mitochondrial dysfunction and alcohol-associated liver disease: a novel pathway and therapeutic target. Signal Transduction and Targeted Therapy 2020, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Malińska, D.; Więckowski, M.R.; Michalska, B.; Drabik, K.; Prill, M.; Patalas-Krawczyk, P.; Walczak, J.; Szymański, J.; Mathis, C.; Van der Toorn, M.; et al. Mitochondria as a possible target for nicotine action. J Bioenerg Biomembr 2019, 51, 259–276. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U S A 2017, 114, E761–e770. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J. Mitochondria-Fundamental to Life and Health. Integr Med (Encinitas) 2014, 13, 8–15. [Google Scholar] [PubMed]
- Burtscher, J.; Romani, M.; Bernardo, G.; Popa, T.; Ziviani, E.; Hummel, F.C.; Sorrentino, V.; Millet, G.P. Boosting mitochondrial health to counteract neurodegeneration. Progress in Neurobiology 2022, 215, 102289. [Google Scholar] [CrossRef]
- Fendt, L.; Fazzini, F.; Weissensteiner, H.; Bruckmoser, E.; Schönherr, S.; Schäfer, G.; Losso, J.L.; Streiter, G.A.; Lamina, C.; Rasse, M.; et al. Profiling of Mitochondrial DNA Heteroplasmy in a Prospective Oral Squamous Cell Carcinoma Study. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat Commun 2017, 8, 656. [Google Scholar] [CrossRef]
- Kloss-Brandstätter, A.; Schäfer, G.; Erhart, G.; Hüttenhofer, A.; Coassin, S.; Seifarth, C.; Summerer, M.; Bektic, J.; Klocker, H.; Kronenberg, F. Somatic mutations throughout the entire mitochondrial genome are associated with elevated PSA levels in prostate cancer patients. Am J Hum Genet 2010, 87, 802–812. [Google Scholar] [CrossRef]
- McMahon, S.; LaFramboise, T. Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 2014, 35, 1046–1054. [Google Scholar] [CrossRef]
- Qi, Y.; Wei, Y.; Wang, Q.; Xu, H.; Wang, Y.; Yao, A.; Yang, H.; Gao, Y.; Zhou, F. Heteroplasmy of mutant mitochondrial DNA A10398G and analysis of its prognostic value in non-small cell lung cancer. Oncol Lett 2016, 12, 3081–3088. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.-S.; Wang, H.-S.; Mugaka, B.P.; Yang, G.-J.; Ding, Y. Mitochondria: promising organelle targets for cancer diagnosis and treatment. Biomaterials Science 2018, 6, 2786–2797. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid Med Cell Longev 2017, 2017, 8060949. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gong, L.; Liu, X.; Chen, X.; Yang, S.; Luo, Y. Mitochondrial DNA genomes revealed different patterns of high-altitude adaptation in high-altitude Tajiks compared with Tibetans and Sherpas. Scientific Reports 2020, 10, 10592. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wong, L.J.; Mims, M.P. Mitochondrial inheritance and cancer. Transl Res 2018, 202, 24–34. [Google Scholar] [CrossRef]
- Grasso, D.; Zampieri, L.X.; Capelôa, T.; Van de Velde, J.A.; Sonveaux, P. Mitochondria in cancer. Cell Stress 2020, 4, 114–146. [Google Scholar] [CrossRef]
- Lajbner, Z.; Pnini, R.; Camus, M.F.; Miller, J.; Dowling, D.K. Experimental evidence that thermal selection shapes mitochondrial genome evolution. Sci Rep 2018, 8, 9500. [Google Scholar] [CrossRef]
- Ma, L.; Fu, Q.; Xu, B.; Zhou, H.; Gao, J.; Shao, X.; Xiong, J.; Gu, Q.; Wen, S.; Li, F.; et al. Breast cancer-associated mitochondrial DNA haplogroup promotes neoplastic growth via ROS-mediated AKT activation. Int J Cancer 2018, 142, 1786–1796. [Google Scholar] [CrossRef]
- Motoi, M.; Nishimura, T.; Egashira, Y.; Kishida, F.; Watanuki, S. Relationship between mitochondrial haplogroup and physiological responses to hypobaric hypoxia. J Physiol Anthropol 2016, 35, 12. [Google Scholar] [CrossRef]
- Toncheva, D.; Serbezov, D.; Karachanak-Yankova, S.; Nesheva, D. Ancient mitochondrial DNA pathogenic variants putatively associated with mitochondrial disease. PLoS One 2020, 15, e0233666. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Li, M.; Wang, J.; Liu, J.; Li, J.; Fang, H.; Lyu, J.; Shen, L. Association between mitochondrial DNA haplogroup variation and coronary artery disease. Nutr Metab Cardiovasc Dis 2020, 30, 960–966. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Lechuga-Vieco, A.V.; Latorre-Pellicer, A.; Johnston, I.G.; Prota, G.; Gileadi, U.; Justo-Méndez, R.; Acín-Pérez, R.; Martínez-de-Mena, R.; Fernández-Toro, J.M.; Jimenez-Blasco, D.; et al. Cell identity and nucleo-mitochondrial genetic context modulate OXPHOS performance and determine somatic heteroplasmy dynamics. Sci Adv 2020, 6, eaba5345. [Google Scholar] [CrossRef] [PubMed]
- Crooks, D.R.; Maio, N.; Lang, M.; Ricketts, C.J.; Vocke, C.D.; Gurram, S.; Turan, S.; Kim, Y.Y.; Cawthon, G.M.; Sohelian, F.; et al. Mitochondrial DNA alterations underlie an irreversible shift to aerobic glycolysis in fumarate hydratase-deficient renal cancer. Sci Signal 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lee, J.H.; Kim, D.K.; Keum, D.Y. Nuclear and mitochondrial DNAs microsatellite instability and mitochondrial DNA copy number in adenocarcinoma and squamous cell carcinoma of lung: a pilot study. Apmis 2015, 123, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Ru, G.; Mao, Z.; Wang, C.; Nie, Z.; Li, Q.; Huang-Yang, Y.; Zhu, L.; Liang, X.; Yu, J.; et al. Mitochondrial DNA depletion, mitochondrial mutations and high TFAM expression in hepatocellular carcinoma. Oncotarget 2017, 8, 84373–84383. [Google Scholar] [CrossRef]
- Shen, J.; Wan, J.; Song, R.; Zhao, H. Peripheral blood mitochondrial DNA copy number, length heteroplasmy and breast cancer risk: a replication study. Carcinogenesis 2015, 36, 1307–1313. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Zorzano, A. Mitochondrial Dynamics and Liver Cancer. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Ray, K.S.; Mukherjee, S. Mitophagy in Carcinogenesis and Tumour Progression- A New Paradigm with Emerging Importance. Anti-Cancer Agents in Medicinal Chemistry 2021, 21, 2130–2141. [Google Scholar] [CrossRef]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, L.X.; Silva-Almeida, C.; Rondeau, J.D.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Amado, C.J.; Bazan-Cordoba, A.; Hidalgo-Miranda, A.; Jiménez-Morales, S. Mitochondrial Heteroplasmy Shifting as a Potential Biomarker of Cancer Progression. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, T.; Jauregui, C.E.; Teng, Y. Friend or foe? Mitochondria as a pharmacological target in cancer treatment. Future Med Chem 2017, 9, 2197–2210. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Frattaruolo, L.; Brindisi, M.; Curcio, R.; Marra, F.; Dolce, V.; Cappello, A.R. Targeting the Mitochondrial Metabolic Network: A Promising Strategy in Cancer Treatment. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
Figure 1.
Schematic model of mitochondria’s role in cancer survival, immune evasion, progression, and treatment resistance. The white boxes depict the mitochondria regulation outcomes. ATP indicates adenosine triphosphate; CA IX, carbonic anhydrase IX; EMT, epithelial–mesenchymal transition; GLUT-1, Glucose transporter-1; GTP, guanosine triphosphate; G6PD, glucose 6-phosphate dehydrogenase; HIF, hypoxia-inducible factor;; IFN, interferon; IL-10, interleukin-10; MDSC, myeloid-derived suppressor cell; MHC-1, major histocompatibility complex class I; mtDNA, mitochondrial DNA; PD-1, programmed cell death protein-1; PD-L1, programmed cell death protein-ligand 1; ROS, reactive oxygen species; TGF-β,transforming growth factor-beta; TME, tumor microenvironment; VEGF, vascular endothelial growth factor;. (retrieved from Taghizadeh-Hesary et al., 2022) [7].
Figure 1.
Schematic model of mitochondria’s role in cancer survival, immune evasion, progression, and treatment resistance. The white boxes depict the mitochondria regulation outcomes. ATP indicates adenosine triphosphate; CA IX, carbonic anhydrase IX; EMT, epithelial–mesenchymal transition; GLUT-1, Glucose transporter-1; GTP, guanosine triphosphate; G6PD, glucose 6-phosphate dehydrogenase; HIF, hypoxia-inducible factor;; IFN, interferon; IL-10, interleukin-10; MDSC, myeloid-derived suppressor cell; MHC-1, major histocompatibility complex class I; mtDNA, mitochondrial DNA; PD-1, programmed cell death protein-1; PD-L1, programmed cell death protein-ligand 1; ROS, reactive oxygen species; TGF-β,transforming growth factor-beta; TME, tumor microenvironment; VEGF, vascular endothelial growth factor;. (retrieved from Taghizadeh-Hesary et al., 2022) [7].
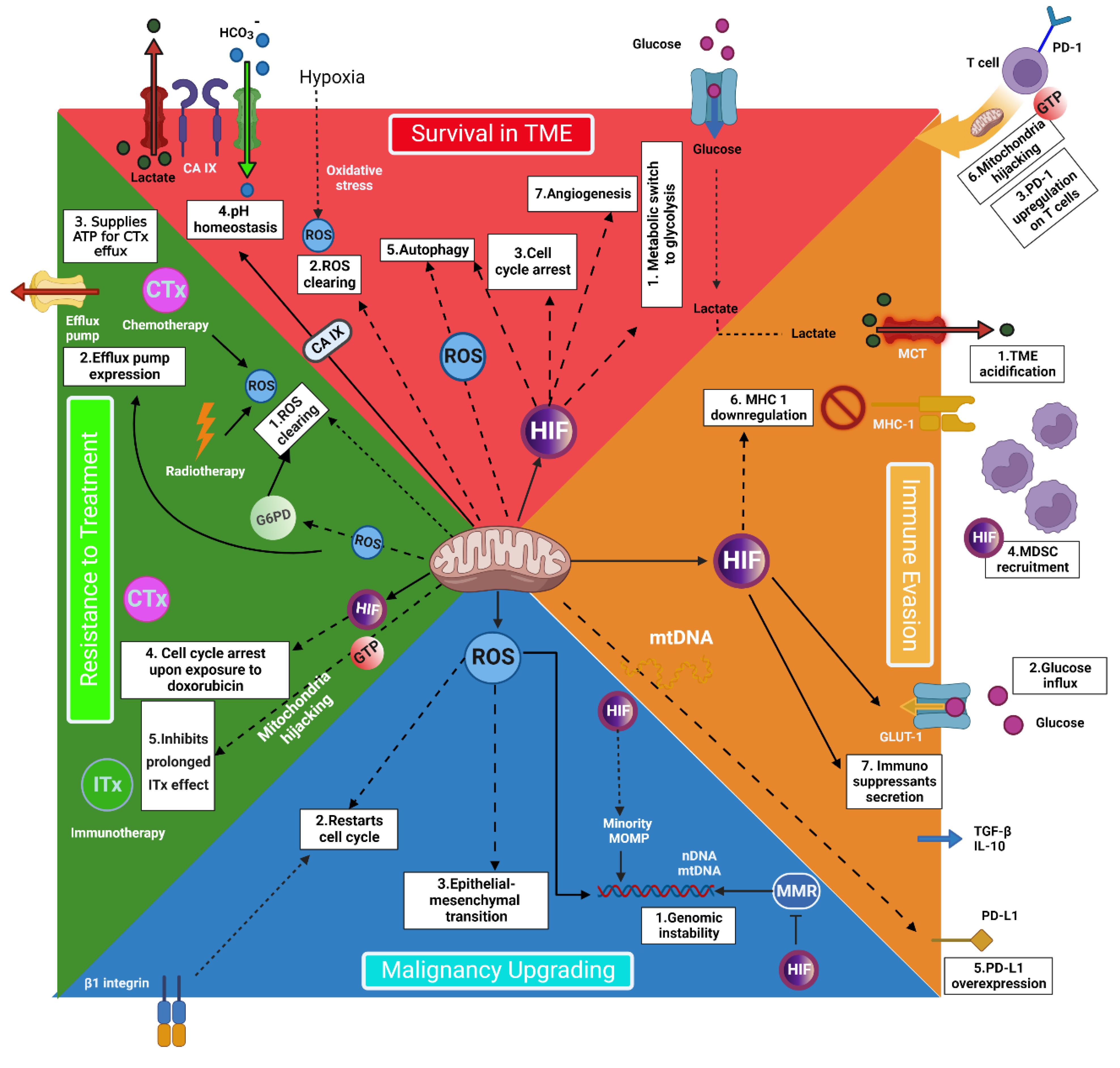
Figure 2.
The contribution of mitochondrial metabolism to 6Rs of radiobiology. (retrieved from Taghizadeh-Hesary et al. study [20]).
Figure 2.
The contribution of mitochondrial metabolism to 6Rs of radiobiology. (retrieved from Taghizadeh-Hesary et al. study [20]).
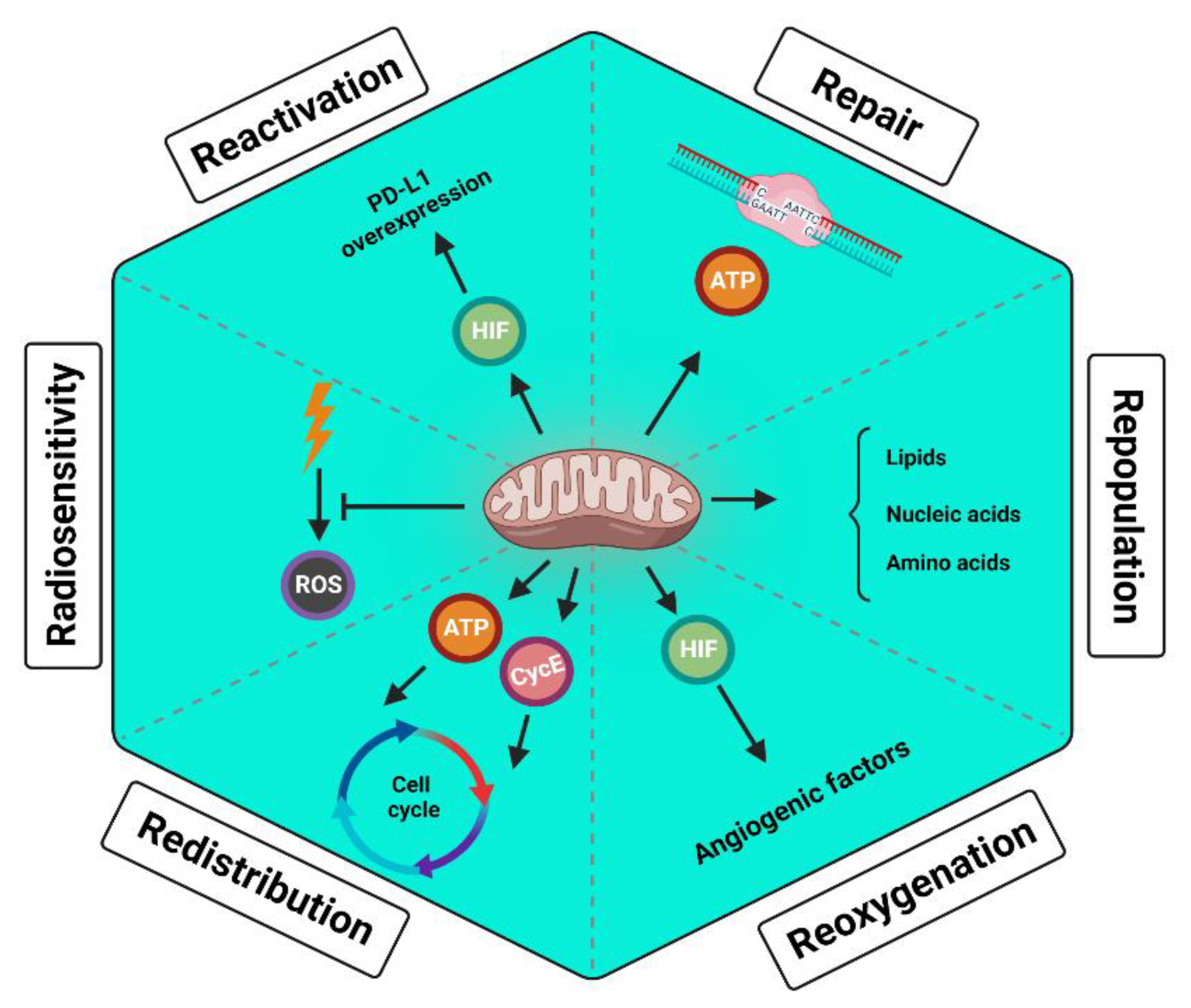

Table 1.
The biological factors of radioresistance from the mitochondria perspective.
| Factors | Cancer | Ref. | Interaction with mitochondria | Ref. |
| Increasing radioresistance | ||||
| Mutated P53 | Various | [32] | - Mutated p53 preserves mtDNA integrity - Mutated p53 improves mt capacity (PGC1α-mediated) - More functional mt scavenge more RT-induced ROS |
[33] [34] [7] |
| TGF-β | HCC | [35] | - TGF-β signalling in CAFs mediates reverse Warburg effect - CAFs’ lactate and pyruvate feed cancer cells’ mt OxPhos - Activated OxPhos helps to restore NADPH - NADPH supports the antioxidant defense system |
[36] [37] [38] [39] |
| IDH1 | Glioblastoma | [40] | - Mutated IDH1 enhances mt OxPhos (ROS generation) - Mutated IDH1 downregulates cytochrome c - Cytochrome c can nullify ROS - Thus, IDH1 mutation disrupts the ROS balance |
[41] [42] [43] |
| PARP | Breast Ovarian Prostate Pancreatic HCC |
[44] [45] |
- PARP requires RAD51 for HR - BRCA2 regulates RAD51 function - BRCA2 requires mt support - Thus, functional mt improves radioresistance by mediating HR |
[46] [46] [47] |
| PI3K/Akt/mTOR pathway | Prostate | [48] | - mTOR upregulates mt proteins responsible for mt metabolism - More functional mt scavenge more RT-induced ROS |
[49] [7] |
| Wnt/β-catenin pathway | Esophageal SCC | [50] | - Wnt upregulates HMGB1 - HMGB1 activates mitochondria - More functional mt scavenge more RT-induced ROS |
[50] [51] [7] |
| NF-κB pathway | Breast Glioma HCC Melanoma NSCLC |
[52] | - Enhances mt respiration - Regulates mt dynamics - Regulates mt gene expression |
[53] |
| 8-oxo-dG | Esophageal Gastric |
[54] | - Serum 8-oxo-DG level represents cellular ROS - Cellular ROS is dependent on mt metabolism |
[54] [7] |
| ATM | Glioma | [55] | - Preserves mtDNA | [56] |
| XRCC1 | NSCLC HNC |
[57] | - Preserves mt respiratory chain | [58] |
| NOTCH2 | NSCLC | [59] | - Regulates mitochondrial function | [60] |
| KEAP1 | NSCLC | [59] | - Regulates mitochondrial function - Regulates mitophagy |
[61] [62] |
| FGFR1/3 | NSCLC | [59] | - Regulates mitochondrial energy metabolism | [63] |
| HOTAIR | Breast | [64] | - Regulates mitochondrial function | [65], [66] |
| AMPK | Glioblastoma | [67] | - Preserves mt biogenesis upon energy stress | [68] |
| RPA1 | Glioblastoma | [69] | - Preserves mtDNA | [70] |
| RSK2 | NSCLC | [71] | - Stimulates mt OxPhos | [72] |
| LAPTM4B | NPC | [73] | - Activates mTOR - mTOR upregulates mt proteins responsible for mt metabolism - More functional mt scavenge more RT-induced ROS |
[74] [49] [7] |
| Decreasing radioresistance | ||||
| TNFα | NSCLC | [75] | - Impairs mt complex I and III - Complex III is essential for NADPH activity - Thus, reduces mt capacity to scavenge RT-induced ROS |
[76] [77] |
| Note: This table is retrieved from Taghizadeh-Hesary et al. study [20]. Abbreviations:8-oxo-dG, 8-hydroxy-2′-deoxyguanosine; Akt, protein kinase B; AMPK, serine/threonine kinase AMP-activated protein kinase; ATM, ataxia-telangiectasia mutated; BRCA2, breast cancer gene 2; CAF, cancer-associated fibroblasts; FGFR1/3, fibroblast growth factor 1/3; HCC, hepatocellular carcinoma; HMGB1, high mobility group box 1; HOTAIR, HOX transcript antisense RNA; HR, homologous recombination; IDH1, Isocitrate dehydrogenase 1; KEAP1, Kelch-like ECH-associated protein; LAPTM4B, lysosome-associated transmembrane protein 4B; mt, mitochondrial; mTOR, mammalian target of rapamycin; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor κB; NOTCH2, neurogenic locus notch homolog protein 2; NPC, nasopharyngeal carcinoma; NSCLC, non-small cell lung cancer; OxPhos, oxidative phosphorylation; PARP, poly (ADP-ribose) polymerase; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator 1α; PI3K, phosphoinositide 3-kinases; ROS, reactive oxygen species; RPA1, replication protein A1; RSK2, ribosomal S6 kinase; RT, radiotherapy; SCC, squamous cell carcinoma; TGF-β, transforming growth factor β; TNFα, tumor necrosis factor α; XRCC1, X-ray repair cross complementing 1. | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



